

Abstract
Cardiac rhythm disorders reflect failures of impulse generation and/or conduction. With the exception of ablation methods that yield selective endocardial destruction, present therapies are nonspecific and/or palliative. Progress in understanding the underlying biology opens up prospects for new alternatives. This article reviews the present state of the art in gene- and cell-based therapies to correct cardiac rhythm disturbances. We begin with the rationale for such approaches, briefly discuss efforts to address aspects of tachyarrhythmia, and review advances in creating a biological pacemaker to cure bradyarrhythmia. Insights gained bring the field closer to a paradigm shift away from devices and drugs, and toward biologics, in the treatment of rhythm disorders.
Keywords: bradycardia, tachycardia, gene therapy, cell therapy
Present therapy for cardiac arrhythmias relies on pharmacology, ablation, or electronic devices, with varying success. Antiarrhythmic drugs have a propensity to provoke new arrhythmias while suppressing others.1-4 Catheter ablation selectively destroys endocardial tissue and thus is curative in destroying inborn wiring errors such as accessory tracts, but falls short in treating more complex cases, such as atrial fibrillation (AF) or ventricular tachycardia (VT).5,6 Electronic devices can sustain heart rate, or deliver shocks to terminate tachycardias. However, the high cost of devices, and complications such as pulmonary collapse, hemorrhage, bacterial infection, and lead/generator failure7 represent limitations of the technology.
Given these shortfalls, we and others have begun to develop biological alternatives (gene therapy, cell therapy, or combinations of the two) or adjuncts to conventional treatment. Work reviewed here indicates that biological therapy can impact on a diverse variety of arrhythmias, at least at the proof-of-principle level. Figure 1 summarizes schematically the regions of the heart, and the rhythm disorders, which have been targeted by various biological therapies with a view to modifying impulse initiation, conduction or repolarization. Bradyarrhythmia is the clinical target which has received the greatest attention. Biological pacemaking can be achieved with astonishing simplicity, as will be discussed below. A major challenge in the field, and one touched on throughout the review, is the gap between conceptual innovation and realistic efforts at translation.
Figure 1.

Principles of biological approaches (in italics) recruited to treat major cardiac arrhythmias, and sites within the heart where the various strategies have been tested.
Gene Therapy Approach to Control Ventricular Rate in AF
Accepted therapy for AF includes either antiarrhythmic drugs to maintain sinus rhythm or drugs that slow conduction in the atrioventricular (AV) node to control the ventricular rate. Pharmacological therapies to control heart rate during AF have targeted the conduction properties of the AV node by suppressing the calcium current (calcium channel blockers) or by affecting the balance of adrenergic and cholinergic tone (β-blockers and digitalis compounds, respectively). For patients with normal ventricular function, AV nodal suppressing drugs reduce the heart rate by 15% to 30%.8 However, the frequent side effects of these drugs include bronchospasm, hypotension, depression, fatigue, and constipation.9 When pharmacological interventions are not tolerated or not effective, and the rhythm itself cannot be controlled, the only remaining option is radiofrequency ablation of the AV node and implantation of an electronic pacemaker.
In the first use of gene therapy to treat an arrhythmia, the α subunit of inhibitory G protein (Gαi2) packaged into adenovirus was infused into the AV nodal artery of pigs,10 in an effort to control ventricular rate during AF. The idea was to suppress basal adenylate cyclase activity in the AV node, which in turn would suppress calcium channel activity focally. With the aid of nitroglycerin, vascular endothelial growth factor, and sildenafil to enhance vascular permeability,11,12 almost 50% of cells in the AV node showed evidence of gene transfer, whereas transduction outside the AV node was minimal. Gαi2 overexpression caused clear phenotypic consequences: during sinus rhythm, the transgene decreased conduction velocity and increased refractoriness in the AV node. In acute AF, the heart rate was reduced by 20% when compared to controls, and this relative reduction persisted after β-adrenergic stimulation. In a follow-up study,13 a constitutively active mutant of Gαi2 (Gαi2-Q205L) was overexpressed in the AV node of animals with persistent AF and tachycardia-induced cardiomyopathy. Whereas the wild type suppressed heart rate only during sleep, the constitutively active mutant decreased the ventricular rate by 15% to 25% in awake as well as inactive animals, resulting in modest recovery of cardiac function.
Direct suppression of L-type calcium current (ICa, L) in the AV node, independent of adrenergic signal transduction, provides an alternative route for rate control in AF. A GTP-binding protein, Gem, which inhibits the trafficking of calcium channel α subunits to the plasma membrane,14 was used as a genetic suppressor of ICa, L in the AV node.15 Adenoviral gene transfer of Gem in the porcine heart slowed conduction velocity in the AV node, and achieved ≈20% reduction in the ventricular rate during acute AF. Pharmacological blockade of calcium channels for the treatment of AF is fraught with side effects that are attributable to the drug action outside the AV node. Such localized gene delivery may, in principle, circumvent the problem of nonspecificity inherent to drug therapy.
As a cell therapy approach, Bunch et al injected autologous skin fibroblasts into peri-AV nodal region of dogs.16 This created a focal scar within the conduction tissue, slowing conduction velocity. When the fibroblasts were pretreated with transforming growth factor-β, conduction velocity was slowed further compared to the nontreated fibroblasts. However, the clinical relevance of this study is diminished by their protocol of injecting the cells in 80 mL of solution in 320 sites, and the method has no apparent advantage to conventional radiofrequency ablation/modification of the AV node.
Use of clinically available catheters and equipment in these studies suggests that the logistics of translating these concepts to human therapy would be relatively straightforward. Major steps between the present state of development and clinical applicability include the use of longer-lasting, less-inflammatory vectors (such as adeno-associated virus), and tests of safety and efficacy in animal models of chronic AF. The impetus for development of such approaches for rate control has waned, however, with the emergence and increasing popularity of potentially curative catheter ablation methods targeted at isolating irritable foci in the pulmonary veins.17
Genetic Interventions to Influence Ventricular Repolarization
Repolarization abnormalities increase the likelihood of early afterdepolarizations (EADs) and heighten the susceptibility to arrhythmogenesis.18 The concept of using somatic gene transfer to alter repolarization was pioneered by Johns et al,19 who overexpressed K+ channels in cardiac myocytes and thereby abbreviated action potential (AP) duration (APD). The idea was subsequently applied in vitro to a disease model (cardiomyocytes from pacing tachycardia heart failure).20 In Long QT syndrome, the prolonged ventricular repolarization and its clinically identified, specific ion channel genes provide rational targets for gene therapy. In this regard, overexpression of HERG, the primary gene encoding the rapid component of delayed rectifier K+ current (IKr), accelerated repolarization, increased refractoriness, diminished beat-to-beat APD variability, and reduced the occurrence of EADs by more than 4-fold in rabbit ventricular myocytes.21 By contrast, when a slow component of delayed rectifier current (IKs) was augmented by wild-type KCNE1 overexpression (which encodes minK, an ancillary subunit of IKs) in guinea pig ventricular myocardium, no phenotypic change was observed.22 The mechanism of HERG and the function of KCNE1 channels in repolarization was further examined with their respective dominant-negative mutant channel proteins. HERG-G628S overexpression decreased refractoriness, increased beat-to-beat AP variability, increased EAD occurrence, but without significantly delaying repolarization. In contrast, KCNE1-D76N suppressed IKs and markedly slowed repolarization, leading to frequent EADs and QT prolongation on the ECG. The drastic undermining of repolarization by KCNE1-D76N is in contrast with the HERG mutant, which facilitated the genesis and propagation of premature beats without prolonging repolarization.21,22 These in vitro studies have recently been extended to intact large-animal studies: molecular ablation of ventricular arrhythmia in vivo was achieved by a focal overexpression of HERG-G628S mutant in the infarct scar border area of a porcine model of VT.23
Calcium-independent transient outward current (Ito1) is another important component of membrane repolarizing currents. Using nonviral as well as viral approaches, the significance of Ito1 during early repolarization was investigated. Guinea pig cardiomyocytes, which normally lack Ito1, were fused with CHO cells overexpressing Kv4.3. This cell fusion instantly reconstituted Kv4.3 channel proteins to the membrane of myocytes. The resultant heterokaryons demonstrated acceleration of early repolarization velocity and abbreviated the APD.24 Cardiomyocytes undergo extensive time-dependent changes of their membrane properties.25 The use of cell fusion avoided culture-related complications by introducing preformed membrane proteins to native cells. In addition, adenoviral delivery of Kv4.3 to guinea pig myocardium complemented the cell fusion approach; ecdysone-mediated, graded expression of Kv4.3 demonstrated that the plateau potential of guinea pig ventricular myocytes is progressively suppressed with increasing density of Ito1. When a dominant-negative form of Kv4.3 channels (W362F) was expressed in rat ventricular myocardium, the results mirrored those observed with wild-type Kv4.3; W362F suppressed peak Ito1, elevated plateau potential, prolonged APD, and increased the QT interval by 30% on ECG.26 Genetic suppression of Ito1 may conceivably be useful in the treatment of Brugada syndrome, where haploinsufficiency of Na+ channels causes a tug-of-war with Ito1 to render repolarization dangerously unstable.27
A transgenic mouse model of the long QT syndrome has been used to test the feasibility of gene transfer to correct the phenotype. Wild-type Kv1.5 was delivered in either adenoviral28 or adeno-associated viral vector29 into the left ventricular base of transgenic mice bearing a dominant negative of Kv1.1 channel. In both studies, Kv1.5 overexpression and the resultant IKs led to shortened APD and QT interval. However, the sham-operated animals exhibited a negligible incidence of VT, and the effect of therapy on arrhythmia prevention could not be tested.
Taken together, these studies demonstrate the feasibility of generating a disease-specific animal model by somatic gene transfer. On the flip side, the studies, although mostly limited to proof-of-concept, do highlight the potential utility of gene transfer in blunting ventricular arrhythmogenesis by overexpressing K+ channels (to accelerate repolarization), or blocking them selectively23 to achieve focal class III30 antiarrhythmic effects.
Biological Pacemaker: Rationale and Clinical Target
The sinoatrial (SA) node initiates the heartbeat, sustains the circulation and sets the rate and rhythm of cardiac contraction.31 Its spontaneous pacemaker activity excites the myocardium, setting the stage for orchestration of cardiac excitation and contraction. Loss of pacemaker activity from the SA node, therefore, results in circulatory collapse, necessitating the implantation of an electronic pacemaker.32 Though effective as a palliative measure, such devices are expensive33 and associated with risks such as device or lead failure, and chronic infection.
Although the notion of a biological pacemaker replacing an electronic pacemaker is attractive, it must be recognized that, despite their limitations, electronic pacemakers generally work quite well; indeed, they have withstood the test of time for more than 50 years. One approach would target patients with “soft” indications for a pacemaker, such as sick sinus syndrome. If a biological pacemaker fails over time in such patients, life is less likely at risk than it would be in complete heart block, for example. On the other hand, perhaps it is neither necessary nor wise to aspire to create a permanent, durable alternative to electronic pacemakers; instead, temporary, “niche” applications may turn out to be more clinically compelling. Consider patients that desperately need an electronic pacemaker, but have strong contraindications to indwelling hardware. Biological pacemakers may be ideally suited for the management of pacemaker-dependent bradycardia complicated by bacterial infection of the device, in which the contraindication to reinfectable metal/plastic leads creates a unique advantage for short-term biological pacing.34 When a patient presents with an infected system requiring revision and antibiotics, the biological pacemaker could be used to create a hardware-free interval sufficient to effectively treat the bacterial infection before reimplanting a permanent device. Such “bridge-to-device” applications constitute a logical niche for the initial clinical development of biological pacemakers, given the proven efficacy of electronic devices for bradycardia not complicated by an infectious substrate.
An initial gene therapy approach to enhance chronotropy used overexpression of human β2-adrenergic receptor. Edelberg et al reported that heart rate could be increased by 40% in mouse right atria35 and by 50% in porcine atria on exogenous β2-adrenergic receptor overexpression.36 These studies demonstrated the ability to upregulate existing sinus rate without introducing rhythm change, but the approach was not designed to convert tissue from quiescent to automatic in terms of electrogenesis.
The first biological approach to create a de novo cardiac pacemaker rendered ventricular myocardium spontaneously active.37 The strategy posits that normally quiescent ventricular myocardium has an innate ability to pace, but is suppressed from pacing by “electric brakes” that stabilize the resting membrane potential. Among the inhibitory currents that are logical candidates to serve as electric brakes, the inward rectifier potassium current (IK1) is notable for its intense expression in nonpacing atria and ventricle and its absence in nodal pacemaker cells.38 IK1, encoded by the Kir2 gene family,39 imparts and stabilizes a strongly negative resting potential and thereby suppresses excitability. We explored the possibility that inhibition of IK1 by overexpressing a Kir2.1 dominant-negative channel (Kir2.1AAA)40 in the ventricle would cripple IK1 and suffice to produce spontaneous, rhythmic electric activity. Kir2.1AAA and GFP were packaged into a bicistronic adenoviral vector and injected into the coronary circulation of guinea pigs during transient cross-clamp of the great vessels.37 Nontransduced left ventricular myocytes isolated from Kir2.1AAA-injected animals, as well as green cells from control hearts, exhibited no spontaneous activity, but fired single APs in response to depolarizing external stimuli (Figure 2A). In contrast, Kir2.1AAA-transduced myocytes exhibited either (1) a stable resting potential from which prolonged APs could be elicited by external stimuli or (2) spontaneous activity (Figure 2B). The spontaneous activity, which was seen in all cells in which IK1 was suppressed below 0.4 pA/pF, resembles that of genuine pacemaker cells: the maximum diastolic potential is relatively depolarized, with repetitive, regular and incessant electric activity initiated by gradual “phase 4” depolarization and a slow upstroke.31,41 More importantly, premature beats of ventricular origin could be distinguished by their broad amplitude, and “marched through” to a beat independent of, and more rapid than, that of the physiological sinus pacemaker (Figure 2D). In these proof-of-concept experiments, ectopic beats, arising from foci of induced biological pacemakers, caused the entire heart to be paced from the ventricle. The focally intense expression was achieved by chance rather than by design.
Figure 2.

Suppression of Kir2.1 channels unmasks latent pacemaker activity in ventricular cells. A, APs evoked by depolarizing external stimuli in control ventricular myocytes. B, Spontaneous APs in Kir2.1AAA-transduced myocytes with depressed IK1. C, Baseline electrocardiograms in normal sinus rhythm. D, Ventricular rhythms 72 hours after gene transfer of Kir2.1AAA. P waves (A and arrow) and wide QRS complexes (V and arrow) march through to their own rhythm. A through D are reproduced from Miake et al with permission.37 E, Guinea pig ventricular myocytes in which Kir2.1AAA was overexpressed (left) and exposed acutely to 1 μmol/L isoproterenol (J. Miake, H. B. Nuss, and E.M., unpublished data, 2002).
A full-fledged biological pacemaker would sustain a wide range of frequencies responding to physiological needs of sleep, exercise, fever, etc. under neurohumoral chronotropic control. β-Adrenergic receptor activation-mediated sympathetic stimulation has a positive chronotropic effect on the native pacemaker. Acetylcholine and vagal stimulation, in contrast, negatively affect chronotropy. If and ICa,L are examples of ionic currents regulated by this neurohumoral influence, accelerating/decelerating the rate of phase 4 depolarization and the AP upstroke.42,43 Figure 2E shows that biological pacemakers engineered by suppression of IK1 respond to the β-adrenergic agonist isoproterenol with an increase in rate. These effects presumably reflect upregulation of L-type Ca2+ channels. Even finer tuning of autonomic responsiveness may be possible when HCN channels are overexpressed (see below), as these channels contain cAMP-response elements.44
Thus, specific suppression of Kir2 channels sufficed to unleash pacemaker activity in ventricular myocytes. The conclusions of Miake et al37 have recently been confirmed and extended by Sekar et al,45 who mapped the origin of focal pacemaker activity in monolayers of rat ventricular myocytes to islands of suppressed IK1 (Figure 3). The crucial factor for pacing was the absence of the strongly polarizing IK1, rather than overexpression of exogenous ionic current(s) that are absent or sparse in ventricular cardiomyocytes (although such currents may play an important modulatory role in genuine pacemaker cells,46 and can suffice to induce pacing if overexpressed in atria and ventricles [next section]).
Figure 3.

Dynamics of induced reentrant spiral waves in 21-mm-diameter monolayers with a central island of Kir2.1 suppression. The mapping region is 17 mm in diameter. A, Island of Kir2.1-suppressed NRVMs (indicated by white circles) acting as a source of spontaneous activity, as seen by the spread of spontaneous cardiac impulses (shown by black arrows) from an interior region of the island. B, An induced spiral wave anchored momentarily to the region of IK1 suppression (shown by white circle). C, Transition of initial spiral wave into a multi-armed spiral wave. D, Transition of initial spiral wave into a 2-armed spiral wave. E, Transition of the 2-armed spiral wave of D into a figure-of-eight reentry that later drifted to the edge of the monolayer and terminated. Reproduced from Sekar et al with permission.45
In a follow-up study,47 Miake et al found that Kir2.1AAA overexpression not only destabilized the resting membrane potential but also led to prolongation of APD. Conditions that prolong APD could potentiate EADs, thereby predisposing to ventricular arrhythmias.48,49 The need for potent suppression of IK1 to unleash pacemaker activity without prolonging APs represents a drawback of the Kir2.1AAA strategy. In this regard, the APD prolonging “side effect” of IK1 suppression may be obviated by intense focal expression of the Kir2.1AAA transgene, or by coexpression of HERG, in a variation of the previously described dual gene therapy approach.50 HERG overexpression would keep the APD short, but not interfere with the resting membrane potential destabilizing effect of Kir2.1AAA at −80 mV.
Gene Therapies to Create Biological Pacemaking in Myocardium
HCN (hyperpolarization-activated, cyclic nucleotide–gated) cation channels have been argued to play an important role in cardiac pace-making.51 Rather than inhibiting K+ channels, Rosen and colleagues overexpressed HCN2 channels to elicit ectopic pacemaker activity in a canine model.52,53 Although HCN4 is the major isoform in nodal tissues,51 Qu et al chose to use HCN2,53 which is the dominant isoform in ventricular myocytes, with cAMP responsiveness comparable to that of HCN4. On suppressing sinus rhythm by vagal stimulation, the authors observed spontaneous rhythms from dogs injected with adenovirus encoding HCN2 into left atrium. When the same construct was injected into the left bundle branch, they observed left ventricle-originated ectopic beats at a rate exceeding that of controls.52 Similarly, an HCN1 channel with a modified gating property has been used to demonstrate ectopic pacing when the biological was focally injected into left atrium in a SA node-ablated porcine model.54 These works were performed in large-animal models, bringing the biological pacemaker closer to clinical application.
As an alternative to wild-type HCN channels, we have created synthetic pacemaker channels by site-directed mutagenesis on a canonical voltage-dependent K channel backbone.55 Such channels activate on hyperpolarization and have nonspecific cation selectivity; moreover, the fact that they do not multimerize with endogenous HCN channels may be advantageous. However, the engineered protein contains extracellularly facing epitopes which may be immunogenic, limiting long-term expression and translational potential.
Gene–Cell Hybrid Approach to Biological Pacemakers
A general hurdle of gene therapies described above lies in the use of viral delivery vehicles. Routine (ie, nongutted) adenoviral vectors elicit acute and prolonged host immune responses, restricting their utility for long-term therapy.56 Adeno-associated virus and lentivirus suffer from small packaging capacity and random insertion of transgenes in the host genome, respectively.57 Used as an alternative delivery vehicle with genetic transduction by nonviral methods, cells could circumvent at least some of the obstacles faced by somatic gene transfer approaches. Human mesenchymal stem cells (hMSCs) have attracted attention as they can be obtained in a large quantities and are claimed to be immuneprivileged.58,59 Mesenchymal stem cells have been used as vehicles to deliver a mouse HCN2 channel. The idea was to impart If to myocardium through electrotonic coupling of cardiomyocytes with hMSCs that overexpress HCN2 channels through routine plasmid transfection.60 Subepicardial injection of HCN2-overexpressing hMSCs into the canine left ventricular wall induced spontaneous rhythms of left-sided origin on chronic AV block in 5 of 6 animals. The pacemaking activity required 10 to 12 days to become stable, then persisted for at least 6 weeks.61 A key prerequisite to this approach is a high degree of gap-junctional coupling between the donor hMSCs and the host myocardium. Such gap junctional coupling, however, may not be stable over time, a problem which must be considered in any pacemaker applications. Indeed, major forms of heart disease that carry increased arrhythmic risk often involve gap junction remodeling and decreased cell–cell coupling.62 In addition, the donor cells may not stay at the specific site of injection as intended; when quantum dot-labeled hMSCs were injected into a rat left ventricular free wall, only 30% of the total injected hMSCs were found to be in the whole heart at 24 hours.63 Migration of injected cells could affect long-term consistency of the biological pacemaker function or present a substrate for arrhythmia.64
A fusion between host myocardial cells and the donor cells, creating heterokaryons, provides a potential solution to uncontrolled migration of injected cells, and obviates the need for gap junctional coupling to deliver the excitatory signal to the host myocardium. We used syngeneic fibroblasts engineered to overexpress a pacemaker ion channel, HCN1, and induced cell–cell fusion using polyethylene glycol.65 The polyethylene glycol–induced membrane fusion events have served as a model system to create mouse and human hybridomas,66 to study eukaryotic cell–cell fusion events,67 and to deliver outward K+ currents into myocytes.24 Focal injection of HCN1-overexpressing fibroblasts suspended in 50% polyethylene glycol into the apex of guinea pig hearts yielded in vivo cell fusion. Heterokaryons of myocytes and HCN1-fibroblasts exhibited spontaneously oscillating APs with slow phase 4 depolarization (Figure 4B, bottom). In vivo biological pacemaker activities were demonstrated by electrocardiography as early as 1 day after cell injection and were stable for at least 2 weeks (Figure 4C).
Figure 4.

A, In vitro and in vivo fusion of myocytes with HCN1-fibroblasts. GFP-positive heterokaryons after in vitro (top) or in vivo (bottom) fusion of myocytes with HCN1-fibroblasts expressing GFP as a reporter. B, Heterokaryon formed by in vivo fusion of HCN1-fibroblast and a myocyte displays spontaneous AP oscillations in normal Tyrode’s (top). Presence of 1 mmol/L isoproterenol in the external solution increased the frequency of spontaneous AP oscillation in the same heterokaryon (bottom). C, ECGs from guinea pig hearts injected with HCN1-fibroblast cells. Top, Ectopic ventricular beats (diagonal arrows) are unleashed on slowing of the heart rate, which share the same polarity and morphology as the electrode-paced ECGs recorded at the site of HCN1-fibroblast injection. Bottom, In another animal, junctional escape rhythms (horizontal arrows) were overtaken by ectopic ventricular beats (diagonal arrows, 16 days after cell injection). Reproduced from Cho et al with permission.65
In contrast to the direct cell injection approaches, the fusion technology implants the biological pacemakers to the site of injection by somatic cell fusion, creating biological pacing at a specific site by design. Previous studies suggest that the in vivo fusion-induced heterokaryons can maintain the nuclei from each fusion partner separately and stably for at least several months.68-70 To develop this technology for clinical application, future steps would logically include large-animal testing to examine toxicity and efficacy.
Use of Stem Cells as Stand-Alone Biological Pacemakers
Human embryonic stem cells (hESCs) are among the most versatile agents for the development of cell-based therapies; they are pluripotent, clonogenic, and self-propagating.71 Human ESCs can differentiate in vitro, forming embryoid bodies (EBs) composed of derivatives of all 3 embryonic germ layers. Human EBs (hEBs) are inherently heterogeneous, and some EBs begin to contract spontaneously, containing cardiac myocytes that exhibit phase 4 depolarization.72-75 We and others reasoned that the hEBs could be used as stand-alone biological pacemakers with no genetic modification.76,77 The first step in this strategy is to verify that hEBs can electronically couple with the host myocardium.
An in vitro transplantation model was developed in which a single hESC-derived, spontaneously beating hEB (≈500 μm in diameter) was transplanted on top of a quiescent monolayer of neonatal rat ventricular myocytes (NRVMs) serving as the recipient. Coculturing a spontaneously beating hEB on a monolayer of quiescent NRVMs demonstrated expression of gap junction at the hEB-NRVM boundary and synchronous rhythmic contractions of the hEB on the NRVM monolayer. Furthermore, multielectrode and optical mapping recordings revealed that the spontaneous APs were initiated from the hEB and propagated to the NRVMs.
Spontaneously beating hEBs were then injected into the left ventricular anterior wall of a guinea pig in vivo. On cryoablating the sinus rhythm of an animal, ex vivo optical mapping displayed spontaneous APs from the left ventricular epicardial surface, which were not present in control hearts (uninjected or saline-injected). Because the hESC-derived biological pacemakers beat on their own before the tissue transplantation, they could prove to be a quick way to generate a spontaneous rhythm in the bradycardic setting. However, the immune response from the host remains a major hurdle in reducing this technology to clinic, a limitation which may eventually be overcome by induced pluripotent stem cell technologies.78 The potential for tumorigenesis, however, plagues both hESC and induced pluripotent stem approaches. Moreover, it is possible, if not likely, that ESC- and induced pluripotent stem–derived cardiac implants would lose their pacing ability as they engraft and mature in vivo.
In recent years, the view that the number of cardiomyocytes we are born stays unchanged has been challenged and overturned.79-81 This shift in paradigm was supported by the discovery of cardiac stem cells. Human adult cardiac stem cells readily form self-aggregating, 3D multicellular structures named cardiospheres.82 Methods that allow retrieval of >106 human adult cells from a single endomyocardial biopsy specimen were found to yield cardiomyocytes with cardiac specific markers, and some of these cells can differentiate into spontaneously contracting cardiac tissue with innate pacemaker function.83 The use of adult stem cells circumvents complications associated with hESCs including ethical concerns,84 immunogenic reactions against the donor cells,85 and teratoma formation.86 Clinical trials of autologous cardiac stem cells for cardiac regeneration have already begun (eg, CADUCEUS; see http://www.clinicaltrials.gov for details). Cardiac stem cells may be viable candidates for stand-alone cell therapy, although the stability of resultant pacemaker function has yet to be established.
Conversion of Nonexcitable Cells to Self-Contained Biological Pacemakers
Equipped with what we have learned from the above studies, we asked if expressing a minimal complement of ion channels would convert a nonexcitable cell into a self-contained, entirely engineered pacemaker cell. We reasoned that a minimal pacing apparatus would require a hyperpolarizing current to produce a negative diastolic potential, a depolarizing current to induce excitability, and a repolarizing current to reprime the system. Inexcitable HEK293 cells were engineered to express (1) an inward rectifier current through Kir2.1 to achieve a polarized membrane potential39 (Figure 5A, left); (2) a repolarizing current provided by human ether-a-go-go channel (HERG, Figure 5A, middle)87; and (3) an excitatory current to provide the upstroke of an AP. A Na+ channel from bacteria88 (NaChBac, Figure 5A, right) was chosen for the excitatory current, taking advantage of its slow gating kinetics. In the HEK293 cells expressing all 3 channel proteins, APs could be generated (n=5/31) on stimulation with a brief depolarizing current (0.3 to 0.7 nA) (Figure 5B). The stimulated APs displayed a large variation in their phenotypes, reflecting the randomness of the amount of cDNA taken up by a cell via routine transfection. At room temperature, the maximum diastolic potentials were −78±7 mV with an APD at 90% repolarization (APD90) value of 475±33 ms (n=5). Our own observations and data from others89,90 suggested that HEK293 cells express endogenous voltage-gated outward currents. This allowed us to omit HERG, and include HCN1 channel in our assay to test whether additional expression of IHCN1 would suffice to generate spontaneously oscillating APs in HEK293 cells. In addition, the compact cDNA of NaChBac allowed us to construct a tricistronic vector containing all 3 transgenes in one plasmid separated by 2 IRES segments under a CMV promoter. Whole-cell recordings from HEK293 cells expressing CMV-HCN1-NaChBac-Kir2.1 revealed spontaneous APs (Figure 5C). The spontaneous APs displayed a maximum diastolic potential of −81.5±11.8 mV, maximum rate of rise (dV/dtmax) of 21.6±8.6 V/sec, APD90 of 660±189 ms, and a rather slow frequency of 3±1 bpm, presumably resulting from the slow gating kinetics of NaChBac channels (n=4/85).91
Figure 5.
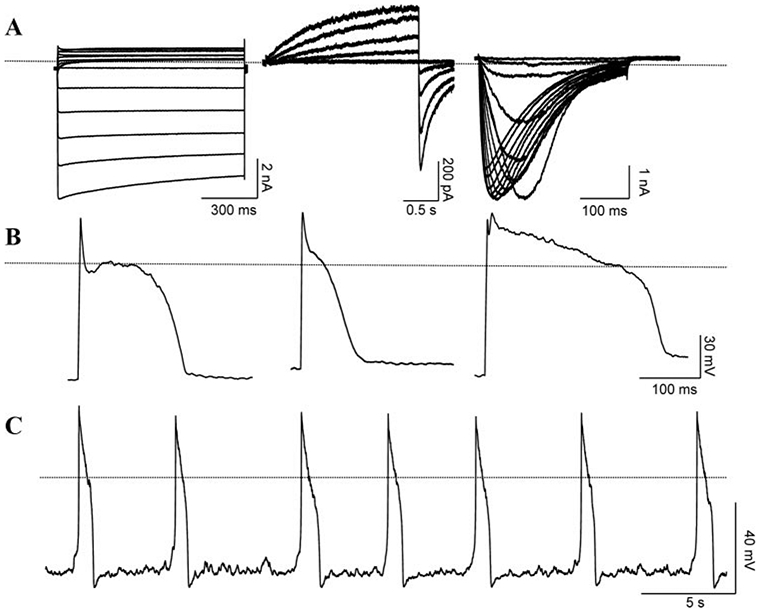
Representative raw recordings from HEK293 cells transfected with the following constructs. A, Voltage-clamp recordings from HEK293 cells transfected with either NaChBac (left), HERG (middle), or Kir2.1 (right). Dotted line indicates zero current level. B, APs from 3 different cells during current-clamp recordings. Each cell expressed all 3 channels, NaChBac, HERG, and Kir2.1. Dotted line indicates zero mV potential. C, Spontaneous APs recorded from a representative cell transfected with single plasmid expressing HCN1, NaChBac, and Kir2.1. Adapted from Cho et al.91
Recently, a body of evidence has emphasized the role of spontaneous, local calcium release (LCR) events from sarcoplasmic reticulum in the pacing mechanism of SA nodal cells.92,93 These studies revisit earlier observations that intracellular Ca2+ oscillators produce spontaneous inward currents,94 and implicate rhythmic LCRs as effectors of normal SA nodal automaticity. Although they may be important physiologically, LCRs are not required for pacing: the work in Figure 5 shows that an inexcitable cell, lacking sarcoplasmic reticulum, can be engineered to pace solely via voltage-dependent mechanisms. Moreover, the cellular work of Miake et al37 was performed in myocytes heavily buffered by internal EGTA, such that LCRs would have been suppressed.
Although conceptually interesting, de novo pacemaker cells are not likely to proceed rapidly to translation. Practical development of biological pacemakers would most logically begin with the straightforward single-transgene strategies (Kir2.1AAA or HCN channels) discussed earlier, as these would seem to be more likely to meet regulatory approval.
Genetic Alterations of Electric Conduction
In arrhythmias such as VT, modifying electric conduction would be a useful principle for treatment. Such is the rationale underlying catheter ablation of zones of slow conduction to suppress VT.95 Gene transfer, however, offers greater engineering versatility than endocardial destruction, not to mention the conceptual advantage that the targeted heart tissue is modified rather than destroyed. Gap junctional intercellular communication is an intuitive target to alter electric conduction. To this end, an internal loop mutant gene of connexin 43 was delivered in a lentiviral vector to uncouple electric connection in NRVMs.96 Transduced cells displayed delayed calcium transients as well as slowed conduction velocity. These data validate a molecular tool targeting gap junctions as a way to modulate electric conduction.
The myocardial infarct border zone is characterized by depolarized membrane potential with low AP upstroke velocity (Vmax) and reduced conduction. To correct the low Vmax by gene transfer, a skeletal Na+ channel gene (SkM1) has been ectopically expressed in the epicardial border zones of a canine infarct model. Adenoviral delivery of SkM1 restored Vmax and reduced the incidence of inducible sustained ventricular tachycardia/fibrillation.97 A skeletal (SkM1) rather than a cardiac (SCN5A) isoform was chosen to take advantage of the positively shifted voltage-dependent activation kinetics of SkM1, which would permit more channel opening than with Nav1.5 channels at depolarized membrane potentials.98 Together with gap-junctional modification, these studies begin to develop a novel toolkit for altering electric conduction by focal gene transfer.
Future Directions
Efforts to create biological alternatives to present antiarrhythmic therapies, particularly biological pacemakers, have led to multiple innovations. Figure 6 summarizes the distinct approaches that have been deployed to initiate automaticity in usually quiescent working myocardium. The genes-only strategy (top row) either overexpresses an excitatory gene, or suppresses IK1, to induce automaticity. The hybrid gene–cell approaches (middle rows) rely on gap-junctional coupling or cell fusion to relay the excitatory effects of an HCN gene. Finally, the use of stem cells (bottom row) relies on the endogenous complement of ion channels in hESC-derived cardiomyocytes to drive the enhanced automaticity.
Figure 6.

A summary of different approaches to creating a biological pacemaker. First approach (top row) is a strict gene therapy in which Kir2.1AAA, HCN, or synthetic pacemaker channel genes are overexpressed in myocytes via adenoviral delivery. Kir2.1 dominant negative proteins suppress repolarizing, outward currents whereas pacemaker channels directly contribute to diastolic membrane potential depolarization. Delivering If by MSCs requires gap-junctional coupling between myocytes and MSCs (second row). In the cell fusion approach (third row), If and the pacemaker activity arise from the HCN channels expressed on the cell membrane of the heterokaryon, without the need for gap-junctional coupling. Spontaneously beating human EBs and cardiospheres transduce their pacemaker activity to cardiomyocytes via electrotonic cell–cell coupling (fourth row).
In addition to new conceptual insights that merit consideration in the further development of biological pacing, much more work will be required on focal delivery of constructs/cells to the myocardium to reduce these concepts to practice. For example, injection of biological agents into the endocardium via an intracardiac injection catheter represents a potentially attractive percutaneous delivery route. Likewise, localized coronary circulation may allow isolated delivery to small regions of the heart (as was achieved with the AV node10). Highly localized biological delivery will be particularly effective for arrhythmias in which focal modifications of electric properties suffice for effective treatment. Because the amount of exogenous biological material delivered can be correspondingly reduced, potential problems attributable to widespread dissemination may be more readily averted. A singular advantage of focal modification is reversibility: if needed, implanted biological pacemakers can be destroyed by conventional electrophysiological ablation, followed by electronic pacemaker implantation.
Different clinical scenarios may call for different gene delivery vectors. Adenoviral vectors, for example, achieve the peak expression earlier than other vectors albeit in a transient manner. This would be ideal for the temporary pacing needs to treat infected pacemaker devices as discussed earlier. In this regard, the high immunogenicity of these vectors could be circumvented by using helper-dependent adenovirus.99 On the other hand, adeno-associated virus or lentivirus vectors would be more suitable for long-term applications.100
Will biological therapies replace drugs and devices? It is too early to tell. Biological pacemakers seem to have a reasonable chance of eventual success. It is our conjecture that the first application will likely be as an alternative to temporary electronic pacemakers in bradycardic patients with pacemaker infections, but more development work in large-animal models will be required before clinical testing can realistically be contemplated. Likewise, biological approaches targeting conduction or repolarization have shown great promise at various levels of preclinical development, but none is ready for prime time. What is certain is that the more we push the envelope, the more likely it is that we will devise creative alternatives to the present palliative approaches.
Further extensive work will be required to optimize the functional effects of biological therapies, to evaluate the toxicology, to exclude potential tumorigenicity in the case of stem cells, and to establish the long-term stability of therapy (in anything other than a purposely temporary indication). Yet, given the promise, the effort to develop biological alternatives to the present therapies appears more justified than ever.
Acknowledgments
We thank Peihong Dong for technical support in plasmid constructions and Michelle K. Leppo for animal procedures.
Sources of Funding
Supported by the Donald W. Reynolds Foundation (to E.M.) and by Heart Rhythm Society fellowships (to H.C.C.).
Non-standard Abbreviations and Acronyms
- AF
atrial fibrillation
- AP
action potential
- APD
action potential duration
- AV
atrioventricular
- EAD
early afterdepolarization
- HCN
hyperpolarization-activated, cyclic nucleotide–gated
- hEB
human embryoid body
- hESC
human embryonic stem cell
- hMSC
human mesenchymal stem cell
- LCR
local calcium release
- NRVM
neonatal rat ventricular myocyte
- SA
sinoatrial
- VT
ventricular tachycardia
Footnotes
Disclosures
E.M. owns stock in Excigen, Inc which licensed intellectual property on biological pacemakers. No research funding was provided by Excigen. H.C.C. reports no conflict of interest.
References
- 1.Coplen SE, Antman EM, Berlin JA, Hewitt P, Chalmers TC. Efficacy and safety of quinidine therapy for maintenance of sinus rhythm after cardioversion. A meta-analysis of randomized control trials. Circulation. 1990;82:1106–1116. [DOI] [PubMed] [Google Scholar]
- 2.Echt DS, Liebson PR, Mitchell LB, Peters RW, ObiasManno D, Barker AH, Arensberg D, Baker A, Friedman L, Green HL, Huther ML, Richardson DW. Mortality and morbidity in patients receiving encainide, flecainide, or placebo. The Cardiac Arrhythmia Suppression Trial. N Engl J Med. 1991;324:781–788. [DOI] [PubMed] [Google Scholar]
- 3.Siebels J, Cappato R, Ruppel R, Schneider MA, Kuck KH. Preliminary results of the Cardiac Arrest Study Hamburg (CASH). CASH Investigators. Am J Cardiol. 1993;72:109F–113F. [DOI] [PubMed] [Google Scholar]
- 4.Waldo AL, Camm AJ, deRuyter H, Friedman PL, MacNeil DJ, Pauls JF, Pitt B, Pratt CM, Schwartz PJ, Veltri EP. Effect of d-sotalol on mortality in patients with left ventricular dysfunction after recent and remote myocardial infarction. The SWORD Investigators. Survival With Oral d-Sotalol. Lancet. 1996;348:7–12. [DOI] [PubMed] [Google Scholar]
- 5.Falk RH. Atrial fibrillation. N Engl J Med. 2001;344:1067–1078. [DOI] [PubMed] [Google Scholar]
- 6.Richardson AW, Josephson ME. Ablation of ventricular tachycardia in the setting of coronary artery disease. Curr Cardiol Rep. 1999;1:157–164. [DOI] [PubMed] [Google Scholar]
- 7.Bernstein AD, Parsonnet V. Survey of cardiac pacing and implanted defibrillator practice patterns in the United States in 1997. Pacing Clin Electrophysiol. 2001;24:842–855. [DOI] [PubMed] [Google Scholar]
- 8.Khand AU, Rankin AC, Kaye GC, Cleland JG. Systematic review of the management of atrial fibrillation in patients with heart failure. Eur Heart J. 2000;21:614–632. [DOI] [PubMed] [Google Scholar]
- 9.Pelargonio G, Prystowsky EN. Rate versus rhythm control in the management of patients with atrial fibrillation. Nat Clin Pract Cardiovasc Med. 2005;2:514–521. [DOI] [PubMed] [Google Scholar]
- 10.Donahue JK, Heldman AW, Fraser H, McDonald AD, Miller JM, Rade JJ, Eschenhagen T, Marban E. Focal modification of electrical conduction in the heart by viral gene transfer. Nat Med. 2000;6:1395–1398. [DOI] [PubMed] [Google Scholar]
- 11.Donahue JK, Kikkawa K, Thomas AD, Marban E, Lawrence JH. Acceleration of widespread adenoviral gene transfer to intact rabbit hearts by coronary perfusion with low calcium and serotonin. Gene Ther. 1998;5:630–634. [DOI] [PubMed] [Google Scholar]
- 12.Nagata K, Marban E, Lawrence JH, Donahue JK. Phosphodiesterase inhibitor-mediated potentiation of adenovirus delivery to myocardium. J Mol Cell Cardiol. 2001;33:575–580. [DOI] [PubMed] [Google Scholar]
- 13.Bauer A, McDonald AD, Nasir K, Peller L, Rade JJ, Miller JM, Heldman AW, Donahue JK. Inhibitory G protein overexpression provides physiologically relevant heart rate control in persistent atrial fibrillation. Circulation. 2004;110:3115–3120. [DOI] [PubMed] [Google Scholar]
- 14.Beguin P, Nagashima K, Gonoi T, Shibasaki T, Takahashi K, Kashima Y, Ozaki N, Geering K, Iwanaga T, Seino S. Regulation of Ca2+ channel expression at the cell surface by the small G-protein kir/Gem. Nature. 2001;411:701–706. [DOI] [PubMed] [Google Scholar]
- 15.Murata M, Cingolani E, McDonald AD, Donahue JK, Marban E. Creation of a genetic calcium channel blocker by targeted gem gene transfer in the heart. Circ Res. 2004;95:398–405. [DOI] [PubMed] [Google Scholar]
- 16.Bunch TJ, Mahapatra S, Bruce GK, Johnson SB, Miller DV, Horne BD, Wang XL, Lee HC, Caplice NM, Packer DL. Impact of transforming growth factor-beta1 on atrioventricular node conduction modification by injected autologous fibroblasts in the canine heart. Circulation. 2006;113:2485–2494. [DOI] [PubMed] [Google Scholar]
- 17.Wright M, Haissaguerre M, Knecht S, Matsuo S, O’Neill MD, Nault I, Lellouche N, Hocini M, Sacher F, Jais P. State of the art: catheter ablation of atrial fibrillation. J Cardiovasc Electrophysiol. 2008;19:583–592. [DOI] [PubMed] [Google Scholar]
- 18.Antzelevitch C, Shimizu W. Cellular mechanisms underlying the long QT syndrome. Curr Opin Cardiol. 2002;17:43–51. [DOI] [PubMed] [Google Scholar]
- 19.Johns DC, Nuss HB, Chiamvimonvat N, Ramza BM, Marban E, Lawrence JH. Adenovirus-mediated expression of a voltage-gated potassium channel in vitro (rat cardiac myocytes) and in vivo (rat liver). A novel strategy for modifying excitability. J Clin Invest. 1995;96:1152–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nuss HB, Johns DC, Kaab S, Tomaselli GF, Kass D, Lawrence JH, Marban E. Reversal of potassium channel deficiency in cells from failing hearts by adenoviral gene transfer: a prototype for gene therapy for disorders of cardiac excitability and contractility. Gene Ther. 1996;3:900–912. [PubMed] [Google Scholar]
- 21.Nuss HB, Marban E, Johns DC. Overexpression of a human potassium channel suppresses cardiac hyperexcitability in rabbit ventricular myocytes. J Clin Invest. 1999;103:889–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hoppe UC, Marban E, Johns DC. Distinct gene-specific mechanisms of arrhythmia revealed by cardiac gene transfer of two long QT disease genes, HERG and KCNE1. Proc Natl Acad Sci U S A. 2001;98:5335–5340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sasano T, McDonald AD, Kikuchi K, Donahue JK. Molecular ablation of ventricular tachycardia after myocardial infarction. Nat Med. 2006;12:1256–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hoppe UC, Johns DC, Marban E, O’Rourke B. Manipulation of cellular excitability by cell fusion: effects of rapid introduction of transient outward K+ current on the guinea pig action potential. Circ Res. 1999;84:964–972. [DOI] [PubMed] [Google Scholar]
- 25.Mitcheson JS, Hancox JC, Levi AJ. Cultured adult cardiac myocytes: future applications, culture methods, morphological and electrophysiological properties. Cardiovasc Res. 1998;39:280–300. [DOI] [PubMed] [Google Scholar]
- 26.Hoppe UC, Marban E, Johns DC. Molecular dissection of cardiac repolarization by in vivo Kv4.3 gene transfer. J Clin Invest. 2000;105:1077–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ruan Y, Liu N, Priori SG. Sodium channel mutations and arrhythmias. Nat Rev Cardiol. 2009;6:337–348. [DOI] [PubMed] [Google Scholar]
- 28.Brunner M, Kodirov SA, Mitchell GF, Buckett PD, Shibata K, Folco EJ, Baker L, Salama G, Chan DP, Zhou J, Koren G. In vivo gene transfer of Kv1.5 normalizes action potential duration and shortens QT interval in mice with long QT phenotype. Am J Physiol. 2003;285:H194–H203. [DOI] [PubMed] [Google Scholar]
- 29.Kodirov SA, Brunner M, Busconi L, Koren G. Long-term restitution of 4-aminopyridine-sensitive currents in Kv1DN ventricular myocytes using adeno-associated virus-mediated delivery of Kv1.5. FEBS Lett. 2003;550:74–78. [DOI] [PubMed] [Google Scholar]
- 30.Singh BN, Wadhani N. Antiarrhythmic and proarrhythmic properties of QT-prolonging antianginal drugs. J Cardiovasc Pharmacol Ther. 2004;9(suppl 1):S85–S97. [DOI] [PubMed] [Google Scholar]
- 31.Brooks CM, Lu H-H. The Sinoatrial Pacemaker of the Heart. Springfield, Ill: Thomas Charles C.; 1972. [Google Scholar]
- 32.Kusumoto FM, Goldschlager N. Cardiac pacing. N Engl J Med. 1996;334:89–97. [DOI] [PubMed] [Google Scholar]
- 33.Rosamond W, Flegal K, Furie K, Go A, Greenlund K, Haase N, Hailpern SM, Ho M, Howard V, Kissela B, Kittner S, Lloyd-Jones D, McDermott M, Meigs J, Moy C, Nichol G, O’Donnell C, Roger V, Sorlie P, Steinberger J, Thom T, Wilson M, Hong Y. Heart disease and stroke statistics—2008 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2008;117:e25–e146. [DOI] [PubMed] [Google Scholar]
- 34.Henrikson CA, Brinker JA. How to prevent, recognize, and manage complications of lead extraction. Part I: avoiding lead extraction–infectious issues. Heart Rhythm. 2008;5:1083–1087. [DOI] [PubMed] [Google Scholar]
- 35.Edelberg JM, Aird WC, Rosenberg RD. Enhancement of murine cardiac chronotropy by the molecular transfer of the human beta2 adrenergic receptor cDNA. J Clin Invest. 1998;101:337–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Edelberg JM, Huang DT, Josephson ME, Rosenberg RD. Molecular enhancement of porcine cardiac chronotropy. Heart. 2001;86:559–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miake J, Marban E, Nuss HB. Biological pacemaker created by gene transfer. Nature. 2002;419:132–133. [DOI] [PubMed] [Google Scholar]
- 38.Schram G, Pourrier M, Melnyk P, Nattel S. Differential distribution of cardiac ion channel expression as a basis for regional specialization in electrical function. Circ Res. 2002;90:939–950. [DOI] [PubMed] [Google Scholar]
- 39.Kubo Y, Baldwin TJ, Jan YN, Jan LY. Primary structure and functional expression of a mouse inward rectifier potassium channel. Nature. 1993;362:127–133. [DOI] [PubMed] [Google Scholar]
- 40.Tinker A, Jan YN, Jan LY. Regions responsible for the assembly of inwardly rectifying potassium channels. Cell. 1996;87:857–868. [DOI] [PubMed] [Google Scholar]
- 41.Irisawa H, Brown HF, Giles W. Cardiac pacemaking in the sinoatrial node. Physiol Rev. 1993;73:197–227. [DOI] [PubMed] [Google Scholar]
- 42.Boyett MR, Honjo H, Kodama I. The sinoatrial node, a heterogeneous pacemaker structure. Cardiovasc Res. 2000;47:658–687. [DOI] [PubMed] [Google Scholar]
- 43.DiFrancesco D. Serious workings of the funny current. Prog Biophys Mol Biol. 2006;90:13–25. [DOI] [PubMed] [Google Scholar]
- 44.Baruscotti M, Difrancesco D. Pacemaker channels. Ann N Y Acad Sci. 2004;1015:111–121. [DOI] [PubMed] [Google Scholar]
- 45.Sekar RB, Kizana E, Cho HC, Molitoris JM, Hesketh GG, Eaton BP, Marban E, Tung L. IK1 heterogeneity affects genesis and stability of spiral waves in cardiac myocyte monolayers. Circ Res. 2009;104:355–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brown HF, Kimura J, Noble D, Noble SJ, Taupignon A. The ionic currents underlying pacemaker activity in rabbit sino-atrial node: experimental results and computer simulations. Proc R Soc Lond B Biol Sci. 1984;222:329–347. [DOI] [PubMed] [Google Scholar]
- 47.Miake J, Marban E, Nuss HB. Functional role of inward rectifier current in heart probed by Kir2.1 overexpression and dominant-negative suppression. J Clin Invest. 2003;111:1529–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brugada P, Wellens HJ. Early afterdepolarizations: role in conduction block, “prolonged repolarization-dependent reexcitation,” and tachyarrhythmias in the human heart. Pacing Clin Electrophysiol. 1985;8:889–896. [DOI] [PubMed] [Google Scholar]
- 49.January CT, Moscucci A. Cellular mechanisms of early afterdepolarizations. Ann N Y Acad Sci. 1992;644:23–32. [DOI] [PubMed] [Google Scholar]
- 50.Ennis IL, Li RA, Murphy AM, Marban E, Nuss HB. Dual gene therapy with SERCA1 and Kir2.1 abbreviates excitation without suppressing contractility. J Clin Invest. 2002;109:393–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Baruscotti M, Bucchi A, Difrancesco D. Physiology and pharmacology of the cardiac pacemaker (“funny”) current. Pharmacol Ther. 2005;107:59–79. [DOI] [PubMed] [Google Scholar]
- 52.Plotnikov AN, Sosunov EA, Qu J, Shlapakova IN, Anyukhovsky EP, Liu L, Janse MJ, Brink PR, Cohen IS, Robinson RB, Danilo P Jr, Rosen MR. Biological pacemaker implanted in canine left bundle branch provides ventricular escape rhythms that have physiologically acceptable rates. Circulation. 2004;109:506–512. [DOI] [PubMed] [Google Scholar]
- 53.Qu J, Plotnikov AN, Danilo P Jr, Shlapakova I, Cohen IS, Robinson RB, Rosen MR. Expression and function of a biological pacemaker in canine heart. Circulation. 2003;107:1106–1109. [DOI] [PubMed] [Google Scholar]
- 54.Tse HF, Xue T, Lau CP, Siu CW, Wang K, Zhang QY, Tomaselli GF, Akar FG, Li RA. Bioartificial sinus node constructed via in vivo gene transfer of an engineered pacemaker HCN Channel reduces the dependence on electronic pacemaker in a sick-sinus syndrome model. Circulation. 2006;114:1000–1011. [DOI] [PubMed] [Google Scholar]
- 55.Kashiwakura Y, Cho HC, Barth AS, Azene E, Marban E. Gene transfer of a synthetic pacemaker channel into the heart: a novel strategy for biological pacing. Circulation. 2006;114:1682–1686. [DOI] [PubMed] [Google Scholar]
- 56.Campos SK, Barry MA. Current advances and future challenges in Adenoviral vector biology and targeting. Curr Gene Ther. 2007;7:189–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vinge LE, Raake PW, Koch WJ. Gene therapy in heart failure. Circ Res. 2008;102:1458–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pittenger MF, Martin BJ. Mesenchymal stem cells and their potential as cardiac therapeutics. Circ Res. 2004;95:9–20. [DOI] [PubMed] [Google Scholar]
- 59.Uccelli A, Moretta L, Pistoia V. Mesenchymal stem cells in health and disease. Nat Rev. 2008;8:726–736. [DOI] [PubMed] [Google Scholar]
- 60.Potapova I, Plotnikov A, Lu Z, Danilo P Jr, Valiunas V, Qu J, Doronin S, Zuckerman J, Shlapakova IN, Gao J, Pan Z, Herron AJ, Robinson RB, Brink PR, Rosen MR, Cohen IS. Human mesenchymal stem cells as a gene delivery system to create cardiac pacemakers. Circ Res. 2004;94:952–959. [DOI] [PubMed] [Google Scholar]
- 61.Plotnikov AN, Shlapakova I, Szabolcs MJ, Danilo P Jr, Lorell BH, Potapova IA, Lu Z, Rosen AB, Mathias RT, Brink PR, Robinson RB, Cohen IS, Rosen MR. Xenografted adult human mesenchymal stem cells provide a platform for sustained biological pacemaker function in canine heart. Circulation. 2007;116:706–713. [DOI] [PubMed] [Google Scholar]
- 62.van der Velden HM, Jongsma HJ. Cardiac gap junctions and connexins: their role in atrial fibrillation and potential as therapeutic targets. Cardiovasc Res. 2002;54:270–279. [DOI] [PubMed] [Google Scholar]
- 63.Rosen AB, Kelly DJ, Schuldt AJ, Lu J, Potapova IA, Doronin SV, Robichaud KJ, Robinson RB, Rosen MR, Brink PR, Gaudette GR, Cohen IS. Finding fluorescent needles in the cardiac haystack: tracking human mesenchymal stem cells labeled with quantum dots for quantitative in vivo three-dimensional fluorescence analysis. Stem Cells. 2007;25:2128–2138. [DOI] [PubMed] [Google Scholar]
- 64.Chang MG, Tung L, Sekar RB, Chang CY, Cysyk J, Dong P, Marban E, Abraham MR. Proarrhythmic potential of mesenchymal stem cell transplantation revealed in an in vitro coculture model. Circulation. 2006;113:1832–1841. [DOI] [PubMed] [Google Scholar]
- 65.Cho HC, Kashiwakura Y, Marban E. Creation of a biological pacemaker by cell fusion. Circ Res. 2007;100:1112–1115. [DOI] [PubMed] [Google Scholar]
- 66.Shirahata S, Katakura Y, Teruya K. Cell hybridization, hybridomas, and human hybridomas. Methods Cell Biol. 1998;57:111–145. [DOI] [PubMed] [Google Scholar]
- 67.Lentz BR, Lee JK. Poly(ethylene glycol) (PEG)-mediated fusion between pure lipid bilayers: a mechanism in common with viral fusion and secretory vesicle release? Mol Membr Biol. 1999;16:279–296. [DOI] [PubMed] [Google Scholar]
- 68.Alvarez-Dolado M, Pardal R, Garcia-Verdugo JM, Fike JR, Lee HO, Pfeffer K, Lois C, Morrison SJ, Alvarez-Buylla A. Fusion of bone-marrow-derived cells with Purkinje neurons, cardiomyocytes and hepatocytes. Nature. 2003;425:968–973. [DOI] [PubMed] [Google Scholar]
- 69.Gussoni E, Bennett RR, Muskiewicz KR, Meyerrose T, Nolta JA, Gilgoff I, Stein J, Chan YM, Lidov HG, Bonnemann CG, Von Moers A, Morris GE, Den Dunnen JT, Chamberlain JS, Kunkel LM, Weinberg K. Long-term persistence of donor nuclei in a Duchenne muscular dystrophy patient receiving bone marrow transplantation. J Clin Invest. 2002;110:807–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Weimann JM, Johansson CB, Trejo A, Blau HM. Stable reprogrammed heterokaryons form spontaneously in Purkinje neurons after bone marrow transplant. Nat Cell Biol. 2003;5:959–966. [DOI] [PubMed] [Google Scholar]
- 71.Hoffman LM, Carpenter MK. Characterization and culture of human embryonic stem cells. Nat Biotechnol. 2005;23:699–708. [DOI] [PubMed] [Google Scholar]
- 72.He JQ, Ma Y, Lee Y, Thomson JA, Kamp TJ. Human embryonic stem cells develop into multiple types of cardiac myocytes: action potential characterization. Circ Res. 2003;93:32–39. [DOI] [PubMed] [Google Scholar]
- 73.Kehat I, Kenyagin-Karsenti D, Snir M, Segev H, Amit M, Gepstein A, Livne E, Binah O, Itskovitz-Eldor J, Gepstein L. Human embryonic stem cells can differentiate into myocytes with structural and functional properties of cardiomyocytes. J Clin Invest. 2001;108:407–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mummery C, Ward-van Oostwaard D, Doevendans P, Spijker R, van den Brink S, Hassink R, van der Heyden M, Opthof T, Pera M, de la Riviere AB, Passier R, Tertoolen L. Differentiation of human embryonic stem cells to cardiomyocytes: role of coculture with visceral endoderm-like cells. Circulation. 2003;107:2733–2740. [DOI] [PubMed] [Google Scholar]
- 75.Xu C, Police S, Rao N, Carpenter MK. Characterization and enrichment of cardiomyocytes derived from human embryonic stem cells. Circ Res. 2002;91:501–508. [DOI] [PubMed] [Google Scholar]
- 76.Kehat I, Khimovich L, Caspi O, Gepstein A, Shofti R, Arbel G, Huber I, Satin J, Itskovitz-Eldor J, Gepstein L. Electromechanical integration of cardiomyocytes derived from human embryonic stem cells. Nat Biotechnol. 2004;22:1282–1289. [DOI] [PubMed] [Google Scholar]
- 77.Xue T, Cho HC, Akar FG, Tsang SY, Jones SP, Marban E, Tomaselli GF, Li RA. Functional integration of electrically active cardiac derivatives from genetically engineered human embryonic stem cells with quiescent recipient ventricular cardiomyocytes: insights into the development of cell-based pacemakers. Circulation. 2005;111:11–20. [DOI] [PubMed] [Google Scholar]
- 78.Zhang J, Wilson GF, Soerens AG, Koonce CH, Yu J, Palecek SP, Thomson JA, Kamp TJ. Functional cardiomyocytes derived from human induced pluripotent stem cells. Circ Res. 2009;104:e30–e41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Anversa P, Kajstura J, Leri A, Bolli R. Life and death of cardiac stem cells: a paradigm shift in cardiac biology. Circulation. 2006;113:1451–1463. [DOI] [PubMed] [Google Scholar]
- 80.Beltrami AP, Barlucchi L, Torella D, Baker M, Limana F, Chimenti S, Kasahara H, Rota M, Musso E, Urbanek K, Leri A, Kajstura J, Nadal-Ginard B, Anversa P. Adult cardiac stem cells are multipotent and support myocardial regeneration. Cell. 2003;114:763–776. [DOI] [PubMed] [Google Scholar]
- 81.Bergmann O, Bhardwaj RD, Bernard S, Zdunek S, Barnabe-Heider F, Walsh S, Zupicich J, Alkass K, Buchholz BA, Druid H, Jovinge S, Frisen J. Evidence for cardiomyocyte renewal in humans. Science. 2009;324:98–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Messina E, De Angelis L, Frati G, Morrone S, Chimenti S, Fiordaliso F, Salio M, Battaglia M, Latronico MV, Coletta M, Vivarelli E, Frati L, Cossu G, Giacomello A. Isolation and expansion of adult cardiac stem cells from human and murine heart. Circ Res. 2004;95:911–921. [DOI] [PubMed] [Google Scholar]
- 83.Smith RR, Barile L, Cho HC, Leppo MK, Hare JM, Messina E, Giacomello A, Abraham MR, Marban E. Regenerative potential of cardiosphere-derived cells expanded from percutaneous endomyocardial biopsy specimens. Circulation. 2007;115:896–908. [DOI] [PubMed] [Google Scholar]
- 84.Robertson JA. Human embryonic stem cell research: ethical and legal issues. Nat Rev Genet. 2001;2:74–78. [DOI] [PubMed] [Google Scholar]
- 85.Gerecht-Nir S, Itskovitz-Eldor J. Cell therapy using human embryonic stem cells. Transpl Immunol. 12:203–209, 2004. [DOI] [PubMed] [Google Scholar]
- 86.Odorico JS, Kaufman DS, Thomson JA. Multilineage differentiation from human embryonic stem cell lines. Stem Cells. 2001;19:193–204. [DOI] [PubMed] [Google Scholar]
- 87.Sanguinetti MC, Jiang C, Curran ME, Keating MT. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell. 1995;81:299–307. [DOI] [PubMed] [Google Scholar]
- 88.Ren D, Navarro B, Xu H, Yue L, Shi Q, Clapham DE. A prokaryotic voltage-gated sodium channel. Science. 2001;294:2372–2375. [DOI] [PubMed] [Google Scholar]
- 89.Kurejova M, Uhrik B, Sulova Z, Sedlakova B, Krizanova O, Lacinova L. Changes in ultrastructure and endogenous ionic channels activity during culture of HEK 293 cell line. Eur J Pharmacol. 567:10–18, 2007. [DOI] [PubMed] [Google Scholar]
- 90.Varghese A, Tenbroek EM, Coles J Jr, Sigg DC. Endogenous channels in HEK cells and potential roles in HCN ionic current measurements. Prog Biophys Mol Biol. 90:26–37, 2006. [DOI] [PubMed] [Google Scholar]
- 91.Cho HC, Kashiwakura Y, Marban E. Conversion of non-excitable cells to self-contained biological pacemakers. Circulation. 2005;112:II–307. Abstract. [Google Scholar]
- 92.Lakatta EG, Maltsev VA, Bogdanov KY, Stern MD, Vinogradova TM. Cyclic variation of intracellular calcium: a critical factor for cardiac pacemaker cell dominance. Circ Res. 2003;92:e45–e50. [DOI] [PubMed] [Google Scholar]
- 93.Lakatta EG, Vinogradova TM, Maltsev VA. The missing link in the mystery of normal automaticity of cardiac pacemaker cells. Ann N Y Acad Sci. 2008;1123:41–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kass RS, Tsien RW. Fluctuations in membrane current driven by intracellular calcium in cardiac Purkinje fibers. Biophys J. 1982;38:259–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Das MK, Dandamudi G, Steiner H. Role of ablation therapy in ventricular arrhythmias. Cardiol Clin. 2008;26:459–479. [DOI] [PubMed] [Google Scholar]
- 96.Kizana E, Chang CY, Cingolani E, Ramirez-Correa GA, Sekar RB, Abraham MR, Ginn SL, Tung L, Alexander IE, Marban E. Gene transfer of connexin43 mutants attenuates coupling in cardiomyocytes: novel basis for modulation of cardiac conduction by gene therapy. Circ Res. 2007;100:1597–1604. [DOI] [PubMed] [Google Scholar]
- 97.Lau DH, Clausen C, Sosunov EA, Shlapakova IN, Anyukhovsky EP, Danilo P Jr, Rosen TS, Kelly C, Duffy HS, Szabolcs MJ, Chen M, Robinson RB, Lu J, Kumari S, Cohen IS, Rosen MR. Epicardial border zone overexpression of skeletal muscle sodium channel SkM1 normalizes activation, preserves conduction, and suppresses ventricular arrhythmia: an in silico, in vivo, in vitro study. Circulation. 2009;119:19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Protas L, Dun W, Jia Z, Lu J, Bucchi A, Kumari S, Chen M, Cohen IS, Rosen MR, Entcheva E, Robinson RB. Expression of skeletal but not cardiac Na+ channel isoform preserves normal conduction in a depolarized cardiac syncytium. Cardiovasc Res. 2009;81:528–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mitani K, Graham FL, Caskey CT, Kochanek S. Rescue, propagation, and partial purification of a helper virus-dependent adenovirus vector. Proc Natl Acad Sci U S A. 1995;92:3854–3858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gray SJ, Samulski RJ. Optimizing gene delivery vectors for the treatment of heart disease. Expert Opin Biol Ther. 2008;8:911–922. [DOI] [PubMed] [Google Scholar]