

Abstract
The critical role of sialyltransferase (ST) enzymes in tumour cell growth and metastasis, as well as links to multi-drug and radiation resistance, has seen STs emerge as a target for potential antimetastatic cancer treatments. One promising class of ST inhibitors that improve upon the pharmacokinetic issues of previous inhibitors is the 1,2,3-triazole-linked transition-state analogues. Herein, we present the design and synthesis of a new generation of 1,2,3-triazole-linked sialyltransferase inhibitors, along with their biological evaluation demonstrating increased potency for phosphonate bearing compounds. The six most promising inhibitors presented in this work exhibited a greater number of binding modes for hST6Gal I over hST3Gal I, with Ki ranging from 3–55 μM. This work highlights phosphonate bearing triazole-linked compounds as a promising class of synthetically accessible ST inhibitors that warrant further investigation.
Key modifications of previous sialyltransferase inhibitors increased their activity against hST6Gal I and has further implications for synthetically accessible ST inhibitor design.
Introduction
N-Acetylneuraminic acid (Neu5Ac, or sialic acid) is a 9-carbon, charged monosaccharide located at the terminus of many human cell-surface glycans. Increased cell surface sialylation due to overexpression of sialyltransferase enzymes (STs) in tumoural cells is a well-established hallmark of cancer, making the development of ST inhibitors a potential route for new cancer treatments and biomarkers.1–3 Hypersialylation has been implicated in increased metastatic potential through integrin and selectin mediated processes,4–6 immune evasion by binding to Siglecs on immune cells,7,8 and improving tumour cell survival by inhibition of apoptotic signalling.9 Increased surface sialylation has also been linked to resistance to chemotherapeutics such as cisplatin and paclitaxel,10,11 and radioresistance in colorectal cancers.12,13 Sialylation also plays a key role in viral infections.14
Sialyltransferase enzymes belong to the glycosyltransferase family and facilitate the transfer of Neu5Ac to an acceptor glycan chain terminated by a galactose (Gal), N-acetylgalactosamine (GalNAc) or another sialic acid (Sia) residue.15,16 Eukaryotic sialyltransferases (ST) are a glycosyltransferase subset including six beta-galactoside α2-3-sialyltransferases (ST3Gal I–VI), two beta-galactoside α2-6-sialyltransferases (ST6Gal I–II), six GalNAc α2-6-sialyltransferases (ST6GalNAc-I–VI), and six α2-8-sialyltransferases (ST8Sia-I–VI, among which ST8Sia-II and ST8Sia-IV are polysialyltransferases).17–19 In humans, STs all utilise the common donor cytidine monophosphate Neu5Ac (CMP-Neu5Ac) making selective inhibitor design a challenge. The various ST subtypes are highly overexpressed up to 50% or more in several cancers including colorectal,20,21 breast,22 and pancreatic cancer.23 Selective targeting of the specific ST subtypes over-expressed in a particular cancer is crucial in order to avoid off-target effects such as liver and kidney dysfunction exhibited for pan ST inhibitors.24 As part of an ongoing study into ovarian and pancreatic cancer, this study is aimed at selective inhibition of human ST3Gal I and ST6Gal I as two of the most well studied STs in terms of their potential as anti-metastatic targets.1–3
Previous studies into ST inhibitors have identified transition-state analogues as the most potent inhibitors to date (Fig. 1).2 These compounds mimic the planar oxocarbenium-like transition-state of the ST mechanism. One of the most potent ST inhibitors in the literature is the phosphodiester-linked cytidine compound (R)-1 (Ki = 19 nM, hST6GAL I) inspired by the donor molecule CMP-Neu5Ac, with an aromatic m-phenoxy system replacing the sialic acid, and a phosphonate group replacing the carboxylate of CMP-Neu5Ac.25,26 More recent research into ST inhibitors has uncovered the highly potent cyclopentyl (R-2, Ki = 28 nM, hST6Gal I), and amide (3Ki = 19 nM, hST6Gal I) phosphodiester-linked compounds. Potential pharmacokinetic issues with the phosphodiester linkage (due to metabolism by phosphatases in vivo)27 has led to the investigation by computational methods of carbamate and 1,2,3-triazole linkers as bioisosteric replacements.28–30
Fig. 1. Structures of literature sialyltransferase inhibitors 1–5, and the general structure of the target triazole-linked compounds of this study.

A study into the synthesis and biological evaluation of carbamate linked ST inhibitors produced the p-fluorinated aryl derivative (4), with a carbamate moiety replacing the phosphate linker (Fig. 1).31 While less active than the parent compound, the carbamate-based derivative showed promising low micromolar activity (4, Ki = 1.1 μM against hST6GAL I), alongside improved drug-likeness and synthetic accessibility as a result of replacing the cytidine moiety with uridine and the charged phosphodiester with a carbamate group. Following on from this, a series of neutral 1,2,3-triazole linked-compounds such as 5 have also been developed and evaluated against hST6Gal I (84% inhibition at 100 μM).32 As the phosphonate moiety is often linked to potency in ST inhibitors,2 herein a series of novel phosphonate-bearing, ether-linked 1,2,3-triazole based compounds have been synthesised aimed at generating a selective, high affinity and accessible ST6Gal I inhibitor for further large-scale biological studies (Fig. 1).
Results and discussion
To probe the effect of increased linker length on ST inhibition, we synthesised a novel series of 14 ether-linked, 1,2,3-triazole based derivatives, examined their comparative binding energies and evaluated their selectivity and inhibitory activity towards human ST6Gal I and ST3Gal I in a luminescence-based microplate assay. Synthesis of the target ether-linked molecules was achieved by the coupling of 5′-O-propargyluridine with an α-azidophosphonate. The 5′-O-propargyluridine fragments were synthesised starting from commercially available uridine (6) via a similar method to Sun et al. (Scheme 1).33 First, the 2′,3′-diol was protected as an acetonide, allowing for successful propargyl addition at the 5′-alcohol utilising sodium hydride and propargyl bromide, to yield the protected compound 7. The acetonide was removed via the method of Golden et al., (ref. 34) utilising an indium triflate catalyst at reflux to give the deprotected 5′-O-progargyluridine (8) in quantitative yield. The overall yield of 8 from uridine was 69%.
Scheme 1. Synthesis of 5′-O-propargyluridine (8). Reagents and conditions: (a) (i) acetone, H2SO4, 0 °C to r.t, overnight, >99%; (ii) propargyl bromide, NaH, THF, 0 °C to r.t, overnight, 70%; (b) In(OTf)3, ACN/H2O (9 : 1), reflux, 6 h, >99%.

The α-azidophosphonates were synthesised from a series of α-hydroxyphosphonates (10a–g, Scheme 2), prepared by reacting an aromatic aldehyde (9) and dibenzyl phosphite.35 The α-hydroxyphosphonate then underwent a Mitsunobu reaction to yield the α-azidophosphonate (11a–g).36
Scheme 2. Synthesis of novel dibenzyl α-azidophosphonates 11a–g, their CuAAC coupling to alkynyluridine 8, and purification to form ether-linked 1,2,3-triazoles 13a–g. Reagents and conditions: (a) dibenzyl phosphite, TEA, DCM, r.t., overnight, 60–85%; (b) HN3, toluene, DIAD, PPh3, THF, 0 °C to r.t., 80–99%; (c) 8, Cu(OAc)2, sodium ascorbate, r.t., overnight; (d) (i) Pd/C, H2, MeOH, 4 h; (ii) RP-HPLC, IR120 Na+. R1 groups and yields are shown in Table 1.

Yields for protected triazoles 12a–g from the copper azide-alkyne cyclisation coupling of 5′-O-propargyluridine (8) and α-azidophosphonates (11a–g), followed by their deprotection to form the final deprotected 1,2,3-triazole series in their Na+ salt form (13a–g).
| R1 | Click yield (R2: OBn)a | Deprotection yield (R2: O−Na+)b |
|---|---|---|
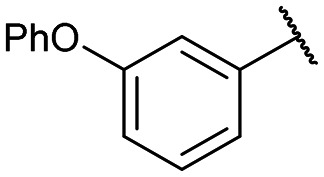
|
12a: 51% | 13a-(s): 99% |
| 13a-(l): 91% | ||
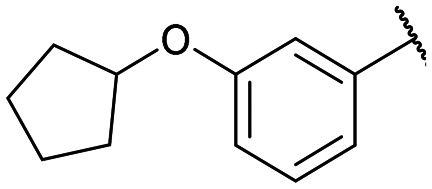
|
12b: 52% | 13b-(s): 94% |
| 13b-(l): 99% | ||
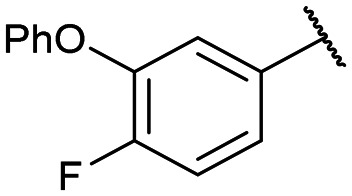
|
12c: 66% | 13c-(s): 97% |
| 13c-(l): 91% | ||
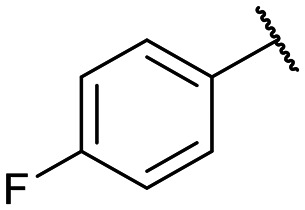
|
12d: 57% | 13d-(s): 92% |
| 13d-(l): 62% | ||
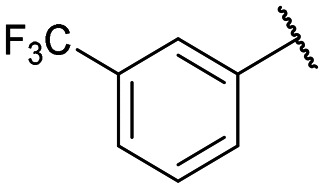
|
12e: 60% | 13e-(s): 92% |
| 13e-(l): 85% | ||
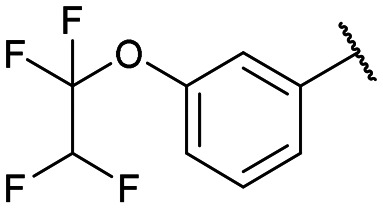
|
12f: 52% | 13f-(s): 95% |
| 13f-(l): 92% | ||
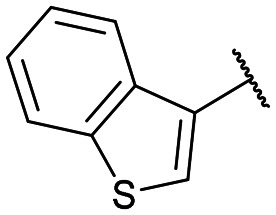
|
12g: 57% | 13g-(s): 96% |
| 13g-(l): 92% |
These two key fragments were then coupled via a copper azide-alkyne cycloaddition (CuAAC) in the presence of Cu(i), generated from Cu(OAc)2 reduced with sodium ascorbate. A wide range of solvent systems have been utilised in the literature with these CuAAC “click” reactions, although many are quite polar (such as ACN/H2O)37,38 and would not be suitable for the highly lipophilic α-azidophosphonates. The solvent system used for this click reaction was 1 : 1 THF/H2O, with the copper and sodium ascorbate dissolved in water and then added to the nucleoside and phosphonate fragments dissolved in neat THF, followed by vigorous stirring to aid solubility of the reagents in the reaction. Formation of the 1,4-disubstituted product was confirmed by the presence of the characteristic proton peak at around 8.5–8.8 ppm, attributed to the proton at C5 of the 1,2,3-triazole ring and showing correlations to both C5 and the chiral carbon.
Once coupled, the fully protected triazole compounds (12a–g) were deprotected via catalytic hydrogenation with a Pd/C catalyst and hydrogen gas in methanol over 4 hours. The fully deprotected compounds (13a–g) were purified and the diastereomers separated via C18 reverse phase HPLC using a mixture of methanol or acetonitrile and a triethylammonium bicarbonate buffer, which yielded the purified compounds as their triethylammonium salts. These were further exchanged with sodium by shaking with Amberlite IR120 Na+ form in MilliQ water for 1 hour (repeated three times). The resulting aqueous solution was lyophilised to yield the final product as an amorphous white powder. A small amount of styrene-divinylbenzene impurity (leached from the ion exchange resin) was observed in the 1H NMR spectra of compounds 13a/b as small peaks at 7.9–8.2 ppm.
Computational analysis
To assess the comparative binding of the triazole compounds of our previous study and the effects of increased linker length on binding, a series of molecular docking simulations were undertaken. Docking was performed with literature phosphodiester-linked compounds (Fig. 1) developed by Skropeta et al. (R-1),25 Li et al. (R-2),26 and Guo et al. (R-3, S-3).39 The S diastereomer of the amide compound 3 was included as that study did not include a determination of stereochemistry, similar to the synthesis seen here. Five 1,2,3-triazoles were also included to test the effects on binding of replacing cytidine (R-1) with uridine (R-14), the presence of a phosphonate (R-5 and R-15), and increasing linker length (R-16, R-17vs.R-13a), as shown in Fig. 2.
Fig. 2. The key compounds used in the docking study against hST3Gal I and hST6Gal I to probe the effects of linker length on binding, along with compound 5.

These compounds were docked into six snapshots from 100 ns of molecular dynamics simulations performed on ST6Gal I (PDB ID: 4JS2) as described by Montgomery et al.29 In addition to the docking into hST6Gal I, molecular dynamics simulations were also conducted with a homology model of hST3Gal I from SWISS-MODEL (UNIPROT ID: Q11201, model version 3.0.0),40 for 100 ns with the phosphodiester lead compound (R)-1, and snapshots taken at 0, 20, 40, 60, 80 and 100 ns timepoints for docking.
Docking was performed with AutoDock Vina (AD Vina) version 1.1.2 (ref. 41) for a box of 30 Å × 30 Å × 30 Å centred at the active site of each enzyme. Receptor structures were prepared for docking using AutoDockTools (ADT) version 4.2.6.42 In the docking procedure, the receptor is treated as rigid and no explicit waters have been included. The top ranked models for each compound tested based on the binding energies calculated by AD Vina were evaluated based upon comparison to the position of the phosphodiester lead compound (R-1) in the simulated snapshots VMD v1.9.4.43 The mean binding energies for all compounds across the snapshots are reported in Table 2.
Mean binding energies of 1,2,3-triazole linked and literature ST inhibitors. See Fig. 1 and 2 for structures.
| Compound | Binding energya (kcal mol−1) | Ref | ||||
|---|---|---|---|---|---|---|
| Phosphodiester | Nucl. | ST3Gal I | n b | ST6Gal I | n b | |
| (R)-1 | Cyt | −9.2 ± 0.3 | 3 | −9.6 ± 0.2 | 6 | 25 |
| (R)-2 | Cyt | −9.5 ± 0.3 | 3 | −9.0 ± 0.1 | 6 | 26 |
| (R)-3 | Cyt | −9.2 ± 0.2 | 3 | −9.4 ± 0.2 | 6 | 40 |
| (S)-3 | Cyt | −9.8 ± 0.3 | 3 | −9.8 ± 0.2 | 6 | 40 |
| (R)-14 | Uri | −9.8 ± 0.2 | 3 | −9.8 ± 0.2 | 6 | — |
| Triazole | ||||||
| (R)-5 | Uri | −9.9 ± 0.1 | 5 | −10.1 ± 0.1 | 6 | 32 |
| (R)-13a | Uri | −10.1 ± −0.7 | 4 | −10.0 ± 0.2 | 5 | — |
| (R)-15 | Uri | −10.1 ± 0.3 | 6 | −10.1 ± 0.2 | 6 | — |
| (R)-16 | Uri | −9.9 ± 0.3 | 5 | −10.2 ± 0.3 | 6 | — |
| (R)-17 | Uri | −9.9 ± 0.4 | 5 | −10.4 ± 0.3 | 6 | — |
Arithmetic mean of the binding energy ± SEM obtained from docking into six snapshots of the ST3Gal I (UNIPROT ID: Q11201) and ST6Gal I (PDB ID: 4JS2) simulations with the phosphodiester lead compound (R)-1.
Number of snapshots (max. possible is 6) with acceptable binding modes that were used to calculate the binding energy in the column immediately to the left.
The docking against ST6Gal I gave the greatest number of binding modes (n = 6) that were used to calculate the binding energies. Overall, the three literature compounds (R)-1, (R)-2, and (R)-3 all had the lowest calculated binding energies against ST6Gal I (of −9.6, −9.0 and −9.4 kcal mol−1 respectively), with the cyclopentyl compound (R)-2 having the lowest binding energy by a statistically significant margin. This may be due to fewer possible protein-ligand interactions identified by AD Vina for these compounds. The docking against ST6Gal I also suggests that increasing the length of the linker in these ST inhibitors may not negatively affect binding, as there was no statistical significance in the results of the triazole compounds, regardless of linker length. As shown in Fig. 3, this may be due to the large size of the binding pocket of ST6Gal I, which normally needs to accommodate both the CMP-Neu5Ac donor substrate and an acceptor glycan chain. A free energy perturbation study with hST6Gal I by Montgomery et al.30 has indicated that the flexibility of this binding pocket also compensates for the added rigidity of the triazole group, allowing the triazole to effectively mimic the phosphodiester linker of traditional ST inhibitors.
Fig. 3. Comparison of binding modes in ST6Gal I (PDB ID: 4JS2) between the (A) co-crystallised substrate CMP; (B) phosphodiester lead compound (R)-1; and (C) ether-linked 1,2,3-triazole based compound (R)-13a. The surface representation highlights the presence of H-bond donors in pink and H-bond acceptors in green. Figure made with Discovery Studio Visualiser.44.

Potential selectivity for ST6Gal I over ST3Gal I is observed, as more acceptable binding modes (n) were obtained for the compounds assessed against ST6Gal I, particularly for the ether-linked 1,2,3-triazole compound (R)-13a, as an example of the compounds synthesised herein. In some cases, no calculated binding modes correlated with the results for the molecular dynamics simulation. In part due to the high variance of binding conformations and averaging of only snapshots where there was deemed an acceptable binding mode, there was no statistical significance between the obtained binding energies against ST3Gal I. Due to these higher errors, there was no significant difference for any of the compounds between their average binding energies against ST3Gal I and ST6Gal I.
Fig. 4 shows the ligand-receptor interactions for phosphodiester (R)-1 and triazole-linked (R)-13a for their binding modes in the crystal structure of hST6Gal I. In particular, the hydrogen bonding interaction between the Tyr354 OH and the ligand phosphonate group is substituted for a hydrophobic interaction between the Tyr354 ring and the triazole CH, which could potentially contribute to the higher exhibited binding energy from the docking calculations.
Fig. 4. Predominant interactions from binding modes of the phosphodiester-linked lead compound (R)-1 and ether-linked triazole compound (R)-13a with the hST6Gal I crystal structure (PDB ID: 4JS2), analysed with Discovery Studio Visualizer.44 Hydrogen bonds are shown in red, and hydrophobic contacts in blue.

When compared to results from our previous investigation with the crystal structure of hST6Gal I, the observed interactions are largely similar.31 Here, we also see importance of hydrophobic contacts for binding of the ether-linked triazole compounds, as the aromatic structures of the phenyl rings and triazole itself participate in interactions with hydrophobic residues. These results highlight the importance of the acceptor binding region of the ST6Gal I active site for inhibitor design.
Biological evaluation
Biological assessment of the activity of these compounds against recombinant human ST3Gal I and ST6Gal I was carried out based on the CMP-Glo™ assay developed by Promega and detailed by Das et al.45 CMP-Neu5Ac was used as the donor and Gal-β1,3-GalNAc and LacNAc as the acceptors for hST3Gal I and hST6Gal I, respectively. CMP produced as a by-product of the ST reaction was detected using a luciferase reaction producing luminescence with a linear response to the concentration of CMP. With acceptor concentration set at 1 mM, the Km for CMP-Neu5Ac was calculated as 37.16 ± 5.42 μM for hST6Gal I, comparable to previously reported values.26 The percentage inhibition of all compounds at 10 and 100 μM (with 1 mM acceptor and 100 μM CMP-Neu5Ac) was initially calculated, and for those compounds with over 50% inhibition at 10 μM, Ki's were determined. Inhibition data for compounds against hST3Gal I is only shown for an inhibitor concentration of 100 μM as they were either only mildly or completely inactive against the enzyme. A summary of the inhibitor screening is shown in Table 3, with the previously described carbamate (4) and triazole (5) compounds included as references.31,32
Inhibitory activity of compounds against hST3Gal I and hST6Gal I.
| Compound | hST6Gal I | hST3Gal I | ||
|---|---|---|---|---|
| Inhibition at 10 μM (%) | K m or Ki (μM) | K m/Ki | Inhibition at 100 μM (%) | |
| CMP-Neu5Ac | — | 37 ± 5 | — | — |
| 4-(l) | 59 | 1.1 ± 0.1 | 32.5 | 38 |
| 5-(s/l) | 84a | — | — | 43 |
| 13a-(s) | 62 | 40 ± 10 | 1.0 | 28 |
| 13a-(l) | 26 | 50 ± 10 | 0.7 | 2 |
| 13b-(s) | 41 | — | — | — |
| 13b-(l) | 10 | — | — | — |
| 13c-(s) | 57 | 6 ± 1 | 6.0 | 43 |
| 13c-(l) | 59 | 34 ± 6 | 1.1 | 9 |
| 13d-(s) | 44 | — | — | — |
| 13d-(l) | 45 | — | — | — |
| 13e-(s) | 39 | — | — | — |
| 13e-(l) | 23 | — | — | — |
| 13f-(s) | 65 | 6.7 ± 0.6 | 5.6 | 49 |
| 13f-(l) | 78 | 3.4 ± 0.6 | 11.0 | 39 |
| 13g-(s) | 25 | — | — | — |
| 13g-(l) | 21 | — | — | — |
Inhibition for this compound was recorded at 100 μM inhibitor concentration.
Broadly, this series of compounds exhibited promising activity against hST6Gal I, with the m-phenoxy, m-phenoxy-p-fluoro, and m-1,1,2,2-tetrafluroethoxy (13a, c, and f, resp.) having Ki's in the mid-to-low micromolar range. The reduced activity of the compounds from this study against hST3Gal I correlates with the reduced binding affinity with the homology model observed in the docking study. Interestingly, it was noted that the compounds that showed activity against hST3Gal I in the range of 28–49% inhibition at 100 uM were also the most active against hST6Gal I in the range of 57–78% at 10 uM (e.g. see compounds 4, 13a, 13c, 13f). Due to this increased activity against hST6Gal I over hST3Gal I, it appears that these compounds show a degree of selectivity towards hST6Gal I.
Consistent with previous literature results on ST inhibitors, there are relatively small differences in activity between diastereomers of the most active inhibitors against hST6Gal I, for example 13f-(s) and 13f-(l) (Table 3). These results also confirm the positive impact of the phosphonate on potency, as shown by comparison of the ‘sans-phosphonate’ compound 5, which showed 84% inhibition at 100 μM, compared with the phosphonate bearing 13a-(s) and 13a-(l) (Ki = 40 and 50 μM, resp.). The higher activities of the m-phenoxy compounds (13a and 13c) are consistent with previous studies, with compounds such as (R)-1, (R)-2, and 3 all possessing a phenyl group in this position.
The most potent inhibitor in the series investigated here is the tetrafluoro compound 13f-(l) (Ki = 3.4 ± 0.6 μM; hST6Gal I), which could be due to the fluorinated substituent participating in similar binding interactions with the enzyme as the glycerol chain on the natural ST donor molecule CMP-Neu5Ac, as fluorine is known to participate in hydrogen binding-like interactions.46 The fluorine moiety could also participate in hydrophobic interactions with the enzyme binding site, similar to the m-phenoxy moiety on the literature compound (R)-1 and the other active triazole compounds 13a and 13c.
A wide array of assays has been used to evaluate the effectiveness of ST inhibitors based on HPLC, MS, fluorescence or luminescence activity readouts and on a range of both bacterial and non-human mammalian ST enzymes, with varying sequence homology. This makes it difficult to compare inhibitor activity values across studies. Herein, the three most active compounds exhibited Ki values of <10 μM in a luminescence-based assay over a single hour incubation period. Recent studies on a murine breast cancer model showed effectiveness in vivo, inhibiting tumour growth, reducing angiogenesis, and delaying metastasis of MDA-MB-231 cells. In this case, the inhibitors also showed activity in the 1–10 μM range against human ST6Gal I and rat ST3Gal III in an enzymatic assay.47
Conclusions
In conclusion, a series of 14 phosphonate-bearing, uridine-based 1,2,3-triazole compounds (13a–g) were synthesised and investigated for their activity as ST inhibitors and to probe any potential selectivity between two of the most therapeutically relevant STs (hST3Gal I and hST6Gal I). To achieve this, a series of novel α-azidophosphonates (11a–g) were synthesised and coupled to 5′-O-propargyluridine (8) via CuAAC chemistry, followed by deprotection and purification to yield a series of 14 phosphonate-bearing triazole compounds (13a–g). These transition-state analogues were then tested for activity against both hST3Gal I and hST6Gal I in vitro. The series appeared to exhibit selectivity for hST6Gal I over ST3Gal I in terms of a larger number of binding modes, with 6 compounds – 13a-(s),13a-(l), 13c-(s),13c-(l), 13f-(s) and 13f-(l) – showing promising activities (Ki = 3.4–53.7 μM; hST6Gal I) compared to other reported ST inhibitors.31,47
These experimental results support previous computational work that suggested 1,2,3-triazoles would be a suitable non-charged linker to replace the phosphodiester linker of lead literature compounds such as (R)-1. The ether-triazole series is also highly synthetically accessible, with only six steps required to yield final compounds, with an overall yield from uridine of up to 44%. With this encouraging activity against hST6Gal I, further investigation is underway on this series evaluating the effect of inhibiting sialylation on cell migration, invasion and tumour spheroid formation in pancreatic, ovarian and hepatic cancer. While panels of all 20 human ST enzymes are not yet commercially available, greatly expanded ST inhibitor screening capabilities in recent years19,48,49 enables the screening of our inhibitors against a wider panel of human ST enzymes in the future, along with computational studies on newly released ST crystal structures.50 Overall, the series herein is a promising step forward in the development of more metabolically stable, synthetically accessible and selective ST inhibitors.
Materials and methods
General docking procedure
Ligands were docked into the binding site of the hST3Gal I homology model (UNIPROT ID: Q11201, model version 3.0.0)40 and hST6Gal I crystal structure (PDB ID: 4JS2) with AutoDock Vina (AD-Vina) version 1.1.2.41 Receptor structures were prepared for docking using AutoDockTools (ADT) version 4.2.6.43 The three-dimensional structure of the inhibitors were prepared utilising ChemDraw 19.0 and Avogadro v1.1.1.51 ADT was used to assign both rigid and rotatable bonds and to remove non-polar hydrogens. Docking was performed for a box of 30 Å × 30 Å × 30 Å centred at the active site. These conditions have been used in our previous computational work.28,29 The receptor is treated as rigid and no explicit waters have been included. The top ranked models for each compound tested based on the binding energies calculated by AD-Vina were evaluated based upon comparison to the position of the ligand in the molecular dynamics simulations of hST3Gal I and hST6Gal I with VMD v1.9.2.43 Statistical analysis was undertaken on the calculated binding energies for each compound to determine if they were statistically different from one another, using a two-tailed students t-test. Results were deemed significant for P < 0.05.
Molecular dynamics simulations
For molecular dynamics simulations of the hST3Gal I homology model with compound (R)-1, the ligand was docked into the crystal structure using AD Vina according to the method above. The system was prepared via the same method as in previous work, with the same forcefields and conditions on the simulation.28 Molecular dynamic simulations were conducted with NAMD 2.12.52
General chemistry
All chemicals were purchased from Sigma Aldrich/Merck (USA), Carbosynth (UK) or ChemSupply (SA) and used as supplied or purified by standard methods. All reactions were performed under anhydrous conditions unless stated otherwise and monitored by TLC with Merck silica gel 60 F254 aluminium backed plates and visualised by UV or staining. Column chromatography was performed under ‘flash’ conditions on silica gel 60 (40–63 μm mesh). 1H, 13C, 19F, 31P, and 2D NMR spectra were acquired on Bruker Avance Neo 500 and 400 MHz NMR instruments. Chemical shifts for 1H and 13C NMR are given in ppm relative to residual solvent peaks or TMS. 19F NMR shifts are reported against an external reference of 0.05% α,α,α-trifluorotoluene in CDCl3, and 31P shifts against an external reference of 85% H3PO4 in D2O. Optical rotation was measured using a Jasco P-2000 polarimeter with specific rotation reported in degrees, and concentration (c) reported in g/100 mL. High resolution electrospray ionisation mass spectrometry (HRMS) was performed on a Waters XEVO G2 Q-TOF spectrometer with leucine enkephalin (LeuEnk) as an internal standard. Analytical RP-HPLC was performed with a Shimadzu Prominence-I LC2030C system with a PDA detector (190–800 nm), with a Luna C18 (2) 100 Å (Phenomenex, 3 μm, 4.6 × 150 mm) column. Preperative RP-HPLC was performed with a Shimadzu Prominence LC-20AP system with a PDS detector (190–800 nm) using a Prep C18 (Shimadzu, 5 μm, 20 × 150 mm) column. The purity of all final compounds was determined to be ≥95% based on 1H NMR data (see ESI†). Synthesis and characterisation of novel compounds not shown here (10e–f, 11a–g, and 12a–g) are detailed in the ESI.†
Deprotection of triazoles
Protected triazole compounds (1 equiv.) was dissolved in 10 mL MeOH and stirred with 10% (w/w) Pd/C catalyst under an atmosphere of hydrogen gas. After completion (judged by ESI-MS), the reaction mixture was filtered over celite and dried on a rotary evaporator to give a crude mixture of deprotected diastereomers. The diastereomers were separated via RP-HPLC, converted to their sodium salts by ion-exchange (IR 120 Na+) in water, and lyophilised to give the purified compounds. 1H, 13C, 31P, and 19F NMR spectra for these compounds are provided in the ESI.†
Disodium 5′-O-[1-(phosphonato-3-phenoxyphenylmethyl)-1,2,3-triazol-4-yl]methyluridine (13a)
From 12a (40.4 mg, 52.6 μmol): purification by RP-HPLC, converted to sodium salts by ion-exchange (IR 120 Na+) in water, and lyophilised to give the purified compounds 13a-(s) (16.0 mg, 99%), 13a-(l) (15.2 mg, 91%). 13a-(s): HPLC (prep. RP-C18, 34–39% MeOH): tR: 17.1 min. [α]25D = +8.4 (c = 0.14, H2O). 1H NMR (400 MHz, D2O): 8.62 (s, 1H), 7.60 (d, 1H, J = 8.0 Hz), 7.17 (t, 1H, J = 7.4 Hz), 4.85–4.72 (m, 2H), 5.38 (d, 1H, J = 8.3 Hz), 7.39–7.35 (m, 4H), 6.94–6.88 (m, 3H), 6.83 (s, 1H), 5.90 (d, 1H, J = 18.3 Hz), 5.89 (d, 1H, J = 4.5 Hz), 4.25 (m, 1H), 4.21 (t, 1H, J = 4.9 Hz), 4.17 (t, 1H, J = 4.7 Hz), 3.93–3.91 (m, 1H), 3.81–3.78 (m, 1H). 13C NMR (100 MHz, D2O): 162.2, 157.9, 157.7, 154.1, 145.0, 142.0, 141.3, 131.24, 131.15, 126.6, 124.9, 124.65, 124.61, 119.6, 119.0, 103.5, 90.0, 84.6, 75.5, 71.5, 70.1, 66.4 (d, J = 124.1 Hz), 64.7. 31P NMR (162 MHz, D2O): 8.36. ESI-HRMS (m/z): calculated for C25H26N5O10P− [M − H]−: 586.1339, found 586.1350. 13a-(l): HPLC (prep. RP-C18, 34–39% MeOH): tR: 19.9 min. [α]25D = −9.9 (c = 0.17, H2O). 1H NMR (500 MHz, D2O): 8.68 (s, 1H), 7.45–7.42 (m, 3H), 7.39–7.38 (m, 1H), 7.35–7.33 (m, 1H), 7.23 (t, 1H, J = 7.4 Hz), 7.01 (d, 2H, J = 8.0 Hz), 6.95 (d, 1H, J = 8.4 Hz), 6.92 (s, 1H), 5.91 (d, 1H, J = 18.5 Hz), 5.90 (d, 1H, J = 3.8 Hz), 5.53 (d, 1H, J = 7.9 Hz), 4.83–4.80 (m, 2H), 4.26 (m, 1H), 4.24–4.21 (m, 2H), 4.01–3.99 (m, 1H), 3.91–3.88 (m, 1H). 13C NMR (125 MHz, D2O): 162.6, 158.0, 157.8, 154.6, 144.8, 141.8, 141.3, 131.29, 131.19, 130.5, 126.5, 125.0, 124.8, 119.6, 119.1, 119.0, 103.5, 90.1, 84.3, 75.4, 71.1, 70.2, 66.5 (d, J = 124.5 Hz), 64.6. 31P NMR (162 MHz, D2O): 10.77. ESI-HRMS (m/z): calculated for C25H26N5O10P− [M − H]−: 586.1339, found 586.1329.
Disodium 5′-O-[1-(phosphonato-3-cyclopentoxyphenyl methyl)-1,2,3-triazol-4-yl]methyluridine (13b)
From 12b (172 mg, 0.163 mmol): purification by RP-HPLC, converted to sodium salts by ion-exchange (IR 120 Na+) in water, and lyophilised to give the purified compounds 13b-(s) (20.2 mg, 94%), 13b-(l) (21.3 mg, 99%). 13b-(s): HPLC (prep. RP-C18, 34–39% MeOH): tR: 17.6 min. [α]25D = −19.0 (c = 0.1, H2O). 1H NMR (500 MHz, D2O): 8.67 (s, 1H), 7.64 (d, 1H, J = 8.2 Hz), 7.30 (t, 1H, J = 7.9 Hz), 7.13 (d, 1H, J = 7.9 Hz), 6.89–6.86 (m, 2H), 5.96 (d, 1H, J = 4.3 Hz), 5.84 (d, 1H, 2J(H,P) = 18.3 Hz), 5.33 (d, 1H, J = 8.5 Hz), 4.86–4.75 (m, 2H), 4.81 (m, 1H), 4.28–4.20 (m, 3H), 3.96 (m, 1H), 3.84 (m, 1H), 1.95–1.86 (m, 2H), 1.71–1.61 (m, 6H). 13C NMR (125 MHz, D2O): 170.4, 162.5, 158.4, 155.4, 144.9, 142.0, 140.9, 130.9, 130.5, 127.2, 126.5, 122.2, 116.4, 116.1, 103.7, 90.1, 84.6, 81.8, 75.6, 71.6, 70.4, 64.9, 62.0 (d, 1J(C,P) = 125.0 Hz), 33.6, 24.9. 31P NMR (162 MHz, D2O): 7.90. ESI-HRMS (m/z): calculated for C24H30N5O10P [M − H]−: 578.1646, found 578.1652. 13b-(s): HPLC (prep. RP-C18, 34–39% MeOH): tR: 20.2 min. [α]25D = −14.8 (c = 0.1, H2O). 1H NMR (500 MHz, D2O): 8.72 (s, 1H), 7.36–7.22 (m, 2H), 7.11–7.07 (m, 1H), 6.91–6.85 (m, 2H), 5.89 (m, 1H), 5.84 (d, 1H, 2J(H,P) = 18.4 Hz), 5.34 (d, 1H, J = 8.4 Hz), 4.86–4.75 (m, 3H), 4.27–4.21 (m, 3H), 4.04–3.85 (m, 2H), 2.00–1.89 (m, 2H), 1.76–1.61 (m, 6H). 13C NMR (125 MHz, D2O): 168.7, 161.3, 157.0, 143.5, 140.3, 139.4, 129.5, 125.9, 125.1, 120.9, 114.9, 114.8, 102.2, 88.8, 82.9, 80.5, 74.2, 69.8, 69.0, 65.5 (d, 1J(C,P) = 121.9 Hz), 63.2, 32.8, 32.2, 23.5. 31P NMR (162 MHz, D2O): 7.98. ESI-HRMS (m/z): calculated for C24H30N5O10P [M − H]−: 578.1649, found 578.1652.
Disodium 5′-O-[1-(phosphonato-3-phenoxy-4-fluorophenyl methyl)-1,2,3-triazol-4-yl]methyluridine (13c)
From 12c (100 mg, 0.120 mmol): purification by RP-HPLC, converted to sodium salts by ion-exchange (IR 120 Na+) in water, and lyophilised to give the purified compounds 13c-(s) (38.1 mg, 97%), 13c-(l) (35.8 mg, 91%). 13c-(s): HPLC (prep. RP-C18, 34–39% MeOH): tR: 17.2 min. [α]25D = +1.5 (c = 0.1, H2O). 1H NMR (500 MHz, D2O): 8.58 (s, 1H), 7.54 (d, 1H, J = 7.9 Hz), 7.38–7.35 (m, 1H), 7.30 (t, 2H, J = 8.1 Hz), 7.20 (dd, 1H, J = 11.1, 8.7 Hz), 7.09 (t, 1H, J = 7.4 Hz), 6.89 (d, 2H, J = 8.0 Hz), 6.86 (d, 1H, J = 8.0 Hz), 5.84 (d, 1H, J = 4.2 Hz), 5.79 (d, 1H, 2J(H,P) = 18.5 Hz), 5.29 (d, 1H, J = 7.8 Hz), 4.71 (m, 2H), 4.16 (m, 1H), 4.09 (m, 2H), 3.80 (m, 2H). 13C NMR (125 MHz, D2O): 162.9, 158.4, 155.8, 145.1, 141.9, 138.0 (d, 1J(C,F) = 283.8 Hz), 131.3, 127.3, 126.8, 124.8, 118.5, 117.9, 103.7, 90.4, 84.5, 75.8, 71.5, 70.2, 66.0 (d, 1J(C,P) = 125.2 Hz), 64.9. 31P NMR (162 MHz, D2O): 7.68. 19F NMR (376 MHz, D2O): −132.8. ESI-HRMS (m/z): calculated for C25H24F4N5O10P− [M − H]−: 604.1245, found 604.1232. 13c-(l): HPLC (prep. RP-C18, 34–39% MeOH): tR: 19.8 min. [α]25D = −17.2 (c = 0.11, H2O). 1H NMR (500 MHz, D2O): 8.16 (s, 1H), 7.86 (d, 1H, J = 8.2 Hz), 6.90–6.88 (m, 2H), 6.54 (d, 1H, 2J(H,P) = 22.2 Hz), 5.88 (d, 1H, J = 4.5 Hz), 5.53 (d, 1H, J = 8.3 Hz), 5.03–4.94 (m, 4H), 4.68 (m, 2H), 4.12 (m, 3H), 3.79 (m, 2H). 13C NMR (125 MHz, D2O): 162.8, 158.3, 154.7, 143.6, 141.8, 137.9 (d, 1J(C,F) = 275.5 Hz), 131.3, 130.5, 127.3, 126.5, 124.8, 122.5, 118.0, 116.8, 102.3, 90.2, 84.3, 75.6, 71.1, 70.2, 66.0 (d, 1J(C,P) = 124.1 Hz), 64.7 31P NMR (162 MHz, D2O): 7.89. 19F NMR (376 MHz, D2O): −133.5. ESI-HRMS (m/z): calculated for C25H24F4N5O10P− [M − H]−: 604.1245, found 604.1238.
Disodium 5′-O-[1-(phosphonato-4-fluorophenylmethyl)-1,2,3-triazol-4-yl]methyluridine (13d)
From 12d (69.0 mg, 0.0995 mmol): purification by RP-HPLC, converted to sodium salts by ion-exchange (IR 120 Na+) in water, and lyophilised to give the purified compounds 13d-(s) (25.5 mg, 92%), 13d-(l) (17.0 mg, 62%). 13d-(s): HPLC (prep. RP-C18, 15–20% MeOH): tR: 27.2 min. [α]25D = −21.8 (c = 0.1, H2O). 1H NMR (500 MHz, D2O): 8.68 (s, 1H), 7.51–7.46 (m, 3H), 7.13–7.08 (m, 2H), 6.01 (m, 1H), 5.89 (d, 1H, J = 17.8 Hz), 5.36 (d, 1H, J = 7.9 Hz), 4.89–4.87 (m, 2H), 4.31–4.27 (m, 3H), 3.97 (m, 1H), 3.88 (m, 1H). 13C NMR (125 MHz, D2O): 172.5, 163.2 (d, J = 242.9 Hz), 156.9, 145.1, 141.6, 135.1, 131.04 (d, J = 4.6 Hz), 130.97 (d, J = 4.6 Hz), 126.4, 116.5 (d, J = 21.7 Hz), 103.7, 90.0, 84.7, 75.6, 72.0, 70.5, 66.1 (d, J = 126.6 Hz), 64.9. 31P NMR (162 MHz, D2O): 8.44. 19F NMR (376 MHz, D2O): −112.9. ESI-HRMS (m/z): calculated for C19H21FN5O9P− [M − H]−: 512.0983, found 512.1000. 13d-(l): HPLC (prep. RP-C18, 15–20% MeOH): tR: 30.4 min. [α]25D = −6.4 (c = 0.16, H2O). 1H NMR (500 MHz, D2O): 8.68 (s, 1H), 7.39–7.37 (m, 2H), 7.01 (m, 3H), 5.88 (d, 1H, J = 4.0 Hz), 5.83 (d, 1H, J = 18.2 Hz), 5.44 (d, 1H, J = 7.4 Hz), 4.80–4.77 (m, 2H), 4.20 (m, 1H), 4.17 (t, 1H, J = 4.6 Hz), 4.14 (t, 1H, J = 5.1 Hz), 3.99 (m, 1H), 3.87 (m, 1H). 13C NMR (125 MHz, D2O): 177.4, 163.2 (d, J = 242.4 Hz), 160.3, 144.8, 140.7, 135.0, 131.0 (d, J = 5.3 Hz), 130.9 (d, J = 4.4 Hz), 126.5, 116.4 (d, J = 22.3 Hz), 104.0, 90.5, 83.7, 75.4, 71.1, 70.5, 66.2 (d, J = 128.4 Hz), 64.6. 31P NMR (162 MHz, D2O): 8.33. 19F NMR (376 MHz, D2O): −115.4. ESI-HRMS (m/z): calculated for C19H21FN5O9P− [M − H]−: 512.0983, found 512.0982.
Disodium 5′-O-[1-(phosphonato-3-trifluoromethylphenyl methyl)-1,2,3-triazol-4-yl]methyluridine (13e)
From 12e (189 mg, 0.186 mmol): purification by RP-HPLC, converted to sodium salts by ion-exchange (IR 120 Na+) in water, and lyophilised to give the purified compounds 13e-(s) (52.1 mg, 92%), 13e-(l) (48.1 mg, 85%). 13e-(s): HPLC (prep. RP-C18, 26–31% MeOH): tR: 16.2 min. [α]25D = −2.6 (c = 0.3, H2O). 1H NMR (500 MHz, D2O): 8.80 (s, 1H), 7.79 (d, 1H, J = 8.0 Hz), 7.71 (d, 1H, J = 7.5 Hz), 7.66–7.62 (m, 2H), 7.64 (d, 1H, J = 8.0 Hz), 6.05 (d, 1H, J = 21.8 Hz), 6.03 (d, 1H, J = 4.3 Hz), 5.19 (d, 1H, J = 8.2 Hz), 4.97–4.87 (m, 2H), 4.42 (m, 1H), 4.39 (t, 1H, J = 4.5 Hz), 4.35 (t, 1H, J = 4.8 Hz), 4.10 (m, 1H), 3.97 (m, 1H). 13C NMR (125 MHz, D2O): 166.8, 152.8, 148.2, 145.2, 142.3, 139.8, 133.0, 130.5, 126.6, 125.8, 125.0, 102.9, 90.0, 84.9, 75.7, 71.6, 70.2, 66.1 (d, J = 125.7 Hz), 64.7. 31P NMR (162 MHz, D2O): 8.01. 19F NMR (376 MHz, D2O): −62.2. ESI-HRMS (m/z): calculated for C20H21F3N5O9P− [M − H]−: 562.0951, found 562.0961. 13e-(l): HPLC (prep. RP-C18, 26–31% MeOH): tR: 20.7 min. [α]25D = +4.9 (c = 0.25, H2O). 1H NMR (400 MHz, D2O): 8.81 (s, 1H), 7.78 (d, 1H, J = 7.4 Hz), 7.73 (d, 1H, J = 7.0 Hz), 7.72 (s, 1H), 7.63 (t, 1H, J = 7.6 Hz), 7.36 (d, 1H, J = 8.1 Hz), 6.08 (d, 1H, J = 18.2 Hz), 5.96 (d, 1H, J = 3.9 Hz), 5.44 (d, 1H, J = 8.0 Hz), 4.97–4.88 (m, 2H), 4.36–4.31 (m, 3H), 4.12 (m, 1H), 4.02 (m, 1H). 13C NMR (100 MHz, D2O): 166.7, 152.6, 145.0, 142.0, 139.6, 132.8, 130.4, 126.5, 125.7, 125.3, 103.2, 89.8, 84.4, 75.2, 71.1, 70.3, 66.1 (d, J = 123.3 Hz), 64.4. 31P NMR (162 MHz, D2O): 8.15. 19F NMR (376 MHz, D2O): −62.1. ESI-HRMS (m/z): calculated for C20H21F3N5O9P− [M − H]−: 562.0951, found 562.0932.
Disodium 5′-O-[1-(phosphonato-3-(1,1,2,2-tetrafluoroethoxy) phenylmethyl)-1,2,3-triazol-4-yl]methyluridine (13f)
From 12f (86.4 mg, 0.110 mmol): purification by RP-HPLC, converted to sodium salts by ion-exchange (IR 120 Na+) in water, and lyophilised to give the purified compounds 13f-(s) (34.1 mg, 95%), 13f-(l) (33.0 mg, 92%). 13f-(s): HPLC (prep. RP-C18, 12–16% ACN): tR: 16.3 min. [α]25D = −7.0 (c = 0.16, H2O). 1H NMR (400 MHz, D2O): 8.67 (s, 1H), 7.57 (d, 1H, J = 8.4 Hz), 7.43 (d, 1H, J = 7.6 Hz), 7.38 (t, 1H, J = 7.8 Hz), 7.15 (d, 1H, J = 7.7 Hz), 7.08 (s, 1H), 6.21 (tt, 1H, 2J(H,F) = 52.4 Hz, 3J(H,F) = 2.7 Hz), 5.87 (d, 1H, J = 4.4 Hz), 5.86 (d, 1H, 2J(H,P) = 18.0 Hz), 5.07 (d, 1H, J = 8.0 Hz),4.81–4.72 (m, 2H), 4.24–4.15 (m, 3H), 3.88 (m, 2H). 13C NMR (100 MHz, D2O): 162.5, 155.5, 149.1, 144.3, 141.0, 140.7, 130.5, 127.2, 126.0, 121.4, 120.9, 116.6 (t, 1J(C,F) = 271.0 Hz), 108.0 (t, 1J(C,F) = 245.8 Hz), 102.6, 89.7, 83.6, 75.1, 70.5, 69.3, 65.6 (d, 1J(C,P) = 123.6 Hz), 64.1. 31P NMR (162 MHz, D2O): 7.59. 19F NMR (376 MHz, D2O): −88.4, −137.7. ESI-HRMS (m/z): calculated for C21H21F4N5O10P [M − H]−: 610.0962, found 610.0961. 13f-(l): HPLC (prep. RP-C18, 12–16% ACN): tR: 19.4 min. [α]25D = −3.8 (c = 0.11, H2O). 1H NMR (400 MHz, D2O): 8.54 (s, 1H), 7.23–7.18 (m, 2H), 7.04 (d, 1H, J = 7.9 Hz), 7.00 (d, 1H, J = 7.1 Hz), 6.93 (s, 1H), 6.08 (tt, 1H, 2J(H,F) = 52.6 Hz, 3J(H,F) = 3.0 Hz), 5.70 (d, 1H, 2J(H,P) = 15.9 Hz), 5.67 (d, 1H, J = 5.6 Hz), 5.15 (d, 1H, J = 7.7 Hz), 4.62 (m, 2H), 4.03–3.99 (m, 3H), 3.82–3.68 (m, 2H). 13C NMR (100 MHz, D2O): 161.7, 156.8, 148.4, 143.5, 140.9, 140.1, 129.7, 126.3, 125.3, 120.5, 120.4, 116.6 (t, 1J(C,F) = 271.0 Hz), 107.8 (t, 1J(C,F) = 245.0 Hz), 102.2, 88.9, 82.7, 74.1, 69.7, 68.9, 65.0 (d, 1J(C,P) = 126.0 Hz), 63.1. 31P NMR (162 MHz, D2O): 7.60. 19F NMR (376 MHz, D2O): −88.3, −137.7. ESI-HRMS (m/z): calculated for C21H21F4N5O10P [M − H]−: 610.0962, found 610.0958.
Disodium 5′-O-[1-(phosphonatobenzothiophen-3-ylmethyl)-1,2,3-triazol-4-yl]methyluridine (13g)
From 12g (42.9 mg, 58.6 μmol): purification by RP-HPLC, converted to sodium salts by ion-exchange (IR 120 Na+) in water, and lyophilised to give the purified compounds 13g-(s) (16.8 mg, 96%), 13g-(l) (16.2 mg, 92%). 13g-(s): HPLC (prep. RP-C18, 26–31% MeOH): tR: 22.1 min. [α]25D = −13.9 (c = 0.1, H2O). 1H NMR (500 MHz, CDCl3): 8.31 (s, 1H), 8.08 (s, 1H), 8.01 (s, 1H), 7.67 (d, 1H, J = 7.9 Hz), 7.35 (m, 1H), 7.10 (m, 2H), 6.96 (d, 1H, J = 7.9 Hz), 6.04 (d, 1H, J = 17.0 Hz), 5.47 (d, 1H, J = 3.7 Hz), 4.94 (d, 1H, J = 8.1 Hz), 4.61–4.43 (m, 2H), 3.93 (m, 1H), 3.86 (t, 1H, J = 5.1 Hz), 3.81 (t, 1H, J = 4.2 Hz), 3.74–3.72 (m, 1H), 3.58–3.56 (m, 1H). 13C NMR (125 MHz, CDCl3): 162.9, 161.3, 144.5, 140.6, 139.0, 131.2, 130.2, 126.8, 125.7, 125.2, 124.0, 122.1, 102.5, 89.7, 83.5, 75.3, 70.3, 68.4, 63.8, 59.3 (d, J = 122.7 Hz).31P NMR (162 MHz, CDCl3): 8.19 ESI-HRMS (m/z): calculated for C21H22N5O9PS [M − H]−: 550.0798, found 550.0789. 13g-(l): HPLC (prep. RP-C18, 26–31% MeOH): tR: 25.8 min. [α]25D = −18.7 (c = 0.1, H2O). 1H NMR (500 MHz, CDCl3): 8.28 (s, 1H), 8.17 (s, 1H), 7.86 (m, 1H), 7.56 (m, 1H), 7.28 (m, 2H), 6.93 (d, 1H, J = 7.9 Hz), 6.20 (d, 1H, J = 17.1 Hz), 5.71 (d, 1H, J = 4.4 Hz), 5.06 (d, 1H, J = 7.9 Hz), 4.82–4.68 (m, 2H), 4.11 (m, 1H), 3.98 (t, 1H, J = 4.7 Hz), 3.93 (t, 1H, J = 4.8 Hz), 3.86–3.83 (m, 1H), 3.77–3.73 (m, 1H). 13C NMR (125 MHz, CDCl3): 163.5, 155.8, 145.1, 140.8, 139.5, 139.4, 131.8, 129.3, 126.0, 125.8, 125.7, 124.5, 122.3, 103.3, 89.9, 84.4, 75.3, 71.5, 70.4, 64.6, 59.9 (d, J = 120.1 Hz). 31P NMR (162 MHz, CDCl3): 8.38. ESI-HRMS (m/z): calculated for C21H22N5O9PS [M − H]−: 550.0798, found 550.0783.
CMP-Glo™ based sialyltransferase assay
Biological assay method and conditions were identical to those described in our previous work.31 Details are given in the ESI.†
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
We thank Phil Clingan, Maxine Stewart and the Illawarra Cancer Carers for financial support including a PhD scholarship for C. D. and R. S. matched by the University of Wollongong. H. Y. is the recipient of an Australian Research Council (ARC) Future Fellowship (Project number FT110100034) and A. M. received an Australian Government Research Training Program Award. This research was in part supported by an ARC Discovery Project (DP170101773), and with resources at the NCI National Facility at the Australian National University through the National Computational Merit Allocation Scheme. We also thank Dr Andrew Tague for his technical support.
Electronic supplementary information (ESI) available. See DOI: 10.1039/d1md00079a
Notes and references
- Munkley J. Elliott D. J. Oncotarget. 2016;7:35478–35489. doi: 10.18632/oncotarget.8155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo R. Skropeta D. Med. Res. Rev. 2017;37:219–270. doi: 10.1002/med.21407. [DOI] [PubMed] [Google Scholar]
- Dobie C. Skropeta D. Br. J. Cancer. 2021;124:76–90. doi: 10.1038/s41416-020-01126-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y. Pan D. Bellis S. L. Song Y. Proteins. 2008;73:989–1000. doi: 10.1002/prot.22126. [DOI] [PubMed] [Google Scholar]
- Chiang C. H. Wang C. H. Chang H. C. More S. V. Li W. S. Hung W. C. J. Cell. Physiol. 2010;223:492–499. doi: 10.1002/jcp.22068. [DOI] [PubMed] [Google Scholar]
- Alessandro N. Mariah L. F. Sophie H. Carolyne F. Lucy K.-M. Matthew S. M. Michaela R. R. Michael O. D. Haematologica. 2020;105:457–467. doi: 10.3324/haematol.2018.212266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macauley M. S. Crocker P. R. Paulson J. C. Nat. Rev. Immunol. 2014;14:653–666. doi: 10.1038/nri3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues E. Macauley M. S. Cancers. 2018;10:207–226. doi: 10.3390/cancers10060207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swindall A. F. Bellis S. L. J. Biol. Chem. 2011;286:22982–22990. doi: 10.1074/jbc.M110.211375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz M. J. Swindall A. F. Wright J. W. Sztul E. S. Landen C. N. Bellis S. L. J. Ovarian Res. 2013;6:25. doi: 10.1186/1757-2215-6-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X. Zhao J. Ruan Y. Sun L. Xu C. Jiang H. Cell Death Discovery. 2018;9:1102. doi: 10.1038/s41419-018-1101-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Punch P. R. Irons E. E. Manhardt C. T. Marathe H. Lau J. T. Y. Glycobiology. 2020;30:446–453. doi: 10.1093/glycob/cwz108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M. Lee H. J. Bae S. Lee Y. S. Mol. Cancer Res. 2008;6:1316–1325. doi: 10.1158/1541-7786.MCR-07-2209. [DOI] [PubMed] [Google Scholar]
- Steele H. T. Tague A. Skropeta D. Curr. Med. Chem. 2021 doi: 10.2174/0929867328666210201153901. [DOI] [PubMed] [Google Scholar]
- Li Y. Chen X. Appl. Microbiol. Biotechnol. 2012;94:887–905. doi: 10.1007/s00253-012-4040-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lairson L. L. Henrissat B. Davies G. J. Withers S. G. Annu. Rev. Biochem. 2008;77:521–555. doi: 10.1146/annurev.biochem.76.061005.092322. [DOI] [PubMed] [Google Scholar]
- Harduin-Lepers A. Vallejo-Ruiz V. Krzewinski-Recchi M. A. Samyn-Petit B. Julien S. Delannoy P. Biochimie. 2001;83:727–737. doi: 10.1016/S0300-9084(01)01301-3. [DOI] [PubMed] [Google Scholar]
- Audry M. Jeanneau C. Imberty A. Harduin-Lepers A. Delannoy P. Breton C. Glycobiology. 2011;21:16–26. doi: 10.1093/glycob/cwq189. [DOI] [PubMed] [Google Scholar]
- Moremen K. W. Ramiah A. Stuart M. Steel J. Meng L. Forouhar F. Moniz H. A. Gahlay G. Gao Z. Chapla D. Wang S. Yang J. Y. Prabhakar P. K. Johnson R. Rosa M. D. Geisler C. Nairn A. V. Seetharaman J. Wu S. C. Tong L. Gilbert H. J. LaBaer J. Jarvis D. L. Expression system for structural and functional studies of human glycosylation enzymes. Nat. Chem. Biol. 2018;14:156–162. doi: 10.1038/nchembio.2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seales E. C. Jurado G. A. Brunson B. A. Wakefield J. K. Frost A. R. Bellis S. L. Cancer Res. 2005;65:4645–4652. doi: 10.1158/0008-5472.CAN-04-3117. [DOI] [PubMed] [Google Scholar]
- Park J.-J. Lee M. Gut Liver. 2013;7:629–641. doi: 10.5009/gnl.2013.7.6.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bos P. D. Zhang X. H. Nadal C. Shu W. Gomis R. R. Nguyen D. X. Minn A. J. van de Vijver M. J. Gerald W. L. Foekens J. A. Massague J. Nature. 2009;459:1005–1009. doi: 10.1038/nature08021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Garay M. Arteta B. Llop E. Cobler L. Pages L. Ortiz R. Ferri M. J. de Bolos C. Figueras J. de Llorens R. Vidal-Vanaclocha F. Peracaula R. Int. J. Biochem. Cell Biol. 2013;45:1748–1757. doi: 10.1016/j.biocel.2013.05.015. [DOI] [PubMed] [Google Scholar]
- Macauley M. S. Arlian B. M. Rillahan C. D. Pang P.-C. Bortell N. Marcondes M. C. G. Haslam S. M. Dell A. Paulson J. C. J. Biol. Chem. 2014;289:35149–35158. doi: 10.1074/jbc.M114.606517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skropeta D. Schworer R. Haag T. Schmidt R. R. Glycoconjugate J. 2004;21:205–219. doi: 10.1023/B:GLYC.0000045093.96413.62. [DOI] [PubMed] [Google Scholar]
- Li W. Niu Y. Xiong D.-C. Cao X. Ye X.-S. J. Med. Chem. 2015;58:7972–7990. doi: 10.1021/acs.jmedchem.5b01181. [DOI] [PubMed] [Google Scholar]
- Kumar R. Nasi R. Bhasin M. Huan Khieu N. Hsieh M. Gilbert M. Jarrell H. Zou W. Jennings H. J. Carbohydr. Res. 2013;378:45–55. doi: 10.1016/j.carres.2012.12.017. [DOI] [PubMed] [Google Scholar]
- Dobie C. Montgomery A. P. Szabo R. Skropeta D. Yu H. J. Mol. Recognit. 2018;31:e2684. doi: 10.1002/jmr.2684. [DOI] [PubMed] [Google Scholar]
- Montgomery A. Szabo R. Skropeta D. Yu H. J. Mol. Recognit. 2016;29:210–222. doi: 10.1002/jmr.2520. [DOI] [PubMed] [Google Scholar]
- Montgomery A. P. Skropeta D. Yu H. Sci. Rep. 2017;7:14428. doi: 10.1038/s41598-017-14560-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery A. P. Dobie C. Szabo R. Hallam L. Ranson M. Yu H. Skropeta D. Bioorg. Med. Chem. 2020;28:115561. doi: 10.1016/j.bmc.2020.115561. [DOI] [PubMed] [Google Scholar]
- Szabo R., Doctor of Philosophy, University of Wollongong, 2017, https://ro.uow.edu.au/theses1/290 [Google Scholar]
- Sun J. Liu R. Fu Q. Zang J. Tao Q. Wu J. Zhu H. Helv. Chim. Acta. 2014;97:733–743. doi: 10.1002/hlca.201300289. [DOI] [Google Scholar]
- Golden K. C. Gregg B. T. Quinn J. F. Tetrahedron Lett. 2010;51:4010–4013. doi: 10.1016/j.tetlet.2010.05.116. [DOI] [Google Scholar]
- Wong O. A. Shi Y. J. Org. Chem. 2009;74:8377–8380. doi: 10.1021/jo901553t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- But T. Y. S. Toy P. H. Chem. – Asian J. 2007;2:1340–1355. doi: 10.1002/asia.200700182. [DOI] [PubMed] [Google Scholar]
- Hein J. E. Fokin V. V. Chem. Soc. Rev. 2010;39:1302–1315. doi: 10.1039/B904091A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meldal M. Tornøe C. W. Chem. Rev. 2008;108:2952–3015. doi: 10.1021/cr0783479. [DOI] [PubMed] [Google Scholar]
- Guo J. Li W. Xue W. Ye X.-S. J. Med. Chem. 2017;60:2135–2141. doi: 10.1021/acs.jmedchem.6b01644. [DOI] [PubMed] [Google Scholar]
- Waterhouse A. Bertoni M. Bienert S. Studer G. Tauriello G. Gumienny R. Heer F. T. de Beer T. A. P. Rempfer C. Bordoli L. Lepore R. Schwede T. Nucleic Acids Res. 2018;46:W296–W303. doi: 10.1093/nar/gky427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott O. Olson A. J. J. Comput. Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris G. M. Huey R. Lindstrom W. Sanner M. F. Belew R. K. Goodsell D. S. Olson A. J. J. Comput. Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W. Dalke A. Schulten K. J. Mol. Graphics. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]; 27–38
- BIOVIA D. S., Discovery Studio, v20.1, Dassault Systèmes, San Diego, 2020 [Google Scholar]
- Das D. Walvoort M. T. Lukose V. Imperiali B. Sci. Rep. 2016;6:33412. doi: 10.1038/srep33412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillis E. P. Eastman K. J. Hill M. D. Donnelly D. J. Meanwell N. A. J. Med. Chem. 2015;58:8315–8359. doi: 10.1021/acs.jmedchem.5b00258. [DOI] [PubMed] [Google Scholar]
- Fu C.-W. Tsai H.-E. Chen W.-S. Chang T.-T. Chen C.-L. Hsiao P.-W. Li W.-S. J. Med. Chem. 2021;64:527–542. doi: 10.1021/acs.jmedchem.0c01477. [DOI] [PubMed] [Google Scholar]
- Sheikh M. O. Halmo S. M. Patel S. Middleton D. Takeuchi H. Schafer C. M. West C. M. Haltiwanger R. Avci F. Y. Moremen K. W. Wells L. Rapid screening of sugar-nucleotide donor specificities of putative glycosyltransferases. Glycobiology. 2017;27:206–212. doi: 10.1093/glycob/cww114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noel M. Gilormini P. A. Cogez V. Lion C. Biot C. Harduin-Lepers A. Guérardel Y. Bioconjugate Chem. 2018;29:3377–3384. doi: 10.1021/acs.bioconjchem.8b00529. [DOI] [PubMed] [Google Scholar]
- Harrus D. Harduin-Lepers A. Glumoff T. J. Struct. Biol. 2020;212:107628. doi: 10.1016/j.jsb.2020.107628. [DOI] [PubMed] [Google Scholar]
- Hanwell M. D. Curtis D. E. Lonie D. C. Vandermeersch T. Zurek E. Hutchison G. R. Aust. J. Chem. 2012;4:17. doi: 10.1186/1758-2946-4-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips J. C. Braun R. Wang W. Gumbart J. Tajkhorshid E. Villa E. Chipot C. Skeel R. D. Kale L. Schulten K. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.