

Summary
The synovial sarcoma X breakpoint 2 (SSX2) belongs to a multigene family of cancer-testis antigens and can be found overexpressed in multiple malignancies. Its restricted expression in immune-privileged normal tissues suggest that SSX2 may be a relevant target antigen for chimeric antigen receptor (CAR) therapy. We have developed a T cell receptor (TCR)-like antibody (Fab/3) that binds SSX2 peptide 41-49 (KASEKIFYV) in the context of HLA-A∗-0201. The sequence of Fab/3 was utilized to engineer a CAR with the CD3 zeta intra-cellular domain along with either a CD28 or 4-1BB costimulatory endodomain. Human T cells from HLA-A2+ donors were transduced to mediate anti-tumor activity against acute myeloid leukemia (AML) tumor cells. Upon challenge with HLA-A2+/SSX2+ AML tumor cells, CAR-expressing T cells released interferon-γ and eliminated the tumor cells in a long-term co-culture assay. Using the HLA-A2+ T2 cell line, we demonstrated a strong specificity of the single-chain variable fragment (scFv) for SSX2 p41-49 and the closely related SSX3 p41-49, with no response against the others SSX-homologous peptides or unrelated homologous peptides. Since SSX3 has not been observed in tumor cells and expression cannot be induced by pharmacological intervention, SSX241-49 represents an attractive target for CAR-based cellular therapy to treat multiple types of cancer.
Keywords: chimeric antigen receptor, CAR, T cell receptor-like antibody, TCR-like antibody, cancer-testis antigen, CTA, synovial sarcoma X breakpoint, SSX, acute myeloid leukemia, AML, cellular therapy
Graphical abstract
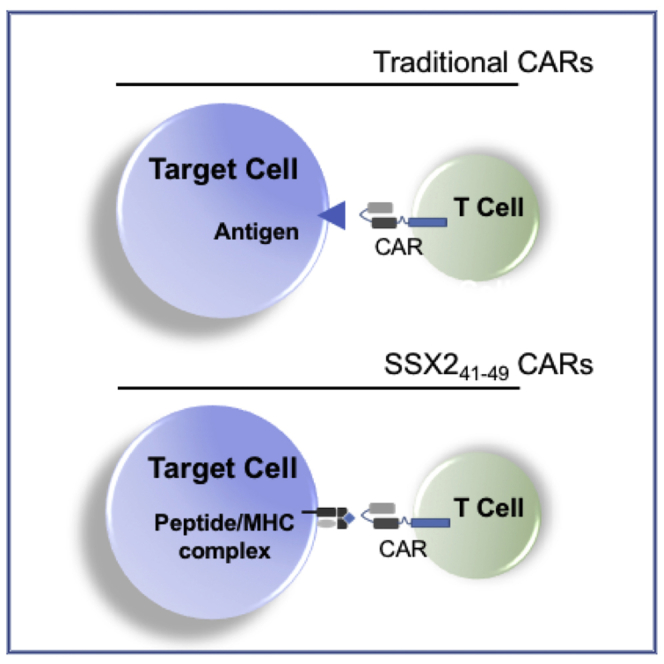
The TCR-like antibody engineered as a chimeric antigen receptor (CAR) is able to target the cancer-testis antigen (CTA) SSX2 peptide 41-49 in an HLA-A∗0201 context. Because of the SSX2 expression pattern in cancers, a cell-therapy approach using T cells expressing the SSX241-49 CAR can be readily deployed to treat patients.
Introduction
Cancer-testis antigens (CTAs) are a subgroup of tumor proteins with normal expression, found almost exclusively in testis germline tissues with aberrant expression in many types of cancer.1,2 The expression of CTAs is usually silenced by promoter hypermethylation, but in cancers, genes could be demethylated and CTAs re-expressed on the cell surface.3 As a result, testis-specific proteins could potentially be recognized as neo-antigens when expressed ectopically in tumor tissue.1 In addition to having a cancer-restricted pattern of expression, CTAs have been shown to be associated with an alteration of the host immune response and therefore represent ideal targets for cancer immunotherapy.4, 5, 6, 7
Of interest, among the CTAs is the synovial sarcoma X breakpoint (SSX) proteins. The SSX genes are located on the X chromosome and encode a family of ten highly homologous nuclear proteins, SSX1–10.8 Like other CTAs, expression of SSX mRNA has been shown to be predominantly restricted to testis germline tissues.8 Multiple SSX family members have been demonstrated to be ectopically expressed in many types of cancer.9 Originally identified as part of a genomic translocation present in synovial sarcoma, SSX2 (also called HOM-Mel-40) is known to induce spontaneous antibody and cellular response in patients with melanoma.10, 11, 12 SSX2 mRNA was also found to be expressed, albeit heterogeneously, in colorectal carcinoma, prostate cancer, breast cancer, hepatocellular carcinoma, glioma, lymphoma, leukemia, gastric carcinoma, and thyroid carcinoma tissue samples.9 In addition, SSX2 interacting protein (SSX2IP), which regulates the function of SSX2 in the testes and malignant cells, has been shown to be elevated in approximately one third of acute myeloid leukemia (AML) patient samples at presentation.13,14 Further, in AML patients, induction of SSX2 is observed after demethylating treatment.15 Because cellular and humoral responses against immunogenic SSX2 peptides have been observed in cancer patients, it is not surprising that SSX2 has also been exploited as a target for immune-based therapy.9 Therapeutic strategies against SSX2 include vaccines,16,17 the expansion of antigen-specific T cells (CTLs),18,19 and the generation of T cell receptor (TCR) alpha- and beta-chains that specifically target the HLA-A∗0201-restricted epitope 41-49 of SSX2.20 An alternative approach to target CTAs expressed on tumor cells, albeit not yet explored for SSX2 targeting, is the development of chimeric antigen receptor (CAR) with TCR-like specificity.21 These TCR-like CARs combined a single-chain variable fragment (scFv) that recognize peptides presented in the context of the major histocompatibility complex (MHC) to endodomains that contain costimulatory molecules (either CD28 or CD137 in combination with CD3ζ, for instance). While traditional CARs typically target proteins expressed on the surface of a tumor cell, TCR-like CARs can target a wider variety of intracellular antigens through their presentation by MHC molecules and can quickly be manufactured and applied to patients.
AML is the most common leukemia occurring in adults and the second most common leukemia of childhood.22 For patients with high-risk AML, relapse-free and overall survival still remain poor even following hematopoietic stem cell transplantation (HSCT).23,24 While CAR T cell therapy has experienced exciting successes for the treatment of patients with ALL, the lack of well-characterized and tumor-specific surface antigens in AML has necessitated consideration of CAR T cell strategies that may also affect normal tissues.25,26 One such example is the well-known surface antigen CD123, which is expressed on the majority of AML blasts, but is also expressed on many normal hematopoietic progenitor cells (HPCs).26,27 Re-expression of CTAs, as well as tumor suppressor genes by inhibiting DNA methyltransferase, has been used with success for the treatment of hematologic malignancies including AML.28,29 Because SSX2 expression in tumor cells has been reported to occur either as naturally expressed or having its expression induced by demethylation agents in tumor cells, our group sought to evaluate a novel scFv specific for SSX2 and develop a CAR with specific activity against AML tumors.15
CARs (SSX241-49 28ζ and SSX241-49 BBζ) that specifically recognized the p41-49 epitope of SSX2 were developed by our group. These CARs successfully targeted SSX2 peptide p41-49 when presented by HLA-A2∗ on transporter associated with antigen processing (TAP)-deficient T2 cells but also endogenously expressed SSX2 on AML tumor cells. Moreover, we demonstrated that our novel SSX241-49 CAR can eliminate HLA-A2+/SSX2+ AML tumor cells in vitro. Hence, the broad expression of SSX2 across multiple cancer types, combined with low and restricted expression in normal tissues, make this CTA an attractive target for CAR-based immunotherapy.
Results
SSX241-49 CARs design
An antibody antigen-binding fragment (Fab), called Fab/3, was selected for specificity against the HLA-A∗0201 epitope SSX241-49. Fab/3 was derived from an immunized Fab phage display library and the nucleotide sequence of the selected phage was analyzed.30 The results of a homology search using IMGT/V-QUEST showed that the variable heavy (VH) chain shares 99% and 88% homology in amino acid sequences with IGHV3-53∗02 and IGHJ3∗02, respectively, for the closest human immunoglobulin (Ig) heavy chain germline variable region.31 Searches for the variable light (VL) chain showed a 96% and 94% homology in amino acid sequences with IGLV6-57∗03 and IGLJ3∗02, respectively, for the human Ig light chain germline variable region.31 The variable regions of the heavy and light chains of Fab/3 shown in Figure 1A were cloned as an scFv fragment into validated CAR templates that include: the CD8a hinge and transmembrane domain with the CD34 epitope recognized by the QBEnd-10 antibody embedded in the hinge, the CD28 or 4-1BB endodomain, and the CD3ζ-signaling domain. The SSX241-49.CAR cassettes were cloned into a retroviral SFG backbone plasmid to generate retroviral vectors that code either for the SSX241-49.28ζ or SSX241-49.BBζ CAR (Figure 1B).
Figure 1.

Amino acid sequence of Fab/3 and SSX241-49-specific CAR design
(A) The amino acid sequence of Fab/a using the IMGT numbering system is presented with framework regions (FRs; FR1, FR2, and FR3) and complementarity-determining regions (CDRs; CDR1, CDR2, and CDR3) identified for the variable regions of both light (VL) and heavy (VH) chains, respectively. (B) The schematic structure of the SSX241-49 CARs SFG retroviral vectors is shown. Both constructs have the scFv with the VL-VH orientation and the QBEnd/10 epitope inserted within the CD8α hinge. Each CAR includes either the CD28 (SSX241-49 28ζ) or the 4-1BB (SSX241-49 BBζ) co-stimulatory domain in addition to the CD3ζ chain.
SSX241-49 CARs show high expression in HLA-A2+ T cells after transduction
HLA-A2 positive donor-derived T cells were transduced with each of the SSX241-49 CAR constructs. On day 5 post-transduction, expression was detected on the T cell surface using an anti-human Fab antibody and the anti-CD34, clone QBend-10 antibody (Figure 2A). A consistently high CAR expression (>60%) was obtained using our retroviral constructs (Figure 2B), and expression of either of the SSX241-49 CARs did not affect the expansion of the transduced T cells. However, expansion of T cells expressing the SSX241-49.BBζ was lower compared to either non-transduced (UnTx) T cells or T cells transduced with the SSX241-49.28ζ retroviral vector (Figure 2C). Similar observations have been made by other groups, but a two-way ANOVA of the cell expansions did not show that these differences were statistically different.32 Finally, the CD4+ and CD8+, CD45RA+CCR7+ (naive), and CD45RA-CCR7- (effector memory) T cell content was similar between the cultures (UnTx, SSX241-49.28ζ, and SSX241-49.BBζ) with no skewing to naive versus memory T cell subsets (Figure S1).
Figure 2.

Expression of SSX241-49-specific CARs in T cells
(A) Representative expression of the SSX241-49 CARs demonstrated by the co-detection of the scFv using an anti-human Fab antibody and the QBEnd/10 epitope using an anti-CD34 (clone QBEnd/10) antibody in transduced human HLA-A2+ T cells. Histogram analysis shows expression of either the scFv or the QBEnd/10 epitope, and co-expression of scFv and QBEnd/10 epitope is presented by dot plots analysis. (B) Summary of the transduction efficiency of SSX241-49 CARs transduction efficiency (n = 5). (C) Total expansion of untransduced (UnTx), SSX241-49 28ζ transduced, and SSX241-49 BBζ transduced T cells after 14 days of culture (n = 5).
T cells expressing SSX241-49 CAR show cross-reactivity for SSX341-49 but minimal/no reactivity against other homologous SSX and unrelated peptides
Since the SSX2 epitope targeted by the CARs lies in a region with high homology between the different members of the SSX family, we evaluated the reactivity of the new SSX241-49.CAR against homologous peptides derived from the 9 others highly homologous SSX proteins. As shown in Figure 3A, homologous SSX peptides differ from SSX241-49 by 1 to 3 residues: peptides from SSX2, SSX3, SSX5, and SSX10 can be considered strong binders to HLA-class I (i.e., <50 nM binding affinity), while SSX4, SSX7, and SSX9 are weak binders (>50 nM and <500 nM binding affinity) and SSX1, SSX6, and SSX8 are non-binders (>500 nM binding affinity).33 We looked at the response of UnTx versus SSX241-49.BBζ, and SSX241-49.28ζ transduced T cells against TAP-deficient T2 cells pulsed with SSX241-49 homologous peptides (10,000 ng, 100 ng, 1 ng, or 0.01 ng per mL) in the presence of β2 microglobulin. The reactivity of SSX241-49-specific CAR T cells was measured by the release of interferon (IFN)-γ after 24 h of culture. Both CARs (SSX241-49.28ζ and SSX241-49.BBζ) responded to SSX2 homologous peptides in a similar way (Figure 3A). SSX241-49 CAR T cells secreted IFN-γ upon exposure to SSX2-, SSX3-, and SSX7-derived peptides. While the response against the SSX7 peptide was only observed at the highest concentration (10,000 ng of peptide/mL), responses against SSX2 and SSX3 peptides, albeit identical, were detected at a lower concentration (100 ng/mL). Secretion of IFN-γ by SSX241-49 CAR T cells after exposure to SSX2- and SSX3-derived peptides was associated with cytolysis of the T2 cells as measured by flow cytometry (data not shown). No response was observed against the SSX1-, SSXX4-, SSX5-, SSX6-, SSX8-, SSX9-, and SSX10-derived peptides, as shown by the absence of IFN-γ in the culture media after 24 h of culture (Figure 3B). The lack of response against these SSX homologous peptides and the difference in reactivity toward SSX2-, SSX3-, and SSX7-derived peptides suggest that recognition of SSX homologous peptides, except for SSX3, is improbable. Cross-reactivity against non-SSX, partially homologous peptides encoded by the IGSF22 and TCOF1 genes was not observed, confirming the specificity of the SSX241-49 CARs (data not shown). In a separate experiment, we examined the reactivity of SSX241-49 CAR T cells against 2 alternate SSX2 peptides (5-13 and 103-111) that are known to be immunogenic.9 T cells expressing the SSX241-49 CAR were not cross-reactive with those 2 peptides, as evidenced by a lack of IFN-γ production even after 24 h co-culture with p5-13 or p103-111 peptide-loaded T2 cells. In contrast, responses to SSX2 p41-49 were confirmed by SSX241-49 CAR-expressing T cell production of IFN-γ and elimination of the T2 cells loaded with p41-49 in a 72 h co-culture experiment, further demonstrating their specificity (Figure S2).
Figure 3.

Recognition of homologous SSX peptides by SSX241-49 CAR T cells
SSX241-49 28ζ and SSX241-49 BBζ transduced T cells were co-cultured with T2 cells pulsed overnight with serial dilution of indicated peptides. (A) SSX homologous peptides sequence and binding affinity (KD, nM) to HLA-A∗0201. (B) Culture supernatant was collected after 24 h of co-culture, IFN-γ production was analyzed by ELISA, and results are expressed as means of the duplicates.
SSX241-49 CAR T cells are cytolytic against AML tumor cells
To test the ability of the SSX241-49 CARs to recognize and kill tumor cells that endogenously express SSX2, T cells transduced with the SSX241-49.28ζ or SSX241-49.BBζ CAR were co-cultured with the AML tumor cell lines THP-1 and KG1a. Flow analysis confirmed that both cell lines are SSX2-positive as demonstrated by flow analysis (Figure 4A), while the SR cell line did not display expression for SSX2. The extended characterization of both cell lines confirmed that the THP-1 cell line is HLA-A2+ while the cell line KG1a is negative for HLA-A2 expression (Figure 4B), and both cell lines showed similar expression levels for CD33 and the co-stimulatory molecules CD80 and CD86 (Figure 4B).
Figure 4.

SSX2 expression on the AML cell lines THP-1 and KG1a
(A) Expression of SSX2 by the KG1a and THP1 AML cell lines was detected by flow cytometry analysis. The SR cell line was used as a negative control for SSX2 expression. (B) Phenotypic analysis of the THP-1 cell line shows HLA-A2 expression, as well as expression of CD33 and the costimulatory molecules CD80 and CD86. The KG1a cell line displays a similar phenotypic profile with THP1 but lacks expression of HLA-A2.
To discover if the SSX241-49.28ζ or SSX241-49.BBζ CAR T cells were functional against the AML cell lines THP1 and KG1a, EGFP-labeled tumor cells were co-cultured with CAR-T cells at an effector to tumor ratio of 0.5:1 in the absence of cytokines. After 4 days of culture, we observed that the THP1 tumor cells were eradicated from the co-cultures with T cells expressing either of the two SSX41-49 CARs but not in co-culture with UnTx T cells. As expected, the HLA-A2– KG1a tumor cells were not eliminated in the 4-day co-culture experiment, demonstrating that SSX2 peptide presentation in a major histocompatibility complex (MHC)-restricted manner is essential for the activity of the CAR-SSX41-49-expressing T cells (Figure 5). Upon stimulation with SSX2+/HLA-A2+ THP1 tumor cells, CAR-SSX41-49-expressing T cells released IFN-γ, tumor necrosis factor (TNF)-α, and interleukin (IL)-2. Figure 6A shows that SSX241-49.28ζ CAR T cells release a higher amount of IFN-γ, TNF-α, and IL-2 compared to the T cells expressing the SSX241-49.BBζ CAR (1,282 ± 23 pg/mL versus 101 ± 34 pg/mL, 668 ± 134pg/mL versus 77 ± 23pg/mL, and 267 ± 63 versus 0 pg/mL, respectively). As expected, no IFN-γ, TNF-α, or IL-2 production was detected in cultures where UnTx T cells were challenged with tumor cells. In addition, TNF-α or IL-2 were undetected, and only a minimal amount of IFN-γ was found in cultures where SSX241-49.28ζ. or SSX241-49.BBζ CAR T cells were challenged with the HLA-A2– KG1a tumor cell line (Figure 6B).
Figure 5.

T cells expressing a SSX241-49 CAR eliminate HLA-A2+/SSX2+ AML tumor cells in vitro
To evaluate the ability of SSX241-49 CAR cells to eliminate HLA-A2+/SSX2+ AML tumor cells, UnTx, SSX241-49 28ζ-, or SSX241-49 BBζ-expressing T cells were plated at 0.5:1 effector to target ratio for 4 days without cytokines added to the cultures. (A) Representative flow cytometry analysis shows residual AML tumor cells (EGFP+) in co-culture experiments. (B) Summaries of 5 co-cultures experiments are shown. Mean values are shown with error bars denoting the standard deviation (SD).
Figure 6.

SSX241-49-specific CARs T cells produce cytokines in response to HLA-A2+/SSX2+ AML tumor cells
To evaluate the ability of SSX241-49 CAR cells to produce cytokines in response to HLA-A2+/SSX2+ AML tumor cells, UnTx, SSX241-49 28ζ-, or SSX241-49 BBζ-expressing T cells were plated at 1:1 effector to target ratio. After 24 h co-culture, levels of IFN-γ, TNF-α, and IL-2 were analyzed by ELISA. Summaries of 3 co-cultures experiments are shown. Mean values are shown with error bars denoting SD. (A) Release of IFN-γ, TNF-α, and IL-2 cytokines were detected when SSX241-49-specific CARs T cells were challenged with the HLA-A2+/SSX2+ THP1 tumor cell line. (B) No cytokines were detected in culture supernatant when the same SSX241-49-specific CARs T cells were cultured with the HLA-A2–/SSX2+ KG1a cell line. As expected, no cytokines were produced by UnTx T cells irrespective of the target used. Summaries of 3 separate experiments are shown with means values and error bars that denote SD.
Discussion
The restricted expression of CTAs in immune-privileged normal tissues and their expression (natural or induced) in tumor cells make them ideal targets for the development of cell-based therapies for cancer treatment.1 SSX2, like many other CTAs, is regarded as a potential target since its expression has been observed in multiple tumor types.9 Our group evaluated an scFv that specifically targets the immunogenic SSX2 peptides, peptide 41-49, in an MHC-restricted context. Using this newly discovered scFv, we successfully generated two CARs, SSX241-49.28ζ and SSX241-49.BBζ, that were able to target the SSX241-49 epitope when expressed on HLA-A2+ tumor cells. Both CARs displayed strong killing activity against AML tumor cells in long-term co-culture experiments, and CAR-expressing T cells specifically released the IFN-γ, TNF-α, and IL-2 cytokines when stimulated with HLA-A2+/SSX2+ AML tumor cells.
Abate-Daga and collaborators20 have previously isolated a TCR that recognizes the identical SSX2 epitope in the same MHC context and demonstrated anti-tumor activity against various type of HLA-A2+/SSX2+ tumor cells. Isolation of the TCR was possible because SSX2 can naturally induce an immune response, and the presence of SSX2-specific CD8+ T cells has been observed in patients with melanoma or hepatocellular carcinoma (HCC).12,34 There are obvious advantages to by-passing the steps required for the specific activation and expansion of SSX241-49-specific cytotoxic T cells by transducing T cells with a transgenic TCR. However, the potential pairing of introduced TCR chains with endogenous TCR chains remains a concern as it may alter the specificity of the transferred TCR.35 Off-target reactivity of the selected TCR, either with or without enhanced affinity for the target, has also been observed with dramatic consequences and highlights potential safety concerns associated with the use of T cells engineered to express transgenic TCR chains.36, 37, 38 Various strategies have been developed in the last 15 years to eliminate TCR mispairing, including the addition of a cysteine residue in the α and β constant regions to promote the formation of disulfide bridges and the engineering of Vα-Vβ single chain TCR (scTv).39,40
An alternative approach for the targeting of peptides bound to the MHC (class I or class II molecules) is to develop antibodies specific for the peptide/MHC complex (pMHC complex). Such antibodies, called TCR-mimic or TCR-like antibodies, were first observed in the early 1980s and can be generated using either a hybridoma-based method or a phage display library-based method.41 The TCR-like antibodies are able to bind their targets (pMHC complexes) with higher affinity than their respective TCR counterparts and can be readily engineered into a CAR. This approach represents an attractive technology for cellular therapy and has been tested against neoantigens, minor hystocompatibility antigens (mHAgs), CTAs, and other pMHC complexes.21 The high-affinity property of TCR-like CARs is important when the targeted peptide is present at low copy numbers on the tumor cells, but off-target toxicity remains a concern as previously observed in clinical trials that utilized an affinity-matured TCR specific for MAGE-A3.37
In contrast, we have developed a novel TCR-like CAR-targeting peptide 41-49 of SSX2, an immunogenic CTA, and show that T cells transduced with this CAR construct can kill AML tumor cells in vitro with high efficacy and specificity.16 Hence, T cells armed with an SSX241-49 CAR can be applied for the treatment of HLA-A∗0201 patients with SSX2-expressing tumors, and modifiable endodomains included in the CAR constructs could provide different quantitative responses of CARs expressing T cells when exposed to the target cells. Indeed, we observed that T cells expressing the SSX241-49 CAR containing 4-1BB plus CD3ζ signaling domains were able to eliminate HLA-A2+ (but not HLA-A2–) SSX2 expressing AML tumor cells in co-culture but released lower amounts of cytokines (IFN-γ, TNF-α, and IL-2) compared to their CD28 plus CD3ζ counterparts. Given these in vitro findings it is critical to further evaluate both CAR constructs in vivo in both liquid tumor (e.g., AML) as well as solid tumor models.
The scFv described in this study has shown a difference in reactivity against homologous peptides, demonstrating a high specificity for SSX241-49 but also for SSX341-49. These two SSX homologous peptides differ by only 2 amino acids and have a similar binding affinity to HLA-A∗0201. However, SSX3 expression has not been reported in normal tissues as well as tumors and cannot be induced by a demethylation agent.9 Responses against the non-SSX and SSX homologous peptides were not detected with only a measurable response against SSX741-49 but with a difference in reactivity of 2 orders of magnitude. Careful screening of SSX2 41-49 peptide expression in HLA-A2+ normal and cancer tissues is therefore necessary for the development of a safe clinical application using TCR-like CAR-expressing T cells.
Future plans with this novel SSX241-49 CAR T cell product includes an in vivo study using mice bearing HLA A0201+ and A0201– AML tumors of variable haplotypes. Such studies will provide valuable information in terms of evaluating the efficacy and safety of our novel SSX241-49 CAR T cell product prior to clinical translation. Therefore, while SSX2 is not known to be expressed on normal HPCs, we will still need to evaluate for possible toxicity against HPCs in clonogenic assays. Because expression of SSX2 in AML tumors (regardless of the stage) can be strongly induced using demethylating agents such as decitabine and 5-azacytidine, combination therapy that includes treatment with a demethylation agent followed by SSX241-49 CAR T cells will be considered. This would be a similar approach to that already used in our current clinical trial evaluating tumor-associated antigen (TAA)-specific T cell therapies for AML (ClinicalTrials.gov: NCT002203902). Finally, we will develop a Fab/3-based antibody to discover, using immunohistochemistry (IHC) or immunofluorescence (IF), if SSX2 41-49 peptide is expressed on normal tissues using various microarrays. The expression of SSX2 on a wide range of tumor types makes it an attractive target for the application of SSX241-49 CAR T cells even beyond the AML setting, which warrants future study.
In summary, we have developed a novel TCR-like CAR-targeting SSX2, which has potential for the treatment of patients with relapsed/refractory AML without targeting normal progenitor hematopoietic stem cells, as observed with the CD123-specific CARs. Hence, we are now developing a phase I clinical trial to evaluate the safety and anti-tumor effects of this CAR T cell therapeutic.
Materials and methods
Cells and cell lines
Steady-state apheresis units were purchased from AllCells and peripheral blood mononuclear cells (PBMCs) were isolated using lymphoprep density gradient. Before cryopreserving the PBMCs, expression of HLA-A2 was assessed by flow cytometry. In this study, T cells were cultivated in T cell medium composed of 50% RPMI (HyClone, GE Healthcare), 40% Click’s media (Millipore Sigma), 10% heat-inactivated fetal bovine serum (FBS; HyClone, GE Healthcare), 2 mM Glutamax (GIBCO, Thermo Fisher Scientific), and penicillin/streptomycin (P/S; GIBCO, Thermo Fisher Scientific). The AML cell lines KG1a and THP-1 and the large cell immunoblastic lymphoma cell line SR were purchased from American Type Culture Collection (ATCC). KG1a was cultured in Iscove’s Modified Dulbecco’s Medium (IMDM; HyClone, GE Healthcare) base medium and RPMI base medium was utilized to culture the THP1 and SR cell lines, with both mediun containing 10% FBS plus P/S and 2mM Glutamax. For in vitro and in vivo experiments, KG1a and THP-1 cell lines were transduced with retroviral vector to express EGFP. The HLA-A∗0201 TAP-deficient cell line T2 was purchased from ATCC and maintained in growth culture media composed of IMDM plus 20% of FBS and 2 mM Glutamax.
Antibodies and flow cytometry analysis
Flow cytometry analysis was performed using the following antibodies: CD3-FITC (HIT3a), CD3-APC (HIT3a), CD4-APC (RPA-T4), CD8-PerCP/Cy5.5 (SK1), CD8-PE (SK1), CD62L-APC (DREG-56), CD45RA-PE/Cy7 (HI100), and CCR7-PE (G043H7) were purchased from BioLegend; CD4-PE (L200), HLA-A2-FITC (BB7.2), CD33-PE (WM53), CD33-APC (WM53), CD123-FITC (7G3), CD56-APC (B159), CD80-FITC (L307.4), and CD86-FITC (2331) were purchased from Becton Dickinson; CD34-PE (QBEnd/10) was purchased from Invitrogen. CAR expression was detected using an AffiniPure F(ab’)2 goat anti-human IgG (H+L)-Alexa Fluor 647 antibody purchased from Jackson ImmunoResearch Laboratories as well as by using the anti-CD34 (QBEND/10) antibody. Cells were acquired using a Beckman Coulter system (CytoFLEX S) and data analyzed using the FlowJo software.
Detection of SSX2 was performed by flow cytometry analysis using the polyclonal anti-SSX2 antibody (PA5-77089) and anti-rabbit PE (P-2771MP) secondary antibody, both from ThermoFisher Scientific. Briefly, SR, THP1, and KG1a tumor cells were collected, washed in PBS-1X, and viability dye (LIVE/DEAD Fixable Aqua Dead Cell Stain Kit from ThermoFisher Scientific) was added to the cell’s pellet per manufacturer recommendation along with a FcR blocking reagent. After 15-min incubation at room temperature, cells were washed twice in PBS-1X and resuspended in cold 4% paraformaldehyde solution for 15 min at 4°C. After 2 washes in Perm/Wash buffer (BD, Biosciences), cells were incubated with anti-SSX2 primary antibody at 4°C for 30 min, washed twice in Perm/Wash buffer, and incubated for 30 min with the secondary antibody. Both primary and secondary antibodies were titrated for optimal staining. Cells were then acquired using a Beckman Coulter system (CytoFLEX S) and data analyzed using the FlowJo software.
Retroviral constructs and retrovirus preparation
The scFv of Fab/3 was cloned into a retroviral SFG vector in frame with the human CD8α hinge containing the QBEnd/10-specific epitope and transmembrane domain, the CD28 or 4-1BB costimulatory intra-cellular domain, and the CD3-zeta intra-cellular signaling domain.42 The CAR containing the CD28/CD3-ζ-signaling domains was named SSX241-49 28ζ, and the SSX241-49 BBζ CAR contains the 4-1BB/CD3-ζ-signaling domains.
The generation of retroviral supernatant for T cell transduction was previously described.43 Briefly, a mixture of the Peg-Pam plasmid encoding the MoMLV gag-pol, the RDF plasmid encoding the RD114 envelop, and either the SSX241-49 28ζ or SSX241-49 BBζ plasmid were transfected in 293-T cells using GeneJuice transfection reagent (Millipore Sigma). Retroviral supernatants were collected at 48 and 72 h after transfection, passed through a 0.45 μm filter, snap frozen, and stored at −80°C for further use.
Synthetic peptides and loading onto T2 cells
The following SSX2 peptides are known to be processed by the proteasome and correspond to amino acids 41 to 49 (KASEKIFYV), 103 to 111 (RLQGISPKI), and 5 to 13 (DAFARRPTV). The SSX homologous peptides corresponding to amino acids 41 to 49 were derived from SSX1 (KYSEKISYV), SSX3 (KVSEKIVYV), SSX4 (KSSEKIVYV), SSX5 (KASEKIIYV), SSX6 (KFSEKISCV), SSX7 (KSLEKISYV), SSX8 (KYSEKISYV), SSX9 (KSSEKIIYV), and SSX10 (KASEKILYV), as well as unrelated peptides derived from IGSF22 (KESAKIFYD), and TCOF1 (KASEKILQV) were synthesized by Genemed Synthesis (San Antonio, TX, USA). Peptides were first dissolved in DMSO and further diluted in RPMI before being loaded onto T2 cells. Binding affinity of each of the peptides listed above to HLA-A∗0201 was determined using NetMHC-4.0 (http://www.cbs.dtu.dk/services/NetMHC/). Peptides loaded onto T2 cells have been previously described.44 Briefly, T2 were washed twice in serum-free culture media (CM) and resuspended at 1 × 106 per mL in serum-free CM containing 3 μg/mL of β2-microglobulin (Millipore Sigma) and various amounts (10,000 ng, 100 ng, and 1 ng or 0.01 ng per mL) of each of the peptides. After 18 h of incubation at 37°C in 5% CO2, peptide-loaded T2 cells were washed once in culture media and co-cultured with non-transduced (UnTx) or CAR-transduced T cells at a 1:1 ratio.
T cell transduction and expansion
After thawing, PBMCs were activated by plate-bound OKT3 and anti-CD28 antibodies, both at 1 μg/mL. At 24 h after activation, IL-7 at 10 ng/mL plus IL-15 at 5n g/mL were added to the activated T cell culture, and a non-treated culture 24-well plate was coated overnight at 4°C with 7 μg/mL of Retronectin. Retroviral vector supernatants were then loaded onto the Retronectin-coated plate, and T cell transduction was carried out as previously described.33 T cell cultures were fed with fresh media and cytokines every 2 to 3 days for 14 days.
Co-culture experiments and cytokine release measurement
In a 96-well plate, 1 × 105 tumor cells expressing EGFP were co-cultured with 0.5 × 105 T cells (E:T ratio of 0.5:1) in T cell medium without exogenous cytokines added. After 4 days of co-culture, cells were collected from the wells, and the contents of T cells (CD3+) and tumor cells (EGFP) were measured by fluorescence-activated cell sorting (FACS) analysis. UnTx and SSX241-49 CAR-expressing T cells were tested for specific response to the KG1a or THP-1 tumor cells or to T2 cells loaded with peptide by cytokine release assay. Briefly, T cells were cultured against AML tumor cells at a 0.5:1 E:T ratio or against peptide-loaded T2 cells at a 1:1 E:T ratio in T cell media not supplemented with IL-7 and IL-15. Culture supernatants were collected 24 h after initiation of the co-culture and stored at −80°C until processing for IFN-γ, TNF-α, and IL-2 content by ELISA (R&D Systems) according to manufacturer’s instructions.
Statistical analysis
To determine statistical significance of the observed differences between experimental groups, Student’s t test was used. Measurements were presented as mean ± SD as noted in the figure legends. One-way and two-way ANOVA were used for multiple group comparisons to determine statistical significance. p values < 0.05 were considered statistically significant. Statistical analysis was performed using GraphPad Prism software.
Acknowledgments
This work was supported in part by a grant from the National Institutes of Health (NIH) National Cancer Institute (P01 CA148600 and P01 CA225618 to C.M.B.), the Board of Visitors of the Children’s National Health System, and The Katzen Foundation. The antigen-binding fragment (Fab/3) was a kind gift from Dr. Michael Pfreundschuh and colleagues from the Saarland University Medical School.
Author contributions
E.Y. and C.M.B. developed the concept, designed, analyzed, and interpreted the data, and wrote the manuscript; S.V.P., K.T., H.D., and P.B.B. performed experiments; S.R. performed experiments and participated in analyzing the data and writing the manuscript.
Declaration of interests
C.M.B. has stock or ownership in Cabaletta Bio, Catamaran Bio, Repertoire Immune Medicines, Mana Therapeutics, and Neximmune. E.Y, C.M.B, S.R., and S.V.P have filed a patent application based on the findings in this paper.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2021.09.008.
Supplemental information
References
- 1.Salmaninejad A., Zamani M.R., Pourvahedi M., Golchehre Z., Hosseini Bereshneh A., Rezaei N. Cancer/Testis Antigens: Expression, Regulation, Tumor Invasion, and Use in Immunotherapy of Cancers. Immunol. Invest. 2016;45:619–640. doi: 10.1080/08820139.2016.1197241. [DOI] [PubMed] [Google Scholar]
- 2.Costa F.F., Le Blanc K., Brodin B. Concise review: cancer/testis antigens, stem cells, and cancer. Stem Cells. 2007;25:707–711. doi: 10.1634/stemcells.2006-0469. [DOI] [PubMed] [Google Scholar]
- 3.Heninger E., Krueger T.E.G., Thiede S.M., Sperger J.M., Byers B.L., Kircher M.R., Kosoff D., Yang B., Jarrard D.F., McNeel D.G., Lang J.M. Inducible expression of cancer-testis antigens in human prostate cancer. Oncotarget. 2016;7:84359–84374. doi: 10.18632/oncotarget.12711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thomas R., Al-Khadairi G., Roelands J., Hendrickx W., Dermime S., Bedognetti D., Decock J. NY-ESO-1 Based Immunotherapy of Cancer: Current Perspectives. Front. Immunol. 2018;9:947. doi: 10.3389/fimmu.2018.00947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gordeeva O. Cancer-testis antigens: Unique cancer stem cell biomarkers and targets for cancer therapy. Semin. Cancer Biol. 2018;53:75–89. doi: 10.1016/j.semcancer.2018.08.006. [DOI] [PubMed] [Google Scholar]
- 6.Chi Soh J.E., Abu N., Jamal R. The potential immune-eliciting cancer testis antigens in colorectal cancer. Immunotherapy. 2018;10:1093–1104. doi: 10.2217/imt-2018-0044. [DOI] [PubMed] [Google Scholar]
- 7.Shires K., Van Wyk T. The role of Cancer/Testis Antigens in Multiple Myeloma pathogenesis and their application in disease monitoring and therapy. Crit. Rev. Oncol. Hematol. 2018;132:17–26. doi: 10.1016/j.critrevonc.2018.09.010. [DOI] [PubMed] [Google Scholar]
- 8.Gure A.O., Türeci O., Sahin U., Tsang S., Scanlan M.J., Jäger E., Knuth A., Pfreundschuh M., Old L.J., Chen Y.T. SSX: a multigene family with several members transcribed in normal testis and human cancer. Int. J. Cancer. 1997;72:965–971. doi: 10.1002/(sici)1097-0215(19970917)72:6<965::aid-ijc8>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 9.Smith H.A., McNeel D.G. The SSX family of cancer-testis antigens as target proteins for tumor therapy. Clin. Dev. Immunol. 2010;2010:150591. doi: 10.1155/2010/150591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crew A.J., Clark J., Fisher C., Gill S., Grimer R., Chand A., Shipley J., Gusterson B.A., Cooper C.S. Fusion of SYT to two genes, SSX1 and SSX2, encoding proteins with homology to the Kruppel-associated box in human synovial sarcoma. EMBO J. 1995;14:2333–2340. doi: 10.1002/j.1460-2075.1995.tb07228.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Türeci O., Sahin U., Schobert I., Koslowski M., Scmitt H., Schild H.J., Stenner F., Seitz G., Rammensee H.G., Pfreundschuh M. The SSX-2 gene, which is involved in the t(X;18) translocation of synovial sarcomas, codes for the human tumor antigen HOM-MEL-40. Cancer Res. 1996;56:4766–4772. [PubMed] [Google Scholar]
- 12.Ayyoub M., Rimoldi D., Guillaume P., Romero P., Cerottini J.C., Valmori D., Speiser D. Tumor-reactive, SSX-2-specific CD8+ T cells are selectively expanded during immune responses to antigen-expressing tumors in melanoma patients. Cancer Res. 2003;63:5601–5606. [PubMed] [Google Scholar]
- 13.Denniss F.A.K., Breslin A., Ingram W., Hardwick N.R., Mufti G.J., Guinn B.A. The leukaemia-associated antigen, SSX2IP, is expressed during mitosis on the surface of myeloid leukaemia cells. Br. J. Haematol. 2007;138:668–669. doi: 10.1111/j.1365-2141.2007.06706.x. [DOI] [PubMed] [Google Scholar]
- 14.Guinn B.A., Bullinger L., Thomas N.S.B., Mills K.I., Greiner J. SSX2IP expression in acute myeloid leukaemia: an association with mitotic spindle failure in t(8;21), and cell cycle in t(15;17) patients. Br. J. Haematol. 2008;140:250–251. doi: 10.1111/j.1365-2141.2007.06892.x. [DOI] [PubMed] [Google Scholar]
- 15.Atanackovic D., Luetkens T., Kloth B., Fuchs G., Cao Y., Hildebrandt Y., Meyer S., Bartels K., Reinhard H., Lajmi N. Cancer-testis antigen expression and its epigenetic modulation in acute myeloid leukemia. Am. J. Hematol. 2011;86:918–922. doi: 10.1002/ajh.22141. [DOI] [PubMed] [Google Scholar]
- 16.Smith H.A., McNeel D.G. Vaccines targeting the cancer-testis antigen SSX-2 elicit HLA-A2 epitope-specific cytolytic T cells. J. Immunother. 2011;34:569–580. doi: 10.1097/CJI.0b013e31822b5b1d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smith H.A., Rekoske B.T., McNeel D.G. DNA vaccines encoding altered peptide ligands for SSX2 enhance epitope-specific CD8+ T-cell immune responses. Vaccine. 2014;32:1707–1715. doi: 10.1016/j.vaccine.2014.01.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gerdemann U., Katari U., Christin A.S., Cruz C.R., Tripic T., Rousseau A., Gottschalk S.M., Savoldo B., Vera J.F., Heslop H.E. Cytotoxic T lymphocytes simultaneously targeting multiple tumor-associated antigens to treat EBV negative lymphoma. Mol. Ther. 2011;19:2258–2268. doi: 10.1038/mt.2011.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lulla P.D., Tzannou I., Vasileiou S., Carrum G., Ramos C.A., Kamble R., Wang T., Wu M., Bilgi M., Gee A.P. The safety and clinical effects of administering a multiantigen-targeted T cell therapy to patients with multiple myeloma. Sci. Transl. Med. 2020;12:eaaz3339. doi: 10.1126/scitranslmed.aaz3339. [DOI] [PubMed] [Google Scholar]
- 20.Abate-Daga D., Speiser D.E., Chinnasamy N., Zheng Z., Xu H., Feldman S.A., Rosenberg S.A., Morgan R.A. Development of a T cell receptor targeting an HLA-A∗0201 restricted epitope from the cancer-testis antigen SSX2 for adoptive immunotherapy of cancer. PLoS ONE. 2014;9:e93321. doi: 10.1371/journal.pone.0093321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Akatsuka Y. TCR-Like CAR-T Cells Targeting MHC-Bound Minor Histocompatibility Antigens. Front. Immunol. 2020;11:257. doi: 10.3389/fimmu.2020.00257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.De Kouchkovsky I., Abdul-Hay M. ‘Acute myeloid leukemia: a comprehensive review and 2016 update’. Blood Cancer J. 2016;6:e441. doi: 10.1038/bcj.2016.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.DiNardo C.D., Cortes J.E. Mutations in AML: prognostic and therapeutic implications. Hematology (Am. Soc. Hematol. Educ. Program) 2016;2016:348–355. doi: 10.1182/asheducation-2016.1.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.DiNardo C.D., Wei A.H. How I treat acute myeloid leukemia in the era of new drugs. Blood. 2020;135:85–96. doi: 10.1182/blood.2019001239. [DOI] [PubMed] [Google Scholar]
- 25.Pehlivan K.C., Duncan B.B., Lee D.W. CAR-T Cell Therapy for Acute Lymphoblastic Leukemia: Transforming the Treatment of Relapsed and Refractory Disease. Curr. Hematol. Malig. Rep. 2018;13:396–406. doi: 10.1007/s11899-018-0470-x. [DOI] [PubMed] [Google Scholar]
- 26.Baroni M.L., Sanchez Martinez D., Gutierrez Aguera F., Roca Ho H., Castella M., Zanetti S.R., Velasco Hernandez T., Diaz de la Guardia R., Castaño J., Anguita E. 41BB-based and CD28-based CD123-redirected T-cells ablate human normal hematopoiesis in vivo. J. Immunother. Cancer. 2020;8:e000845. doi: 10.1136/jitc-2020-000845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Muñoz L., Nomdedéu J.F., López O., Carnicer M.J., Bellido M., Aventín A., Brunet S., Sierra J. Interleukin-3 receptor alpha chain (CD123) is widely expressed in hematologic malignancies. Haematologica. 2001;86:1261–1269. [PubMed] [Google Scholar]
- 28.Navada S.C., Steinmann J., Lübbert M., Silverman L.R. Clinical development of demethylating agents in hematology. J. Clin. Invest. 2014;124:40–46. doi: 10.1172/JCI69739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Almstedt M., Blagitko-Dorfs N., Duque-Afonso J., Karbach J., Pfeifer D., Jäger E., Lübbert M. The DNA demethylating agent 5-aza-2′-deoxycytidine induces expression of NY-ESO-1 and other cancer/testis antigens in myeloid leukemia cells. Leuk. Res. 2010;34:899–905. doi: 10.1016/j.leukres.2010.02.004. [DOI] [PubMed] [Google Scholar]
- 30.Shen Y., Yang X., Dong N., Xie X., Bai X., Shi Y. Generation and selection of immunized Fab phage display library against human B cell lymphoma. Cell Res. 2007;17:650–660. doi: 10.1038/cr.2007.57. [DOI] [PubMed] [Google Scholar]
- 31.Giudicelli V., Brochet X., Lefranc M.-P. IMGT/V-QUEST: IMGT standardized analysis of the immunoglobulin (IG) and T cell receptor (TR) nucleotide sequences. Cold Spring Harb. Protoc. 2011;2011:695–715. doi: 10.1101/pdb.prot5633. [DOI] [PubMed] [Google Scholar]
- 32.Du H., Hirabayashi K., Ahn S., Kren N.P., Montgomery S.A., Wang X., Tiruthani K., Mirlekar B., Michaud D., Greene K. Antitumor Responses in the Absence of Toxicity in Solid Tumors by Targeting B7-H3 via Chimeric Antigen Receptor T Cells. Cancer Cell. 2019;35:221–237.e8. doi: 10.1016/j.ccell.2019.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhao W., Sher X. Systematically benchmarking peptide-MHC binding predictors: From synthetic to naturally processed epitopes. PLoS Comput. Biol. 2018;14:e1006457. doi: 10.1371/journal.pcbi.1006457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bricard G., Bouzourene H., Martinet O., Rimoldi D., Halkic N., Gillet M., Chaubert P., Macdonald H.R., Romero P., Cerottini J.C., Speiser D.E. Naturally acquired MAGE-A10- and SSX-2-specific CD8+ T cell responses in patients with hepatocellular carcinoma. J. Immunol. 2005;174:1709–1716. doi: 10.4049/jimmunol.174.3.1709. [DOI] [PubMed] [Google Scholar]
- 35.van Loenen M.M., de Boer R., Amir A.L., Hagedoorn R.S., Volbeda G.L., Willemze R., van Rood J.J., Falkenburg J.H.F., Heemskerk M.H.M. Mixed T cell receptor dimers harbor potentially harmful neoreactivity. Proc. Natl. Acad. Sci. USA. 2010;107:10972–10977. doi: 10.1073/pnas.1005802107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cameron B.J., Gerry A.B., Dukes J., Harper J.V., Kannan V., Bianchi F.C., Grand F., Brewer J.E., Gupta M., Plesa G. Identification of a Titin-derived HLA-A1-presented peptide as a cross-reactive target for engineered MAGE A3-directed T cells. Sci. Transl. Med. 2013;5:197ra103. doi: 10.1126/scitranslmed.3006034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Morgan R.A., Chinnasamy N., Abate-Daga D., Gros A., Robbins P.F., Zheng Z., Dudley M.E., Feldman S.A., Yang J.C., Sherry R.M. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J. Immunother. 2013;36:133–151. doi: 10.1097/CJI.0b013e3182829903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van den Berg J.H., Gomez-Eerland R., van de Wiel B., Hulshoff L., van den Broek D., Bins A., Tan H.L., Harper J.V., Hassan N.J., Jakobsen B.K. Case report of a fatal serious adverse event upon administration of T cells transduced with a MART-1-specific T-cell receptor. Mol. Ther. 2015;23:1541–1550. doi: 10.1038/mt.2015.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kuball J., Dossett M.L., Wolfl M., Ho W.Y., Voss R.-H., Fowler C., Greenberg P.D. Facilitating matched pairing and expression of TCR chains introduced into human T cells. Blood. 2007;109:2331–2338. doi: 10.1182/blood-2006-05-023069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aggen D.H., Chervin A.S., Schmitt T.M., Engels B., Stone J.D., Richman S.A., Piepenbrink K.H., Baker B.M., Greenberg P.D., Schreiber H., Kranz D.M. Single-chain VαVβ T-cell receptors function without mispairing with endogenous TCR chains. Gene Ther. 2012;19:365–374. doi: 10.1038/gt.2011.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wylie D.E., Sherman L.A., Klinman N.R. Participation of the major histocompatibility complex in antibody recognition of viral antigens expressed on infected cells. J. Exp. Med. 1982;155:403–414. doi: 10.1084/jem.155.2.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Philip B., Kokalaki E., Mekkaoui L., Thomas S., Straathof K., Flutter B., Marin V., Marafioti T., Chakraverty R., Linch D. A highly compact epitope-based marker/suicide gene for easier and safer T-cell therapy. Blood. 2014;124:1277–1287. doi: 10.1182/blood-2014-01-545020. [DOI] [PubMed] [Google Scholar]
- 43.Yvon E., Del Vecchio M., Savoldo B., Hoyos V., Dutour A., Anichini A., Dotti G., Brenner M.K. Immunotherapy of metastatic melanoma using genetically engineered GD2-specific T cells. Clin. Cancer Res. 2009;15:5852–5860. doi: 10.1158/1078-0432.CCR-08-3163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Molldrem J., Dermime S., Parker K., Jiang Y.Z., Mavroudis D., Hensel N., Fukushima P., Barrett A.J. Targeted T-cell therapy for human leukemia: cytotoxic T lymphocytes specific for a peptide derived from proteinase 3 preferentially lyse human myeloid leukemia cells. Blood. 1996;88:2450–2457. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.