

Abstract
Genome editing is potentially a curative technique available to all individuals with β-hemoglobinopathies, including sickle cell disease (SCD). Fetal hemoglobin (HbF) inhibits sickle hemoglobin (HbS) polymerization, and it is well described that naturally occurring hereditary persistence of HbF (HPFH) alleviates disease symptoms; therefore, reawakening of developmentally silenced HbF in adult red blood cells (RBCs) has long been of interest as a therapeutic strategy. Recent advances in genome editing platforms, particularly with the use of CRISPR-Cas9, have paved the way for efficient HbF induction through the creation of artificial HPFH mutations, editing of transcriptional HbF silencers, and modulating epigenetic intermediates that govern HbF expression. Clinical trials investigating BCL11A enhancer editing in patients with β-hemoglobinopathies have demonstrated promising results, although follow-up is short and the number of patients treated to date is low. While practical, economic, and clinical challenges of genome editing are well recognized by the scientific community, potential solutions to overcome these hurdles are in development. Here, we review the recent progress and obstacles yet to be overcome for the most effective and feasible HbF reactivation practice using CRISPR-Cas9 genome editing as a curative strategy for patients with SCD.
Keywords: HbF, genome editing, globins, hemoglobinopathies, base editors, gene therapy
Graphical abstract
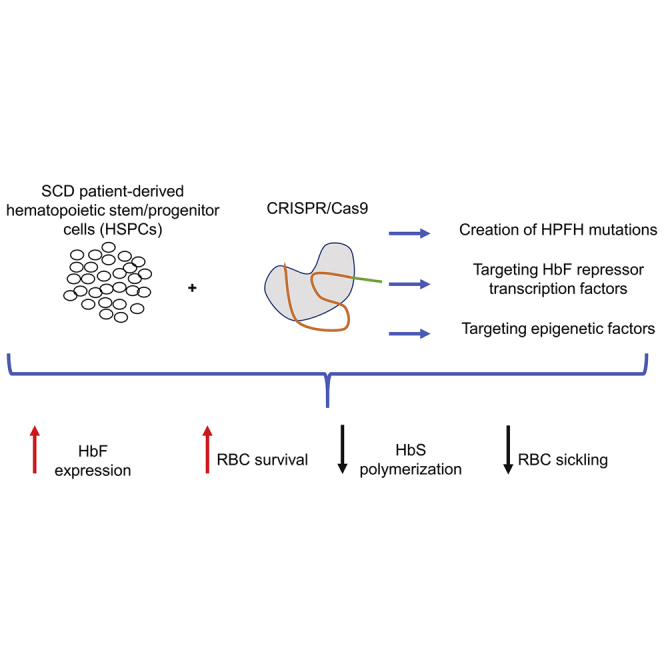
Given the ability of fetal hemoglobin (HbF) to inhibit sickle hemoglobin polymerization, HbF reactivation by the creation of naturally occurring HbF-associated mutations, editing HbF repressors/their binding site, or epigenetic intermediates using CRISPR-Cas9 are promising. Recent clinical data are encouraging; nevertheless, long-term follow-up is lacking, and genome editing safety and efficacy concerns remain.
Introduction
Sickle cell disease (SCD) is a collection of disorders characterized by the inheritance of a single base substitution (replacement of hydrophilic glutamic acid by hydrophobic valine) in the first exon of the β-globin gene (HBB). Whether inherited in a homozygous manner or with another mutation in HBB, the sickle hemoglobin (α2βs2, HbS) substitution significantly alters the function of hemoglobin, resulting in polymerization and aggregation upon deoxygenation. This distortion results in decreased red blood cell (RBC) survival and the downstream clinical manifestations of the disorder, including chronic anemia, pain events, stroke, multiorgan damage and failure, and premature mortality.1
In the setting of deoxygenation, the intracellular hemoglobin concentration and the presence of fetal hemoglobin (α2γ2, HbF) have profound impacts on the rate of polymerization of HbS. The delay period before fibers appear is known as the “delay time” and is inversely dependent to the 30th power of the deoxy-HbS concentration.2 This phenomenon is consistent with what is observed clinically: (1) newborns are protected from SCD complications in the first 6 months of life when HbF levels are naturally elevated,3 and (2) patients who co-inherit large deletions, small insertions/deletions (INDELs), or point mutations within the HBB gene cluster resulting in persistently elevated HbF (hereditary persistence of HbF [HPFH]) are relatively asymptomatic.4, 5, 6 Of note, patients with β-thalassemia, characterized by diminished or absent β-globin protein due to mutations in the β-globin gene, inheriting HPFH mutations also dramatically ameliorate their clinical symptoms, as γ-globin can compensate reduced β-globin levels and decrease globin chain imbalance and toxic-free intracellular α-globin.7
Hydroxyurea (HU), a US Food and Drug Administration (FDA)-approved drug for SCD patients, induces HbF and is the most widely used disease-modifying therapy, although other HbF-inducing pharmacologic agents are being investigated.8 Despite being the mainstay in therapy for SCD for nearly 30 years, the mechanism for HbF induction is not fully understood and its use does not fully eliminate the consequences of the disease, particularly given a variable response.9,10 Unlike HbS/HPFH where there is pancellular distribution of HbF among RBCs, there is heterocellular distribution with the use of HU, as well as person-to-person variation in the distribution of HbF, and therefore continued disease manifestations despite well-established clinical improvements.10, 11, 12, 13, 14 HPFH variants can be present as heterozygotes or homozygotes and can be distributed in a pancellular or heterocellular manner. Pancellular HPFH is caused either by large deletions in the HBB cluster (deletional HPFH) or point mutation and small deletions in the γ-globin gene promoter (non-deletional HPFH). Even when pancellularly distributed, the concentration of HbF among F cells is not always uniform. When heterocellularly distributed, increases in HbF are usually more modest and some RBCs may have no detectable HbF; this may be the result of epigenetic influences or potentially the mere insensitivity of HbF measurement below the level of detection. The optimal strategy to ensure elimination of clinical complications would thus involve a pharmacologic agent or gene therapy method that creates a pancellular distribution of elevated HbF mimicking deletional or non-deletional pancellular HbS/HPFH.
Current investigations of genetic modification of autologous hematopoietic stem cells (HSCs) are focused on two broad strategies: (1) expression of normal adult hemoglobin (HbA) or an HbA variant either by addition of an anti-sickling variant or direct correction of the causative point mutation by clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas9), or (2) HbF induction through gene addition, editing of HbF regulatory elements, or knockdown of HbF repressors to increase HbF expression.15, 16, 17 Classical CRISPR-Cas9 editing relies on double-strand breaks (DSBs) followed by a subsequent cellular repair of chromosomal breaks either through non-homologous end joining (NHEJ) or homology-directed repair (HDR). NHEJ is an error-prone process where broken DNA ends are ligated with no regard for homology, whereas HDR is a precise repair mechanism given that a donor template is provided where desired mutations/corrections can be introduced/corrected. Clinical trials for gene editing in SCD are focused first on editing to induce HbF expression rather than direct correction of the sickle mutation, as the former does not require HDR, which is difficult to achieve in HSCs. As of early 2021, there were three trials investigating genetic strategies in SCD studying the safety and efficacy of HbF induction (ClinicalTrials.gov: NCT03745287, NCT04443907, and NCT03653247). Two trials are investigating the use of CRISPR-Cas9-modified human HSCs (ClinicalTrials.gov: NCT03745287 and NCT04443907), with preliminary data recently made available in the first two participants from ClinicalTrials.gov: NCT03745287 showing HbF induction and clinical improvement.18,19 Here, we review the current strategies of HbF derepression using CRISPR-Cas9 for a potential universal cure for patients with SCD.
Generation of de novo HPFH mutations
Numerous approaches have been tested to reactivate developmentally silenced HbF in adult patient-derived hematopoietic stem and progenitor cells (HSPCs). Pancellular HbF expression (20%–30%) in patients with SCD and β-thalassemia alleviates deleterious effects of the severe disease phenotypes.20,21 Thus, genome editing tools, particularly CRISPR-Cas9, are highly effective and widely available to achieve these therapeutic HbF levels.
Various deletional HPFH mutations in the β-globin locus (7.2- to 105-kb deletions) share overlapping sequences; however, there is not a consistent elevation of HbF in patients despite having the same mutations, suggesting that different HbF silencing mechanisms and additional factors are involved in the expression of globin genes. A comparison of deletional mutations in patients with varying HbF levels revealed that a 3.5-kb γ-δ intergenic region is crucial for HbF silencing.22 CRISPR-Cas9-mediated deletion of a 13.6-kb region (similar to the naturally occurring 12.9-kb HPFH-5 deletion) encompassing the δ- and β-globin genes and the δ-γ intergenic genomic region derepressed HbF expression in erythroblasts and ameliorated RBC sickling.23 This work emphasized the importance of not only disrupting regulatory sequences but also chromosomal configuration given that the deletion or inversion of the 13.6-kb area caused robust reactivation of HbF synthesis greater than disruption of just the putative HbF silencer alone due to epigenetic modifications and changes in chromatin contacts within the β-globin locus.
Creating large deletional mutations ex vivo theoretically requires simultaneous cleavage in at least two separate DNA regions and is not as efficient as generating INDELs that theoretically require a single DNA target. Substantial focus has therefore been directed toward mimicking nondeletional HPFH mutations (point mutations) using either catalytically active Cas9 to create DSBs or using base editing strategies. Some of these current targets for high-level HbF expression include generating mutations at the γ-globin promoters at positions of −200, −175, −158, and −115 that either disrupt HbF repressor binding sequences6,24 or create de novo transcription activator binding sites. While creation of exact HPFH mutation through HDR would allow predictable HbF expression, HDR is not as efficient as NHEJ in long-term engrafting stem cells;25 therefore, INDEL generation to disrupt repressor DNA binding motifs in the targeted area is a primary strategy to induce HbF.
The BCL11A binding site (−115) is a critical region in the HBG promoter (Figure 1).26 CRISPR-Cas9-mediated editing of the binding region induced robust γ-globin expression in both progenitor cells and an immortalized human erythroid cell line (HUDEP-2).24,27 In addition to the BCL11A binding region, disruption of the ZBTB7A (also known as LRF) binding site (−200 region, Figure 1) provided substantial γ-globin reactivation in HUDEP-2 cells as well as in erythroid cells from SCD patient-derived HSPCs. Sickling of edited cell-derived RBCs was inhibited, and modified healthy HSPCs persisted in immunocompromised NSG mice over the long-term.6 In addition, targeting of the LRF binding site through INDEL creation induced HbF expression in HUDEP-2 and erythroid cells from β039-thalassemia patient-derived HSPCs; interestingly, F cell percentages were comparable in both large deletion clones (4.9-kb deletion eliminating HGB2 gene due to the sequence homology of the target areas in HBG1 and HBG2 promoters) and small INDEL clones.28 In addition to interrupting repressor binding sites, the creation of de novo activator sequences in the promoters are correlated with γ-globin reactivation as well. Of those, generation of −113A>G HPFH mutation, an artificial GATA site, using CRISPR-Cas9 and a donor oligonucleotide sequence containing the desired mutation induced substantial levels of γ-globin expression by recruiting GATA1 and outcompeting BCL11A for binding, while a control −113A>C substitution did not significantly alter the protein levels.29 Altogether these reports provide a viable option for patients with SCD by the creation of de novo HFPH-like mutations to reactivate HbF in adult RBCs.
Figure 1.

Schematic representation of β-globin locus constituting locus control region and β-like globin genes on chromosome 11
DNA binding sequences of ZBTB7A (also known as LRF, red), BCL11A (brown), and NF-Y (pink) in the identical HBG1 and HBG2 promoters are shown below.
Targeting HbF transcription factors
HbF expression is controlled by a series of epigenetic factors and DNA binding activators/repressors in a tightly controlled and well-orchestrated manner. Mutations in the HBG promoter provide either an artificial activator binding site or disruption in the repressor binding region as described above. Other than disrupting the repressor binding site, direct knockout of HbF silencers is a theoretical therapeutic option to reactivate HbF in adult RBCs. Genome-wide association studies (GWASs) exploring natural human mutations revealed that polymorphisms in the BCL11A gene, intergenic region between the genes HBS1L and MYB and β-globin locus are associated with elevated HbF expression.30,31 Extensive genetic analyses along with human cell line and mouse model investigations confirmed BCL11A as a master HbF repressor.32, 33, 34 Although robust HbF induction is seen after BCL11A silencing, BCL11A expression is also required for some non-erythroid lineages, including B lymphocytes, HSCs, and dendritic, breast, and pancreatic cells, as well as cells within the central nervous system.35 Moreover, BCL11A null mice are postnatally lethal,36 and haploinsufficiency in mice causes impaired cognition, abnormal social behavior, and microcephaly.37 In humans, along with elevated levels of HbF expression, an intellectual developmental disorder (Dias-Logan syndrome) and developmental delay, including weak motor coordination skills, are reported in those with BCL11A deletions and haploinsufficiency.38,39
In 2013, a specific region in intron-2 of the BCL11A gene was reported as an erythroid-specific enhancer, making it a more clinically relevant and applicable target.40 Upon editing with a series of guide RNAs, critical sequences were determined in the enhancer region that provided robust RBC-specific silencing of BCL11A expression and HbF induction without affecting erythropoiesis.40, 41, 42 Furthermore, the expression levels of BCL11A in other lineages, and HSC function and engraftment potential in immunocompromised NSG mice were not perturbed. To validate the clinical importance of the approach, we recently evaluated the BCL11A enhancer-edited HSPCs in a rhesus macaque model where we demonstrated long-term persistence of the edited cells (up to 80% edited cells in the bone marrow and peripheral blood of rhesus macaques) and elevated HbF induction (4.3%–28.6%) in peripheral blood RBCs without anemia or other apparent hematologic toxicity.43 More interestingly, we observed a positive interaction between biallelic gene editing and stress erythropoiesis with regard to γ-globin induction. While INDEL ratios in edited rhesus in stressed conditions were stable, stress erythropoiesis amplified γ-globin induction effect of BCL11A enhancer editing, proposing that the HbF inductive effect of BCL11A might be controlled differentially in stable and stressed conditions.43 This stress interaction is yet to be elucidated, particularly what, if any, impact stress erythropoiesis has on transcription factor expression/binding. As such, a recent report demonstrated an unknown globin expression regulation mechanism where transcription factor NF-Y and BCL11A compete at the HBG promoter for control of globin transcription output. BCL11A binding at the distal “TGACCA” motif constitutes a steric barrier to co-activator NF-Y binding to the proximal “CCAAT” motif (Figure 1).44 Thus, BCL11A-nucleosome remodeling and deacetylase (NuRD) complex binding to the promoter blocks NF-Y-coactivator recruitment, resulting in γ-globin repression. High prevalence of the NuRD complex in adult erythroid cells compared to co-activators maintains the competitive advantage for HbF repression.45
Overall, more research is warranted to fully understand the regulatory mechanism of globin expression to enhance the success rate in clinical settings, as erythropoietic stress and chronic inflammation in patients with SCD may overestimate the short-term effect of BCL11A enhancer editing in clinical trials. Pre-clinical evaluation led to the opening of clinical trials evaluating CRISPR-Cas9 editing to disrupt the GATA1 binding site in the BCL11A enhancer to reactivate HbF expression in adult RBCs of patients with transfusion-dependent β-thalassemia (ClinicalTrials.gov: NCT03655678) and severe SCD (ClinicalTrials.gov: NCT03745287). Short-term follow-up for the first two patients infused with edited autologous HSPCs revealed pancellular, elevated, and stable HbF expression providing transfusion independence and elimination of vaso-occlusive episodes.18 Similarly, although CRISPR-Cas9 editing was not utilized, another clinical trial (ClinicalTrials.gov: NCT03282656) utilizing short hairpin RNA (shRNA) targeting BCL11A mRNA embedded in a microRNA (shmiR), which is expressed in an erythroid-specific fashion, for SCD patients demonstrated similar results,46 indicating the importance and feasibility of BCL11A silencing for clinical success. However, long-term follow-up data are required to demonstrate the safety and efficacy of these approaches in terms of improved hematological parameters without any significant genotoxicity or cytotoxicity.
Novel HbF regulator editing
Comprehensive studies on globin expression regulation revealed several master regulators, including TAL1, GATA1, SOX6, KLF1, and MYB, that are required for multiprotein complex formation to arrange appropriate chromosomal reorganization in globin switching (Figure 2).47,48 Although significant HbF induction has been demonstrated either by direct gene editing49, 50, 51 or disruption of regulatory binding sites,24,29,52,53 none of these particular targets has been given focus considering the myriad functions of these transcription factors in erythropoiesis, including cell proliferation, differentiation, and maturation. In a head-to-head comparison of KLF1, BCL11A, and HBG1/2 promoter targeting in human CD34+ HSPCs, there was comparable F cell and HbF induction; however, KLF1 editing resulted in several dysregulated genes involved in cell cycle, apoptosis, and immune pathways.54 Another critical HbF regulator, LRF, displays essential anti-apoptotic activity during terminal erythroid differentiation.55 Therefore, unlike BCL11A as a potential therapeutic target, none of these additional regulators has made its way into large-animal models or clinical trials at this time.
Figure 2.

Schematic representation of transcriptional fetal hemoglobin regulation
One powerful approach to determine novel HbF regulators has been using CRISPR libraries, particularly in HUDEP-2 cells. A domain-focused single guide RNA (sgRNA) library approach has identified an erythroid-specific kinase, heme-regulated inhibitor (HRI, also known as EIF2AK1 phosphorylating eIF2α), regulating transcription factor ATF4 that directly stimulates BCL11A expression.49,56 A recent study not only corroborated the HbF induction potential of HRI kinase depletion but also demonstrated synergistic activity when combined with pharmacological inducers in healthy and SCD patient-derived HSPCs.57 When exploring therapeutic approaches targeting HRI for SCD, one critical aspect that should be taken into consideration is that HRI is essential for translational regulation of α- and β-globins and the survival of erythroid progenitors, and it is activated by a number of cytoplasmic stresses other than heme deficiency, including oxidative stress and heat shock. The lack of HRI adversely modifies the phenotype of two known human RBCs disorders, erythropoietic protoporphyria (EPP) and β-thalassemia.58 Similar data in SCD are lacking; however, elevation of HbF with reduction of stress erythropoiesis could mitigate symptoms as described in other disorders and could be another potential approach by using novel kinase pharmacological inducers.
Two independent groups have identified the transcription factor ZNF410 as uniquely regulating the NuRD component CHD4 to suppress γ-globin transcription in erythroid cells using CRISPR-Cas9 screening in HUDEP-2 cells.59,60 In these reports, as expected, genome-wide screening identified NuRD complex subunits, including CHD4, MTA2, GATAD2A, MBD2, and HDAC2, as well as known HbF repressors such as BCL11A and LRF as HbF regulators. In mammalian cells, conventional transcription factors control a large number of genes by binding to thousands of sites genome-wide. Therefore, transcription factors have always been considered “undruggable” targets due to potential undesired effects. Interestingly, ZNF410 was reported to target only CHD4 in human cells without affecting hematopoiesis, stem cell activity, or erythropoiesis.59,60 These observations suggest that this novel transcription factor could be tested as a potential treatment option for SCD through genome editing or, potentially, small molecule inhibitors. Genome editing tools to induce HbF for patients with SCD are highly efficient with encouraging clinical trial data;18 however, this approach is currently available to only a small portion of patients living in developed countries. Therefore, establishing the effectiveness of a HbF inducing cost-effective drug repressing one of the main HbF repressors such as ZNF410 is a more practical and suitable strategy to address the global burden of disease.
Targeting epigenetic factors
In addition to transcription factors, epigenetic mediators play a critical role in HbF repression. Epigenetic factors coordinate with transcription factors to repress HbF, although the exact molecular mechanism is poorly understood. CpG-rich sequences in the HBG promoters are recognized by the methyl-CpG binding domain (MBD)-2 protein that recruits other members of the co-repressor NuRD complex to repress HbF expression.61,62 Transcription factors including BCL11A, LRF, and ETO-2 recruit the NuRD complex to the HBG promoters to silence γ-globin expression. The NuRD complex consists of six core proteins, each of which has multiple paralog proteins, including HDAC1/2, MTA1/2/3, RBBP4/7, P66a/b, MBD2/3, and CHD3/4, which display dual enzymatic functionality with its deacetylation (HDAC1/2) and chromatin remodeling units (CHD3/4).63
Using CRISPR-Cas9 comprehensive mutagenesis in HUDEP-2 cells, it was shown that only five members of the NuRD complex (CHD4, GATAD2A, HDAC2, MBD2, and MTA2) affect γ-globin expression.64 Editing of these genes offers a potential approach to induce HbF for the treatment of SCD. However, one major challenge in epigenetic editing is to apply editing in a cell-specific manner to reduce possible hematological side effects such as neutropenia, thrombocytopenia, or thrombophilia that have been reported in clinical trials using pharmacological inhibitors targeting epigenetic-modifying enzymes.65 Interestingly, mice with knockout of MBD2, which was proven to induce significant γ-globin expression in HUDEP-2 cells66 and β-YAC transgenic mice,67 exhibited a mild phenotype and were viable and fertile.68 However, to explore the full potential, large animal models are required to confirm the safety and efficacy of MBD2 and other epigenetic players as well as other NuRD complex members.
Another known epigenetic regulator, DNA methyltransferase 1 (DNMT1), cooperates with BCL11A and Myb to repress γ-globin expression by maintaining promoter CpG methylation.69,70 A recent report demonstrated that a naturally occurring missense N-terminal regulatory domain mutation in the DNMT1 gene (S878F) diminished the association of DNMT1 with BCL11A, GATA1, and histone deacetylase 1/2 (HDAC1/2), and it reactivated γ-globin in adult erythroid cells.71 Similarly, euchromatic histone lysine methyltransferase (EHMT)-1, EHMT-2, and lysine-specific demethylase 1 (LSD1/KDM1A) inhibition was associated with robust HbF induction in human adult erythroid cells.72,73 RBCs from both primary HSPCs and HUDEP-2 cells with CRISPR-Cas9-targeted depletion of the ETS2 repressor factor (ERF) displayed marked HbF induction, and edited HSPCs demonstrated engraftment in immunocompromised mice.74 This observation was also confirmed by enrichment of DNMT3 through dead Cas9 (dCas9), leading to high HbF levels, implying that DNMT3A-mediated hypermethylation of ERF elevates γ-globin expression. Although systemic targeting of epigenetic factors does not seem feasible at the moment, understanding the regulatory mechanisms would be helpful particularly when persistence of editing HbF levels are not maintained over time in clinical approaches.
Base editing to induce HbF
The safety of the CRISPR-Cas9 system, including DSBs and creating potential off-target edits, remains a major concern. To date, no malignant transformation or clonal expansion of CRISPR-Cas9-edited cells has been reported in non-human primate models43,75,76 or in clinical trials, although follow-up data are much shorter.18,77 Safety assessment, however, should be conducted case by case, as specificity of the editing is mainly dependent on the specificity of guide RNAs used. Current clinical trials (ClinicalTrials.gov: NCT03653247, NCT03432364, NCT03745287, and NCT03655678) are using genome-editing tools to create DSBs in the erythroid-specific BCL11A enhancer to induce HbF levels as a potential treatment option for SCD and β-thalassemia. Although no significant adverse events have been reported to date from these clinical trials, it has been well documented that DSBs can lead to large deletions, chromosomal rearrangements, and genetic instability, as well as p53 response activation.78, 79, 80 As an alternative approach, CRISPR-Cas9 base editors can modify the genome without the need for DSBs.81
Two base editors, cytosine base editors (CBEs convert C:G to A:T base pair) and adenine base editors (ABEs convert A:T to C:G base pair), include catalytically dead Cas9 or a nickase Cas9 (nCas9) fused with a DNA deaminase enzyme.82 Applicability of base editors to induce HbF was demonstrated using an ABE7.10 evolved base editor (ABE8e) to modify the BCL11A enhancer in HEK293T cells, leading to significant editing levels (∼54%) in the GATA1 binding site.83 To confirm the HbF inducing effect of base editors, it was further demonstrated that correction of the HBB−28A>G promoter mutation and/or BCL11A enhancer disruption using A3A (N57Q)-BE3 base editor reversed the pathological phenotypes of HSPCs derived from patients with SCD and β-thalassemia.84 Robust editing ratios in SCD patient-derived HSPCs (86%–91%) provided 27% –32% HbF, indicating the promising potential of the method. The edited cells persisted in primary and secondary immunocompromised NBSGW mice. Although C>T base-edited allele frequencies were comparable in the infusion product and engrafted cells, a significant loss was noted in the C>G/A edits, suggesting that HSCs compared to non-engrafting progenitor cells favor C>T editing.
Similar approaches have been tested to create HPFH mutations to induce HbF without creating DSBs. The feasibility of the idea was first demonstrated in HEK293 using ABE7.10 by simultaneously converting −198 C to T in the HBG1 promoter and −198 T to C in the HBG2 promoter with 29%–30% editing efficiency.85 The same group optimized cytidine (BE4) and adenine (ABE7.10) base editors by modifying the nuclear localization signals (NLSs) and codon usage, as well as ancestral reconstruction of the deaminase component. Multiple regions in the γ-globin promoters were targeted (−175T>C creating the TAL1 binding site, −116A>G disrupting the BCL11A binding site, and A>G−113 creating the GATA1 binding site) with up to 52% editing efficiency.86 In an attempt to narrow the editing window and avoid off-target edits in the activity window of the base editor (bystander editing), a variant of the HUDEP-2 cell line consisting of only one γ-globin gene was edited at the position of −117G>A by a base editor transferred by lentivirus (hyA3A-BE4) resulting in substantially elevated γ-globin expression.87 Directed evolution of ABE7.10 revealed ABE8 with higher editing efficiency (up to 60%) at the −198 position and greater HbF induction (3.5-fold more compared to mock-treated primary HSPCs).88 When applying a dual adenine and cytosine base editor (A&C-BEmax) system to create C to T and A to G conversions at the same target site in HUDEP-2 cells, there was simultaneous −113A/−114C conversion (disrupting the BCL11A binding site and creating the GATA1 binding site) providing robust γ-globin mRNA expression.89 Although the current approach is to mimic naturally occurring HFPH mutations, a recent bioRxiv report claimed more targets to be investigated and demonstrated that screening of the HBG promoters using ABE and CBEs revealed novel HbF regulatory sites of which HbF inducing activity was comparable to known mutations in yet-to-be peer-reviewed data.90
In some preclinical studies, INDEL frequency decreased at the early post-transplantation,43,76,91 and low HbF induction and INDEL percentages were reported in some non-human primates despite having high editing rates in the infusion products.43 To overcome possible limited HbF expression after edited cell infusion, an in vivo transduction and selection system was designed in which a helper-dependent adenovirus vector (HDAd5/35++) expressing a base editor targeted the BCL11A binding site (–113A>G) and co-expressed a P140K mutant of the human O-6-methylguanine-DNA methyltransferase (MGMT) gene. This co-expression of a mutant MGMT gene granted drug resistance and selective expansion of gene-modified cells upon selection with an MGMT inhibitor.92 After selection, they reported 21% γ-globin expression in peripheral blood and 20% edited cells in the bone marrow of β-YAC mice without significant hematological toxicity. As base editors do not result in DSBs (or minimal DSBs), only 0.8% large deletion (4.9-kb intergenic deletion including the whole HBG2 gene) were detected as opposed to the use of catalytically active Cas9 targeting leading to significant deletion levels.29,76,93 Similarly, a base editor (hA3A-BE3) targeting −115C and −114C did not result in intergenic region deletion in healthy donor- or β-thalassemia patient-derived HSPCs.94 Although the consensus of this large deletion is not yet fully understood, it has been demonstrated that loss of 4.9-kb region was not deleterious to engrafting HSCs and did not alter HbF levels.95 Recently, thousands of computationally predicted cis-regulatory elements (CREs) with base pair resolution in the BCL11A, MYB-HBS1L, KLF1, and β-like globin gene loci were investigated using adenine base editors that exposed previously unknown genetic complexities of globin silencing, potentially paving the way for new therapeutic modalities to treat SCD.96
Challenges and future perspective
Clinical trials investigating editing of the BCL11A enhancer to reactivate HbF for patients with SCD and β-thalassemia (ClinicalTrials.gov: NCT03655678 and NCT03745287)18 have preliminarily demonstrated disease symptom alleviation; however, for lifelong benefit, the persistence of elevated HbF expression is required. Data are insufficient to determine the long-term consequences of gene-edited stem cells and the ability to reactivate HbF for long-term benefit. Although the studies in mice indicated a significant reduction of the gene-edited cells in primary and in secondary transplants (reviewed by Maganti et al.91), non-human primate studies43,76 and clinical trial data demonstrated persistence of edited cells (>1 year). Of note, persistence of NHEJ repaired alleles and selective reduction of microhomology-mediated end joining (MMEJ) repaired alleles have been demonstrated in both mice97 and rhesus macaques,43 indicating that progenitor cells favor MMEJ repair and quiescent HSCs, a rare subpopulation within the HSPC product containing the long-term engrafting HSCs, and preferentially use NHEJ repair. Therefore, as MMEJ repair is mostly seen in progenitors, caution should be taken to maintain long-term HbF induction when establishing MMEJ-based strategies such as the creation of a 13-bp HPFH deletion.98
Understanding HbF switching and silencing mechanisms will bring more targetable and druggable candidates to the table. Because CRISPR-Cas systems need a recognition site (the protospacer adjacent motif [PAM]), targetable sequences in the genome for specific Cas enzymes can be limited despite the discovery of various Cas9 and Cas12 orthologs that recognize various PAM sequences.99 Numerous engineered or evolved Cas variants with greater PAM compatibilities have been identified (reviewed in Anzalone et al.82). However, broader PAM recognition ability could potentially enhance off-target effects that remain a realistic concern for CRISPR-Cas9 editing. Base editors that avoid the creation of toxic DSBs (few or none) allow more precise editing and eliminate some genotoxicity from catalytically active Cas enzymes;16 however, these tools are not without their drawbacks, including low-rate DSBs, off-target editing, and bystander edits in the editing window.82 More sequence-specific base editors with narrower editing windows eliminating bystander edits would enhance the safety of the practice. Available methods for off-target analysis coupling genome-wide next-generation sequencing including cell-based or in vitro assays can determine unintended edits at a detection limit of 0.01%–1% allele frequency. In theory, oncogenic mutations could arise at low frequencies that are outside the detection limits of current analytic techniques, which might lead mutated cells to proliferate in vivo and result in dysplastic or malignant transformation. These unintended dangerous edits could be overlooked in mice with short-term monitoring of engraftment (<6 months). Therefore, establishment of methods with more sensitive off-target analysis are needed in the development of safer genome editing techniques.
In an autologous transplantation setting, the patient needs to be conditioned using either radiation or chemotherapy such as busulfan to favor edited cell engraftment. Current strategies employ myeloablation of the marrow space, eliminating stem cells to make room for the edited cells, but also eliminating beneficial bone marrow cells and disrupting the matrix organization, which decreases the efficiency of the engraftment and increases the risk for secondary malignancy. In a clinical trial investigating adult β-globin gene transfer to SCD patients’ HSPCs, myelodysplastic syndrome (MDS) developed in a patient after gene therapy.100 Comprehensive genome integration analysis demonstrated the absence of vector integration in the blasts and vector-mediated oncogenesis as the cause of MDS. The authors concluded that hematopoietic stress in SCD patients and conditioning with alkylating agents might have enhanced the genomic instability of the patient’s remaining HSCs that potentially resulted in MDS.100,101 This trial was recently placed on a temporary suspension due to a reported the suspected unexpected serious adverse reaction (SUSAR) of AML.102 However, investigations again appear to exonerate vector-mediated dyserythropoiesis as a result of integration into transcriptional or mechanistic changes that would likely contribute to oncogenesis, and multiple factors are known to contribute to increased risk for post-HCT malignancy, particularly in patients with SCD.103 One possible strategy to avoid toxic conditioning involves antibody (CD117104 and CD45105)-drug conjugates with selective depletion of HSCs. Antibody conditioning has demonstrated multilineage engraftment in mice, although safety and efficacy need to be confirmed in larger animal models.
Current editing protocols in clinical trials for SCD rely on electroporation routed delivery of ribonucleoprotein (RNP) assembling catalytically active Cas9 and guide RNA due to its superior efficiency in terms of editing and fast kinetics. However, electroporation toxicity to HSCs due to the high-voltage shock required for cell permeabilization might limit the approach for patients with SCD, particularly for poor HSC mobilizers, whose bone marrow environment is inflammatory and where HSCs are less prevalent.106,107 Successful gene editing approach currently relies on obtaining a high number of HSCs in order to account for cell loss during the manufacturing process, including potential damage from electroporation. Engraftment of edited cells is highly correlated with infused cell numbers;43 therefore, less cytotoxic delivery methods are needed to enhance the success rate and offer the approach for all.
While methods are improved to increase HbF expression and conditioning is made safer, the largest hurdle for the adoption of this as a universal curative approach remains the exorbitant economic cost and the logistical demands required. Costs for gene therapy are suggested to be as high as US $900,000–$2.1 million,108 severely limiting real-world applicability. In the current market, most expenses for gene therapy occur in the pre-transplant period given the complexity of drug product collection, the requirement for good manufacturing practice (GMP) facilities, and compliance with safety and regulatory standards. If all facilities in the US focus on bone marrow transplantation for SCD patients, just 5,000 transplantations can be performed in a year even though it is not possible to manufacture 5,000 infusion products,109 nor is it economically feasible in the current healthcare system. Based on initial pricing experience with gene therapy in Europe, with an estimated upfront treatment price of more than US $1 million per patient, the cumulative budget impact is not sustainable. According to health policy experts, “the cumulative budget impact at that price could rise to $3 trillion, as much as is currently spent in a year on all health care in the USA.”110
Future therapies that could skip issues surrounding cell collection, expansion, editing, and reinfusion would involve direct infusion of the editing constructs. To do so would require improving the efficiency of deliverable in vivo genome editing toolboxes with targeted delivery and editing. Currently, in vivo editing is being used for limited diseases, including mucopolysaccharidosis type I (ClinicalTrials.gov: NCT02702115), hemophilia B (ClinicalTrials.gov: NCT02695160), and congenital blindness (ClinicalTrials.gov: NCT03872479). For SCD, in vivo editing resulted in 30% γ-globin expression and complete phenotypic correction using the HDAd5/35++ adenovirus vector system, which drives both transposon-mediated γ-globin addition and γ-globin reactivation by targeting the BCL11A binding site in the HBG promoter in healthy CD46/β-YAC mice and an SCD mouse model (CD46/Townes).111 Similarly, ABE editors targeting the BCL11A binding site delivered via HDAd5/35++ vectors provided 21% γ-globin expression in β-YAC mice.92 Although the results are encouraging and provide the proof of concept for substantial HbF induction using in vivo editing, the immune response against adenoviruses might hinder the translation of these approaches. Thus, safer viral or non-viral delivery of editing components for SCD-derived HSCs are not yet viable alternatives.
Conclusions
Creating pancellular, therapeutically elevated HbF expression that is sustained over the lifetime of a patient is a key strategy for the cure of SCD. Advances in genome editing have provided a tremendous understanding of transcriptional globin control, and targets of HbF regulation have shown clinical promise. At this time, various candidates investigated to induce HbF have provided variable levels of HbF expression in ex vivo cell culture models, mice, and non-human primate models, and it remains to be determined whether these methods will provide stable, lifelong therapeutic HbF induction in humans. The simplest, most efficient, and, importantly, cost-effective editing strategy for therapeutic levels of HbF induction in adult RBCs is yet to be defined but offers a potentially curative therapy for millions living with SCD.
Acknowledgments
The authors thank Dr. Gerd A. Blobel for valuable comments on the figures.
Author contributions
Conceptualization, S.D., A.L., and J.F.T.; writing – original draft, S.D. and A.L.; writing – review & editing, S.D., A.L., K.E., and J.F.T; figures, S.D. and K.E.
Declaration of interests
The authors declare no competing interests.
Contributor Information
Selami Demirci, Email: selami.demirci@nih.gov.
John F. Tisdale, Email: johntis@nhlbi.nih.gov.
References
- 1.Paulukonis S.T., Eckman J.R., Snyder A.B., Hagar W., Feuchtbaum L.B., Zhou M., Grant A.M., Hulihan M.M. Defining sickle cell disease mortality using a population-based surveillance system, 2004 through 2008. Public Health Rep. 2016;131:367–375. doi: 10.1177/003335491613100221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hofrichter J., Ross P.D., Eaton W.A. Kinetics and mechanism of deoxyhemoglobin S gelation: A new approach to understanding sickle cell disease. Proc. Natl. Acad. Sci. USA. 1974;71:4864–4868. doi: 10.1073/pnas.71.12.4864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Watson J., Stahman A.W., Bilello F.P. The significance of the paucity of sickle cells in newborn Negro infants. Am. J. Med. Sci. 1948;215:419–423. doi: 10.1097/00000441-194804000-00008. [DOI] [PubMed] [Google Scholar]
- 4.Jacob G.F., Raper A.B. Hereditary persistence of foetal haemoglobin production, and its interaction with the sickle-cell trait. Br. J. Haematol. 1958;4:138–149. doi: 10.1111/j.1365-2141.1958.tb03844.x. [DOI] [PubMed] [Google Scholar]
- 5.Forget B.G. Molecular basis of hereditary persistence of fetal hemoglobin. Ann. N Y Acad. Sci. 1998;850:38–44. doi: 10.1111/j.1749-6632.1998.tb10460.x. [DOI] [PubMed] [Google Scholar]
- 6.Weber L., Frati G., Felix T., Hardouin G., Casini A., Wollenschlaeger C., Meneghini V., Masson C., De Cian A., Chalumeau A. Editing a γ-globin repressor binding site restores fetal hemoglobin synthesis and corrects the sickle cell disease phenotype. Sci. Adv. 2020;6:eaay9392. doi: 10.1126/sciadv.aay9392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cui S., Engel J.D. Reactivation of fetal hemoglobin for treating β-thalassemia and sickle cell disease. Adv. Exp. Med. Biol. 2017;1013:177–202. doi: 10.1007/978-1-4939-7299-9_7. [DOI] [PubMed] [Google Scholar]
- 8.Telen M.J. Beyond hydroxyurea: New and old drugs in the pipeline for sickle cell disease. Blood. 2016;127:810–819. doi: 10.1182/blood-2015-09-618553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Steinberg M.H., Lu Z.H., Barton F.B., Terrin M.L., Charache S., Dover G.J., Multicenter Study of Hydroxyurea Fetal hemoglobin in sickle cell anemia: Determinants of response to hydroxyurea. Blood. 1997;89:1078–1088. [PubMed] [Google Scholar]
- 10.Ware R.E. How I use hydroxyurea to treat young patients with sickle cell anemia. Blood. 2010;115:5300–5311. doi: 10.1182/blood-2009-04-146852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Steinberg M.H., Barton F., Castro O., Pegelow C.H., Ballas S.K., Kutlar A., Orringer E., Bellevue R., Olivieri N., Eckman J. Effect of hydroxyurea on mortality and morbidity in adult sickle cell anemia: Risks and benefits up to 9 years of treatment. JAMA. 2003;289:1645–1651. doi: 10.1001/jama.289.13.1645. [DOI] [PubMed] [Google Scholar]
- 12.Steinberg M.H., Chui D.H., Dover G.J., Sebastiani P., Alsultan A. Fetal hemoglobin in sickle cell anemia: A glass half full? Blood. 2014;123:481–485. doi: 10.1182/blood-2013-09-528067. [DOI] [PubMed] [Google Scholar]
- 13.Wang W.C., Ware R.E., Miller S.T., Iyer R.V., Casella J.F., Minniti C.P., Rana S., Thornburg C.D., Rogers Z.R., Kalpatthi R.V., BABY HUG investigators Hydroxycarbamide in very young children with sickle-cell anaemia: A multicentre, randomised, controlled trial (BABY HUG) Lancet. 2011;377:1663–1672. doi: 10.1016/S0140-6736(11)60355-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zimmerman S.A., Schultz W.H., Burgett S., Mortier N.A., Ware R.E. Hydroxyurea therapy lowers transcranial Doppler flow velocities in children with sickle cell anemia. Blood. 2007;110:1043–1047. doi: 10.1182/blood-2006-11-057893. [DOI] [PubMed] [Google Scholar]
- 15.Demirci S., Leonard A., Tisdale J.F. Genome editing strategies for fetal hemoglobin induction in beta-hemoglobinopathies. Hum. Mol. Genet. 2020;29(R1):R100–R106. doi: 10.1093/hmg/ddaa088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Doerfler P.A., Sharma A., Porter J.S., Zheng Y., Tisdale J.F., Weiss M.J. Genetic therapies for the first molecular disease. J. Clin. Invest. 2021;131:e146394. doi: 10.1172/JCI146394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Demirci S., Uchida N., Tisdale J.F. Gene therapy for sickle cell disease: An update. Cytotherapy. 2018;20:899–910. doi: 10.1016/j.jcyt.2018.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Frangoul H., Altshuler D., Cappellini M.D., Chen Y.-S., Domm J., Eustace B.K., Foell J., de la Fuente J., Grupp S., Handgretinger R. CRISPR-Cas9 gene editing for sickle cell disease and β-thalassemia. N. Engl. J. Med. 2021;384:252–260. doi: 10.1056/NEJMoa2031054. [DOI] [PubMed] [Google Scholar]
- 19.Frangoul H., Bobruff Y., Cappellini M.D., Corbacioglu S., Fernandez C., De La Fuente J., Grupp S., Handgretinger R., Ho T.W., Imren S. Safety and efficacy of CTX001 in patients with transfusion-dependent β-thalassemia or sickle cell disease: Early results from the CLIMB THAL-111 and CLIMB SCD-121 studies of autologous CRISPR-CAS9-modified CD34+ hematopoietic stem and progenitor cells. Blood. 2020;131(Suppl 1):3–4. [Google Scholar]
- 20.Bauer D.E., Kamran S.C., Orkin S.H. Reawakening fetal hemoglobin: prospects for new therapies for the β-globin disorders. Blood. 2012;120:2945–2953. doi: 10.1182/blood-2012-06-292078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Powars D.R., Weiss J.N., Chan L.S., Schroeder W.A. Is there a threshold level of fetal hemoglobin that ameliorates morbidity in sickle cell anemia? Blood. 1984;63:921–926. [PubMed] [Google Scholar]
- 22.Sankaran V.G., Xu J., Byron R., Greisman H.A., Fisher C., Weatherall D.J., Sabath D.E., Groudine M., Orkin S.H., Premawardhena A., Bender M.A. A functional element necessary for fetal hemoglobin silencing. N. Engl. J. Med. 2011;365:807–814. doi: 10.1056/NEJMoa1103070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Antoniani C., Meneghini V., Lattanzi A., Felix T., Romano O., Magrin E., Weber L., Pavani G., El Hoss S., Kurita R. Induction of fetal hemoglobin synthesis by CRISPR/Cas9-mediated editing of the human β-globin locus. Blood. 2018;131:1960–1973. doi: 10.1182/blood-2017-10-811505. [DOI] [PubMed] [Google Scholar]
- 24.Martyn G.E., Wienert B., Yang L., Shah M., Norton L.J., Burdach J., Kurita R., Nakamura Y., Pearson R.C.M., Funnell A.P.W. Natural regulatory mutations elevate the fetal globin gene via disruption of BCL11A or ZBTB7A binding. Nat. Genet. 2018;50:498–503. doi: 10.1038/s41588-018-0085-0. [DOI] [PubMed] [Google Scholar]
- 25.Genovese P., Schiroli G., Escobar G., Tomaso T.D., Firrito C., Calabria A., Moi D., Mazzieri R., Bonini C., Holmes M.C. Targeted genome editing in human repopulating haematopoietic stem cells. Nature. 2014;510:235–240. doi: 10.1038/nature13420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu N., Hargreaves V.V., Zhu Q., Kurland J.V., Hong J., Kim W., Sher F., Macias-Trevino C., Rogers J.M., Kurita R. Direct promoter repression by BCL11A controls the fetal to adult hemoglobin switch. Cell. 2018;173:430–442.e17. doi: 10.1016/j.cell.2018.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Masuda T., Wang X., Maeda M., Canver M.C., Sher F., Funnell A.P., Fisher C., Suciu M., Martyn G.E., Norton L.J. Transcription factors LRF and BCL11A independently repress expression of fetal hemoglobin. Science. 2016;351:285–289. doi: 10.1126/science.aad3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mingoia M., Caria C.A., Ye L., Asunis I., Marongiu M.F., Manunza L., Sollaino M.C., Wang J., Cabriolu A., Kurita R. Induction of therapeutic levels of HbF in genome-edited primary β039-thalassaemia haematopoietic stem and progenitor cells. Br. J. Haematol. 2021;192:395–404. doi: 10.1111/bjh.17167. [DOI] [PubMed] [Google Scholar]
- 29.Martyn G.E., Wienert B., Kurita R., Nakamura Y., Quinlan K.G.R., Crossley M. A natural regulatory mutation in the proximal promoter elevates fetal globin expression by creating a de novo GATA1 site. Blood. 2019;133:852–856. doi: 10.1182/blood-2018-07-863951. [DOI] [PubMed] [Google Scholar]
- 30.Menzel S., Garner C., Gut I., Matsuda F., Yamaguchi M., Heath S., Foglio M., Zelenika D., Boland A., Rooks H. A QTL influencing F cell production maps to a gene encoding a zinc-finger protein on chromosome 2p15. Nat. Genet. 2007;39:1197–1199. doi: 10.1038/ng2108. [DOI] [PubMed] [Google Scholar]
- 31.Uda M., Galanello R., Sanna S., Lettre G., Sankaran V.G., Chen W., Usala G., Busonero F., Maschio A., Albai G. Genome-wide association study shows BCL11A associated with persistent fetal hemoglobin and amelioration of the phenotype of β-thalassemia. Proc. Natl. Acad. Sci. USA. 2008;105:1620–1625. doi: 10.1073/pnas.0711566105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bauer D.E., Orkin S.H. Hemoglobin switching’s surprise: The versatile transcription factor BCL11A is a master repressor of fetal hemoglobin. Curr. Opin. Genet. Dev. 2015;33:62–70. doi: 10.1016/j.gde.2015.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu J., Sankaran V.G., Ni M., Menne T.F., Puram R.V., Kim W., Orkin S.H. Transcriptional silencing of γ-globin by BCL11A involves long-range interactions and cooperation with SOX6. Genes Dev. 2010;24:783–798. doi: 10.1101/gad.1897310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu J., Peng C., Sankaran V.G., Shao Z., Esrick E.B., Chong B.G., Ippolito G.C., Fujiwara Y., Ebert B.L., Tucker P.W., Orkin S.H. Correction of sickle cell disease in adult mice by interference with fetal hemoglobin silencing. Science. 2011;334:993–996. doi: 10.1126/science.1211053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Papizan J.B., Porter S.N., Sharma A., Pruett-Miller S.M. Therapeutic gene editing strategies using CRISPR-Cas9 for the β-hemoglobinopathies. J. Biomed. Res. 2020;35:115–134. doi: 10.7555/JBR.34.20200096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu P., Keller J.R., Ortiz M., Tessarollo L., Rachel R.A., Nakamura T., Jenkins N.A., Copeland N.G. Bcl11a is essential for normal lymphoid development. Nat. Immunol. 2003;4:525–532. doi: 10.1038/ni925. [DOI] [PubMed] [Google Scholar]
- 37.Dias C., Estruch S.B., Graham S.A., McRae J., Sawiak S.J., Hurst J.A., Joss S.K., Holder S.E., Morton J.E., Turner C., DDD Study BCL11A haploinsufficiency causes an intellectual disability syndrome and dysregulates transcription. Am. J. Hum. Genet. 2016;99:253–274. doi: 10.1016/j.ajhg.2016.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Basak A., Hancarova M., Ulirsch J.C., Balci T.B., Trkova M., Pelisek M., Vlckova M., Muzikova K., Cermak J., Trka J. BCL11A deletions result in fetal hemoglobin persistence and neurodevelopmental alterations. J. Clin. Invest. 2015;125:2363–2368. doi: 10.1172/JCI81163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wessels M.W., Cnossen M.H., van Dijk T.B., Gillemans N., Schmidt K.L.J., van Lom K., Vinjamur D.S., Coyne S., Kurita R., Nakamura Y. Molecular analysis of the erythroid phenotype of a patient with BCL11A haploinsufficiency. Blood Adv. 2021;5:2339–2349. doi: 10.1182/bloodadvances.2020003753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bauer D.E., Kamran S.C., Lessard S., Xu J., Fujiwara Y., Lin C., Shao Z., Canver M.C., Smith E.C., Pinello L. An erythroid enhancer of BCL11A subject to genetic variation determines fetal hemoglobin level. Science. 2013;342:253–257. doi: 10.1126/science.1242088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brendel C., Guda S., Renella R., Bauer D.E., Canver M.C., Kim Y.-J., Heeney M.M., Klatt D., Fogel J., Milsom M.D. Lineage-specific BCL11A knockdown circumvents toxicities and reverses sickle phenotype. J. Clin. Invest. 2016;126:3868–3878. doi: 10.1172/JCI87885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Canver M.C., Smith E.C., Sher F., Pinello L., Sanjana N.E., Shalem O., Chen D.D., Schupp P.G., Vinjamur D.S., Garcia S.P. BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature. 2015;527:192–197. doi: 10.1038/nature15521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Demirci S., Zeng J., Wu Y., Uchida N., Shen A.H., Pellin D., Gamer J., Yapundich M., Drysdale C., Bonanno J. BCL11A enhancer-edited hematopoietic stem cells persist in rhesus monkeys without toxicity. J. Clin. Invest. 2020;130:6677–6687. doi: 10.1172/JCI140189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu N., Xu S., Yao Q., Zhu Q., Kai Y., Hsu J.Y., Sakon P., Pinello L., Yuan G.-C., Bauer D.E., Orkin S.H. Transcription factor competition at the γ-globin promoters controls hemoglobin switching. Nat. Genet. 2021;53:511–520. doi: 10.1038/s41588-021-00798-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gillespie M.A., Palii C.G., Sanchez-Taltavull D., Shannon P., Longabaugh W.J., Downes D.J., Sivaraman K., Espinoza H.M., Hughes J.R., Price N.D. Absolute quantification of transcription factors reveals principles of gene regulation in erythropoiesis. Mol. Cell. 2020;78:960–974.e11. doi: 10.1016/j.molcel.2020.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Esrick E.B., Lehmann L.E., Biffi A., Achebe M., Brendel C., Ciuculescu M.F., Daley H., MacKinnon B., Morris E., Federico A. Post-transcriptional genetic silencing of BCL11A to treat sickle cell disease. N. Engl. J. Med. 2021;384:205–215. doi: 10.1056/NEJMoa2029392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Love P.E., Warzecha C., Li L. Ldb1 complexes: The new master regulators of erythroid gene transcription. Trends Genet. 2014;30:1–9. doi: 10.1016/j.tig.2013.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wienert B., Martyn G.E., Funnell A.P.W., Quinlan K.G.R., Crossley M. Wake-up sleepy gene: Reactivating fetal globin for β-hemoglobinopathies. Trends Genet. 2018;34:927–940. doi: 10.1016/j.tig.2018.09.004. [DOI] [PubMed] [Google Scholar]
- 49.Grevet J.D., Lan X., Hamagami N., Edwards C.R., Sankaranarayanan L., Ji X., Bhardwaj S.K., Face C.J., Posocco D.F., Abdulmalik O. Domain-focused CRISPR screen identifies HRI as a fetal hemoglobin regulator in human erythroid cells. Science. 2018;361:285–290. doi: 10.1126/science.aao0932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shariati L., Khanahmad H., Salehi M., Hejazi Z., Rahimmanesh I., Tabatabaiefar M.A., Modarressi M.H. Genetic disruption of the KLF1 gene to overexpress the γ-globin gene using the CRISPR/Cas9 system. J. Gene Med. 2016;18:294–301. doi: 10.1002/jgm.2928. [DOI] [PubMed] [Google Scholar]
- 51.Shariati L., Rohani F., Heidari Hafshejani N., Kouhpayeh S., Boshtam M., Mirian M., Rahimmanesh I., Hejazi Z., Modarres M., Pieper I.L., Khanahmad H. Disruption of SOX6 gene using CRISPR/Cas9 technology for gamma-globin reactivation: An approach towards gene therapy of β-thalassemia. J. Cell. Biochem. 2018;119:9357–9363. doi: 10.1002/jcb.27253. [DOI] [PubMed] [Google Scholar]
- 52.Fanis P., Kousiappa I., Phylactides M., Kyrri A., Hadjigavriel M., Christou S., Sitarou M., Kleanthous M. A novel mutation in the erythroid transcription factor KLF1 is likely responsible for ameliorating β-thalassemia major. Hum. Mutat. 2019;40:1768–1780. doi: 10.1002/humu.23817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li B., Zhu X., Hossain M.A., Guy C.R., Xu H., Bungert J., Pace B.S. Fetal hemoglobin induction in sickle erythroid progenitors using a synthetic zinc finger DNA-binding domain. Haematologica. 2018;103:e384–e387. doi: 10.3324/haematol.2017.185967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lamsfus-Calle A., Daniel-Moreno A., Antony J.S., Epting T., Heumos L., Baskaran P., Admard J., Casadei N., Latifi N., Siegmund D.M. Comparative targeting analysis of KLF1, BCL11A, and HBG1/2 in CD34+ HSPCs by CRISPR/Cas9 for the induction of fetal hemoglobin. Sci. Rep. 2020;10:10133. doi: 10.1038/s41598-020-66309-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Maeda T., Ito K., Merghoub T., Poliseno L., Hobbs R.M., Wang G., Dong L., Maeda M., Dore L.C., Zelent A. LRF is an essential downstream target of GATA1 in erythroid development and regulates BIM-dependent apoptosis. Dev. Cell. 2009;17:527–540. doi: 10.1016/j.devcel.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huang P., Peslak S.A., Lan X., Khandros E., Yano J.A., Sharma M., Keller C.A., Giardine B., Qin K., Abdulmalik O. The HRI-regulated transcription factor ATF4 activates BCL11A transcription to silence fetal hemoglobin expression. Blood. 2020;135:2121–2132. doi: 10.1182/blood.2020005301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Peslak S.A., Khandros E., Huang P., Lan X., Geronimo C.L., Grevet J.D., Abdulmalik O., Zhang Z., Giardine B.M., Keller C.A. HRI depletion cooperates with pharmacologic inducers to elevate fetal hemoglobin and reduce sickle cell formation. Blood Adv. 2020;4:4560–4572. doi: 10.1182/bloodadvances.2020002475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Han A.-P., Fleming M.D., Chen J.-J. Heme-regulated eIF2α kinase modifies the phenotypic severity of murine models of erythropoietic protoporphyria and β-thalassemia. J. Clin. Invest. 2005;115:1562–1570. doi: 10.1172/JCI24141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lan X., Ren R., Feng R., Ly L.C., Lan Y., Zhang Z., Aboreden N., Qin K., Horton J.R., Grevet J.D. ZNF410 uniquely activates the NuRD component CHD4 to silence fetal hemoglobin expression. Mol. Cell. 2021;81:239–254.e8. doi: 10.1016/j.molcel.2020.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vinjamur D.S., Yao Q., Cole M.A., McGuckin C., Ren C., Zeng J., Hossain M., Luk K., Wolfe S.A., Pinello L., Bauer D.E. ZNF410 represses fetal globin by singular control of CHD4. Nat. Genet. 2021;53:719–728. doi: 10.1038/s41588-021-00843-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cramer J.M., Scarsdale J.N., Walavalkar N.M., Buchwald W.A., Ginder G.D., Williams D.C., Jr. Probing the dynamic distribution of bound states for methylcytosine-binding domains on DNA. J. Biol. Chem. 2014;289:1294–1302. doi: 10.1074/jbc.M113.512236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ramírez J., Dege C., Kutateladze T.G., Hagman J. MBD2 and multiple domains of CHD4 are required for transcriptional repression by Mi-2/NuRD complexes. Mol. Cell. Biol. 2012;32:5078–5088. doi: 10.1128/MCB.00819-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Allen H.F., Wade P.A., Kutateladze T.G. The NuRD architecture. Cell. Mol. Life Sci. 2013;70:3513–3524. doi: 10.1007/s00018-012-1256-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sher F., Hossain M., Seruggia D., Schoonenberg V.A.C., Yao Q., Cifani P., Dassama L.M.K., Cole M.A., Ren C., Vinjamur D.S. Rational targeting of a NuRD subcomplex guided by comprehensive in situ mutagenesis. Nat. Genet. 2019;51:1149–1159. doi: 10.1038/s41588-019-0453-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rivers A., Molokie R., Lavelle D. A new target for fetal hemoglobin reactivation. Haematologica. 2019;104:2325–2327. doi: 10.3324/haematol.2019.230904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yu X., Azzo A., Bilinovich S.M., Li X., Dozmorov M., Kurita R., Nakamura Y., Williams D.C., Jr., Ginder G.D. Disruption of the MBD2-NuRD complex but not MBD3-NuRD induces high level HbF expression in human adult erythroid cells. Haematologica. 2019;104:2361–2371. doi: 10.3324/haematol.2018.210963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rupon J.W., Wang S.Z., Gaensler K., Lloyd J., Ginder G.D. Methyl binding domain protein 2 mediates γ-globin gene silencing in adult human βYAC transgenic mice. Proc. Natl. Acad. Sci. USA. 2006;103:6617–6622. doi: 10.1073/pnas.0509322103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hendrich B., Guy J., Ramsahoye B., Wilson V.A., Bird A. Closely related proteins MBD2 and MBD3 play distinctive but interacting roles in mouse development. Genes Dev. 2001;15:710–723. doi: 10.1101/gad.194101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Roosjen M., McColl B., Kao B., Gearing L.J., Blewitt M.E., Vadolas J. Transcriptional regulators Myb and BCL11A interplay with DNA methyltransferase 1 in developmental silencing of embryonic and fetal β-like globin genes. FASEB J. 2014;28:1610–1620. doi: 10.1096/fj.13-242669. [DOI] [PubMed] [Google Scholar]
- 70.Xu J., Bauer D.E., Kerenyi M.A., Vo T.D., Hou S., Hsu Y.-J., Yao H., Trowbridge J.J., Mandel G., Orkin S.H. Corepressor-dependent silencing of fetal hemoglobin expression by BCL11A. Proc. Natl. Acad. Sci. USA. 2013;110:6518–6523. doi: 10.1073/pnas.1303976110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gong Y., Zhang X., Zhang Q., Zhang Y., Ye Y., Yu W., Shao C., Yan T., Huang J., Zhong J. A natural DNMT1 mutation elevates the fetal hemoglobin level via epigenetic derepression of the γ-globin gene in β-thalassemia. Blood. 2021;137:1652–1657. doi: 10.1182/blood.2020006425. [DOI] [PubMed] [Google Scholar]
- 72.Renneville A., Van Galen P., Canver M.C., McConkey M., Krill-Burger J.M., Dorfman D.M., Holson E.B., Bernstein B.E., Orkin S.H., Bauer D.E., Ebert B.L. EHMT1 and EHMT2 inhibition induces fetal hemoglobin expression. Blood. 2015;126:1930–1939. doi: 10.1182/blood-2015-06-649087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shi L., Cui S., Engel J.D., Tanabe O. Lysine-specific demethylase 1 is a therapeutic target for fetal hemoglobin induction. Nat. Med. 2013;19:291–294. doi: 10.1038/nm.3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bao X., Zhang X., Wang L., Wang Z., Huang J., Zhang Q., Ye Y., Liu Y., Chen D., Zuo Y. Epigenetic inactivation of ERF reactivates γ-globin expression in β-thalassemia. Am. J. Hum. Genet. 2021;108:709–721. doi: 10.1016/j.ajhg.2021.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Humbert O., Peterson C.W., Norgaard Z.K., Radtke S., Kiem H.-P. A nonhuman primate transplantation model to evaluate hematopoietic stem cell gene editing strategies for β-hemoglobinopathies. Mol. Ther. Methods Clin. Dev. 2017;8:75–86. doi: 10.1016/j.omtm.2017.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Humbert O., Radtke S., Samuelson C., Carrillo R.R., Perez A.M., Reddy S.S., Lux C., Pattabhi S., Schefter L.E., Negre O. Therapeutically relevant engraftment of a CRISPR-Cas9-edited HSC-enriched population with HbF reactivation in nonhuman primates. Sci. Transl. Med. 2019;11:eaaw3768. doi: 10.1126/scitranslmed.aaw3768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Stadtmauer E.A., Fraietta J.A., Davis M.M., Cohen A.D., Weber K.L., Lancaster E., Mangan P.A., Kulikovskaya I., Gupta M., Chen F. CRISPR-engineered T cells in patients with refractory cancer. Science. 2020;367:eaba7365. doi: 10.1126/science.aba7365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kosicki M., Tomberg K., Bradley A. Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 2018;36:765–771. doi: 10.1038/nbt.4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Shin H.Y., Wang C., Lee H.K., Yoo K.H., Zeng X., Kuhns T., Yang C.M., Mohr T., Liu C., Hennighausen L. CRISPR/Cas9 targeting events cause complex deletions and insertions at 17 sites in the mouse genome. Nat. Commun. 2017;8:15464. doi: 10.1038/ncomms15464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Haapaniemi E., Botla S., Persson J., Schmierer B., Taipale J. CRISPR-Cas9 genome editing induces a p53-mediated DNA damage response. Nat. Med. 2018;24:927–930. doi: 10.1038/s41591-018-0049-z. [DOI] [PubMed] [Google Scholar]
- 81.Komor A.C., Kim Y.B., Packer M.S., Zuris J.A., Liu D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016;533:420–424. doi: 10.1038/nature17946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Anzalone A.V., Koblan L.W., Liu D.R. Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat. Biotechnol. 2020;38:824–844. doi: 10.1038/s41587-020-0561-9. [DOI] [PubMed] [Google Scholar]
- 83.Richter M.F., Zhao K.T., Eton E., Lapinaite A., Newby G.A., Thuronyi B.W., Wilson C., Koblan L.W., Zeng J., Bauer D.E. Phage-assisted evolution of an adenine base editor with improved Cas domain compatibility and activity. Nat. Biotechnol. 2020;38:883–891. doi: 10.1038/s41587-020-0453-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zeng J., Wu Y., Ren C., Bonanno J., Shen A.H., Shea D., Gehrke J.M., Clement K., Luk K., Yao Q. Therapeutic base editing of human hematopoietic stem cells. Nat. Med. 2020;26:535–541. doi: 10.1038/s41591-020-0790-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gaudelli N.M., Komor A.C., Rees H.A., Packer M.S., Badran A.H., Bryson D.I., Liu D.R. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature. 2017;551:464–471. doi: 10.1038/nature24644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Koblan L.W., Doman J.L., Wilson C., Levy J.M., Tay T., Newby G.A., Maianti J.P., Raguram A., Liu D.R. Improving cytidine and adenine base editors by expression optimization and ancestral reconstruction. Nat. Biotechnol. 2018;36:843–846. doi: 10.1038/nbt.4172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhang X., Chen L., Zhu B., Wang L., Chen C., Hong M., Huang Y., Li H., Han H., Cai B. Increasing the efficiency and targeting range of cytidine base editors through fusion of a single-stranded DNA-binding protein domain. Nat. Cell Biol. 2020;22:740–750. doi: 10.1038/s41556-020-0518-8. [DOI] [PubMed] [Google Scholar]
- 88.Gaudelli N.M., Lam D.K., Rees H.A., Solá-Esteves N.M., Barrera L.A., Born D.A., Edwards A., Gehrke J.M., Lee S.J., Liquori A.J. Directed evolution of adenine base editors with increased activity and therapeutic application. Nat. Biotechnol. 2020;38:892–900. doi: 10.1038/s41587-020-0491-6. [DOI] [PubMed] [Google Scholar]
- 89.Zhang X., Zhu B., Chen L., Xie L., Yu W., Wang Y., Li L., Yin S., Yang L., Hu H. Dual base editor catalyzes both cytosine and adenine base conversions in human cells. Nat. Biotechnol. 2020;38:856–860. doi: 10.1038/s41587-020-0527-y. [DOI] [PubMed] [Google Scholar]
- 90.Ravi N.S., Wienert B., Wyman S.K., Vu J., Pai A.A., Balasubramanian P., Nakamura Y., Kurita R., Marepally S., Thangavel S. Identification of novel HPFH-like mutations by CRISPR base editing that elevates the expression of fetal hemoglobin. bioRxiv. 2020 doi: 10.1101/2020.06.30.178715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Maganti H.B., Bailey A.J.M., Kirkham A.M., Shorr R., Pineault N., Allan D.S. Persistence of CRISPR/Cas9 gene edited hematopoietic stem cells following transplantation: A systematic review and meta-analysis of preclinical studies. Stem Cells Transl. Med. 2021;10:996–1007. doi: 10.1002/sctm.20-0520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Li C., Georgakopoulou A., Mishra A., Gil S., Hawkins R.D., Yannaki E., Lieber A. In vivo HSPC gene therapy with base editors allows for efficient reactivation of fetal γ-globin in β-YAC mice. Blood Adv. 2021;5:1122–1135. doi: 10.1182/bloodadvances.2020003702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Li C., Psatha N., Sova P., Gil S., Wang H., Kim J., Kulkarni C., Valensisi C., Hawkins R.D., Stamatoyannopoulos G., Lieber A. Reactivation of γ-globin in adult β-YAC mice after ex vivo and in vivo hematopoietic stem cell genome editing. Blood. 2018;131:2915–2928. doi: 10.1182/blood-2018-03-838540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wang L., Li L., Ma Y., Hu H., Li Q., Yang Y., Liu W., Yin S., Li W., Fu B. Reactivation of γ-globin expression through Cas9 or base editor to treat β-hemoglobinopathies. Cell Res. 2020;30:276–278. doi: 10.1038/s41422-019-0267-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Métais J.-Y., Doerfler P.A., Mayuranathan T., Bauer D.E., Fowler S.C., Hsieh M.M., Katta V., Keriwala S., Lazzarotto C.R., Luk K. Genome editing of HBG1 and HBG2 to induce fetal hemoglobin. Blood Adv. 2019;3:3379–3392. doi: 10.1182/bloodadvances.2019000820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Cheng L., Li Y., Qi Q., Xu P., Feng R., Palmer L., Chen J., Wu R., Yee T., Zhang J. Single-nucleotide-level mapping of DNA regulatory elements that control fetal hemoglobin expression. Nat. Genet. 2021;53:869–880. doi: 10.1038/s41588-021-00861-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wu Y., Zeng J., Roscoe B.P., Liu P., Yao Q., Lazzarotto C.R., Clement K., Cole M.A., Luk K., Baricordi C. Highly efficient therapeutic gene editing of human hematopoietic stem cells. Nat. Med. 2019;25:776–783. doi: 10.1038/s41591-019-0401-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Traxler E.A., Yao Y., Wang Y.-D., Woodard K.J., Kurita R., Nakamura Y., Hughes J.R., Hardison R.C., Blobel G.A., Li C., Weiss M.J. A genome-editing strategy to treat β-hemoglobinopathies that recapitulates a mutation associated with a benign genetic condition. Nat. Med. 2016;22:987–990. doi: 10.1038/nm.4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Cebrian-Serrano A., Davies B. CRISPR-Cas orthologues and variants: optimizing the repertoire, specificity and delivery of genome engineering tools. Mamm. Genome. 2017;28:247–261. doi: 10.1007/s00335-017-9697-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hsieh M.M., Bonner M., Pierciey F.J., Jr., Uchida N., Rottman J., Demopoulos L., Schmidt M., Kanter J., Walters M.C., Thompson A.A. Myelodysplastic syndrome unrelated to lentiviral vector in a patient treated with gene therapy for sickle cell disease. Blood Adv. 2020;4:2058–2063. doi: 10.1182/bloodadvances.2019001330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Pagano L., Pulsoni A., Tosti M.E., Annino L., Mele A., Camera A., Martino B., Guglielmi C., Cerri R., Di Bona E. Acute lymphoblastic leukaemia occurring as second malignancy: Report of the GIMEMA archive of adult acute leukaemia. Gruppo Italiano Malattie Ematologiche Maligne dell’Adulto. Br. J. Haematol. 1999;106:1037–1040. doi: 10.1046/j.1365-2141.1999.01636.x. [DOI] [PubMed] [Google Scholar]
- 102.Business Wine . 2021. bluebird bio announces temporary suspension on phase 1/2 and phase 3 studies of LentiGlobin gene therapy for sickle cell disease.https://www.businesswire.com/news/home/20210216005442/en/ [Google Scholar]
- 103.Leonard A., Tisdale J.F. A pause in gene therapy: Reflecting on the unique challenges of sickle cell disease. Mol. Ther. 2021;29:1355–1356. doi: 10.1016/j.ymthe.2021.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Li Z., Czechowicz A., Scheck A., Rossi D.J., Murphy P.M. Hematopoietic chimerism and donor-specific skin allograft tolerance after non-genotoxic CD117 antibody-drug-conjugate conditioning in MHC-mismatched allotransplantation. Nat. Commun. 2019;10:616. doi: 10.1038/s41467-018-08202-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Palchaudhuri R., Saez B., Hoggatt J., Schajnovitz A., Sykes D.B., Tate T.A., Czechowicz A., Kfoury Y., Ruchika F., Rossi D.J. Non-genotoxic conditioning for hematopoietic stem cell transplantation using a hematopoietic-cell-specific internalizing immunotoxin. Nat. Biotechnol. 2016;34:738–745. doi: 10.1038/nbt.3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Leonard A., Bonifacino A., Dominical V.M., Luo M., Haro-Mora J.J., Demirci S., Uchida N., Pierciey F.J., Jr., Tisdale J.F. Bone marrow characterization in sickle cell disease: Inflammation and stress erythropoiesis lead to suboptimal CD34 recovery. Br. J. Haematol. 2019;186:286–299. doi: 10.1111/bjh.15902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Leonard A., Sharma A., Uchida N., Stroncek D., Panch S.R., West K., Molloy E., Hughes T.E., Hauffe S., Taylor T. Disease severity impacts plerixafor-mobilized stem cell collection in patients with sickle cell disease. Blood Adv. 2021;5:2403–2411. doi: 10.1182/bloodadvances.2021004232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Jensen K. 2019. Bluebird proposes installment plan for LentiGlobin gene therapy.https://www.biopharmadive.com/news/jpm19-bluebird-proposes-installment-plan-for-lentiglobin-gene-therapy/545646/ [Google Scholar]
- 109.Urnov F.D. The Cas9 hammer—and sickle: A challenge for genome editors. CRISPR J. 2021;4:6–13. doi: 10.1089/crispr.2021.29120.fur. [DOI] [PubMed] [Google Scholar]
- 110.Hampson G., Towse A., Pearson S.D., Dreitlein W.B., Henshall C. Gene therapy: Evidence, value and affordability in the US health care system. J. Comp. Eff. Res. 2018;7:15–28. doi: 10.2217/cer-2017-0068. [DOI] [PubMed] [Google Scholar]
- 111.Li C., Wang H., Georgakopoulou A., Gil S., Yannaki E., Lieber A. In vivo HSC gene therapy using a bi-modular HDAd5/35++ vector cures sickle cell disease in a mouse model. Mol. Ther. 2021;29:822–837. doi: 10.1016/j.ymthe.2020.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]