

Abstract
Protein tyrosine phosphatases (PTPs) counteract the enzymatic activity of protein tyrosine kinases to modulate levels of both normal and disease-associated protein tyrosine phosphorylation. Aberrant activity of PTPs has been linked to the progression of a number of disease states, yet no PTP inhibitors are currently clinically available. PTPs are without a doubt a difficult drug target. Despite this, a number of selective, potent, and bioavailable PTP inhibitors have been described, suggesting PTPs should once again be looked at as viable therapeutic targets. Herein, we summarize recently discovered PTP inhibitors and their use in the functional interrogation of PTPs in disease states. In addition, an overview of the therapeutic targeting of PTPs is described using SHP2 as a representative target.
Introduction
Post-translational phosphorylation of tyrosine residues regulates a wide variety of cellular processes including cell survival, growth, and migration among other roles [1]. Tyrosine phosphorylation status is kept in check by the contrasting enzymatic activity of protein tyrosine kinases (PTKs) and protein tyrosine phosphatases (PTPs) that append or remove phosphate, respectively. Considering the scope of cellular functions that PTKs and PTPs are associated with, it comes as no surprise that dysregulation of tyrosine phosphorylation is implicated in a number of disease states including cancers, diabetes, and immune disfunction [2,3]. While therapeutic agents targeting PTKs are already available in the clinic, no such compounds exist for PTPs. PTPs represent a promising class of therapeutic targets for a number of reasons. Treatment of patients using kinase inhibitors initially shows promise though these molecules are eventually rendered ineffective through a variety of resistance mechanisms.[4] Given the interwoven nature of PTKs and PTPs, it is tempting to envision PTP inhibition as a novel route to modify the same cellular pathways that may be targeted by PTK inhibitors. Furthermore, PTPs remain relatively unexplored in the context of disease treatment in humans and might represent a treasure trove of novel therapeutic opportunities.
Among major challenges of targeting PTPs for drug discovery are the limited understanding of PTPs in disease biology and the general lack of PTP-specific small molecule probes for functional interrogation, target validation, and therapeutic development. Chemical probes can help address these issues by illuminating PTP druggability and providing the foundation for the development of novel therapeutics.
Genetic approaches have been widely employed to study the role of protein targets in disease state pathogenesis. The recent advent of CRISPR gene editing supplemented the already powerful genetic engineering tools such as RNA interference and gene knockout / overexpression techniques, providing scientists with a multitude of ways to quickly probe the role of proteins in a cellular environment [5]. However, these genetic approaches are limited in their ability to provide information on the temporal, spatial, and dynamic roles of enzymes in cellular signaling processes. Genetic manipulation of proteins may also result in confounding variables such as cellular compensation, resulting in unintended and unexpected cellular phenotypes. Furthermore, since these techniques usually result in complete ablation of proteins, the role of targets with multiple functions (e.g., enzymatic activity vs. protein-protein interactions) cannot be accurately delineated, limiting the applicability of this type of approach.
The availability of quality chemical probes is therefore of paramount importance [6]. In addition to complementing the variety of genetic techniques, small molecule probes are rapid and reversible, allowing for temporal interrogation of protein targets. In contrast to common genetic techniques, pharmacological probes must overcome potency and selectivity issues that are often time consuming and represent a significant hurdle in the development of quality probes.
Widely considered an “undruggable” class of enzymes, probe development for PTPs is hindered by two key factors [7]. First, residues composing the active site create a positively charged binding surface (Figure 1), and the high-affinity inhibitors that exploit this characteristic are thus negatively charged, reducing cell permeability. Second, the enzyme active site (pTyr pocket) is highly conserved among PTP enzymes, making selective PTP inhibitors difficult to develop. Despite these difficulties a number of potent and/or selective inhibitors have been developed for a variety of therapeutically relevant PTPs [8–10] including Phosphatase of regenerating liver (PRL), Mycobacterium tuberculosis PTP B (mPTPB), Low-molecular-weight protein tyrosine phosphatase (LMW-PTP), Mitogen-activated protein kinase phosphatase 5 (MKP5), and Src homology domain containing phosphatase 2 (SHP2) [11–17]. This minireview will focus on recently developed selective PTP inhibitors and their use in the functional interrogation of PTPs, as well as provide an understanding of the therapeutic value of PTP inhibitors using SHP2 as an example. While many quality inhibitors exist for PTPs, our goal is not to provide a comprehensive review of all inhibitors, but rather to showcase representative PTP inhibitors with various mechanisms of action (MOAs) including both allosteric and active site targeted inhibition (Table 1).
Figure 1.
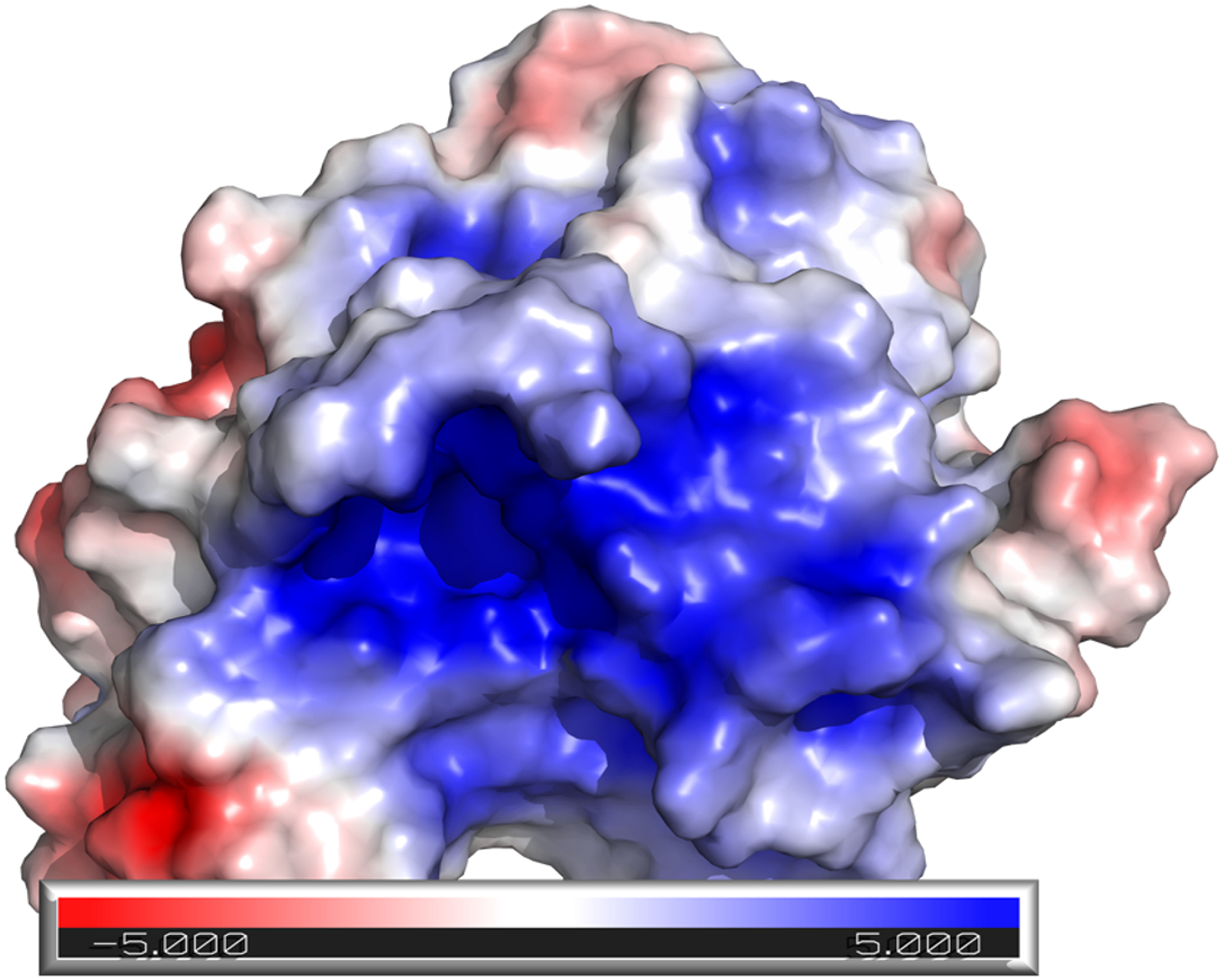
The active site of Src homology domain containing phosphatase 2 (SHP2) (PDB ID 3ZM0) is positively charged (blue surface), a characteristic that is common among many protein tyrosine phosphatases.
Table 1.
PTP inhibitors described in this review
| Target (Inhibitor) | Chemical Structure | Method of Discovery | Binding Site | Supporting Evidence |
|---|---|---|---|---|
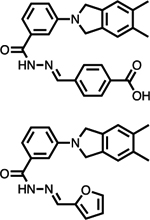
| PRL-1 (Cmpd 43 and Analog 3) |
|
Virtual HTS | Allosteric site - PRL-1 trimer interface | Crystal structure and biochemical data |
| mPTPB (4t) |
|
Fragment based and rational design | Active site | Docking and biochemical data |
| LMW-PTP (18 and 23) |
|
HTS | Active site | Crystal structure and biochemical data |
| MKP5 (Compound 1) |
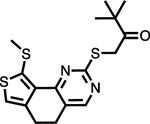
|
HTS | Allosteric site | Crystal structure and biochemical data |
| SHP2 (See Figure 3) | See Figure 3 | HTS, then rational design | Allosteric site | Crystal structure and biochemical data |
Functional interrogation of PTPs using small molecule inhibitors
Phosphatase of Regenerating Liver
Phosphatase of regenerating liver (PRL) enzymes consist of three homologous members, PRL 1, 2, and 3 [18–20]. Exogenous expression of PRLs promotes cell proliferation, and constitutive PRL expression increases the metastatic capabilities of cancer cells by activating the ERK1/2 and AKT pathways [21–23]. Furthermore, cells overexpressing PRL form highly metastatic tumors upon injection into mice, while knockdown of PRL reduces tumorigenesis in vivo [24]. In humans, elevated levels of PRL expression are observed in a number of cancers including colon [25], liver [26], ovarian [27], and others [20,28,29]. Additionally, PRL overexpression is strongly correlated to late-stage metastasis and poor clinical outcomes, providing further clinical significance. Taken together, these data implicate PRLs as oncogenes that are viable therapeutic targets [30].
PRLs can form homotrimers in both protein crystals and in solution, a property that is unique among phosphatases and has been shown to contribute to PRL-1 mediated cell growth and migration [31]. Leveraging this characteristic, it was hypothesized by Bai et al. that a selective inhibitor could be developed by binding to the interface of the PRL monomers, avoiding the drawbacks of active site inhibitors [11].
With three monomers forming the trimer complex in the PRL-1 crystal structure, it was rationalized that disrupting the interface between any one of these three monomers might preclude the formation of the trimer, thus reducing the PRL-1 mediated cell growth and migration. A structure-based virtual screen was carried out targeting the two different trimer interfaces on the PRL-1 monomer. Hits were validated biochemically, and the most effective detrimerizer of the hits was Cmpd-43. A structurally related molecule, Analog 3, was crystallographically confirmed to disrupt PRL-1 trimerization by binding to the interface between PRL-1 monomers. Cmpd-43 was shown to have no effect on the phosphatase activity of a large panel of phosphatases including PRL-1 at 20 μM (Figure 2A). Cmpd-43 was confirmed to attenuate ERK1/2 and AKT activity and reduce PRL-mediated cell proliferation and migration, while an inactive analog could not. Furthermore, Cmpd-43 had no effect on a cell line transfected with the trimerization deficient PRL1/G97R loss of function mutant. Taken together, these data provided strong evidence that Cmpd-43 selectively inhibits PRL-1 trimerization, resulting in an abrogation of PRL-1 mediated cellular signaling and a reduction in cell proliferation and migration. Furthermore, Cmpd-43 was able to reduce tumor volume and weight in a mouse xenograft model in a manner consistent with the previously observed cellular results.
Figure 2.
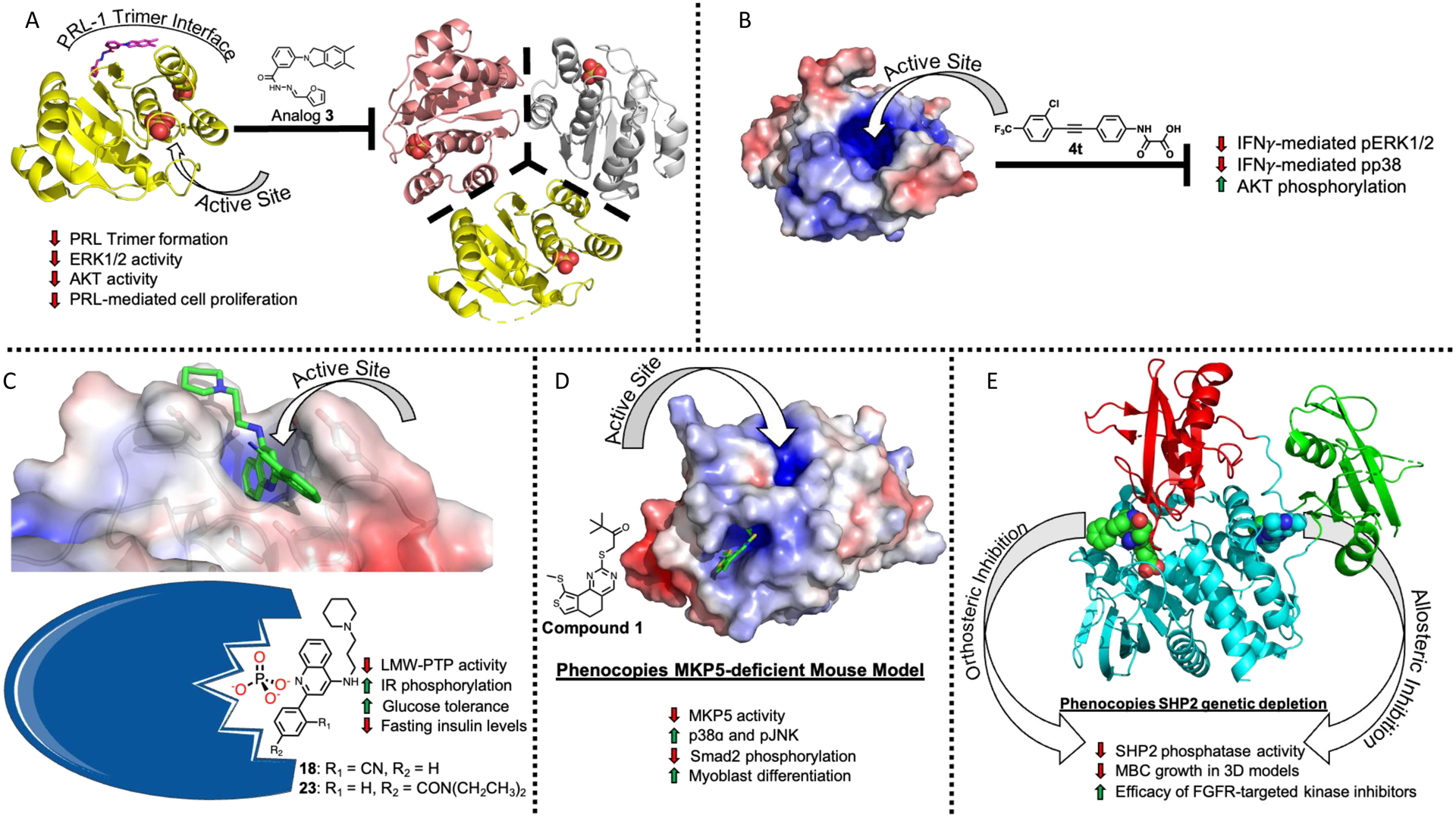
A) Analog 3 (magenta sticks) (PDB ID 5BX1) and Cmpd 43 (not shown) both prevent PRL-1 (yellow) from forming a homotrimer, resulting in decreased ERK1/2 activity, decreased AKT activity, and decreased cell proliferation. B) Inhibition of mPTPB by compound 4t prevents the interferon gamma (IFNγ) mediated phosphorylation of ERK1/2 and p38 and increase AKT phosphorylation. C) Uncompetitive inhibitor 18 (green sticks) reduces phosphatase activity of LMW-PTP (Cartoon depiction adapted from Servier Medical Art under the Creative Commons Attribution 3.0), resulting in increased insulin receptor (IR) phosphorylation, increased glucose tolerance, and decreased fasting insulin levels, linking LMW-PTP activity to diabetes. D) Allosteric inhibition of MPK5 by compound 1 (green sticks) results in decreased MPK5 activity and increased levels of p38ɑ and pJNK, leading to decreased Smad2 phosphorylation levels and increased myoblast differentiation. E) Active site inhibitors (green spheres) and allosteric inhibition (blue spheres) phenocopy genetic depletion of SHP2, decreasing growth of metastatic breast cancer tumors in 3D models and increasing efficacy of FGFR-targeted kinase inhibitors.
Protein-protein interactions (PPIs) are inherently difficult to target, characterized by large, flat, highly hydrophobic surfaces.[32,33] Thus, it is difficult to generate high-affinity small molecule inhibitors for these types of binding sites. However, with the crystal structure available for the PRL-1 detrimerizer, it is possible that structure-based design may be employed to optimize Cmpd-43 to overcome this issue of potency.
Since the discovery of these detrimerizing compounds, a number of PRL-1 active site inhibitors have also been disclosed, confirming the importance of this target.[34,35] Collectively these observations provide strong evidence for the functional requirement for PRL trimerization and suggest that inhibition of PRL trimerization is a viable mechanism for developing novel therapeutics for PRL-associated diseases.
Mycobacterium tuberculosis protein tyrosine phosphatases
Mycobacterium tuberculosis (Mtb), the pathogenic cause of tuberculosis (Tb) infected approximately 10 million people in 2019 alone (WHO, 2020 tuberculosis report), causing 1.4 million deaths according to the World Health Organization (WHO). Tb is the leading cause of death worldwide from a single agent, and while substantial efforts have been made to reduce the morbidity and mortality of Tb, progress is hampered by a number of factors including drug resistance [36]. Thus, novel Tb targets are of great interest in the fight against Tb.
Tb encodes for a phosphatase, mPTPB, which is secreted into the cytoplasm of host macrophages, promoting bacterial survival by altering the host macrophage signaling [37]. Mechanistically, mPTPB has been shown to attenuate the innate immune responses by blocking the ERK1/2 and p38 kinase mediated IL-6 production and prevent cell death through activation of the AKT pathway in the macrophage [38]. Therefore, mPTPB represents a promising target for anti-Tb drug development.
A recently published potent and selective inhibitor of mPTPB was used to confirm previously observed cellular effects from pharmacological inhibition [12]. Briefly, compound 4t increased the phosphorylation status of ERK1/2 and p38 and decreased AKT phosphorylation (Figure 2B). These observations are similar to those invoked by structurally unrelated small molecule mPTPB inhibitors [39,40], indicating that the noted cellular effects of 4t in macrophages are indeed from specific mPTPB inhibition. With corroborating results between distinct probe series and genetic approaches, it is clear that pharmacological inhibition of mPTPB represents a viable route towards the development of novel Tb therapeutic agents, and other groups are continuing drug discovery efforts targeting this protein, denoting the importance of this target for the development of anti-Tb agents.[41] Existence of these inhibitors also supports the concept that selective, potent, and drug-like active site inhibitors are possible to develop.
Low molecular weight protein tyrosine phosphatase
Low-molecular-weight protein tyrosine phosphatase (LMW-PTP) is a ubiquitously expressed PTP whose function is not fully understood and appears to be disease state dependent [13]. The 18 kDa LMW-PTP is found in the cytosol of both prokaryotes and eukaryotes and has been shown to regulate a wide variety of signaling cascades and receptors, including the insulin receptor (IR) [42]. As such, LMW-PTP has been proposed as a therapeutically relevant target for type 2 diabetes (T2D). Studies have shown that decreased LMW-PTP activity is protective against increased blood sugar levels in both diabetic and nondiabetic groups [43] and genetic manipulation of LMW-PTP levels decreases insulin resistance in mice and increases IR phosphorylation in murine liver and fat cells [44], suggesting that LMW-PTP is directly responsible for IR phosphorylation status through its phosphatase activity [42,45]. Taken together, these data suggest that LMW-PTP represents a promising therapeutic target for modulating T2D and insulin resistance.
In a recent study by Stanford et al. a global knockout (KO) murine model was used to demonstrate that global deletion of LMW-PTP reduced diabetes in LMW-PTP KO mice that were fed a high-fat diet compared to the wildtype (WT) control group [14]. LMW-PTP is not tissue specific, so the authors sought to determine if results from the global KO mouse model may be attributed to certain tissues. Indeed, the phenotypic results from the global LMW-PTP KO model were recapitulated only when LMW-PTP was deleted in the liver. Further investigation provided evidence that IR tyrosine phosphorylation was increased in the LMW-PTP KO mice compared to the control group, with increased phosphorylation levels of AKT and ERK, downstream proteins of the IR pathway.
To identify novel LMW-PTP inhibitors without highly charged functional groups, a high-throughput screen (HTS) was carried out to find inhibitors that did not display a competitive mode of inhibition, yielding a number of hits after a robust series of counter-screening and validation [14]. Structure-activity relationship (SAR) experiments for one hit resulted in the uncompetitive inhibitor 18, with a half-maximal inhibitor concentration (IC50) value of 0.46 μM and significant selectivity towards both isoforms of LMW-PTP. Compound 18 was determined to form a ternary complex with LMW-PTP and vanadate via crystallography, corroborating the uncompetitive mechanism of inhibition. The selectivity of 18 was explained by the structural information; compared to other PTPs, 18 is able to prevent hydrolysis of only the LMW-PTP phsophocysteine. As an example, the authors point to PTP1B, where the active site could accommodate both 18 and water molecules simultaneously, reducing the ability of 18 to prevent phosphocysteine hydrolysis.
With this novel and selective probe series in hand, the functional interrogation of LMW-PTP’s role in IR phosphorylation and diabetes was carried out [14]. Treatment of human hepatocytes with the orally bioavailable and selective analog 23 resulted in increased IR phosphorylation, matching the observed effect of the LMW-PTP KO model (Figure 2C) [14]. Compound 23 was well tolerated by diabetes-induced obese mice that were fed a high-fat diet, with no change in body weight being observed. Additionally, both glucose tolerance and decreased fasting insulin levels were improved. Similar to the observed results in the cellular experiments, treatment of mice with 23 for two weeks resulted in an increased level of IR tyrosine phosphorylation, reducing phosphorylation of downstream AKT and ERK; these results were not observed in a liver-specific knockout mouse model treated with 23, suggesting that the phenotypic effects of 23 are specifically from LMW-PTP inhibition. Taken together, these results provide critical evidence supporting the role of LMW-PTP as a novel therapeutic target in diabetes that is worthy of further exploration.
Mitogen-activated protein kinase phosphatase 5
Mitogen-activated protein kinases (MAPKs) are regulated by MAPK phosphatases (MKPs), a family of dual-specificity phosphatases (DUSPs) [46]. MAPKs are involved in critical signal transduction pathways, and abnormal activity of MAPKs is integral to a number of diseases including cancer [47], diabetes [48], and others [49]. MKPs are able to regulate the MAPKs by removing phosphate groups from threonine and tyrosine side chain hydroxyls on MAPK activation loops [50]. The specificity of the MPKs is attributed to a kinase interaction motif that binds to specific MAPK substrates [51,52].
MKP5 has emerged as a potential therapeutic target for Duchenne muscular dystrophy (DMD) [53], a genetic disorder that prevents the ability of skeletal muscle to regenerate. This results in the replacement of skeletal muscle with fibrotic tissue, which is inevitably fatal [54–56]. The transforming growth factor-β1 (TGF- β1) pathway is partially responsible for the development of fibrosis, and as such represents a strategic route for the treatment of DMD [57]. Interestingly, mice that lack MKP5 expression show an enhanced ability to regenerate muscle tissue, and a MKP5 KO mouse model reduces the DMD phenotype [53]. This suggests a function for MKP5 in DMD, though the mechanistic role was not clear.
To this end, Gannam et al. developed an HTS biased to identify allosteric inhibitors using a phosphopeptide instead of a small molecule substrate [15]. After retesting and triaging compounds, the resulting 27 molecules were tested for selectivity against a panel of DUSPs. The most interesting molecule, compound 1, displayed an IC50 value of 3.9 μM and displayed a 51-fold and 25-fold selectivity window against STEP-46 and PTP1B. Kinetic characterization of compound 1 revealed a mixed mechanism of inhibition by slowing down the rate of enzymatic catalysis. Co-crystallization of MKP5 and compound 1 provided insight into the results from the kinetic experiments. Compound 1 was determined to bind to a previously unrecognized allosteric site on MKP5. Interestingly, binding of compound 1 results in a conformational shift in the backbone of residues that form the active site, reducing the volume of the active site by 18%. Mutational analysis of the allosteric binding pocket revealed that the amino acids responsible for the interactions between compound 1 and MKP5 are required for catalysis and are not conserved across MKPs, explaining the selectivity of compound 1 for MKP5.
Discovery of compound 1 provided the ability to explore the role of MKP5 in disease states [15]. Corroborating previously observed results, treatment of mouse myoblasts (C2C12 cells) with compound 1 resulted in a dose-dependent increase in p38ɑ and pJNK while having no effect on pERK1/2. Compound 1 was able to phenocopy MKP5-deficient mice and significantly increase myoblast differentiation, suggesting that selective inhibition of MKP5 by compound 1 increases levels of pJNK and pMAPK (Figure 2D). Hypothesizing that MKP5 is involved in the TGF-β1 driven progression of fibrosis, the authors investigated how a MKP5 knockout mouse model effects enzymes downstream of the TGF-β1 pathway. Mice lacking MKP5 (MKP5−/−) had reduced Smad2 phosphorylation levels compared to wild-type control mice. Smad2 phosphorylation was significantly decreased in the TGF-β1 MKP5−/− embryonic fibroblasts compared to the MKP5+/+ control cells while pJNK and pp38MAPK levels were increased with no change in pERK1/2. Furthermore, treatment of MKP5−/− cells with compound 1 displayed no additional effects on Smad2 phosphorylation levels or on pJNK, pMAPK, and pERK1/2, suggesting that compound 1 selectively targets MKP5 to counter TGF-β1 signaling in DMD. While more work needs to be done to further the understanding of MKP5’s role in muscular dystrophy, these results clearly indicate that MKP5 is a viable therapeutic target for the treatment of DMD. Furthermore, the authors suggest that allosteric inhibition may be applicable to target other members of the MKP family, opening an avenue for the development of additional therapeutic agents.
Therapeutic targeting of SHP2
As one of the most highly studied PTPs, Src homology domain containing phosphatase 2 (SHP2) is considered a bona fide oncogene [7]. An in-depth analysis of all SHP2 inhibitors is beyond the scope of this mini review, thus we will focus on the recent use of SHP2 inhibitors and the therapeutic targeting of SHP2 through the use of these molecules. Reviews covering the variety of different SHP2 inhibitors are available elsewhere [58–61].
SHP2 resides upstream of RAS and functions as a shared signaling node [62–65]. In its basal state, SHP2 is autoinhibited by interactions between its N-SH2 and PTP domains, blocking the catalytic site from binding its substrate [66]. As such, the phosphatase activity of full length wild-type SHP2 is 5% of the catalytic domain alone [67]. However, upon binding to specific pTyr motifs on receptor PTKs, SHP2 is concomitantly localized to its intracellular substrates and brought into an active conformation [68]. The N-SH2 and C-SH2 domains interact with the pTyr motifs, weakening the intramolecular N-SH2/PTP interaction and revealing the active site, a requirement for the full activation of the RAS and ERK1/2 pathway [7]. Inhibition of SHP2 by both orthosteric and allosteric mechanisms phenocopy genetic depletion, resulting in decreased phosphatase activity and decreased growth of metastatic breast cancer cells in 3D models (Figure 2E) [69].
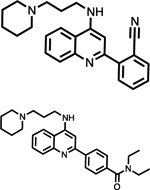
In 2016, scientists from Novartis revolutionized the PTP inhibitor field with their discovery of the first allosteric SHP2 inhibitors [70]. Assay design was key to this discovery; it was reasoned that screening compounds against SHP2 in an equilibrium between the open and closed conformation would bias results towards allosteric rather than orthosteric hits. The resulting molecule was determined to inhibit the full length SHP2 construct with greater efficacy than towards the PTP domain alone. Indeed, as was their intention, the inhibitor was shown to bind at the interface between all three domains, stabilizing the inactive conformation of SHP2. Structure-based optimization resulted in the first generation allosteric SHP2 inhibitor SHP099 (Figure 3), with IC50 values of 0.07 μM and 0.62 μM against SHP2 and pERK respectively. In an attempt to optimize SHP099, Bagdanoff et al. employed scaffold hopping in combination with structural information from both previous and novel SHP2 allosteric inhibitors [71]. This resulted in SHP389 (Figure 3), an improved analog of SHP099 whose development was ultimately terminated due to sub-optimal drug-like properties. However, this study provided critical information about the key structural features required for binding to the allosteric site of SHP2. Eventually, Lamarche et al. described the development of TNO155 (Figure 3), an orally available inhibitor of SHP2 (IC50 = 0.011 μM) that is currently in phase 1 clinical trials [72]. Inspired by this series, a number of SHP2 allosteric inhibitors were described by other groups; an in-depth review of these molecules, their SAR, and efficacies is described by Yuan et al. and will not be discussed here [58]. Of these SHP2 allosteric inhibitors, multiple are currently being tested in clinical trials as either a monotherapy or in combination with other approved anticancer therapies (Table 2) [73].
Figure 3.
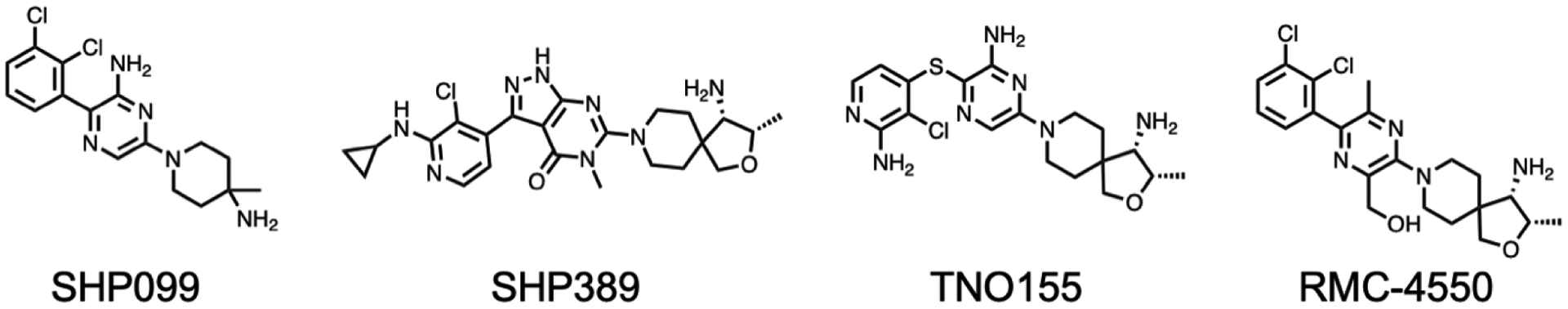
Structures of select SHP2 allosteric inhibitors mentioned in this text.
Table 2.
Allosteric SHP2 inhibitors in clinical trials
| Drug | Company | Status | Indication | Combination | NCT ID |
|---|---|---|---|---|---|
| JAB-3068 | Jacobio Pharmaceuticals | Phase I (Ongoing) |
|
Monotherapy | NCT03518554 |
| JAB-3068 | Jacobio Pharmaceuticals | Phase I/II (Ongoing) |
|
Monotherapy | NCT03565003 |
| JAB-3312 | Jacobio Pharmaceuticals | Phase I (Ongoing) |
|
Monotherapy | NCT04121286 |
| JAB3312 | Jacobio Pharmaceuticals | Phase I/II (Ongoing) |
|
Combination therapy with Pembrolizumab or Binimetinib | NCT04720976 |
| JAB-3312 | Jacobio Pharmaceuticals | Phase I (Ongoing) |
|
Monotherapy | NCT04045496 |
| TNO155 | Novartis Pharmaceuticals | Phase I (Ongoing) |
|
Mono therapy or combined with EGF816 (Nazartinib) | NCT03114319 |
| TNO155 | Novartis Pharmaceuticals | Phase I (Ongoing) |
|
Combination therapy with Ribociclib or Spartalizumab | NCT04000529 |
| TNO155 | Mirati Therapeutics Inc. and Novartis Pharmaceuticals | Phase I/II (Ongoing) |
|
Combination therapy of MRTX849 with TNO155 | NCT04330664 |
| TNO155 | Novartis Pharmaceuticals | Phase I (Ongoing) |
|
Multiple of Dabrafenib with other drugs, including TNO155 | NCT04294160 |
| RMC-4630 | Revolution Medicines, Inc. and Sanofi | Phase I (Ongoing) |
|
Monotherapy | NCT03634982 |
| RMC-4630 | Revolution Medicines, Inc. and Sanofi | Phase I (Ongoing) |
|
Combination of RMC-4630 with Cobimetinib or Osimertinib | |
| RLY-1971 | Relay Therapeutics, Inc. | Phase I (Ongoing) |
|
Monotherapy | NCT04252339 |
| BBP-398 | Navire Pharmaceuticals | Phase I (Ongoing) |
|
Monotherapy | NCT04528836 |
| ERAS-601 | Erasca, Inc. | Phase I (Ongoing) |
|
Monotherapy or in combination with undescribed MEK inhibitor | NCT04670679 |
| N/A | Amgen | Phase I (Ongoing) |
|
Sotorasib in combination with other therapies, including undescribed SHP2 inhibitor. | NCT04185883 |
All of these trials are in progress at the time of this writing, so very little information regarding efficacy, safety, and tolerability is available. However, preliminary data regarding the efficacy of RMC-4630 (Structure unavailable) in patients with KRAS-mutant non-small cell lung cancer has been published. In this study, 56 patients are being treated with RMC-4630, and 19/23 patients diagnosed with non-small cell lung cancer (NSCLC) harbor a KRAS mutation [74]. The disease control rate for patients with KRASG12C mutations is 71%, and tumor volume was reduced in three of these patients, suggesting preliminary efficacy. Additionally, one patient with a KRASG12D oncogenic mutation displayed antitumor activity when treated with RMC-4630. Evaluation of blood cells and tumor biopsies suggested on-target efficacy according to pERK levels.
Though information regarding the efficacy of SHP2 inhibitors in clinical trials is sparce, the preclinical observations appear promising. Two groups independently identified SHP2 as a target for KRAS-mutant NSCLC cells, with synergism occurring when both MEK and SHP2 were targeted concomitantly using selumetinib or trametinib and SHP099, supporting the concept of the MEK/SHP2 combination therapies in clinical trials [75,76]. Another group used RMC-4550 (Figure 3) to show the reliance of BRAF, NF1, and KRAS oncogenic mutants on the catalytic activity of SHP2 [77]. These RAS pathway oncogenes are often observed in NSCLC and, until very recently were not treatable with pharmacological intervention[78], suggesting SHP2 may function as a novel therapeutic target for patients with cancers reliant on these proteins [77]. An additional study from Wong et. al. provided further support for the combinatorial targeting of MEK and SHP2, where it was determined that pharmacological inhibition of SHP2 proved an effective therapeutic option for cancers where MEK inhibitors were no longer viable due to KRAS amplification [79]. Further support for the clinical evaluation of SHP2 inhibitors was detailed by Ahmed et al., where SHP2 was determined to reduce the efficacy of BRAF and MEK inhibitors in ERK-dependent tumors [80]. The combined inhibition of MEK and SHP2 was efficacious against a variety of cancer cell lines, though the success of this approach was dependent on a variety of factors and may not be broadly applicable [80]. Very recently, Liu et al. provided yet further supporting evidence for the utility of SHP2 inhibitor combination therapies. Inhibition of SHP2 using TNO155 was able to overcome resistance to EGFR inhibitors osimertinib and nazartinib by preventing RTK-mediated MAPK pathway reactivation [81]. Additionally, because of RAS-MAPK feedback pathway regulation, combination of TNO155 with MEK inhibitors was also shown to be synergistic, though this approach may be limited due to insufficient tolerability. The most effective combination during this study was seen when using both TNO155 and KRASG12C inhibitors, a promising result that is supportive of the potential success for clinical trials using this approach [81]. Taken together, it is apparent that there is abundant preclinical data to support the successful outcomes of these SHP2 mono and combination therapy based clinical trials.
Conclusion
Despite the obvious importance of PTPs in disease state pathogenesis, many have given up on this class of targets, describing them as “undruggable”. From a drug discovery standpoint, the difficulty of this class stems from the high sequence homology and highly charged active site. Many of the high-affinity inhibitors that were a result of early drug discovery campaigns often had multiple negative charges resulting in reduced bioavailability and cell permeability. However, recent developments have shown that not only is it possible to develop potent and selective PTP inhibitors, but these inhibitors can also be very drug-like. Moreover, allosteric PTP inhibitors that modulate unique regulatory mechanisms represent another productive avenue for probe and drug development. Whether or not the clinical trials for the allosteric SHP2 inhibitors are successful is yet to be determined. However, it is clear that the discovery of these inhibitors has reinvigorated the field and PTPs should be once again considered highly valuable therapeutic targets.
Perspective.
Protein tyrosine phosphatases represent a heavily underleveraged family of enzymes for the development of novel therapeutic agents. However, novel probes are required to bolster the confidence of the community that these compounds are druggable, enabling studies to validate specific PTPs in a disease-dependent context.
Widely considered “undruggable” and given up on as viable drug targets, it is apparent that PTPs are high-value targets that may be targeted with drug-like small molecules by targeting allosteric binding sites in addition to the active site. Novel mechanisms of action have been utilized to develop PTP inhibitors that are effective and drug-like.
Additional efforts are necessary to expand the PTP inhibitor toolbox. We hypothesize that new and re-emerging techniques such as proteolysis targeting chimeras and targeted covalent inhibitors will be utilized in the development of novel PTP probes, reinvigorating the PTP inhibitor field.
Funding:
This work was supported in part by NIH R01CA69202 and R01CA207288.
Footnotes
Conflicts of Interest: The authors declare no conflict of interest.
References
- [1].Tonks NK (2006) Protein tyrosine phosphatases: From genes, to function, to disease. Nat. Rev. Mol. Cell. Biol 7, 833–846 10.1038/nrm2039 [DOI] [PubMed] [Google Scholar]
- [2].Yu Z-H and Zhang Z-Y (2018) Regulatory Mechanisms and Novel Therapeutic Targeting Strategies for Protein Tyrosine Phosphatases. Chem. Rev 118, 1069–1091 10.1021/acs.chemrev.7b00105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Tonks NK (2013) Protein tyrosine phosphatases – from housekeeping enzymes to master regulators of signal transduction. FEBS J. 280, 346–378 10.1111/febs.12077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Lovly CM, Shaw AT (2014) Molecular pathways: Resistance to kinase inhibitors and implications for therapeutic strategies. Clin. Cancer Res 20, 2249–2256 doi: 10.1158/1078-0432.CCR-13-1610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Jiang F and Doudna JA (2017) CRISPR – Cas9 Structures and Mechanisms. Annu. Rev Biophys 46, 505–531. 10.1146/annurev-biophys-062215-010822 [DOI] [PubMed] [Google Scholar]
- [6].Arrowsmith CH, Audia JE, Austin C, Baell J, Bennett J, Blagg J et al. (2015) The promise and peril of chemical probes. Nat. Chem. Biol 11, 536–541. 10.1038/nchembio.1867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Frankson R, Yu Z-H, Bia Y, Li Q, Zhang R-Y and Zhang Z-Y, (2017) Therapeutic Targeting of Oncogenic Tyrosine Phosphatases. Cancer Res. 77, 5701–5705 10.1158/0008-5472.CAN-17-1510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Zhang Z-Y (2017) Drugging the Undruggable: Therapeutic Potential of Targeting Protein Tyrosine Phosphatases. Acc. Chem. Res 50, 122–129 10.1021/acs.accounts.6b00537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Stanford SM and Bottini N (2017) Targeting Tyrosine Phosphatases: Time to End the Stigma. Trends Pharmacol. Sci 38, 524–540 10.1016/j.tips.2017.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Krishnan N, Koveal D, Miller DH, Xue B, Akshinthala SD, Kragelj J et al. (2014) Targeting the disordered C terminus of PTP1B with an allosteric inhibitor. Nat. Chem. Biol 10, 558–566. 10.1038/nchembio.1528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Bai Y, Yu ZH, Liu S, Zhang L, Zhang RY, Zeng LF et al. (2016) Novel anticancer agents based on targeting the trimer interface of the PRL phosphatase. Cancer Res. 76, 4805–4815 DOI: 10.1158/0008-5472.CAN-15-2323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Ruddraraju KV, Aggarwal D, Niu C, Baker EA, Zhang RY, Wu L et al. (2020) Highly Potent and Selective N-Aryl Oxamic Acid-Based Inhibitors for Mycobacterium tuberculosis Protein Tyrosine Phosphatase B. J. Med. Chem 63, 9212–9227 10.1021/acs.jmedchem.0c00302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].He R, Wang J, Yu Z-H, Zhang R-Y, Liu S, Wu L et al. (2016) Inhibition of low molecular weight protein tyrosine phosphatase by an induced-fit mechanism. J. Med. Chem 59, 9094–9106 doi: 10.1021/acs.jmedchem.6b00993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Stanford SM, Aleshin AE, Zhang V, Ardecky RJ, Hedrick MP, Zou J et al. (2017) Diabetes reversal by inhibition of the low-molecular-weight tyrosine phosphatase. Nat. Chem. Biol 13, 624–632 10.1038/nchembio.2344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Gannam ZTK, Min K, Shillingford SR, Zhang L, Herrington J, Abriola L et al. (2020) An allosteric site on MKP5 reveals a strategy for small-molecule inhibition. Sci. Signal 13, eaba3043 10.1126/scisignal.aba3043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Garcia Fortanet J, Chen CHT, Chen YNP, Chen Z, Deng Z, Firestone B et al. (2016) Allosteric Inhibition of SHP2: Identification of a Potent, Selective, and Orally Efficacious Phosphatase Inhibitor. J. Med. Chem 59, 7773–7782 10.1021/acs.jmedchem.6b00680 [DOI] [PubMed] [Google Scholar]
- [17].Zeng LF, Zhang RY, Yu ZH, Li S, Wu L, Gunawan AM et al. (2014) Therapeutic potential of targeting the oncogenic SHP2 phosphatase. J. Med. Chem 57, 6594–6609 10.1021/jm5006176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Bessette DC, Qiu D and Pallen CJ (2008) PRL PTPs: Mediators and markers of cancer progression. Cancer Metastasis Rev. 27, 231–252 10.1007/s10555-008-9121-3 [DOI] [PubMed] [Google Scholar]
- [19].Rios P, Li X and Köhn M (2013) Molecular mechanisms of the PRL phosphatases. FEBS J. 280, 505–524 10.1111/j.1742-4658.2012.08565.x [DOI] [PubMed] [Google Scholar]
- [20].Stephens BJ, Han H, Gokhale V and von Hoff DD (2005) PRL phosphatases as potential molecular targets in cancer. Mol. Cancer Ther 4, 1653–1661 10.1158/1535-7163.MCT-05-0248 [DOI] [PubMed] [Google Scholar]
- [21].Mu N, Gu J, Liu N, Xue X, Shu Z, Zhang K et al. (2018) PRL-3 is a potential glioblastoma prognostic marker and promotes glioblastoma progression by enhancing MMP7 through the ERK and JNK pathways. Theranostics. 8, 1527–1539 10.7150/thno.22699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Zhang Y, Li Z, Fan X, Xiong J, Zhang G, Luo X et al. (2018) PRL-3 promotes gastric cancer peritoneal metastasis via the PI3K/AKT signaling pathway in vitro and in vivo. Oncol. Lett 15, 9069–9074 10.3892/ol.2018.8467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Stephens B, Han H, Hostetter G, Demeure MJ and von Hoff DD (2008) Small interfering RNA-mediated knockdown of PRL phosphatases results in altered Akt phosphorylation and reduced clonogenicity of pancreatic cancer cells. Mol. Cancer Ther 7, 202–210 10.1158/1535-7163.MCT-07-0542 [DOI] [PubMed] [Google Scholar]
- [24].Hardy S, Wong NN, Muller WJ, Park M and Tremblay ML (2010) Overexpression of the protein tyrosine phosphatase PRL-2 correlates with breast tumor formation and progression. Cancer Res. 70, 8959–8967 10.1158/0008-5472.CAN-10-2041 [DOI] [PubMed] [Google Scholar]
- [25].Peng L, Ning J, Meng L and Shou C (2004) The association of the expression level of protein tyrosine phosphatase PRL-3 protein with liver metastasis and prognosis of patients with colorectal cancer. J. Cancer Res. Clin. Oncol 130, 521–526 10.1007/s00432-004-0563-x [DOI] [PubMed] [Google Scholar]
- [26].Jin S, Wang K, Xu K, Xu J, Sun J, Chu Z et al. (2014) Oncogenic function and prognostic significance of protein tyrosine phosphatase PRL-1 in hepatocellular carcinoma. Oncotarget. 5, 3685–3696 10.18632/oncotarget.1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ren T, Jiang B, Xing X, Dong B, Peng L, Meng L et al. (2009) Prognostic significance of phosphatase of regenerating liver-3 expression in ovarian cancer. Pathol. Oncol. Res 15, 555–560 10.1007/s12253-009-9153-1 [DOI] [PubMed] [Google Scholar]
- [28].Wang Q, Holmes DIR, Powell SM, Lu QL and Waxman J (2002) Analysis of stromal-epithelial interactions in prostate cancer identifies PTPCAAX2 as a potential oncogene. Cancer Lett. 175, 63–69 10.1016/s0304-3835(01)00703-0 [DOI] [PubMed] [Google Scholar]
- [29].Radke I, Götte M, Kersting C, Mattsson B, Kiesel L and Wülfing P (2006) Expression and prognostic impact of the protein tyrosine phosphatases PRL-1, PRL-2, and PRL-3 in breast cancer. Br. J. Cancer 95, 347–354 10.1038/sj.bjc.6603261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Li Q, Bai Y, Tiffany Lyle L, Yu G, Amarasinghe O, Meke FN et al. (2020) Mechanism of PRL2 phosphatase-mediated PTEN degradation and tumorigenesis. Proc. Nat. Acad. Sci. U S A 117, 20538–20548 10.1073/pnas.2002964117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Sun JP, Luo Y, Yu X, Wang WQ, Zhou B, Liang F et al. (2007) Phosphatase activity, trimerization, and the C-terminal polybasic region are all required for PRL1-mediated cell growth and migration. J. Biol Chem 282, 29043–29051 10.1074/jbc.M703537200 [DOI] [PubMed] [Google Scholar]
- [32].Mabonga L, Kappo AP (2019) Protein-protein interaction modulators: advances, successes and remaining challenges. Biophys. Rev 11, 559–581. 10.1007/s12551-019-00570-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Lu H, Zhou Q, He J, Jiang Z, Peng C, Tong R et al. (2020) Recent advances in the development of protein–protein interactions modulators: mechanisms and clinical trials. Signal Transduction and Targeted Therapy. Signal. Transduct. Target Ther 5(1), 213 10.1038/s41392-020-00315-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Lazo JS, Blanco IK, Tasker NR, Rastelli EJ, Burnett JC, Garrott SR et al. (2019) Next-generation cell-active inhibitors of the undrugged oncogenic PTP4A3 phosphatase. J. Pharmacol. Exp. Ther 371, 652–662. 10.1124/jpet.119.262188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Rastelli EJ, Sannino S, Hart DJ, Sharlow ER, Lazo JS, Brodsky JL et al. (2021) Synthesis and evaluation of bifunctional PTP4A3 phosphatase inhibitors activating the ER stress pathway. Bioorg. Med. Chem. Lett 46, 128167. 10.1016/j.bmcl.2021.128167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Schön T, Miotto P, Köser CU, Viveiros M, Böttger E and Cambau E (2017) Mycobacterium tuberculosis drug-resistance testing: challenges, recent developments and perspectives. Clin. Microbiol. Infect 23, 154–160 10.1016/j.cmi.2016.10.022 [DOI] [PubMed] [Google Scholar]
- [37].Ruddraraju KV, Aggarwal D and Zhang Z-Y (2021) Therapeutic targeting of protein tyrosine phosphatases from mycobacterium tuberculosis. Microorganisms. 9, 1–13 10.3390/microorganisms9010014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Zhou B, He Y, Zhang X, Xu J, Luo Y, Wang Y et al. (2010) Targeting mycobacterium protein tyrosine phosphatase B for antituberculosis agents. Proc. Nat. Acad. Sci. U S A 107, 4573–4578 10.1073/pnas.0909133107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].He Y, Xu J, Yu ZH, Gunawan AM, Wu L, Wang L et al. (2013) Discovery and evaluation of novel inhibitors of mycobacterium protein tyrosine phosphatase B from the 6-hydroxy-benzofuran-5-carboxylic acid scaffold. J. Med. Chem 56, 832–842 10.1021/jm301781p [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].He R, Yu ZH, Zhang RY, Wu L, Gunawan AM and Zhang Z-Y (2015) Cefsulodin Inspired Potent and Selective Inhibitors of mPTPB, a Virulent Phosphatase from Mycobacterium tuberculosis. ACS Med. Chem. Lett 6, 1231–1235 10.1021/acsmedchemlett.5b00373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Vickers CF, Silva APG, Chakraborty A, Fernandez P, Kurepina N, Saville C et al. (2018) Structure-Based Design of MptpB Inhibitors That Reduce Multidrug-Resistant Mycobacterium tuberculosis Survival and Infection Burden in Vivo. J. Med. Chem 61, 8337–8352. 10.1021/acs.jmedchem.8b00832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Chiarugi P, Cirri P, Marra F, Raugei G, Camici G, Manao G et al. (1997) LMW-PTP is a negative regulator of insulin-mediated mitotic and metabolic signalling. Biochem. Biophys. Res. Commun 238, 676–682 10.1006/bbrc.1997.7355 [DOI] [PubMed] [Google Scholar]
- [43].Iannaccone U, Bergamaschi A, Magrini A, Marino G, Bottini N, Lucarelli P et al. (2005) Serum glucose concentration and ACP1 genotype in healthy adult subjects. Metabolism. 54, 891–894 10.1016/j.metabol.2005.02.002 [DOI] [PubMed] [Google Scholar]
- [44].Pandey SK, Yu XX, Watts LM, Michael MD, Sloop KW, Rivard AR et al. (2007) Reduction of low molecular weight protein-tyrosine phosphatase expression improves hyperglycemia and insulin sensitivity in obese mice. J. Biol. Chem 282, 14291–14299 10.1074/jbc.M609626200 [DOI] [PubMed] [Google Scholar]
- [45].Stefani M, Caselli A, Bucciantini M, Pazzagli L, Dolfi F, Camici G et al. (1993) Dephosphorylation of tyrosine phosphorylated synthetic peptides by rat liver phosphotyrosine protein phosphatase isoenzymes. FEBS Lett. 326, 131–134 10.1016/0014-5793(93)81776-v [DOI] [PubMed] [Google Scholar]
- [46].Caunt CJ and Keyse SM (2013) Dual-specificity MAP kinase phosphatases (MKPs): Shaping the outcome of MAP kinase signalling. FEBS J. 280, 489–504 10.1111/j.1742-4658.2012.08716.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Braicu C, Buse M, Busuioc C, Drula R, Gulei D, Raduly L et al. (2019) A Comprehensive Review on MAPK: A Promising Therapeutic Target in Cancer. Cancers (Basel). 11, 1–25 10.3390/cancers11101618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Sidarala V and Kowluru A (2016) The Regulatory Roles of Mitogen-Activated Protein Kinase (MAPK) Pathways in Health and Diabetes: Lessons Learned from the Pancreatic β-Cell. Recent Pat. Endocr. Metab. Immune Drug Discov 10, 76–84 10.2174/1872214810666161020154905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Lawrence MC, Jivan A, Shao C, Duan L, Goad D, Zaganjor E et al. (2008) The roles of MAPKs in disease. Cell Res. 18, 436–442 10.1038/cr.2008.37 [DOI] [PubMed] [Google Scholar]
- [50].Farooq A and Zhou MM (2004) Structure and regulation of MAPK phosphatases. Cell. Signal 16, 769–779 10.1016/j.cellsig.2003.12.008 [DOI] [PubMed] [Google Scholar]
- [51].Camps M, Nichols A, Gillieron C, Antonsson B, Muda M, Chabert C et al. (1998) Catalytic activation of the phosphatase MKP-3 by ERK2 mitogen-activated protein kinase. Science. 280, 1262–1265 10.1126/science.280.5367.1262 [DOI] [PubMed] [Google Scholar]
- [52].Zhang YY, Wu JW and Wang ZX (2011) A distinct interaction mode revealed by the crystal structure of the kinase p38a with the MAPK binding domain of the phosphatase MKP5. Sci. Signal 4, 1–10 10.1126/scisignal.2002241 [DOI] [PubMed] [Google Scholar]
- [53].Shi H, Verma M, Zhang L, Dong C, Flavell RA and Bennett AM (2013) Improved regenerative myogenesis and muscular dystrophy in mice lacking Mkp5. J. Clin. Invest 123, 2064–2077 10.1172/JCI64375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Hoffman EP, Brown RH and Kunkel LM (1987) Dystrophin: The protein product of the duchenne muscular dystrophy locus. Cell. 51, 919–928 10.1016/0092-8674(87)90579-4 [DOI] [PubMed] [Google Scholar]
- [55].Serrano AL and Muñoz-Cánoves P (2010) Regulation and dysregulation of fibrosis in skeletal muscle. Exp. Cell Res 316, 3050–3058 10.1016/j.yexcr.2010.05.035 [DOI] [PubMed] [Google Scholar]
- [56].Lieber RL and Ward SR (2013) Cellular mechanisms of tissue fibrosis. 4. structural and functional consequences of skeletal muscle fibrosis. Am. J. Physiol. Cell Physiol 305 10.1152/ajpcell.00173.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Tang LY, Heller M, Meng Z, Yu LR, Tang Y, Zhou M et al. (2017) Transforming growth factor-β (TGF-β) directly activates the JAK1-STAT3 axis to induce hepatic fibrosis in coordination with the SMAD pathway. J. Biol. Chem 292, 4302–4312 10.1074/jbc.M116.773085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Yuan X, Bu H, Zhou J, Yang CY and Zhang H (2020) Recent Advances of SHP2 Inhibitors in Cancer Therapy: Current Development and Clinical Application. J. Med. Chem 63, 11368–11396 10.1021/acs.jmedchem.0c00249 [DOI] [PubMed] [Google Scholar]
- [59].Liu Q, Qu J, Zhao M, Xu Q and Sun Y (2020) Targeting SHP2 as a promising strategy for cancer immunotherapy. Pharmacol Res. 152, 104595 10.1016/j.phrs.2019.104595 [DOI] [PubMed] [Google Scholar]
- [60].Song Z, Wang M, Ge Y, Chen XP, Xu Z, Sun Y et al. (2020) Tyrosine phosphatase SHP2 inhibitors in tumor-targeted therapies. Acta Pharm. Sin. B 11, 13–29 10.1016/j.apsb.2020.07.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Tripathi RKP and Ayyannan SR (2021) Emerging chemical scaffolds with potential SHP2 phosphatase inhibitory capabilities – A comprehensive review. Chem. Biol. Drug Des 97, 721–773 10.1111/cbdd.13807 [DOI] [PubMed] [Google Scholar]
- [62].Bunda S, Burrell K, Heir P, Zeng L, Alamsahebpour A, Kano Y et al. (2015) Inhibition of SHP2-mediated dephosphorylation of Ras suppresses oncogenesis. Nat. Commun 6, 8859 10.1038/ncomms9859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Liu W, Yu W-M, Zhang J, Chan RJ, Loh ML, Zhang Z et al. (2017) Inhibition of the Gab2/PI3K/mTOR signaling ameliorates myeloid malignancy caused by Ptpn11 (Shp2) gain-of-function mutations. Leukemia. 31, 1415–1422 10.1038/leu.2016.326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Ran H, Tsutsumi R, Araki T and Neel BG (2016) Sticking It to Cancer with Molecular Glue for SHP2. Cancer Cell. 30, 194–196 10.1016/j.ccell.2016.07.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Agazie YM and Hayman MJ (2003) Molecular Mechanism for a Role of SHP2 in Epidermal Growth Factor Receptor Signaling. Mol. Cell Biol 23, 7875–7886 10.1128/MCB.23.21.7875-7886.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Hof P, Pluskey S, Dhe-Paganon S, Eck MJ and Shoelson SE (1998) Crystal structure of the tyrosine phosphatase SHP-2. Cell. 92, 441–450 10.1016/s0092-8674(00)80938-1 [DOI] [PubMed] [Google Scholar]
- [67].Yu ZH, Xu J, Walls CD, Chen L, Zhang S, Zhang R et al. (2013) Structural and mechanistic insights into LEOPARD syndrome-associated SHP2 mutations. J. Biol. Chem 288, 10472–10482 10.1074/jbc.M113.450023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Mohi MG and Neel BG (2007) The role of Shp2 (PTPN11) in cancer. Curr. Opin. Genet. Dev 17, 23–30 10.1016/j.gde.2006.12.011 [DOI] [PubMed] [Google Scholar]
- [69].Chen H, Libring S, Ruddraraju KV, Miao J, Solorio L, Zhang Z-Y et al. (2020) SHP2 is a multifunctional therapeutic target in drug resistant metastatic breast cancer. Oncogene. 7166–7180 10.1038/s41388-020-01488-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Chen YNP, Lamarche MJ, Chan HM, Fekkes P, Garcia-Fortanet J, Acker MG et al. (2016) Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature. 535, 148–152 10.1038/nature18621 [DOI] [PubMed] [Google Scholar]
- [71].Bagdanoff JT, Chen Z, Acker M, Chen YN, Chan H, Dore M et al. (2019) Optimization of Fused Bicyclic Allosteric SHP2 Inhibitors. J. Med. Chem 62, 1781–1792 10.1021/acs.jmedchem.8b01725 [DOI] [PubMed] [Google Scholar]
- [72].Lamarche MJ, Acker M, Argintaru A, Bauer D, Boisclair J, Chan H et al. (2020) Identification of TNO155, an Allosteric SHP2 Inhibitor for the Treatment of Cancer. J. Med. Chem 63, 13578–13594 10.1021/acs.jmedchem.0c01170 [DOI] [PubMed] [Google Scholar]
- [73].Mullard A (2018) Phosphatases start shedding their stigma of undruggability. Nat. Rev. Drug Discov 17, 847–849 10.1038/nrd.2018.201 [DOI] [PubMed] [Google Scholar]
- [74].Ou SI, Koczywas M, Ulahannan S, Janne P, Pacheco J, Burris H et al. (2020) The SHP2 Inhibitor RMC-4630 in Patients with KRAS-Mutant Non-Small Cell Lung Cancer: Preliminary Evaluation of a First-in-Man Phase 1 Clinical Trial. J. Thorac. Oncol 15, S15–S16 10.1016/j.jtho.2019.12.041 [DOI] [Google Scholar]
- [75].Mainardi S, Mulero-Sánchez A, Prahallad A, Germano G, Bosma A, Krimpenfort P et al. (2018) SHP2 is required for growth of KRAS-mutant non-small-cell lung cancer in vivo letter. Nat. Med 24, 961–967 10.1038/s41591-018-0023-9 [DOI] [PubMed] [Google Scholar]
- [76].Ruess DA, Heynen GJ, Ciecielski KJ, Ai J, Berninger A, Kabacaoglu D et al. (2018) Mutant KRAS-driven cancers depend on PTPN11/SHP2 phosphatase. Nat. Med 24, 954–960 10.1038/s41591-018-0024-8 [DOI] [PubMed] [Google Scholar]
- [77].Nichols RJ, Haderk F, Stahlhut C, Schulze CJ, Hemmati G, Wildes D et al. (2018) RAS nucleotide cycling underlies the SHP2 phosphatase dependence of mutant BRAF-, NF1- and RAS-driven cancers. Nat. Cell Biol 20, 1064–1073 10.1038/s41556-018-0169-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Cancer.gov. [Internet] (2021) FDA Approval of KRAS Inhibitor Sotorasib for Lung Cancer Hailed as Milestone. Cancer Currents Blog. [updated 2021 June 25; cited 2021 July 26] Available from: https://www.cancer.gov/news-events/cancer-currents-blog/2021/fda-sotorasib-lung-cancer-kras [Google Scholar]
- [79].Wong GS, Zhou J, Liu JB, Wu Z, Xu X, Li T et al. (2018) Targeting wild-type KRAS-amplified gastroesophageal cancer through combined MEK and SHP2 inhibition. Nat. Med 24, 968–977 10.1038/s41591-018-0022-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Ahmed TA, Adamopoulos C, Karoulia Z, Wu X, Sachidanandam R, Aaronson SA et al. (2019) SHP2 Drives Adaptive Resistance to ERK Signaling Inhibition in Molecularly Defined Subsets of ERK-Dependent Tumors. Cell Rep. 26, 65–78.e5 10.1016/j.celrep.2018.12.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Liu C, Lu H, Wang H, Loo A, Zhang X, Yang G, et al. (2021) Combinations with allosteric SHP2 inhibitor TNO155 to block receptor tyrosine kinase signaling. Clin. Cancer Res 27, 342–354 10.1158/1078-0432.CCR-20-2718 [DOI] [PubMed] [Google Scholar]