

Abstract
Bacteriophages infecting pathogenic hosts play an important role in medical research, not only as potential treatments for antibiotic-resistant infections but also offering novel insights into pathogen genetics and evolution. A prominent example is cluster K mycobacteriophages infecting Mycobacterium tuberculosis, a causative agent of tuberculosis in humans. However, as handling M. tuberculosis as well as other pathogens in a laboratory remains challenging, alternative nonpathogenic relatives, such as Mycobacterium smegmatis, are frequently used as surrogates to discover therapeutically relevant bacteriophages in a safer environment. Consequently, the individual host ranges of the majority of cluster K mycobacteriophages identified to date remain poorly understood. Here, we characterized the complete genome of Stinson, a temperate subcluster K1 mycobacteriophage with a siphoviral morphology. A series of comparative genomic analyses revealed strong similarities with other cluster K mycobacteriophages, including the conservation of an immunity repressor gene and a toxin/antitoxin gene pair. Patterns of codon usage bias across the cluster offered important insights into putative host ranges in nature, highlighting that although all cluster K mycobacteriophages are able to infect M. tuberculosis, they are less likely to have shared an evolutionary infection history with Mycobacterium leprae (underlying leprosy) compared to the rest of the genus’ host species. Moreover, subcluster K1 mycobacteriophages are able to integrate into the genomes of Mycobacterium abscessus and Mycobacterium marinum—two bacteria causing pulmonary and cutaneous infections which are often difficult to treat due to their drug resistance.
Keywords: mycobacteriophages, cluster K, de novo assembly, genome annotation, phylogeny, codon usage bias
Introduction
First discovered by Frederick Twort and Félix d'Herelle in the early 20th century, bacteriophages (i.e., viruses that infect bacteria) are one of the most abundant biological entities on our planet (Rohwer 2003). Their study can provide important insights, not only into their own genetics and evolution (Hatfull 2010) but also into that of their (often) pathogenic hosts, many of which are intimately linked to human health and disease (Hatfull 2012). Due to the challenges involved in the handling of pathogenic strains in laboratories, fast-growing nonpathogenic strains (e.g., Mycobacterium smegmatis mc2155) are frequently used as surrogates (Mizuguchi 1984). These strains are also natural choices for course-based undergraduate research experiences, such as Howard Hughes Medical Institute’s Science Education Alliance—Phage Hunters Advancing Genomics and Evolutionary Science (HHMI SEA-PHAGES; Hatfull 2006). Undergraduate researchers involved in the program—now running in its 13th year—have isolated ∼18,000 bacteriophages, including 2,015 mycobacteriophages (https://phagesdb.org; last accessed August 3, 2021; Russell and Hatfull 2017). Based on the similarity in the nucleotide sequence, these can be grouped into 29 clusters (A-Z, AA, AB, and AC) which are further divided into several subclusters with common genomic architectures, as well as a few individual outliers without any close relatives yet defined (Hatfull 2010; Pope et al. 2011b). Among these, cluster K mycobacteriophages are of particular interest to the scientific community due to their ability to infect Mycobacterium tuberculosis (Pope et al. 2011a)—the primary bacterium underlying tuberculosis which causes more than 1 million deaths per year (World Health Organization 2020). Cluster K mycobacteriophages have successfully been used to diagnose tuberculosis infections (e.g., Jain et al. 2011), genetically manipulate M. tuberculosis (e.g., by transferring foreign DNA via shuttle plasmids; Jacobs et al. 1987), and assess drug susceptibility (e.g., Jacobs et al. 1993; Piuri et al. 2009). Moreover, they hold great promise for the treatment of drug-resistant tuberculosis strains via bacteriophage therapy, as well as prophylaxis aiding in the prevention of M. tuberculosis infections (see review by Allué-Guardia et al. 2021 and references therein). Although studies remain limited to date (Sula et al. 1981; Peng et al. 2009; and see review by Azimi et al. 2019), research into these medical and therapeutic applications will likely accelerate in the near future, spearheaded by the recently FDA-approved Center for Innovative Phage Applications and Therapeutics.
Many cluster K mycobacteriophages have the ability to infect a diverse range of hosts—from slow growing (e.g., M. tuberculosis) to fast growing (e.g., M. smegmatis) mycobacteria (Pope et al. 2011a), yet detailed insights into individual host ranges remain largely lacking due to the fact that the majority of known mycobacteriophages were isolated using M. smegmatis mc2155. As most bacteriophages utilize the translational machinery of their host, their protein synthesis is more efficient if their codon usage patterns (i.e., preferences in the usage of synonymous codons) are in agreement with those of their hosts (Carbone 2008; Lucks et al. 2008; Esposito et al. 2016). Therefore, a better understanding of bacteriophage codon usage biases can provide important clues toward both their mycobacterial host range in nature (Hassan et al. 2009) as well as candidates for future bacteriophage therapies. Here, we characterize the complete genome sequence of Stinson, a temperate subcluster K1 mycobacteriophage, and, through computational analysis of codon usage bias patterns, infer putative host ranges of 129 genomically characterized cluster K mycobacteriophages to aid future medical and therapeutic investigations.
Materials and methods
Sample collection, isolation, purification, and amplification of Stinson followed the HHMI SEA-PHAGES Phage Discovery Guide (https://seaphagesphagediscoveryguide.helpdocsonline.com/home; last accessed August 3, 2021); sequencing, de novo assembly, and genome annotation followed the HHMI SEA-PHAGES Bioinformatics Guide (https://seaphagesbioinformatics.helpdocsonline.com/home; last accessed August 3, 2021).
Sample collection and isolation
Stinson was obtained from a soil sample collected at the Hope College Pine Grove (42.787471 N, 86.102762 W) on a warm, humid morning. The sample was enriched by adding a bacterial host culture of M. smegmatis mc2155, submerging in 7H9 liquid medium [Middlebrook 7H9 broth base, 0.5% glycerol 10% AD supplement (145 mM NaCl, 5% Albumin Fraction V, 2% dextrose), 50 μg/mL carbenicillin stock, 10 μg/mL cycloheximide stock, 1 mM CaCl2], and incubating at 32°C for 2–5 days. After incubation, the enriched culture was centrifuged and filtered through a 0.22-μm filter. A spot test was performed by adding 250 μL of M. smegmatis mc2155 culture to 3 mL of molten top agar (1 mM CaCl2, 7H9 liquid media, agar), then immediately plating on an L-Agar plate [Luria broth base (10 g peptone, 5 g yeast extract, 10 g NaCl), agar, 50 μg/mL CB, 10 μg/mL CHX]. Ten microliters of bacteriophage filtrate was transferred to the plate, which was incubated at 32°C for 24–48 h before checking for plaques.
Purification and amplification
Bacteriophages were purified by selecting a single, well-isolated (1.5 cm apart) plaque to resuspend in phage buffer (10 mM pH 7.5 Tris, 10 mM MgSO4, 68 mM NaCl, 1 mM CaCl2). A series of 10-fold dilutions were performed and each dilution was dispensed into a tube containing 250 μL of host bacteria. The sample was incubated at room temperature for 10 min, then mixed with 3 mL of molten top agar, and immediately plated on an agar plate. The plates were incubated at 32°C for 24–48 h. This step was repeated until countable plates showed plaques of similar morphology that suggested that each grew from a single bacteriophage.
Bacteriophage lysate was harvested by flooding a plate containing a large number of purified bacteriophage plaques with 8 mL of phage buffer. The plate was set at room temperature for an hour, and then lysate was collected and filtered through a 0.22-μm filter. Tenfold serial dilutions were made with the bacteriophage lysate and then plated for a spot titer assay. Based on the spot titer results, a full titer assay was completed and the bacteriophage lysate titer was calculated to be 4.2 × 1010 PFU/mL.
Sequencing, de novo assembly, and genome annotation
Genomic DNA, extracted using a Promega Wizard® DNA Clean-Up kit, was used to prepare a single-indexed (TruSeq) sequencing library, which was subsequently sequenced on an Illumina MiSeq instrument, resulting in 2,197,235 high-quality single-end 150-bp reads (>5500X coverage; Supplementary Figure S1). Following Russell (2018), reads were de novo assembled using Newbler v.2.9, resulting in a fully assembled genome (length: 59,918 base pairs). The resulting assembly was checked for completeness, accuracy, and genomic termini using Consed v.29.0 (Gordon et al. 1998).
The genome of bacteriophage Stinson was annotated using DNA Master v.5.23.6 following Pope and Jacobs-Sera (2018), along with GLIMMER v.3.02 (Delcher et al. 1999) and GeneMark v.2.5 (Lukashin and Borodovsky 1998) embedded within. Start codons of putative genes were chosen to maximize coding potential (as reported by GeneMark) and confirmed using Starterator v.1.2 reports from closely related bacteriophages (https://seaphages.org/software/#Starterator; last accessed August 3, 2021). Predicted genes as well as any gaps larger than 10 bp were blasted both within the DNA Master environment using BLASTp v.2.9 (Altschul et al. 1990) as well as on NCBI (https://blast.ncbi.nlm.nih.gov/Blast.cgi; last accessed August 3, 2021). Aragorn v.1.1 (embedded in DNA Master) was utilized to locate tRNAs in the genome and Aragorn v1.2.38 (Laslett and Canback 2004) was applied for improved recognition of the ends of the identified tRNA. In addition, the software tRNAscan-SE v.2.0 (Lowe and Eddy 1997) was employed to search for noncanonical tRNAs. The protein product sequence from DNA Master was processed on HHPred (Söding et al. 2005), as well as on NCBI BLAST, utilizing the nonredundant protein sequences database (Marchler-Bauer et al. 2015), which assisted in the determination of putative gene functions. Genes with no known function were refined through TMHMM v.2.0 (Krogh et al. 2001) and SOSUI v.1.11 (Hirokawa et al. 1998) to predict membrane proteins. Lastly, Phamerator (Cresawn et al. 2011) was used to assess synteny among closely related bacteriophages. All software was executed using default settings.
Comparative genomic analyses
To characterize phylogenetic relationships and patterns of codon usage bias, genomic data for 128 Mycobacterium cluster K bacteriophages was downloaded from the NCBI Sequence Read Archive (Supplementary Table S1). A multiple-sequence alignment was generated with MAFFT v.7 (Katoh and Standley 2013) through the online EMBL-EBI bioinformatics toolkit (Madeira et al. 2019) and used to construct a neighbor-joining tree in MEGA X (Kumar et al. 2018) using a bootstrap test of phylogeny with 10,000 replicates. Closely related bacteriophages were then visually compared using Phamerator (Cresawn et al. 2011). In addition, dot plots, generated using Gepard v.1.40 (Krumsiek et al. 2007), were used to compare nucleotide sequence relatedness among the bacteriophages. Pairwise Average Nucleotide Identities (ANIs) were calculated using the DNA Master v.5.23.6 Genome Comparison Tool and plotted using the heatmap.2 function in R v.4.0.2. To determine codon usage bias within and across genomes, the COdon Usage Similarity INdex (COUSIN59) was calculated for each of the 129 mycobacteriophages (Supplementary Table S1) across 14 putative mycobacterial host species (Supplementary Table S2) using the software COUSIN (Bourret et al. 2019). All software was executed using default settings.
Identification of prophages within putative bacterial host genomes
PHASTER (https://phaster.ca; last accessed August 3, 2021; Arndt et al. 2016) was utilized to identify prophage sequences within the 14 putative mycobacterial host species (Supplementary Table S2). Next, MUSCLE v.3.8 (https://www.ebi.ac.uk/Tools/msa/muscle/; last accessed August 3, 2021; Edgar 2004) was used to study the evolutionary relationship between the immunity repressor gene of Stinson and the subcluster K1 mycobacteriophages able to integrate in these bacterial hosts. All software was executed using default settings.
Results and discussion
Stinson is a cluster K mycobacteriophage with a Siphoviridae morphology (i.e., nonenveloped with an icosahedral head and a noncontractile tail) and a temperate lifestyle. The fully sequenced and annotated genome is 59,918 base pairs long, with a GC-content of 66.6%—similar to that of its host, M. smegmatis mc2 155 (67.4%). Stinson’s genome contains 96 tightly packed protein-coding genes as well as a single tRNA, corresponding to a gene density of 1.60 genes/kb (Figure 1)—within the previously reported range for cluster K mycobacteriophages (1.34–1.74 genes/kb; Supplementary Table S1). Synteny is preserved with other cluster K mycobacteriophages, with the left arm of the genome encoding for structural and assembly proteins (i.e., terminase, portal protein, MuF-like minor capsid protein, scaffolding protein, major capsid protein, head-to-tail adaptor, head-to-tail stopper, tail terminator, major tail protein, tail assembly chaperones expressed via a translational frameshift (Xu et al. 2004), tape measure protein, and eight minor tail protein subunits), followed by the lysis cassette (lysin A and lysin B), holin, and genes responsible for integration into the host (Figure 1). The right arm of the genome contains nonstructural genes (Hatfull 2014), several of which are of unknown function. An important exception is the immunity repressor (gene 44), a downregulator of lytic gene expression, suggesting that Stinson might exhibit lysogenic maintenance and superinfection immunity (Petrova et al. 2015). Also of note is the toxin/antitoxin (TA) system (genes 92 and 93) which shows similarity to hicAB, a TA system found in Pseudomonas aeruginosa and Escherichia coli (Yamaguchi and Inouye 2011; Li et al. 2016). Overall, 41 out of the 96 genes could be assigned a putative function and an additional three genes were identified as membrane proteins. Similar to other cluster K mycobacteriophages, Stinson encodes a tRNAtrp (gene 5) as well as an RtcB-like RNA ligase (gene 86), likely serving to protect bacteriophages against tRNA cleavage by the host (Labrie et al. 2010; Pope et al. 2011a).
Figure 1.

Phamerator map of the whole genomes of Stinson and two closely related Mycobacterium bacteriophages, LaterM (Gaballa et al. 2019) and Murucutumbu (Pope et al. 2015). In this Phamerator map, protein-coding genes with their putative functional assignments (if available) are displayed above or below a ruler, signifying genes on forward or reverse strands, respectively. The numbers shown above each gene indicate the protein family (pham) and, in parenthesis, the number of members in the pham family. Coloring between genomes represents nucleotide similarity with areas of highest similarity shown in purple (BLAST e-value = 0), followed by red (BLAST e-value of ∼10−4) and white (no significant similarity). Stinson’s lysis cassette (lysin A and lysin B), holin, the immunity repressor gene, and the TA system (highlighted by blue boxes) show strong similarities between the three mycobacteriophages.
To characterize phylogenetic relationships as well as patterns of codon usage bias amongst cluster K mycobacteriophages, comparative analyses focused on Stinson and 128 previously characterized mycobacteriophages, including 67 members of subcluster K1, 9 of subcluster K2, 6 of subcluster K3, 14 of subcluster K4, 15 of subcluster K5, 16 of subcluster K6, and 1 of subcluster K7. The constructed neighbor-joining tree suggests that Stinson is a member of the K1 subcluster—a subcluster that shares a common ancestor with Marshawn (GenBank accession number: MN284895), a member of the paraphyletic subcluster K6 (Figure 2). This classification is supported by both the dot plot analysis (Figure 3 and Supplementary Figures S2–S8) as well as the pairwise ANI (Figure 4), which also highlights the strong relatedness between Stinson and other cluster K1 mycobacteriophages on the sequence level. Among the 67 subcluster K1 mycobacteriophages, Stinson is most closely related to LaterM (Gaballa et al. 2019) and Murucutumbu (Pope et al. 2015), with a nucleotide percent identity of 98.34% and 97.73%, respectively.
Figure 2.

Neighbor-joining tree generated using a multiple-sequence alignment of 129 Mycobacterium bacteriophage cluster K genomes (Supplementary Table S1) with 10,000 bootstrap replicates. Colors highlight membership in subclusters K1–K7.
Figure 3.

Dot plot of Stinson and one representative from each of the Mycobacterium bacteriophage cluster K subclusters: LaterM (K1), TM4 (K2), Pixie (K3), Cheetobro (K4), Collard (K5), Unicorn (K6), and Aminay (K7). Detailed information regarding each mycobacteriophage’s genome is provided in Supplementary Table S1.
Figure 4.
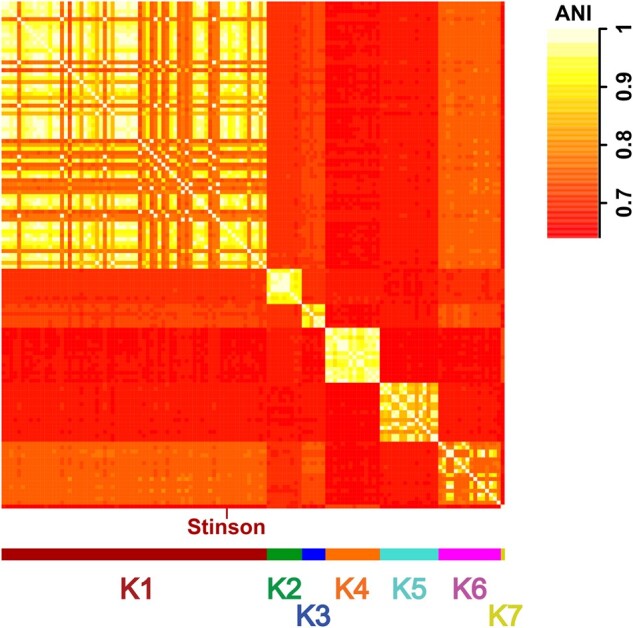
Heatmap of ANI values of the 129 Mycobacterium bacteriophage genomes (Supplementary Table S1). Colors highlight membership in subclusters K1–K7.
Patterns of codon usage bias amongst the 129 mycobacteriophages highlight strong similarities to the preferred usage of synonymous codons of 13 out of the 14 putative Mycobacterium host species (Figure 5), as may be expected from similarities in genomic GC-content (64–69%; Supplementary Tables S1 and S2). An important exception is the bacterium Mycobacterium leprae (GC-content: 57.8%; Supplementary Table S2), the primary causative agent of leprosy in humans. These results are in agreement with more limited experimental evidence highlighting that all cluster K mycobacteriophages are able to infect M. tuberculosis (Pope et al. 2011a) but suggest that they are less likely to have shared an evolutionary infection history with M. leprae compared to the rest of the genus’ host species.
Figure 5.

COdon Usage Similarity INdex (COUSIN59) of 129 Mycobacterium bacteriophage genomes (Supplementary Table S1) across 14 Mycobacterium host species (Supplementary Table S2), ordered by GC-content of the mycobacteriophage genomes. Colors highlight membership in subclusters K1–K7; shapes refer to the Mycobacterium host species.
To investigate whether Stinson or any closely related subcluster K1 mycobacteriophages might exhibit lysogenic properties, prophage sequences within the genomes of the putative hosts (Supplementary Table S2) were computationally predicted. The genome of 3 out of the 14 putative host species include intact prophages (Supplementary Table S3): Mycobacterium abscessus (GC-content: 64.2%) contains three intact and two incomplete prophages, Mycobacterium marinum (GC-content: 65.2%) contains one intact and one incomplete prophage, and M. smegmatis (GC-content: 67.4%) contains one intact and nine incomplete prophages. Although none of the prophages contained in the genome of M. smegmatis were predicted to have arisen from the integration of mycobacteriophages, the genomes of M. abscessus (Figure 6A) and M. marinum (Figure 6B) harbor prophages from several subcluster K1 (Adephagia, Amelie, Beezoo, and LastHope) as well as other cluster K mycobacteriophages (Supplementary Table S3). Given the similarity between Stinson and the subcluster K1 mycobacteriophages (see Figure 2 for a genome-wide representation and Supplementary Figure S9 for the immunity repressor gene specifically), it is expected that Stinson will also be able to integrate in these bacterial hosts. As M. abscessus and M. marinum play important roles in human health and disease, causing pulmonary (Winthrop and Roy 2020) and cutaneous (Aubry et al. 2000) infections which are often difficult to treat due to drug resistance, cluster K mycobacteriophages might provide important future avenues toward improved medical and therapeutic management strategies.
Figure 6.

Identification of prophages within (A) M. abscessus and (B) M. marinum. Regions displayed in green contain an intact and regions in red an incomplete prophage. Within a region, phage-like proteins are shown in dark green above or below a ruler, signifying proteins on forward or reverse strands, respectively.
Data availability
Supplementary Figure S1 displays the quality control of Stinson’s raw reads. Supplementary Figures S2–S8 show the dot plots of Stinson and mycobacteriophages from subclusters K1–K7, respectively. Supplementary Figure S9 displays the multiple-sequence alignment of the immunity repressor gene of Stinson and the subcluster K1 mycobacteriophages able to integrate in the putative bacterial hosts. Mycobacteriophages and putative mycobacterial hosts included in the comparative analyses are listed in Supplementary Tables S1 and S2, respectively. Prophages contained within the mycobacterial genomes are listed in Supplementary Table S3. Whole-genome sequence data are available through the NCBI Sequence Read Archive (BioProject accession number PRJNA488469). The annotated genome assembly is available through NCBI GenBank (accession number MZ355721).
Supplementary material is available at G3 online.
Supplementary Material
Acknowledgments
Bacteriophage isolation, library preparation, and sequencing were performed at Hope College, Holland, MI. De novo assembly was performed at the University of Pittsburgh, Pittsburgh, PA. Computations were performed at Arizona State University's High Performance Computing facility, Tempe, AZ. The authors are grateful to Suhail Ghafoor for IT support, Daniel A. Russell and Rebecca A. Garlena for de novo assembly, Vassie C. Ware for guidance regarding our genome annotation, as well as Billy Biederman, Graham Hatfull, Deborah Jacobs-Sera, and Vic Sivanathan for training and continued support in the SEA-PHAGES program.
Funding
This work was supported by a National Science Foundation CAREER grant to S.P.P. (DEB-2045343), Howard Hughes Medical Institute's SEA-PHAGES program, and Arizona State University's School of Life Sciences.
Conflicts of interest
The authors declare that there is no conflict of interest.
Literature cited
- Allué-Guardia A, Saranathan R, Chan J, Torrelles JB.. 2021. Mycobacteriophages as potential therapeutic agents against drug-resistant tuberculosis. Int J Mol Sci. 22:735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ.. 1990. Basic local alignment search tool. J Mol Biol. 215:403–410. [DOI] [PubMed] [Google Scholar]
- Arndt D, Grant J, Marcu A, Sajed T, Pon A, et al. 2016. PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44:W16–W21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubry A, Jarlier V, Escolano S, Truffot-Pernot C, Cambau E.. 2000. Antibiotic susceptibility pattern of Mycobacterium marinum. Antimicrob Agents Chemother. 44:3133–3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azimi T, Mosadegh M, Nasiri MJ, Sabour S, Karimaei S, et al. 2019. Phage therapy as a renewed therapeutic approach to mycobacterial infections: a comprehensive review. Infect Drug Resist. 12:2943–2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourret JS, Alizon IG., Bravo. 2019. COUSIN (COdon Usage Similarity INdex): a normalized measure of codon usage preferences. Genome Biol Evol. 11:3523–3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbone A. 2008. Codon bias is a major factor explaining phage evolution in translationally biased hosts. J Mol Evol. 66:210–223. [DOI] [PubMed] [Google Scholar]
- Cresawn SG, Bogel M, Day N, Jacobs-Sera D, Hendrix RW, et al. 2011. Phamerator: a bioinformatic tool for comparative bacteriophage genomics. BMC Bioinformatics. 12: 395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delcher AL, Harmon D, Kasif S, White O, Salzberg SL.. 1999. Improved microbial gene identification with GLIMMER. Nucleic Acids Res. 27:4636–4641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. 2004. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 5:113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito LA, Gupta S, Streiter F, Prasad A, Dennehy JJ.. 2016. Evolutionary interpretations of mycobacteriophage biodiversity and host-range through the analysis of codon usage bias. Microb Genom. 2:e000079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaballa JM, Dabrian K, Desai R, Ngo R, Park D, et al. 2019. Genome sequences of cluster K mycobacteriophages Deby, LaterM, LilPharaoh, Paola, SgtBeansprout, and Sulley. Microbiol Resour Announc. 8:e01481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon D, Abajian C, Green P.. 1998. Consed: a graphical tool for sequence finishing. Genome Res. 8:195–202. [DOI] [PubMed] [Google Scholar]
- Hassan S, Mahalingam V, Kumar V.. 2009. Synonymous codon usage analysis of thirty two mycobacteriophage genomes. Adv Bioinformatics. 2009:316936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatfull GF. 2006. Mycobacteriophages. In: Calendar R, editor. The Bacteriophages. New York, NY: Oxford University Press. p. 602–620. [Google Scholar]
- Hatfull GF. 2010. Mycobacteriophages: genes and genomes. Annu Rev Microbiol. 64:331–356. [DOI] [PubMed] [Google Scholar]
- Hatfull GF. 2012. The secret lives of mycobacteriophages. In: Łobocka M, Szybalski WT, editors. Advances in Virus Research, Vol. 82. Burlington Academic Press. p. 178–288. [DOI] [PubMed] [Google Scholar]
- Hatfull GF. 2014. Molecular genetics of mycobacteriophages. Microbiol Spectr. 2:1–36. [PMC free article] [PubMed] [Google Scholar]
- Hirokawa T, Boon-Chieng S, Mitaku S.. 1998. SOSUI: classification and secondary structure prediction system for membrane proteins. Bioinformatics. 14:378–379. [DOI] [PubMed] [Google Scholar]
- Jacobs WR Jr, Barletta RG, Udani R, Chan J, Kalkut G, et al. 1993. Rapid assessment of drug susceptibilities of Mycobacterium tuberculosis by means of luciferase reporter phages. Science. 260:819–822. [DOI] [PubMed] [Google Scholar]
- Jacobs WR Jr, Tuckman M, Bloom BR.. 1987. Introduction of foreign DNA into mycobacteria using a shuttle phasmid. Nature. 327:532–535. [DOI] [PubMed] [Google Scholar]
- Jain P, Thaler DS, Maiga M, Timmins GS, Bishai WR, et al. 2011. Reporter phage and breath tests: emerging phenotypic assays for diagnosing active tuberculosis, antibiotic resistance, and treatment efficacy. J Infect Dis. 204(Suppl 4):S1142–S1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Standley DM.. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30:772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh A, Larsson B, von Heijne G, Sonnhammer ELL.. 2001. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 305:567–580. [DOI] [PubMed] [Google Scholar]
- Krumsiek J, Arnold R, Rattei T.. 2007. Gepard: a rapid and sensitive tool for creating dotplots on genome scale. Bioinformatics. 23:1026–1028. [DOI] [PubMed] [Google Scholar]
- Kumar S, Stecher G, Li M, Knyaz C, Tamura K.. 2018. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol Biol Evol. 35:1547–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labrie SJ, Samson JE, Moineau S.. 2010. Bacteriophage resistance mechanisms. Nat Rev Microbiol. 8:317–327. [DOI] [PubMed] [Google Scholar]
- Laslett D, Canback B.. 2004. ARAGON, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 32:11–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Shen M, Lu S, Le S, Tan Y, et al. 2016. Identification and characterization of the hicAB toxin-antitoxin system in the opportunistic pathogen Pseudomonas aeruginosa. Toxins (Basel). 8:113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe TM, Eddy SR.. 1997. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25:955–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucks JB, Nelson DR, Kudla GR, Plotkin JB.. 2008. Genome landscapes and bacteriophage codon usage. PLoS Comput Biol. 4:e1000001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukashin AV, Borodovsky M.. 1998. GeneMark.hmm: new solutions for gene finding. Nucleic Acids Res. 26:1107–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madeira F, Park YM, Lee J, Buso N, Gur T, et al. 2019. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 47:W636–W641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchler-Bauer A, Derbyshire MK, Gonzales NR, Lu S, Chitsaz F, et al. 2015. CDD: NCBI's conserved domain database. Nucleic Acids Res. 43(Database issue):D222–D226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuguchi Y. 1984. Mycobacteriophages. In: Kubica GP, Wayne LG, editors. The Mycobacteria: A Sourcebook. New York, NY: Marcel Dekker. p. 641–662. [Google Scholar]
- Peng L, Luo Y, Chen B, Li Y, Shen X, et al. 2009. Therapeutic effect of bacteriophage D29 in the treatment for guinea pigs infected with sensitive strain of Mycobacterium tuberculosis. Chin J Zoonoses. 25:733–736. [Google Scholar]
- Petrova ZO, Broussard GW, Hatfull GF.. 2015. Mycobacteriophage-repressor-mediated immunity as a selectable genetic marker: Adephagia and BPs repressor selection. Microbiology (Reading). 161:1539–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piuri M, Jacobs WR Jr, Hatfull GF.. 2009. Fluoromycobacteriophages for rapid, specific, and sensitive antibiotic susceptibility testing of Mycobacterium tuberculosis. PLoS One. 4:e4870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pope WH, Bowman CA, Russell DA, Jacobs-Sera D, Asai DJ, et al. ; Mycobacterial Genetics Course. 2015. Whole genome comparison of a large collection of mycobacteriophages reveals a continuum of phage genetic diversity. Elife. 4:e06416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pope WH, Ferreira CM, Jacobs-Sera D, Benjamin RC, Davis AJ, et al. 2011a. Cluster K mycobacteriophages: insights into the evolutionary origins of mycobacteriophage TM4. PLoS One. 6:e26750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pope WH, Jacobs-Sera D.. 2018. Annotation of bacteriophage genome sequences using DNA Master: an overview. Methods Mol Biol. 1681:217–229. [DOI] [PubMed] [Google Scholar]
- Pope WH, Jacobs-Sera D, Russell DA, Peebles CL, Al-Atrache Z, et al. 2011b. Expanding the diversity of mycobacteriophages: insights into genome architecture and evolution. PLoS One. 6:e16329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohwer F. 2003. Global phage diversity. Cell. 113:141. [DOI] [PubMed] [Google Scholar]
- Russell DA. 2018. Sequencing, assembling, and finishing complete bacteriophage genomes. Methods Mol Biol. 1681:109–125. [DOI] [PubMed] [Google Scholar]
- Russell DA, Hatfull GF.. 2017. PhagesDB: the actinobacteriophage database. Bioinformatics. 33:784–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Söding J, Biegert A, Lupas AN.. 2005. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 33(Web Server issue):W244–W248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sula L, Sulova J, Stolcpartova M.. 1981. Therapy of experimental tuberculosis in guinea pigs with mycobacterial phages DS-6A, GR-21 T, My-327. Czech Med. 4:209–214. [PubMed] [Google Scholar]
- Winthrop KL, Roy EE.. 2020. Mycobacteria and immunosuppression. In: Atzeni F, Galloway JB, Gomez-Reino JJ, Galli M, editors. Handbook of Systemic Autoimmune Diseases, Vol. 16. Elsevier Ltd. p. 83–107. [Google Scholar]
- World Health Organization. 2020. Global Tuberculosis Report. Geneva, Switzerland: World Health Organization. [Google Scholar]
- Xu J, Hendrix RW, Duda RL.. 2004. Conserved translational frameshift in dsDNA bacteriophage tail assembly genes. Mol Cell. 16:11–21. [DOI] [PubMed] [Google Scholar]
- Yamaguchi Y, Inouye M.. 2011. Regulation of growth and death in Escherichia coli by toxin-antitoxin systems. Nat Rev Microbiol. 9:779–790. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Supplementary Figure S1 displays the quality control of Stinson’s raw reads. Supplementary Figures S2–S8 show the dot plots of Stinson and mycobacteriophages from subclusters K1–K7, respectively. Supplementary Figure S9 displays the multiple-sequence alignment of the immunity repressor gene of Stinson and the subcluster K1 mycobacteriophages able to integrate in the putative bacterial hosts. Mycobacteriophages and putative mycobacterial hosts included in the comparative analyses are listed in Supplementary Tables S1 and S2, respectively. Prophages contained within the mycobacterial genomes are listed in Supplementary Table S3. Whole-genome sequence data are available through the NCBI Sequence Read Archive (BioProject accession number PRJNA488469). The annotated genome assembly is available through NCBI GenBank (accession number MZ355721).
Supplementary material is available at G3 online.