

Abstract
Background:
Overweight and obesity are significant risk factors for deep vein thrombosis (DVT). Cellular fibronectin containing extra domain A (Fn-EDA), an endogenous ligand for toll-like-receptor 4 (TLR4), contributes to thrombo-inflammation. The role of Fn-EDA in the modulation of DVT is not elucidated yet.
Objective:
To determine whether Fn-EDA promotes DVT in the context of diet-induced obesity.
Methods:
Wild-type (WT) and Fn-EDA-deficient mice were either fed control or high-fat diet (HF-diet) for 12-weeks. DVT was induced by inferior vena cava (IVC) stenosis and evaluated after 48 hours. Cellular Fn-EDA levels in the plasma of venous thromboembolism (VTE) patients were measured by sandwich enzyme-linked immunosorbent assay (ELISA).
Results:
We found that cellular Fn-EDA levels were significantly elevated in VTE patients’ plasma and positively correlated with body mass index. HF diet-fed WT mice exhibited increased DVT susceptibility compared with control diet-fed WT mice. In contrast, HF diet-fed Fn-EDA-deficient mice exhibited significantly reduced thrombus weight and decreased incidence (%) of DVT compared with HF diet-fed WT mice concomitant with reduced neutrophil content and citrullinated histone H3-positive cells (a marker of NETosis) in IVC thrombus. Exogenous cellular Fn-EDA potentiated NETosis in neutrophils stimulated with thrombin-activated platelets via TLR4. Genetic deletion of TLR4 in Fn-EDA+ mice (constitutively express Fn-EDA in plasma and tissues), but not in Fn-EDA-deficient mice, reduced DVT compared with respective controls.
Conclusion:
These results demonstrate a previously unknown role of Fn-EDA in the DVT exacerbation, which may be an essential mechanism promoting DVT in the setting of diet-induced obesity.
Keywords: Fibronectin, Deep Vein Thrombosis, diet-induced obesity, NETosis
1. INTRODUCTION
Obesity is prevalent and associated with a prothrombotic and hypercoagulable state.[1] Epidemiological studies suggest that obesity is a significant risk factor for deep vein thrombosis (DVT) even after adjusting major risk factors including age, race, hypertension, and hyperlipidemia.[2–4] Obesity is characterized by the state of chronic low-grade inflammation and increased plasma levels of many prothrombotic proteins, including von Willebrand factor, fibrinogen, and fibronectin.[5, 6] Despite this, the underlying mechanisms for increased DVT risk in the preexisting comorbid condition of obesity are not well understood. Understanding the mechanisms that promote DVT in the context of overweight or obesity may significantly impact the development of novel and safe therapeutic strategies for DVT.
The plasma fibronectin (Fn) is known to modulate thrombosis.[7, 8] Two primary forms of Fn isoforms exist in human and mice: 1) plasma Fn, which does not contain alternatively spliced extra domain A (EDA) or extra domain B (EDB) and 2) cellular Fn, which contains EDA (Fn-EDA) or EDB or both in different proportions. Fn-EDA is an endogenous ligand for TLR4. Increased plasma levels of Fn-EDA have been reported in overweight, diabetic patients, and patients with atherosclerosis and vascular injury.[9–11] We have previously reported that cellular Fn-EDA contributes to arterial thrombosis [12, 13] and promotes thrombo-inflammation in various experimental models.[14–18] The role of Fn-EDA in the modulation of DVT remains unexplored. An animal model, such as diet-induced obesity, which displays several features of human obesity, could be used as a model to study mechanisms for DVT exacerbation in the context of obesity. Previously it was shown that diet-induced obese mice are more susceptible to arterial thrombosis [19] and display impaired DVT resolution through altered inflammatory and fibrinolytic responses.[20] In the current study, we report that plasma levels of cellular Fn-EDA are elevated in VTE patients that positively correlated with body mass index. Utilizing mutant mouse models, we demonstrate that Fn-EDA promotes DVT in the preexisting comorbid condition of diet-induced obesity.
2. METHODS
2.1. Mice
The generation of the mice devoid of regulated splicing at the EDA exon has been previously described.[21] Briefly, to obtain constitutive inclusion of the EDA exon into the Fn mRNA (EDA+ allele), both splice sites of the EDA exon were optimized: the 5’ splice site was mutated to match that of the consensus sequence, and the 3’ splice site was replaced by that of the constitutively spliced-in second exon of the apolipoprotein AI gene, which matches precisely the 3’ splice site consensus. Additionally, lox P sites were inserted in the EDA flanking introns to make cell-specific EDA null allele. Mice homozygous for EDA+/+ or EDAfl/fl (inclusion of the modified EDA exon in both the Fn alleles) were obtained by heterozygous mating progenitors (EDA+/wt or EDAfl/wt) and maintained as a separate line. EDAfl/wt mice were crossed with Cre-recombinase transgenic mice to obtain heterozygous EDA−/wt progeny that were subsequently mated to obtain EDA−/− animals. EDAwt/wt were obtained by the mating of heterozygous progenitors (EDA-/wt) and maintained as a separate line. EDA+/+TLR4−/− mice were obtained by crossing heterozygous EDA+/wtTLR4−/− mice. Similarly, EDA−/−TLR4−/− mice were obtained by crossing heterozygous EDA-/wtTLR4−/− mice. Previously, all these modified mice strains have been described.[12, 14, 21] For simplification in the remainder of the manuscript: 1) EDAwt/wt mice are controls and will be referred to as WT mice. 2) EDA−/− mice lack EDA domain in the Fn both in the plasma and tissues and be referred to as Fn-EDA− mice. 3) EDA+/+ mice constitutively express EDA exon in both of the Fn alleles in the plasma and tissues and will be referred to as Fn-EDA+ mice. Male WT and Fn-EDA− mice were fed on control or high-fat diet (Cat # TD.06414, Envigo, 60 % Kcal from fat) for 12-week starting from the age of 6-week. Mice were kept in standard animal house conditions with controlled temperature and humidity and had ad libitum access to standard chow diet and water, except otherwise noted. All the mice used in the present study were on the C57BL/6J background. The University of Iowa Animal Care and Use Committee approved all the procedures.
2.2. Quantification of cellular Fn-EDA in plasma samples
Plasma samples from the patients with VTE were obtained from Dr. Anetta Undas (Institute of Cardiology, Jagiellonian University, Kraków, Poland). Cellular Fn-EDA levels in the plasma were measured by sandwich enzyme-linked immunosorbent assay (ELISA) as described.[14–16] Briefly, microtiter plates were coated overnight at 4°C with primary antibody for Fn-EDA (3E2, 10 μg/mL, Sigma, catalog # F6140) diluted in 50 mM sodium carbonate buffer. 10 μl of plasma samples (diluted 1:1 in PBS) were incubated for 2 h in the coated wells at 37°C. After five washes, biotinylated secondary antibody to Fn (2 μg/ml diluted in blocking buffer, Sigma, catalog # F3648) was added to wells and incubated for 1 hour at room temperature. The avidin HRP solution (1:1000) in the blocking buffer was added to wells and incubated for 30 minutes following five washes. Micro titer plates were washed five times before adding 3, 3’, 5, 5’-tetramethylbenzidine substrate solution (Sigma, catalog # T0440) to the wells, and the colorimetric reaction was stopped with 2 M H2SO4 after 10 min. Results were read in an ELISA microplate reader at A450 nm. Human cellular Fn (Sigma, catalog # F2518) was used for standards.
2.3. IVC stenosis model for DVT
DVT was induced as per previously published methods with slight modifications.[22] Briefly, for stenosis, a space holder (30-gauge, 3-mm long needle) was positioned on the outside of the exposed IVC, and a permanent narrowing ligature was placed below the left renal vein. Next, the needle was removed to restrict blood flow to 80-90%. Due to the high peritoneal fat content in obese mice, side branches were not ligated. This protocol allowed us to reduce surgery time and subsequently improved post-surgical survival significantly. IVC thrombi were harvested 48-hour post-stenosis, detached from the vessel wall, dried, and weighed in a microbalance, and imaged.
2.4. Laser speckle contrast imaging
To assess post-stenosis venous blood flow, we used a laser speckle contrast imager (moorFLPI-2 from Moor instruments), which provides real-time, high-resolution blood flow images, as described.[18] Briefly, mice were anesthetized using isoflurane (2.5% induction, 1 % maintenance), and an incision was made to visualize IVC at the ligation site. Speckle imaging was obtained using a temporal filter (250 frames, 10 secs/frame) at 0.1 Hz at baseline, during stenosis, and 4-hour post-stenosis. Blood fluxes were measured in the pre-stenosis regions, and fluxes were expressed in arbitrary units using a 12-color palette. The representative image for each group was selected based upon the mean value.
2.5. Immunocytochemistry
IVC thrombi were harvested 48-hour post-stenosis, detached from the vessel wall, dried and weighed in a microbalance followed by embedded in optimal cutting temperature compound, frozen at −80°C, and was cut with a cryotome (CryoStar NX70 Cryostat; ThermoFisher Scientific) into 5-μm sections. Following blocking with 5% BSA for 1 hour, samples were incubated with antibodies specific for, Neutrophils (Ly6GB.2, Abcam, catalog # ab210204), anti-Histone H3 antibody (Abcam, catalog # ab61251, diluted 1:100) overnight at 4° C. The samples were washed and labeled with the FITC-conjugated secondary antibody was from Southern Biotech (catalog #4052-02, diluted 1:250) and Alexa Fluor-568-conjugated appropriate secondary antibodies for 1hr at room temperature. Sections were counterstained with Hoechst (5 μg/mL) before mounting, and images were acquired using an Olympus fluorescent microscope (BX51) equipped with a UPlanFLN 20x/0.5 NA objective lens and DP71 color CCD camera. Image analysis was done using ImageJ software. Staining and image acquisition was performed in parallel for the entire set.
2.6. NETosis assay
Freshly isolated neutrophils from bone marrow were seeded on poly-L lysine-coated coverslips (1 X 104 cells/coverslip). Cells were incubated for 60 minutes in a CO2 incubator. NETosis assay was performed using cellular Fn (Sigma, catalog # F2518, 5 μg/mL) and thrombin (0.1 U/ml) activated platelets (1 X107), isolated from whole blood. Cells were incubated for 4 hours in a CO2 incubator at 37°C. 500 μl ice-cold PBS was added to stop the reaction, and the coverslips were placed on ice for 10 minutes. Coverslips were gently drained to discard liquid, and cells were fixed for 15 minutes in ice-cold PBS containing 2% paraformaldehyde at room temperature. The fixed cells were then washed with ice-cold PBS. For specific staining of extracellular nuclear structures, cells were then incubated at room temperature with PlaNET Green (1:10 dilution: #PLANET-001, Sunshine antibodies) dye for 60 minutes at 40°C. Coverslips were washed with PBS and mounted onto glass slides using a drop of mounting medium containing DAPI (Vector Labs, Cat. #H-1200.), prior to fluorescence microscopy analysis. Samples were analyzed using an Olympus BX51 microscope. For quantitation, two fields at 20x magnification were counted (coverslip edges were avoided). In total 350-400 neutrophils were counted/slide. Neutrophils releasing only extracellular structures (PlaNET Green-positive) were counted per field. Measurements were obtained from 2 different slides (experiment), and the mean was calculated per animal.
2.7. Plasma insulin quantification
Plasma insulin levels were quantified by ELISA in overnight fasted mice after 12-week of control or high-fat (HF) diet feeding as per the kit manufacturer’s instruction (Sigma, catalog # EZRMI-13K).
2.8. Oral glucose tolerance test
After baseline blood glucose measurement, overnight fasted mice were administered glucose load (1.5 g/kg) by oral gavage and blood glucose levels were measured at 15, 30, 60- and 120-min post glucose load.
2.9. Statistical analysis
For analysis, GraphPad Prism software (7.04) was used. Shapiro-Wilk test was used to check normality, and Bartlett’s test was used to check equal variance. The results were considered significant at P<0.05. Normally distributed data were analyzed by Student’s t-test or two-way ANOVA followed by Sidak’s multiple comparisons test, and non-normally distributed data were analyzed using the Mann Whitney test (for two-group) or non-parametric two-way ANOVA followed by Fischer’s LSD test. Bodyweight and blood glucose levels (during oral glucose tolerance test) were analyzed using repeated measure ANOVA followed by Sidak’s multiple comparisons test. Thrombosis incidence data were analyzed using the chi-square test. Correlation of plasma Fn-EDA with body mass index (BMI) was analyzed using Pearson’s Correlation Coefficient.
3. RESULTS
3.1. Plasma levels of cellular fibronectin (Fn-EDA) are elevated in patients with VTE
Increased plasma Fn-EDA levels are reported in patients with cardiovascular disorders[10, 23] and in overweight, diabetic patients.[9] We first determined the Fn-EDA levels in the plasma of VTE patients using ELISA. Patients characteristics are provided in Table 1 in the Data Supplement. In the control group, 54% of subjects were males, and 46% of subjects were females, while in the VTE group, 35% of subjects were males and 65% of subjects were females. We found significantly elevated plasma Fn-EDA levels (5.8 ± 0.5 μg/mL) in VTE patients compared with controls (0.8 ± 0.03 μg/mL, P<0.05, Figure 1A). Next, we evaluated whether the body mass index (BMI) of VTE patients correlates with Fn-EDA levels. We found that plasma Fn-EDA levels were positively correlated with BMI (unadjusted Pearson’s Correlation Coefficient, R2 0.48, Figure 1B).
Figure 1: Plasma Fn-EDA was elevated in VTE patients that positively correlated with BMI.
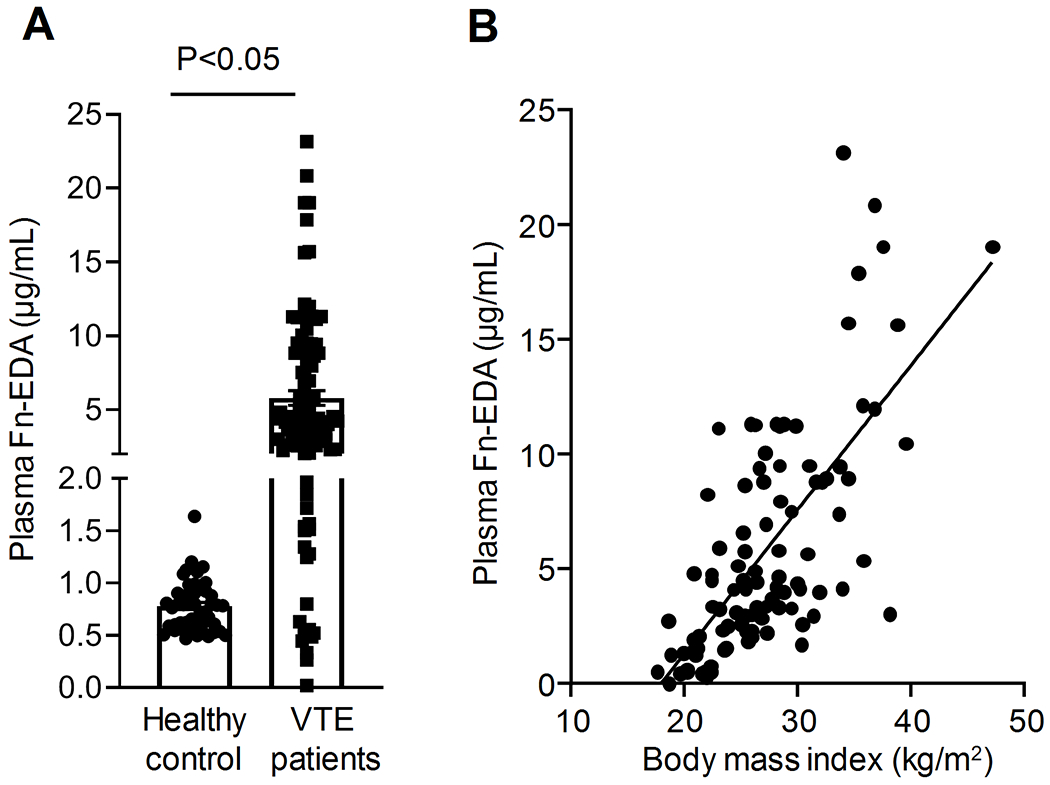
A, Plasma Fn-EDA levels in VTE patients and healthy controls as determined by ELISA (n=48, 100). Data are mean ± SEM. B, Correlation of plasma Fn-EDA levels with BMI in VTE patients (Pearson’s Correlation Coefficient R2 0.4845) using the data in A.
3.2. Fn-EDA mediates DVT in the preexisting diet-induced obese condition
To investigate whether Fn-EDA plays a causative role in modulation of DVT in the context of obesity, we utilized the diet-induced obesity model. WT and Fn-EDA− mice were fed either a control or high-fat diet (HF) diet for 12-weeks. Bodyweight gain was comparable between HF diet-fed WT and HF diet-fed Fn-EDA− mice suggesting that Fn-EDA does not contribute to obesity (Figure 1 in the Data Supplement). Both WT and Fn-EDA− mice, developed a similar degree of obesity and similar T2DM features such as increased fasting blood glucose and worsen glucose tolerance (Figure 1 in the Data Supplement). Complete blood cell counts were similar in both groups (Table 2 in the Data Supplement). Plasma Fn-EDA levels were significantly elevated in HF diet-fed WT mice when compared to control diet-fed mice (Figure 2A). WT and Fn-EDA− mice were subjected to IVC stenosis to evaluate DVT susceptibility. In this model, closure of ≥80% of the IVC lumen for 48 hours results in the development of thrombi structurally similar to those observed in humans.[24] We found a significantly increased incidence of DVT in HF diet-fed WT mice (82% versus 50% in control diet-fed mice). In contrast, HF diet-fed Fn-EDA− mice exhibited significantly decreased incidence of DVT (58% versus 82% in HF diet-fed WT mice, Figure 2B). Next, we evaluated thrombus weight in the mice that exhibited thrombosis following IVC stenosis. The decreased incidence of DVT in the HF diet-fed Fn-EDA− mice was associated with reduced thrombus weight at 48 hours (Figure 2C). DVT incidence and thrombus weight was comparable between WT and Fn-EDA− mice fed a control diet because a minimal amount of Fn-EDA is present in the plasma of WT mice. Previously, it was shown that 25-50% of WT mice develop thrombi within 2 to 6 hours in the IVC stenosis model.[22, 25] Utilizing laser-speckle imaging, we determined early changes in the IVC blood flow following stenosis. We found that HF diet-fed Fn-EDA− mice exhibited significantly increased IVC blood flow (~30%), 4-hour after stenosis compared with HF diet-fed WT mice (Figure 2 in the data supplement). Together, these results suggest that Fn-EDA promotes venous thrombosis in the context of diet-induced thrombosis.
Figure 2. Fn-EDA promotes venous thrombosis in the context of diet-induced obesity.
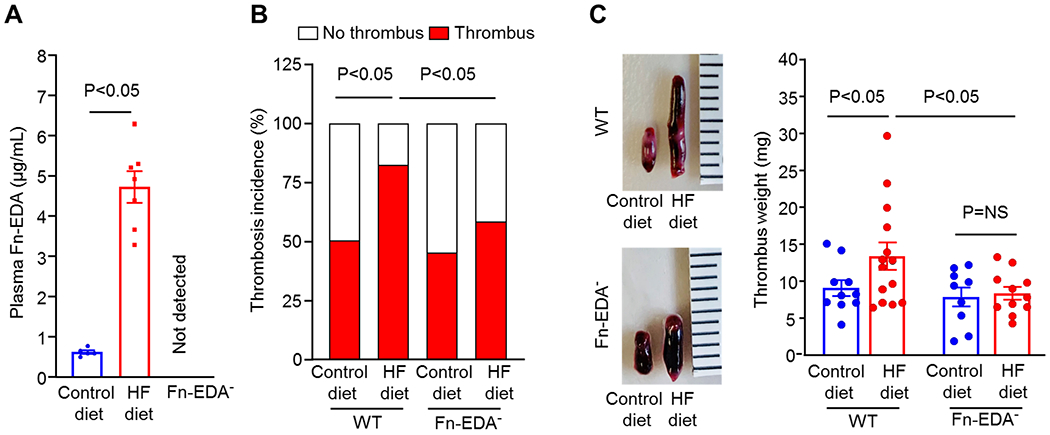
A, Plasma Fn-EDA levels in WT mice fed either a control diet or HF diet for 12-weeks (n=5,7, unpaired t-test). B, Thrombosis incidence (n=20,18, 20,19, chi-square test). C, Left, Representative IVC thrombus harvested 48-hour post-stenosis from each group. Right, thrombus weight (non-parametric two-way ANOVA followed by Fisher’s LSD test). Only mice that exhibited thrombosis were included to quantify the thrombus weight. Each dot represents a single mouse. Data are mean ± SEM. HF indicates high-fat.
3.3. Fn-EDA− mice exhibit reduced neutrophil influx and NETosis
Previously studies have found that Fn-EDA potentiates neutrophil influx in experimental animal models.[14, 16] Evidence from animal models suggest that neutrophils contribute to the pathophysiology of DVT.[24, 26] To determine whether Fn EDA promotes neutrophil influx in the DVT model, we performed immunohistochemistry of an IVC thrombus. We found a significantly reduced number of neutrophils within the thrombi HF diet-fed Fn-EDA− mice when compared with HF diet-fed WT mice (Figure 3A). Total platelet content, as analyzed by immunohistochemistry (CD41-positive), was comparable within thrombi of HF diet-fed Fn-EDA− mice and HF diet-fed WT mice (Figure 3 in data supplement). Because neutrophils are known to potentiate DVT through several mechanisms, including the formation of neutrophil extracellular traps (NETs),[25], we quantified NETs within thrombi. We found a significant decrease in citrullinated histone H3-positive cells (a marker of NETs) in HF diet-fed Fn-EDA− mice compared with HF diet-fed WT mice (Figure 3A), suggesting that Fn-EDA may exacerbate DVT by potentiating neutrophil infiltration and NETosis. Fn-EDA is an endogenous ligand for TLR4. To determine whether TLR4 contributes to Fn-EDA-mediated NETosis, we performed an in vitro NET assay using thrombin activated platelets as stimuli. Neutrophils isolated from WT and TLR4−/− mice were stimulated with thrombin (0.1 U/mL)-activated platelets (1X107), and NETosis was evaluated in the presence or absence of cFn containing EDA. We found that exogenous cFn potentiated the release of NETs in neutrophils isolated from WT mice, which was significantly reduced in the neutrophils isolated from TLR4−/− mice, suggesting that TLR4 contributes to Fn-EDA mediated NETosis in vitro (Figure 3B).
Figure 3. TLR4 contributes to Fn-EDA-mediated NETosis.
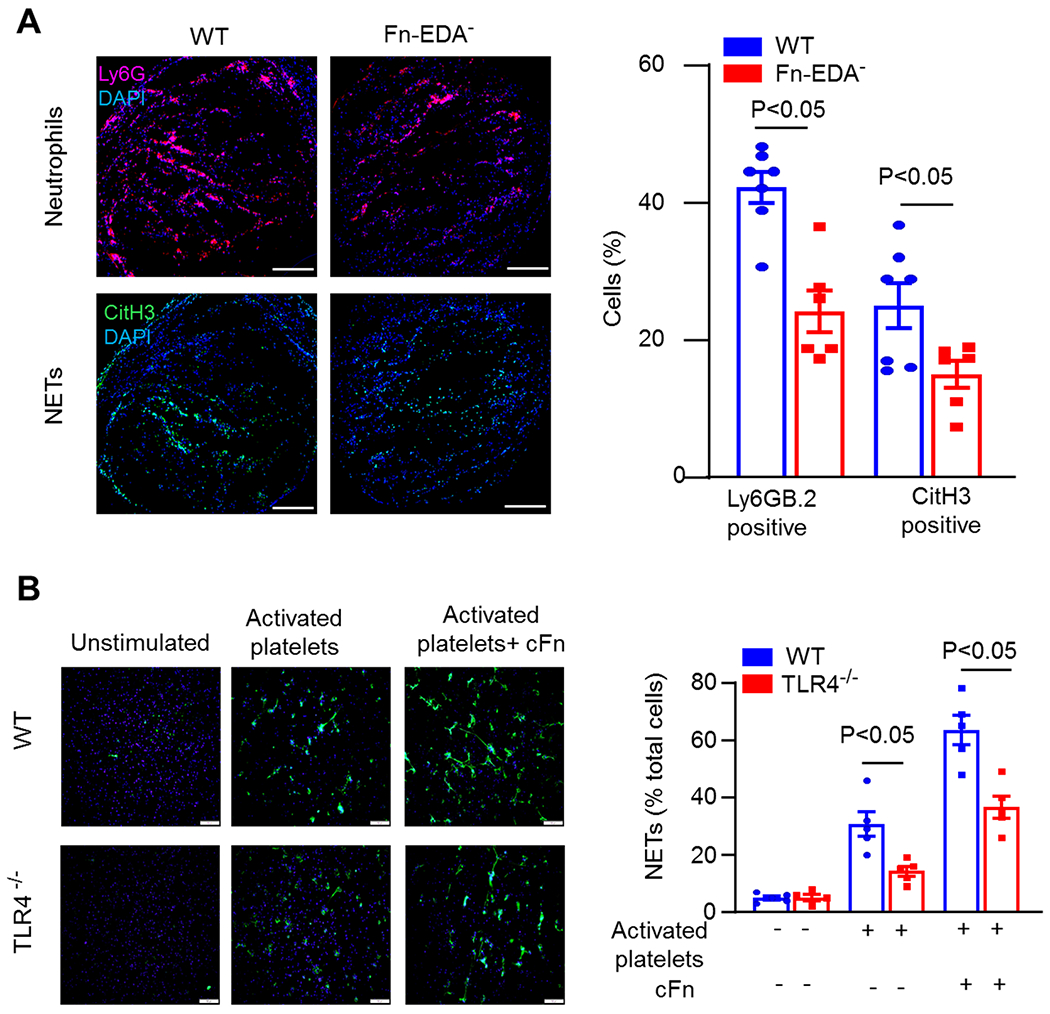
A, Left, Representative cross-sectional immunofluorescence image of the isolated IVC (on day-2 post-stenosis) from 12-week HF diet-fed WT and EDA− mice stained for Ly6G (neutrophils, red) and anti-histone H3 (citrulline R2 + R8 + R17) (NETs, green) and DAPI (blue). Scale bar 200 μm. Right, Quantification of the % Ly6G and % CitH3 positive cells (n=7, 6, 7, 6, unpaired t-test). Data are mean ± SEM. B, NETs assay was performed by stimulating bone marrow-derived neutrophils from either WT or TLR4−/− mice with thrombin (0.1 U/mL)-activated platelets (1X107), in the presence or absence of cFn (5 μg/mL). Left, Representative microphotographs of NETs stained with PlaNET green (stains extracellular DNA, green) and counterstained with Hoechst (stains nuclei, blue). Scale bar 100 μm. Right, Quantification of the percentage of cells releasing NETs (n =5 mice/group, two-way ANOVA followed by Sidak’s multiple comparisons test). The value for each mouse represents a mean from 2 fields. cFn, cellular fibronectin, NETs, neutrophil extracellular traps.
3.4. TLR4 contributes to Fn-EDA mediated deep vein thrombosis
To evaluate the functional in vivo significance of the Fn-EDA/TLR4 axis in the modulation of DVT, we utilized Fn-EDA+TLR4−/− mice, which constitutively express Fn-EDA in both tissues and plasma.[12, 21], and Fn-EDA−TLR4−/− mice. These mice strains were fed a control diet but not HF diet because the genetic deletion of TLR4 reduces body weight [27], which could confound the DVT outcome in the context of diet-induced obesity. We found a reduced incidence of DVT in Fn-EDA+TLR4−/− mice when compared with Fn-EDA+ mice (62% versus 91%, Figure 4A), but no significant effect was observed in Fn-EDA−TLR4−/− mice when compared with Fn-EDA− mice (36% versus 42%, Figure 4A). In the line of these results, we observed a significant reduction in thrombus weight in Fn-EDA+TLR4−/− mice compared with Fn-EDA+ mice; however, no change in thrombus weight was observed in Fn-EDA−TLR4−/− mice compared with Fn-EDA− mice (Figure 4B).
Figure 4. TLR4 contributes to Fn-EDA-mediated DVT.
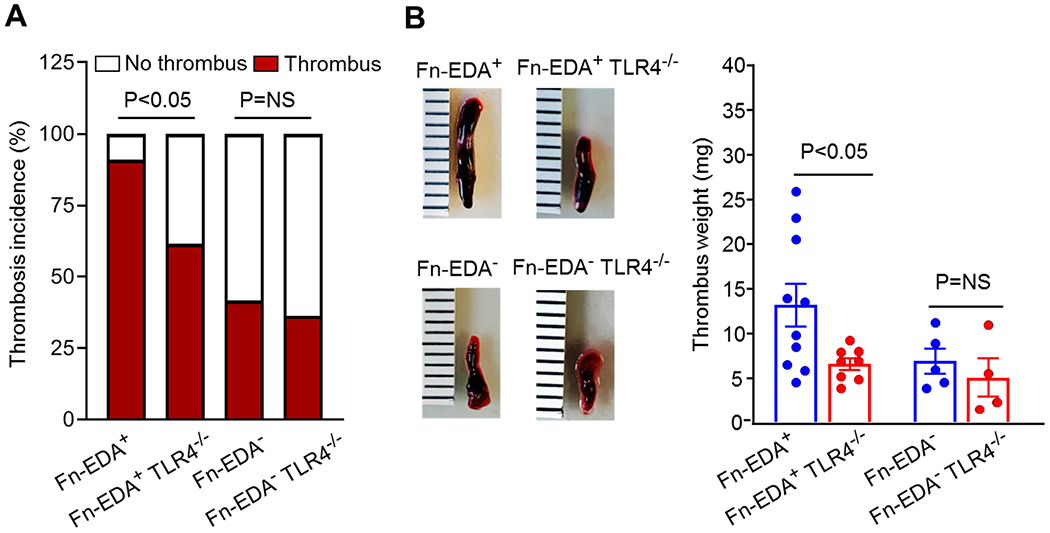
A, Thrombosis incidence (n=11,13,12,11, chi-square test). B, Left, Representative IVC thrombus harvested 48-hour post-stenosis from each genotype. Right, Thrombus weight (non-parametric two-way ANOVA followed by Fisher’s LSD test). Only mice with thrombosis were included to quantify thrombosis weight. Data are mean ± SEM. Each dot represents a single mouse.
4. DISCUSSION
The current study demonstrates a novel role for Fn-EDA in the modulation of DVT in diet-induced obese conditions. We believe that the findings may have clinical significance for the following reasons. First, obesity and overweight are a significant risk factor for VTE.[3, 4] We found significantly elevated plasma Fn-EDA levels in VTE patients that positively correlated with BMI. Second, we provide evidence that the genetic ablation of Fn-EDA results in reduced susceptibility to DVT in the preexisting comorbid diet-induced obese condition. Finally, our mechanistic data suggest that Fn-EDA/TLR4 axis may promote NETosis and, thereby, DVT in mice.
Epidemiological studies suggest that obese patients have an increased risk of VTE.[2–4] Obese patients display sustained endothelial activation [28], which can promote the release of cellular fibronectin.[29, 30] Increased levels of plasma cellular fibronectin (Fn-EDA) have been reported in overweight and diabetic patients.[9] Based on these reports, we hypothesized that enhanced plasma Fn-EDA levels in obese conditions might promote DVT. In order to mimic this clinical situation, we used a diet-induced obesity mouse model because of the following reasons. First, it is a good model mimicking many features of metabolic syndromes, such as hyperglycemia and hyperlipidemia, that are observed with human obesity.[31] Second, other than HF diet feeding, this model does not require genetic manipulation, for example, in the case of ob/ob mice or db/db mice. Third, we observed significantly elevated Fn-EDA levels in the plasma of HF diet-fed mice. Bodyweight and type-2 diabetes, including increased fasting blood glucose and worsen glucose tolerance, were comparable in HF diet WT and Fn-EDA− mice, suggesting that Fn-EDA by itself does not contribute to diet-induced obesity. In contrast, HF diet-fed Fn-EDA− mice exhibited significantly reduced DVT compared with HF diet-fed WT mice, suggesting that Fn-EDA promotes DVT exacerbation in the context of diet-induced obesity. Previous studies have suggested a role for Fn-EDA in arterial thrombosis. In the flow chamber assay, the presence of Fn-EDA in whole blood produced larger thrombi [12] and accelerated thrombosis in FeCl3 injury-induced mesenteric arterial thrombosis model.[12] Furthermore, the mice constitutively expressing Fn-EDA exhibited increased mortality in the collagen-epinephrine induced pulmonary thromboembolism model.[12] The results of this study further confirm the prothrombotic role of Fn-EDA in the setting of DVT.
Coordinated interactions between neutrophil, platelet, and endothelial cell contribute to the DVT.[32–34] Neutrophils potentiate DVT by forming neutrophil-platelet aggregates, secreting inflammatory mediators, releasing tissue factor, generating free radicals, and producing neutrophil extracellular traps (NETs).[24, 25] In the current study, we found that neutrophils and citrullinated histone H3 positive cells were significantly reduced in HF diet-fed Fn-EDA− mice compared to HF diet-fed WT mice. Previous studies have found that Fn-EDA/TLR4 axis plays a mechanistic role in modulation of thrombo-inflammation.[14, 17, 35] In line with these studies, we found that exogenous cFn potentiated the NETs release, a process that was TLR4 dependent. Furthermore, we demonstrated that TLR4 contributes to Fn-EDA mediated DVT in the IVC stenosis model. Previously, it was shown that EDA, but not other domains of Fn, activates human TLR4 expressed in HEK293 cells.[36] Like lipopolysaccharide, EDA activation of TLR4 requires MD-2, an accessory protein associated with extracellular domain A of TLR4 and required for TLR4-dependent LPS response.[36] Additionally, TLR4 was shown to modulate microvascular as well as venous thrombosis in response to endotoxemia in experimental models.[37, 38] Although with the experimental conditions described herein, we found that TLR4 contributes to Fn-EDA mediated DVT, it remains possible that some of the effects of Fn-EDA are TLR4-independent, perhaps mediated by binding sites for leukocyte integrins α4β1 and α9β1 in the EDA domain. Additional studies are warranted to determine whether disrupting Fn-EDA-integrin interactions in vivo reduces NETosis and subsequent DVT.
Despite its strength, our study has a few limitations. First, we did not evaluate the role of tissue factor, other coagulation factors, or total plasma Fn levels in the modulation of DVT in the context of diet-induced obesity. It is known that all these factors are elevated in obesity [6, 39], and may contribute to DVT exacerbation in addition to Fn-EDA.[8, 39] Second, we did not determine whether endothelial cell-derived Fn-EDA modulates DVT in obese mice. Third, we used global TLR4 deficient mice for our mechanistic studies. Future studies are warranted to determine the cell-specific role of TLR4 in modulating Fn-EDA mediated DVT in the context of diet-induced obesity. In summary, these findings identify a novel role of cellular Fn-EDA in modulating DVT. The clinical implications of the current data remain to be explored, but increased plasma levels of Fn-EDA may be an important mechanism promoting DVT in the setting of diet-induced obesity.
Supplementary Material
Essentials.
Cellular Fn-EDA levels were significantly elevated in the plasma of VTE patients and positively correlated with BMI.
The role of Fn-EDA in the modulation of DVT remains unclear.
Using mutant mice, we demonstrate that Fn-EDA promotes experimental DVT in the setting of diet-induced obesity.
Fn-EDA potentiates NETosis via TLR4.
Funding:
The work is supported by grants from the National Institutes of Health grants (R35HL139926, R01NS109910 & U01NS113388) and by Established Investigator Award 18EIA33900009 from the American Heart Association to A. KC ND is supported by the ASH Scholar award from the American Society of Hematology.
Footnotes
CONFLICT OF INTEREST
The author (s) declare (s) no conflict of interest.
References:
- 1.Campello E, Zabeo E, Radu CM, Spiezia L, Gavasso S, Fadin M, Woodhams B, Vettor R, Simioni P. Hypercoagulability in overweight and obese subjects who are asymptomatic for thrombotic events. Thromb Haemost. 2015; 113: 85–96. 10.1160/TH14-02-0156. [DOI] [PubMed] [Google Scholar]
- 2.Yang G, De Staercke C, Hooper WC. The effects of obesity on venous thromboembolism: A review. Open J Prev Med. 2012; 2: 499–509. 10.4236/ojpm.2012.24069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stein PD, Beemath A, Olson RE. Obesity as a risk factor in venous thromboembolism. Am J Med. 2005; 118: 978–80. 10.1016/j.amjmed.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 4.Klovaite J, Benn M, Nordestgaard BG. Obesity as a causal risk factor for deep venous thrombosis: a Mendelian randomization study. J Intern Med. 2015; 277: 573–84. 10.1111/joim.12299. [DOI] [PubMed] [Google Scholar]
- 5.Mertens I, Van Gaal LF. Obesity, haemostasis and the fibrinolytic system. Obes Rev. 2002; 3: 85–101. 10.1046/j.1467-789x.2002.00056.x. [DOI] [PubMed] [Google Scholar]
- 6.Andersen T, Dejgaard A, Astrup A, Gluud C. Increased plasma fibronectin concentrations in obesity: normalization during weight loss. Acta Med Scand. 1987; 222: 275–9. 10.1111/j.0954-6820.1987.tb10670.x. [DOI] [PubMed] [Google Scholar]
- 7.Wang Y, Carrim N, Ni H. Fibronectin orchestrates thrombosis and hemostasis. Oncotarget. 2015; 6: 19350–1. 10.18632/oncotarget.5097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang Y, Reheman A, Spring CM, Kalantari J, Marshall AH, Wolberg AS, Gross PL, Weitz JI, Rand ML, Mosher DF, Freedman J, Ni H. Plasma fibronectin supports hemostasis and regulates thrombosis. J Clin Invest. 2014; 124: 4281–93. 10.1172/JCI74630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kanters SD, Banga JD, Algra A, Frijns RC, Beutler JJ, Fijnheer R. Plasma levels of cellular fibronectin in diabetes. Diabetes Care. 2001; 24: 323–7. 10.2337/diacare.24.2.323. [DOI] [PubMed] [Google Scholar]
- 10.Lemanska-Perek A, Krzyzanowska-Golab D, Pupek M, Klimeczek P, Witkiewicz W, Katnik-Prastowska I. Analysis of Soluble Molecular Fibronectin-Fibrin Complexes and EDA-Fibronectin Concentration in Plasma of Patients with Atherosclerosis. Inflammation. 2016; 39: 1059–68. 10.1007/s10753-016-0336-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peters JH, Maunder RJ, Woolf AD, Cochrane CG, Ginsberg MH. Elevated plasma levels of ED1+ (“cellular”) fibronectin in patients with vascular injury. J Lab Clin Med. 1989; 113: 586–97. [PubMed] [Google Scholar]
- 12.Chauhan AK, Kisucka J, Cozzi MR, Walsh MT, Moretti FA, Battiston M, Mazzucato M, De Marco L, Baralle FE, Wagner DD, Muro AF. Prothrombotic effects of fibronectin isoforms containing the EDA domain. Arteriosclerosis, thrombosis, and vascular biology. 2008; 28: 296–301. 10.1161/ATVBAHA.107.149146. [DOI] [PubMed] [Google Scholar]
- 13.Prakash P, Kulkarni PP, Lentz SR, Chauhan AK. Cellular fibronectin containing extra domain A promotes arterial thrombosis in mice through platelet Toll-like receptor 4. Blood. 2015; 125: 3164–72. 10.1182/blood-2014-10-608653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dhanesha N, Ahmad A, Prakash P, Doddapattar P, Lentz SR, Chauhan AK. Genetic Ablation of Extra Domain A of Fibronectin in Hypercholesterolemic Mice Improves Stroke Outcome by Reducing Thrombo-Inflammation. Circulation. 2015; 132: 2237–47. 10.1161/CIRCULATIONAHA.115.016540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dhanesha N, Chorawala MR, Jain M, Bhalla A, Thedens D, Nayak M, Doddapattar P, Chauhan AK. Fn-EDA (Fibronectin Containing Extra Domain A) in the Plasma, but Not Endothelial Cells, Exacerbates Stroke Outcome by Promoting Thrombo-Inflammation. Stroke. 2019; 50: 1201–9. 10.1161/STROKEAHA.118.023697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chorawala MR, Prakash P, Doddapattar P, Jain M, Dhanesha N, Chauhan AK. Deletion of Extra Domain A of Fibronectin Reduces Acute Myocardial Ischaemia/Reperfusion Injury in Hyperlipidaemic Mice by Limiting Thrombo-Inflammation. Thromb Haemost. 2018; 118: 1450–60. 10.1055/s-0038-1661353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Doddapattar P, Jain M, Dhanesha N, Lentz SR, Chauhan AK. Fibronectin Containing Extra Domain A Induces Plaque Destabilization in the Innominate Artery of Aged Apolipoprotein E-Deficient Mice. Arterioscler Thromb Vasc Biol. 2018; 38: 500–8. 10.1161/ATVBAHA.117.310345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dhanesha N, Jain M, Tripathi AK, Doddapattar P, Chorawala M, Bathla G, Nayak MK, Ghatge M, Lentz SR, Kon S, Chauhan AK. Targeting Myeloid-Specific Integrin alpha9beta1 Improves Short- and Long-Term Stroke Outcomes in Murine Models With Preexisting Comorbidities by Limiting Thrombosis and Inflammation. Circ Res. 2020; 126: 1779–94. 10.1161/CIRCRESAHA.120.316659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nagai N, Hoylaerts MF, Cleuren AC, Van Vlijmen BJ, Lijnen HR. Obesity promotes injury induced femoral artery thrombosis in mice. Thromb Res. 2008; 122: 549–55. 10.1016/j.thromres.2007.12.017. [DOI] [PubMed] [Google Scholar]
- 20.Bouzeghrane F, Zhang X, Gevry G, Raymond J. Deep vein thrombosis resolution is impaired in diet-induced type 2 diabetic mice. J Vasc Surg. 2008; 48: 1575–84. 10.1016/j.jvs.2008.07.050. [DOI] [PubMed] [Google Scholar]
- 21.Muro AF, Chauhan AK, Gajovic S, Iaconcig A, Porro F, Stanta G, Baralle FE. Regulated splicing of the fibronectin EDA exon is essential for proper skin wound healing and normal lifespan. J Cell Biol. 2003; 162: 149–60. 10.1083/jcb.200212079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Payne H, Brill A. Stenosis of the Inferior Vena Cava: A Murine Model of Deep Vein Thrombosis. J Vis Exp. 2017. 10.3791/56697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pupek M, Krzyzanowska-Golab D, Kotschy D, Witkiewicz W, Kwiatkowska W, Kotschy M, Katnik-Prastowska I. Time-dependent changes in extra-domain A-fibronectin concentration and relative amounts of fibronectin-fibrin complexes in plasma of patients with peripheral arterial disease after endovascular revascularisation. Int Wound J. 2018; 15: 649–59. 10.1111/iwj.12909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Budnik I, Brill A. Immune Factors in Deep Vein Thrombosis Initiation. Trends Immunol. 2018; 39: 610–23. 10.1016/j.it.2018.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fuchs TA, Brill A, Wagner DD. Neutrophil extracellular trap (NET) impact on deep vein thrombosis. Arteriosclerosis, thrombosis, and vascular biology. 2012; 32: 1777–83. 10.1161/ATVBAHA.111.242859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kapoor S, Opneja A, Nayak L. The role of neutrophils in thrombosis. Thromb Res. 2018; 170: 87–96. 10.1016/j.thromres.2018.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pierre N, Deldicque L, Barbe C, Naslain D, Cani PD, Francaux M. Toll-like receptor 4 knockout mice are protected against endoplasmic reticulum stress induced by a high-fat diet. PLoS One. 2013; 8: e65061. 10.1371/journal.pone.0065061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Caballero AE. Endothelial dysfunction in obesity and insulin resistance: a road to diabetes and heart disease. Obes Res. 2003; 11: 1278–89. 10.1038/oby.2003.174. [DOI] [PubMed] [Google Scholar]
- 29.Odekon LE, Frewin MB, Del Vecchio P, Saba TM, Gudewicz PW. Fibronectin fragments released from phorbol ester-stimulated pulmonary artery endothelial cell monolayers promote neutrophil chemotaxis. Immunology. 1991; 74: 114–20. [PMC free article] [PubMed] [Google Scholar]
- 30.Vincent PA, Del Vecchio PJ, Saba TM. Release of fibronectin fragments from endothelial cell monolayers exposed to activated leukocytes: relationship to plasma fibronectin levels after particle infusion. Exp Mol Pathol. 1988; 48: 403–18. 10.1016/0014-4800(88)90074-3. [DOI] [PubMed] [Google Scholar]
- 31.Wang CY, Liao JK. A mouse model of diet-induced obesity and insulin resistance. Methods Mol Biol. 2012; 821: 421–33. 10.1007/978-1-61779-430-8_27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.von Bruhl ML, Stark K, Steinhart A, Chandraratne S, Konrad I, Lorenz M, Khandoga A, Tirniceriu A, Coletti R, Kollnberger M, Byrne RA, Laitinen I, Walch A, Brill A, Pfeiler S, Manukyan D, Braun S, Lange P, Riegger J, Ware J, Eckart A, Haidari S, Rudelius M, Schulz C, Echtler K, Brinkmann V, Schwaiger M, Preissner KT, Wagner DD, Mackman N, Engelmann B, Massberg S. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med. 2012; 209: 819–35. 10.1084/jem.20112322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van Montfoort ML, Stephan F, Lauw MN, Hutten BA, Van Mierlo GJ, Solati S, Middeldorp S, Meijers JC, Zeerleder S. Circulating nucleosomes and neutrophil activation as risk factors for deep vein thrombosis. Arteriosclerosis, thrombosis, and vascular biology. 2013; 33: 147–51. 10.1161/ATVBAHA.112.300498. [DOI] [PubMed] [Google Scholar]
- 34.Schulz C, Engelmann B, Massberg S. Crossroads of coagulation and innate immunity: the case of deep vein thrombosis. Journal of thrombosis and haemostasis : JTH. 2013; 11 Suppl 1: 233–41. 10.1111/jth.12261. [DOI] [PubMed] [Google Scholar]
- 35.Bhattacharyya S, Tamaki Z, Wang W, Hinchcliff M, Hoover P, Getsios S, White ES, Varga J. FibronectinEDA promotes chronic cutaneous fibrosis through Toll-like receptor signaling. Sci Transl Med. 2014; 6: 232ra50. 10.1126/scitranslmed.3008264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Okamura Y, Watari M, Jerud ES, Young DW, Ishizaka ST, Rose J, Chow JC, Strauss JF 3rd. The extra domain A of fibronectin activates Toll-like receptor 4. J Biol Chem. 2001; 276: 10229–33. 10.1074/jbc.M100099200. [DOI] [PubMed] [Google Scholar]
- 37.Stark RJ, Aghakasiri N, Rumbaut RE. Platelet-derived Toll-like receptor 4 (Tlr-4) is sufficient to promote microvascular thrombosis in endotoxemia. PLoS One. 2012; 7: e41254. 10.1371/journal.pone.0041254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Obi AT, Andraska E, Kanthi Y, Kessinger CW, Elfline M, Luke C, Siahaan TJ, Jaffer FA, Wakefield TW, Henke PK. Endotoxaemia-augmented murine venous thrombosis is dependent on TLR-4 and ICAM-1, and potentiated by neutropenia. Thromb Haemost. 2017; 117: 339–48. 10.1160/TH16-03-0218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Blokhin IO, Lentz SR. Mechanisms of thrombosis in obesity. Curr Opin Hematol. 2013; 20: 437–44. 10.1097/MOH.0b013e3283634443. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.