

Abstract
Opening of the CFTR channel is coupled to the motion of its two nucleotide-binding domains (NBDs): they form a heterodimer sandwiching two functionally distinct ATP binding sites (site 1 and 2). While active ATP hydrolysis in site 2 triggers rapid channel closure, the functional role of stable ATP binding in the catalysis-incompetent (or degenerate) site 1, a feature conserved in many other ABC transporter proteins, remains elusive. Here, we found that CFTR loses its prompt responsiveness to ATP after the channel is devoid of ATP for tens to hundreds of seconds. Mutants with weakened ATP binding in site 1 and the most prevalent disease-causing mutation F508del are more vulnerable to ATP depletion. In contrast, strengthening ligand binding in site 1 with N6-(2-phenylethyl)-ATP, a high-affinity ATP analog, or abolishing ATP hydrolysis in site 2 by the mutation D1370N helps sustain a durable function of the otherwise unstable mutant channels. Thus, tight binding of ATP in the degenerate ATP binding site is crucial to the functional stability of CFTR. Small molecules targeting site 1 may bear therapeutic potential to overcome membrane instability of F508del-CFTR.
Keywords: anion channel, gating, ABC transporter, cystic fibrosis, ATP hydrolysis
Introduction
Unlike most other ATP-binding cassette (ABC) proteins that function as active transporters, the cystic fibrosis transmembrane conductance regulator (CFTR), mutations of which lead to the genetic disease cystic fibrosis (CF), is a bona fide ion channel (Riordan et al., 1989; Bear et al., 1992). CFTR inherits the canonical structural motifs evolutionarily conserved in ABC transporters: two transmembrane domains (TMDs) that craft a gated anion-selective pore, and two cytosolic nucleotide-binding domains (NBD1 and NBD2) coalescing into a head-to-tail dimer upon ATP binding to form two ATP binding sites (site 1 and site 2) at the dimer interface (Zhang & Chen, 2016; Liu et al., 2017; Zhang et al., 2017; Zhang et al., 2018). In addition, CFTR possesses a regulatory (R) domain whose phosphorylation by protein kinase A (PKA) enables ATP to open the channel (Cheng et al., 1991; Picciotto et al., 1992). In an unphosphorylated CFTR, the R domain is wedged between the TMDs and the NBDs, preventing NBD dimerization (Liu et al., 2017). The phosphorylated R domain is relocated to allow the interaction between the two halves of CFTR (Zhang et al., 2018). In short, the opening and closing of phosphorylated CFTR are driven respectively by ATP binding-induced NBD dimerization and hydrolysis-elicited separation of the NBD dimer (Fig. 1A; Vergani et al., 2005).
Figure 1. Closed and open CFTR structures may not suffice to explain CFTR’s response to ATP following prolonged channel closure.
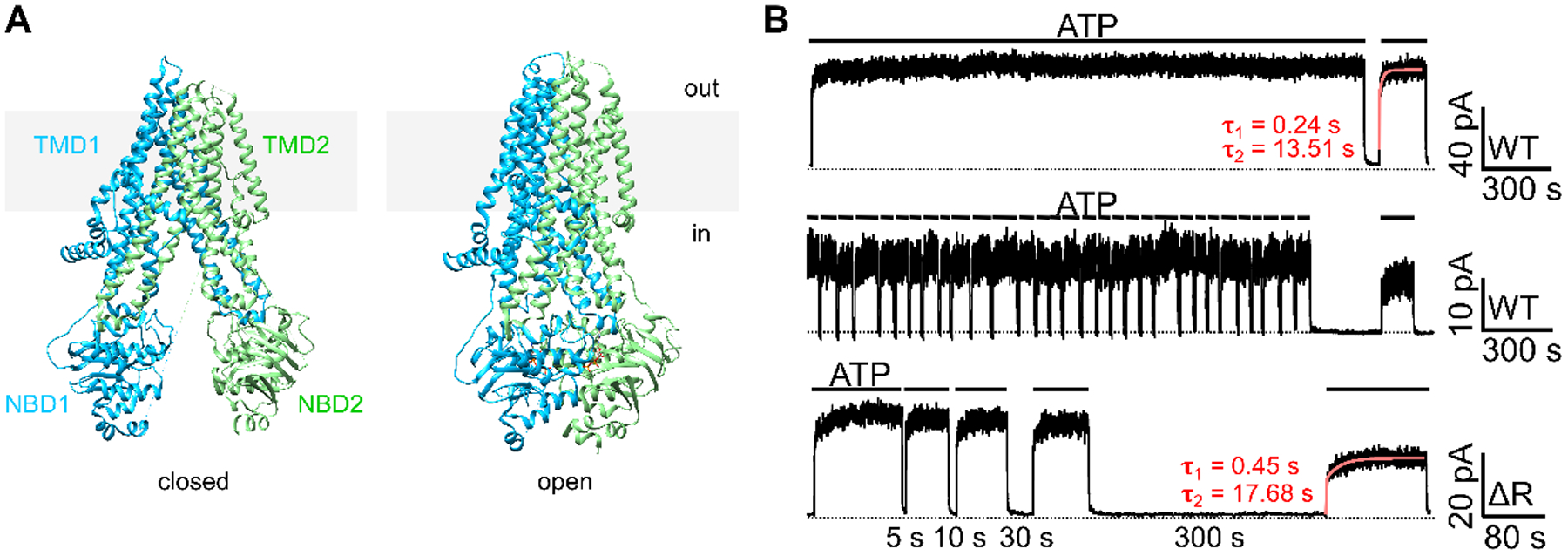
(A) CryoEM structures of the presumed closed (left) and open (right) channel conformations of CFTR. The closed conformation is represented by the structure of unphosphorylated, ATP-free CFTR (left; PDB: 5UAK; Liu et al., 2017); whereas the open conformation is represented by phosphorylated, ATP-bound CFTR (right; PDB: 6MSM; Zhang et al., 2018). Of note, although the pore of the ATP-bound CFTR structure is not wide enough for the passage of a dehydrated chloride, the overall architecture should nonetheless closely resemble an open conformation (Zhang et al., 2018). For clarity, the R domain (a.a. 637–845) was removed from the structures. Grey rectangles represent the lipid bilayer. In: intracellular side. Out: extracellular side. (B) Prolonged channel closure alters the response of WT- and ΔR-CFTR to ATP. Once activated by PKA and ATP, WT-CFTR currents remained stable in the continuous presence of ATP for 30 minutes (top trace). Repeated brief removal of ATP (10 seconds, 30 times) did not affect the current amplitude in the presence of ATP (middle trace), yet a 300-second prolonged withdrawal of ATP resulted in irreversible loss of the currents (~25% in this experiment). Like WT-CFTR, ΔR-CFTR, after prolonged depletion of ATP, failed to achieve the initial current amplitude prior to the 300-second removal of ATP (bottom trace). The red curves mark the double-exponential fit, yielding a fast (τ1) and a slow (τ2) time constants. Similar observations were made in 4 patches with WT-CFTR and 5 patches with ΔR-CFTR. Inward currents are displayed as upward deflections in all figures.
The two ATP binding sites are biochemically asymmetric and contribute differently to CFTR gating. Structurally, site 1 lacks the catalytic glutamate and the conserved H-loop histidine, two amino acids critical for ATP hydrolysis (Lewis et al., 2004). Photolabeling experiments using 8-Azido-32P-labeled nucleotides identified site 1 as a high-affinity binding site with negligible catalytic activity, in contrast to site 2 which has a lower binding affinity but rapidly hydrolyzes ATP (Aleksandrov et al., 2002; Basso et al., 2003). Ligand exchange experiments using ATP and the high-affinity ATP analog N6-(2-phenylethyl)-ATP (P-ATP) in inside-out patches further suggested that ATP stays bound in site 1 for tens of seconds (Tsai et al., 2010b). This stable binding of ATP in site 1 allows CFTR’s dimeric NBDs (or full dimer) to be converted—upon ATP hydrolysis and release of the hydrolytic products—to a state where site 1 remains occupied but site 2 becomes vacated. Multiple cycles of ATP binding and hydrolysis can therefore repeatedly occur in site 2 without the dissociation of the tightly bound ATP in site 1 (Tsai et al., 2010b; Szollosi et al., 2011; Jih et al., 2012).
While gating of CFTR is mainly governed by binding and hydrolysis of ATP in the NBDs, other factors may affect CFTR channel function. It has been reported that the activity of phosphorylated CFTR could deteriorate over time even when the supply of ATP remains constant in electrophysiological experiments (Weinreich et al., 1999; Szellas & Nagel, 2003; Wang et al., 2014). This gradual loss of channel activity is known as “channel rundown,” a phenomenon not unique to CFTR (Becq, 1996; Tóth & Csanády, 2012; Proks et al., 2016). Some early reports showed that membrane-associated phosphatases, by dephosphorylating PKA-activated CFTR, play a role in rundown in excised membrane patches; however, this functional decay caused by dephosphorylation can be prevented by phosphatase inhibitors or rescued by re-application of PKA (Berger et al., 1993; Becq et al., 1994). In our own experiments as well as others (Fig. 1 in Szellas & Nagel, 2003; Fig. 2 in Tsai et al., 2010b; Fig. 1 in Mihályi et al., 2016; Fig. 4 in Mihályi et al., 2020), we noticed phosphorylation-independent rundown: After CFTR channels are deprived of ATP for a prolonged period of time, upon re-addition of ATP, CFTR currents rise in two phases—a fast one completed within seconds followed by a slower rising phase that can last for tens to hundreds of seconds (e.g., Fig. 1B top trace). This observation indicates that when CFTR channels are deprived of ATP for extended time, there exist at least two distinct populations of closed channels with different responsiveness to ATP. Of note, cryo-EM studies so far have resolved only one closed conformation (Fig. 1A), which do not explain these functional data.
Figure 2. Relationship between ATP depletion time and the degree of rundown for WT-CFTR.

(A) A real-time macroscopic current trace of WT-CFTR pre-activated by PKA plus 2 mM ATP at −30 mV in response to ATP removal and re-addition. Red lines represent double-exponential fit of the current rising phase upon additions of ATP, yielding the first fast time constant (τ1) and the second slow time constant (τ2). Dashed line indicates the baseline. The duration of each ATP removal was labeled at the bottom of the trace. The boxed areas are expanded in B. (B) Expanded view of the recovery of currents after 10-second and 300-second washout. (C) Relationships between the two time constants (τ1, left y-axis; τ2, right y-axis) and the duration of ATP depletion. Each τ was compared to the first τ after 5-second washout. *p (τ2) = 0.007 and < 0.001. n = 4 – 20. (D) Relationships between the magnitudes of reversible/irreversible rundown and the duration of ATP removal. Each measured current amplitude was normalized to the initial steady-state current amplitude before the first ATP washout in the same recording. The fraction of the currents without rundown and that recovering from reversible rundown was determined respectively by the first and second components of the double-exponential fit in (B). Compared to the initial currents, the lost activity after ATP re-application was ascribed to irreversible rundown (see Materials and Methods for details). The segments in each bar represent the fraction of irreversible, reversible, and no rundown, stacked on the top, middle, and bottom of the bar, respectively. Error bars represent standard deviation (SD). Each group is compared to the first 5-second group: *p (reversible) = 0.028, 0.027, and < 0.001. #p (irreversible) = 0.001 and 0.002. n = 3 – 20.
Figure 4. Micromolar concentration of ATP is sufficient to suppress reversible and irreversible rundowns of WT-CFTR.
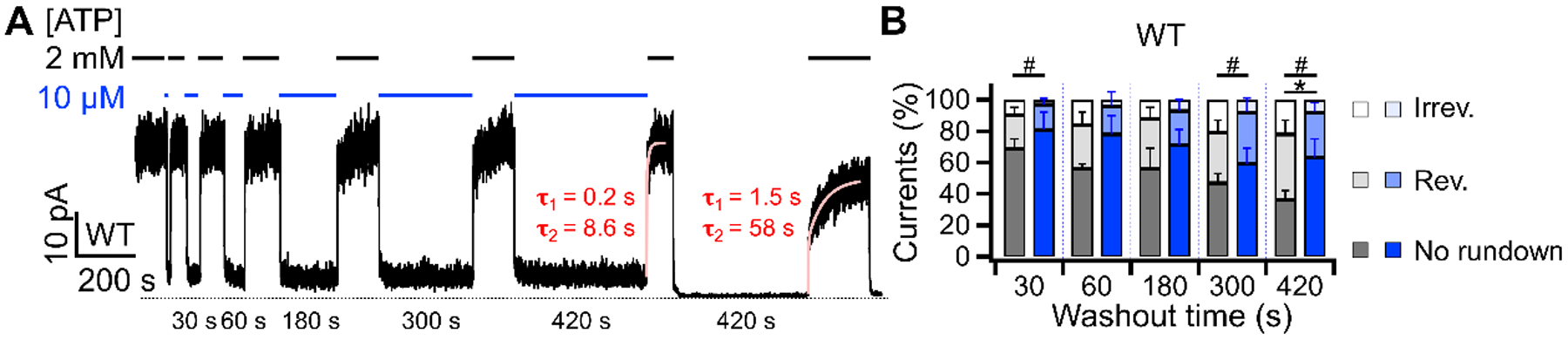
(A) A representative experiment showing effective suppression of rundown by 10 μM ATP. Macroscopic WT-CFTR currents, pre-activated with PKA and ATP, were elicited with 2 mM ATP (black) and the perfusion solution was switched to one with 10 μM ATP (blue) for different durations. Notice a significant residual activity at 10 μM ATP compared to that in the absence of ATP. The last 420 seconds of ATP deprivation resulted in a ~25% irreversible loss of currents. (B) Proportions of reversible and irreversible rundown of WT-CFTR currents in the absence of ATP (black, from Fig. 2D) or in the presence of 10 μM ATP (blue). *p (reversible) = 0.001. #p (irreversible) = 0.022, 0.010, and 0.038 respectively. n = 4 – 9.
Apart from reversible rundown, an irreversible loss of CFTR function was also seen particularly with the most prevalent disease-associated mutation F508del (Schultz et al., 1999). Since the irreversible rundown in F508del-CFTR is aggravated with increasing temperature, thermal instability caused by the mutation may be responsible (Wang et al., 2011; Aleksandrov et al., 2012; Liu et al., 2012). However, we have also observed that WT-CFTR channels undergo irreversible rundown at room temperature (Fig. 1B, middle trace). Physiological and clinical importance of CFTR rundown lies in the observation that the CFTR potentiator VX-770 (aka ivacaftor in clinics) accentuates the rundown of F508del-CFTR (Cholon et al., 2014; Veit et al., 2014; Wang et al., 2014; but compare Froux et al., 2019).
In this study, we investigated the mechanisms responsible for phosphorylation-independent, reversible and irreversible rundowns caused by depletion of ATP. We provided evidence that stable ATP binding in site 1 plays a critical role in maintaining CFTR’s rapid response to ATP, and that ATP hydrolysis at site 2 is attributable to CFTR’s vulnerability to ATP depletion. Structural and functional implications of our results will be discussed.
Materials and Methods
Mutagenesis and channel expression
CFTR mutants were constructed with QuikChange XL kit (Agilent) and sequenced by the DNA Core Facility at University of Missouri-Columbia. Chinese hamster ovary (CHO) cells were grown at 37°C in Dulbecco’s modified Eagle’s medium supplemented with 10% (vol/vol) fetal bovine serum (Sigma). Transient expression of CFTR in CHO Cells was performed with PolyFect transfection reagent (Quiagen) and pcDNA plasmids carrying CFTR constructs. A plasmid containing green fluorescence protein (GFP) cDNA (pEGFP-C3, Takara Bio) was used to co-transfect cells so that cells expressing CFTR and GFP can be identified under a fluorescent microscope. Cells were incubated at 37°C for 6 hours (macroscopic recording) or 3 hours (microscopic recording) for the transient transfection, and were transferred to 27°C for 2 – 6 days before patch-clamp recordings.
Electrophysiological recordings
Patch-clamp pipettes made from borosilicate capillary glass were pulled with a two-stage micropipette puller (PP-81; Narishige) and the pipette tips were polished with a homemade microforge. The pipette resistance was 2 – 4 MΩ in the bath solution. In all patch-clamp experiments, glass chips carrying transfected cells were transferred to a chamber filled with bath solution on the stage of an inverted microscope (IX51; Olympus). Membrane patch was excised to an inside-out configuration with a seal resistance > 40 GΩ. 11 nM PKA and 2 mM ATP were perfused to the patch until the current reached a steady state. Data were recorded with a patch-clamp amplifier (EPC9; HEKA) at room temperature, filtered through an eight-pole Bessel filter (LPF-8; Warner Instruments) at 100 Hz, and digitized to a computer at a sampling rate of 500 Hz. The membrane potential was held at −30 mV and −50 mV for macroscopic and microscopic current recordings, respectively. Solution exchange was achieved by a fast solution change system (SF-77B; Warner Instruments) with a dead time of ~30 ms (Tsai et al., 2009). For presentation purposes, inward currents are displayed as upward deflections in all figures.
Chemicals and solution compositions
For all patch-clamp experiments, pipette solution contained (mM): 140 NMDG-Cl (N-methyl-D-glucamine-chloride), 5 CaCl2, 2 MgCl2, and 10 HEPES, pH 7.4 with NMDG. Before patch excision, cells were perfused with a bath solution containing (mM): 145 NaCl, 2 MgCl2, 5 KCl, 1 CaCl2, 5 glucose, 5 HEPES, and 20 sucrose, pH 7.4 with NaOH. The bath solution was switched to a standard perfusion solution containing (mM): 150 NMDG-Cl, 2 MgCl2, 10 EGTA, 8 Tris, and 10 HEPES, pH 7.4 with NMDG, in which the patch was excised to an inside-out configuration.
Purified PKA catalytic subunit was purchased from Sigma-Aldrich (P-2645). MgATP (Sigma-Aldrich) was stored as a 500 mM stock at −20°C. The [MgATP] used in this study was 2 mM unless otherwise indicated. P-ATP [N6-(2-phenylethyl)-adenosine-5’-O-triphosphate] was custom-synthesized by Biolog Life Science Institute (Bremen, Germany) and stored as a 10 mM stock at −80°C. All chemicals were diluted to the concentrations indicated in each figure with the standard perfusion solution (pH adjusted to 7.4 with NMDG).
Data analysis and statistics
Data were analyzed using the Igor Pro program (version 8.0; WaveMetrics) with built-in single and double exponential fittings. Single-channel kinetic analysis was done with a program developed by Csanády (2000). Unpaired Student’s t tests assuming equal variance were used for comparisons between two groups. One-way ANOVA followed by Tukey’s range test were used for comparison among multiple groups. The exact p values were stated in the text. P values below 0.001 are presented as p < 0.001 to improve readability. All results were presented as “mean ± (standard deviation, SD).” Error bars in all figure represent the SD. N represents the number of excised patches, each from an individual cell. The raw dataset is available in the Supporting Information.
In Fig. 2B–D where we determined the fraction of WT-CFTR currents undergoing rundown, we ascribed the first component of the double exponential fit to channels without rundown, the second component to reversible rundown, and the reduction in total currents to irreversible rundown. Here, we assumed that the first group of channels with their rapid response to ATP has the normal WT-CFTR’s Po and that the Po of the second group of channels slowly recovered back to normal. Under these assumptions, we asserted that the reduced portion of the currents results from channels completely losing their function due to irreversible rundown. Since the time constant τ1 for the rapid current-rising phase upon re-addition of ATP remains fairly constant with the periods of ATP removal ≤ 180 seconds (multiple comparisons for the time constants τ1 among groups show no significant difference, see Supporting information for each p value), our assumptions and hence the analyses are validated. The τ1 seems slightly prolonged over longer washout time (e.g., > 300 seconds), though statistical significance was not reached; we cannot exclude the possibility that the first group of channels are no longer in the same state as they are prior to any ATP washout. Then, the irreversible loss of currents could instead be due to a lower Po for all remaining channels (the first and the second groups combined). Nonetheless, this more complicated scenario, demanding more complex mechanisms, still indicates the existence of irreversible rundown—just a different kind.
In Results and Discussion, the term closed channel indicates the interburst closure, rather than the intraburst flickery state, unless otherwise specified. The term open channel is equivalent to the channel in an opening burst.
Results
ATP depletion causes reversible and irreversible rundowns on WT-CFTR.
In excised inside-out patches, after removal of ATP for a prolonged period, WT-CFTR currents respond to ATP re-application with a biphasic time course: an instantaneous increase followed by a slower rise that could last for tens or hundreds of seconds (Fig. 2A and B). While this phenomenon has not been characterized intensively, it is a consistent observation in both our and others’ reports (see Introduction). As seen in Fig. 2A, upon a short (e.g., 5 seconds) exposure to ATP-free solution, nearly all currents are recovered rapidly (within a second or two) upon re-application of ATP. In contrast, as the ATP washout time increases to 30 seconds, a clear double-exponential current rise is seen as if a subset of channels fails to respond to ATP expediently, and it takes tens of seconds for those channels to recover. Since the addition of PKA bears little effect once the ATP-induced currents reach the plateau, dephosphorylation of CFTR by membrane-associated phosphatases is unlikely the cause. Of note, the overall ATP-dependent current amplitude remains little changed after a 30-second ATP removal. Thus, following 30 seconds of ATP deprivation, those channels that respond to ATP sluggishly could eventually recover completely. We observed a similar biphasic current response to ATP in a CFTR construct whose R domain is deleted (i.e., ΔR-CFTR, residues 634–836 were removed, Fig. 1B). Since the ATP-dependent channel activity of ΔR-CFTR does not require prior phosphorylation (Bompadre et al., 2005), the slow recovery of the ΔR-CFTR current is not a result of phosphorylation, further supporting the idea that this “rundown” of WT-CFTR is not caused by dephosphorylation. We hereby define the fraction of the slowly-recovered currents phosphorylation-independent reversible rundown, which will be referred to as reversible rundown unless otherwise specified.
In our experimental setting, the activity of WT-CFTR can remain stable without any forms of rundown in the continuous presence of ATP for at least thirty minutes (Fig. 1B, upper trace), whereas discernable, reversible rundown happened once the ATP was withdrawn for tens of seconds in the same patch. Experimental protocol shown in Fig. 2A allows us to quantitatively examine the relationship between rundown and ATP washout time. The time course of current recovery upon ATP re-application was fitted with a double exponential function, yielding two time constants (τ1 and τ2 in Fig. 2B and C) representing at least two populations of channels: one with and the other without reversible rundown respectively. As the time of ATP depletion is prolonged, the time constant τ2 representing the speed of recovery from rundown increases. In addition, a larger fraction of the currents attributed to reversible rundown is seen with longer durations of ATP withdrawal (Fig. 2D).
Although the WT-CFTR currents can almost fully recover to the original level after 30 seconds of ATP deprivation, prolonged ATP depletion lasting longer than 30 seconds resulted in a permanent loss of a fraction of the currents that neither ATP nor PKA plus ATP can restore (Fig. 2A and 2D). This phosphorylation-independent irreversible rundown is also positively correlated with the durations of ATP depletion: The longer the channels stayed closed in the absence of ATP, the more of them lose their responsiveness to ATP. For example, after 420 seconds of ATP depletion, 22 ± 9 % of the original currents became oblivious to ATP re-application (Fig. 2D). Of note, irreversible rundown occurred only when the channels endured a long stretch of continuous absence of ATP. Fig. 1B shows that brief (10 seconds), repeated withdrawals of ATP for 30 times did not cause irreversible rundown. However, in the same patch, a one-time removal of ATP lasting for 300 seconds caused significant irreversible rundown. Thus, in the absence of ATP, WT-CFTR channels undergo two types of rundown: reversible and irreversible (see more details in Materials and Methods). The transition to the rundown states appears to be state-dependent, as keeping the channels functionally active with millimolar ATP prevents rundown. In addition, since rundown occurs upon a continuous absence of ATP for tens of seconds, the rate of conformational changes to the rundown states is slow—relative to the CFTR gating rate of 1 – 2/second.
To further investigate the process of the slow recovery from reversible rundown, we recorded the change in microscopic activity of a few WT-CFTR channels in response to ATP removal and re-application. Fig. 3 shows a real-time recording of a membrane patch containing at least three WT-CFTR channels fully activated by PKA plus ATP. The open probabilities (Po) before and after 10-second ATP washout are similar and remain stable (Po = 0.48, 0.32, and 0.47 for sweep I, II, and III, respectively). However, after a 180-second ATP removal, the re-application of ATP induced an activity markedly lower than that before ATP depletion. Note there is only one channel opening step immediately upon ATP re-addition (i.e., sweep IV). It takes tens of seconds to see the second and the third opening steps before they recovered to the initial activity (Po = 0.43, sweep V). This finding is consistent with the idea that prolonged ATP depletion causes WT-CFTR channels to enter a rundown state that responds to ATP more slowly. It also supports the idea that the transition to the rundown state is a slow process as a short period of ATP depletion induces minimal rundown.
Figure 3. Reversible rundown of WT-CFTR at the microscopic level.
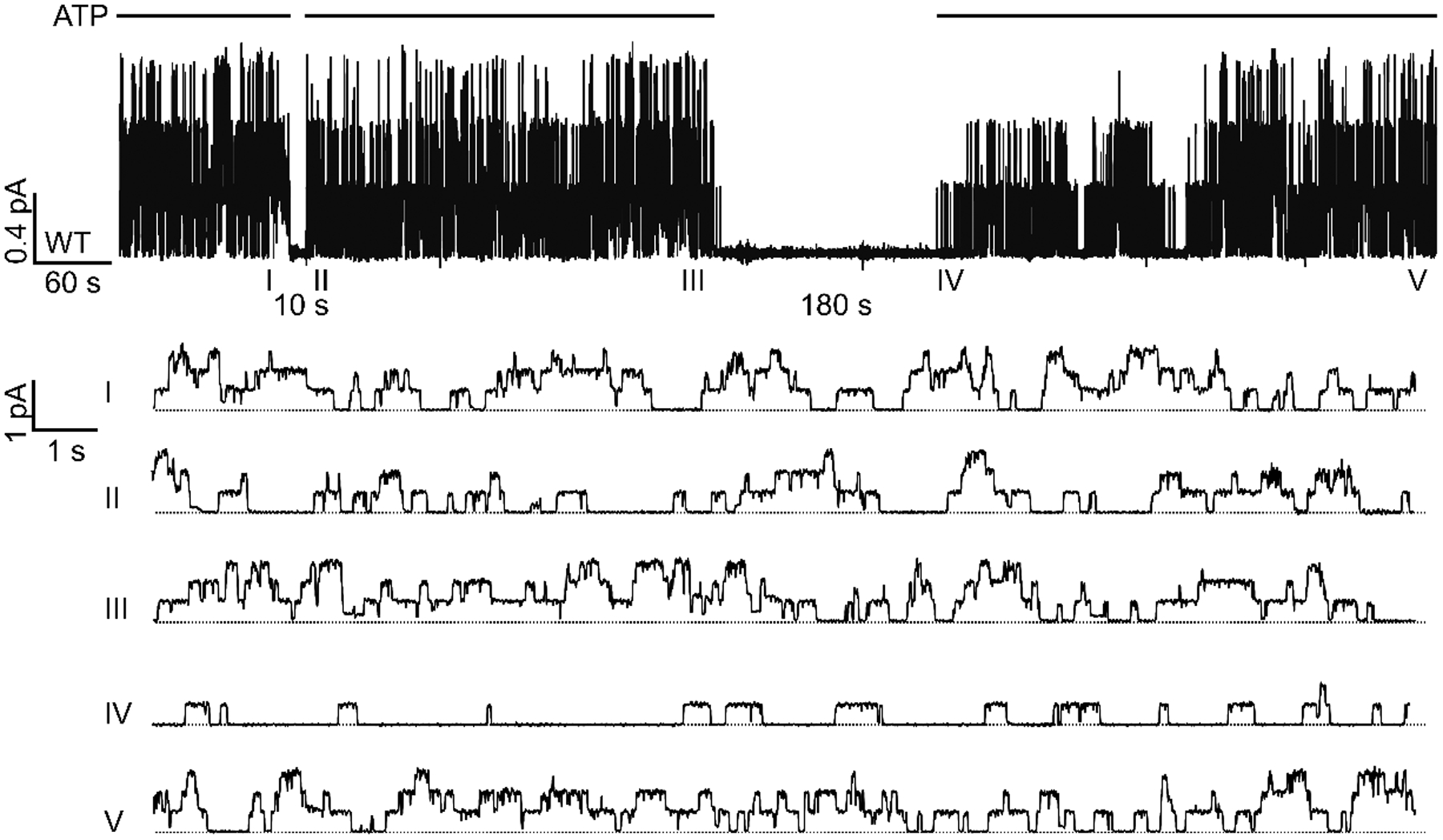
The patch containing three phosphorylated WT-CFTR channels was deprived of ATP for 10 seconds or 180 seconds. Notice that after 10 seconds of ATP washout, the activity was restored almost immediately upon re-application of ATP (sweep II). In the following 5 minutes (from II to III), all three channel opening steps could be readily discerned. In contrast, after a 180-second long washout, only one opening step was seen upon addition of ATP (sweep IV), followed by an incremental recovery of activity to eventual all three opening steps (sweep V). The five expanded sweeps were extracted from the top trace at the marked time points. Each sweep is 20-second long. Dashed lines mark the zero-current level where all channels are closed. Similar observations were made in 3 patches.
Roles of Site 1 ATP in CFTR rundown.
It is generally agreed that the duration of WT-CFTR’s gating cycles, spanning several hundred milliseconds to a second or so, primarily reflects the ATP turnover rate at site 2 (Hwang et al., 2018; Csanády et al., 2019). On the other hand, ATP stays bound in CFTR’s catalysis-incompetent site 1 upon ATP washout for tens of seconds presumably because of the formation of a stable partial NBD dimer before complete dissociation of the two NBDs (Tsai et al., 2010b; Szollosi et al., 2011; but compare Chaves & Gadsby, 2015). As both types of rundown are exacerbated by ATP depletion lasting long enough for ATP to dissociate from site 1, we reasoned that channels enter the rundown state after two NBDs separate completely to afford ATP dissociation from site 1. This hypothesis leads to the following three predictions. First, maintaining ATP occupancy at site 1 is sufficient to prevent rundown. Second, increasing/reducing the ATP binding affinity to site 1 would ameliorate/aggravate rundown. Third, for mutants with decreased ATP binding affinity at site 1, reversible rundown would occur even in the presence of ATP.
We tested the first prediction by supplying a lower concentration of ATP that in theory will keep site 1 occupied but assume minimum occupancy at site 2. Fig. 4A showed that WT-CFTR currents were subject to the same protocol as in Fig. 2A, with a twist that instead of removing all ATP, we kept 10 μM ATP in the perfusion solution. Once 2 mM ATP recovered the currents after a 420-second application of 10 μM ATP, a complete withdrawal of all ATP for 420 seconds was performed for comparison. Remarkably, both types of rundown were largely suppressed by 10 μM ATP (Fig. 4B), even though a > 90% reduction in Po with 10 μM ATP suggests the cumulative closed time must exceed 300 seconds during that 420-second interval. This result is consistent with the idea that CFTR visits the rundown state after a complete separation of the NBDs and subsequent dissociation of ATP from site 1. This experiment also suggests that the conclusion that keeping channels active (open) with ATP prevents rundown (Fig. 1 and 2) should be further refined: keeping channels in the presence of even micromolar ATP, regardless of its conducting states, prevents rundown.
To test the second prediction that reducing ATP binding affinity to site 1 would aggravate rundown, we examined the severity of rundown on CFTR mutants K464A and W401G whose binding of ATP in site 1 is compromised (Vergani et al., 2003; Zhou et al., 2006). In Fig. 5A, macroscopic K464A-CFTR lost 55 ± 21 % of currents after 300 seconds of ATP washout, which is significantly higher than the irreversible rundown of WT-CFTR (21 ± 8 %, p = 0.025, Fig. 2D). In addition, the second time constant τ2 representing the rate of recovery from reversible rundown was prolonged in K464A-CFTR (p < 0.001 vs WT-CFTR after 300-second washout, Fig. 5B). The exacerbation of rundown by mutations at site 1 is unlikely due to nonspecific effects because the worsened rundown on K464A-CFTR can be rescued by the high-affinity ATP analog P-ATP. For example, K464A-CFTR gated by P-ATP recovered faster from reversible rundown (i.e. shorter τ2) than those gated by ATP (Fig. 5B). K464A-CFTR opened by P-ATP also experienced less irreversible rundown after 300-second long washout (22 ± 9 % with P-ATP vs 45 ± 21 % with ATP, p = 0.034, Fig. 5C). Similarly, rundown of W401G-CFTR is more severe than WT-CFTR, reflected by a prolonged τ2 (Fig. 5D and E) and larger magnitude of irreversible rundown following 300 seconds of ATP removal (54 ± 14 %, p < 0.001). Consistent with K464A-CFTR, rundown of W401G-CFTR can be alleviated by P-ATP (Fig. 5D–F). These results again support the idea that the stability of ATP binding at CFTR’s site 1 determines the degree of rundown.
Figure 5. Roles of site 1 ATP binding in reversible and irreversible rundown.
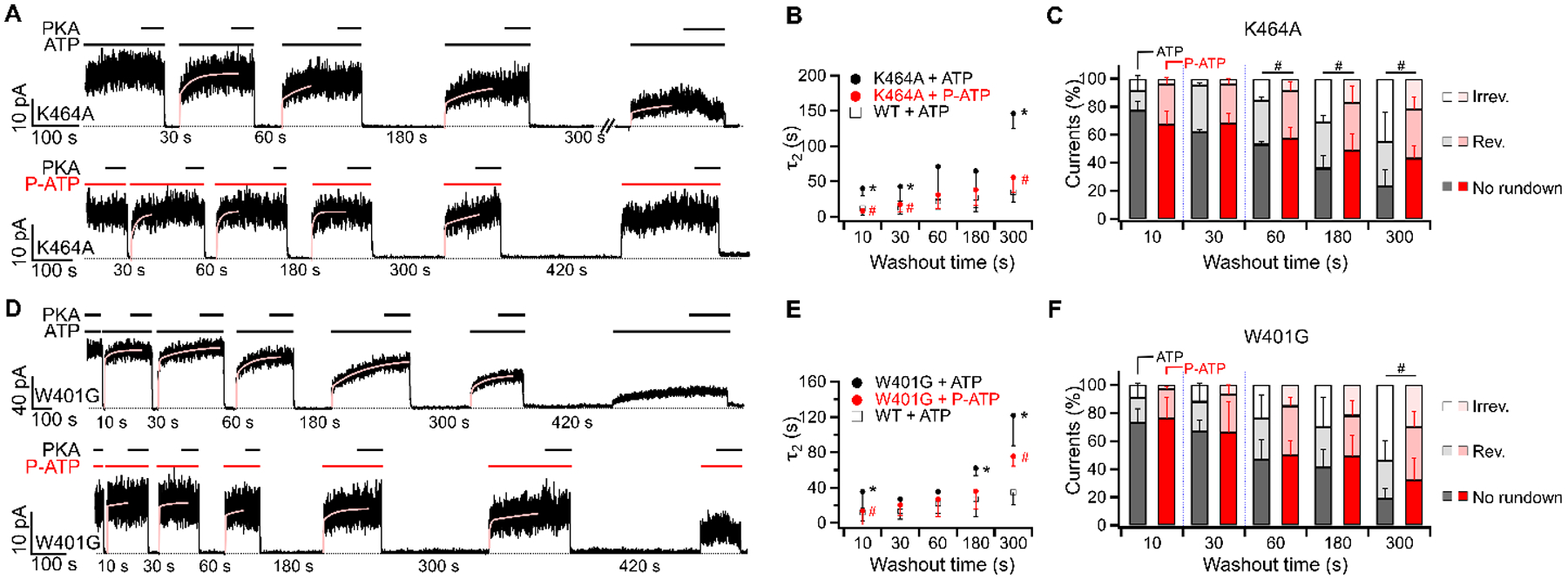
(A and D) Rundown of macroscopic K464A-CFTR (A) or W401G-CFTR (D) currents in experiments using 2 mM ATP (upper) or 50 μM P-ATP (lower). PKA was added to minimize the effect of dephosphorylation. Of note, P-ATP is a poor substrate for PKA-dependent phosphorylation on CFTR (Mihályi et al., 2020). The recovery of currents upon application of ATP or P-ATP was fitted with a double-exponential function as in Fig. 2A and the slow time constant (τ2) was summarized in (B and E). (B and E) The relationship between τ2 measuring the recovery from reversible rundown and the duration of ATP (or P-ATP) removal in K464A-CFTR (B) or W401G-CFTR (E). The currents after 420 seconds of ATP washout were too small for accurate quantification and therefore were not presented. Data of WT-CFTR was extracted from Fig. 2C. *p (K464A vs WT) = < 0.001, 0.008 and < 0.001 respectively. #p (K464A in ATP vs P-ATP) = < 0.001, 0.042, and < 0.001. *p (W401G vs WT) = 0.009, 0.006 and < 0.001. #p (W401G in ATP vs P-ATP) = 0.037 and 0.023. (C and F) Reversible and irreversible rundown after removal of ATP (black) or P-ATP (red) for different durations in K464A-CFTR (C) or W401G-CFTR (F). #p (K464A, irreversible) = 0.047, 0.034 and 0.034 respectively. #p (W401G, irreversible) = 0.035. n = 3 – 7 for K464A. n = 2 – 8 for W401G.
The third prediction is that even in the presence of ATP, mutant channels with lower ATP binding affinity to site 1 (i.e., K464A and W401G) would undergo rundown, which is manifested as stretches of low channel activity amongst the otherwise normal activity in a recording, because each time the mutant channel closes, ATP in site 1 has a higher probability to dissociate, rendering the channel more vulnerable to rundown. Fig. 6A shows a continuous recording of a K464A-CFTR channel in the presence of 2 mM ATP. As predicted, the whole trace appears as high-activity bursts interrupted by periods of low activity. Specifically, the first 30 seconds of recording (sweep I) shows a Po of 0.17 with apparent long-lasting closed events. But, during the next 60 seconds (sweep II and III), the Po increased to 0.35. Then, long closed events were seen at the beginning and the end of sweep IV (Po = 0.25), followed by a robust activity in the final 30 seconds (Po = 0.41, sweep V). W401G-CFTR behaves similarly (Fig. 6B), although stretches of high activity interrupted by low Po behavior are not as apparent as K464A-CFTR. Nonetheless, one can still see a long stretch of low-Po activity (Po = 0.21) in sweep IV followed by 30 seconds of robust activity in sweep V (Po = 0.47). Kinetic analysis on each sweep of K464A- and W401G-CFTR shows that the decrease in Po is primarily due to longer closed times (Table 1).
Figure 6. Continuous microscopic recordings of K464A-CFTR, W401G-CFTR, and WT-CFTR.
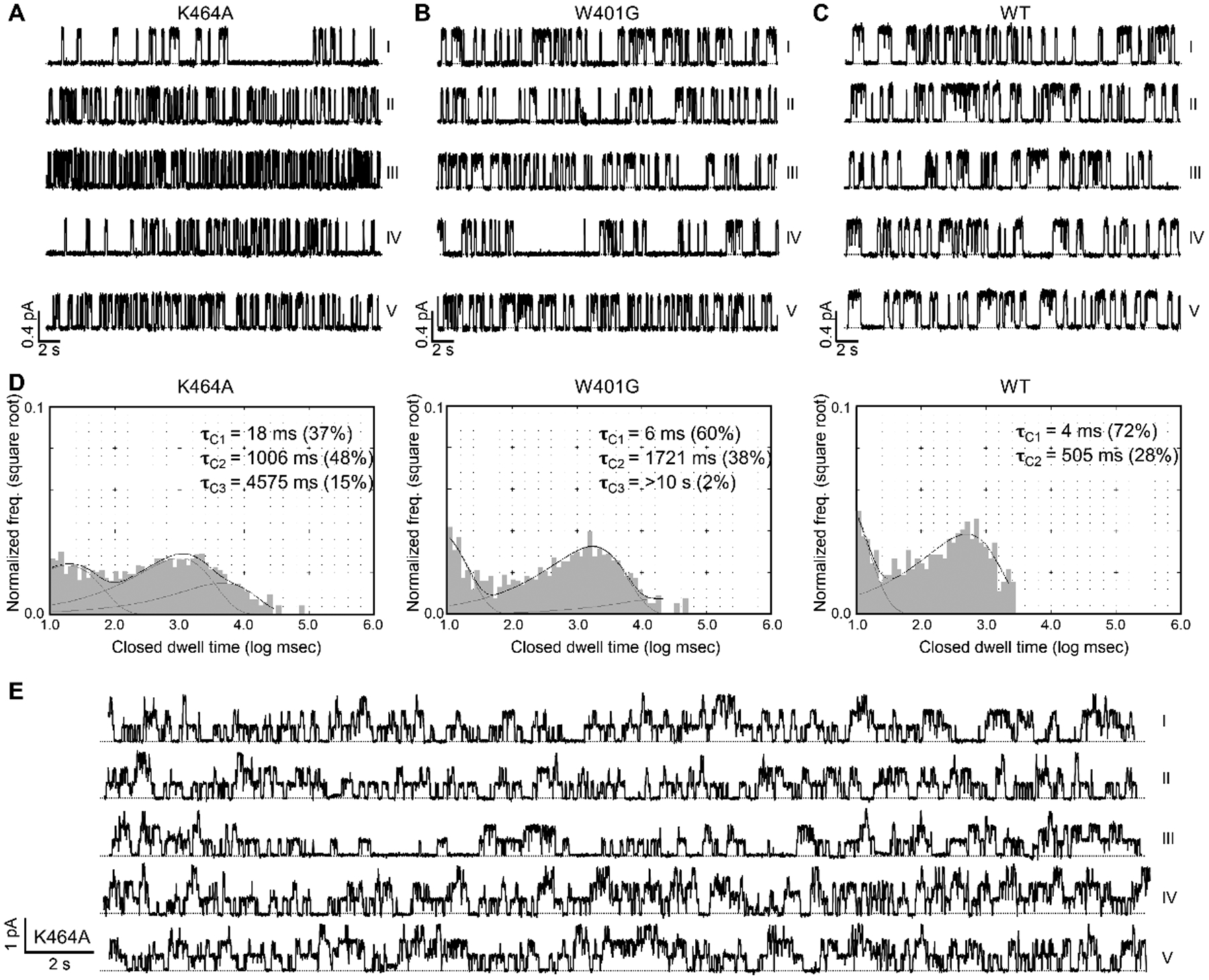
(A) A continuous single-channel recording of K464A-CFTR at 2 mM ATP. The Po of K464A-CFTR is 0.24 ± 0.07 (n = 6). Notice the long closure in sweep I and the decreased activity at the beginning of sweep IV. Each sweep is 30-second long. The dashed line indicates the baseline. Kinetic parameters are summarized in Table 1. (B) A continuous recording of a W401G-CFTR channel in the presence of 2 mM ATP. The Po of W401G-CFTR is 0.3 ± 0.06 (n = 3). Long interburst closures were observed in sweep IV. Kinetic parameters are summarized in Table 1. (C) A single-channel recording of WT-CFTR in the continuous presence of 2 mM ATP. (D) Single-channel dwell-time analysis of ATP-dependent gating of K464A-, W401G-, and WT-CFTR. To collect sufficient events for dwell-time analysis, data from different patches were pooled: four patches containing K464A-CFTR with a total of 2109 seconds; two patches containing W401G-CFTR with a total of 1664 seconds; data for WT-CFTR were adopted from Yu et al. (2016). Closed time constants (τC) based on curve fitting were marked. Note a major difference between WT and site 1 mutants is the lack of closed events longer than 3 seconds (log 3000 ≈ 3.5 on the x-axis). (E) A continuous recording of three K464A-CFTR channels in the presence of 2 mM ATP. Notice the decreased activity in sweep III (Po = 0.22; also visibly long closures of all three channels) flanked by relatively higher activities in the other sweeps. Po (I, II, IV, and V) = 0.33, 0.28, 0.38, and 0.37, respectively.
Table 1.
| P o | Open time (ms) | Closed time (ms) | |
|---|---|---|---|
| K464A | |||
| I | 0.17 | 258 | 1157 |
| II | 0.35 | 143 | 230 |
| III | 0.38 | 145 | 221 |
| IV | 0.25 | 180 | 398 |
| V | 0.41 | 166 | 219 |
| W401G | |||
| I | 0.41 | 279 | 389 |
| II | 0.27 | 194 | 501 |
| III | 0.38 | 237 | 374 |
| IV | 0.21 | 304 | 918 |
| V | 0.47 | 240 | 262 |
| WT | |||
| I | 0.40 | 319 | 466 |
| II | 0.45 | 381 | 449 |
| III | 0.35 | 370 | 624 |
| IV | 0.41 | 370 | 517 |
| V | 0.47 | 478 | 520 |
In contrast, the activity of WT-CFTR remains stable throughout the experimental timeframe (Fig. 6C). Single-channel closed-time analyses demonstrated a clear difference between the mutants and WT channels (Fig. 6D). For WT-CFTR, a very brief closure (τC1) and a closed time constant of several hundred milliseconds (τC2) represent the intra- and inter-burst closures, respectively. For K464A- and W401G-CFTR, however, the histograms cover a much wider range of closed times with long right tails. Exponential curve fittings yield a third time constant lasting for several seconds long. These results indicate that both mutant channels are occasionally trapped in a much more stable closed state that WT-CFTR almost never visited at 2 mM ATP. The temporary decrease in Po amongst the otherwise normal activity can also be seen with a recording containing three active K464A channels (Fig. 6E), where a 30-second stretch of recording with lower Po (sweep III) is sandwiched by relatively robust activities in the continuous presence of ATP.
Data presented so far suggest that closed CFTR channels with both ATP binding sites vacated are prone to rundown; whereas a tighter binding of ATP analogs at site 1 renders the channels more resistant to rundown.
Abolishing ATP hydrolysis in site 2 deters rundown
For WT-CFTR, a complete separation of NBDs upon deprivation of ATP begins with ATP hydrolysis and the release of hydrolytic products from site 2, followed by a slower dissociation of ATP from site 1 (Csanády et al., 2019). In contrast, in CFTR mutants whose ATP hydrolysis is abolished, hydrolysis-independent channel closure takes place presumably through a separation of the NBD dimer by the thermal energy RT (0.593 kcal/mol, at 25°C), which is an order of magnitude smaller than the free energy released from ATP hydrolysis. We thus wondered whether the rundown process described above is affected by ATP hydrolysis. The hydrolysis-deficient mutant D1370N-CFTR is an ideal subject to address this issue as, unlike most hydrolysis-deficient mutations such as E1371S or K1250A, this mutant has a Po similar to WT-CFTR (Csanády et al., 2010). Fig. 7A shows that macroscopic D1370N-CFTR currents were resistant to irreversible rundown: the magnitude of irreversible rundown in D1370N-CFTR was markedly reduced (5 ± 5 % vs 22 ± 9 % for WT-CFTR after a 420-second washout, p = 0.026). However, the fraction of reversible rundown remained similar to that of WT-CFTR (Fig. 7B). The time course for the recovery from reversible rundown (τ2) was surprisingly longer for D1370N- than WT-CFTR after 300- and 420-second washout (p < 0.001 and = 0.047 respectively.) The effect of the D1370N mutation on suppressing irreversible rundown was even more evident in the double mutant W401G/D1370N-CFTR (Fig. 7C). With the additional D1370N mutation, the double mutant channels preserved 83 ± 9 % of activity after a 300-second washout of ATP (including currents with reversible and no rundown), while for the single mutation W401G-CFTR, only 46 ± 14 % of the channels survived the same treatment (Fig. 7D, p < 0.001). Thus, ATP hydrolysis plays a role in the conformational changes associated with CFTR rundown (see Discussion for details).
Figure 7. Prevention of rundown by the hydrolysis-deficient mutation D1370N.
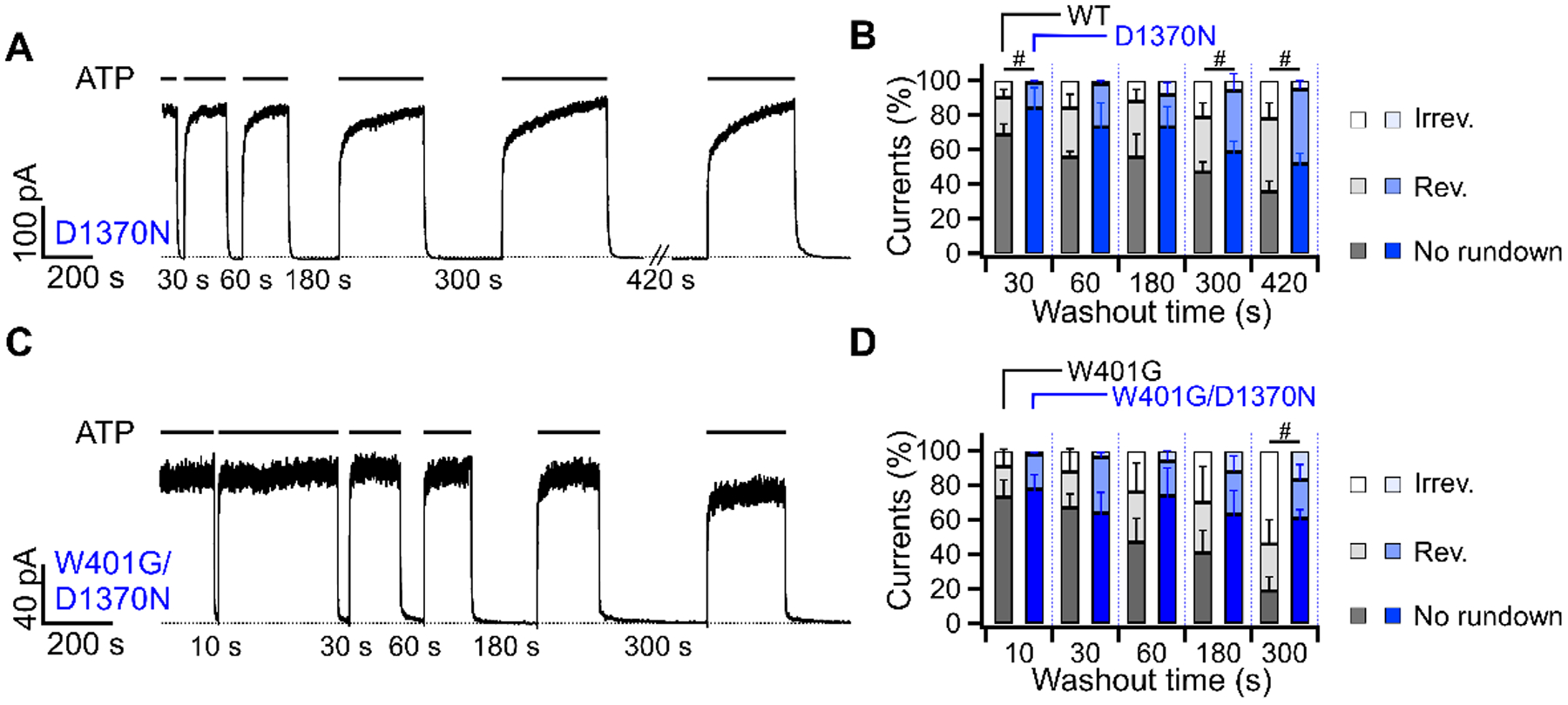
(A) Irreversible rundown is averted by mutation that abolishes ATP hydrolysis in site 2. Macroscopic D1370N-CFTR currents was subjected to the same washout protocol shown in previous figures. Notice a complete recovery of currents after 420 seconds of ATP removal. The second time constants (τ2) for the current recovery from reversible rundown are 32 ± 24 s, 40 ± 25 s, 63 ± 47 s, 221 ± 117 s, 241 ± 155 s for the 30, 60, 180, 300, and 420-second washout respectively. n = 4. (B) Comparison of reversible and irreversible rundown between WT-CFTR (black, from Fig. 2D) and D1370N-CFTR (blue). #p (irreversible) = 0.004, 0.038 and 0.026 respectively. n = 3 – 4. (C) Rundown of W401G-CFTR currents mitigated by an additional D1370N mutation. (D) Rundown in the single mutant W401G-CFTR (black, from Fig. 5F) and the double mutant W401G/D1370N-CFTR (blue). Currents for W401G-CFTR after 420 seconds of ATP removal were too small for reliable analysis. #p (irreversible) = 0.001. n = 5 – 6.
Rundown of F508del-CFTR can be partially restored by P-ATP.
The disease-associated mutation F508del is known to accelerate channel rundown (Schultz et al., 1999; Wang et al., 2011; Liu et al., 2012). The activity of a fully activated F508del-CFTR at 23 °C can sustain over at least 9 minutes, but the Po declines nearly to zero within 7 minutes at 37 °C, suggesting that rundown of F508del-CFTR occurs even in the continuous presence of ATP at 37 °C (Wang et al., 2014). We asked if ATP depletion can facilitate rundowns of F508del-CFTR at the room temperature. Indeed, less than 22 ± 15 % of the F508del-CFTR activity remained after just 180 seconds of ATP depletion (Fig. 8A vs 88 ± 7 % of WT-CFTR currents in Fig. 2D, p < 0.001). The irreversible rundown of F508del-CFTR was not due to dephosphorylation as adding PKA at the end of each ATP application did not restore the currents (Fig. 8A, top trace). Like K464A- and W401G-CFTR, F508del-CFTR recovered from reversible rundown faster with P-ATP as a ligand, and the fraction of irreversible rundown was significantly reduced with P-ATP (Fig. 8B and 8C). Unexpectedly, abolishing ATP hydrolysis by the D1370N mutation does not prevent irreversible rundown of F508del-CFTR as effectively as it does on WT- or W401G-CFTR (Fig. 8).
Figure 8. Rundown of F508del-CFTR is partially rectified by P-ATP.
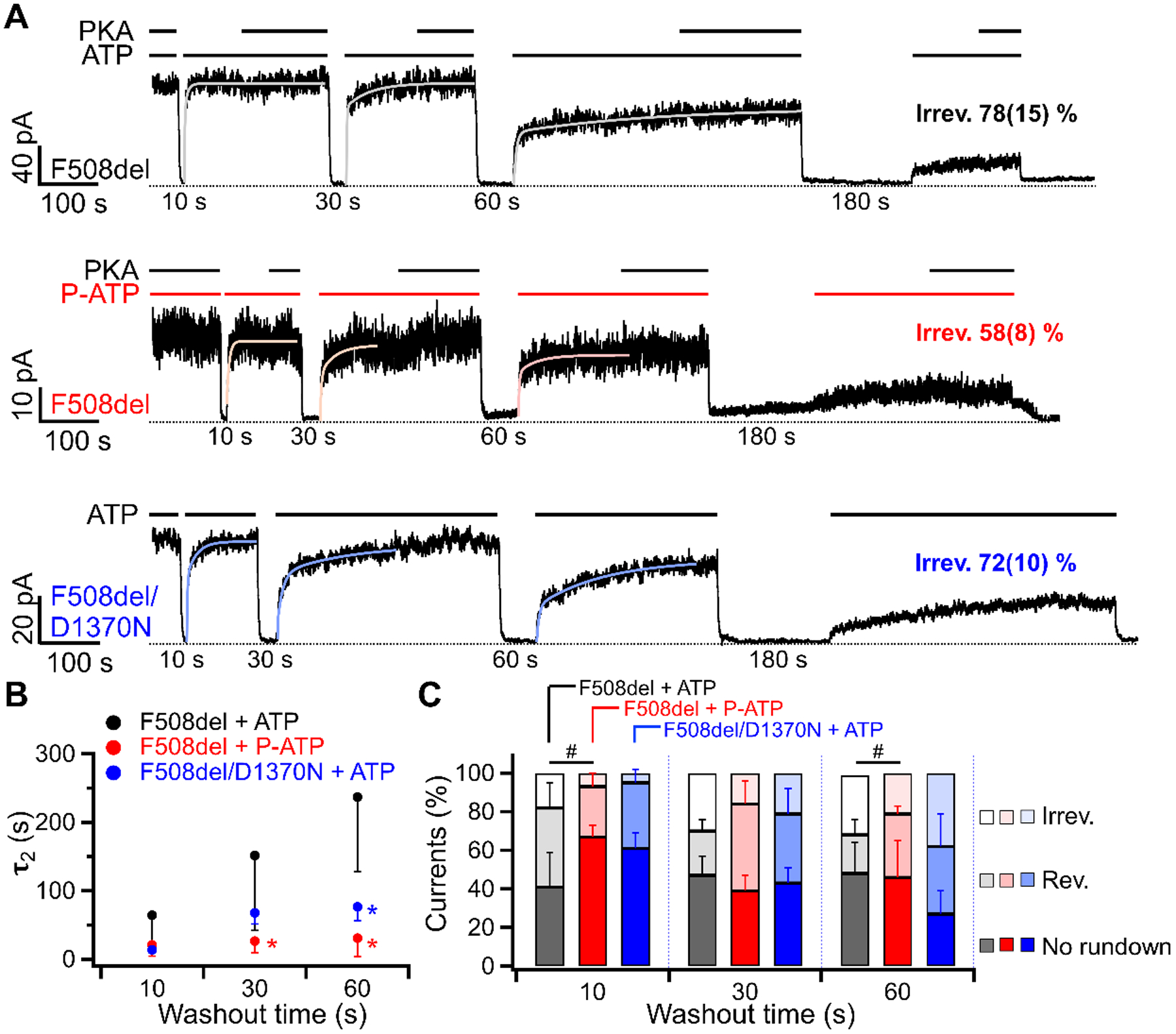
(A) Macroscopic recordings of F508del-CFTR or F508del/D1370N currents in response to the washout of 2 mM ATP (top and bottom) or 50 μM P-ATP (middle) for different durations. F508del-CFTR lost 78 ± 15 % (n = 4) of the initial currents after 180 seconds of ATP washout, similar to F508del/D1370N-CFTR, but 58 ± 8 % (n = 6) reduction was seen when the ligand was P-ATP (ATP vs P-ATP: p = 0.024). Of note, PKA can not use P-ATP for phosphorylation of CFTR. (Mihályi et al., 2020) (B) The relationship between τ2 measuring the recovery from reversible rundown and the duration of ATP (or P-ATP) removal. The τ2 after 180 seconds of washout was not included as the residual currents were too small for a reliable double-exponential fit. Each data point is compared to the F508del + ATP group at the same washout time. *p = 0.032 (P-ATP, 30 s), 0.007 (P-ATP, 60 s), and 0.003 (F508del/D1370N, 60 s) respectively. (C) A bar graph summarizing the fraction of reversible and irreversible rundown for F508del-CFTR opened by ATP (black), P-ATP (red), or F508del/D1370N-CFTR opened by ATP (blue). The proportion of irreversible rundown was reduced by P-ATP, but the percentage of reversible rundown remained similar. Each bar is compared to F508del-CFTR opened by ATP (black). #p (irreversible) = 0.042 (P-ATP, 10 s) and 0.041 (P-ATP, 60 s). n = 4 – 5 for F508del. n = 4 – 6 for F508del/D1370N.
Discussion
Kinetic relationships between the reversible and irreversible rundown states
We have previously proposed a gating model for CFTR where WT-CFTR can assume a partial dimer configuration with site 1 occupied by ATP but site 2 vacated (Jih & Hwang, 2012). The NBDs would either dimerize upon the replenishment of ATP in site 2 or separate completely to allow the ATP in site 1 to dissociate. Since ATP in site 1 can stay bound for tens of seconds to minutes (Tsai et al., 2010b), the two NBDs of WT-CFTR are unlikely to dissociate completely in the presence of ATP. We found that depletion of ATP for extended time, a condition allowed for complete separation of the two NBDs, results in a significant proportion of channels losing their ability to respond to ATP at a fast speed. As the time constant of the fast phase of current rise upon re-addition of ATP (τ1 in Fig. 2C) is relatively insensitive to the duration of the ATP removal, we can safely assume that the currents are generated by functional channels not yet undergoing rundown. The slow responders then should represent a population of rundown channels, which slowly recover in the presence of ATP (i.e., the slow rising phase upon ATP re-application). These observations lead us to propose a transition from the closed state (C0), where ATP remains bound in site 1, to the reversible rundown state (CR) with site 1 vacated and a complete separation of the NBDs.
That WT-CFTR channels do not run down in the continuous presence of ATP suggests that the transition from C0 to CR is a slow process and that with a closed time of several hundred milliseconds in a normal gating cycle, the closed WT-CFTR barely visits the CR state before it is opened by ATP again. As the closed time is drastically prolonged by ATP depletion, channels start entering the CR state, where they can still open in response to ATP, albeit with a much slower rate. The slow, stepwise restoration of the microscopic activity by ATP in Fig. 3 demonstrates the recovery from CR. Interestingly, 10 μM ATP effectively prevents rundown, suggesting that when the high-affinity ATP binding site (site 1) is occupied, WT-CFTR channels can stay in the C0 state without proceeding to the CR state. Consistent with this picture, we found that mutations (i.e., W401G and K464A) weakening the affinity of site 1 for ATP exacerbate rundown, and slow down the recovery from reversible rundown as manifested by prolonging the slow time constant (τ2). The importance of ATP binding in site 1 to prevent rundown is further supported by the observation that the high-affinity ATP analog P-ATP can partly rectify the heightened rundown of W401G and K464A and expedite the recovery from rundown in these mutants (Fig. 5).
However, the model “open state ⇌ C0 ⇌ CR” described above does not explain the permanent loss of channel activity in extended ATP washout. The channels that irreversibly (within the time frame of our experiments) lose their responsiveness to ATP are in a condition distinct from the C0 and CR states. Similar to the CR, this irreversible rundown state (CIR) is only seen in continuous ATP depletion, as channels entering the C0 state only briefly due to intermittent ATP removals do not end up in the CIR state (Fig. 1B). Here, CIR could emerge from a pathway independently of CR (i.e., C0 ⇀ CIR, Fig. 9A), or it follows the CR state, forming a sequential transition (C0 ⇌ CR ⇀ CIR, Fig. 9B). In both cases, the irreversible process takes place when ATP is depleted for a long time, allowing the channels to leave C0. It is challenging to differentiate between these two models (or an even more complex scenario, e.g., CR can sojourn to CIR, dashed arrow in Fig. 9A). Channels visiting either the CR state or CIR state are only revealed upon re-application of ATP, leaving any transition steps between CR and or CIR “invisible”.
Figure 9. Simplified schemes depicting the relationships between reversible and irreversible rundown states.
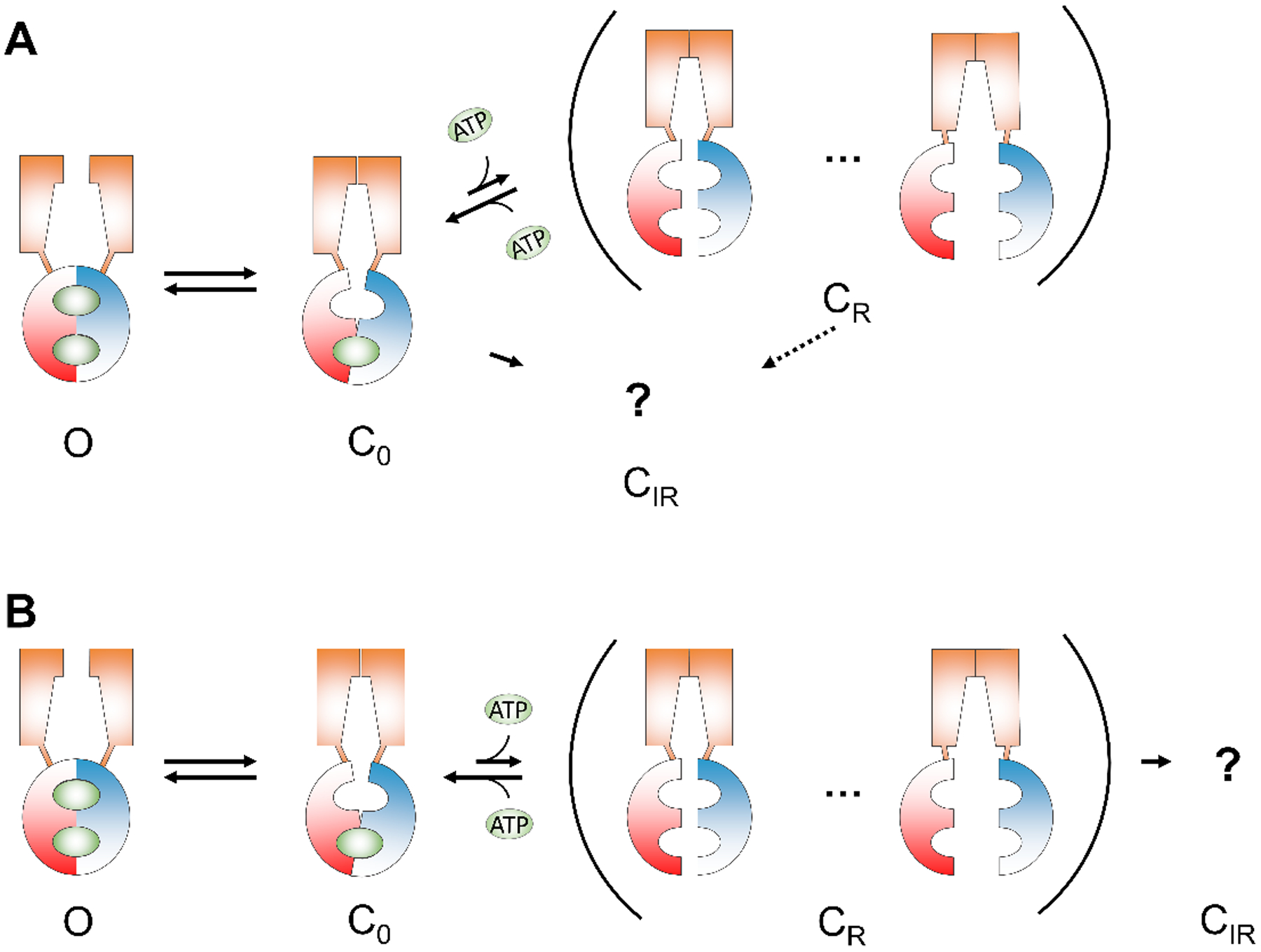
(A) Reversible and irreversible rundown states emerge independently from the closed state. The dashed arrow indicates possible transitions from CR to CIR. (B) A sequential transition from reversible to irreversible rundown states. The ellipsis in the parenthesis indicates multiple reversible rundown states with different degree of NBD separation. The question mark represents the unknown structure of irreversible rundown state. Note that both schemes are overly simplified as multiple open states with different configurations of the NBDs (Jih & Hwang, 2012) are not included. Possible direct transitions between CR and other open state(s) are also omitted due to lack of supporting structural evidence. O: open state; C0: closed state with ATP in site 1; CR: reversible rundown state; CIR: irreversible rundown state. Orange trapezoid: TMD; Red and blue hemispheres: NBDs; Green oval: ATP.
Although we cannot exclude the possibility of direct conformational changes from C0 to CIR, we favor the sequential model as whenever the process involving CR is altered, the population of channels visiting CIR is also affected: First, when the proportion of reversible rundown in WT-CFTR becomes larger with longer ATP washout; same is the severity of irreversible rundown (Fig. 2D). Second, mutations that slow down the recovery from CR (τ2) also aggravate irreversible rundown (Fig. 5). Third, interventions that ameliorate reversible rundown can lessen irreversible rundown as well. For instance, P-ATP shortens the recovery time constant τ2 and reduces irreversible rundown for W401G, K464A (Fig. 5), and F508del-CFTR (Fig. 8).
Here we also caution our readers that the proposed model is overly simplified. The increasingly longer τ2 (i.e., prolonged recovery from rundown) with an increased duration of ATP depletion (Fig. 2C) implies that there should be multiple CR states where the channels require longer time to recover from the more deeply entrenched rundown state(s). We thus conclude that a model containing multiple states (C0 ⇌ CR …⇀ CIR; Fig. 9) is likely to be more realistic than the simplified three states scenario (C0 ⇌ CR ⇀ CIR). In addition, the proposed model only allows one route for channel opening (O ⇌ C0) where the open state has dimerized NBDs. We omit the more complex scenarios that C0 could transit to multiple open states with different configurations of NBDs (Jih & Hwang, 2012) and that channels may open directly from the CR state(s). When we examined the single-channel recordings of K464A- and W401G-CFTR (Fig. 6), we noticed that the activity following the long interburst closure, which presumably corresponds to the CR state, does not immediately recover to the high Po gating. If the channel behaved like the simplified model (O ⇌ C0 ⇌ CR ⇀ CIR), it would have shown a stretch of consistent high Po interrupted by an isolated long closed event. Instead, a stretch of low Po is observed and the recovery to high Po gating is delayed. It is therefore likely that different open states are involved, a subject demanding further investigation.
Possible structural mechanisms for CFTR rundown
As described above, the transition from C0 to CR likely happens after ATP has dissociated from site 1, leading to a complete separation of the NBDs. In theory, the distance between the two separated NBDs, by imposing a physical hurdle in the process of ATP-mediated NBD dimerization, may determine the time required to complete this reaction that triggers gate opening. It follows that channels in the CR state should react to ATP much slower than those in the C0 state. Furthermore, compared to the C0 state where the NBDs are partially connected by ATP in site 1 and therefore are more conformationally constrained, the CR state, on the contrary, may carry less-constrained NBDs with a wider range of separation. While presently no direct structural evidence suggests that CFTR’s NBDs assume multiple conformations in the absence of ATP, extreme conformational flexibility of certain ABC transporters’ NBDs has been reported. The lipid-linked oligosaccharide flippase PgIK, for instance, has been crystallized in two distinct apo states that show various degrees of NBD separation as well as different orientations at the NBD interface (Perez et al., 2015). Another lipid flippase MsbA also has two crystallographic nucleotide-free structures, known as the open-apo and closed-apo conformations, of which the NBDs exhibit a wide difference in separation despite a similar orientation (Ward et al., 2007; Zou et al., 2009). The physiological significance of these differences in the NBDs in ABC transporters remains debated (Locher, 2016), as cellular ATP concentrations are normally above the Kd for ATP binding. Nevertheless, this structural flexibility of the NBDs raises the possibility that CFTR, as a member of the ABC transporter family, could adopt conformations featuring different degrees of NBD separation when the condition permits.
Interjecting the structural mechanism described in the last paragraph into the sequential model (C0 ⇌ CR… ⇀ CIR), we propose that multiple CR states may represent a continuum of conformations with incrementally larger degrees of NBD separation and more diverse orientations. As each NBD deviates from its original orientation that is most efficient for dimerization (i.e. C0 state), recovery of rundown becomes increasingly difficult. When the NBDs re-dimerize after large degree of separation, the configuration of the dimer, or even the head and tail subdomains within one NBD, might no longer be identical to the one that never experiences rundown before. In other words, when the rundown channel opens again, the conformation of its NBDs might not resemble a “healthy” channel. Moreover, if the NBDs remain in a nucleotide-free solution for long enough time, the distance between the two NBDs could grow so apart that the reunion of the NBDs is thermodynamically too unfavorable and hence irreversible rundown (CIR) occurs.
Role of the R domain in CFTR rundown
If the ATP-free closed states are prone to rundown, one has to wonder if the unphosphorylated CFTR channels, which assume seemingly similar nucleotide-free NBDs and inward-facing TMDs as the closed conformation, also undergo rundown. We have no answer to this question at this juncture, but we speculate that the unphosphorylated CFTR might have less conformational flexibility than the phosphorylated closed states that are vulnerable to rundown for the following reason. The cryo-EM structure of the unphosphorylated, ATP-free CFTR reveals that the R domain is wedged between the two TMDs and extended into the interface between the two NBDs (Liu et al., 2017). These extensive contacts between parts of the R domain and CFTR’s two halves may serve to minimize the tendency of NBD separation, and thus prevent rundown of unphosphorylated channels.
In the phosphorylated ATP-bound structures (Zhang et al., 2018), the largely unstructured region of the R domain moves to the peripheral surface of NBD1 with a helical segment now interacting with the so-called lasso motif. It is unclear whether these newly-formed interactions serve any functional purposes. Here, we speculate that the phosphorylated R domain may impose some structural constraints to limiting large separation of the NBDs. For example, by clinging to the surface of NBD1 and connecting to the TMD2, the R domain limits the range of NBD1’s movement and thereby deterring the undesirable transition to the CIR state. We indeed noted that the irreversible rundown of ΔR-CFTR is more severe than WT-CFTR (Fig. 1B). It is perhaps also worth noting that members in the ABCE and ABCF subfamilies of mammalian ABC proteins consist of only NBDs without the accompanying TMDs, a linker region evolved to keep the two NBDs from falling apart via physical tethering (Ford & Hellmich, 2020). We therefore anticipate more extensive studies on the potential role of CFTR’s R domain in preventing rundown.
New function of the degenerate nucleotide binding site
Despite being a channel and possessing an R domain, CFTR shares a similar overall molecular architecture with other ABC transporters. Contrary to the prokaryotic ABC transporters containing two identical NBDs, many human ABC proteins, including CFTR, are heterodimeric and carry a degenerate nucleotide binding site where ATP hydrolysis either slows down (present in most ABCB and ABCC members) or is completely diminished, e.g., the bile salt export pump ABCB11 and CFTR (Sohail et al., 2017; Szöllősi et al., 2018). The degenerate site likely acquires some functional advantages since its sequence does not further diverge amidst millions of years of evolutionary pressure. It has been proposed that the degenerate site may assume a mechanical role, acting as a stable hinge to guide the conformational changes during each transport cycle (Stockner et al., 2020). With ATP staying bound in the degenerate site for multiple cycles, a complete separation of the NBDs and the consequent extensive motions of TMDs could be prevented. Thus, the degenerate site promotes the efficiency of energy utilization by deterring large structural change in both the NBDs and the TMDs. That some primordial transporters with two identical NBDs alternate the hydrolysis function of the two sites seems to suggest a division of labor: when one site hydrolyzes ATP to power the substrate transport, the other site keeps the NBD in contact throughout the catalytic cycle (Jones et al., 2009; Szöllősi et al., 2018). Our data with K464A-, W401G-CFTR and the effects of P-ATP indeed support the idea that tight ATP binding at the degenerate site plays a critical role in maintaining a consistent channel function of CFTR. Of note, in the framework of the schemes in Fig. 9, the effect of P-ATP on accelerating recovery of rundown seems to imply that dimerization of site 1 is facilitated by P-ATP. Without structural evidence, we do not have a definitive answer to the question of how P-ATP facilitates recovery from rundown. One possibility is that by staying bound to site 1 for longer time, P-ATP helps other currently unknown rate-limiting step(s) following binding of nucleotide to take place.
Even if we accept the argument that disabling one of the sites grants some functional advantage for heterodimeric ABC proteins, we have yet to answer the question why this is necessary. Our results (Fig. 7) that the hydrolysis-deficient mutant D1370N exhibits minimal irreversible rundown may provide a clue. One unique biochemical characteristic of ABC proteins is that ATP hydrolysis takes place at the NBD dimer interface. Thus, one can imagine that the tremendous free energy released upon ATP hydrolysis during each gating/transport cycle could exert a fierce force that may disrupt the structural assembly of the protein. A tight ATP binding at the degenerate site may just provide the resistive force to avoid structural destabilization accompanying ATP hydrolysis. We therefore posit that in heterodimeric transporters such as CFTR, evolution further optimizes the specialized tasks of the two NBDs by assigning one site to constantly connect two NBDs and the other to power the transport cycle via hydrolysis.
Pathophysiological and therapeutic implications on the F508del-CFTR
The most common disease-associated mutation F508del causes functional defects at multiple levels (Dalemans et al., 1991; Wang et al., 2000; Wang et al., 2011; Lukacs & Verkman, 2012; Kopeikin et al., 2014). Besides well-known trafficking and gating defects, functional F508del-CFTR channels also assume a shortened half-life when they reside in the plasma membrane (Lukacs et al., 1993). If CFTR’s site 1 is responsible for stabilizing the overall architecture of the channel and hence preventing rundown, one immediate question is how the severe rundown of F508del-CFTR is attributed to its degenerate site. The crystal structure of NBD1 with the F508del mutation shows little structural perturbation (Lewis et al., 2005), although the additional solubilizing mutations present in the structure might have altered the conformation and thus the construct might not fully represent the disease-causing F508del-CFTR (Pissarra et al., 2008). Nonetheless, recent cryo-EM structures of WT-CFTR reveal that the side-chain of F508 is positioned at the junction of the fourth intracellular loop (ICL4) and TM11, distant from the dimer interface (Zhang et al., 2018). The apparent affinity for ATP, as measured with an assay assessing the function of the channel, is not altered in F508del-CFTR, either (Wang et al., 2000). On the other hand, our previous reports suggest that strengthening the binding affinity for ATP in site 1 improves the activity of F508del-CFTR (Miki et al., 2010; Tsai et al., 2010a), and that the F508del mutation destabilizes both the full and partial NBD dimer states (Jih et al., 2011). The result that P-ATP effectively slows down F508del-CFTR rundown is consistent with the idea of impaired stability of F508del-CFTR’s NBD dimer (Fig. 8). Nevertheless, this mechanism is not likely the sole determinant as, unlike what we observed in K464A-CFTR, P-ATP-treated F508del-CFTR still showed worse rundown than WT-CFTR. Moreover, that abolishing hydrolysis is ineffective in preventing F508del-CFTR rundown indicates that other structural perturbations must contribute to rundown of F508del-CFTR. Several studies have demonstrated that at 37 degrees, the pore of F508del-CFTR becomes unstable and the channels open to subconductance states (Aleksandrov et al., 2010; Aleksandrov et al., 2012; Meng et al., 2017), suggesting an allosteric effect of the F508del mutation on the structural integrity of the pore-forming TMDs.
It has long been postulated that the poor function of F508del-CFTR results from the disruption of coupling between the TMDs and NBD1 (Mornon et al., 2008; Serohijos et al., 2008; Mendoza et al., 2012; Rabeh et al., 2012), which seems plausible in light of the location of F508 revealed in the cryo-EM structures (Liu et al., 2017; Zhang et al., 2018). Based on this defective coupling hypothesis, the coupling helices play a critical role in positioning the two NBDs in a dimerization-competent manner. In principle, a weakened TMD-NBD connection would result in sideway displacement and/or rotation of the NBDs, hampering dimerization-driven channel gating. While direct structural evidence for the defects in F508del-CFTR awaits future study, this hypothesis provides another plausible explanation for the severe rundown seen in F508del-CFTR. In addition to the unstable dimer states described in the last paragraph, the NBD1 of closed F508del-CFTR can adopt more flexible conformations as it is less constrained by the ICL4. Then, a larger displacement of the NBD1 in F508del-CFTR accelerates the transition from CR to CIR, resulting in the grave loss of channel function.
Regardless of the specific structural mechanisms for F508del-CFTR rundown, our findings suggest a therapeutic strategy to rectifying the functional instability of F508del-CFTR. In theory, developing potentiators that strengthen the interaction between the NBD dimer could not only enhance gating but also protect the channel from rundown, an advantage that current potentiators targeting the TMDs such as VX-770 apparently lack (Cholon et al., 2014; Veit et al., 2014).
In conclusion, our study reveals two types of CFTR rundown and provides insight into the structural basis of rundown. Pharmacological interventions that target the NBD interface could potentially rescue the deleterious effect of F508del on the stability of the channel.
Supplementary Material
Key Points Summary.
During evolution, many ATP-binding cassette transporters—including the CFTR chloride channel whose dysfunction causes cystic fibrosis (CF)—lose the ability to hydrolyze ATP in one of the two ATP-binding sites.
Here we show that tight ATP binding at this degenerate site in CFTR is central for maintaining stable, robust function of normal CFTR.
We also demonstrate that membrane instability of the most common CF-causing mutant F508del-CFTR can be rescued by strengthening ATP binding at CFTR’s degenerate site.
Our data thus explain an evolutionary puzzle and offer a potential therapeutic strategy for CF.
Acknowledgments
We thank Cindy Chu and Shenghui Hu for their technical assistance.
Funding source statement
This work is supported by the National Institutes of Health (grant NIHR01DK55835), the Cystic Fibrosis Foundation (grant Hwang19G0), and Ministry of Science and Technology, Taiwan (109-2320-B-010-049-MY2) to T. -C. Hwang.
Footnotes
Competing interests
None of the authors has any conflicts of interests
Data availability
The raw dataset is included in the Supporting Information document.
References
- Aleksandrov AA, Kota P, Aleksandrov LA, He L, Jensen T, Cui L, Gentzsch M, Dokholyan NV & Riordan JR. (2010). Regulatory insertion removal restores maturation, stability and function of DeltaF508 CFTR. Journal of molecular biology 401, 194–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aleksandrov AA, Kota P, Cui L, Jensen T, Alekseev AE, Reyes S, He L, Gentzsch M, Aleksandrov LA, Dokholyan NV & Riordan JR. (2012). Allosteric modulation balances thermodynamic stability and restores function of ΔF508 CFTR. Journal of molecular biology 419, 41–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aleksandrov L, Aleksandrov AA, Chang XB & Riordan JR. (2002). The First Nucleotide Binding Domain of Cystic Fibrosis Transmembrane Conductance Regulator Is a Site of Stable Nucleotide Interaction, whereas the Second Is a Site of Rapid Turnover. The Journal of biological chemistry 277, 15419–15425. [DOI] [PubMed] [Google Scholar]
- Basso C, Vergani P, Nairn AC & Gadsby DC. (2003). Prolonged nonhydrolytic interaction of nucleotide with CFTR’s NH2-terminal nucleotide binding domain and its role in channel gating. The Journal of general physiology 122, 333–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bear CE, Li CH, Kartner N, Bridges RJ, Jensen TJ, Ramjeesingh M & Riordan JR. (1992). Purification and functional reconstitution of the cystic fibrosis transmembrane conductance regulator (CFTR). Cell 68, 809–818. [DOI] [PubMed] [Google Scholar]
- Becq F (1996). Ionic channel rundown in excised membrane patches. Biochimica et biophysica acta 1286, 53–63. [DOI] [PubMed] [Google Scholar]
- Becq F, Jensen TJ, Chang XB, Savoia A, Rommens JM, Tsui LC, Buchwald M, Riordan JR & Hanrahan JW. (1994). Phosphatase inhibitors activate normal and defective CFTR chloride channels. Proc Natl Acad Sci 91, 9160–9164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger HA, Travis SM & Welsh MJ. (1993). Regulation of the cystic fibrosis transmembrane conductance regulator Cl- channel by specific protein kinases and protein phosphatases. The Journal of biological chemistry 268, 2037–2047. [PubMed] [Google Scholar]
- Bompadre SG, Ai T, Cho JH, Wang X, Sohma Y, Li M & Hwang T-C. (2005). CFTR Gating I : Characterization of the ATP-dependent Gating of a Phosphorylation-independent CFTR Channel (ΔR-CFTR). Journal of General Physiology 125, 361–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaves LA & Gadsby DC. (2015). Cysteine accessibility probes timing and extent of NBD separation along the dimer interface in gating CFTR channels. The Journal of general physiology 145, 261–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng SH, Rich DP, Marshall J, Gregory RJ, Welsh MJ & Smith AE. (1991). Phosphorylation of the R domain by cAMP-dependent protein kinase regulates the CFTR chloride channel. Cell 66, 1027–1036. [DOI] [PubMed] [Google Scholar]
- Cholon DM, Quinney NL, Fulcher ML, Esther CR Jr., Das J, Dokholyan NV, Randell SH, Boucher RC & Gentzsch M. (2014). Potentiator ivacaftor abrogates pharmacological correction of ΔF508 CFTR in cystic fibrosis. Science translational medicine 6, 246ra296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csanády L (2000). Rapid Kinetic Analysis of Multichannel Records by a Simultaneous Fit to All Dwell-Time Histograms. Biophysical Journal 78, 785–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csanády L, Vergani P & Gadsby DC. (2010). Strict coupling between CFTR’s catalytic cycle and gating of its Cl ion pore revealed by distributions of open channel burst durations. Proc Natl Acad Sci 107, 1241–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csanády L, Vergani P & Gadsby DC. (2019). STRUCTURE, GATING, AND REGULATION OF THE CFTR ANION CHANNEL. Physiological reviews 99, 707–738. [DOI] [PubMed] [Google Scholar]
- Dalemans W, Barbry P, Champigny G, Jallat S, Dott K, Dreyer D, Crystal RG, Pavirani A, Lecocq JP & Lazdunski M. (1991). Altered chloride ion channel kinetics associated with the delta F508 cystic fibrosis mutation. Nature 354, 526–528. [DOI] [PubMed] [Google Scholar]
- Ford RC & Hellmich UA. (2020). What monomeric nucleotide binding domains can teach us about dimeric ABC proteins. FEBS letters 594, 3857–3875. [DOI] [PubMed] [Google Scholar]
- Froux L, Coraux C, Sage E & Becq F. (2019). Short-term consequences of F508del-CFTR thermal instability on CFTR-dependent transepithelial currents in human airway epithelial cells. Scientific reports 9, 13729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang TC, Yeh JT, Zhang J, Yu YC, Yeh HI & Destefano S. (2018). Structural mechanisms of CFTR function and dysfunction. The Journal of general physiology 150, 539–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jih KY & Hwang TC. (2012). Nonequilibrium gating of CFTR on an equilibrium theme. Physiology (Bethesda, Md) 27, 351–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jih KY, Li M, Hwang TC & Bompadre SG. (2011). The most common cystic fibrosis-associated mutation destabilizes the dimeric state of the nucleotide-binding domains of CFTR. The Journal of physiology 589, 2719–2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jih KY, Sohma Y, Li M & Hwang TC. (2012). Identification of a novel post-hydrolytic state in CFTR gating. The Journal of general physiology 139, 359–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones PM, O’Mara ML & George AM. (2009). ABC transporters: a riddle wrapped in a mystery inside an enigma. Trends in Biochemical Sciences 34, 520–531. [DOI] [PubMed] [Google Scholar]
- Kopeikin Z, Yuksek Z, Yang HY & Bompadre SG. (2014). Combined effects of VX-770 and VX-809 on several functional abnormalities of F508del-CFTR channels. Journal of cystic fibrosis : official journal of the European Cystic Fibrosis Society 13, 508–514. [DOI] [PubMed] [Google Scholar]
- Lewis HA, Buchanan SG, Burley SK, Conners K, Dickey M, Dorwart M, Fowler R, Gao X, Guggino WB, Hendrickson WA, Hunt JF, Kearins MC, Lorimer D, Maloney PC, Post KW, Rajashankar KR, Rutter ME, Sauder JM, Shriver S, Thibodeau PH, Thomas PJ, Zhang M, Zhao X & Emtage S. (2004). Structure of nucleotide-binding domain 1 of the cystic fibrosis transmembrane conductance regulator. The EMBO journal 23, 282–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis HA, Zhao X, Wang C, Sauder JM, Rooney I, Noland BW, Lorimer D, Kearins MC, Conners K, Condon B, Maloney PC, Guggino WB, Hunt JF & Emtage S. (2005). Impact of the deltaF508 mutation in first nucleotide-binding domain of human cystic fibrosis transmembrane conductance regulator on domain folding and structure. The Journal of biological chemistry 280, 1346–1353. [DOI] [PubMed] [Google Scholar]
- Liu F, Zhang Z, Csanády L, Gadsby DC & Chen J. (2017). Molecular Structure of the Human CFTR Ion Channel. Cell 169, 85–95.e88. [DOI] [PubMed] [Google Scholar]
- Liu X, O’Donnell N, Landstrom A, Skach WR & Dawson DC. (2012). Thermal instability of ΔF508 cystic fibrosis transmembrane conductance regulator (CFTR) channel function: protection by single suppressor mutations and inhibiting channel activity. Biochemistry 51, 5113–5124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locher KP. (2016). Mechanistic diversity in ATP-binding cassette (ABC) transporters. Nature Structural & Molecular Biology 23, 487–493. [DOI] [PubMed] [Google Scholar]
- Lukacs GL, Chang XB, Bear C, Kartner N, Mohamed A, Riordan JR & Grinstein S. (1993). The delta F508 mutation decreases the stability of cystic fibrosis transmembrane conductance regulator in the plasma membrane. Determination of functional half-lives on transfected cells. The Journal of biological chemistry 268, 21592–21598. [PubMed] [Google Scholar]
- Lukacs GL & Verkman AS. (2012). CFTR: folding, misfolding and correcting the ΔF508 conformational defect. Trends in molecular medicine 18, 81–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendoza JL, Schmidt A, Li Q, Nuvaga E, Barrett T, Bridges RJ, Feranchak AP, Brautigam CA & Thomas PJ. (2012). Requirements for efficient correction of ΔF508 CFTR revealed by analyses of evolved sequences. Cell 148, 164–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng X, Wang Y, Wang X, Wrennall JA, Rimington TL, Li H, Cai Z, Ford RC & Sheppard DN. (2017). Two Small Molecules Restore Stability to a Subpopulation of the Cystic Fibrosis Transmembrane Conductance Regulator with the Predominant Disease-causing Mutation. The Journal of biological chemistry 292, 3706–3719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihályi C, Iordanov I, Töröcsik B & Csanády L. (2020). Simple binding of protein kinase A prior to phosphorylation allows CFTR anion channels to be opened by nucleotides. Proc Natl Acad Sci 117, 21740–21746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihályi C, Töröcsik B & Csanády L. (2016). Obligate coupling of CFTR pore opening to tight nucleotide-binding domain dimerization. eLife 5, e18164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki H, Zhou Z, Li M, Hwang TC & Bompadre SG. (2010). Potentiation of disease-associated cystic fibrosis transmembrane conductance regulator mutants by hydrolyzable ATP analogs. The Journal of biological chemistry 285, 19967–19975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mornon JP, Lehn P & Callebaut I. (2008). Atomic model of human cystic fibrosis transmembrane conductance regulator: membrane-spanning domains and coupling interfaces. Cellular and molecular life sciences : CMLS 65, 2594–2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez C, Gerber S, Boilevin J, Bucher M, Darbre T, Aebi M, Reymond JL & Locher KP. (2015). Structure and mechanism of an active lipid-linked oligosaccharide flippase. Nature 524, 433–438. [DOI] [PubMed] [Google Scholar]
- Picciotto MR, Cohn JA, Bertuzzi G, Greengard P & Nairn AC. (1992). Phosphorylation of the cystic fibrosis transmembrane conductance regulator. The Journal of biological chemistry 267, 12742–12752. [PubMed] [Google Scholar]
- Pissarra LS, Farinha CM, Xu Z, Schmidt A, Thibodeau PH, Cai Z, Thomas PJ, Sheppard DN & Amaral MD. (2008). Solubilizing mutations used to crystallize one CFTR domain attenuate the trafficking and channel defects caused by the major cystic fibrosis mutation. Chemistry & biology 15, 62–69. [DOI] [PubMed] [Google Scholar]
- Proks P, Puljung MC, Vedovato N, Sachse G, Mulvaney R & Ashcroft FM. (2016). Running out of time: the decline of channel activity and nucleotide activation in adenosine triphosphate-sensitive K-channels. Philosophical transactions of the Royal Society of London Series B, Biological sciences 371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabeh WM, Bossard F, Xu H, Okiyoneda T, Bagdany M, Mulvihill CM, Du K, di Bernardo S, Liu Y, Konermann L, Roldan A & Lukacs GL. (2012). Correction of both NBD1 energetics and domain interface is required to restore ΔF508 CFTR folding and function. Cell 148, 150–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou JL & et al. (1989). Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science (New York, NY) 245, 1066–1073. [DOI] [PubMed] [Google Scholar]
- Schultz BD, Frizzell RA & Bridges RJ. (1999). Rescue of dysfunctional deltaF508-CFTR chloride channel activity by IBMX. The Journal of membrane biology 170, 51–66. [DOI] [PubMed] [Google Scholar]
- Serohijos AW, Hegedus T, Aleksandrov AA, He L, Cui L, Dokholyan NV & Riordan JR. (2008). Phenylalanine-508 mediates a cytoplasmic-membrane domain contact in the CFTR 3D structure crucial to assembly and channel function. Proc Natl Acad Sci 105, 3256–3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohail MI, Schmid D, Wlcek K, Spork M, Szakács G, Trauner M, Stockner T & Chiba P. (2017). Molecular Mechanism of Taurocholate Transport by the Bile Salt Export Pump, an ABC Transporter Associated with Intrahepatic Cholestasis. Molecular pharmacology 92, 401–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockner T, Gradisch R & Schmitt L. (2020). The role of the degenerate nucleotide binding site in type I ABC exporters. FEBS letters 594, 3815–3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szöllősi D, Rose-Sperling D, Hellmich UA & Stockner T. (2018). Comparison of mechanistic transport cycle models of ABC exporters. Biochimica et biophysica acta Biomembranes 1860, 818–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szellas T & Nagel G. (2003). Apparent affinity of CFTR for ATP is increased by continuous kinase activity. FEBS letters 535, 141–146. [DOI] [PubMed] [Google Scholar]
- Szollosi A, Muallem DR, Csanády L & Vergani P. (2011). Mutant cycles at CFTR’s non-canonical ATP-binding site support little interface separation during gating. The Journal of general physiology 137, 549–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tóth B & Csanády L. (2012). Pore collapse underlies irreversible inactivation of TRPM2 cation channel currents. Proc Natl Acad Sci 109, 13440–13445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai MF, Jih KY, Shimizu H, Li M & Hwang TC. (2010a). Optimization of the degenerated interfacial ATP binding site improves the function of disease-related mutant cystic fibrosis transmembrane conductance regulator (CFTR) channels. The Journal of biological chemistry 285, 37663–37671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai MF, Li M & Hwang TC. (2010b). Stable ATP binding mediated by a partial NBD dimer of the CFTR chloride channel. The Journal of general physiology 135, 399–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai MF, Shimizu H, Sohma Y, Li M & Hwang TC. (2009). State-dependent modulation of CFTR gating by pyrophosphate. The Journal of general physiology 133, 405–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veit G, Avramescu RG, Perdomo D, Phuan PW, Bagdany M, Apaja PM, Borot F, Szollosi D, Wu YS, Finkbeiner WE, Hegedus T, Verkman AS & Lukacs GL. (2014). Some gating potentiators, including VX-770, diminish ΔF508-CFTR functional expression. Science translational medicine 6, 246ra297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergani P, Lockless SW, Nairn AC & Gadsby DC. (2005). CFTR channel opening by ATP-driven tight dimerization of its nucleotide-binding domains. Nature 433, 876–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergani P, Nairn AC & Gadsby DC. (2003). On the mechanism of MgATP-dependent gating of CFTR Cl- channels. The Journal of general physiology 121, 17–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Zeltwanger S, Hu S & Hwang TC. (2000). Deletion of phenylalanine 508 causes attenuated phosphorylation-dependent activation of CFTR chloride channels. The Journal of physiology 524 Pt 3, 637–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Okeyo GO, Tao B, Hong JS & Kirk KL. (2011). Thermally unstable gating of the most common cystic fibrosis mutant channel (ΔF508): “rescue” by suppressor mutations in nucleotide binding domain 1 and by constitutive mutations in the cytosolic loops. The Journal of biological chemistry 286, 41937–41948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Liu J, Loizidou A, Bugeja LA, Warner R, Hawley BR, Cai Z, Toye AM, Sheppard DN & Li H. (2014). CFTR potentiators partially restore channel function to A561E-CFTR, a cystic fibrosis mutant with a similar mechanism of dysfunction as F508del-CFTR. British journal of pharmacology 171, 4490–4503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward A, Reyes CL, Yu J, Roth CB & Chang G. (2007). Flexibility in the ABC transporter MsbA: Alternating access with a twist. Proc Natl Acad Sci 104, 19005–19010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinreich F, Riordan JR & Nagel G. (1999). Dual Effects of Adp and Adenylylimidodiphosphate on Cftr Channel Kinetics Show Binding to Two Different Nucleotide Binding Sites. Journal of General Physiology 114, 55–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu YC, Sohma Y & Hwang TC. (2016). On the mechanism of gating defects caused by the R117H mutation in cystic fibrosis transmembrane conductance regulator. The Journal of physiology 594, 3227–3244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z & Chen J. (2016). Atomic Structure of the Cystic Fibrosis Transmembrane Conductance Regulator. Cell 167, 1586–1597.e1589. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Liu F & Chen J. (2017). Conformational Changes of CFTR upon Phosphorylation and ATP Binding. Cell 170, 483–491.e488. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Liu F & Chen J. (2018). Molecular structure of the ATP-bound, phosphorylated human CFTR. Proc Natl Acad Sci 115, 12757–12762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Wang X, Liu HY, Zou X, Li M & Hwang TC. (2006). The two ATP binding sites of cystic fibrosis transmembrane conductance regulator (CFTR) play distinct roles in gating kinetics and energetics. The Journal of general physiology 128, 413–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou P, Bortolus M & McHaourab HS. (2009). Conformational cycle of the ABC transporter MsbA in liposomes: detailed analysis using double electron-electron resonance spectroscopy. Journal of molecular biology 393, 586–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw dataset is included in the Supporting Information document.