

Abstract
Objectives
During chronic human immunodeficiency virus (HIV)‐1 infection, inhibitory molecules upregulated on lymphocytes contribute to effector cell dysfunction and immune exhaustion. People living with HIV (PLWH) are at greater risk for age‐related morbidities, an issue magnified by human cytomegalovirus (CMV) coinfection. As CMV infection modifies natural killer (NK) cell properties and NK cells contribute to protection against HIV‐1 infection, we considered the role of T‐cell immunoreceptor with immunoglobulin and intracellular tyrosine inhibitory motif domains (TIGIT) in NK cell‐based HIV‐1 immunotherapy and elimination strategies.
Methods
We measured TIGIT expression on immune cell subsets of 95 PLWH and assessed its impact on NK cell function, including elimination of autologous CD4+ T cells infected through reactivation of endogenous HIV‐1.
Results
TIGIT was expressed on CD4+ T cells, CD8+ T cells and NK cells from PLWH. Although TIGIT levels on T cells correlated with HIV‐1 disease progression, the extent of TIGIT expression on NK cells more closely paralleled adaptation to CMV. TIGIT interacts with its predominant ligand, poliovirus receptor (PVR), to inhibit effector cell functions. Circulating CD4+ T cells from PLWH more frequently expressed PVR than HIV‐seronegative controls, and PVR expression was enriched in CD4+ T cells replicating HIV‐1 ex vivo. Treatment with anti‐TIGIT monoclonal antibodies increased NK cell HIV‐1‐specific antibody‐dependent cytotoxicity in vitro and ex vivo.
Conclusion
Blocking TIGIT may be an effective strategy to invigorate antibody‐dependent NK cell activity against HIV‐1 activated in cellular reservoirs for cure or treatment strategies.
Keywords: ADCC, checkpoint inhibitor, CMV, HIV‐1, NK cells, PVR, TIGIT
In this study, we found positive correlations between the frequency of TIGIT expression on natural killer (NK) cells from people living with human immunodeficiency virus (HIV)‐1 and markers of NK cell adaptation to cytomegalovirus infection. The main ligand for TIGIT, poliovirus receptor (PVR), was expressed on circulating CD4+ T cells of people living with HIV‐1 and its expression increased with ex vivo reactivation of HIV‐1 replication. Preventing NK cell TIGIT interaction with PVR augments NK cell effector functions against autologous HIV‐1 infected CD4+ T cells.
Introduction
Chronic viral infection imposes a persistent burden on human health. Human immunodeficiency virus (HIV)‐1 remains a pandemic, despite tremendous improvements to combination antiretroviral therapies (cARTs) that have extended the health and lifespan of people living with HIV (PLWH). Active HIV‐1 infection drives immune dysfunction with generalised T‐cell and natural killer (NK) cell exhaustion in untreated PLWH. This dysfunctional immune phenotype is punctuated by increased expression of inhibitory immune checkpoint receptors, including PD‐1, CTLA‐4, TIM‐3 and LAG‐3. 1 , 2 , 3 Early intervention with cART attenuates this expression, but levels remain elevated compared with people not living with HIV. 3
In this context, some checkpoint inhibitors currently used in cancer therapy have potential relevance in controlling viral infections, reducing viral load and revitalising host immunity. T‐cell immunoreceptor with immunoglobulin (Ig) and intracellular tyrosine inhibitory motif domains (TIGIT) is differentially expressed on T cells and NK cells and negatively regulates effector function when engaged by its ligands, PVR (CD155) or PVRL2 (CD112). 4 , 5 , 6 These ligands can also be recognised by killer cell Ig‐like receptor (KIR)2DL5 and members of the nectin and nectin‐like family of receptors that include DNAM‐1 (CD226), TACTILE (CD96) and PVR‐related Ig domain (PVRIG). 7 , 8 , 9 , 10 Broad dysregulation of TIGIT expression on CD4+ and CD8+ T cells in HIV‐1 infection has been extensively noted; however, a role for TIGIT in modulating NK cell function in HIV‐1 infection remains controversial. 11 , 12 , 13 , 14 , 15
Natural killer cells are among the first cells to respond to virus infection and as such can contribute to control of HIV‐1. 16 , 17 , 18 Natural killer cell function is scrupulously regulated through aggregate signals transmitted from interactions between distinct cellular and/or foreign ligands and activating and/or inhibitory receptors. Ligands can include self, altered and induced self, virus‐encoded proteins and IgG antibodies bound to cells (reviewed in Kielczewska et al., 19 Moretta et al. 20 and Pegram et al. 21 ). NK cells recognise cell‐bound IgG antibodies specifically and independently through interactions with CD16 (FcγRIIIa) receptors and transmit signals through FcRγ and CD3ζ to perform antibody‐dependent cell‐mediated cytotoxicity (ADCC). 22 In contrast, natural cytotoxicity is mediated by a wide array of activating receptors including receptors directly interacting with PVR (e.g. DNAM‐1) or altered self (e.g. NKG2D). 23 , 24 , 25 , 26 Restraint over these mechanisms is imposed through inhibitory receptors, including inhibitory KIR, NKG2A and TIGIT. 27 , 28 , 29 , 30 , 31
Previous reports outlined a role for HIV‐1‐based modulation of TIGIT expression on both CD8+ T cells and NK cells with NK cell TIGIT levels increasing with HIV‐1 disease progression, as measured by CD4+ T cell nadir. 12 , 13 , 14 Dysregulated NK cell TIGIT expression in the context of chronic infection suggests targeting TIGIT could be an attractive option for HIV‐1 immunotherapy and cure strategies; however, whether monoclonal antibodies (mAbs) can improve effector function for TIGIT‐expressing NK cells and enhance antiviral responses in vivo remains unclear. 13 , 14 , 32 Moreover, inflammation, immune dysfunction and immune senescence in PLWH are further accentuated in relation to inflated immune responses against human cytomegalovirus (CMV). 33 The high worldwide prevalence of CMV (40–100% seroprevalence) increases with ageing and is especially high in PLWH (80–100% seroprevalence), with the immune deficit caused by HIV‐1 infection allowing a higher frequency of CMV reactivation in PLWH. 34 , 35 , 36 , 37
Our conventional baseline NK cell receptor repertoire is distorted by CMV infection and the subsequent immune response, generating a collection of NK cells with distinct phenotypic features. 38 Large fractions of the NK cell repertoire of PLWH coinfected with CMV commonly express CD57 and NKG2C together with CD16 receptors that have lost the signalling adaptor subunit FcRγ. 39 , 40 Given the association of CMV with increased inflammation, skewed T‐cell responses, immune senescence and exaggerated NK cell adaptation, CMV coinfection must be considered as a key factor modulating the immune compartment in HIV‐1 infection. 38 , 39 , 41 , 42
Therefore, we addressed whether alterations in NK cell TIGIT expression impact HIV‐1‐specific functions. We measured natural and antibody‐dependent cytotoxicity of NK cells from PLWH against an in vitro HIV‐1‐infected CD4+ T‐cell line in the presence and absence of a blocking anti‐TIGIT mAb and observed that TIGIT blockade consistently potentiated HIV‐1‐specific NK cell responses. Although activated CD4+ T cells upregulate PVR expression, the extent of its modulation in active HIV‐1 infection remains unresolved. 15 , 25 , 43 , 44 , 45 , 46 Here, we demonstrated that PVR is inducible and enriched on endogenous HIV‐1 antigen‐positive CD4+ T cells expanded from PLWH. When HIV‐1 replication was activated in autologous CD4+ T cells ex vivo, NK cell effector responses against the infected cells increased with inclusion of anti‐TIGIT blocking mAb. These data indicate that therapeutic antibodies preventing TIGIT–PVR interactions can benefit HIV‐1 treatment strategies and increase cell‐mediated effector responses against HIV‐1‐infected cells.
Results
TIGIT expression on immune cells from newborns, HIV‐seronegative persons and PLWH
Widespread expression of TIGIT on immune cells makes it an attractive potential target to incorporate into HIV‐1 treatment strategies. We applied an immune cell phenotyping panel to determine the extent TIGIT was expressed on immune cell subsets of 95 PLWH attending the Provincial HIV Clinic in St. John's, Newfoundland and Labrador. All participants were receiving cART, and our selection was representative of the Provincial HIV Cohort of 188 individuals (2019). Their general demographics are outlined in Table 1, and our flow cytometry gating strategy is depicted in Figure 1a. TIGIT was moderately expressed on CD4+ T cells [median (IQR) 22.0% (9.4%)] and, to a greater extent, on both CD8+ T cells [44.9% (21.3%)] and NK cells [73.6% (18.2%); Figure 1b]. To address the significance of TIGIT expression on these immune cell subsets and evaluate whether levels relate to HIV‐1, ‘immune experience’ or other chronic viral infections, we also analysed nascent immune cells [cord blood mononuclear cells (CBMC), n = 15] and peripheral blood mononuclear cells (PBMC) from a cohort of HIV‐seronegative individuals (n = 26). The latter group was matched to the PLWH cohort for age, sex and CMV serostatus (Table 1).
Table 1.
General cohort demographics
| Characteristic | PLWH | HIV‐seronegative |
|---|---|---|
| n [undetectable HIV‐1 a (%)] | 95 (84.2%) | 26 (N/A) |
| Age [years, mean (SD)] | 51.6 (10.8) | 51.1 (13.1) |
| Male [n (%)] | 72 (75.8%) | 19 (73.1%) |
| Years of HIV‐1 infection [mean (SD)] | 15.8 (10.3) | N/A |
| CD4 nadir b [cells μL−1, median (IQR)] | 211 (372.8) | N/A |
| HBV‐positive [n (%)] | 4 (4.2%) | 0 |
| HCV‐positive [n (%)] | 4 (4.2%) | 0 |
| CMV‐positive [n (%)] | 77 (81.0%) | 21 (80.8%) |
| NKG2Cnull [n (%)] | 4 (4.2%) | 1 (3.8%) |
< 40 copies HIV RNA per mL of plasma at time of testing.
Lowest recorded CD4+ T‐cell count per µL of peripheral blood.
Figure 1.
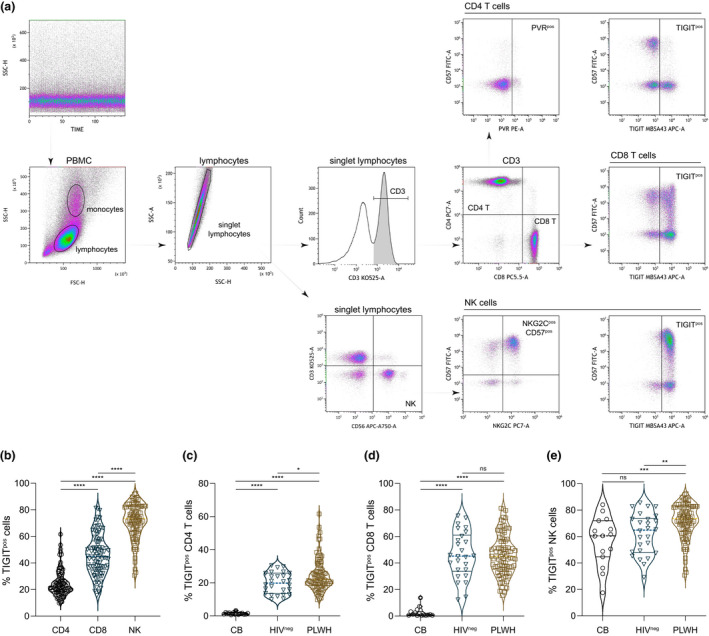
TIGIT expression on immune cell subsets from cord blood and peripheral blood of PLWH and HIV‐seronegative adults. (a) Quality of flow cytometry data acquisition was monitored by side scatter over time. Lymphocytes were identified by scatter characteristics and doublet exclusion. T cells were identified as CD3+ lymphocytes and distinguished by either CD4 or CD8 expression, and NK cells were CD3‐CD56+ lymphocytes. Subsets of CD4+ T cells, CD8+ T cells and NK cells were further demarcated by TIGIT and CD57 expression, and CD4+ T cells were analysed for PVR expression and NK cells for NKG2C and CD57 expression. (b) Compiled TIGIT expression levels on CD4+ T cells, CD8+ T cells and NK cells from PLWH (n = 95). Friedman test ****P < 0.0001. Expression of TIGIT on (c) CD4+ T cells, (d) CD8+ T cells and (e) NK cells from nascent (CB, n = 15), HIV‐seronegative (HIVneg, n = 26) and PLWH (n = 95) was compared. Kruskal–Wallis test *P = 0.0408 **P = 0.0082 ***P = 0.0008 ****P < 0.0001. Horizontal lines bisecting groups represent median with IQR.
Comparison of TIGIT expression on CD4+ T cells (Figure 1c), CD8+ T cells (Figure 1d) and NK cells (Figure 1e) between the three groups indicated significantly higher frequency TIGIT expression on CD4+ T cells of PLWH than on either nascent CD4+ T cells or those from HIV‐seronegative participants (Figure 1c). General ageing/immune experience also elevates TIGIT levels on CD8+ T cells from HIV‐seronegative individuals in comparison to CBMC (Figure 1d). However, there was no significant difference in TIGIT expression on CD8+ T cells from cART‐treated PLWH and the age‐matched HIV‐seronegative group (Figure 1d). In contrast, TIGIT expression was elevated on NK cells from PLWH compared with the NK cells of both nascent controls and the matched HIV‐seronegative participants (Figure 1e). Although TIGIT expression on T cells from HIV‐seronegative adults was elevated compared with cord blood T cells, there was no significant difference in TIGIT expression between cord blood NK cells and NK cells from HIV‐seronegative adults (Figure 1e).
Contribution of HIV‐1 and CMV to CD4+ T‐cell, CD8+ T‐cell and NK cell TIGIT expression
To further determine how increased levels of TIGIT relate to HIV‐1 infection, we assessed correlation between TIGIT expression on each immune cell subtype and CD4+ T cell nadir, a marker for the historical extent of HIV‐1 disease progression. Many participants with lower nadir CD4+ T‐cell counts had higher levels of TIGIT expression on both CD4+ T cells (Figure 2a) and CD8+ T cells (Figure 2b). Although significant, these correlations were relatively weak, and in some instances, participants with high historical CD4+ T‐cell counts had large fractions of CD4+ T cells or CD8+ T cells expressing TIGIT. In contrast, and despite TIGIT being elevated on NK cells from PLWH, there was no significant correlation between NK cell TIGIT expression and CD4+ T‐cell nadir (Figure 2c). Although chronic HIV‐1 seems to contribute to increased levels of TIGIT on CD4+ T cells and both CD4+ T‐cell and CD8+ T‐cell TIGIT expression correlates with HIV‐1 disease progression, other cofactors appear to drive elevated NK cell TIGIT expression in PLWH.
Figure 2.
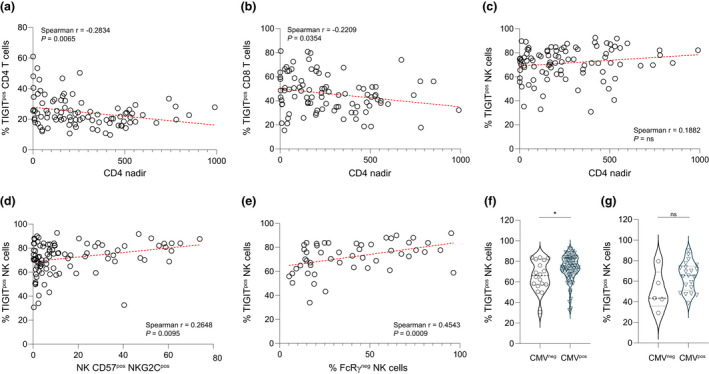
TIGIT expression on immune cell subsets and impact of HIV‐1 and CMV infection. Correlation of TIGIT expression levels on (a) CD4+ T cells, (b) CD8+ T cells and (c) NK cells with CD4+ T‐cell nadir was evaluated for the PLWH cohort (n = 95) to gauge linkage with HIV‐1 disease progression. The extent of TIGIT expression on NK cells was plotted against the percentage of NK cells that were (d) CD57+NKG2C+ (n = 95) or (e) FcRγ‐ (n = 50) and correlation assessed. Individuals within the (f) PLWH or (g) HIV‐seronegative cohort were grouped based on CMV serostatus and NK cell TIGIT expression for those who were CMV‐seronegative (PLWH n = 18; HIV‐seronegative n = 5) was compared with those who were CMV‐seropositive (PLWH n = 77; HIV‐seronegative n = 21). Mann–Whitney U‐test *P = 0.0256. Horizontal lines bisecting groups in f and g represent median with IQR.
Given the high worldwide prevalence of PLWH coinfected with CMV and considering 81% of the PLWH in this study were CMV‐seropositive, we investigated whether known markers of NK cell adaptation to CMV infection related to TIGIT expression. TIGIT was expressed at higher levels on CD57+ NKG2C+ NK cells and on NK cells lacking FcRγ in PLWH (Figure 2d and e). 14 In addition, a higher percentage of NK cells expressed TIGIT in CMV‐seropositive than in CMV‐seronegative PLWH (Figure 2f). There was no significant difference in NK cell TIGIT expression between those who were CMV‐seronegative vs CMV‐seropositive within either the HIV‐seronegative cohort (n = 21) used as controls for the functional studies (Figure 2g) or a group (n = 31) of age‐ and sex‐matched HIV‐seronegative participants expanded to include more CMV‐seronegative individuals for statistical testing (Supplementary figure 1a). In contrast to NK cells, the CMV serostatus of PLWH had no impact on CD4+ T‐cell (Supplementary figure 1b) or CD8+ T‐cell (Supplementary figure 1c) TIGIT expression. The proclivity of CMV infection to accentuate inflammation and immune dysfunction in PLWH may be a cofactor driving increased levels of TIGIT expression on NK cells of PLWH.
PVR expression on circulating CD4+ T cells from PLWH
Interaction between TIGIT and its ligands inhibits immune cell activation. As TIGIT expression is dysregulated in PLWH, we assessed expression of PVR, the predominant ligand for TIGIT, on CD4+ T cells within each study group and compared levels between the groups. PVR was present at low levels on resting peripheral blood CD4+ T cells, with higher expression on CD4+ T cells from PLWH than from other groups (Figure 3a). 12 There was no significant difference in PVR expression on CD4+ T cells from nascent and age‐matched HIV‐seronegative groups (Figure 3a). PLWH who had a higher frequency of CD4+ T cells expressing PVR generally had lower historical CD4+ T‐cell counts (CD4 nadir), linking HIV‐1 disease progression with frequency of circulating CD4+ T cells expressing PVR (Figure 3b). 13 Mirroring the increased TIGIT expression on NK cells from CMV‐seropositive PLWH noted in Figure 2f, CD4+ T cells from CMV‐seropositive PLWH expressed higher levels of PVR than CD4+ T cells of their CMV‐seronegative counterparts (Figure 3c). Although CD4+ T‐cell PVR expression directly correlated with the extent to which TIGIT was expressed on CD4+ T cells (Figure 3d), we did not find similar associations between elevated numbers of PVR+ CD4+ T cells and either TIGIT+ CD8+ T cells (Supplementary figure 1d) or TIGIT+ NK cells (Supplementary figure 1e). These data shape a perspective that progression of HIV‐1 infection contributes to TIGIT expression on CD4+ T cells, that elevated CD8+ T‐cell TIGIT can return to similar levels as in HIV‐seronegative individuals with effective treatment and that other factors underlie increased TIGIT expression on NK cells from PLWH. 12
Figure 3.
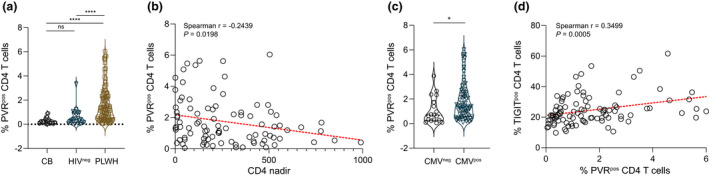
PVR expression on circulating CD4+ T cells from PLWH and HIV‐seronegative controls. (a) The extent of PVR expression on CD4+ T cells from newborns (CB), HIV‐seronegative (HIVneg) adults or PLWH was measured and compared. Kruskal–Wallis test ****P < 0.0001. (b) Correlation between PVR expression on CD4+ T cells from PLWH (n = 95) and HIV‐1 disease progression (CD4 nadir) was assessed and (c) expression of PVR on CD4+ T cells from CMV‐seronegative (n = 18) or CMV‐seropositive (n = 77) PLWH contrasted. Mann–Whitney U‐test *P = 0.0290. (d) Correlation between CD4+ T‐cell TIGIT and PVR expression is depicted. Horizontal lines bisecting groups in a and c represent median with IQR.
Effect of TIGIT blockade on NK cell cytotoxicity against conventional targets
TIGIT is widely expressed on multiple immune cell subsets and has a role in regulating their function, 4 , 5 , 6 , 47 , 48 , 49 yet limited information on its functional role in the context of HIV‐1 infection exists and interpretation of that which is available differs. 12 , 13 , 14 To investigate cytotoxic activity of NK cells from PLWH in the context of TIGIT expression, we selected a subset (n = 26) of the PLWH cohort (n = 95) for functional experiments. These representative individuals were selected based on age, sex, CMV serostatus, years of HIV‐1 infection and CD4+ T‐cell nadir (Table 2). All participants were on cART; however, samples from two individuals used for functional studies were collected when they had detectable viral loads (> 40 copies HIV‐1 RNA mL−1 blood). We considered NK cell TIGIT expression in the selection to ensure the range of TIGIT expression within the entire cohort (Figure 1b; range 43.8–89.7%) was represented within the group studied functionally (Figure 4a, range 49.8–90.0%). PLWH with hepatitis B virus (HBV) or hepatitis C virus (HCV) coinfection, CMV‐seropositive participants with no functional NKG2C gene (NKG2Cnull) and PLWH with < 10% baseline NK cell cytotoxicity against conventional K562 target cells were excluded from functional studies.
Table 2.
Characteristics of PLWH included in functional studies
| Characteristic | PLWH |
|---|---|
| n [undetectable HIV‐1 a (%)] | 26 (92.3%) |
| Age [years, mean (SD)] | 55.4 (9.7) |
| Male [n (%)] | 21 (80.8%) |
| Years of HIV‐1 [years, mean (SD)] | 17 (9.1) |
| CD4 nadir b [cells μL−1, median (IQR)] | 188 (282) |
| CMV‐positive [n (%)] | 16 (61.5%) |
< 40 copies HIV RNA per mL of plasma at time of testing.
Lowest recorded CD4+ T‐cell count per µL of peripheral blood.
Figure 4.
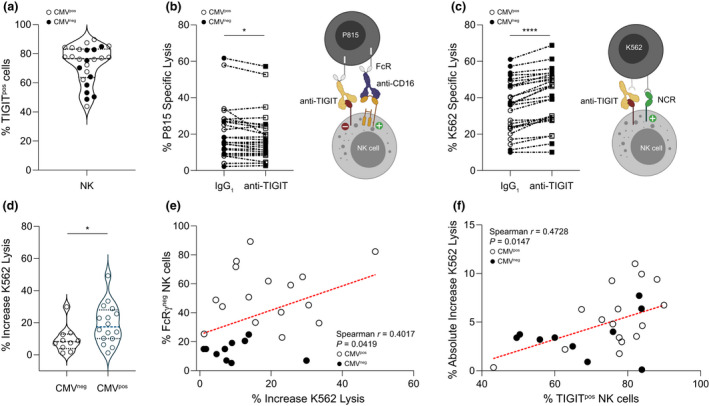
Impact of TIGIT engagement on NK cell CD16‐mediated and natural cytotoxicity. (a) The range of TIGIT expression on NK cells (49.8−90.0%) from PLWH selected for functional studies is depicted (median ± IQR 76.6 ± 19.6%). (b) NK cells were triggered to lyse P815 cells using anti‐CD16, and the overall effect of NK cell TIGIT cross‐linking was measured using IgG1 isotype control (not depicted) or anti‐TIGIT (represented by the yellow antibody). Percent specific lysis of P815 cells mediated through CD16 with either IgG1 or anti‐TIGIT was compared. Wilcoxon signed‐rank test *P = 0.0351. (c) NK cells were pretreated with anti‐TIGIT to prevent interaction with PVR on K562 cells, and lysis was measured and compared with lysis mediated by NK cells pretreated with IgG1. Student's paired t‐test ****P < 0.0001. Percent increase in NK cell cytotoxicity against K562 targets in the presence of anti‐TIGIT vs IgG1 was calculated from raw data in (c) and compared between (d) groups distinguished by CMV serostatus (Mann–Whitney U‐test *P = 0.0309) and (e) correlated with percentage of FcRγ– NK cells. (f) Absolute increase in NK cell lysis of K562 cells in the presence of anti‐TIGIT mAb was correlated with the percentage of TIGIT+ NK cells. For all graphs, filled symbols represent CMV‐seronegative (n = 10) participants, and open symbols depict CMV‐seropositive participants (n = 16). Horizontal lines bisecting groups in a and d represent median with IQR.
To measure the effect of TIGIT engagement on NK cell cytotoxicity, we sensitised IgG Fc receptor (FcR)‐expressing P815 cells for redirected NK cell lysis using monoclonal anti‐CD16 antibody (Figure 4b, right panel). In most cases, engagement of TIGIT decreased NK cell activity against P815 cells, independent of donor CMV infection status (Figure 4b, left panel). Having established that engaging TIGIT inhibited NK cell CD16‐mediated cytotoxicity for a large fraction of the subcohort, we tested whether preventing TIGIT–ligand interactions increased NK cell natural cytotoxicity (Figure 4c, right panel). The K562 cell line is a conventional NK cell target that expresses high levels of PVR. 50 Natural cytotoxicity receptor‐mediated killing was generally more robust than CD16‐directed P815 lysis, and preventing TIGIT–PVR interactions increased NK cell natural cytotoxicity by a mean ± SD of 15.6 ± 11.9% (Figure 4c, left panel).
Although NK cells from both the CMV‐seronegative and seropositive groups responded to TIGIT blockade, the median (IQR) percent increase in natural cytotoxicity was greater for CMV‐seropositive [17.4% (17.9%)] than CMV‐seronegative [8.2% (9.0%)] individuals (Figure 4d) and increased for those with fewer FcRγ+ NK cells (Figure 4e). This functional impact aligns with the observation of higher NK cell TIGIT expression levels in CMV‐seropositive individuals within the greater PLWH cohort (Figure 2f). In this subset of PLWH, the absolute increase in natural cytotoxicity in response to TIGIT blockade directly related to the extent of TIGIT expression on NK cells (Figure 4f).
HIV‐1 infection of PVR‐expressing CEM.NKR‐CCR5 cells
After demonstrating that TIGIT engagement negatively impacted NK cell cytotoxicity through CD16 and that NK cell natural cytotoxicity is increased by blocking TIGIT–PVR interactions, we next determined the impact of TIGIT–PVR interaction on NK cell‐mediated killing of HIV‐1‐infected cells. To do so, we transduced PVR into HIV‐1‐permissive CCR5‐expressing CEM.NKR (CEM.NKR‐CCR5 PVR+) cells (Supplementary figure 2a) as they are resistant to NK cell‐mediated lysis and constitutively express very low levels of CD112. 50 , 51 , 52 Both the parental and PVR‐transduced CEM.NKR‐CCR5 cell lines were permissive for HIV‐1 A17 infection and were infected at comparable levels based on intracellular p24 (Supplementary figure 2b) and surface gp120 (Supplementary figure 2c) detection 96 h postinfection. During this time period, surface PVR mean fluorescence intensity (MFI) increased on CEM.NKR‐CCR5 PVR+ cells from 32 739 ± 3318 on uninfected cells to 109 736 ± 36 409 for HIV‐1 A17‐infected cells, indicating that in vitro HIV‐1 infection has a profound effect on PVR expression of this CD4+ T‐cell line (Supplementary figure 2d). 46 To determine whether TIGIT has an impact on NK cell cytotoxicity in this setting, we used uninfected and HIV‐1 A17‐infected parental and PVR‐transduced CEM.NKR‐CCR5 cell lines in tandem as targets for HIV‐1‐specific cytotoxicity experiments.
TIGIT blockade augments HIV‐1‐specific natural cytotoxicity
Despite their natural resistance to NK cell‐based killing, CEM.NKR‐CCR5 cells infected with HIV‐1 A17 became modestly susceptible to NK cell natural cytotoxicity (Figure 5a, dataset 1 and 2). Introducing PVR rendered uninfected CEM.NKR‐CCR5 targets marginally susceptible to NK cell lysis as we noted slightly increased natural cytotoxicity against these cells compared with the parental line (Figure 5a, dataset 1 and 3). PVR expression also facilitated higher natural killing of HIV‐1 A17‐infected CEM.NKR‐CCR5 PVR+ targets than of the CEM.NKR‐CCR5 cells not expressing PVR (Figure 5a, dataset 2 and 4) or the uninfected CEM.NKR‐CCR5 PVR+ targets (Figure 5a, dataset 3 and 4), likely through NK cell DNAM‐1–PVR interactions. 53 As productive HIV‐1 infection inflates cell surface PVR expression (Supplementary figure 2d), 46 the increased killing noted for the PVR‐transduced CEM.NKR‐CCR5 cells infected with HIV‐1 A17 in Figure 5a may partly reflect an HIV‐1‐dependent increase in PVR levels. Blocking NK cell TIGIT receptors increased natural cytotoxicity against HIV‐1 A17‐infected CEM.NKR‐CCR5 target cells expressing PVR (Figure 5b) but had no effect on NK cell activity against infected parental CEM.NKR‐CCR5, indicating that the ligand for TIGIT must be expressed for inhibition to occur and for anti‐TIGIT mAb blockade to be effective. This supports the notion that PVR is the major inhibitory ligand for TIGIT expressed on these target cells and that its expression can modulate NK cell cytotoxicity against uninfected and HIV‐1‐infected targets.
Figure 5.
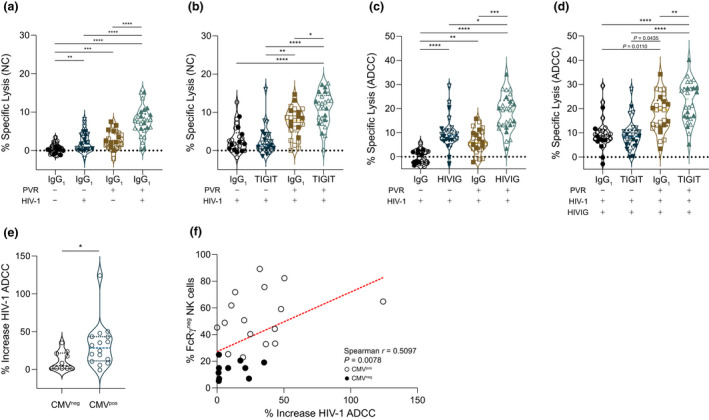
Impact of TIGIT blockade on natural and antibody‐dependent NK cell‐mediated cytotoxicity. Cytotoxicity experiments were performed over 5 h with PBMC from PLWH and uninfected or HIV‐1 A17‐infected CEM.NKR‐CCR5 or CEM.NKR‐CCR5 PVR+ target cells. (a) Natural cytotoxicity (NC) in the absence (−) or presence (+) of PVR was measured against uninfected (−) or HIV‐1 infected (+) targets. One‐way ANOVA with Tukey's multiple comparison **P = 0.0071 ***P = 0.0006 ****P < 0.0001. (b) PBMC pre‐incubated with control IgG1 or anti‐TIGIT mAb were tested for NK cell‐mediated killing against HIV‐1 A17‐infected CEM.NKR‐CCR5 or CEM.NKR‐CCR5 PVR+ targets. Nonparametric ANOVA with Dunn's multiple comparison *P = 0.0158 **P = 0.0035 ****P < 0.0001. (c) Control IgG or HIVIG was used to elicit ADCC against HIV‐1 A17‐infected targets with or without PVR and cytotoxicity was measured. Nonparametric ANOVA with Dunn's multiple comparison *P = 0.0158 **P = 0.0016 ***P = 0.0002 ****P < 0.0001. (d) PBMC were pretreated with control IgG1 or anti‐TIGIT mAb, and the effect of TIGIT blockade on NK cell HIV‐1‐specific ADCC was measured against HIV‐1 A17‐infected CEM.NKR‐CCR5 or CEM.NKR‐CCR5 PVR+ cells in the presence of HIVIG. Nonparametric ANOVA with Dunn's multiple comparison **P = 0.0076 ****P < 0.0001. (e) Percent increase in NK cell cytotoxicity against CEM.NKR‐CCR5 PVR+ targets in the presence of HIVIG and anti‐TIGIT vs IgG1 was calculated from raw data in d and compared between (e) groups distinguished by CMV serostatus (Mann–Whitney U‐test *P = 0.0198) and (f) correlated with the percentage of FcRγ– NK cells. For all graphs, filled symbols represent CMV‐seronegative (n = 10) participants, and open symbols represent CMV‐seropositive participants (n = 16). Horizontal lines bisecting groups represent median with IQR.
TIGIT blockade increases HIV‐1‐specific NK cell ADCC
Data in Figure 4b indicate that TIGIT engagement negatively impacted CD16 signalling. We also noted positive correlation between expression of NK cell TIGIT and markers of NK cell adaptation to CMV (Figure 2d and e). Despite the relationship between CMV infection and loss of FcRγ subunits, the implications for NK cell cytotoxicity triggered by CD16 are unclear 40 , 54 , 55 , 56 , 57 , 58 ; therefore, we examined the impact of TIGIT blockade on HIV‐1‐specific NK cell cytotoxicity triggered specifically by CD16 receptors. To accomplish this, we focussed NK cells onto HIV‐1 A17‐infected CEM.NKR‐CCR5 or CEM.NKR‐CCR5 PVR+ cells using a standard pooled polyclonal anti‐HIV‐1 Ig preparation from PLWH (HIVIG) or pooled Ig from HIV‐seronegative individuals (IgG) as control. For both cell lines, HIVIG elicited NK cell‐mediated ADCC against HIV‐1 A17‐infected targets compared with control IgG (Figure 5c) and robust HIV‐1‐specific ADCC occurred independent of PVR expression (Figure 5c, dataset 2 and 4). TIGIT blockade increased HIV‐1‐specific ADCC responses against infected target cells expressing PVR but not against target cells that did not express PVR (Figure 5d). This again illustrates the selective benefit of TIGIT blockade when target cells, such as HIV‐1‐infected CD4+ T cells, express PVR. The predisposition for NK cells from CMV‐seropositive PLWH to experience greater responses to anti‐TIGIT mAb treatment is evident with a significantly greater overall percent increase in HIV‐1‐specific NK cell ADCC (Figure 5e) and a direct correlation between the percent increase and frequency of FcRγ‐ NK cells (Figure 5f).
Another inhibitory receptor expressed by NK cells, KIR2DL5, also selectively interacts with PVR. 9 , 59 We measured the percentage of NK cells expressing KIR2DL5 to determine whether there was any association between NK cell KIR2DL5 and TIGIT expression or whether the percentage of NK cells expressing KIR2DL5 related to the impact of TIGIT blockade on individual functional responses. Approximately 25% (n = 7) of PLWH in the functionally studied cohort expressed KIR2DL5 on their NK cells with a wide range of frequency (2.7–23.0%; Supplementary figure 3a and b). Within these experimental constraints and cohort studied, KIR2DL5 and TIGIT expression were not linked (Supplementary figure 3c) and KIR2DL5 expression had no bearing on NK cell cytotoxicity in response to TIGIT blockade (Supplementary figure 3d).
TIGIT blockade increases HIV‐1‐specific NK cell cytotoxicity against autologous CD4+ T cells
Whether in vitro disruption of NK cell TIGIT–PVR interactions can translate in vivo to strengthen NK cell activity against HIV‐1 is the key issue for evaluating the immunotherapeutic potential of TIGIT blockade in PLWH. Our data demonstrate TIGIT blockade is effective when target cells express PVR; thus, for anti‐TIGIT mAb to have a meaningful impact on NK cell cytotoxicity against HIV‐1 in vivo, CD4+ T cells harbouring HIV‐1 must express PVR. In PLWH, circulating CD4+ T cells express PVR (Figure 3a), 13 and it is expressed to a greater extent on CD4+ T cells in lymph nodes, the major compartment for HIV‐1 reservoirs. 12 , 60 , 61 , 62 Cell cycle activation induces PVR on primary CD4+ T cells from both HIV‐seronegative individuals and PLWH. 63 , 64 To determine whether PVR was induced on ex vivo CD4+ T cells supporting HIV‐1 replication, we depleted CD8+ T cells from PBMC of PLWH (CD8dep PBMC) and, to control for exogenous factors modulating PVR expression, cultured otherwise unstimulated CD8dep PBMC in interleukin (IL)‐2 and measured intracellular CD4+ T‐cell HIV‐1 p24 (Figure 6a) and surface PVR expression on HIV‐1 p24+ (Figure 6b) and HIV‐1 p24‐ cells (Figure 6c). Data were included for analysis when p24 expression was detected on > 1500 cells within the CD4+ T‐cell gate. With this protocol, 9.8 ± 3.9% (mean ± SD) of CD4+ T cells expressed p24 (Figure 6d, left axis), with 74.8 ± 20.1% (mean ± SD) of this fraction being PVR+ (Figure 6d, right axis). In stark contrast, only 0.3 ± 0.4% (mean ± SD) of CD4+ T cells not expressing p24 (Figure 6c) were PVR+ (Figure 6d, right axis). Therefore, PVR expression is highly enriched on ex vivo CD4+ T cells reactivated to express endogenous HIV‐1 compared with uninfected CD4+ T cells.
Figure 6.
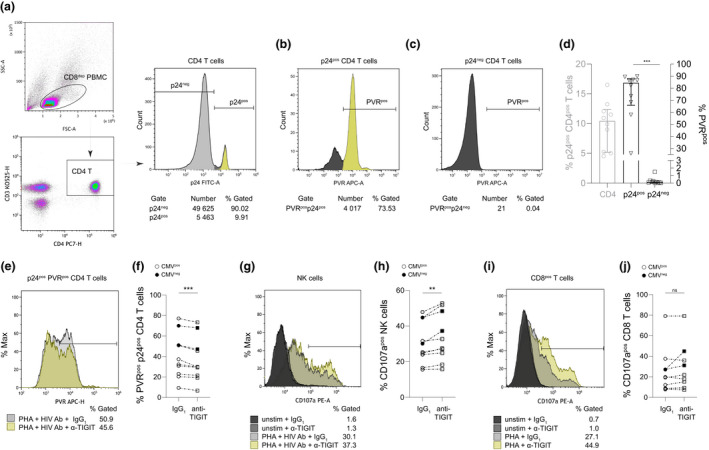
Effect of TIGIT blockade on HIV‐1‐specific NK cell and CD8+ T‐cell activation and elimination of endogenously infected PVR‐expressing CD4+ T cells ex vivo. (a) PBMC from PLWH were depleted of CD8+ T cells, treated with IL‐2 and assessed for HIV‐1 infection by p24 expression. Representative plots depict initial gating on CD3+CD4+CD8dep lymphocytes and determination of the percentage of PVR expression for (b) p24+ or (c) p24‐ cells. (d) Summary data as in a–c from 10 individuals. The left axis depicts percentage of CD4+ T cells that were p24+ 72–144 h after CD8+ T‐cell depletion, and the right axis represents the percentage of PVR expressing cells in either the p24+ or p24− CD4+ T‐cell populations. ANOVA with Tukey's multiple comparison ***P = 0.0001. Error bars in d represent median with IQR. Following 72 h PHA‐P activation of PBMC, (e, f) loss of p24+PVR+CD4+ T cells or (g, h) NK cell and (i, j) CD8+ T‐cell degranulation was evaluated 24 h after treatment with either IgG1 or anti‐TIGIT mAb in the presence of anti‐HIV‐1 antibodies. Histogram plots for each analysis are depicted in e, g, i with data summarised for 10 participants in f, h, j. Degranulation was assessed after gating on lymphocytes and enumerating CD107a‐expressing cells in either CD3‐CD56+ (NK cell) or CD3+CD8+ (CD8+ T cell) populations. P‐values in f, h, j were calculated using the Student's paired t‐test **P = 0.0034 ***P = 0.0007.
Although allowing CD8dep PBMC from PLWH to ‘spontaneously’ express HIV‐1 p24 permitted detection of selective PVR expression on p24+ CD4+ T cells, it did not result in sufficient expression of antigen to measure autologous HIV‐1‐specific killing. Thus, added stimulus with the mitogenic lectin phytohaemagglutinin (PHA)‐P was required to reactivate endogenous HIV‐1 within CD4+ T cells and allow spread to sufficient levels to measure autologous HIV‐1‐specific NK cell and CD8+ T‐cell responses from whole PBMC cultures. HIV‐1 p24 was detected on 40.3 ± 21.8% (mean ± SD) of PVR+ CD4+ T‐cell fractions (Figure 6e and summarised in Figure 6f). Addition of HIV‐1‐specific antibodies to induce ADCC with inclusion of anti‐TIGIT mAb increased NK cell antibody‐dependent activation (Figure 6g and summarised in Figure 6h) and decreased the survival of p24+ PVR+ CD4+ T cells compared with control conditions (Figure 6e and f). Increased HIV‐1‐specific CD8+ T‐cell degranulation against autologous CD4+ T cells reactivated to express endogenous HIV‐1 in the presence of anti‐TIGIT mAb was noted for some individuals; however, overall CD8+ T‐cell degranulation was not significantly affected under these conditions (Figure 6i and summarised in Figure 6j). These data demonstrating ex vivo elimination of autologous p24‐expressing targets corroborate in vitro data using cell lines overexpressing PVR and suggest that anti‐TIGIT mAb treatment is a rational approach to augment NK cell lysis of HIV‐1‐infected targets in vivo.
Discussion
An important role for TIGIT in regulating immune responses was demonstrated in recent studies. 11 , 12 , 13 , 14 The breadth of TIGIT expression on different lymphocyte subsets, together with increased expression of its ligand, PVR, on CD4+ T cells infected with HIV‐1, creates a compelling target for checkpoint inhibition in PLWH. Several studies reported altered regulation of TIGIT expression levels in PLWH and showed that TIGIT modulates the function of immune effector cells, including HIV‐1‐specific CD8+ T cells. 12 , 13 , 14 , 65 However, questions remain as to what factors underlie increased TIGIT expression on different immune cell subsets and what impact TIGIT expression has on effector cell functions in different contexts. We studied TIGIT expression on the immune cell subsets of over 90 PLWH and focused functional studies on NK cells from a representative subset. Our data indicate that upregulation of TIGIT on different immune cell subsets relates to distinct factors and that blockade of NK cell TIGIT interaction with PVR consistently increases natural cytotoxicity and ADCC against HIV‐1‐infected and other target cells.
In PLWH with chronic infection, TIGIT was expressed to the greatest extent on NK cells, followed by CD8+ T cells and CD4+ T cells. Compared with age‐matched HIV‐seronegative controls, TIGIT expression in PLWH was elevated on NK cells and CD4+ T cells but not on CD8+ T cells. Expression of TIGIT on CD4+ T cells increases with age/immune experience and further increases with chronic HIV‐1 infection, despite cART. Considering the mean duration of HIV‐1 infection (15.8 ± 10.3 years) for the PLWH cohort, our data indicate that levels of TIGIT on CD8+ T cells of cART‐treated PLWH return to similar levels as expressed by age‐matched HIV‐seronegative individuals. There was very little TIGIT expression on T cells in CBMC; however, TIGIT was expressed on a substantial proportion of cord blood NK cells, albeit at a lower frequency than on NK cells from PLWH. This indicates that TIGIT expression occurs as a natural aspect of NK cell ontogeny, although its expression on T cells reflects post‐developmental aspects of T‐cell maturation. Levels of TIGIT on both CD4+ and CD8+ T cells of PLWH correlated with extent of HIV‐1 disease progression as defined by CD4+ T‐cell nadir. Although several other studies associated elevated NK cell TIGIT expression with correlates of HIV‐1 disease progression, 11 , 12 , 13 , 14 we found that the frequency of TIGIT expression on NK cells of PLWH correlated with the extent of NK cell adaptation to CMV infection, as indicated by loss of FcRγ or increased expression of NKG2C and CD57.
The influence of CMV infection was also illustrated in functional modulation of NK cells by TIGIT engagement. Experiments using P815 and K562 NK cell targets verified the influence of TIGIT engagement on NK cell cytotoxicity. Engaging NK cell TIGIT compromised signalling through CD16 and blocking TIGIT–PVR interaction effectively augmented NK cell natural cytotoxicity. The NK cells from CMV‐seropositive PLWH with higher levels of TIGIT expression had larger increases in NK cell cytotoxicity in the presence of anti‐TIGIT mAb than NK cells from the CMV‐seronegative group. Considering the caustic association between HIV‐1 and CMV infection and the reciprocal role each virus has contributing to the others' reactivation, the higher prevalence of TIGIT+ NK cells in PLWH than in the HIV‐seronegative group could reasonably be attributed to more frequent CMV reactivation in PLWH. This possibility is consistent with the exaggerated extent of both CMV‐specific CD8+ T‐cell memory inflation and NK cell adaptation to CMV infection reported in PLWH. 42 , 66 Just as an increase in the rate of NK cell adaptation to CMV is accelerated by HIV‐1 infection, so may be the rate of NK cell TIGIT accumulation. These results underscore the need to consider CMV coinfection in the context of NK cell studies with PLWH.
Most relevant to enhancing immune‐mediated strategies to address the HIV‐1 reservoir in PLWH, we demonstrated that TIGIT blockade increases NK cell cytotoxicity and degranulation in response to HIV‐1‐infected CD4+ T cells in vitro and ex vivo. Transduction of an HIV‐1‐permissive CD4+ T‐cell line with PVR showed that the increase in NK cell cytotoxicity induced by TIGIT blockade was dependent upon target cell PVR expression. We further showed that reactivation of HIV‐1 in CD4+ T cells from PLWH selectively increased PVR expression on infected cells, rendering infected cells susceptible to NK cell‐mediated ADCC, and that TIGIT blockade increased NK cell degranulation in response to HIV‐1‐bearing autologous target cells. Moreover, the reduction in HIV‐1 antigen‐positive PVR+ CD4+ T cells in anti‐TIGIT mAb conditions compared with the control demonstrated accelerated elimination of HIV‐1+ cells by NK cells. A previous study reported that TIGIT blockade did not increase autologous NK cell activation against CD4+ T cells infected overnight in vitro with HIV‐1 and that a higher frequency of NK cells expressing TIGIT was activated against the infected CD4+ T cells than those not expressing TIGIT. 14 The same study found that NK cells expressing TIGIT had enhanced functions following stimulation with K562 cells, cytokines or PMA/ionomycin. 14 More recently, Zhang et al. reported conflicting findings in that lower frequencies of TIGIT‐expressing NK cells than those not expressing TIGIT were activated against K562 cells. 32 In both studies, NK cells were activated with IL‐2 under slightly different conditions. Although the difference could relate to the study subjects and their CMV infection status (healthy controls versus a mix of healthy controls and persons with acute and chronic HIV infection), addressing that possibility would require further investigation. In our functional studies, we measured cytotoxicity as an effector function of NK cells without discriminating TIGIT‐expressing and nonexpressing cells. Blocking TIGIT consistently increased NK cell natural cytotoxicity and ADCC, but that does not preclude the possibility that a higher fraction of TIGIT‐expressing NK cells was responding in the absence of TIGIT blockade as the majority of NK cells in most of our subjects did express TIGIT. We also measured cytotoxicity or elimination as a read‐out for effector function against HIV‐1‐infected target cells with a focus on ADCC and consideration of PVR expression levels on the target cells, either manipulated by transduction or in association with p24 expression. In contrast to Vendrame et al., who observed no selective upregulation of PVR on CD4+ T cells activated with combined PHA, anti‐CD3 and anti‐CD28, we observed selective expression of PVR on p24‐expressing CD4+ T cells after removal of CD8+ T cells to allow endogenous HIV‐1 replication. Despite the differences reported, TIGIT‐expressing NK cells can clearly mediate effector functions with appropriate stimulation. Another consistent finding is that TIGIT expression is, to some extent, associated with adaptation/maturation of NK cells, as indicated by their co‐expression of NKG2C and loss of NKG2A, FcRγ and even CD56. 14 , 32 As a substantial fraction of essentially naive NK cells from cord blood already expresses TIGIT, but its expression increases with different forms of stimulation, TIGIT‐expressing NK cells are likely a diverse population with different levels of responsiveness to TIGIT‐mediated inhibition and TIGIT blockade. If so, it will be important to distinguish TIGIT‐expressing NK cells at these different phases in chronically infected individuals to maximise the therapeutic potential of TIGIT blockade.
KIR2DL5 is another inhibitory receptor expressed by NK cells and CD8+ T cells capable of binding PVR independently. 9 In preventing TIGIT–PVR interactions, we expected that PLWH with higher levels of KIR2DL5‐expressing NK cells might not respond to anti‐TIGIT mAb blockade as robustly as those not expressing KIR2DL5. Despite detecting and measuring KIR2DL5 expression on seven of the twenty‐six participants within the functional cohort, we found KIR2DL5 expression to have no significant impact on NK cell natural or antibody‐dependent functions in the presence of anti‐TIGIT mAb. As the total number of participants identified with KIR2DL5+ NK cells in the functional cohort was low, and only two individuals had > 10% KIR2DL5 expression on their NK cells, further investigation that includes a larger cohort of PLWH exhibiting higher levels of KIR2DL5 expression coupled with the consideration of differential NK cell education is required.
Conscripting our own immune system to purge HIV‐1 reservoirs is an attractive approach not yet successfully applied. One major barrier to engaging NK cells in HIV‐1 reservoir elimination is the limited expression of HIV‐1‐specific or HIV‐1‐related cell surface antigens during latency and early reactivation. Without a basis for specifically targeting HIV‐1‐infected cells, no amount of reinvigoration of NK cell activity will address the reservoir. The enriched presence of HIV‐1 in CD4+ T cells expressing PVR provides a means of targeting cells in which HIV‐1 is reactivated and, under appropriate conditions, PVR expression promotes NK cell activity. As DNAM‐1–PVR interactions favor effector cell‐mediated killing and TIGIT–PVR engagement decreases or prevents killing, TIGIT blockade has a dual effect of reducing inhibitory signalling and enhancing activation. 5 , 67
The use of anti‐TIGIT mAb to prevent inhibitory TIGIT–PVR interactions increased NK cell activity against HIV‐1‐infected autologous CD4+ T cells. Circulating CD4+ T cells from PLWH express appreciable levels of TIGIT, and CD4+ T‐cell TIGIT and PVR expression were positively correlated. In relation to HIV‐seronegative controls, PVR expression on CD4+ T cells from PLWH was elevated; however, the relationship between HIV‐1 and CMV infection that encourages sustained inflammation and immune activation is highlighted in our finding that levels of PVR were particularly enriched on circulating CD4+ T cells from CMV‐seropositive PLWH compared with CMV‐seronegative PLWH. Lymph node‐resident follicular helper CD4+ T cells or circulating CD4+ T cells in which the HIV‐1 reservoir is concentrated are more likely to express TIGIT in combination with other inhibitory immune checkpoint receptors, such as PD‐1 and LAG‐3. 4 , 49 , 68 Therefore, TIGIT expression alone or in combination with other inhibitory immune checkpoint receptors identifies CD4+ T cells enriched for HIV‐1 infection. Introducing anti‐TIGIT mAb could have even broader influence by lifting any inhibition of latent HIV‐1 activation imposed through TIGIT–PVR interaction, thereby increasing HIV‐1 antigen expression and the potential for immune recognition. Studies examining the influence of TIGIT blockade on lymph node‐resident TIGIT‐expressing CD4+ T cells harbouring HIV‐1 are warranted. Humanised anti‐TIGIT mAbs are in early trials for use in cancer therapy, and anti‐PD‐1 treatment is being investigated in clinical studies targeting cancer and, to a limited extent, latent HIV‐1 reservoirs expressing PD‐1. 69 , 70 Treatment with anti‐PD‐1 was generally well‐tolerated in PLWH with controlled HIV‐1 viral loads, and future inclusion of PLWH in immunotherapy trials will inform whether anti‐TIGIT mAb is tolerated to the same extent as in HIV‐seronegative participants. 70
In summary, we found that TIGIT expression is upregulated on T cells and NK cells of PLWH. The extent of expression relates to HIV‐1 disease progression in the case of T cells and to CMV coinfection in the case of NK cells. The NK cells of PLWH coinfected with CMV exhibit greater increases in cytotoxicity with TIGIT blockade than NK cells from CMV‐seronegative PLWH, and NK cells of PLWH respond to TIGIT blockade with increased activation against autologous CD4+ T cells when endogenous HIV‐1 replication is stimulated. Thus, TIGIT blockade could have a synergistic effect as a component of immunotherapeutic strategies targeting the HIV‐1 reservoir by releasing immune effector cells from inhibition and favoring HIV‐1 replication in the TIGIT+ reservoirs being targeted. The impact of TIGIT blockade on recognition of HIV‐1‐infected cells supports broader application of NK cell‐based therapies in other chronic conditions exploiting TIGIT–PVR interactions for immune regulation.
Methods
Study subjects
This study was carried out in accordance with recommendations of the Canadian Tri‐Council Policy Statement: Ethical Conduct for Research Involving Humans. Protocols to obtain anonymised umbilical cord blood and peripheral blood from HIV‐seronegative donors and from PLWH recruited through the Newfoundland and Labrador Provincial HIV Clinic were approved by the Health Research Ethics Authority of Newfoundland and Labrador, Canada. Peripheral blood was collected from study subjects after obtaining written informed consent in accordance with the Declaration of Helsinki.
Blood sample processing
Whole blood was collected by venipuncture in acid citrate dextrose vacutainers, after which plasma was collected following 10 min centrifugation at 500 g and stored at −80°C. CBMC and PBMC were isolated using the Canadian Autoimmunity Standardization Core consensus standard operating procedure (version: March 21, 2019). Freshly isolated PBMC were resuspended in a freezing medium consisting of foetal calf serum (FCS) supplemented with 10% dimethyl sulphoxide (Sigma‐Aldrich, St. Louis MO, USA) and cooled at 1°C per minute overnight to −80°C. Frozen PBMC were then kept in liquid nitrogen until use.
PBMC phenotyping
Isolated PBMC (106) were phenotyped using directly conjugated mAb (conjugate, clone in parentheses) against human CD16 (VioBlue, REA423), CD3 (VioGreen, BW264/56), CD4 (PE‐Vio770, REA623), NKG2C (PE‐Vio770, REA205), CD56 (APC‐Vio770, REA196) from Miltenyi Biotec (San Diego CA, USA), CD57 (FITC, TB01), PVR (PE, 2H7CD155), TIGIT (AlexaFluor 647, MBSA43) from Thermo Fisher Scientific (San Diego CA, USA) and CD8 (PerCP, HIT8a) from Biolegend (San Diego, CA, USA) and fixed with 2% paraformaldehyde (Sigma‐Aldrich) prior to data acquisition. Participants selected for functional assays were also phenotyped with anti‐KIR2DL5 (PE, UP‐R1) from Biolegend or polyclonal anti‐FcRγ (FITC) from Millipore Sigma (Oakville ON, Canada) using an Inside Stain Kit (Miltenyi Biotec) as per manufacturer's instructions. Data were acquired using the CytoFLEX flow cytometer and analysed and illustrated using Kaluza software (both Beckman Coulter, Brea CA, USA) and GraphPad Prism version 8.4.3.
Cell culture
K562 (ATCC® CCL 243™), P815 (ATCC® TIB‐64™), H9, CEM.NKR‐CCR5 (NIH HIV Reagent Program, Division of AIDS, NIAID, NIH) and CEM.NKR‐CCR5 PVR+ (see below) cell lines were propagated in lymphocyte medium consisting of RPMI‐1640 with 10% FCS, 200 IU mL−1 penicillin/streptomycin, 0.01 M HEPES, 1% L‐glutamine (all from Invitrogen, Carlsbad CA, USA) and 2.0 × 10−5 M 2‐mercaptoethanol (Sigma‐Aldrich) at 37°C, 5% CO2. The CEM.NKR‐CCR5 cell lines were maintained on a precise passage regimen as outlined. 51 , 52 , 71 Lenti‐X™ 293T cells (TakaRa, Mountain View CA, USA) were propagated in DMEM (Sigma‐Aldrich) with 10% tetracycline‐free FCS (TakaRa) and 1 mm sodium pyruvate (Sigma‐Aldrich) at 37°C, 5% CO2.
PVR gene transfer and expression in CEM.NKR‐CCR5 cells
The recombinant Lenti‐X™ pLVX‐IRES lentiviral vector expression system (TakaRa) was used to introduce PVR into the CEM.NKR‐CCR5 cell line. Briefly, the canonical PVR sequence was obtained from UniProtKB (P15151‐1), synthesised by Invitrogen GeneArt (Thermo Fisher Scientific) and inserted (Rapid DNA Ligation Kit; Roche, Mannheim, Germany) into pLVX‐IRES after SpeI and EcoRI restriction digestion (New England Biolabs, Ipswich, MA, USA) and agarose gel extraction/purification (QIAquick Gel Extraction; Qiagen, Toronto, ON, Canada). The pLVX‐IRES/PVR expression vector was transformed into Stellar™ Competent Cells (TakaRa) from which midi‐scale plasmid DNA was prepared (NucleoBond Xtra Midi, TakaRa), with concentration determined using NanoDrop™ (Thermo Fisher Scientific) and sequenced (positive and negative strands) to ensure authenticity. Lenti‐X™ 293T cells were transfected with the pLVX‐IRES/PVR expression vector using the Lenti‐X™ Single Shot (TakaRa) packaging and transfection system. Lentiviral supernatants were collected 48 h after transfection, filtered through a 0.45‐μm polyethersulphone filter to remove cellular debris, aliquoted and frozen at −80°C. A p24 ELISA [Leidos Biomedical Research, Inc., for the National Cancer Institute (NCI), Frederick, MD, USA], read at 450 nm on a Synergy HT BioTek microplate reader, was used to obtain viral titres, and CEM.NKR‐CCR5 cells were transduced with lentiviral supernatant and 4 μg mL−1 polybrene (Sigma‐Aldrich) in 96‐well round‐bottom plates by 90 min 1200 g spinoculation at 32°C. Transduction medium was replaced 24 h later with lymphocyte medium, and cells were propagated and then sorted for PVR expression (2H7CD155 APC; Thermo Fisher Scientific) using a MoFlo Astrios EQ flow cytometer (Beckman Coulter).
HIV‐1 stock generation
The HIV‐1IIIB A17 variant (lot no. 9/03/92) was obtained through the NIH HIV Reagent Program. 72 The entire aliquot was used to infect 106 H9 cells. The cells were maintained in the minimal volume of lymphocyte medium required for growth, and 7 days after infection, supernatant was collected by 5 min 450 g centrifugation, followed by 0.45‐μm polyethersulphone filtration. To generate high titre stock, 105 H9 cells were infected with 1 mL of the infection supernatant, maintained in the minimal volume of lymphocyte medium required for growth and supernatant collected 14 days after infection as mentioned before. The stocks were split into single‐use vials, and p24 quantity was determined by ELISA as before.
In vitro HIV‐1 infection
CEM.NKR‐CCR5 and CEM.NKR‐CCR5 PVR+ cells (105) split 1:3 the day prior to infection were infected with 200 ng p24 HIV‐1 IIIB A17 by spinoculation in a 15‐mL tube at 32°C, 1200 g for 90 min. Cells were cultured at 37°C 5% CO2 for 1 h before adding 1 mL of lymphocyte medium. Experiments were performed 96 h after infection. HIV‐1 infection was quantified by anti‐p24 FITC [24‐4, Santa Cruz (Dallas, TX, USA)] or gp120 expression [50 ng HIVIG (NIH HIV Reagent Program) per 105 cells, followed by anti‐human IgG Fc (PE, eBioscience)]. Anti‐PVR‐APC (2H7CD1555, Thermo Fisher) was used to assess PVR expression. Cells were fixed with 2% paraformaldehyde (Sigma‐Aldrich) prior to data acquisition using a CytoFLEX (Beckman Coulter).
Chromium release assays
Cryopreserved PBMC were recovered overnight in lymphocyte medium at 37°C, 5% CO2. The cells were recounted after recovery by trypan blue exclusion and used when > 75% viable. Target cells were labelled for 90 min with 100 μCi Na2 51CrO4 (PerkinElmer, Akron, OH, USA) at 37°C, 5% CO2, washed four times in PBS containing 1% FCS before resting in 5 mL of lymphocyte medium for 1 h at 37°C, 5% CO2 to minimise spontaneous release and resuspended at 105 cells mL‐1. Where indicated, PBMC and IgG1 isotype control (11711, R&D Systems) or anti‐TIGIT (MBSA43, Thermo Fisher Scientific) were pre‐incubated for 30 min in a microtitre plate at a final mAb concentration of 5 µg mL−1. NK cell CD16‐mediated cytotoxicity was measured using 51Cr‐labelled P815 cells and 30 ng per well IgG1 isotype control (11711, R&D systems) or anti‐CD16 (3G8, Biolegend). We used HIVIG pooled from inactivated human sera (NIH HIV Reagent Program) or control IgG from human sera (Sigma) at a final concentration of 10 μg mL−1 to measure ADCC. All 51Cr release assays were conducted at E:T 30:1 (Vf = 300 μL), and cytotoxic activity was measured by 51Cr release over 5 h. 51Cr release was measured in 125 μL of supernatant on a Wallac 1480 Wizard gamma counter and percent specific lysis calculated by (experimental 51Cr release – spontaneous 51Cr release)/(maximum 51Cr release – spontaneous 51Cr release) × 100. Where indicated, the percent increase was calculated by (% specific lysis anti‐TIGIT‐treated condition) − (% specific lysis IgG1‐treated condition)/(% specific lysis IgG1‐treated condition) × 100, and absolute increase in cytotoxicity was calculated by (% specific lysis anti‐TIGIT‐treated condition) − (% specific lysis IgG1‐treated condition).
Primary CD4+ T cell HIV‐1 reactivation
Peripheral blood mononuclear cells were depleted of CD8+ T cells (StemCell Technologies, Vancouver BC, Canada) and cultured in lymphocyte medium without phenol red supplemented to 50 IU mL−1 IL‐2 (NCI) for up to 144 h. Aliquots were removed at 24‐h intervals and stained with anti‐human CD4 (PE‐Vio770), CD3 (VioGreen), from Miltenyi, CD8 (PerCP, Biolegend), PVR (APC, Invitrogen) and intracellular anti‐HIV‐1 p24 (FITC, Santa Cruz) using the Inside Stain Kit (Miltenyi).
Primary CD4+ T‐cell HIV‐1 stimulation and antibody‐dependent NK cell activation assay
Peripheral blood mononuclear cells from PLWH were resuspended at 2.5 × 106 cells mL−1 in lymphocyte medium without phenol red supplemented with 50 IU mL−1 IL‐2. PBMC were left unstimulated or stimulated with 5 μg mL−1 PHA‐P (Sigma) and divided equally into three conditions: untreated or treated with 10 μg IgG mL−1 of purified Ab from plasma of HIV‐seronegative individuals (CON Ab) or Ab from plasma of PLWH (HIV Ab). Briefly, Ab was purified by pooling heat‐inactivated (1 h at 56°C) plasma from HIV‐seronegative donors or PLWH (matched for CMV status). Pooled plasma was centrifuged at 10 000 g for 20 min, diluted 1:1 in endotoxin‐free binding buffer (0.2 m Na3PO4, Millipore Sigma) and purified using a 1‐mL Cytiva HiTrap™ Protein G HP column (Millipore Sigma). Purified Ab was adsorbed on CEM.NKR‐CCR5 and PHA‐P‐stimulated CD8dep PBMC from HIV‐seronegative participants to reduce nonspecific binding. Approximately 48 h after stimulation, PBMC from each condition were treated with IgG1 (11711, R&D Systems) or anti‐TIGIT (MBSA43, Thermo Fisher Scientific) at a final mAb concentration of 5 µg mL−1 and (i) incubated with Vf 500 μL IL‐2‐supplemented lymphocyte medium for 24 h or (ii) labelled with 0.25 µg per 106 PBMC of anti‐CD107a (H4A3; BioLegend) for 24 h (Vf 500 μL). Cells in condition (i) were stained using anti‐human CD3 (VioGreen), CD4 (PE‐Vio770) from Miltenyi, PVR (APC 2H7CD155, Thermo Fisher) and intracellular anti‐HIV‐1 p24 (FITC, Santa Cruz) using Inside Stain Kit (Miltenyi) and condition (ii) were assessed using CD3 (VioGreen), CD56 (APC‐Vio770) from Miltenyi and CD8 (PerCP, Biolegend). Data were acquired using a CytoFLEX (Beckman Coulter).
Statistical analysis
Statistical analyses were performed using GraphPad Prism version 8.4.3, with two‐sided P‐values < 0.05 considered significant. Normality of data distributions was assessed using the Shapiro–Wilk test. Significance in correlations was assessed using Spearman's rank correlation coefficient. Differences in means with standard deviation (SD) or medians with interquartile range (IQR, calculated as IQR = Q3−Q1) between groups were compared by using one‐way ANOVA, the Student's t‐test or the Mann–Whitney U‐test, as appropriate, based on normality of data distribution. For paired analyses, the Student's paired t‐test was used when data were normally distributed, and the nonparametric Wilcoxon signed rank test otherwise.
Conflict of interest
The authors declare no conflict of interest.
Author contributions
Kayla Holder: Conceptualization; Data curation; Formal analysis; Investigation; Methodology; Project administration; Writing‐original draft; Writing‐review & editing. Kimberley Burt: Data curation; Investigation; Methodology; Project administration; Resources. Michael Grant: Conceptualization; Funding acquisition; Investigation; Methodology; Project administration; Supervision; Validation; Writing‐review & editing.
Supporting information
Supplementary figures 1–3
Acknowledgments
We thank all participants for providing samples. Research was supported by a research operating grant from the Canadian Institutes of Health Research (CIHR) PJT 361426 awarded to MDG. KAH was supported by a CIHR doctoral fellowship. Adobe Illustrator 24.1.1 was used to construct figures, and illustrations were created using BioRender.com. The authors declare no competing financial interests.
References
- 1. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol 2015; 15: 486–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bi J, Tian Z. NK cell exhaustion. Front Immunol 2017; 8: 760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wykes MN, Lewin SR. Immune checkpoint blockade in infectious diseases. Nat Rev Immunol 2018; 18: 91–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yu X, Harden K, Gonzalez LC et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat Immunol 2009; 10: 48–57. [DOI] [PubMed] [Google Scholar]
- 5. Stanietsky N, Simic H, Arapovic J et al. The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc Natl Acad Sci USA 2009; 106: 17858–17863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Levin SD, Taft DW, Brandt CS et al. Vstm3 is a member of the CD28 family and an important modulator of T‐cell function. Eur J Immunol 2011; 41: 902–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dougall WC, Kurtulus S, Smyth MJ, Anderson AC. TIGIT and CD96: new checkpoint receptor targets for cancer immunotherapy. Immunol Rev 2017; 276: 112–120. [DOI] [PubMed] [Google Scholar]
- 8. Sanchez‐Correa B, Valhondo I, Hassouneh F et al. DNAM‐1 and the TIGIT/PVRIG/TACTILE Axis: Novel Immune Checkpoints for Natural Killer Cell‐Based Cancer Immunotherapy. Cancers (Basel) 2019; 11: 877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Husain B, Ramani SR, Chiang E et al. A platform for extracellular interactome discovery identifies novel functional binding partners for the immune receptors B7–H3/CD276 and PVR/CD155. Mol Cell Proteomics 2019; 18: 2310–2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhang Z, Wu N, Lu Y, Davidson D, Colonna M, Veillette A. DNAM‐1 controls NK cell activation via an ITT‐like motif. J Exp Med 2015; 212: 2165–2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chew GM, Fujita T, Webb GM et al. TIGIT marks exhausted T cells, correlates with disease progression, and serves as a target for immune restoration in HIV and SIV infection. PLoS Pathog 2016; 12: e1005349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tauriainen J, Scharf L, Frederiksen J et al. Perturbed CD8+ T cell TIGIT/CD226/PVR axis despite early initiation of antiretroviral treatment in HIV infected individuals. Sci Rep 2017; 7: 40354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yin X, Liu T, Wang Z et al. Expression of the inhibitory receptor TIGIT is up‐regulated specifically on NK cells with CD226 activating receptor from HIV‐infected individuals. Front Immunol 2018; 9: 2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vendrame E, Seiler C, Ranganath T et al. TIGIT is upregulated by HIV‐1 infection and marks a highly functional adaptive and mature subset of natural killer cells. AIDS 2020; 34: 801–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Holder KA, Grant MD. TIGIT blockade: a multipronged approach to target the HIV reservoir. Front Cell Infect Microbiol 2020; 10: 175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Martin MP, Gao X, Lee JH et al. Epistatic interaction between KIR3DS1 and HLA‐B delays the progression to AIDS. Nat Genet 2002; 31: 429–434. [DOI] [PubMed] [Google Scholar]
- 17. Martin MP, Qi Y, Gao X et al. Innate partnership of HLA‐B and KIR3DL1 subtypes against HIV‐1. Nat Genet 2007; 39: 733–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Alter G, Heckerman D, Schneidewind A et al. HIV‐1 adaptation to NK‐cell‐mediated immune pressure. Nature 2011; 476: 96–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kielczewska A, Pyzik M, Sun T et al. Ly49P recognition of cytomegalovirus‐infected cells expressing H2‐Dk and CMV‐encoded m04 correlates with the NK cell antiviral response. J Exp Med 2009; 206: 515–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Moretta L, Biassoni R, Bottino C et al. Human NK cells and their receptors. Microbes Infect 2002; 4: 1539–1544. [DOI] [PubMed] [Google Scholar]
- 21. Pegram HJ, Andrews DM, Smyth MJ, Darcy PK, Kershaw MH. Activating and inhibitory receptors of natural killer cells. Immunol Cell Biol 2011; 89: 216–224. [DOI] [PubMed] [Google Scholar]
- 22. Lanier LL. Up on the tightrope: natural killer cell activation and inhibition. Nat Immunol 2008; 9: 495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bryceson YT, March ME, Barber DF, Ljunggren HG, Long EO. Cytolytic granule polarization and degranulation controlled by different receptors in resting NK cells. J Exp Med 2005; 202: 1001–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bryceson YT, Ljunggren HG, Long EO. Minimal requirement for induction of natural cytotoxicity and intersection of activation signals by inhibitory receptors. Blood 2009; 114: 2657–2666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Davis ZB, Sowrirajan B, Cogswell A, Ward JP, Planelles V, Barker E. CD155 on HIV‐infected cells is not modulated by HIV‐1 Vpu and Nef but synergizes with NKG2D ligands to trigger NK cell lysis of autologous primary HIV‐infected cells. AIDS Res Hum Retroviruses 2017; 33: 93–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lanier LL. On guard–activating NK cell receptors. Nat Immunol 2001; 2: 23–27. [DOI] [PubMed] [Google Scholar]
- 27. Kim S, Poursine‐Laurent J, Truscott SM et al. Licensing of natural killer cells by host major histocompatibility complex class I molecules. Nature 2005; 436: 709–713. [DOI] [PubMed] [Google Scholar]
- 28. Anfossi N, Andre P, Guia S et al. Human NK cell education by inhibitory receptors for MHC class I. Immunity 2006; 25: 331–342. [DOI] [PubMed] [Google Scholar]
- 29. Johansson S, Johansson M, Rosmaraki E et al. Natural killer cell education in mice with single or multiple major histocompatibility complex class I molecules. J Exp Med 2005; 201: 1145–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fernandez NC, Treiner E, Vance RE, Jamieson AM, Lemieux S, Raulet DH. A subset of natural killer cells achieves self‐tolerance without expressing inhibitory receptors specific for self‐MHC molecules. Blood 2005; 105: 4416–4423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. He Y, Peng H, Sun R et al. Contribution of inhibitory receptor TIGIT to NK cell education. J Autoimmun 2017; 81: 1–12. [DOI] [PubMed] [Google Scholar]
- 32. Zhang X, Lu X, Cheung AKL et al. Analysis of the characteristics of TIGIT‐Expressing CD3‐CD56+ NK cells in controlling different stages of HIV‐1 infection. Front Immunol 2021; 12: 602492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Christensen‐Quick A, Vanpouille C, Lisco A, Gianella S. Cytomegalovirus and HIV persistence: pouring gas on the fire. AIDS Res Hum Retroviruses 2017; 33: S23–S30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Durier N, Ananworanich J, Apornpong T et al. Cytomegalovirus viremia in Thai HIV‐infected patients on antiretroviral therapy: prevalence and associated mortality. Clin Infect Dis 2013; 57: 147–155. [DOI] [PubMed] [Google Scholar]
- 35. Gianella S, Massanella M, Wertheim JO, Smith DM. The sordid affair between human herpesvirus and HIV. J Infect Dis 2015; 212: 845–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bate SL, Dollard SC, Cannon MJ. Cytomegalovirus seroprevalence in the United States: the national health and nutrition examination surveys, 1988–2004. Clin Infect Dis 2010; 50: 1439–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Freeman ML, Lederman MM, Gianella S. Partners in crime: the role of CMV in immune dysregulation and clinical outcome during HIV infection. Curr HIV/AIDS Rep 2016; 13: 10–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Guma M, Angulo A, Vilches C, Gomez‐Lozano N, Malats N, Lopez‐Botet M. Imprint of human cytomegalovirus infection on the NK cell receptor repertoire. Blood 2004; 104: 3664–3671. [DOI] [PubMed] [Google Scholar]
- 39. Guma M, Cabrera C, Erkizia I et al. Human cytomegalovirus infection is associated with increased proportions of NK cells that express the CD94/NKG2C receptor in aviremic HIV‐1‐positive patients. J Infect Dis 2006; 194: 38–41. [DOI] [PubMed] [Google Scholar]
- 40. Hwang I, Zhang T, Scott JM et al. Identification of human NK cells that are deficient for signaling adaptor FcRγ and specialized for antibody‐dependent immune functions. Int Immunol 2012; 24: 793–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lachmann R, Bajwa M, Vita S et al. Polyfunctional T cells accumulate in large human cytomegalovirus‐specific T cell responses. J Virol 2012; 86: 1001–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Heath J, Newhook N, Comeau E, Gallant M, Fudge N, Grant M. NKG2C+CD57+ natural killer cell expansion parallels cytomegalovirus‐specific CD8+ T cell evolution towards senescence. J Immunol Res 2016; 2016: 7470124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Vassena L, Giuliani E, Matusali G, Cohen EA, Doria M. The human immunodeficiency virus type 1 Vpr protein upregulates PVR via activation of the ATR‐mediated DNA damage response pathway. J Gen Virol 2013; 94: 2664–2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Matusali G, Potesta M, Santoni A, Cerboni C, Doria M. The human immunodeficiency virus type 1 Nef and Vpu proteins downregulate the natural killer cell‐activating ligand PVR. J Virol 2012; 86: 4496–4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bolduan S, Reif T, Schindler M, Schubert U. HIV‐1 Vpu mediated downregulation of CD155 requires alanine residues 10, 14 and 18 of the transmembrane domain. Virology 2014; 464–465: 375–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tremblay‐McLean A, Bruneau J, Lebouche B, Lisovsky I, Song R, Bernard NF. Expression profiles of ligands for activating natural killer cell receptors on HIV infected and uninfected CD4+ T cells. Viruses 2017; 9: 295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Boles KS, Vermi W, Facchetti F et al. A novel molecular interaction for the adhesion of follicular CD4 T cells to follicular DC. Eur J Immunol 2009; 39: 695–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wang F, Hou H, Wu S et al. TIGIT expression levels on human NK cells correlate with functional heterogeneity among healthy individuals. Eur J Immunol 2015; 45: 2886–2897. [DOI] [PubMed] [Google Scholar]
- 49. Wu H, Chen Y, Liu H et al. Follicular regulatory T cells repress cytokine production by follicular helper T cells and optimize IgG responses in mice. Eur J Immunol 2016; 46: 1152–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tremblay‐McLean A, Coenraads S, Kiani Z, Dupuy FP, Bernard NF. Expression of ligands for activating natural killer cell receptors on cell lines commonly used to assess natural killer cell function. BMC Immunol 2019; 20: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Trkola A, Matthews J, Gordon C, Ketas T, Moore JP. A cell line‐based neutralization assay for primary human immunodeficiency virus type 1 isolates that use either the CCR5 or the CXCR4 coreceptor. J Virol 1999; 73: 8966–8974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lyerly HK, Reed DL, Matthews TJ et al. Anti‐GP 120 antibodies from HIV seropositive individuals mediate broadly reactive anti‐HIV ADCC. AIDS Res Hum Retroviruses 1987; 3: 409–422. [DOI] [PubMed] [Google Scholar]
- 53. Reymond N, Imbert AM, Devilard E et al. DNAM‐1 and PVR regulate monocyte migration through endothelial junctions. J Exp Med 2004; 199: 1331–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhang T, Scott JM, Hwang I, Kim S. Cutting edge: antibody‐dependent memory‐like NK cells distinguished by FcRγ deficiency. J Immunol 2013; 190: 1402–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wu Z, Sinzger C, Frascaroli G et al. Human cytomegalovirus‐induced NKG2Chi CD57hi natural killer cells are effectors dependent on humoral antiviral immunity. J Virol 2013; 87: 7717–7725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Luetke‐Eversloh M, Hammer Q, Durek P et al. Human cytomegalovirus drives epigenetic imprinting of the IFNG locus in NKG2Chi natural killer cells. PLoS Pathog 2014; 10: e1004441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Luetke‐Eversloh M, Cicek BB, Siracusa F et al. NK cells gain higher IFN‐γ competence during terminal differentiation. Eur J Immunol 2014; 44: 2074–2084. [DOI] [PubMed] [Google Scholar]
- 58. Holder KA, Lajoie J, Grant MD. Natural killer cells adapt to cytomegalovirus along a functionally static phenotypic spectrum in human immunodeficiency virus infection. Front Immunol 2018; 9: 2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wojtowicz WM, Vielmetter J, Fernandes RA et al. A human IgSF cell‐surface interactome reveals a complex network of protein‐protein interactions. Cell 2020; 182: 1027–1043 e1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lindqvist M, van Lunzen J, Soghoian DZ et al. Expansion of HIV‐specific T follicular helper cells in chronic HIV infection. J Clin Invest 2012; 122: 3271–3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Perreau M, Savoye AL, De Crignis E et al. Follicular helper T cells serve as the major CD4 T cell compartment for HIV‐1 infection, replication, and production. J Exp Med 2013; 210: 143–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Banga R, Procopio FA, Noto A et al. PD‐1+ and follicular helper T cells are responsible for persistent HIV‐1 transcription in treated aviremic individuals. Nat Med 2016; 22: 754–761. [DOI] [PubMed] [Google Scholar]
- 63. Ardolino M, Zingoni A, Cerboni C et al. DNAM‐1 ligand expression on Ag‐stimulated T lymphocytes is mediated by ROS‐dependent activation of DNA‐damage response: relevance for NK‐T cell interaction. Blood 2011; 117: 4778–4786. [DOI] [PubMed] [Google Scholar]
- 64. Cella M, Presti R, Vermi W et al. Loss of DNAM‐1 contributes to CD8+ T‐cell exhaustion in chronic HIV‐1 infection. Eur J Immunol 2010; 40: 949–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Hasan MM, Nair SS, O'Leary JG et al. Implication of TIGIT+ human memory B cells in immune regulation. Nat Commun 2021; 12: 1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Naeger DM, Martin JN, Sinclair E et al. Cytomegalovirus‐specific T cells persist at very high levels during long‐term antiretroviral treatment of HIV disease. PLoS One 2010; 5: e8886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Castriconi R, Dondero A, Corrias MV et al. Natural killer cell‐mediated killing of freshly isolated neuroblastoma cells: critical role of DNAX accessory molecule‐1‐poliovirus receptor interaction. Cancer Res 2004; 64: 9180–9184. [DOI] [PubMed] [Google Scholar]
- 68. Fromentin R, Bakeman W, Lawani MB et al. CD4+ T cells expressing PD‐1, TIGIT and LAG‐3 contribute to HIV persistence during ART. PLoS Pathog 2016; 12: e1005761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Gay CL, Bosch RJ, McKahnn A et al. Suspected immune‐related adverse events with an anti‐PD‐1 inhibitor in otherwise healthy people with HIV. J Acquir Immune Defic Syndr 2021; 87: e234–e236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Sahin IH, Kane SR, Brutcher E et al. Safety and efficacy of immune checkpoint inhibitors in patients with cancer living with HIV: a perspective on recent progress and future needs. JCO Oncol Pract 2020; 16: 319–325. [DOI] [PubMed] [Google Scholar]
- 71. Howell DN, Andreotti PE, Dawson JR, Cresswell P. Natural killing target antigens as inducers of interferon: studies with an immunoselected, natural killing‐resistant human T lymphoblastoid cell line. J Immunol 1985; 134: 971–976. [PubMed] [Google Scholar]
- 72. Nunberg JH, Schleif WA, Boots EJ et al. Viral resistance to human immunodeficiency virus type 1‐specific pyridinone reverse transcriptase inhibitors. J Virol 1991; 65: 4887–4892. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary figures 1–3