

Abstract
A dopamine D2 receptor mutation was recently identified in a family with a novel hyperkinetic movement disorder. Compared to the wild type D2 receptor, the novel allelic variant D2-I212F activates a Gαi1β1γ2 heterotrimer with higher potency and modestly enhanced basal activity in human embryonic kidney (HEK) 293 cells, and has decreased capacity to recruit arrestin3. We now report that omitting overexpressed G protein-coupled receptor kinase-2 (GRK2) decreased the potency and efficacy of quinpirole for arrestin recruitment. The relative efficacy of quinpirole for arrestin recruitment to D2-I212F compared to D2-WT was considerably lower without overexpressed GRK2 than with added GRK2. D2-I212F exhibited higher basal activation of GαoA than Gαi1, but little or no increase in the potency of quinpirole relative to D2-WT. Other signs of D2-I212F constitutive activity for G protein-mediated signaling, in addition to basal activation of Gαi/o, were enhanced basal inhibition of forskolin-stimulated cyclic AMP accumulation that was reversed by the inverse agonists sulpiride and spiperone and a ~4-fold increase in the apparent affinity of D2-I212F for quinpirole, determined from competition binding assays. In mouse midbrain slices, inhibition of tonic current by the inverse agonist sulpiride in dopamine neurons expressing D2-I212F was consistent with our hypothesis of enhanced constitutive activity and sensitivity to dopamine relative to D2-WT. Molecular dynamics simulations with D2 receptor models suggested that an ionic lock between the cytoplasmic ends of the third and sixth α-helices that constrains many G protein-coupled receptors in an inactive conformation spontaneously breaks in D2-I212F. Overall, these results confirm that D2-I212F is a constitutively active and signaling-biased D2 receptor mutant, and also suggest that the effect of the likely pathogenic variant in a given brain region will depend on the nature of G protein and GRK expression.
Keywords: dopamine, D2 receptor, allelic variant, constitutive activity, biased signaling, G protein, arrestin
Graphical Abstract
INTRODUCTION
The dopamine D2 receptor is a G protein-coupled receptor that signals through both Gαi/o and arrestin to regulate movement and motivated behavior (1–3). The D2 receptor is a target of virtually all antipsychotic drugs currently in use, and also a frequent drug target in the treatment of movement disorders such as Parkinson’s disease and chorea (4, 5). The D2 receptor has long (D2L) and short (D2S) splice variants; if and in what way the splice variants are functionally distinct is an active area of research (6, 7).
We recently described a four-generation family with an autosomal dominant genetic disorder characterized by chorea and cervical dystonia, in which affected family members carry the novel D2 receptor missense variant DRD2 (c.634A>T;p.I212F) (8). Ile212 (Ile2125.61 according to the Ballesteros-Weinstein numbering scheme)(9) is in the cytoplasmic extension of the 5th transmembrane α-helix, at the N-terminus of the D2 receptor 3rd cytoplasmic loop. A deep mutational analysis of the β2-adrenoceptor identified position 5.61 as being one of the top four mutationally intolerant positions at the β2-Gαs interface, and also in a part of the receptor where many mutations are activating or inactivating (10). Mutations introduced in this region of the D2 receptor decrease the binding of at least three D2 receptor-interacting proteins: arrestin (11, 12), calmodulin (13), and S100B (62, 63). Our initial studies demonstrated that recruitment of arrestin by D2L/S-I212F in human embryonic kidney (HEK) 293 cells is decreased compared to wild type D2L/S (D2 L/S-WT), whereas D2L/S-I212F activation of a Gαi1β1γ2 heterotrimer and inhibition of cAMP accumulation are enhanced (8).
G protein-coupled receptor (GPCR) kinases (GRKs) facilitate arrestin recruitment by phosphorylating serine and threonine residues on the intracellular domains of GPCRs, typically leading to receptor desensitization, internalization, and either degradation or resensitization, and also promoting arrestin-mediated signaling (14). GRK is frequently co-transfected in cellular studies of arrestin recruitment to maximize the signal. GRK2/3 are ubiquitously expressed GRKs that are the major subtypes interacting with the D2 receptor (15–17). Thus, our previous arrestin recruitment studies were performed with overexpressed GRK2.
Similarly, we assessed D2 receptor activation of a G protein heterotrimer containing Gαi1 even though the D2 receptor activates both Gαi and Gαo (18). Gαo is the most abundant Gα subunit in mammalian brain, comprising about 1% of total membrane protein (19). Furthermore, Gαo knockout mice have greatly decreased dopamine-stimulated GTPγS binding and a complete loss of GTP-sensitive dopamine binding in brain, suggesting that Gαo contributes importantly to dopamine signaling (20). Both Gαi and Gαo mediate D2 receptor signaling in brain, with the contribution of specific subtypes varying among brain regions (21).
We now report that D2L/S-I212F receptors have a more stringent requirement than D2-WT for GRKs, so that the novel allelic variant had a more profound loss of arrestin recruitment, compared to D2-WT, in the absence of overexpressed GRK2 than when the kinase was overexpressed. We also describe the effect of the mutation on D2 receptor activation of GαoA, which differed from Gαi1 activation in effects on both agonist potency and basal activity. Furthermore, the mutation increased constitutive inhibition of cyclic AMP accumulation in HEK293 cells and increased the apparent affinity of quinpirole for the D2 receptor. In midbrain dopamine neurons expressing D2-I212F, photoactivated sulpiride inhibited a substantial tonic current, consistent with both the constitutive activity and enhanced agonist potency suggested by studies of the novel variant in HEK293 cells. Molecular dynamics (MD) simulations indicate that these effects of the mutation are associated with the breaking of an “ionic lock” that constrains many unliganded GPCRs in an inactive conformation.
RESULTS AND DISCUSSION
Arrestin3 recruitment by D2-I212F depends heavily on GRK2.
We previously investigated the ability of D2S/L-I212F receptors to recruit arrestin3 under the most favorable conditions by overexpressing GRK2, which enhances arrestin recruitment to the D2 receptor (15). We reported that recruitment of arrestin3 by D2S/L-I212F is reduced by ~30–50% compared to D2S/L-WT receptors, whereas the potency of quinpirole is modestly enhanced at D2S/L-I212F (8). We now describe arrestin3 recruitment in the absence of GRK2. HEK293 cells were transiently co-transfected with D2S/L-WT or D2S/L-I212F fused with RLuc8 (BRET donor) and mVenus-tagged arrestin3 (BRET acceptor). Previous results with overexpressed GRK2 are shown for comparison. Quinpirole-induced recruitment of arrestin3 by both D2 receptor splice variants was substantially decreased for D2L/S-I212F with or without overexpressed GRK2 (Fig. 1A and B; Table 1), whereas the potency of quinpirole was modestly increased for all conditions compared to D2-WT (Table 1). The mutation-induced reduction in maximal recruitment of arrestin was considerably larger in the absence of expressed GRK2 for both splice variants. Thus, with added GRK2, Emax for D2L/S-I212F was decreased by 44% (D2L) or 27% (D2S), but in the absence of overexpressed GRK2, Emax for D2L/S-I212F was decreased by 73% (D2L) and 64% (D2S) compared to the corresponding condition for D2-WT (Table 1). In contrast, omitting GRK2 decreased the potency similarly for all variants, ranging from a 4.2-fold decrease for D2s-WT to a 6.7-fold decrease for D2L-I212F (Table 1).
Figure 1.
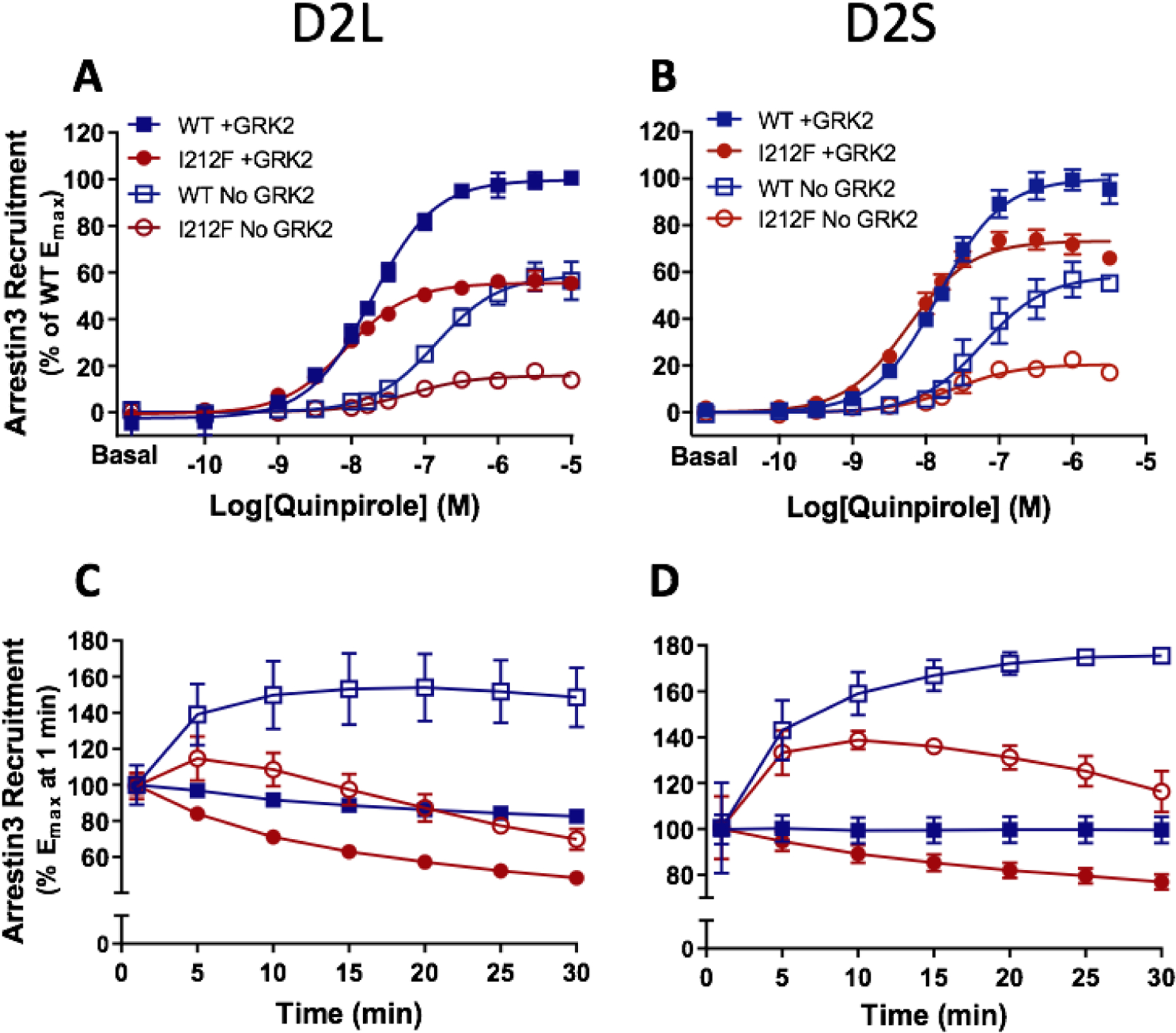
Dose-response curves for quinpirole-induced arrestin3 recruitment mediated by D2L/S-WT and D2L/S-I212F. Arrestin3 recruitment was measured in HEK293 cells co-transfected with GRK2 (+ GRK2) or nonspecific plasmid DNA (No GRK2). Values plotted are the means ± SD of 3–4 independent experiments performed in quadruplicate. A and B, quinpirole concentration-response curves measured at 10 min. Data from each independent experiment were normalized by subtracting the baseline and expressed as a percentage of maximum arrestin3 recruitment by D2-WT+GRK2. Data for + GRK2 are from the dataset described in van der Weijden et al. (8), where results were shown after 20 min of agonist stimulation. C and D, change in Emax values over 30 min, with each condition normalized to Emax for that condition at 1 min. Data for + GRK2 were previously described in van der Weijden et al. (8).
Table 1.
Arrestin recruitment: requirement for overexpressed GRK2
| Receptor | Arrestin Recruitment | |||
|---|---|---|---|---|
| + GRK2a | No GRK2 | |||
| −LogEC50 | Emax (% of WT+GRK2) |
−LogEC50 | Emax (% of WT+GRK2) |
|
| D2L-WT | 7.69 ± 0.05 | 100 ± 2 | 6.85 ± 0.05††† | 59 ± 4††† |
| D2L-I212F | 8.06 ± 0.03** | 56 ± 1*** (−44%) | 7.22 ± 0.08**,††† | 16 ± 1***,††† (−73%) |
| D2S-WT | 7.83 ± 0.01 | 100 ± 3 | 7.20 ± 0.13††† | 59 ± 2††† |
| D2S-I212F | 8.24 ± 0.04** | 73 ± 2*** (−27%) | 7.59 ± 0.09*,††† | 21 ± 0.4***,††† (−66%) |
Quinpirole potency is shown as −logEC50. Emax was calculated by subtracting basal response from maximal response at 10 min after adding the substrate coelenterazine h, and is shown as the percentage of D2-WT with added GRK2. For D2-I212F, the percent reduction compared to the corresponding D2-WT Emax is included in parentheses. N = 3–4 independent experiments for each condition. Data are presented as mean ± SEM.
From the dataset described in van der Weijden et al. (8), except after 10 min instead of 20 min of agonist stimulation. Statistical differences were calculated by 2-way ANOVA followed by Turkey’s post-hoc test (*p<0.05, **p<0.01, ***p<0.001 compared to D2-WT; †††p<0.001 compared to the corresponding +GRK2 condition).
In the presence of overexpressed GRK2, maximal arrestin recruitment peaked by the first measurement, (1 min after adding coelenterazine h, which was approximately 4 minutes after addition of quinpirole), whereas maximal recruitment was delayed without overexpressed GRK2, particularly for D2S/L-WT (Fig. 1C and D). Emax decreased more rapidly for D2L/S-I212F than for D2L/S-WT and more rapidly without overexpressed GRK2 than with GRK2 transfection. Thus, a significant interaction among the three factors of time, GRK2 condition, and genotype was determined by 3-way RM ANOVA (D2L: F (6, 60) = 17.77, p < 0.0001; D2S: F (6, 60) = 6.689, p < 0.0001). This was followed by 2-way RM ANOVAs to assess the interaction between genotype and time (D2L + GRK: F (6, 24) = 69.15, p < 0.0001; D2L No GRK: F (6, 36) = 81.08, p < 0.0001; D2S + GRK: F (6, 36) = 118.9, p < 0.0001; D2S No GRK: F (6, 24) = 12.49, p < 0.0001) and between GRK treatment and time (D2L-WT: F (6, 30) = 103.7, p < 0.0001; D2S-WT: F (6, 30) = 71.26, p < 0.0001; D2L-I212F: F (6, 30) = 16.62, p < 0.0001; D2S-I212F: F (6, 30) = 19.16, p < 0.0001). Consistent with our previous report (8), expression of D2L/S-I212F was only 35–40% of D2L/S-WT (Table S1).
Decreased arrestin3 recruitment by D2-I212F was not due to lower receptor expression.
Based on unpublished data mentioned previously (8), we considered it unlikely that the reduced arrestin recruitment by D2L/S-I212F was simply due to lower receptor expression. Furthermore, the mutation-induced instability of the interaction with arrestin (Fig. 1C and D) and the greater dependence of arrestin recruitment to D2L/S-I212F on GRK2 are difficult to explain as simply due to lower receptor number. Nevertheless, to confirm that reduced arrestin recruitment by D2-I212F was not a consequence of lower receptor expression, we repeated these experiments with D2L under conditions where the wild type and mutant variants were expressed at similar levels and observed a similar mutation-induced decrease in Emax (Fig. S1). At this lower level of D2 receptor expression, GRK2 overexpression had no effect on Emax for D2-WT but continued to regulate quinpirole potency at D2-WT.
Contribution of endogenous GRK to arrestin3 recruitment.
To determine if endogenous GRK2/3 contributes significantly to arrestin recruitment in this assay, we repeated the experiments above with D2L-WT and D2L-I212F, adding a condition in which cells were pretreated with the GRK2/3 inhibitor Compound 101 (Cmpd101). In the absence of Cmpd101 (Table S2), results were indistinguishable from those presented above and replicate prior results for D2L-WT and D2L-I212F with overexpressed GRK2 (Table 1). Inhibiting endogenous GRK2/3 significantly decreased the maximal response for both allelic variants with (Fig. S2A and C) or without (Fig. S2B and C) overexpressed GRK2. Nevertheless, maximal arrestin recruitment by D2L-I212F was always less than the corresponding condition for D2L-WT (Table S2). In contrast, quinpirole potency was decreased by either omitting overexpressed GRK2 or adding Cmpd101, but there was no detectable additivity (Fig. S2D; Table S2).
GRK2 has both phosphorylation-dependent and –independent effects on D2 receptor function (17, 22). Inhibition of arrestin recruitment by the active-site inhibitor Cmpd101 (23) may suggest that at least some of the observed effects of GRK2 require D2 receptor phosphorylation, although it is notable that translocation of GRK2 to the μ-opioid receptor can be inhibited by Compd101 (24, 25). Observed effects of overexpressed GRK2 despite the presence of Cmpd101 may reflect phosphorylation-independent processes.
D2-I212F receptor expression increased basal GαoA protein-activation.
We assessed G protein activation using a Gαo energy donor (GαoA-91-RLuc8), a Gβ1/Gγ2 acceptor (mVenus-Gβ1γ2), and D2S/L-WT or D2S/L-I212F transiently expressed in HEK293 cells. Quinpirole produced a concentration-dependent increase in GαoA protein activation for both D2S/L-I212F and D2S/L-WT receptors (Figure 2A–B). For D2L, no significant difference in the potency of quinpirole at D2L-WT and D2L-I212F receptors was observed, whereas quinpirole was slightly but significantly more potent at D2S-WT (1 nM) than at D2S-I212F (2 nM; Table 2). On the other hand, basal GαoA activation by D2L-I212F (43% of maximal stimulation) or D2S-I212F (57%) was markedly higher than for D2S/L-WT (set as 0%; Fig. 2A–B; Table 2). Enhanced basal activity was observed despite lower expression of D2S/L-I212F than D2S/L-WT (Table S1).
Figure 2.
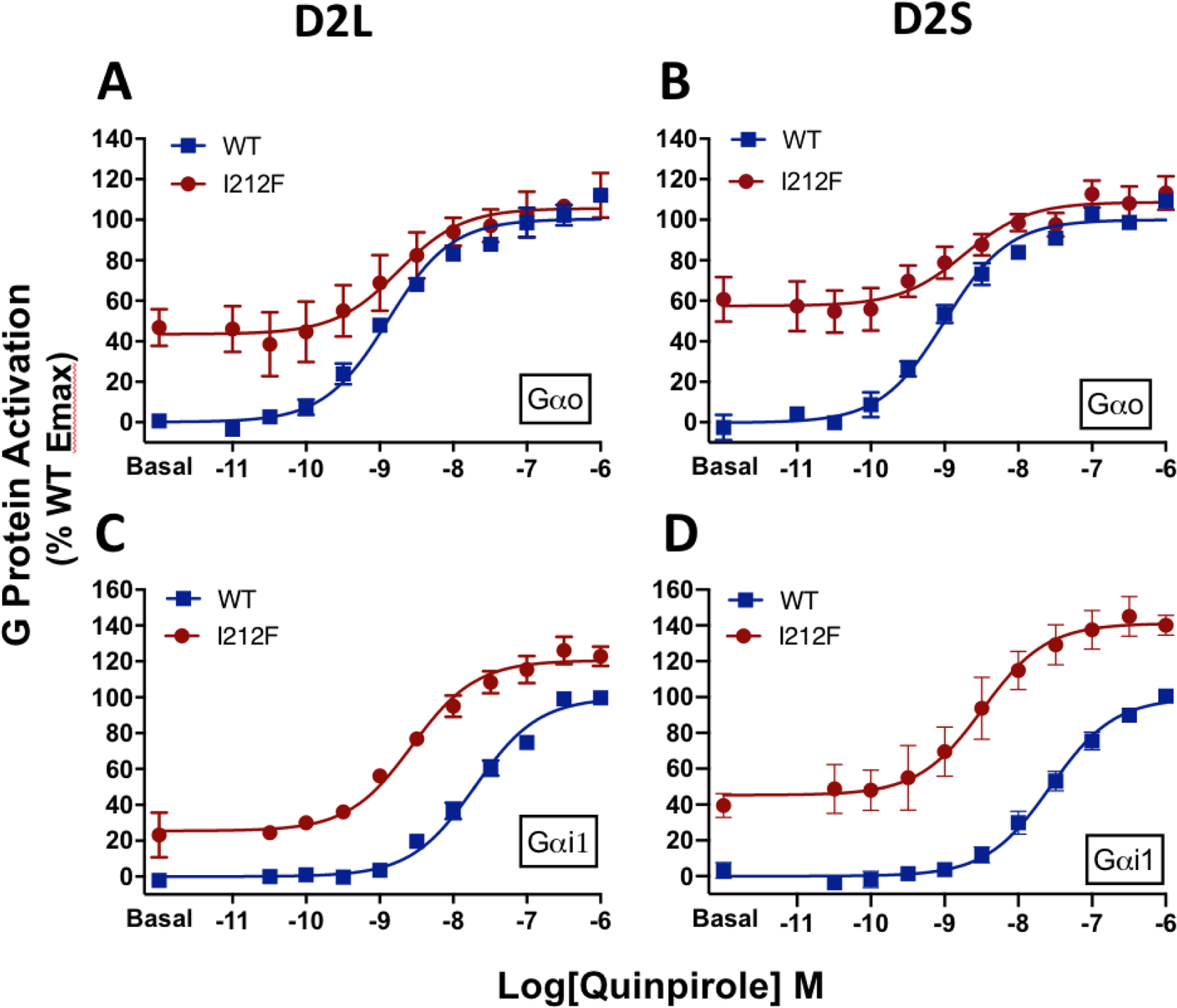
Concentration-response curves for Gαi/o protein activation mediated by D2-WT and D2-I212F in response to stimulation with quinpirole. Results are expressed as the percentage of maximum G protein activation by D2-WT, measured 10 min after adding coelenterazine h. A, Activation of GαoA by D2L-WT/I212F, B, Activation of GαoA by D2S-WT/I212F, C, Activation of Gαi1 by D2L-WT/I212F, and D, Data from van der Weijden et al. (8) for activation of Gαi1 by D2SWT/I212F. Values plotted represent means ± SD of three (panel C) or four (panels A, B, D) independent experiments performed in quadruplicate.
Table 2.
Gα protein activation in HEK293 cells
| Receptor | −LogEC50 | Basal Activity (% of WT Max) |
||
|---|---|---|---|---|
| Gαi1 | GαoA | Gαi1 | GαoA | |
| D2L-WT | 7.7 ± 0.05 | 8.9 ± 0.03 | 0 ± 0.01 | 0 ± 0.02 |
| D2L-I212F | 8.6 ± 0.04*** | 8.8 ± 0.09 | 25 ± 3** | 43 ± 6*** |
| D2S-WT | 7.6 ± 0.05a | 9.0 ± 0.02 | 0 ± 0.01a | 0 ± 0.02 |
| D2S-I212F | 8.4 ± 0.02a*** | 8.7 ± 0.05** | 35 ± 6a*** | 57 ± 5*** |
Quinpirole potency is shown as −logEC50. Basal activity for D2L/S-I212F is expressed as a percentage of the respective D2-WT maximal response. Data are presented as mean ± SEM of three (Gαi1-D2L) or four (Gαo-D2L/S) independent experiments performed in quadruplicate.
Data are from van der Weijden et al. (8). Statistical differences were calculated by 2-way ANOVA followed by Turkey’s post-hoc test (**p<0.01, ***p<0.001 compared to D2-WT).
Because the effect of the I212F mutation on GαoA protein activation differed in several respects from what we observed previously using Gαi1 (8), we repeated those experiments with D2L-WT and D2L-I212F (Fig. 2C; previously published results for D2S shown in Fig. 2D for comparison). Whereas quinpirole potency for activating GαoA was not substantially changed by the I212F mutation, we confirmed our previous observation that the potency of D2L-I212F for activating Gαi1 (3 nM) is markedly increased compared to D2L-WT (21 nM; Fig. 2C and 3A). Basal activity of Gαi1 was enhanced by 25% of WT Emax in cells expressing D2L-I212F (Fig. 2C). This enhanced basal activity associated with D2-I212F expression was significantly lower than that observed for GαoA for both splice variants (Fig. 3B).
Figure 3.
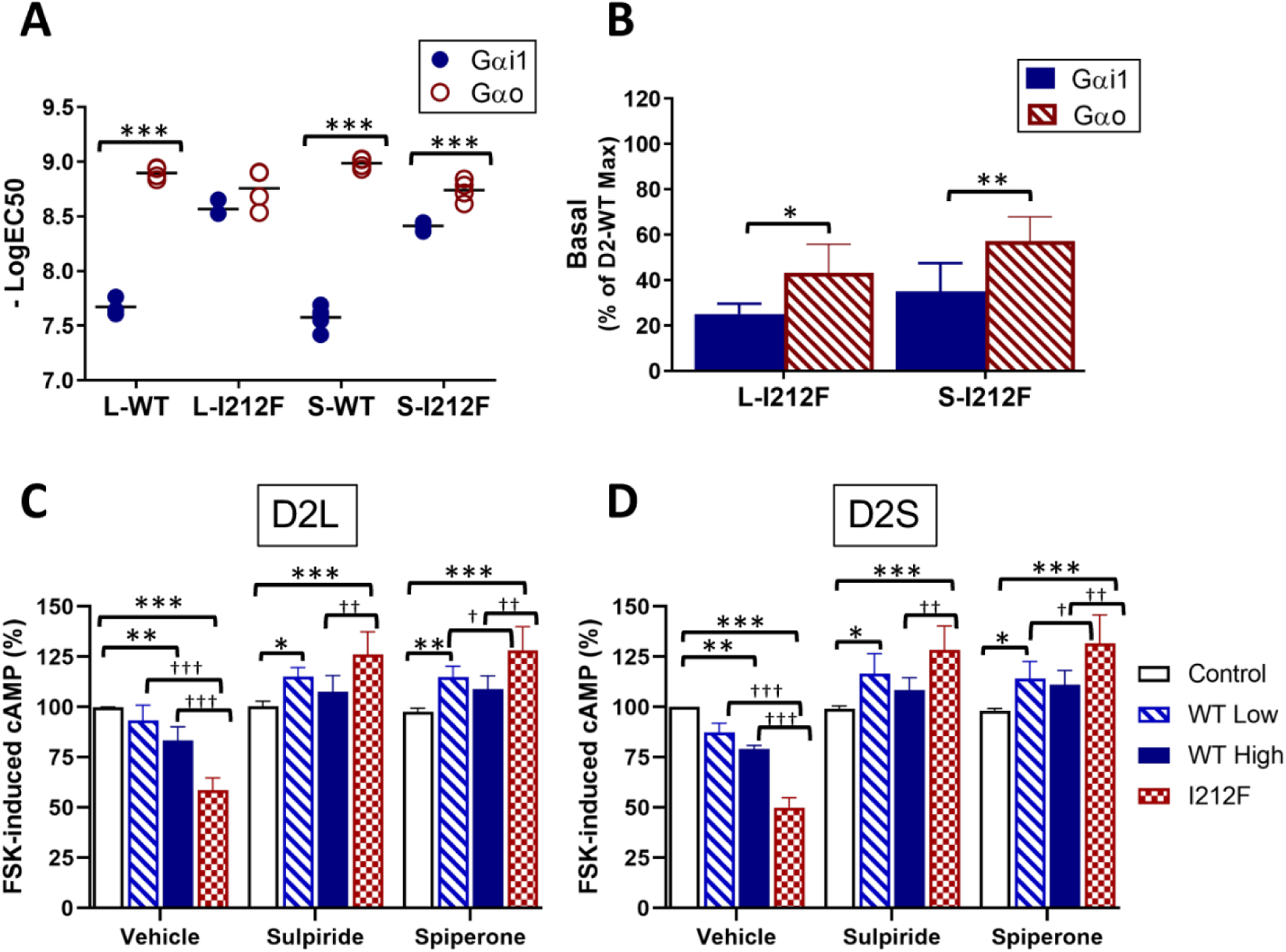
Increased constitutive activity of D2-I212F. A, B Concentration-response curves for Gαi/o protein activation mediated by D2-WT and D2-I212F were analyzed by nonlinear regression to determine quinpirole potency (A), expressed as the −LogEC50, and activation in the absence of quinpirole (B), expressed as the percentage of Emax for D2-WT. Data are from Table 2. Statistical differences were determined as described in Table 2 (*p<0.05, **p<0.01, ***p<0.001). C, D Cyclic AMP accumulation was measured in the presence of 10 μM forskolin (FSK) in HEK293 cells transfected with the cyclic AMP biosensor CAMYEL and either control plasmid DNA (control), a high (0.5 μg, WT High) or low (0.2 μg, WT Low) amount of D2L/S-WT DNA, or D2 L/S-I212F plasmid DNA (0.5 μg, I212F). Measurements were taken 10 min after addition of either vehicle, sulpiride (10 μM) or spiperone (1 μM), FSK (10 μM) and coelenterazine h. Results are expressed as a percentage of cyclic AMP accumulation by control cells treated with the inverse agonist vehicle. Values plotted are mean ± SD of four independent experiments performed in sextuplicate. Statistical differences were determined by 2-way ANOVA followed by Turkey’s post-hoc test (*p<0.05, **p<0.01, ***p<0.001 compared to the corresponding control condition; †p<0.05, ††p<0.01, †††p<0.001 compared to D2-WT).
Quinpirole was considerably more potent at GαoA than at Gαi1 for D2-WT, consistent with prior work (26). The G protein subtype-specific effect of the mutation on agonist potency is particularly interesting in light of recent findings that Gαo mediates a relatively high-affinity response to dopamine in the mouse nucleus accumbens that is eliminated by repeated treatment with cocaine (21). This is in contrast to a lower-affinity, cocaine treatment-insensitive response in the dorsal striatum that is mediated by Gαi. In our results, the mutation-induced shift in potency of quinpirole at Gαi1 eliminated the difference between the G protein subtypes (Fig. 3A; Table 2). Thus, mice expressing D2-I212F might display higher sensitivity responses to dopamine in both nucleus accumbens and dorsal striatum, responses that would perhaps be unaffected by repeated cocaine treatment.
Constitutive inhibition of cyclic AMP accumulation.
Increased basal activation of G proteins by D2-I212F could be indicative of a higher constitutive activity than D2-WT. To test this hypothesis, we measured the ability of D2 receptors to inhibit forskolin-stimulated cyclic AMP accumulation in the absence of agonist. HEK293 cells were transiently co-transfected with the BRET-based cylic AMP sensor, CAMYEL (27) and D2L/S-WT or D2L/S-I212F. Compared to control cells, cells transfected with a higher amount of D2L/S-WT plasmid DNA (0.5 μg) or D2L/S-I212F showed reductions in cyclic AMP accumulation that were greater for D2-I212F (41–50%) than for D2-WT (17–21%) (Fig. 3C–D; D2L: 83 ± 7% of control for WT High vs. 59 ± 6% for D2L-I212F; D2S: 79 ± 2% of control for WT High vs. 50 ± 5% for D2S-I212F). Preincubation with either of the inverse agonists sulpiride and spiperone not only reversed the constitutive inhibition of cyclic AMP accumulation but also yielded significantly enhanced cyclic AMP levels that were highest for D2L/S-I212F (Fig. 3C–D; D2L-I212F: 126 ± 11% of control for sulpiride and 128 ± 12% of control for spiperone; D2S-I212F: 128 ± 12% for sulpiride and 132 ± 14% for spiperone). We hypothesize that increased FSK-stimulated cyclic AMP accumulation in the presence of inverse agonists reflects heterologous sensitization of adenylyl cyclase resulting from prolonged constitutive activation of Gαi/o by D2-I212F (28, 29).
Enhanced affinity of D2-I212F for quinpirole.
Receptor constitutive activity is commonly reflected in increased affinity for agonists (30–32). We carried out competition binding assays to compare the apparent affinity of D2-WT and D2-I212F for quinpirole in membranes prepared from HEK293 cells stably expressing the receptors (Fig. S3). In four experiments, the geometric mean for quinpirole Ki decreased from 1.5 μM to 0.4 μM (D2L-WT and D2L-I212F, respectively; p = 0.0027) and from 1.9 to 0.5 μM (D2S-WT and D2S-I212F, respectively; p = 0.0005). Thus, the affinity of D2L/S-I212F for quinpirole was increased roughly 4-fold compared to D2L/S-WT.
Altered D2-I212F receptor-GIRK currents in mouse midbrain slices.
To characterize effects of the mutation in a native environment for brain D2 receptors, we used AAV-mediated expression of DIO-Flag-D2S-WT or -I212F to restore D2 receptor function in dopamine neurons of auto-D2-KO mice and characterized D2 receptor activation of G protein-regulated inward-rectifying potassium channels (GIRKs). We used D2S for these studies because of evidence that this splice variant might contribute more than D2L to autoreceptor activity (6, 7). The effect of the mutation on D2 receptor function in HEK293 cells was qualitatively similar for both splice variants. Values for both basal G protein activation and maximal arrestin recruitment were greater compared to D2L/S-WT for D2S-I212F than for D2L-I212F, although for most experiments there was neither a significant main effect of splice variant nor an interaction with genotype. The only statistically significant difference among the data presented here was a greater relative efficacy of D2S-I212F for recruitment of arrestin in the presence of GRK2 compared to D2L-I212F (p = 0.001).
In midbrain slices prepared two weeks after AAV injection, the GIRK response to iontophoretically applied dopamine (Fig. 4A) was smaller in amplitude (Fig. 4B), slower in rise time, and longer in duration (Fig. 4C) for D2S-I212F-expressing neurons compared to D2S-WT transduced controls. Despite the smaller peak amplitude, the prolonged duration of the response meant that total charge transfer was higher for D2S-I212F (409 ± 92 pC) than for D2s-WT (207 ± 29 pC; Fig. 4D).
Figure 4.
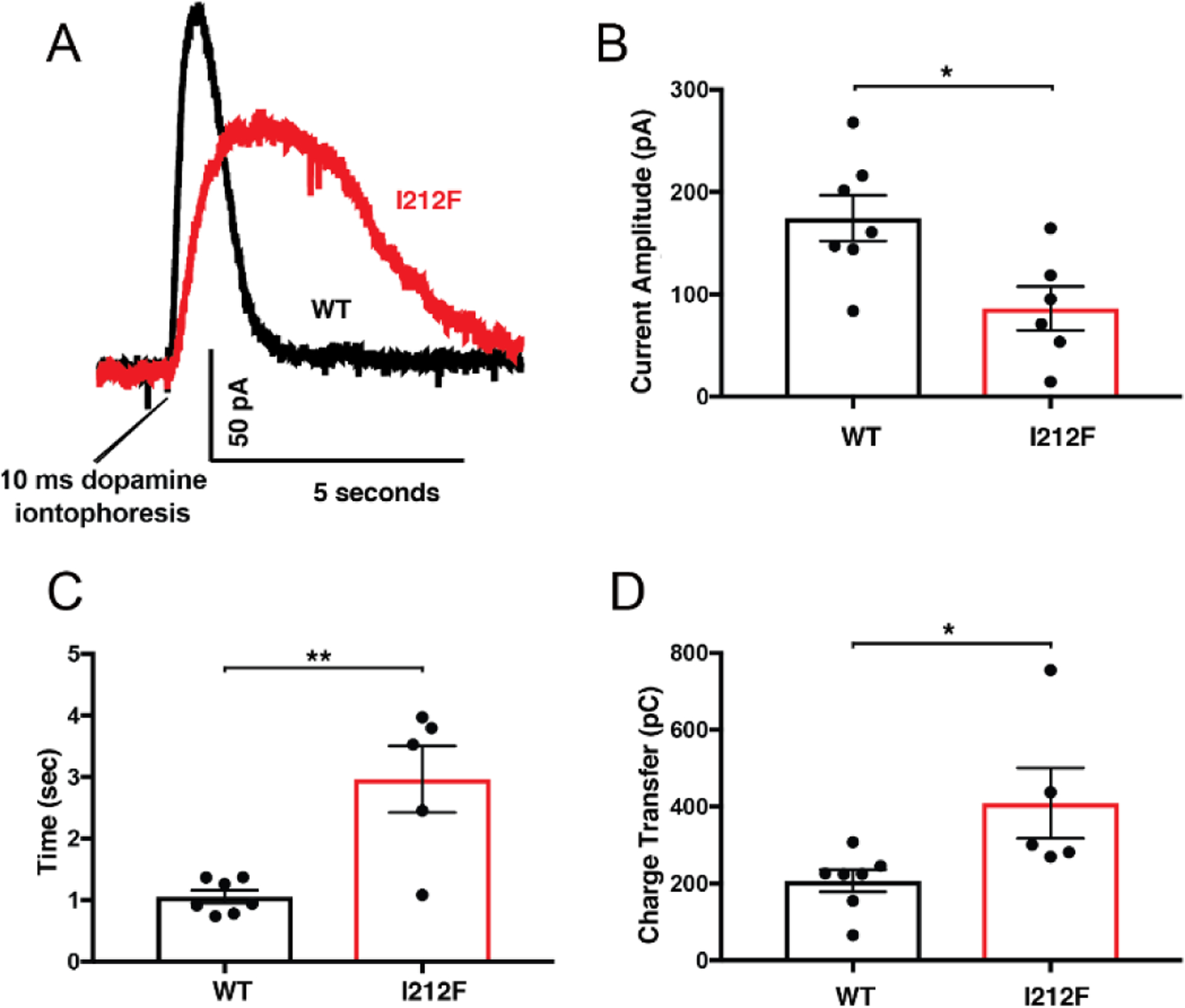
Activation of GIRK currents by dopamine iontophoresis was assessed in mouse midbrain slices. AAV-DIO-D2S-WT or -D2S-I212F was used to restore D2 receptor expression in dopamine neurons of auto-D2-KO mice. A, representative outward currents in response to iontophoresis of dopamine (1 M) for 10 msec. Mean ± SEM is shown for (B) current amplitude, (C) peak half-width, and (D) charge transfer. The number of cells differs among panels for D2-I212F because kinetics in the lowest amplitude response in panel B could not be accurately resolved. Student’s t-test: *p < 0.05, **p < 0.01.
Photolytic release of sulpiride from CyHQ-sulpiride (33) produced a small inhibition of a tonic GIRK current in cells expressing D2-WT (−9 pA), and a much larger inhibition in cells expressing D2-I212F (−62 pA; Fig. 5A and B). The tonic current could reflect constitutive activation of G proteins, similar to what we observed in HEK293 cells (Fig. 2 and Fig. 3D), or it could reflect the heightened sensitivity of D2-I212F to agonist that is suggested by data for activation of Gαi1 (Fig. 2C–D) and inhibition of cAMP accumulation (8). To distinguish between these possibilities, we treated slices with reserpine to deplete endogenous dopamine. Reserpine treatment abolished or greatly decreased the response to sulpiride in cells expressing D2-WT or D2-I212F, respectively (Fig. 5B and C), indicating that most of the tonic current is due to endogenous dopamine to which D2-I212F is more sensitive, but that D2-I212F also displayed some constitutive activity in the presumed absence of dopamine. Because the mutation increased agonist potency for activation of Gαi1 but not Gαo in HEK293 cells, this may indicate that D2 receptor signaling in SNc dopamine neurons is mediated by Gαi. Interestingly, the current decay in response to sulpiride photoactivation was slower for D2-I212F than for D2-WT even following reserpine treatment and the presumed absence of dopamine (Fig. 5D), suggesting that relaxation of the receptor or uncoupling of the signaling machinery is inherently slower for the mutant receptor.
Figure 5.
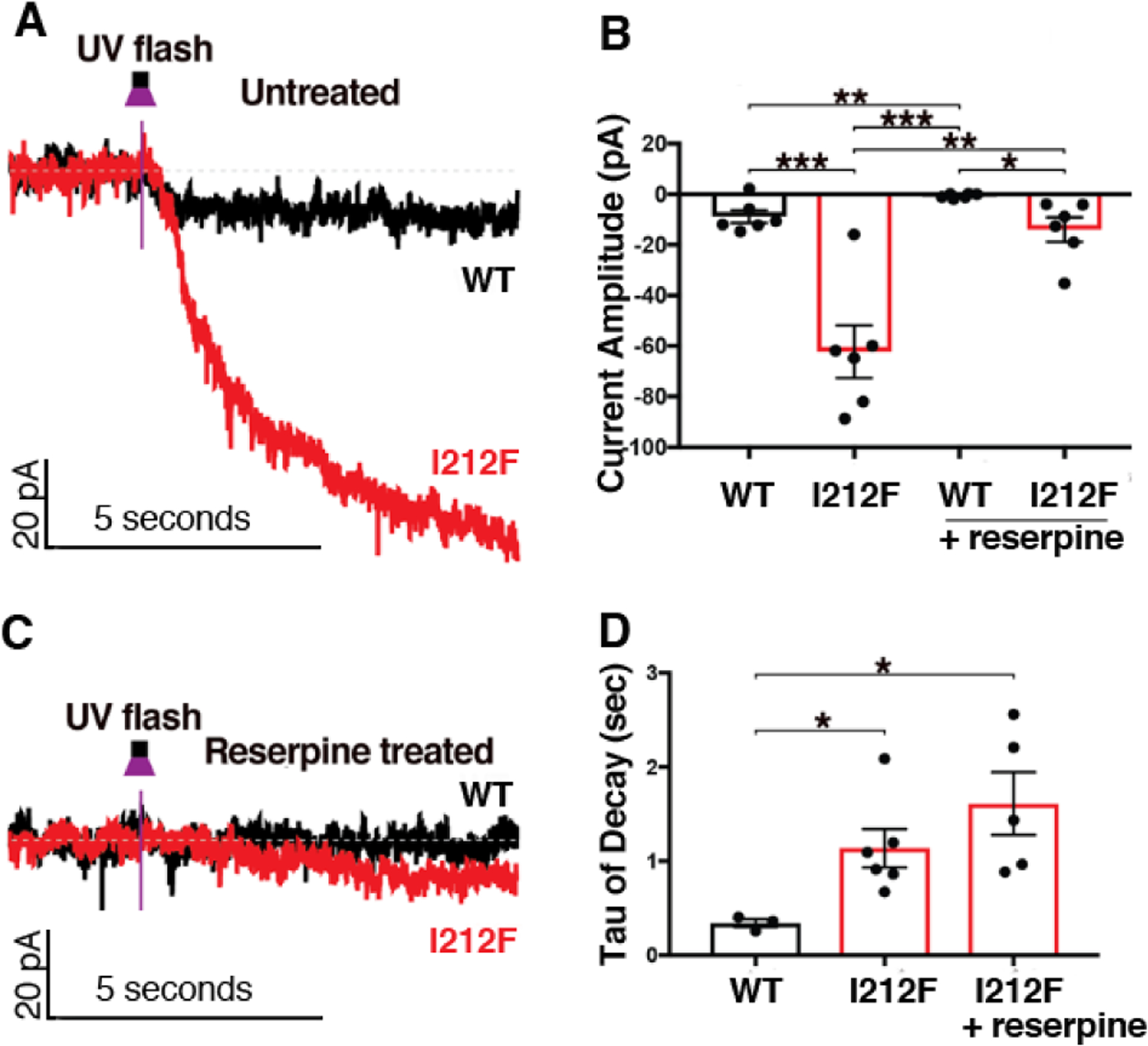
AAV-DIO-D2S-WT or -D2S-I212F was used to restore D2 expression in dopamine neurons of auto-D2-KO mice. CyHQ-sulpiride (5 μM) was circulated over a midbrain slice, and photolysis was by means of a 50 msec flash (365 nm) from a 6.5 mW LED light. The left panels depict representative traces for untreated slices (A) and slices incubated with reserpine to deplete endogenous dopamine (C). The right panels depict the mean ± SEM of the decreased current amplitude (B) and the rate of decay of the current after photorelease of sulpiride (D) in control slices and in slices pretreated with reserpine. It was not possible to calculate a decay rate for reserpine-treated slices from mice expressing D2-WT. For some conditions the number of cells differs between panels B and D because kinetics could not be accurately resolved in the lowest amplitude responses in panel B. Student’s t-test: *p < 0.05, **p < 0.01, ***p < 0.001.
Molecular Dynamics Simulations with D2 Receptor Homology Models.
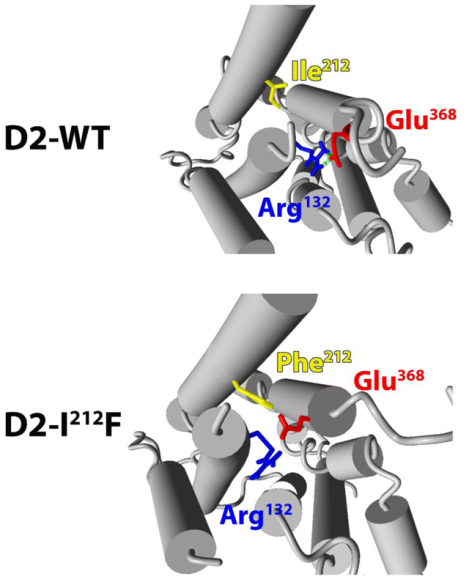
One feature of many GPCRs is an “ionic lock” between an Arg residue at the cytoplasmic end of the 3rd transmembrane domain (TM3, Arg132 in the D2 receptor) and a Glu residue at the cytoplasmic end of TM6 (Glu368 in the human D2L receptor) (34, 35). These residues are Arg3.50 and Glu6.30 in the Ballesteros-Weinstein index (9). This ionic lock contributes to maintaining the unliganded receptor in an inactive conformation; the lock is broken in the agonist-activated receptor and, conversely, breaking the lock frequently creates a constitutively active receptor (34, 36, 37). We used crystal structures of the D2 receptor (38) and other Gαi/o-coupled GPCRs to build homology models of the human D2 receptor in both inactive and active conformations, with either Ile212 or Phe212. As depicted in Figure 6, the side chains of Arg132 and Glu368 are in sufficient proximity to form an ionic bond or salt bridge in both inactive models, but are too distant for salt bridge formation in both active models. After MD simulations for 15 nsec, the Glu368 side chain separated from Arg132 in the inactive D2-I212F model, breaking the ionic lock (Fig. 6 and 7).
Figure 6.
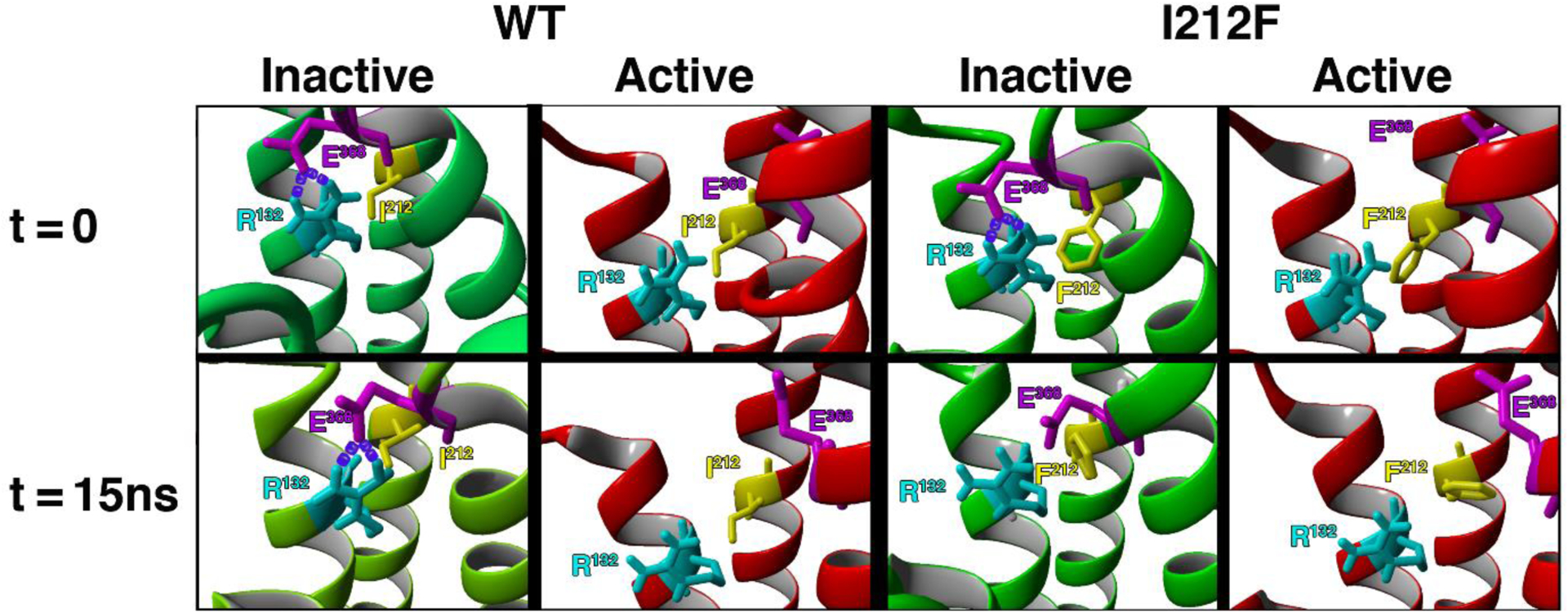
The eight panels show TM3–6 ionic lock residues Arg132 (light blue) and Glu368 (magenta), as well as the variant residue Ile/Phe212 (yellow). Models are shown before (t=0) and after (t=15ns) MD simulations for 15 nsec. At t=0, the distances between OE1 of Glu368 and HH1 and HH2 of Arg132 are short enough to form salt bridges (purple) in both inactive models, but are too far apart in both active models. After 15 nsec MD simulations the WT model is essentially unchanged, whereas the presence of Phe212 separates the side chains of Arg132 and Glu368, preventing maintenance of the ionic lock in inactive D2-I212F. The cytoplasmic face is up.
Figure 7.
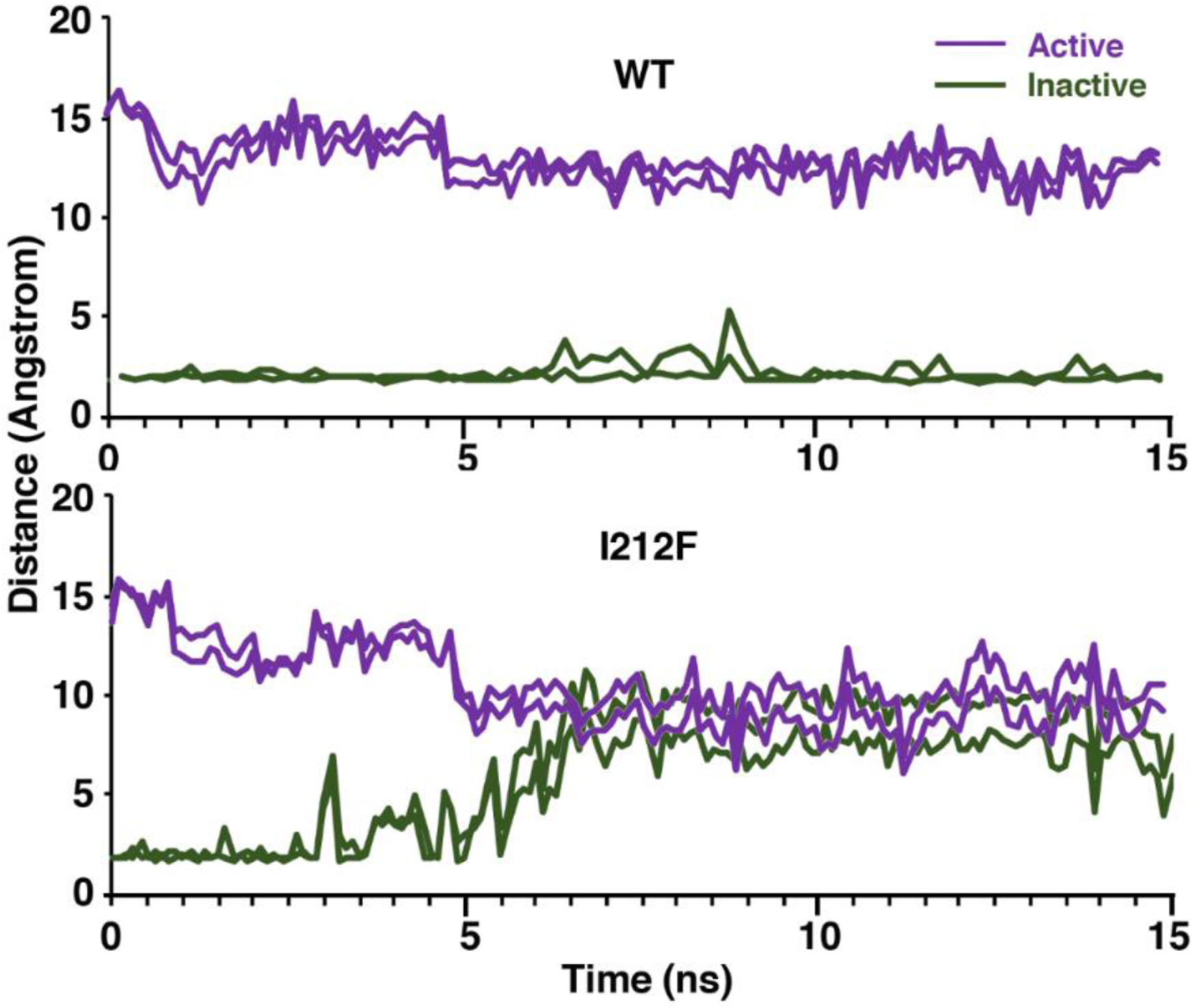
The distances between the atoms that form the two bonds of the ionic lock are shown for all four models during 15 nsec MD simulations. Note that the distances are relatively stable for the active and inactive D2-WT models, whereas the distances increased 6–8 angstroms in the inactive D2-I212F model and decreased ~5 angstroms in the active model.
During the MD simulation with the inactive D2-I212F model (Figure 8), a comparison of snapshots obtained at t=0.5 nsec (left panel) or 7.5 nsec (right panel) shows that initially the Phe212 side chain extends very close to TM3 residue Ser129 (Ser3.47; not shown), whereas Ile212 in the inactive D2-WT model is more distant from TM3 (Figure 6). A steric effect of the close Ser129-Phe212 interaction may provide some of the energy needed to separate the ionic lock residues. As depicted in Figure 8 (right panel), Arg132 appeared to strengthen its interaction with Asp131 (D3.49) likely through ionic interactions (34). Concomitantly, the ionic lock between residues Glu368-Arg132 was disrupted as Glu368 moved away from Arg132 with the rotation and translocation of TM6, perhaps interacting with other residues in TM6. The Phe212 side chain was reoriented towards a possible interaction with Leu216.
Figure 8.
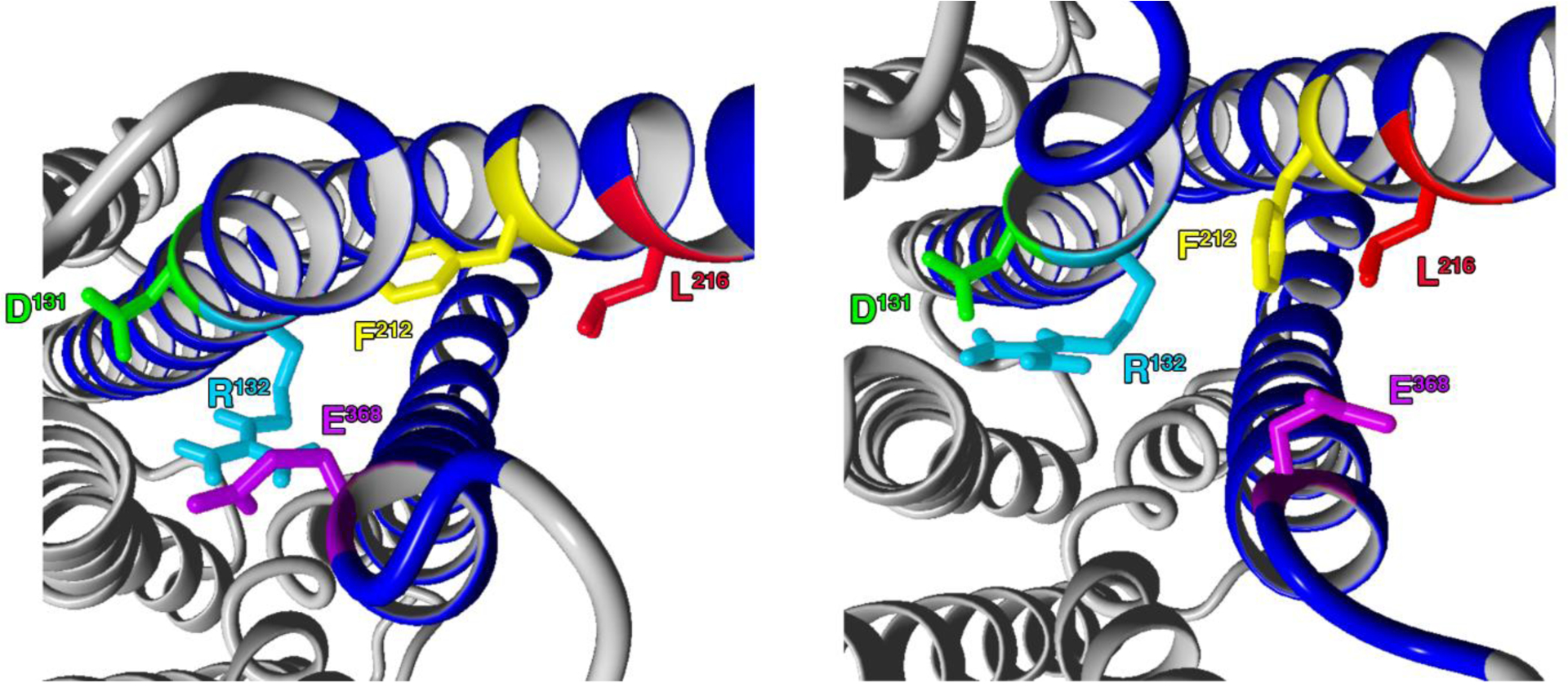
Residues involved in the disruption of the ionic lock are shown at t = 0.5 nsec (left panel) and 7.5 nsec (right panel) during MD simulation with the inactive D2-I212F model. Ballesteros-Weinstein numbering for the colored residues is Asp1313.49 (green), Arg1323.50 (light blue), Phe2125.61 (yellow), Leu2165.65 (red), and Glu3686.30 (magenta).
These results provide a structural rationale for the effects of the mutation on G protein activation; separation of the lock residues would better enable the conformational rearrangement of TM6 that creates space for binding of Gα (34, 35, 39, 40). Activating mutations of the ionic lock residues also render the receptor less stable, which is often reflected in decreased expression (36, 37, 41) as observed here (Table S1).
Constitutive activation of the receptor potentially explains decreased recruitment of arrestin to D2-I212F, particularly if one speculates that enhanced binding of Gα might competitively inhibit binding of arrestin (42–44). However, we have also described concurrent reductions in arrestin recruitment and G protein-mediated signaling for a D2 receptor with a targeted mutation in this part of the receptor (12), in contrast to the reciprocal effects described here, so it may be that distinct mechanisms underlie the observed effects on arrestin and G protein interaction with D2-I212F. For example, abundant data support a model in which arrestin has separate binding determinants for negatively charged phosphorylated residues on the receptor (“phosphorylation sensor”) and for receptor sites that are exposed by receptor activation (“activation sensor”) (45–47). We have proposed that non-natural mutations in this intracellular extension of the fifth α-helical domain selectively affect presentation of the activation sensor (11). Decreased engagement of the activation sensor could increase reliance on the phosphorylation sensor, which might explain why arrestin recruitment by D2-I212F has a greater dependence on GRK2 and GRK2-catalyzed phosphorylation.
CONCLUSIONS
The data presented here are consistent with a model in which substitution of Ile212 with a Phe residue in the dopamine D2 receptor breaks an interhelical salt bridge that constrains the unliganded receptor. As a result, D2-I212F constitutively activates Gαi/o and mediates high-potency agonist activation of at least one Gαi/o subtype in HEK293 cells and in dopamine neurons. D2-I212F is biased toward G protein-mediated signaling, because the enhanced Gαi/o activation was combined with reduced arrestin recruitment that was particularly profound under conditions where GRK2 activity was limited. A hyperactive D2 receptor would be predicted to cause over-inhibition of D2 receptor-expressing medium spiny neurons of the neostriatum and nucleus accumbens. Mice in which the activity of these neurons is genetically inhibited show increased locomotor activity (48). Furthermore, overstimulation of G protein-mediated signaling by the D2 receptor exacerbates, and overexpression of arrestin3 protects against, L-DOPA-induced dyskinesia in mice (49). This D2 receptor variant c.634A>T;p.I212F is carried by patients with a hyperkinetic movement disorder characterized by both chorea and dystonia (8); we speculate that both constitutive activity and G protein bias of D2-I212F contribute to the clinical phenotype, and that an effective treatment should target these characteristics of the receptor.
METHODS
Recombinant cDNA constructs.
All human D2 receptor cDNA constructs contained a signal peptide and a FLAG epitope tag at the receptor N-terminus. The wild type (WT) short (SF-hD2S) and long (SF-hD2L) isoforms of human D2 receptors as well as the SF-hD2L(I212F) and SF-hD2S(I212F) variants in pcDNA3 and the corresponding RLuc8 fusion proteins for arrestin BRET assays were described previously (8). Other plasmids for arrestin3 recruitment BRET assays (human arrestin3 fused to mVenus and human GRK2), for measuring G protein activation (Gαi1-91-RLuc8, V1-Gß1 and V2-Gγ2), and for inhibition of cAMP accumulation (pcDNA3L-His-CAMYEL; ATCC MBA-277) were previously described (12, 27), except for the plasmid GαoA-91-RLuc8 (21, 50) that was obtained from Jonathan Javitch (Columbia University, USA). For animal studies, recombinant adeno-associated viral (AAV8.2) vectors containing a Cre recombinase-dependent double-floxed inverted open reading frame (DIO) for hD2S-WT or -I212F, tagged at their C terminus with a self-cleaving 2A peptide and EGFP were described previously (8) and were produced by Virovek, Inc (Hayward, CA, USA). To generate stable transfected HEK293 cells, pcDNA3.1[SF-hD2S-P2A-EGFP] was obtained from Jonathan Javitch (Columbia University, USA). Plasmids containing wild type and mutated D2L isoforms (pcDNA3.1[SF-hD2L-P2A-EGFP] and pcDNA3.1[SF-hD2L(I212F)-P2A-EGFP], respectively) and mutated D2S isoform (pcDNA3.1[SF-hD2S(I212F)-P2A-EGFP]) were generated by digesting pcDNA3.1[SF-hD2S-P2A-EGFP] with BstEII and PmlI (New England BioLabs, MA, USA). The purified 6.8 kb fragment was ligated to each 782 bp insert generated by BstEII/PmlI digestion of pcDNA3[SF-hD2L] and pcDNA3[SF-hD2L(I212F)], or 695 bp insert of pcDNA3[SF-hD2S(I212F)], using T4 DNA Ligase (New England BioLabs). All new constructs were verified by DNA sequencing at the OHSU Vollum DNA Sequencing Core Facility (Portland, Oregon, USA).
Cell Culture and Transfection Conditions.
HEK293 cells obtained from Caroline Enns (Oregon Health & Science University, USA) were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% FetalClone I serum (FCS; Thermo Fisher Scientific; Waltham, MA, USA) at 37°C in a 5% CO2 atmosphere. New cell cultures were initiated frequently from frozen stocks. Eighteen hours before transfection, HEK293 cells were plated in 100-mm dishes at 60–80% confluence. HEK293 cells were transfected with equal amounts of D2-WT or D2-I212F receptor DNA, except in some arrestin recruitment (Fig S1) and D2-mediated inhibition of cAMP (Fig 3C–D) assays. Transient transfections were performed with polyethylenimine (PEI; MAX 40K reagent, Polysciences, Inc.; Warrington, PA, USA) in Opti-MEM I (Gibco by Life Technologies; Logan, UT, USA). In most cases, two 100-mm petri dishes per condition were transfected to allow carrying out BRET and radioligand binding assays using identically treated cells. Transfections were incubated for 5–6 h at 37°C in the 5% CO2 humidified atmosphere, after which the medium was replaced by fresh DMEM plus 10% FCS. Cells were harvested 48 h post-transfection for BRET studies and frozen for radioligand binding assays.
To study quinpirole affinity of the D2 variants, stable HEK293 cells expressing either SF-hD2L/S-P2A-EGFP or SF-hD2L/S(I212F)-P2A-EGFP were generated by transfecting the respective plasmids with PEI in 12-well plates, as described above. Two days after transfection, cells recovered from each well were plated into two 100-mm dishes in supplemented DMEM containing 500 ug/ml of G-418 (Gold Biotechnology Inc; MO, USA). Colonies were initially screened for EGFP expression by immunoblotting. Then, EGFP-positive clones were screened for Flag tagged-D2 expression by immunoblotting, using a rabbit polyclonal antibody anti-DYKDDDDKC epitope tag (Invitrogen; CA, USA), and radioligand binding assays as described below. Stable transfected cells were maintained in DMEM plus 10% FCS, with 500 ug/ml of G-418.
Bioluminescence Resonance Energy Transfer (BRET) Assays.
For arrestin3 recruitment, cells were co-transfected with plasmids contain mVenus-Arr3 (2.5 μg) and the WT or I212F-mutated D2 receptor fused to RLuc8 (0.25 μg except for Fig. S1, where D2-WT receptor DNA amounts were adjusted to yield similar levels of receptor expression for the allelic variants), with or without hGRK2 (2 μg). For G protein activation, cells were co-transfected with WT or I212F-mutated D2 receptor (0.5 μg), the G protein subunits V1-Gß1 (2 μg) and V2-Gγ2 (2 μg), and the Gα proteins Gαi1-91-RLuc8 (0.2 μg) or GαoA-91-RLuc8 (0.2 μg). For cyclic AMP accumulation, cells were co-transfected with WT (0.2 μg for WT-Low and 0.5 μg for WT-High) or I212F-mutated D2 receptor (0.5 μg), and the cyclic AMP sensor CAMYEL (2.5 μg). Control cells were transfected with CAMYEL and nonspecific plasmid DNA. After 48 h, cells were harvested, washed, resuspended in PBS containing CaCl2, MgCl2, and 11 mM D-glucose, plated at 100,000–150,000 cells/well in 96-well OptiPlates (PerkinElmer Life Sciences), and incubated at 37°C and 5% CO2 atmosphere for 1 h before adding the agonist quinpirole. Compound 101 (Cmpd101; HelloBio, Princeton, NJ) was initially dissolved in DMSO at 100 mM and subsequently diluted in PBS. For GRK2 inhibition during arrestin recruitment-BRET assays, HEK293 cells were pretreated with 30 μM of Cmpd101 or vehicle (DMSO diluted in PBS) 30 minutes before agonist addition. For D2-mediated inhibition of cyclic AMP, HEK293 cells were pretreated with the dopamine D1 receptor antagonist SCH 23390 (1 μM) and the β-adrenoceptor antagonist pindolol (0.1 μM; Sigma-Aldrich; MO, USA) before the addition of inverse agonist (10 μM sulpiride or 1 μM spiperone) and 10 μM forskolin (Sigma-Aldrich; MO, USA). Emission of the donor (460 μm) and acceptor (535 μm) was measured at room temperature several times after adding the luciferase substrate coelenterazine h, and BRET ratios were calculated as previously described (12, 51).
D2 Receptor Radioligand Binding.
Membrane expression of the receptors was evaluated exactly as described previously (8). Cells were lysed in ice-cold hypotonic buffer (1 mM HEPES, 2 mM EDTA, pH 7.4), scraped from the plate, and centrifuged at 17,000 × g at 4°C for 20 min. The resulting pellet was resuspended in Tris-buffered saline (TBS: 50 mM Tris, 120 mM NaCl, pH 7.4) and homogenized for 10 s using a Polytron homogenizer (Brinkmann Instruments, Westbury, NY). Protein determination was performed using the BCA Protein Assay Kit (Thermo Scientific). Samples were incubated in TBS containing 0.002% BSA and [3H]spiperone at 37°C for 1 h in a final volume of 1 ml before addition of ice-cold buffer and vacuum filtration. Nonspecific binding was assessed using (+)-butaclamol (2 μM). Competition binding assays were carried out using membranes prepared from HEK293 cells stably expressing each of the four receptor variants. The ability of various concentrations of quinpirole to inhibit the binding of [3H]spiperone (~85 pM) was analyzed by nonlinear regression. IC50 values were converted to Ki according the method of Cheng and Prusoff (52).
Mice.
All studies were conducted in accordance with the Institutional Animal Care and Use Committees at the VA Portland Health Care System (VAPORHCS) and Oregon Health & Science University (OHSU). Twelve mice (4 male and 8 female, 59–96 days old on day of surgery) were used in this study. Auto-D2-KO mice were bred at the VAPORHCS Veterinary Medical Unit by crossing Drd2loxP/loxP mice (53), obtained from Jonathan Javitch (Columbia University, USA), with heterozygous B6.SJL-Slc6a3tm1.1(cre)Bkmn/J mice (54) obtained from the Jackson Laboratory (JAX stock #006660). All lines are maintained on a C57BL/6 background. Mice were housed in standard plastic containers on a 12 hr light/dark cycle with food and water available ad libitum. For expression of recombinant D2S receptors in dopamine neurons, auto-D2-KO mice were immobilized in a stereotaxic alignment system after injection of an anesthesia cocktail consisting of 7.1 mg/kg xylazine, 71.4 mg/kg ketamine, and 1.4 mg/kg acepromazine (10 ml/kg, i.p.). Mice received bilateral 500 nl injections of AAV-DIO-hD2S-WT or -I212F in the ventral tegmental area, at a rate of 200 nl/min, with the injection needle left in place for an additional 5 min before it was slowly withdrawn. The coordinates for injections were AP −3.26 mm, ML ±1.2 mm, DV −4.0 mm. After injections, mice recovered in individual (male) or group (female) housing for 2–3 weeks to allow for expression.
Slice Electrophysiology.
Mice were deeply anesthetized with isoflurane and euthanized by decapitation. Brains were removed and placed in warm (30°C) physiologically equivalent saline solution (modified Krebs buffer) containing NaCl (126 mM), KCl (2.5 mM), MgCl2 (1.2 mM), CaCl2 (2.4 mM), NaH2PO4 (1.4 mM), NaHCO3 (25 mM), and D-glucose (11 mM) with MK-801 (3 μM), and cut horizontally (222 μm) using a vibrating microtome (Leica). Slices recovered at 30°C in vials with 95/5% O2/CO2 saline with MK801 (10 μM) for at least 30 min prior to recording. For reserpine treatment, slices instead recovered for 15 minutes in MK801 followed by 1 hour in reserpine (1 μM). Slices were mounted in the recording chamber of an upright microscope (Olympus). The temperature was maintained at 34–36 °C, and modified Krebs buffer was perfused over the slices at 1–2 mL/min. Recordings were obtained with large glass electrodes with a resistance of 1.3–1.9 MΩ when filled with an internal solution containing potassium methanesulfonate (75 mM), NaCl (20 mM), MgCl2 (1.5 mM), HEPES potassium salt (5 mM), ATP (2 mM), GTP (0.2 mM), phosphocreatine (10 mM), and BAPTA tetrapotassium salt pH 7.35–7.45 (10 mM) at 275–288 mOsm. Cells were voltage-clamped at −60 mV using an Axopatch 200A integrating patch clamp (Axon Instruments). Recordings were made using Axograph 10 and Chart 5.5. D2 receptor-expressing dopamine neurons in the substantia nigra were identified by location, size, firing properties, and EGFP fluorescence.
CyHQ-sulpiride (33) was kept as a stock solution in DMSO (10 mM) and diluted to a 5 μM working solution. A ThorLabs M365LP1-C1 LED was used to photolyze CyHQ-sulpiride by means of a 50 msec flash (365 nm) at 6.5 mW. Dopamine iontophoresis (1 M) was done using a thin-walled glass electrode (70–110 MΩ) with its tip placed within 10 μm of the soma. Dopamine was kept in place with a 4 nA backing current and ejected with a 10 ms, 100 nA pulse using an Axoclamp-2a amplifier.
D2 Receptor homology models and molecular dynamics simulations.
Homology modeling was performed using YASARA Structure (55) that features a CASP- (Critical Assessment of Structure Prediction) approved protocol (56). The inactive state of the D2 receptor was modeled using inactive structures of the β2-adrenoceptor (2RH1), the A2A adenosine receptor (3EML and 6GT3), the M2 muscarinic receptor (3UON), bovine rhodopsin (1GZM), and the D2 receptor (6CM4) as templates. The D2 receptor active state model was built using active-state structures of the M2 receptor (4MQS), the β2-adrenoceptor (3SN6 and 3P0G), the A2A receptor (2YDV and 5WF6), the CB1 receptor (6N4B), the μ-opioid receptor (6DDE), and rhodopsin (3PQR) as templates. Multiple D2 receptor models for inactive and active states (48 and 45 models, respectively) were obtained, and side chain rotamers were optimized using backbone-dependent probabilities and knowledge-based force fields in YASARA (57). The resulting models were further optimized for hydrogen bonding, refined using short molecular dynamics simulations, and ranked. Residue-specific quality graphs were calculated for each model and a final hybrid model was developed through an iterative process, replacing poorly scoring regions in the best model with the corresponding regions from other models, with the goal of increasing the accuracy beyond each of the contributing models. The stereochemical properties of the homology models were verified using the PROCHECK module (58) of the PDBSum server, which examines protein quality based on parameters such as percentage of residues lying in favored and allowed regions, the number of glycine and proline residues, the orientation of dihedral angles including phi (φ) and psi (ψ), and backbone conformation. The VERIFY3D (59) server was used to check the compatibility of atomic models (3D) with its own primary amino acid sequences (1D). The RSMD of the alpha-helical segments of the resulting active-state homology model of the D2 receptor was 1.62 and 1.63, respectively, relative to 6CM4 and the recently published active-state structure of the D2 receptor 6VMS (60).
To assess the potential functional impact of the I212F substitution on the active and inactive D2 receptor homology models, the system was simulated atomistically for 15 ns using the YASARA software package under an NPT ensemble with the AMBER14 force field (61), with a timestep of 5.0 fs. Simulation conditions were conducted with periodic boundaries, at 0.9% NaCl concentration by mass, pH 7.4, 298K, at atmospheric pressure. The water model employed was TIP3 equivalent. Snapshots were saved every 100 ps. Structures were visualized using YASARA and the distances between the atom OE1 of E368 and HH1 and HH2 atoms of R132 were monitored during the simulation and plotted using Prism GraphPad software.
Data Analysis and Statistics.
Concentration-response curves and radioligand saturation binding curves were analyzed by nonlinear regression using Prism 8 (GraphPad Software Inc.; San Diego, CA, USA). For affinity and potency values, the geometric mean (mean of logKd/i or logEC50) was calculated and used for statistical comparison. Statistical significance between two means was determined using Student’s t-test, and for comparisons of more than two means by 2-way ANOVA followed by Tukey’s multiple comparisons test except for data shown in Fig 1C–D. The effect of genotype and GRK2 condition on maximal arrestin recruitment over time was assessed using 3-way repeated measures (RM) ANOVA with Geisser-Greenhouse correction, followed by 2-way RM ANOVA, with genotype and/or GRK2 condition as matched values and time (7-time points) as the RM factor.
Supplementary Material
Table S1 Bmax values for HEK293 studies
Table S2 Regression analyses for Compound 101 experiment – comparison of +/− GRK2 and D2-WT vs. D2-I212F
Figure S1 Arrestin recruitment with matched receptor expression levels for D2L-WT and D2L-I212F
Figure S2 Concentration-response curves for arrestin recruitment and comparison of +/− Compound 101
Figure S3 Quinpirole competition binding curves
ACKNOWLEDGEMENTS
Support for this research was provided by the National Institute of Neurological Disorders and Stroke (R21NS117713), the National Institute on Drug Abuse (F31DA047007 and R01DA004523), the US Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development, Biomedical Laboratory Research and Development (Merit Review Award BX003279), and New York University Abu Dhabi.
ABBREVIATIONS
- AAV
adeno-associated viral
- BRET
bioluminescence resonance energy transfer
- Cmpd101
GRK2/3 inhibitor Compound 101
- CyHQ-sulpiride
1-((8-cyano-7-hydroxyquinolin-2-yl)methyl)-1-ethyl-2-((2-methoxy-5-sulfamoylbenzamido)methyl)pyrrolidin-1-ium 2,2,2-trifluoroacetate
- DIO
double-floxed inverted open reading frame
- EGFP
green fluorescent protein
- FCS
FetalClone I Serum
- FSK
forskolin
- GIRKs
G protein-coupled inwardly-rectifying potassium channels
- GPCR
G protein-coupled receptor
- GRK
G protein-coupled receptor kinase
- HEK
human embryonic kidney
- MD
molecular dynamics
- RLuc
Renilla luciferase
- RM ANOVA
repeated measures ANOVA
- TM
transmembrane domain
- WT
wild type
Footnotes
The authors declare no competing financial interest.
Contributor Information
Dayana Rodriguez-Contreras, Research Service, VA Portland Health Care System, and Department of Behavioral Neuroscience, Oregon Health & Science University, Portland, Oregon 97239, United States.
Alec F. Condon, Vollum Institute, Oregon Health & Science University, Portland, Oregon 97239, United States.
David C. Buck, Research Service, VA Portland Health Care System, Portland, Oregon 97239, United States
Dineke S. Verbeek, Expertise Center Movement Disorders and Department of Genetics, University of Groningen, 9700 AB Groningen, The Netherlands.
Marina A.J. Tijssen, Expertise Center Movement Disorders and Department of Neurology, University of Groningen, 9700 AB Groningen, The Netherlands.
Ujwal Shinde, Department of Chemical Physiology & Biochemistry, Oregon Health & Science University, Portland, Oregon 97239, United States.
John T. Williams, Vollum Institute, Oregon Health & Science University, Portland, Oregon 97239, United States.
Kim A. Neve, Research Service, VA Portland Health Care System, and Department of Behavioral Neuroscience, Oregon Health & Science University, Portland, Oregon 97239, United States.
REFERENCES
- 1.Beaulieu JM; Sotnikova TD; Marion S; Lefkowitz RJ; Gainetdinov RR; Caron MG, An Akt/β-arrestin 2/PP2A signaling complex mediates dopaminergic neurotransmission and behavior. Cell 2005, 122 (2), 261–273. [DOI] [PubMed] [Google Scholar]
- 2.Donthamsetti P; Gallo EF; Buck DC; Stahl EL; Zhu Y; Lane JR; Bohn LM; Neve KA; Kellendonk C; Javitch JA, Arrestin recruitment to dopamine D2 receptor mediates locomotion but not incentive motivation. Mol Psychiatry 2018, 25, 2086–2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rose SJ; Pack TF; Peterson SM; Payne K; Borrelli E; Caron MG, Engineered D2R variants reveal the balanced and biased contributions of G-protein and β-arrestin to dopamine-dependent functions. Neuropsychopharmacology 2018, 43 (5), 1164–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cepeda C; Murphy KP; Parent M; Levine MS, The role of dopamine in Huntington’s disease. Prog. Brain Res 2014, 211, 235–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moritz AE; Free RB; Sibley DR, Advances and challenges in the search for D2 and D3 dopamine receptor-selective compounds. Cell Signal 2018, 41, 75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gantz SC; Robinson BG; Buck DC; Bunzow JR; Neve RL; Williams JT; Neve KA, Distinct regulation of dopamine D2S and D2L autoreceptor signaling by calcium. eLife 2015, 4, e09358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Radl D; Chiacchiaretta M; Lewis RG; Brami-Cherrier K; Arcuri L; Borrelli E, Differential regulation of striatal motor behavior and related cellular responses by dopamine D2L and D2S isoforms. Proc. Natl. Acad. Sci. USA 2018, 115 (1), 198–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van der Weijden MCM; Rodriguez-Contreras D; Delnooz CCS; Robinson BG; Condon AF; Kielhold ML; Stormezand GN; Ma KY; Dufke C; Williams JT, et al. , A gain-of-function variant in dopamine D2 receptor and progressive chorea and dystonia phenotype. Mov Disord 2020, doi: 10.1002/mds.28385. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ballesteros J; Weinstein H, Integrated methods for modeling G-protein coupled receptors. Methods Neurosci 1995, 25, 366–428. [Google Scholar]
- 10.Jones EM; Lubock NB; Venkatakrishnan AJ; Wang J; Tseng AM; Paggi JM; Latorraca NR; Cancilla D; Satyadi M; Davis JE, et al. , Structural and functional characterization of G protein-coupled receptors with deep mutational scanning. eLife 2020, 9, 10.7554/eLife.54895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lan H; Liu Y; Bell MI; Gurevich VV; Neve KA, A dopamine D2 receptor mutant capable of G protein-mediated signaling but deficient in arrestin binding. Mol Pharmacol 2009, 75, 113–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clayton CC; Donthamsetti P; Lambert NA; Javitch JA; Neve KA, Mutation of three residues in the third intracellular loop of the dopamine D2 receptor creates an internalization-defective receptor. J. Biol. Chem 2014, 289 (48), 33663–33675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu Y; Buck DC; Macey TA; Lan H; Neve KA, Evidence that calmodulin binding to the dopamine D2 receptor enhances receptor signaling. J. Recept. Signal Transduct. Res 2007, 27 (1), 47–65. [DOI] [PubMed] [Google Scholar]
- 14.Moore CA; Milano SK; Benovic JL, Regulation of receptor trafficking by GRKs and arrestins. Annu Rev Physiol 2007, 69, 451–82. [DOI] [PubMed] [Google Scholar]
- 15.Kim KM; Valenzano KJ; Robinson SR; Yao WD; Barak LS; Caron MG, Differential regulation of the dopamine D2 and D3 receptors by G protein-coupled receptor kinases and β-arrestins. Journal of Biological Chemistry 2001, 276 (40), 37409–37414. [DOI] [PubMed] [Google Scholar]
- 16.Gurevich EV; Gainetdinov RR; Gurevich VV, G protein-coupled receptor kinases as regulators of dopamine receptor functions. Pharmacol Res 2016, 111, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Namkung Y; Dipace C; Urizar E; Javitch JA; Sibley DR, G protein-coupled receptor kinase-2 constitutively regulates D2 dopamine receptor expression and signaling independently of receptor phosphorylation. J. Biol. Chem 2009, 284 (49), 34103–34115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Neve KA; Seamans JK; Trantham-Davidson H, Dopamine receptor signaling. J. Recept. Signal Transduct. Res 2004, 24 (3), 165–205. [DOI] [PubMed] [Google Scholar]
- 19.Jiang M; Bajpayee NS, Molecular mechanisms of Go signaling. Neurosignals 2009, 17 (1), 23–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jiang MS; Spicher K; Boulay G; Wang Y; Birnbaumer L, Most central nervous system D2 dopamine receptors are coupled to their effecters by Go. Proc Natl Acad Sci USA 2001, 98 (6), 3577–3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marcott PF; Gong S; Donthamsetti P; Grinnell SG; Nelson MN; Newman AH; Birnbaumer L; Martemyanov KA; Javitch JA; Ford CP, Regional heterogeneity of D2-receptor signaling in the dorsal striatum and nucleus accumbens. Neuron 2018, 98 (3), 575–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Namkung Y; Dipace C; Javitch JA; Sibley DR, G protein-coupled receptor kinase-mediated phosphorylation regulates post-endocytic trafficking of the D2 dopamine receptor. J. Biol. Chem 2009, 284 (22), 15038–15051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thal DM; Yeow RY; Schoenau C; Huber J; Tesmer JJ, Molecular mechanism of selectivity among G protein-coupled receptor kinase 2 inhibitors. Mol Pharmacol 2011, 80 (2), 294–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gondin AB; Halls ML; Canals M; Briddon SJ, GRK mediates μ-opioid receptor plasma membrane reorganization. Front Mol Neurosci 2019, 12, 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miess E; Gondin AB; Yousuf A; Steinborn R; Mösslein N; Yang Y; Göldner M; Ruland JG; Bünemann M; Krasel C, et al. , Multisite phosphorylation is required for sustained interaction with GRKs and arrestins during rapid μ-opioid receptor desensitization. Sci Signal 2018, 11 (539). [DOI] [PubMed] [Google Scholar]
- 26.Gazi L; Nickolls SA; Strange PG, Functional coupling of the human dopamine D2 receptor with Gαi1, Gαi2, Gαi3 and Gαo G proteins: evidence for agonist regulation of G protein selectivity. Br. J. Pharmacol 2003, 138 (5), 775–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jiang LI; Collins J; Davis R; Lin KM; DeCamp D; Roach T; Hsueh R; Rebres RA; Ross EM; Taussig R, et al. , Use of a cAMP BRET sensor to characterize a novel regulation of cAMP by the sphingosine 1-phosphate/G13 pathway. J. Biol. Chem 2007, 282 (14), 10576–10584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Watts VJ; Neve KA, Sensitization of endogenous and recombinant adenylate cyclase by activation of D2 dopamine receptors. Mol. Pharmacol 1996, 50 (4), 966–976. [PubMed] [Google Scholar]
- 29.Watts VJ; Neve KA, Sensitization of adenylate cyclase by Gαi/o-coupled receptors. Pharmacol Ther 2005, 106 (3), 405–421. [DOI] [PubMed] [Google Scholar]
- 30.Parker EM; Ross EM, Truncation of the extended carboxyl-terminal domain increases the expression and regulatory activity of the avian β-adrenergic receptor. J. Biol. Chem 1991, 266 (15), 9987–9996. [PubMed] [Google Scholar]
- 31.Kjelsberg MA; Cotecchia S; Ostrowski J; Caron MG; Lefkowitz RJ, Constitutive activation of the α1B-adrenergic receptor by all amino acid substitutions at a single site: evidence for a region which constrains receptor activation. J. Biol. Chem 1992, 267 (3), 1430–1433. [PubMed] [Google Scholar]
- 32.Wilson J; Lin H; Fu D; Javitch JA; Strange PG, Mechanisms of inverse agonism of antipsychotic drugs at the D2 dopamine receptor: use of a mutant D2 dopamine receptor that adopts the activated conformation. J. Neurochem 2001, 77 (2), 493–504. [DOI] [PubMed] [Google Scholar]
- 33.Asad N; McLain DE; Condon AF; Gore S; Hampton SE; Vijay S; Williams JT; Dore TM, Photoactivatable dopamine and sulpiride to explore the function of dopaminergic neurons and circuits. ACS Chem Neurosci 2020, 11 (6), 939–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ballesteros JA; Jensen AD; Liapakis G; Rasmussen SG; Shi L; Gether U; Javitch JA, Activation of the β2 adrenergic receptor involves disruption of an ionic lock between the cytoplasmic ends of transmembrane segments 3 and 6. J. Biol. Chem 2001, 276, 29171–29177. [DOI] [PubMed] [Google Scholar]
- 35.Kling RC; Clark T; Gmeiner P, Comparative MD simulations indicate a dual role for Arg1323.50 in dopamine-dependent D2R activation. PLoS One 2016, 11 (1), e0146612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rasmussen SGF; Jensen AD; Liapakis G; Ghanouni P; Javitch JA; Gether U, Mutation of a highly conserved aspartic acid in the β2 adrenergic receptor: Constitutive activation, structural instability, and conformational rearrangement of transmembrane segment 6. Molecular Pharmacology 1999, 56 (1), 175–184. [DOI] [PubMed] [Google Scholar]
- 37.Alewijnse AE; Timmerman H; Jacobs EH; Smit MJ; Roovers E; Cotecchia S; Leurs R, The effect of mutations in the DRY motif on the constitutive activity and structural instability of the histamine H(2) receptor. Mol Pharmacol 2000, 57 (5), 890–8. [PubMed] [Google Scholar]
- 38.Wang S; Che T; Levit A; Shoichet BK; Wacker D; Roth BL, Structure of the D2 dopamine receptor bound to the atypical antipsychotic drug risperidone. Nature 2018, 555 (7695), 269–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Farrens DL; Altenbach C; Yang K; Hubbell WL; Khorana HG, Requirement of rigid-body motion of transmembrane helices for light activation of rhodopsin. Science 1996, 274 (5288), 768–770. [DOI] [PubMed] [Google Scholar]
- 40.Rasmussen SG; Choi HJ; Rosenbaum DM; Kobilka TS; Thian FS; Edwards PC; Burghammer M; Ratnala VR; Sanishvili R; Fischetti RF, et al. , Crystal structure of the human β2 adrenergic G-protein-coupled receptor. Nature 2007, 450 (7168), 383–387. [DOI] [PubMed] [Google Scholar]
- 41.Gether U; Ballesteros JA; Seifert R; Sanders-Bush E; Weinstein H; Kobilka BK, Structural instability of a constitutively active G protein-coupled receptor. Agonist-independent activation due to conformational flexibility. J Biol Chem 1997, 272 (5), 2587–90. [DOI] [PubMed] [Google Scholar]
- 42.Mafi A; Kim SK; Goddard WA 3rd, Mechanism of β-arrestin recruitment by the μ-opioid G protein-coupled receptor. Proc Natl Acad Sci U S A 2020, 117 (28), 16346–16355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang W; Masureel M; Qu Q; Janetzko J; Inoue A; Kato HE; Robertson MJ; Nguyen KC; Glenn JS; Skiniotis G, et al. , Structure of the neurotensin receptor 1 in complex with β-arrestin 1. Nature 2020, 579 (7798), 303–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Staus DP; Hu H; Robertson MJ; Kleinhenz ALW; Wingler LM; Capel WD; Latorraca NR; Lefkowitz RJ; Skiniotis G, Structure of the M2 muscarinic receptor-β-arrestin complex in a lipid nanodisc. Nature 2020, 579 (7798), 297–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gurevich VV; Gurevich EV, The structural basis of arrestin-mediated regulation of G-protein-coupled receptors. Pharmacol Ther 2006, 110 (3), 465–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhuang T; Chen Q; Cho MK; Vishnivetskiy SA; Iverson TM; Gurevich VV; Sanders CR, Involvement of distinct arrestin-1 elements in binding to different functional forms of rhodopsin. Proc Natl Acad Sci USA 2013, 110 (3), 942–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hilger D; Masureel M; Kobilka BK, Structure and dynamics of GPCR signaling complexes. Nat Struct Mol Biol 2018, 25 (1), 4–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bateup HS; Santini E; Shen W; Birnbaum S; Valjent E; Surmeier DJ; Fisone G; Nestler EJ; Greengard P, Distinct subclasses of medium spiny neurons differentially regulate striatal motor behaviors. Proc. Natl. Acad. Sci. U. S. A 2010, 107 (33), 14845–14850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Urs NM; Bido S; Peterson SM; Daigle TL; Bass CE; Gainetdinov RR; Bezard E; Caron MG, Targeting β-arrestin2 in the treatment of L-DOPA-induced dyskinesia in Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2015, 112 (19), E2517–E2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Saulière A; Bellot M; Paris H; Denis C; Finana F; Hansen JT; Altié MF; Seguelas MH; Pathak A; Hansen JL, et al. , Deciphering biased-agonism complexity reveals a new active AT1 receptor entity. Nat Chem Biol 2012, 8 (7), 622–30. [DOI] [PubMed] [Google Scholar]
- 51.Pfleger KD; Seeber RM; Eidne KA, Bioluminescence resonance energy transfer (BRET) for the real-time detection of protein-protein interactions. Nat Protoc 2006, 1 (1), 337–345. [DOI] [PubMed] [Google Scholar]
- 52.Cheng Y-C; Prusoff WH, Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol 1973, 22, 3099–3108. [DOI] [PubMed] [Google Scholar]
- 53.Bello EP; Mateo Y; Gelman DM; Noain D; Shin JH; Low MJ; Alvarez VA; Lovinger DM; Rubinstein M, Cocaine supersensitivity and enhanced motivation for reward in mice lacking dopamine D2 autoreceptors. Nat Neurosci 2011, 14 (8), 1033–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Backman CM; Malik N; Zhang Y; Shan L; Grinberg A; Hoffer BJ; Westphal H; Tomac AC, Characterization of a mouse strain expressing Cre recombinase from the 3’ untranslated region of the dopamine transporter locus. Genesis 2006, 44 (8), 383–390. [DOI] [PubMed] [Google Scholar]
- 55.Krieger E; Koraimann G; Vriend G, Increasing the precision of comparative models with YASARA NOVA--a self-parameterizing force field. Proteins 2002, 47 (3), 393–402. [DOI] [PubMed] [Google Scholar]
- 56.Krieger E; Joo K; Lee J; Lee J; Raman S; Thompson J; Tyka M; Baker D; Karplus K, Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in CASP8. Proteins 2009, 77 Suppl 9 (Suppl 9), 114–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Krieger E; Darden T; Nabuurs SB; Finkelstein A; Vriend G, Making optimal use of empirical energy functions: force-field parameterization in crystal space. Proteins 2004, 57 (4), 678–83. [DOI] [PubMed] [Google Scholar]
- 58.Laskowski RA; MacArthur MW; Moss DS; Thornton JM, PROCHECK - a program to check the stereochemical quality of protein structures. J. Appl. Cryst 1993, 26, 283–291. [Google Scholar]
- 59.Eisenberg D; Lüthy R; Bowie JU, VERIFY3D: assessment of protein models with three-dimensional profiles. Methods Enzymol 1997, 277, 396–404. [DOI] [PubMed] [Google Scholar]
- 60.Yin J; Chen KM; Clark MJ; Hijazi M; Kumari P; Bai XC; Sunahara RK; Barth P; Rosenbaum DM, Structure of a D2 dopamine receptor-G-protein complex in a lipid membrane. Nature 2020, 584 (7819), 125–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ponder JW; Case DA, Force fields for protein simulations. Adv Protein Chem 2003, 66, 27–85. [DOI] [PubMed] [Google Scholar]
- 62.Liu Y; Buck DC and Neve KA (2008) Novel interaction of the dopamine D2 receptor and the Ca2+ binding protein S100B: role in D2 receptor function. Mol. Pharmacol 74, 371–378, DOI: 10.1124/mol.108.044925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lee H-J; Rodriguez-Contreras D and Neve KA (2021) Commentary on “Novel interaction of the dopamine D2 receptor and the CA2+ binding protein S100B: role in D2 receptor function”. Mol. Pharmacol DOI: 10.1124/molpharm.121.000284 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Bmax values for HEK293 studies
Table S2 Regression analyses for Compound 101 experiment – comparison of +/− GRK2 and D2-WT vs. D2-I212F
Figure S1 Arrestin recruitment with matched receptor expression levels for D2L-WT and D2L-I212F
Figure S2 Concentration-response curves for arrestin recruitment and comparison of +/− Compound 101
Figure S3 Quinpirole competition binding curves