

Abstract
Despite the important roles played by protein–protein interactions (PPIs) in disease, they have been long considered as ‘undruggable’. However, recent advances have suggested that PPIs are druggable but may not follow conventional rules of ‘drug ability’. Here we explore which physicochemical parameters are essential for a PPI modulator to be a clinical drug by analysing the physicochemical properties of small-molecule PPI modulators in the market, in clinical trials, and published. Our analysis reveals that those compounds currently on the market have a larger range of values for most of the physicochemical parameters, whereas those in clinical trials fit much more stringently to standard drug-like parameters. This observation was particularly true for molecular weight, clog P and topological polar surface area, where aside from a few outliers, most of the compounds in clinical trials fit within standard drug-like parameters. This implies that the newer PPI modulators are more drug-like than those currently on the market, suggesting that designing new PPI-specific screening libraries should remain within standard drug-like parameters in order to obtain a clinical candidate. Taken together, our analysis has important implications for designing future drug discovery campaigns aimed at targeting PPIs.
The physicochemical properties of protein–protein interaction (PPI) modulators vary between those on the market, those in clinical trials, and those in the early drug discovery pipeline.
Introduction
Despite the vast scientific and technological advancements made since the 1950s, drug development costs had shown a seemingly paradoxical twofold increase every nine years with drug approvals per US$ 1 billion global R&D dramatically decreasing.1 Aside from the obvious tightening of regulatory laws, another proposed reason is the “low-hanging fruit” phenomenon, i.e. all the easily druggable targets have been picked. Therefore, alternative strategies in drug discovery have been the focus of scientists in recent years. One such strategy is to broaden the target space for drug discovery to include protein–protein interaction (PPI) inhibitors. This approach should lead to a wide variety of new targets and as such new therapeutics.2,3
There are over 300 000 PPI pairs so far predicted in the genome.4 The plethora of genomic data and advances in computer power have meant that, for the first time, we can begin to understand the complex protein–protein interaction networks that are occurring in the cell.5,6 These advances in computational systems and network biology have confirmed the importance of this network structure and have revealed that many PPIs represent important targets for drug discovery.
In 2020 there were 2374 small-molecule PPI inhibitors registered in iPPI-DB, a manually curated dataset of small-molecule PPI inhibitors.7,8 This is a 48% increase from 2016 and highlights the burgeoning interest in small-molecule drug design for PPIs. Furthermore, in the past year, there have been over 339 publications listed in PubMed related to “protein–protein interaction” AND “inhibitor”, emphasising this significant interest in PPI inhibition.
PPIs are defined as instances of specific, physical contacts that occur between two or more protein molecules in living organisms. Although challenging, recently there have been many advances in our understanding of how to drug protein–protein interfaces,9–11 leading to breakthroughs into discovering small molecules for these targets, which may pave the way for a new surge of marketed drugs.12
PPI modulators can be either inhibitors or stabilisers (both orthosteric and allosteric) and are primarily small-molecule modulators, peptides, and antibodies.13 Small-molecule modulators offer the advantage of high cell membrane permeability, oral administration, and lower research costs, whereas peptides and antibodies have higher specificity and affinity. In terms of disadvantages, the low selectivity of small-molecule modulators makes them more prone to side effects; peptides are susceptible to degradation by hydrolases, leading to instability in vivo, low oral bioavailability and a shorter half-life,14 whereas antibody usage is usually constrained to extracellular targets owing to their inability to cross cell membranes. Antibodies have also been known to activate severe immune reactions.13 Although all modalities are viable therapeutics for PPIs, this paper will focus on the desirable properties of small-molecule modulators of PPIs only.
Although most of the research in this field has been focussed on non-covalent modulation, covalent PPI inhibitors are being recognised in recent years due to smaller risks associated with drug resistance in oncology and infectious diseases.15 Specifically, due to the irreversible warhead nature of covalent inhibitors, they are often able to retain activity in situations where drug-resistant mutations have arisen due to treatment with reversible inhibitors. Evidence suggests that these inhibitors offer higher potency, are less limited by their pharmacokinetics and that the implementation of covalent bifunctional blockers could help reduce side effects.16
Even with numerous triumphs, modulating PPIs represents a significant challenge. This seemingly undruggable nature of PPIs has been attributed to several factors. PPI interfaces, unlike enzymes and ligand-binding domains of receptors, have not evolved to bind to small molecules. Many PPI interfaces tend to be flat and devoid of the usual cavities present on typical proteins that allow binding to ligands.17 The interface area of PPIs also tends to be larger (1500–3000 Å) and more hydrophobic than conventional drug interactions.18
These challenges suggest that small-molecule-based modulators of PPIs may require different design strategies and/or follow a separate set of rules than traditional small-molecule drugs. This is especially true with respect to achieving oral bioavailability, the gold standard of small-molecule drug discovery, due to its non-invasive and convenient nature. Traditional drug discovery approaches have used rules such as Lipinski's rule of 5 (RO5) to predict the suitability of oral drug candidates.19 These rules add strict levels to parameters such as molecular weight (<500 daltons), partition coefficient (<5.0), and hydrogen bond acceptors (<10.0) and donors (<5.0). However, the atypical characteristics of PPI targets suggest that these rules need to be rewritten to tap into the immense potential of protein–protein interaction drugs.
In recent years, PPI specific variants to Lipinski's rule of 5 have been suggested such as the ‘rule of four’.22–25 This rule of four suggests that generic PPI modulators should have a MW >400 Da, clog P >4, number of rings >4 and number of hydrogen bond acceptors >4, a direct contrast to Lipinski's rule of 5. However, whether drug discovery efforts towards orally bioavailable PPI modulators should preferably focus on RO5 space or follow additional trends remains an open question for the field.
To address this question, here we analysed the physicochemical properties of compounds on the market and in clinical trials to determine if the rule-of-four trends hold for the subset of PPI modulators that have advanced beyond preclinical studies and into clinical trials and beyond. Our analysis suggests that although the majority of approved drugs that modulate PPIs fall outside the RO5 parameter space, the compounds currently in clinical trials are more RO5 compliant. This insight may have important implications for informing drug discovery efforts in this area.
Methods
To compile a comprehensive list of small-molecule PPI modulators currently in the market and in clinical trials, a broad search of peer-reviewed journals was conducted. PubMed, RMIT LibrarySearch, Scopus and Google Scholar were explored using key terms such as “protein protein interaction” and its variants (e.g. PPI and 2P2I) combined with the term “drugs” and its variants (e.g. modulators, inhibitors, and stabilizers). Examples of specific Boolean operators are shown in the examples below:
“protein protein interaction” AND pharmacology
“protein protein interaction” OR 2P2I OR PPI OR 2P2Is AND disease OR treatment
(“protein protein interaction” OR 2P2I OR PPI OR 2P2Is)
AND (drugs OR medicine OR pharmaceutical)
This search process uncovered 159 compounds that were PPI drugs or candidates. Antibody-based PPI modulators and peptides were then removed from the dataset and compounds were split based on their clinical trial status (i.e. approved, stage I, II, III or IV).
To obtain a control data set of other drugs on the market, we utilised the DrugBank database,26 downloaded on the 20th June 2021. All PPI modulators were removed.
Compound physicochemical properties were calculated using ChemAxon software (http://chemaxon.com/) to obtain physiochemical properties including molecular weight (MW), partition coefficient (clog P), number of hydrogen bond donors (HBDs), number of hydrogen bond acceptors (HBAs), topological polar surface area (TPSA), number of rotatable bonds (RBs), and the number of rings.
GraphPad Prism v 9.1.0 was used to conduct statistical analyses and construct graphs. Specifically, descriptive statistics were utilised including minimum, maximum, range, mean, standard deviation, standard error of the mean, 25th quartile, median, 75th quartile, 90th quartile, coefficient of variation, skewness and kurtosis, and quadratic mean. To gain insight into the density of data at different values, violin plots were utilised to plot most of the data. Like a box plot, these plots show the range of values with the median and interquartile ranges displayed. Furthermore, these plots allow one to visualise the density of the data, i.e. wider regions correspond to more data points.
Results
PPI inhibitors on the market
Our literature search found a total of 25 approved PPI small-molecule drugs (Table 1). As expected from the analysis of others,21,27,28 76% percent of the small-molecule PPI modulators on the market failed Lipinski's RO5. This observation agrees with the notion that most PPI drugs are RO5 outliers. Specifically, in our list of 25 compounds, only six compounds (colchicine, levetiracetam, selinexor, tafamidis, tirofiban, and vorinostat) did not break the Lipinski RO5 criterion. Two of these drugs (colchicine and tirofiban) are naturally derived products, whereas the remaining four are synthetic. In recent years there has been discussion regarding alternate types of PPI interfaces, with the suggestion that some are easier to target than others.18,25 However, all six RO5 PPI modulators act at alternate PPI interfaces and modalities. Therefore, due to the limited number of PPI modulators on the market, we chose to analyse all small-molecule PPI modulators together irrespective of the type of interaction they modulate.
Physicochemical properties of clinically approved PPI modulators. Shown are the PPI target, method of discovery, i.e. derived from natural products or synthetic, molecular weight (MW), partition coefficient (clog P), topological polar surface area (TPSA), number of hydrogen bond donors (#HBD), number of hydrogen bond acceptors (#HBA), number of rotatable bonds (#RB), number of rings (#ring), number of carbons (#C), number of heteroatoms (#Het), and number of heavy atoms (#HA).
| PPI drug | Target | Method | MW (Da) | clog P | TPSA (Å2) | #HBD | #HBA | #RB | #ring | #C | #Het | #HA |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
Avatrombopag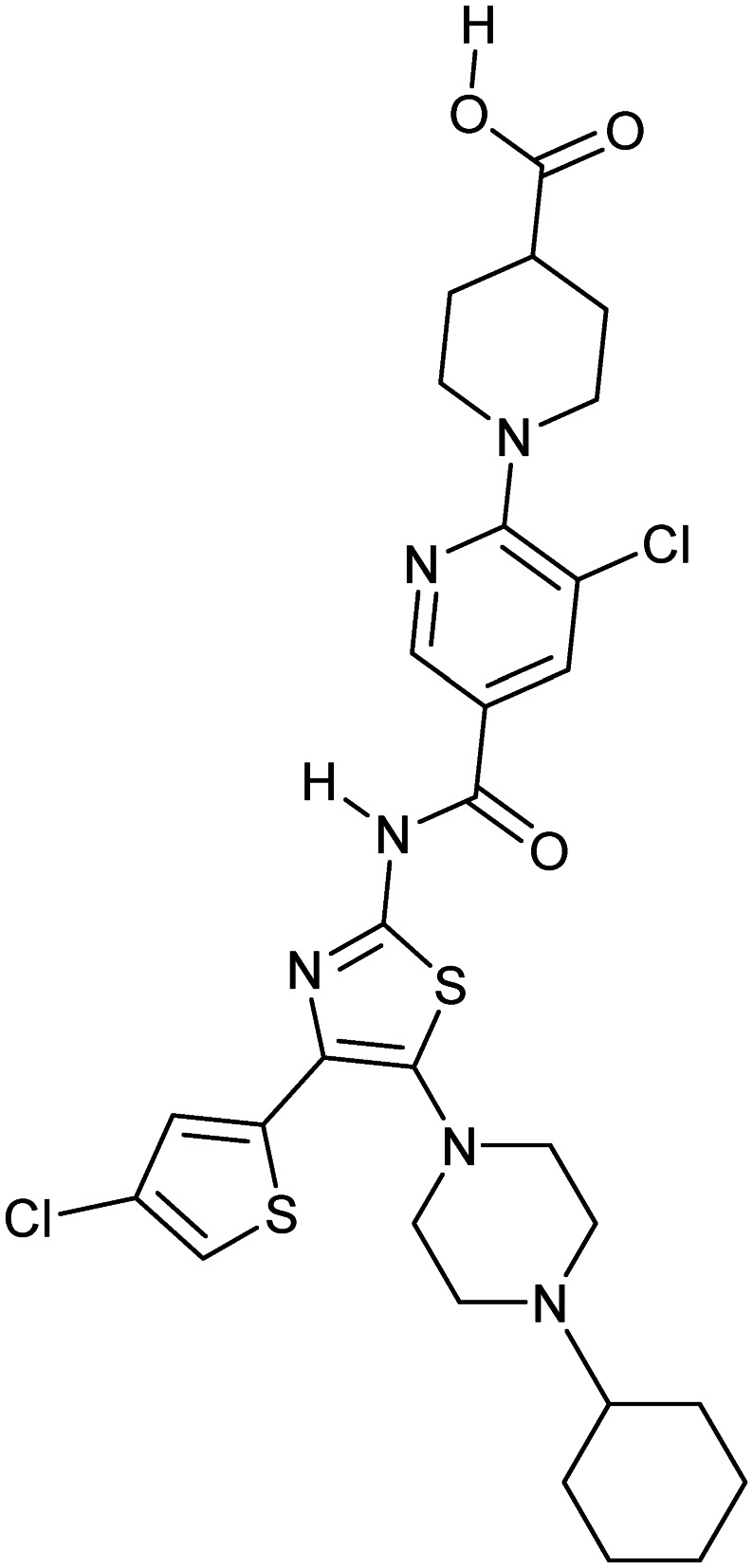
|
MPL/TPO | Synthetic | 649.7 | 4.13 | 158 | 2 | 10 | 7 | 6 | 29 | 13 | 42 |
Cabazitaxel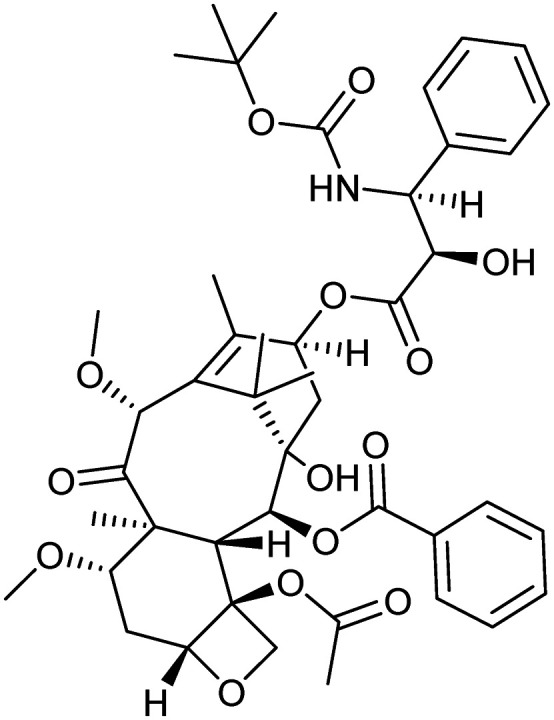
|
Microtubule | Natural | 835.9 | 5.44 | 202 | 3 | 14 | 15 | 5 | 45 | 15 | 60 |
Colchicine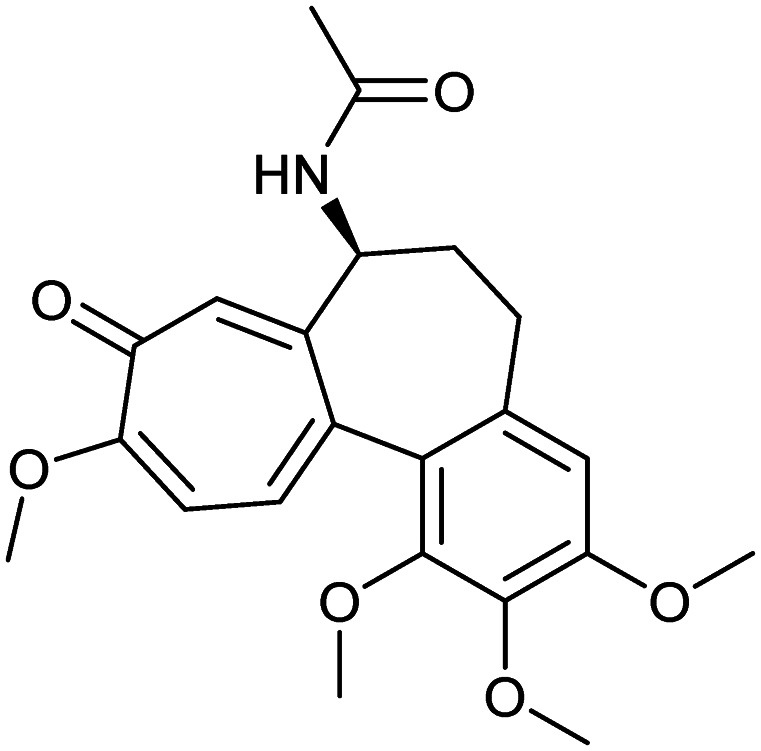
|
Microtubule | Natural | 399.4 | 1.20 | 83 | 1 | 6 | 5 | 3 | 22 | 7 | 29 |
Dimethyl fumarate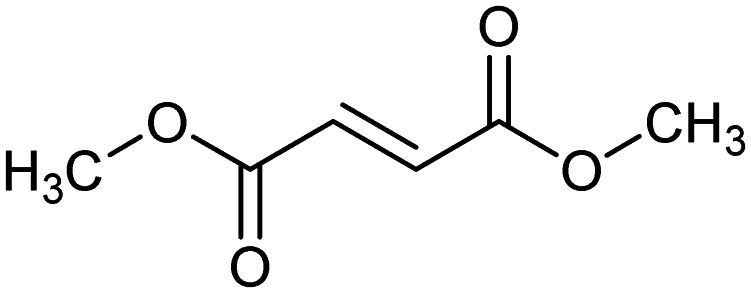
|
KEAP1/NRF2 | Synthetic | 144.1 | 0.78 | 52.6 | 0 | 4 | 4 | 0 | 6 | 4 | 10 |
Docetaxel
|
Microtubule | Synthetic | 807.9 | 4.08 | 224 | 5 | 14 | 13 | 5 | 43 | 15 | 58 |
Eltrombopag
|
MPL/TPO | Synthetic | 442.5 | 5.25 | 115 | 3 | 7 | 5 | 4 | 25 | 8 | 33 |
Eribulin mesylate
|
Microtubule | Natural | 826 | 1.24 | 209 | 3 | 15 | 4 | 8 | 41 | 16 | 57 |
Everolimus
|
FKBP12/MTOR | Natural | 958.2 | 7.10 | 205 | 3 | 14 | 9 | 2 | 53 | 15 | 68 |
Levetiracetam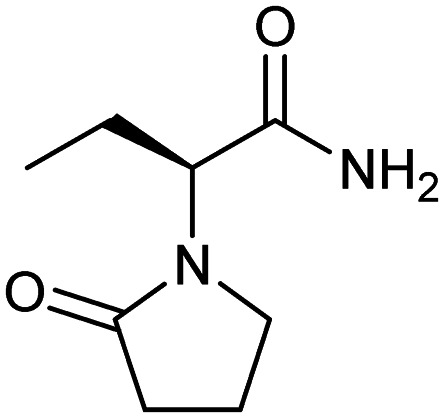
|
CACNA1B | Synthetic | 170.2 | −0.34 | 63.4 | 1 | 2 | 3 | 1 | 8 | 4 | 12 |
Lifitegrast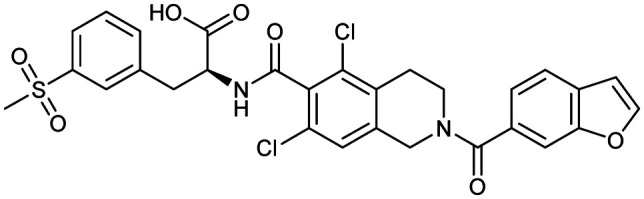
|
ITGAL/ICAM1 | Synthetic | 615.5 | 2.28 | 142 | 2 | 7 | 7 | 5 | 29 | 12 | 41 |
Lusutrombopag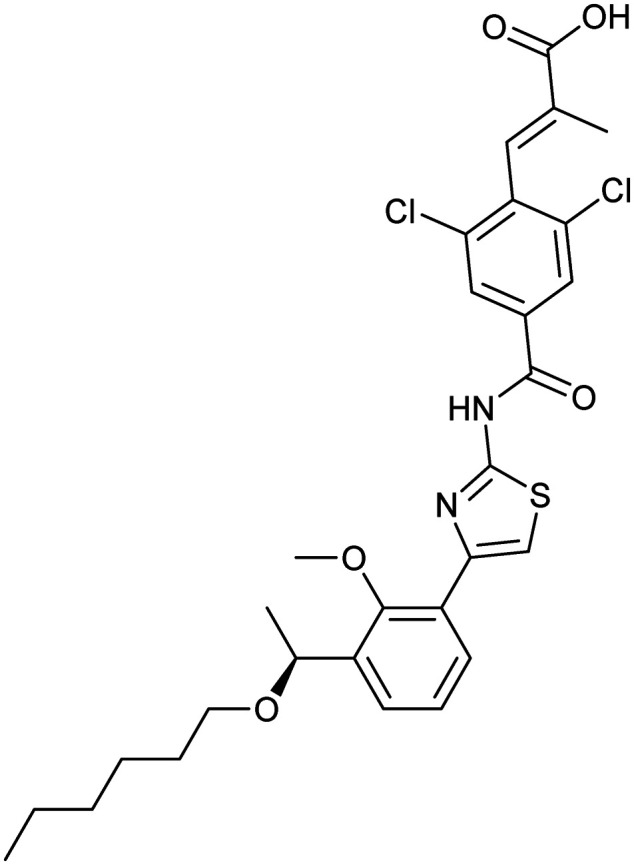
|
MPL/TPO | Synthetic | 591.5 | 7.92 | 126 | 2 | 7 | 13 | 3 | 29 | 10 | 39 |
Maraviroc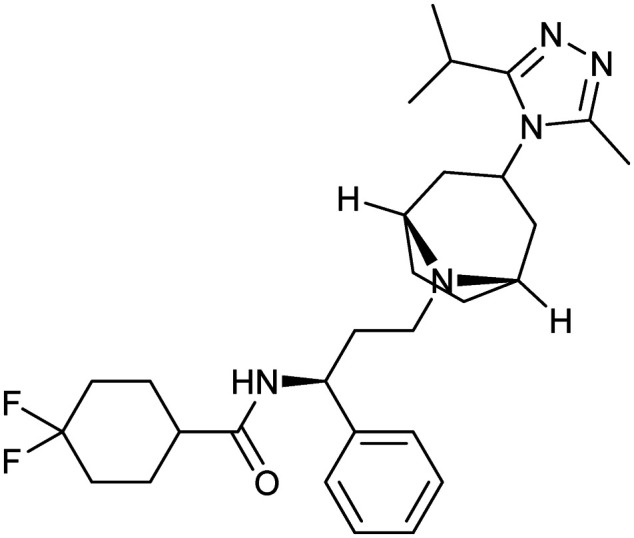
|
CCR5/gp120 | Synthetic | 513.7 | 3.26 | 63 | 1 | 6 | 8 | 6 | 29 | 8 | 37 |
Paclitaxel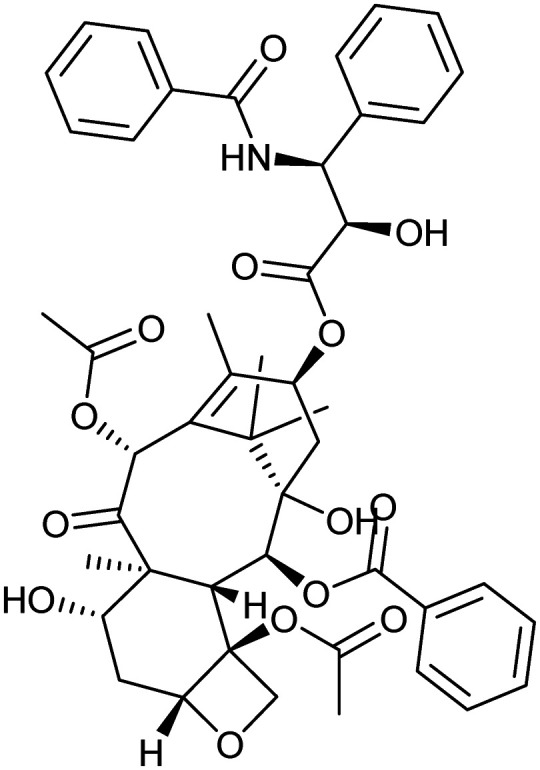
|
Microtubule | Natural | 853.3 | 4.73 | 221 | 4 | 14 | 14 | 9 | 47 | 15 | 62 |
Pimecrolimus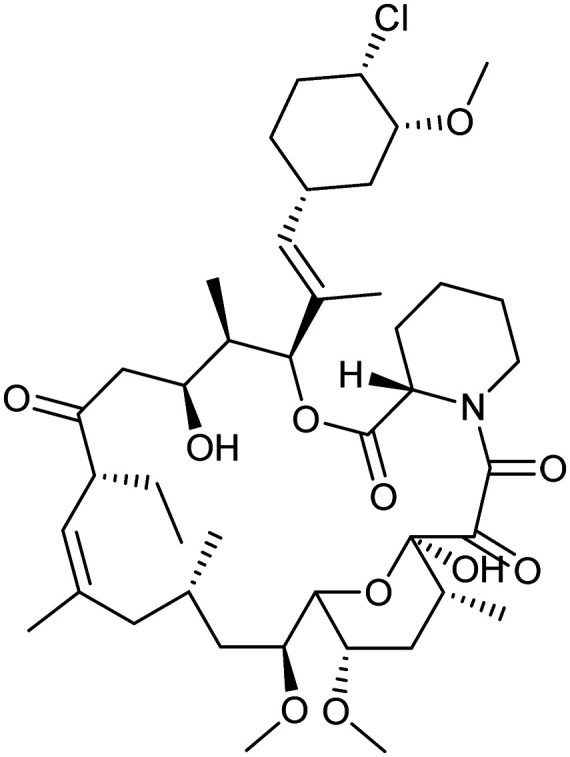
|
FKBP12/CNA/CNB | Natural | 810.4 | 7.10 | 158 | 2 | 11 | 6 | 3 | 43 | 13 | 56 |
Plerixafor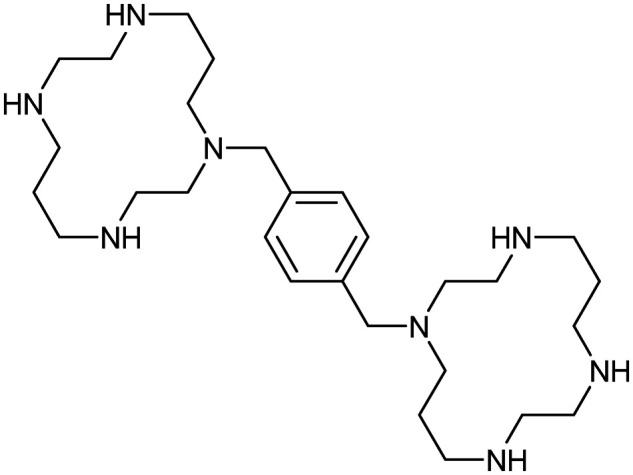
|
CXCR4/CXCL12 | Synthetic | 502.8 | −0.25 | 79 | 6 | 8 | 4 | 3 | 28 | 8 | 36 |
Romidepsin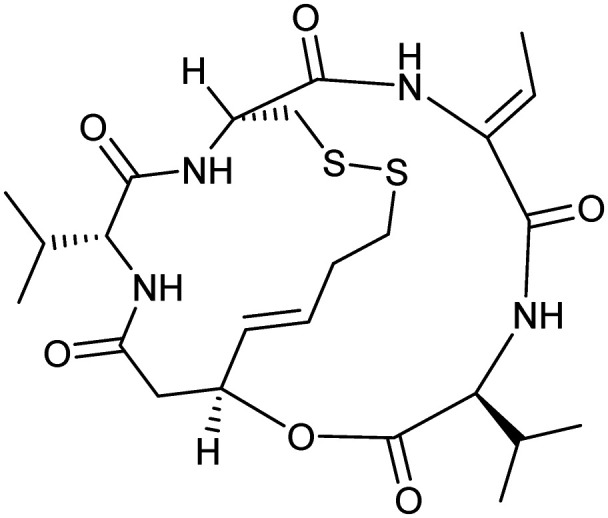
|
HDAC | Natural | 540.7 | 3.44 | 193 | 4 | 8 | 2 | 2 | 24 | 12 | 36 |
Selinexor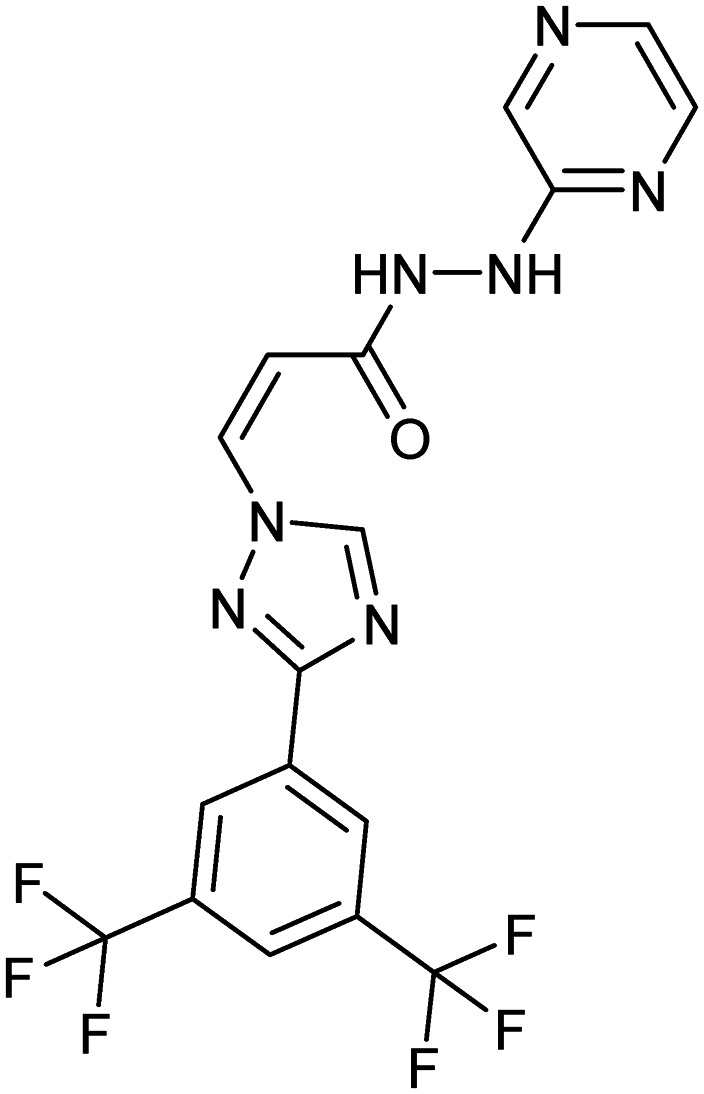
|
CRM1/tumour suppressor proteins | Synthetic | 443.3 | 2.68 | 97.6 | 2 | 12 | 5 | 3 | 17 | 14 | 31 |
Rapamycin (sirolimus)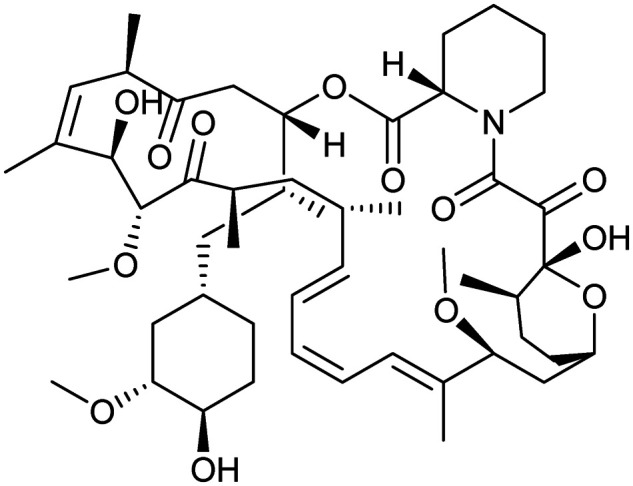
|
FKBP12/MTOR | Natural | 914.2 | 7.04 | 195 | 3 | 13 | 6 | 3 | 51 | 14 | 65 |
Tacrolimus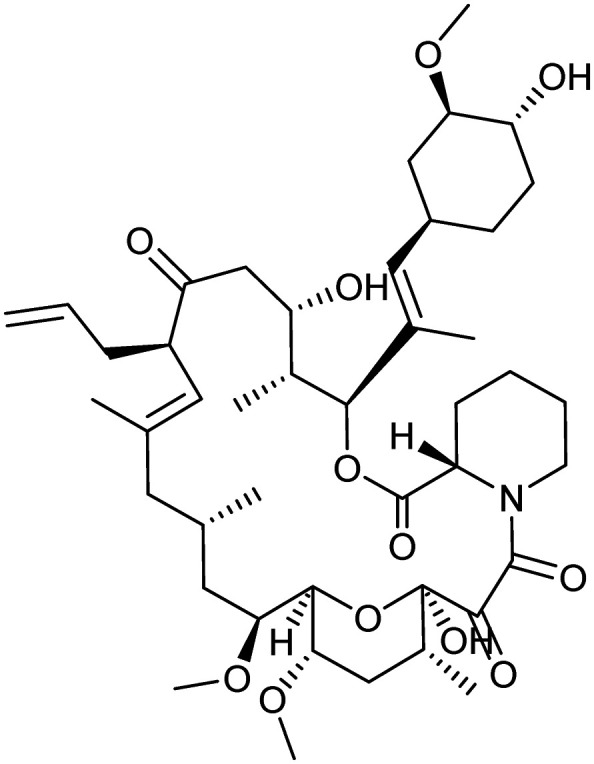
|
FKBP12/CNA/CNB | Natural | 804.0 | 5.78 | 178 | 3 | 12 | 7 | 4 | 44 | 13 | 57 |
Tafamidis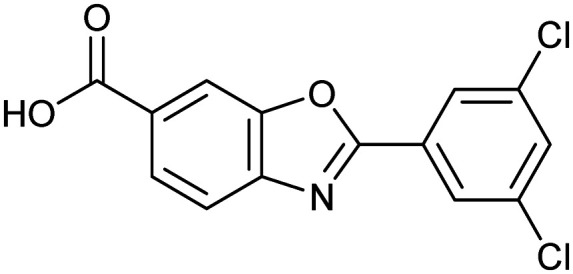
|
TTR tetramer | Synthetic | 308.1 | 5.00 | 63 | 1 | 4 | 2 | 3 | 14 | 6 | 20 |
Temsirolimus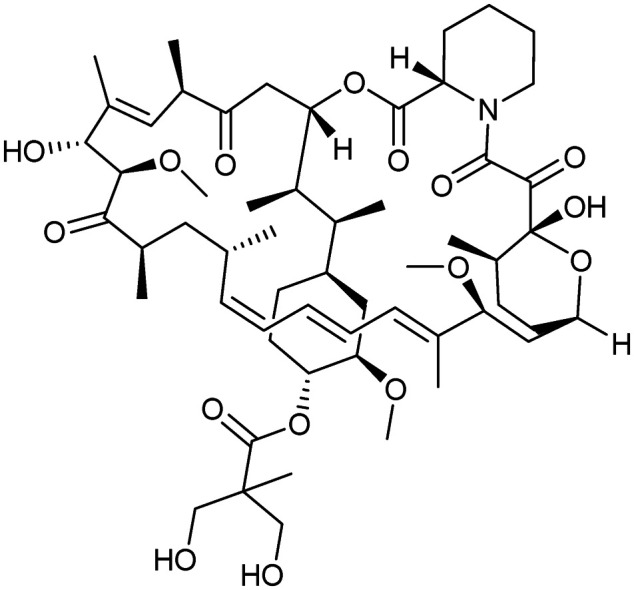
|
FKBP12/MTOR | Natural | 1030.3 | 7.46 | 242 | 4 | 16 | 11 | 3 | 14 | 17 | 73 |
Tirofiban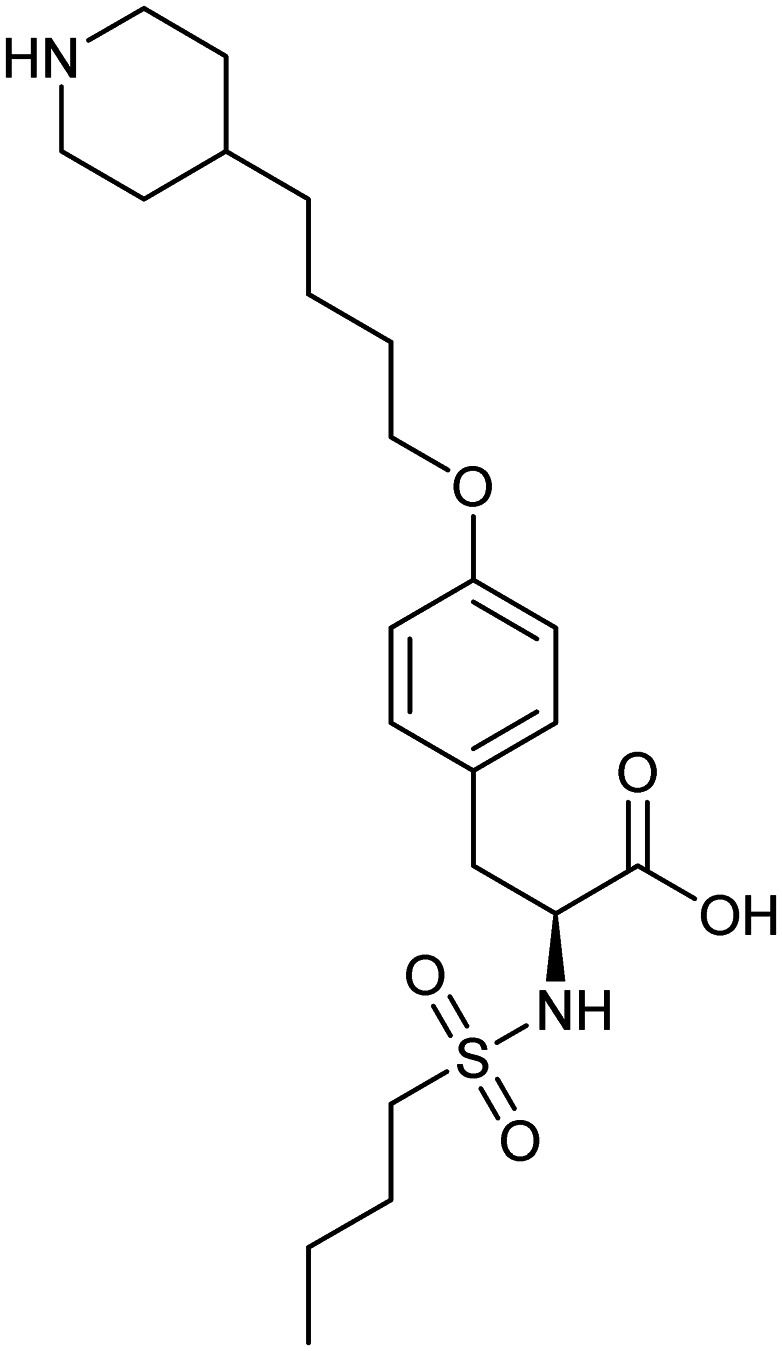
|
FGG/ITGA2B/ITGB3 | Natural | 440.6 | 2.00 | 113 | 3 | 7 | 14 | 2 | 22 | 8 | 30 |
Venetoclax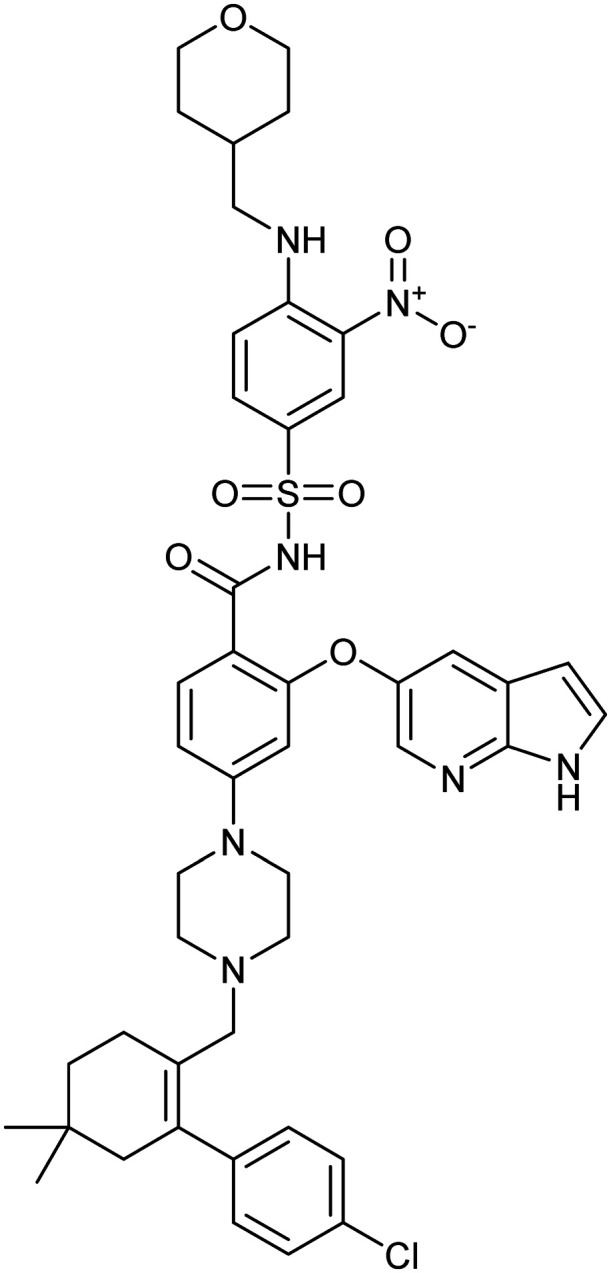
|
BCL2/BAX | Synthetic | 868.4 | 10.31 | 183 | 3 | 11 | 12 | 8 | 45 | 17 | 61 |
Vinblastine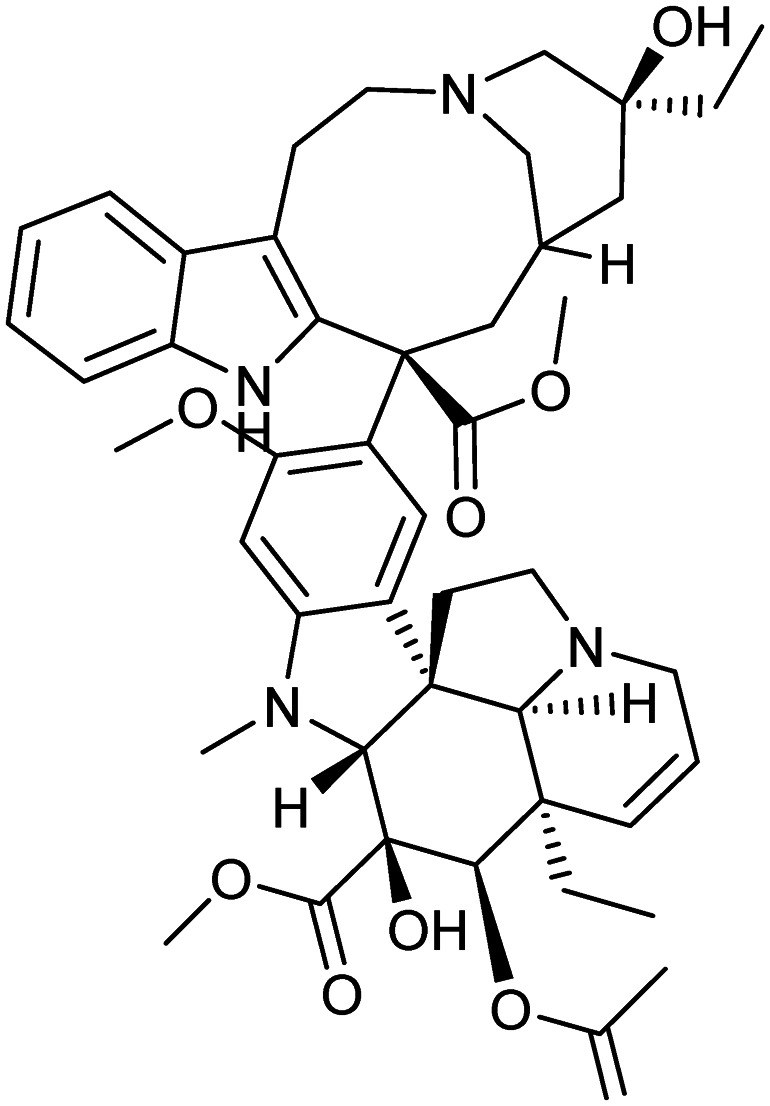
|
Microtubule | Natural | 811.0 | 5.23 | 150 | 3 | 12 | 10 | 9 | 46 | 13 | 59 |
Vorinostat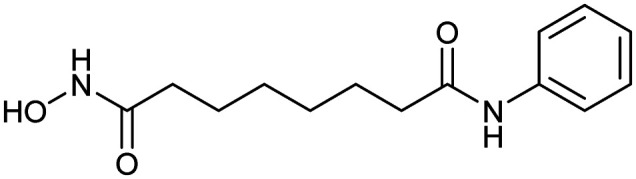
|
HDAC | Synthetic | 264.3 | 0.99 | 78 | 3 | 3 | 8 | 1 | 14 | 5 | 19 |
When analysing all the PPI modulators on the market, one parameter, HBD, remained strictly within Lipinski's RO5. The majority of compounds (24 out of 25) contained ≤5 HBDs, with only plerixafor containing 6 HBDs. Furthermore, the average was only one extra than the mean of all approved drugs (meanPPI = 3.323, meanALL = 2.239, ESI† Tables S1 and S2), suggesting that this is a tight parameter. Others have also found that HBD counts remain relatively invariant when analysing the chemical properties of orally available drugs over time.28 This suggests that the number of HBDs may be an important physicochemical parameter for all drugs, including PPI modulators.
Compared to HBD, HBA in PPIs are often inflated past Lipinski's rule. Twelve of the 25 PPI drugs on the market exceeded the Lipinski guideline of no more than 10 HBA. On average, the mean number of HBA of the drugs in the list approaches this limit and is greater than those of other approved drugs (meanPPI = 9.48, meanALL = 4.91). Increasing the number of HBDs and/or HBAs often leads to an increase in TPSA, which in turn reduces the ability of the small-molecule compounds to pass through cell membranes.
Interestingly, the mean TPSA value we calculated for all PPI modulators on the market was significantly larger (meanPPI = 143.7 Å, ESI† Table S1) than the average of conventional drugs (meanALL = 91 Å, ESI† Table S2). In fact, the mean is also higher than the established drug discovery cut-off of 140 Å (ref. 29) and is higher than reported in other studies on PPI modulator properties.27 As stated above, a higher TPSA reduces the ability of compounds to pass through cell membranes, often leading to low oral bioavailability. Thus, TPSA is thought to be a useful estimation of oral bioavailability and permeability29,30 which have been shown to be inversely related to TPSA.29–31 When we compared the route of administration against TPSA for the current clinical PPI modulators (Fig. 1A), drugs with higher TPSA values were observed to use non-oral routes of administration. Therefore, the use of alternative routes of administration may reflect a need to circumvent oral bioavailability problems. This may be possible for diseases with a significant death burden (e.g. cancer) but is not viable for other diseases (e.g. CNS disorders) for which there is a clear unmet need which could be answered by small-molecule PPI modulators.
Fig. 1. (A) Higher TPSA values are observed in clinical drugs employing non-oral routes of administration. (B) There is a clear positive correlation between MW and TPSA for clinical drugs. (C) A positive correlation also exists for clinical drugs between the number of HBAs and TPSA. (D) A linear regression model of TPSA with HBA and molecular weight as variables for clinical drugs. The linear fit is slightly improved relative to the model presented in (A). However, the p value of HBA being >0.05 suggests that the contribution of the HBA variable is not statistically significant.

Although the polar surface area of PPI drugs is larger than that of conventional drugs, PPI modulators are also larger in mass than conventional drugs. Our analysis showed a significantly higher median/mean MW than other drugs (medianPPI 615.5, meanPPI 621.8, compared to medianALL 335.5, meanALL 384.7, ESI† Tables S1 and S2 and Fig. 2). Therefore, the higher TPSA values may simply be reflective of larger molecules. To assess this further we mapped the molecular weight against the TPSA and found a positive correlation (adjusted r2 = 0.8070, p = 6.7 × 10−10) (Fig. 1B). TPSA and number of HBAs also showed a positive correlation (adjusted r2 = 0.7789, p = 5.34 × 10−9) (Fig. 1C). However, a linear model of TPSA with molecular weight and HBA as variables (Fig. 1D) only improved the fit slightly over the linear TPSA model with only molecular weight as a variable (adjusted r2 = 0.8185), with the HBA contribution in the multivariable model being statistically insignificant (p = 0.118), indicating that the contribution of HBA to TPSA is already accounted for in the molecular weight variable in the model. Although this is a small data set, our analysis suggests that in general, larger PPI inhibitors contain more HBAs, contributing to an increase in TPSA.
Fig. 2. Violin plots displaying the distribution of molecular weight, clog P and topological polar surface area (TPSA). Displayed is the range of values with the median (thick dotted line) and interquartile ranges (thin dotted line). The graph thickens in relation to more data points. All data points are shown for the PPI modulators which have been approved (approved) and those currently in clinical trials (clinical trial). Due to the large data set, these are not displayed for the non-PPI compounds which have been approved for use26 (non-PPI) and the PPI inhibitors collated in the iPPI-DB7,8 (iPPI-db).

These larger compounds also tend to be more flexible, with PPI modulators in the clinic showing on average a larger number of rotatable bonds (mean = 7.760, ESI† Table S1), whereas non-PPI drugs in the clinic show an average of 6.03 rotatable bonds (ESI† Table S2). Although this may seem like a modest increase, studies have shown that compounds with more than 7 rotatable bonds have been associated with poor oral bioavailability, with the effect more pronounced when that number exceeds 10.29 The effect of this increase in rotatable bonds may extend beyond just reduced bioavailability.
One protein can form numerous PPIs, thought to be possible due to flexibility in the protein interfaces themselves,32 and this conformational plasticity at the protein–protein interaction interface has been successfully exploited to generate suitable binding pockets for PPI modulators.33 Since protein interfaces involved in the PPI can be flexible, the addition of conformational constraints to the inhibitors could effectively improve potency as restricting the molecule towards productive conformers would reduce the entropic barriers to binding.
Therefore, a promiscuous protein that has multiple potential binding partners needs a compound that is very target specific to reduce the chance of unwanted side effects. In recent years, higher clog P has been thought to add to the promiscuity of compounds, with an ideal clog P being under 3.38 Specifically, high lipophilicity has been shown to drive binding to unwanted pharmacological targets, including hERG,39,40 leading to the likelihood of reactive metabolites and/or organ toxicity. This is reflected by all non-PPI drugs displaying a very low mean clog P of 1.623 (ESI† Table S2). In contrast, our analysis of the PPI modulators on the market showed a much higher average clog P (mean = 4.2, median = 4.1, ESI† Table S1 and Fig. 2). Although 60% of the PPI modulating compounds on the market had a clog P within Lipinski's RO5, reducing this should be a priority for future studies to reduce unwanted effects.
Of note, venetoclax, an approved PPI inhibitor of Bcl-2 for chronic lymphocytic leukaemia (CLL), has the highest clog P of 10.31 and is also a substrate for cytochrome P450 3A. Therefore it is often co-administered with CYP3A inhibitors to counteract this effect.41 Additionally, venetoclax is also practically insoluble in aqueous solutions and encounters strong food-dependent bioavailability. It displays a 5-fold increase in oral bioavailability following a high-fat meal than when compared to a fasted state42 due to its lipophilicity;43 therefore a meal prior to oral drug administration is required to achieve acceptable oral bioavailability.44
Furthermore, the drug discovery program for venetoclax and its derivative navitoclax were plagued by pharmacokinetic and pharmacodynamic issues.45,46 These issues may be acceptable for first-in-class cancer treatment; however, care would need to be taken if increasing clog P for other diseases. It is also worth noting that calculated log Ps can be very different from experimental log Ps, and log P does not consider ionisable compounds. Regardless, this analysis does highlight the importance of this physicochemical parameter in all aspects of drug discovery.
Many early PPI modulators that have been approved for human use are based on natural products, and as such their physicochemical characteristics may be more varied. Examples, such as vinblastine and paclitaxel, are large, un-druglike compounds derived from natural products. Conversely, tirofiban is also a natural product; however, it is more “drug-like” in terms of its physicochemical parameters.
To explore the potential differences between synthetic and naturally derived PPI modulators, we analysed each group separately. The data set contained an almost identical split between naturally derived molecules (12 compounds) and synthetically derived molecules (13 compounds). When analysed separately, they maintain an almost identical clog P (meanNAT = 3.86, meanSYN = 3.87); however, the MW (meanNAT = 697.4, meanSYN = 528.4) and TPSA (meanNAT = 181.8, meanSYN = 116.7) were both larger for the compounds derived from natural products (Fig. 3). Nonetheless, both sets of PPI modulators still displayed a significantly larger MW and TPSA than those of conventional small-molecule modulators (meanMW = 384.7, meanTPSA = 91.9, ESI† Table S2).
Fig. 3. Column graphs showing the differences between the molecular weight (MW), clog P and topological polar surface area (TPSA) for PPI modulators on the market that are derived from natural products (NAT) or synthetically derived (SYN). The clog P values for both are very similar, whereas natural product derived compounds have slightly higher MW and TPSA. Error bars show the standard deviation.

Taken together, our analysis suggests that PPI modulators approved for human use mostly reside outside the RO5 guidelines as expected based on previous analysis. This is also in agreement with expectations based on differences between PPI properties when compared to enzyme active sites or ligand binding sites of different receptors.
PPI modulators in clinical trials
To expand our analysis beyond the very limited number of PPI drugs on the market, we explored the features of those PPI modulators that are in clinical trials. These compounds have been extensively characterised in a preclinical setting. Therefore, their physicochemical properties should be a good indicator of the parameters needed for a PPI modulator. Our search found 30 such compounds in Phase 1–3 clinical trials (Table 2).
Physicochemical properties of PPI modulators in clinical trials. Shown are the PPI target, clinical trial phase, molecular weight (MW), partition coefficient (clog P), topological polar surface area (TPSA), number of hydrogen bond donors (#HBD), number of hydrogen bond acceptors (#HBA), number of rotatable bonds (#RB), number of rings (#ring), number of carbons (#C), number of heteroatoms (#Het), and number of heavy atoms (#HA).
| PPI drug | Target | Phase | MW (Da) | clog P | TPSA (Å2) | #HBD | #HBA | #RB | #ring | #C | #Het | #HA |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
AMG-232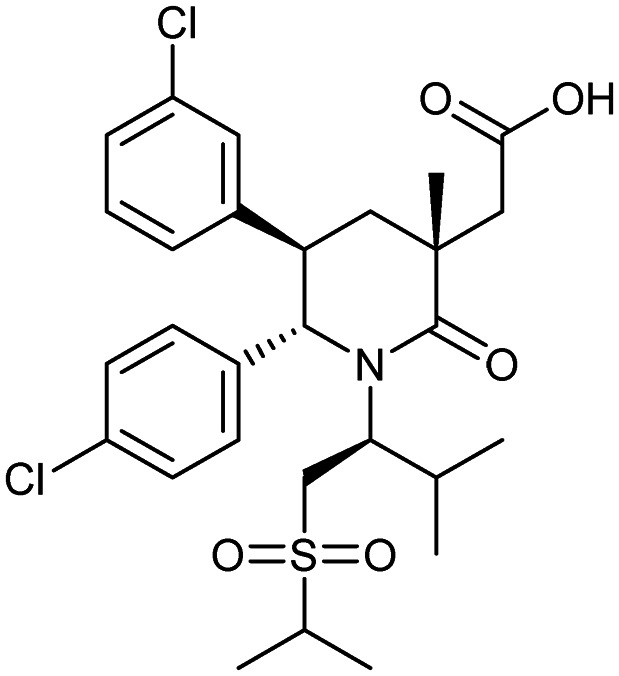
|
MDM2/p53 | 1/2 | 568.6 | 6.49 | 100 | 1 | 5 | 9 | 3 | 28 | 10 | 38 |
Apabetalone (RVX-208)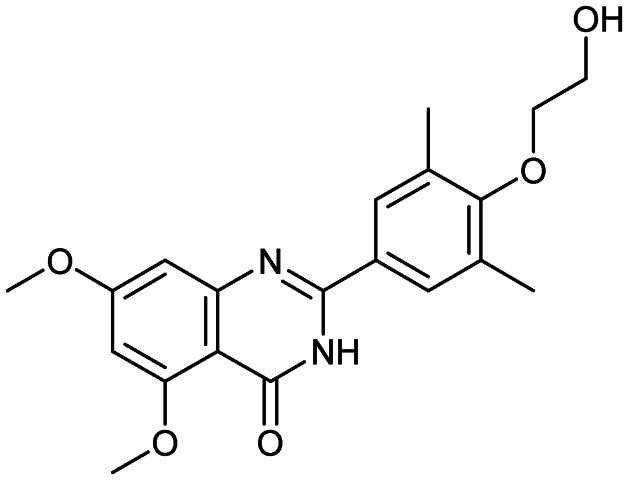
|
Bromodomain/histone | 3 | 370.4 | 3.25 | 89.4 | 2 | 6 | 6 | 3 | 20 | 7 | 27 |
APG-115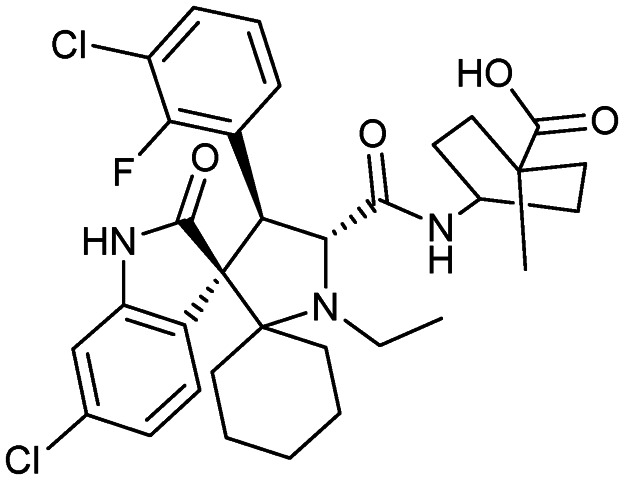
|
MDM2/p53 | 1/2 | 642.6 | 4.36 | 98.7 | 3 | 6 | 5 | 6 | 34 | 10 | 44 |
ASTX660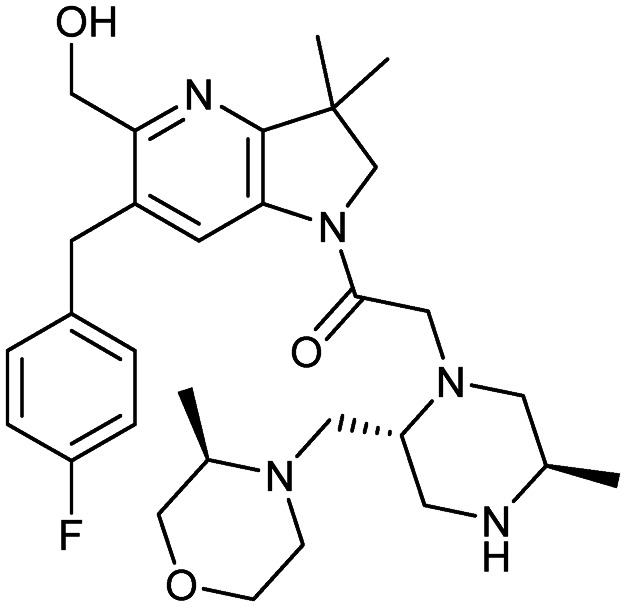
|
XIAP/caspase-9 | 1/2 | 539.7 | 2.85 | 81.2 | 2 | 8 | 7 | 5 | 30 | 9 | 39 |
Birinapant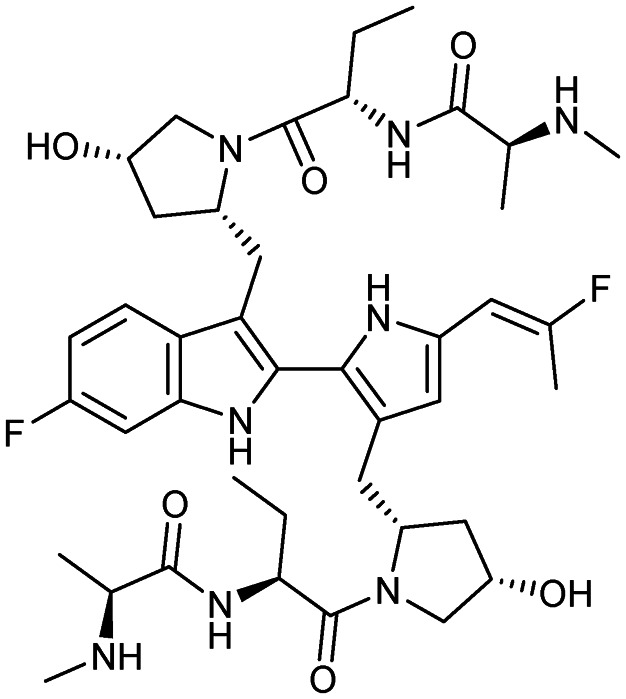
|
XIAP/caspase-9 | 1 | 806.9 | 2.78 | 195 | 8 | 10 | 15 | 6 | 42 | 16 | 58 |
Carotegrast methyl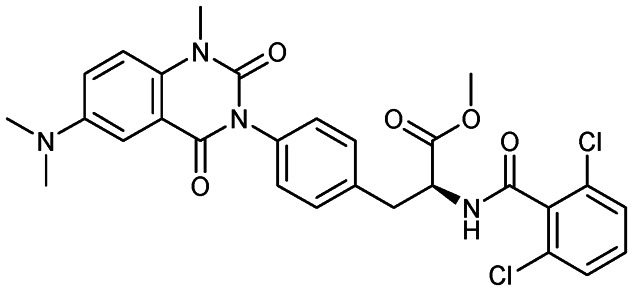
|
α4-Integrin | 3 | 569.4 | 4.61 | 99.3 | 1 | 6 | 8 | 4 | 28 | 11 | 39 |
CCX140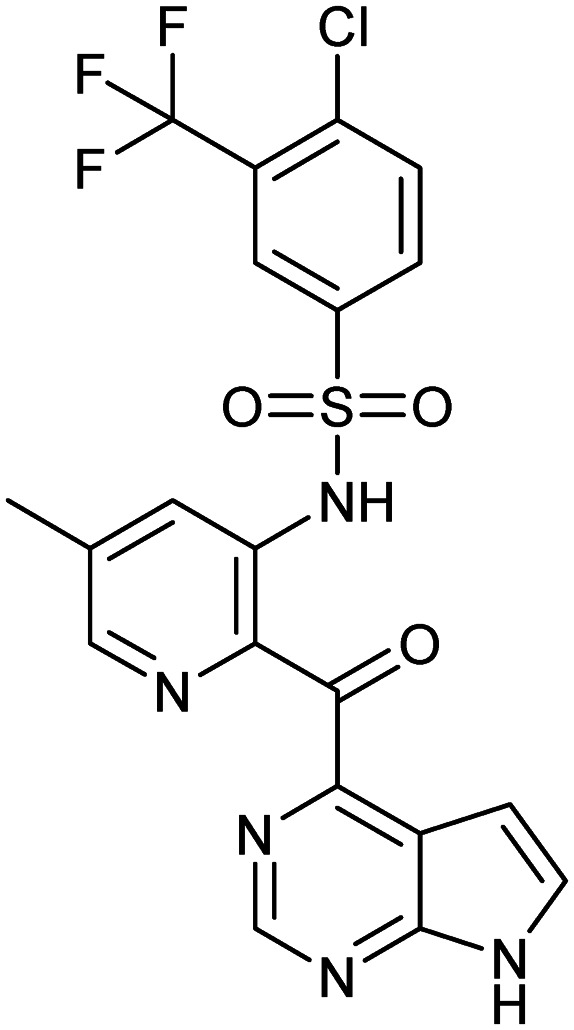
|
CCR2 | 2 | 495.9 | 4.37 | 126 | 2 | 10 | 5 | 4 | 20 | 13 | 33 |
CCX354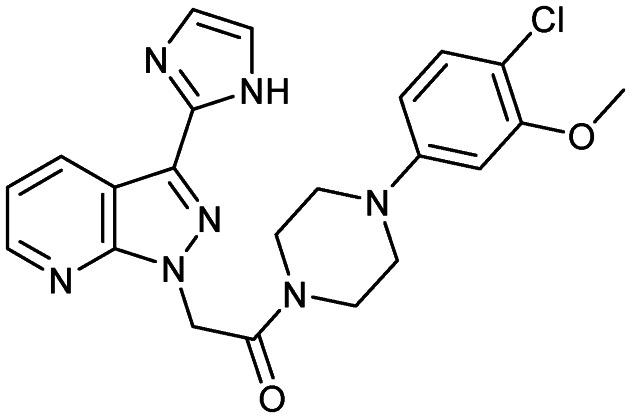
|
CCR1 | 2 | 451.9 | 2.96 | 92.2 | 1 | 6 | 5 | 5 | 22 | 10 | 32 |
Cenicriviroc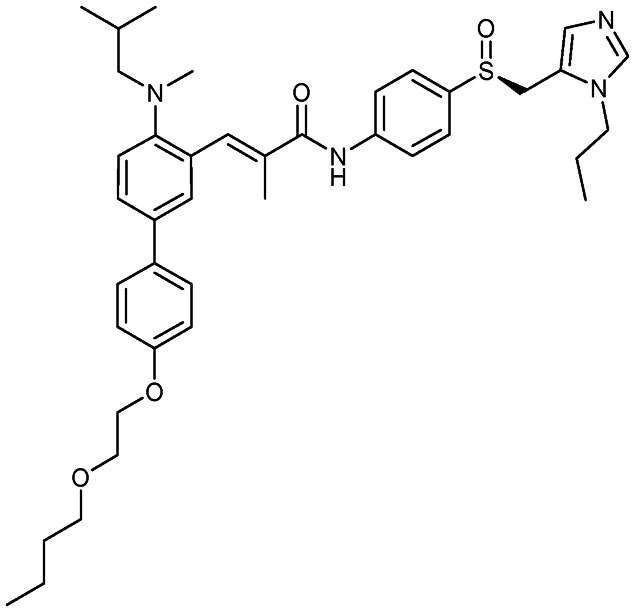
|
CCR2/CCR5 | 3 | 696.9 | 8.78 | 105 | 1 | 7 | 17 | 5 | 41 | 9 | 50 |
CGM097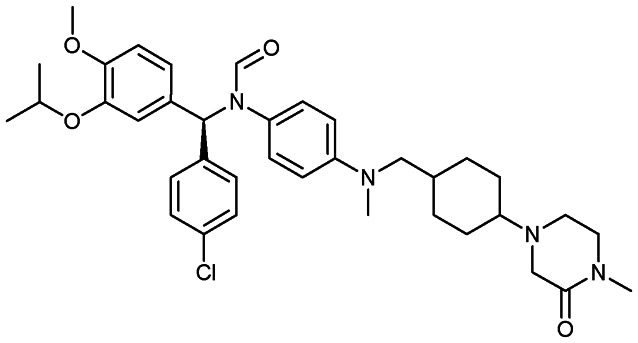
|
MDM2/p53 | 1 | 659.3 | 7.88 | 65.6 | 0 | 6 | 9 | 6 | 38 | 9 | 47 |
CPI-0610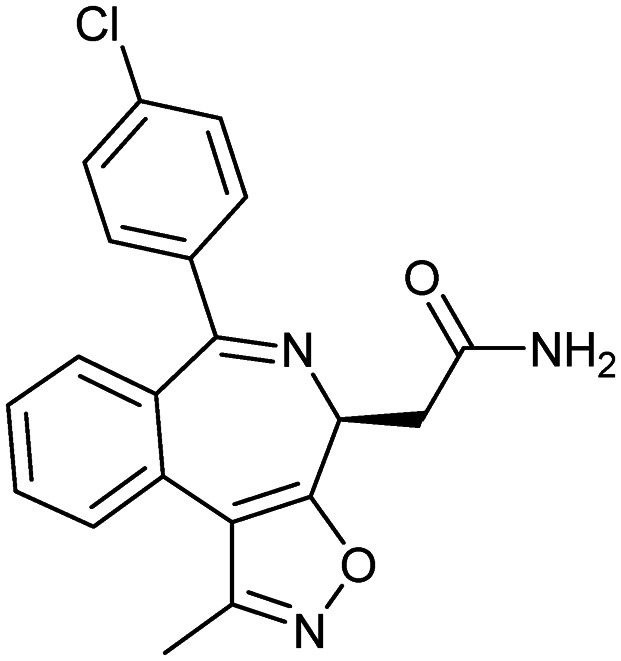
|
BET proteins | 3 | 365.8 | 2.57 | 81.5 | 1 | 4 | 3 | 4 | 20 | 6 | 26 |
CUDC-427 (GDC-0917)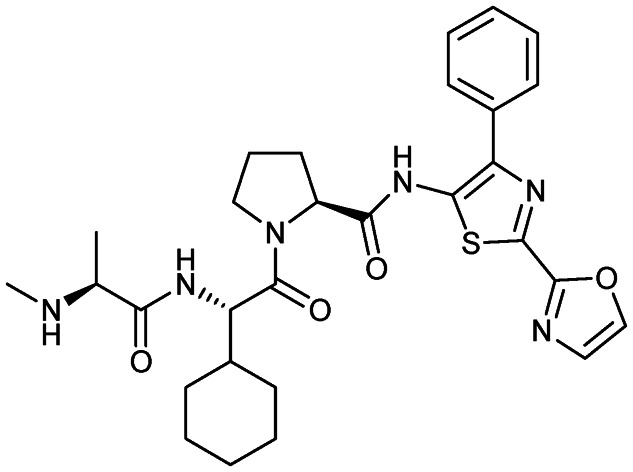
|
XIAP/caspase-9 | 1 | 564.7 | 3.56 | 158 | 3 | 8 | 9 | 5 | 29 | 11 | 40 |
Epothilone B (patupilone)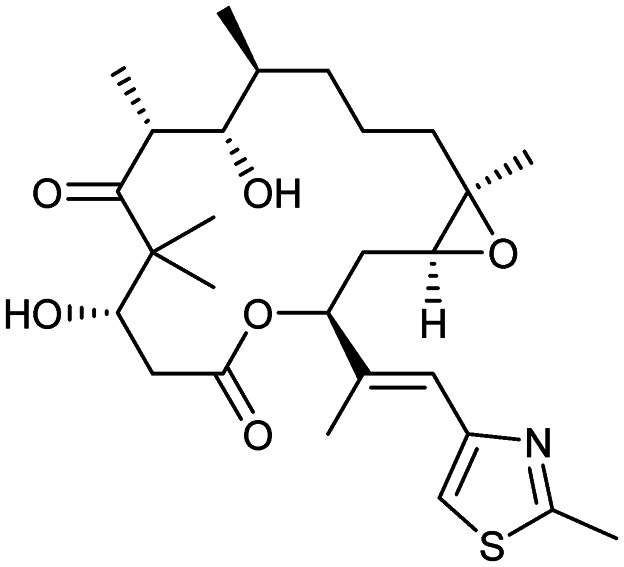
|
Microtubule | 2 | 507.7 | 3.21 | 138 | 2 | 8 | 2 | 2 | 27 | 8 | 35 |
Idasanutlin (RG-7388)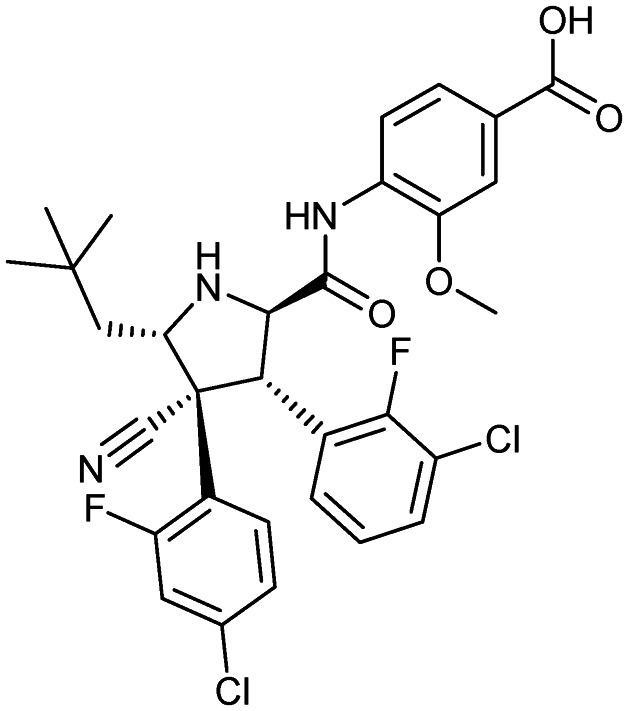
|
MDM2/p53 | 3 | 616.5 | 4.97 | 111 | 3 | 8 | 8 | 4 | 31 | 11 | 42 |
LCL161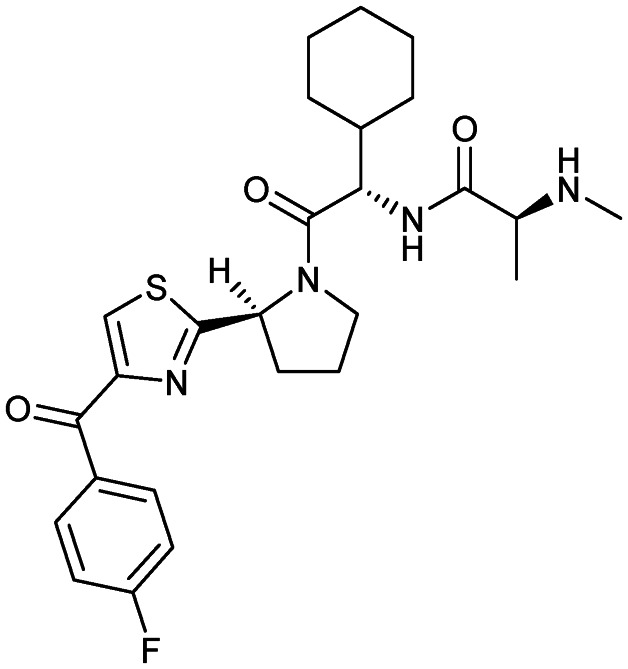
|
XIAP/caspase-9 | 2 | 500.6 | 3.57 | 120 | 2 | 7 | 8 | 4 | 26 | 9 | 35 |
Milademetan (DS-3032)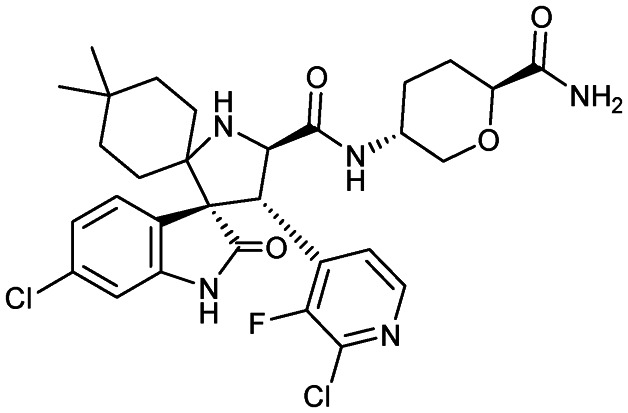
|
MDM2/p53 | 1 | 618.5 | 3.70 | 135 | 4 | 7 | 4 | 6 | 30 | 12 | 42 |
Navarixin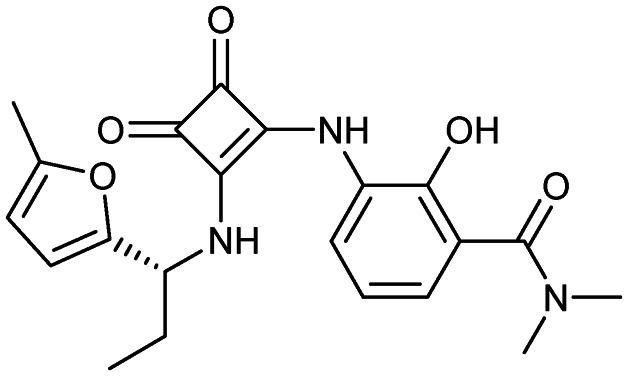
|
CXCR2 | 2 | 397.4 | 1.31 | 112 | 3 | 7 | 7 | 3 | 21 | 8 | 29 |
Navitoclax (ABT-263)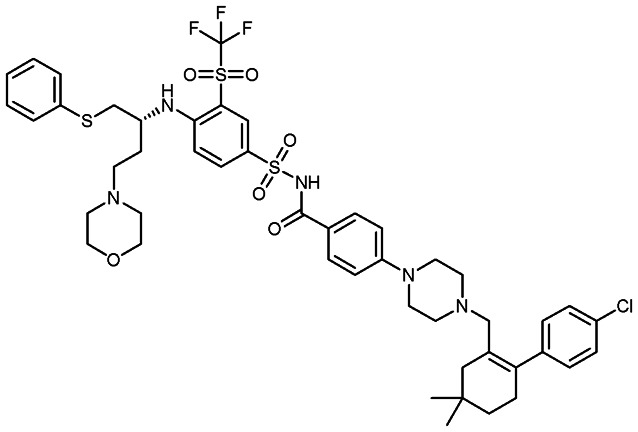
|
BCL-2 family | 2 | 974.6 | 12.39 | 170 | 2 | 14 | 16 | 7 | 47 | 18 | 65 |
Obatoclax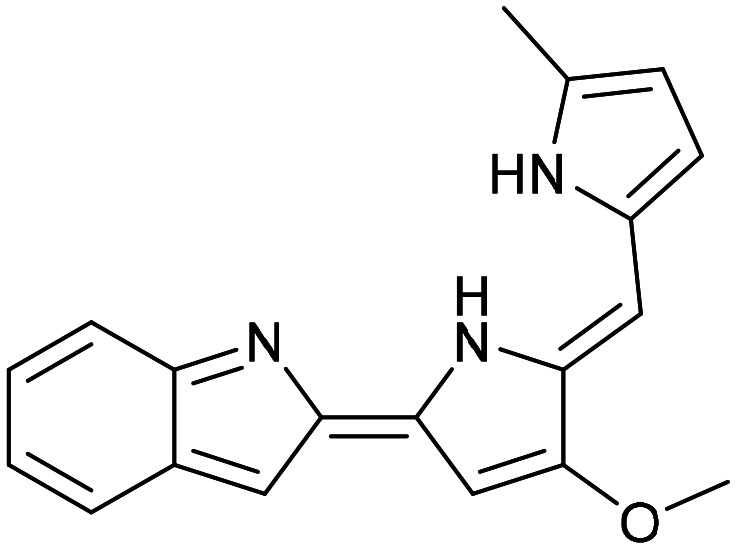
|
MCL1/BAX | 3 | 317.4 | 1.78 | 49.4 | 2 | 3 | 2 | 4 | 20 | 4 | 24 |
| BCL2/BAX | ||||||||||||
| BCLXL/BAK | ||||||||||||
Pevonedistat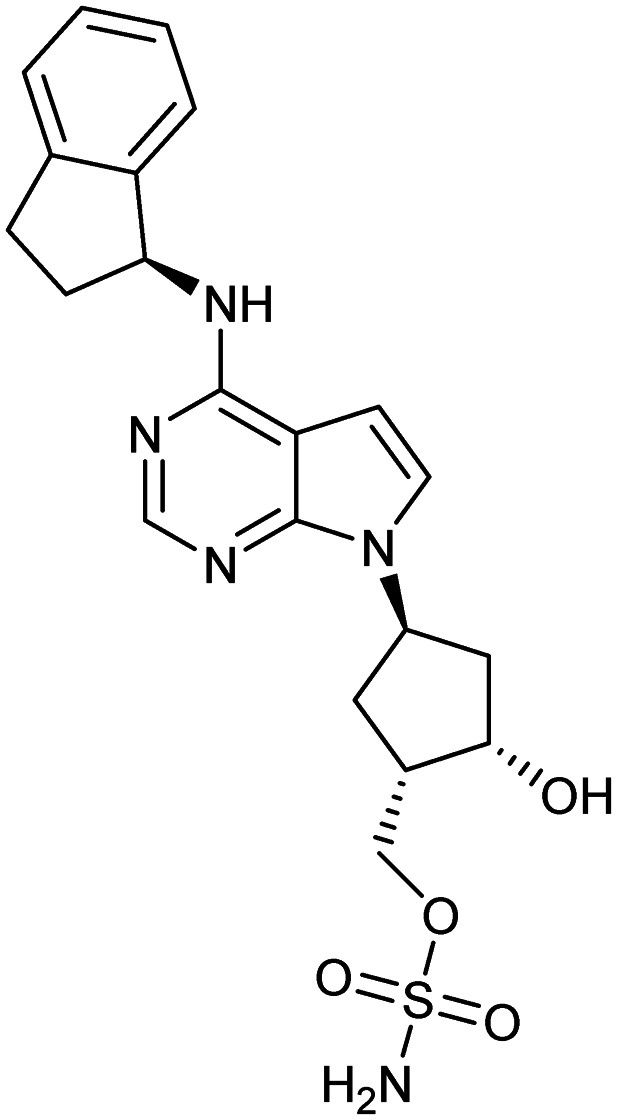
|
NAE | 2 | 443.5 | 1.16 | 141 | 3 | 8 | 6 | 5 | 21 | 10 | 31 |
PF-4136309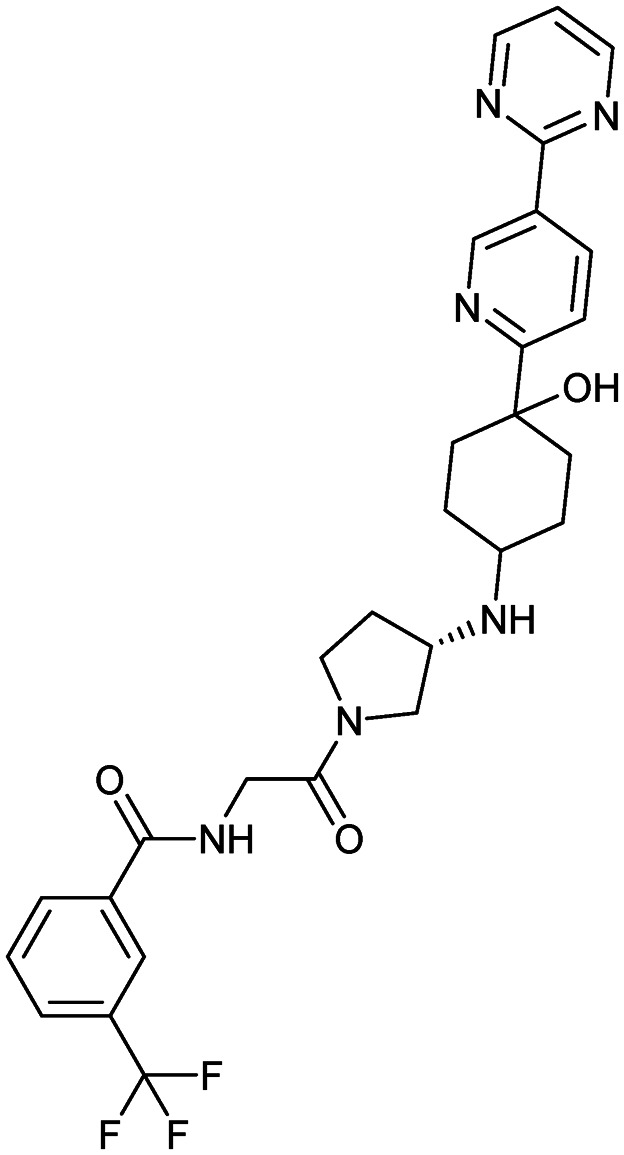
|
CCR2 | 2 | 568.6 | 1.57 | 120 | 3 | 10 | 7 | 5 | 29 | 12 | 41 |
PRI-724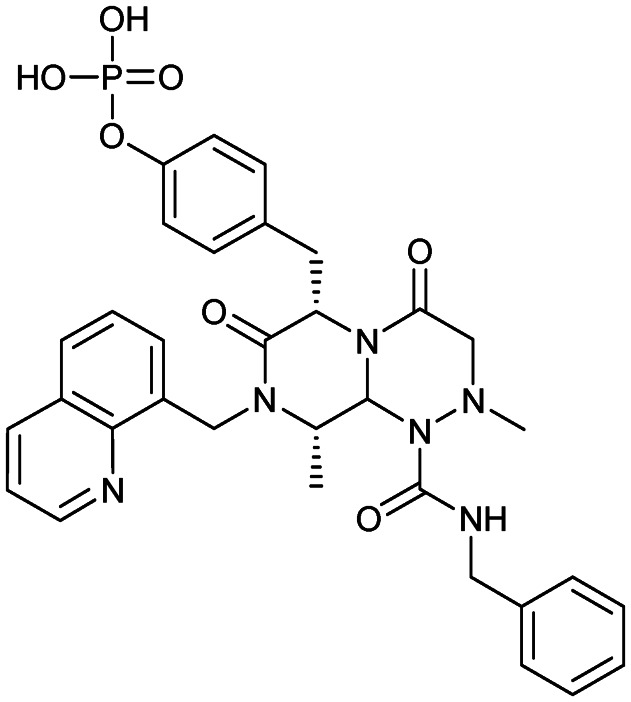
|
β-Catenin/CBP | 1/2 | 658.6 | 3.53 | 156 | 3 | 9 | 8 | 6 | 33 | 14 | 47 |
Reparixin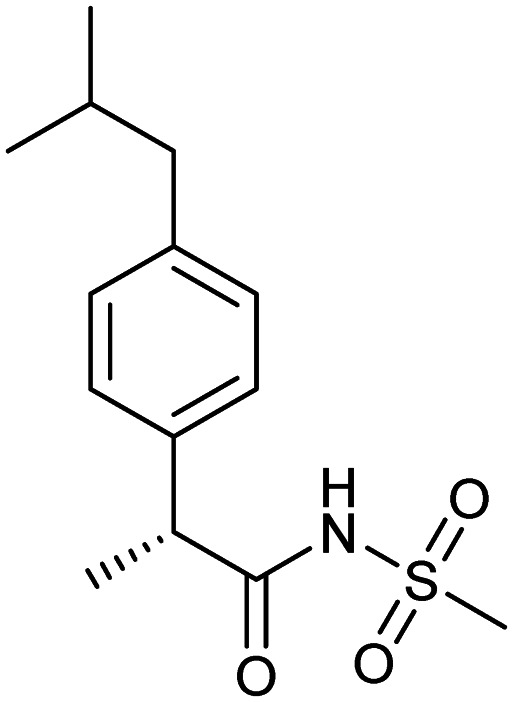
|
CXCR1/2 | 3 | 283.4 | 2.66 | 71.6 | 1 | 3 | 5 | 1 | 14 | 5 | 19 |
RG7112 (RO5045337)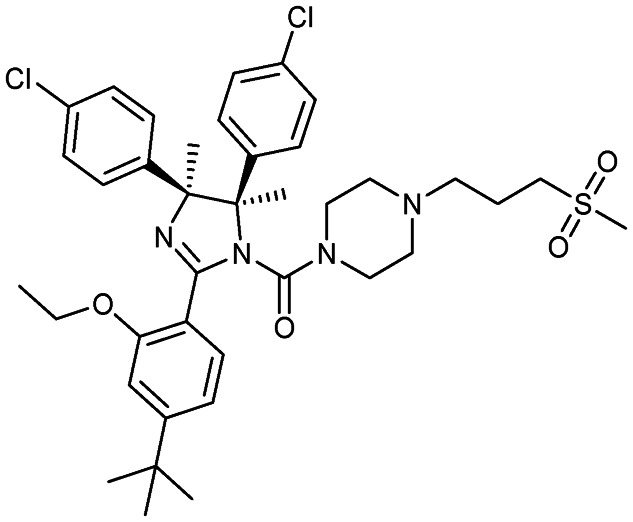
|
MDM2/p53 | 1 | 727.8 | 10.6 | 90.9 | 0 | 6 | 10 | 5 | 38 | 11 | 49 |
RO6870810 (TEN-010)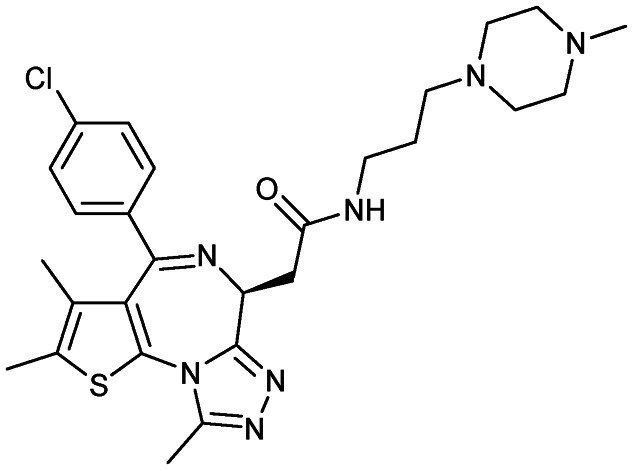
|
BET proteins | 1 | 540.1 | 2.45 | 107 | 1 | 7 | 7 | 5 | 27 | 10 | 37 |
SAR405838 (MI-77301)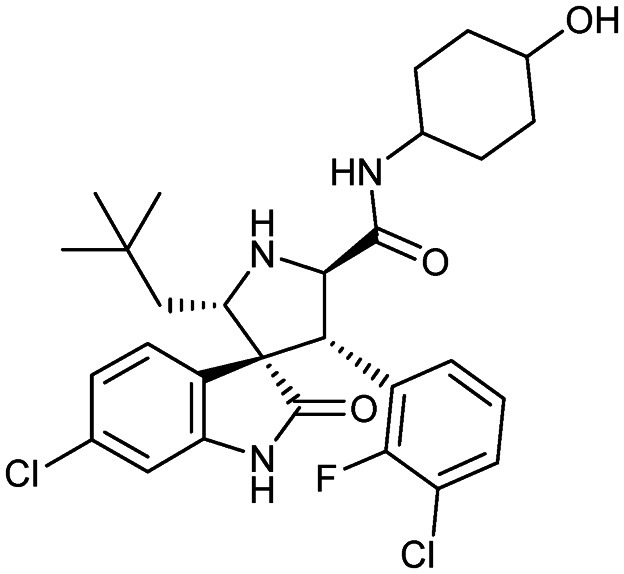
|
MDM2/p53 | 1 | 562.5 | 4.95 | 90.5 | 4 | 5 | 5 | 5 | 29 | 9 | 38 |
Serdemetan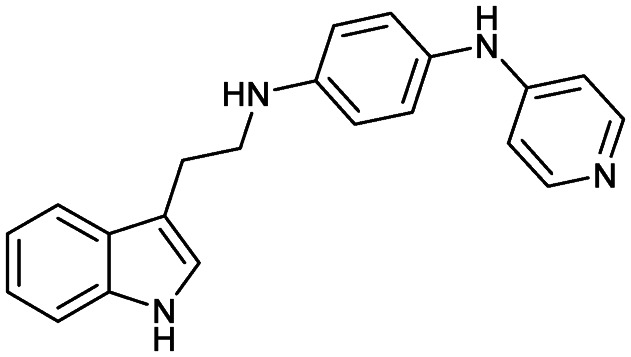
|
MDM2/p53 | 1 | 328.4 | 4.53 | 52.7 | 3 | 3 | 6 | 4 | 21 | 4 | 25 |
Siremadlin (HDM-201)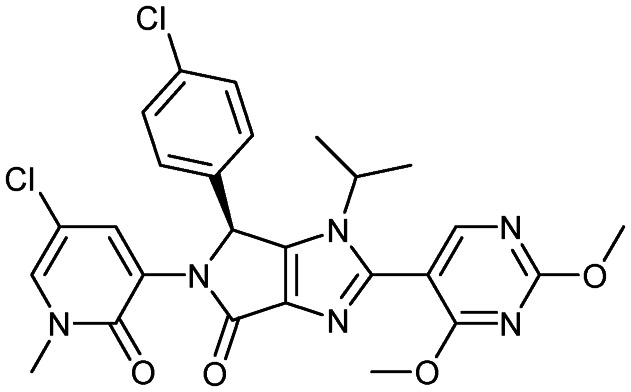
|
MDM2/p53 | 1/2 | 555.4 | 4.11 | 103 | 0 | 7 | 6 | 5 | 26 | 12 | 38 |
Vercirnon (Traficet-EN)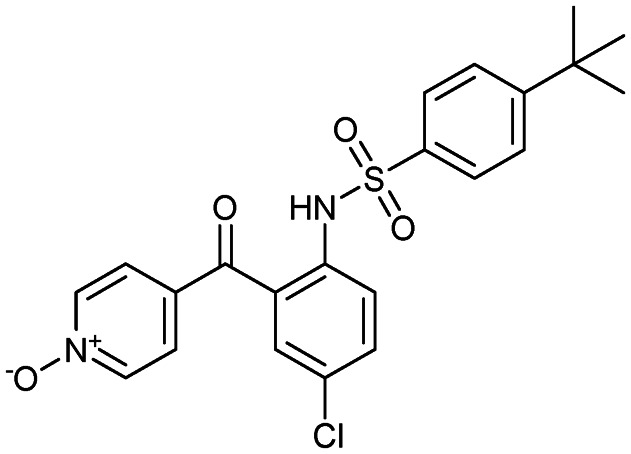
|
CCR9 | 3 | 444.9 | 4.33 | 97.1 | 1 | 5 | 6 | 3 | 22 | 8 | 30 |
Xevinapant (AT-406)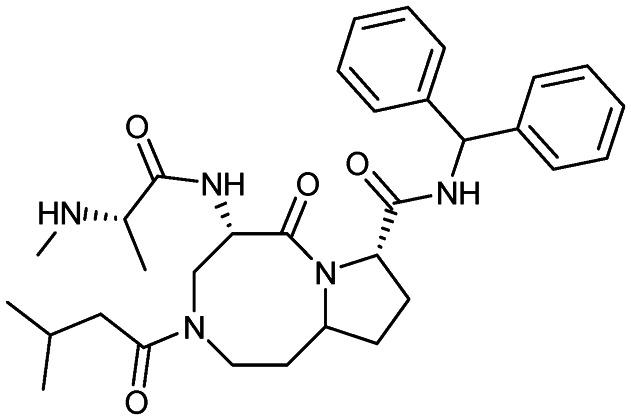
|
IAP | 1/2 | 561.7 | 2.95 | 111 | 3 | 5 | 9 | 4 | 32 | 9 | 41 |
We observed that PPI modulators currently in clinical trials did have differing properties than those already in the market. In particular, the spread/range of the values for each of the calculated properties was tighter. For example, where the MW for PPI drugs on the market had a range of 886.2 Da (144.1–1030) and a mode around 800 Da, those in clinical trials have a much tighter range of 691 Da (283.4–974.6) and a mode around 500 Da (Fig. 2).
A similar trend is seen with almost every other parameter (ESI† Table S3), with the mode of the physicochemical parameters being much higher for those on the market than those in clinical trials.
In our data set, over 90% of the compounds in clinical trials are synthetically derived; therefore we compared our synthetically derived PPI drugs on the market to those in clinical trials. This showed a tighter comparison; however, the range of values for MW and TPSA were still larger for the PPI drugs on the market, with range values of 724.3 and 171.4, respectively, than those in clinical trials (range values of 691.2 and 145.6, respectively, for MW and TPSA) (Fig. 4).
Fig. 4. Violin plots comparing the PPI modulators in clinical trials (clinical trials) compared to those approved for use which are synthetically derived (synthetic) show a much closer distribution of molecular weight and TPSA, although the range is still larger for the PPI modulators which have been approved.

As described above, the clog P did not significantly change regardless of the discovery method (synthetic or natural) for the approved drugs and this remained true for those in clinical trials (Fig. 2, ESI† Table S3). Of note, these clog P values are all still higher than those of the non-PPI drugs (Fig. 2). However, a 2014 analysis showed that clog P values of PPI inhibitors in clinical trials were not higher than those of non-PPI inhibitors in clinical trials.47 This suggests that in coming years, the average clog P may in fact increase for all approved drugs, not just PPI modulators.
Of note, although the clog P mean remained similar for the compounds in clinical trials compared to those on the market, the median was reduced (median = 3.6, mean = 4.3) with navitoclax, a derivative of venetoclax, as an outlier with the highest clog P of any compound in clinical trials (clog P = 12.39). Furthermore, as with venetoclax, the drug discovery program for navitoclax was plagued by pharmacokinetic and pharmacodynamic issues.45,46 The overall trend towards lower clog P may be indicative of a concerted effort to lower clog P and the unwanted side effects mentioned above (Fig. 2).
Like the drugs in the market, most PPI modulators in clinical trials fit within the Lipinski RO5 for HBDs. The only exception was birinapant. Although classed as a small molecule, birinapant is a peptidomimetic, and therefore this is not unexpected. However, the number of HBAs is notably less for those in clinical trials (mean = 6.8, median = 7, ESI† Table S3) compared to those on the market (mean = 9.4, median = 10, ESI† Table S1) which is likely leading to the significantly lower TPSA for modulators in clinical trials (mean = 108.9, median = 104) compared to those on the market (mean = 143.7, median = 150) (Fig. 2). These parameters are notably less even when the clinical trial compounds are compared to just the synthetically derived approved drugs. As described above, TPSA can be used as an estimation of oral bioavailability and permeability;29,30 therefore this may be reflective of a push towards PPI modulators which are orally available.
Overall, the differences between PPI drugs on the market and compounds currently in clinical trials that we observed were unexpected. We observed that PPI modulators in clinical trials are closer to RO5 parameters. This trend was unexpected and might be suggestive of those few on the market being the “low-hanging fruit” of PPIs. This change in distribution of properties may also be reflective of a shift away from natural products towards more synthetic compounds in the clinical trials set compared to the PPI modulators on the market. A third possible explanation may be that chemists have learned from those PPI modulators that have failed to reach the clinic and modified their developmental pipeline to include more conventional “drug-like” parameters.
Preclinical PPI modulator pipeline
To access the properties of PPI modulators currently in the preclinical phase of lead and probe development, we used a publicly available, manually curated data set of small-molecule PPI inhibitors, the iPPI-DB,7 which importantly only includes compounds if the binding activity and target are known. The vast majority (>90%) are investigational drugs. We calculated the same range of physicochemical parameters (MW, TPSA, clog P, HBD and HBA) and observed that PPI inhibitors in the iPPI-DB display the same trend as those in clinical trials, i.e. the physicochemical properties and distribution modes shifted towards more conventional drug-like values (Fig. 2). However, iPPI-DB has a broad coverage and includes not only possible lead compounds but also a number of experimental tool compounds that have not been optimised for drug-like properties. This is reflected in the larger range of some parameters. For example, the clog P had a larger range (Fig. 2) which could perhaps be an indication of the un-drug like nature of some of these compounds.
Taken together, despite the expectation that PPI modulators would routinely exhibit properties outside RO5, we observed that the current space of PPI modulators in clinical trials and preclinical development is enriched for compounds that fall within traditional rules for oral bioavailability.
Discussion
Small molecules dominate the PPI inhibitor drug discovery landscape, representing over 50% of modulators in manually curated lists.48 However, despite the clear unmet need, compared to conventional drugs, a disproportionately small number of PPI modulators have entered the market12 and small-molecule PPI modulators represent approximately 10% of the DrugBank database.26 We conducted the analysis of PPI modulators to better understand the distribution of their physicochemical parameters and the results of our analysis can be used as a starting point to inform future PPI modulator development.
The selection of chemical starting points can be critical to the success of a drug development project. Thus, based on our analysis, we propose that for an initial screening library, the physicochemical properties should be clog P <3.5, MW <500, TPSA <100, HBD <3, and RB <6. Keeping these parameters lower than the average will allow for medicinal chemistry optimisation to occur readily without sacrificing physicochemical properties due to “property inflation”.49 As the lead compound is optimised, properties such as molecular weight and lipophilicity are likely to increase.50 For example, it has been shown that a medicinal chemistry campaign adds on approximately 70–100 Da when progressing from hit to lead51 and starting with these larger compounds may result in difficult optimisation campaigns.
There have been previous attempts to derive physicochemical and pharmacological trends in PPI modulators and use this information to generate targeted/focussed libraries (Table 3). Three publicly available databases are TIMBAL20, iPPI-DB7 and 2P2I21.8,21,55 At the time of writing, TIMBAL contains 8889 small-molecule protein–protein inhibitors, many of which have been defined from the ChEMBL database. The iPPI database contains 2378 compounds and allows the arrangement of data through physicochemical and pharmacological features. Although these databases present an opportunity to mine for trends in PPI properties, it is worth noting that they often do not distinguish between those compounds which have reached clinical trials compared to those which are only at the discovery stage. Furthermore, commercial compound suppliers including Asinex, ChemDiv, LifeChemicals, Otava Chemicals and NQuix also have PPI focussed libraries. These libraries use selection criteria and methods like RO4, similarity analysis, and secondary structure-based design alongside decision trees and machine learning techniques. However, using only the RO4 as the selection criterion skews compounds towards large and more lipophilic compounds, therefore enriching for compounds that may have a greater risk of unfavourable pharmacokinetic and pharmacodynamic properties.53 Furthermore, our analysis showed that although >80% of PPI drugs on the market adhered to the RO4 in terms of the number of H-bond acceptors, almost half of them broke the RO4 parameter associated with the number of ring systems. Therefore, this suggests that the RO4 is not a good starting point for a compound library.
Academic and commercial attempts to generate focussed libraries and/or algorithms to generate focussed libraries targeting PPI interactions.
| Company/academic group | Library | Library construction method | # of compounds | Commercial/academic | Ref. |
|---|---|---|---|---|---|
| Institut Paoli-Calmettes (ISCB) | 2P2I3D | Rule of four and machine learning | 1683 | Academic | 52 |
| Asinex | PPI | Shape analysis with dedicated synthesis | 11 439 | Commercial | 65 |
| ChemDiv | PPI 2.0 | β-Turn, helix, 3D peptidomimetics | 210 000 | Commercial | 66 |
| ChemDiv | Eccentric PPI | “Escape from flatland” shape analysis | 13 000 | Commercial | 67 |
| French consortium | Fr-PPIChem | Machine learning | 10 314 | Academic | 68 |
| Life Chemicals | Machine learning | Decision Tree (machine learning) | 6865 | Commercial | 69 |
| Life Chemicals | Similarity | Similarity search | 17 410 | Commercial | 69 |
| Life Chemicals | Rule of four | Rule of four | 3368 | Commercial | 69 |
| NQuix | N/A | Molecular mimics of secondary structure conformations | N/A | Commercial | 70, 71 |
| Otava Chemicals | iPPI Tree Library | Decision Tree | 1211 | Commercial | 72 |
| Otava Chemicals | iPPI Bayesian Library | Similarity search | 2637 | Commercial | 72 |
| Otava Chemicals | Peptidomimetic libraries | α-Helix and β-turn peptidomimetics | 2288 | Commercial | 73 |
Analysis of the physicochemical properties of these databases had been previously completed for nine of the eleven databases shown in Table 3.54 This analysis concluded that most libraries contained compounds compliant with Lipinski's RO5, ranging from 81% to 92% compliant. This observation may reflect an intent of library developers in keeping PPI modulators drug-like. However, as described above, in a screening library, where one is looking for hit compounds, it is advantageous to keep molecules well within drug-like parameters to allow for follow-up medicinal chemistry optimisation from hit to lead, and from lead to drug.
Another approach may be to utilise fragment screening against a PPI target. This approach promotes ligand efficient compounds with a high ligand binding energy per non-hydrogen atom. In addition, beginning with lower molecular weight molecules reduces the number of pharmacophoric elements, giving larger scope for medicinal chemistry optimisation and can also pinpoint the required interactions necessary to elicit modulation.55
PPIs have been shown to contain hotspot residues which contribute to up to 80% of the binding energy.9,20,56,57 Molecular dynamics modelling has suggested that many existing PPI modulators do not effectively interact with receptor hotspots, and thus improved hotspot engagement by ligands could result in higher ligand efficiency and increased clinical success.58 Hotspots are predominantly hydrophobic but are often surrounded by O-ring residues, i.e. hydrophilic residues, which usually form the salt bridges between the two proteins.20,59 This provides an opportunity to start with a screening library containing small molecules that incorporate hydrophobic cores with hydrophilic termini, which may be a good option. Fragment screens are expected to yield molecules that take full advantage of PPI hotspots, with many PPI targets utilising fragment-based discovery methods including HPV-11 E2:E1, IL2:IL2Rα, MDM2:p53 and BCL-XL:BAK.60 Notably, based on our analysis, we suggest that efforts to elaborate fragments into lead compounds should continue to adhere to the RO5.
For non-PPI drugs, different physicochemical properties are acceptable for different molecular targets. In a study by Morphy,61 on average, the molecular weight of ligands increases for ligands targeting transporters, ion channels, monoamine GPCRs, oxidases, kinases, nuclear receptors, proteases, transferases and peptide GPCRs. This has recently been expanded to include PPI inhibitors.28 Unsurprisingly, PPI inhibitors displayed the highest average mean MW out of all the drug classes. Furthermore, on average, low clog P values were observed for proteases and ion channels compared to ligands for nuclear receptors and PPI inhibitors. This reflects the physical nature of these proteins, i.e. nuclear receptor binding sites are notoriously hydrophobic and, as described above, PPI hotspots are also hydrophobic.
As described above, there have been attempts to delineate PPIs into classes. Specifically, there are narrow groove-like interfaces such as those seen with BCL-XL:BAX, and those which are wide and flat, as seen with IL2:IL2Rα.9 An affinity analysis of PPI inhibitors with respect to their type of interface showed that most inhibitors bound to the narrow interfaces compared to the wide interfaces.18 Therefore, careful analysis of PPI interface properties should also be an important feature of a PPI drug discovery campaign. Optimal targets will likely be narrow and/or contain small hotspot regions which can be readily covered by a small molecule.
Another option would be to take advantage of the plasticity of a PPI interface. There are examples where PPI modulators have been shown to take advantage of conformational changes which can occur when two proteins interact, binding to a dynamic pocket.32 An example of this is BILH434 inhibition of the human papillomavirus 11 E2 transcription factor. Specifically, the E2 transcription factor interacts with the E1 helicase and DNA. BILH434 stops this interaction by binding to the E1 enzyme in a deep hydrophobic pocket generated through a conformational change of residues at the interface region.34 A similar phenomenon was observed in the IL-2 PPI inhibitor SP4206. Normally the IL2 interaction surface with receptor α appears flat. However, in the presence of SP4206, a pocket which is occupied by the inhibitor is observed.35 This method was successfully utilised for the XIAP:SMAC PPI interface. Specifically, a tetrapeptide derived from SMAC (AVPI) was conformationally constrained via the fusion of two amino acids, leading to a greater potency of this tetrapeptide.36 Similar considerations to conformation were applied in structure-based design to generate non-peptide, conformationally constrained, SMAC mimetics.37 One of these mimetics, xevinapant, is in clinical trials. These examples highlight that reducing rotatable bonds may be an area of improvement for future PPI inhibitor design. However, the identification of such pockets is a non-trivial task, in many cases difficult, both computationally and experimentally.62
Finally, orthosteric modulators may be a drug discovery opportunity for PPI inhibition. Although PPI interfaces are often large, orthosteric inhibitors that disrupt the interaction do not necessarily need to be large. Instead they could strongly bind via cryptic orthosteric pockets away from the interface to disrupt the complementarity and prevent protein association.63 The benefit for PPIs is that complete disruption may not be required, as merely disrupting the equilibrium may be sufficient for a therapeutic effect.64
Conclusion
The notion that PPIs are an undruggable target continues to be challenged by new drug successes, especially as new drugs arise from structure-based rational design and fragment screening. However, the number of PPIs in the human proteome is vast, and the number of PPIs which have been modulated by small molecules remains negligible. Therefore, targeting PPIs represents an exciting and mostly untapped opportunity in drug discovery.
Here we analysed the physicochemical characteristics of approved PPI modulators. PPI modulators in clinical trials and experimental PPI modulators were investigated. We show that the majority of PPI modulators in recent years display properties that are more aligned with orally bioavailable small molecules than with PPI drugs that are currently on the market. We believe that these properties need to be tighter (clog P to <3.5, MW <500, TPSA <100, HBD <3, and RB <6) to allow for successful lead-to-drug optimisation, providing a margin for the physicochemical properties' inflation during the final steps of absorption, distribution, metabolism and excretion toxicity (ADMET) optimisation. Furthermore, incorporating fragment-based strategies and knowledge about the target PPI properties, such as hotspot analysis and dynamics of complex formation, will further improve our ability to drug PPIs.
Author contributions
All authors designed, researched and wrote this article.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
J. K. H. is a RMIT Vice Chancellors Research Fellow. This research was in part funded by Cancer Australia/Cure Cancer Project Grants (GNT1184339, GNT1157298).
Electronic supplementary information (ESI) available. See DOI: 10.1039/d1md00213a
References
- Scannell J. W. Blanckley A. Boldon H. Warrington B. Nat. Rev. Drug Discovery. 2012;11:191–200. doi: 10.1038/nrd3681. [DOI] [PubMed] [Google Scholar]
- Chène P. ChemMedChem. 2006;1:400–411. doi: 10.1002/cmdc.200600004. [DOI] [PubMed] [Google Scholar]
- Wells J. A. Mcclendon C. L. Nature. 2007;450:1001–1009. doi: 10.1038/nature06526. [DOI] [PubMed] [Google Scholar]
- Zhang Q. C. Petrey D. Deng L. Qiang L. Shi Y. Thu C. A. Bisikirska B. Lefebvre C. Accili D. Hunter T. Maniatis T. Califano A. Honig B. Nature. 2012;490:556–560. doi: 10.1038/nature11503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn H. Lawrence M. S. Chouinard C. R. Shrestha Y. Hu J. X. Worstell E. Shea E. Ilic N. Kim E. Kamburov A. Kashani A. Hahn W. C. Campbell J. D. Boehm J. S. Getz G. Lage K. Nat. Methods. 2018;15:61–66. doi: 10.1038/nmeth.4514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J. Wang H. L. Ma F. M. Guo H. P. Fang N. N. Wang S. S. Li X. H. Mol. Med. Rep. 2017;16:7907–7914. doi: 10.3892/mmr.2017.7649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labbé C. M. Kuenemann M. A. Zarzycka B. Vriend G. Nicolaes G. A. F. Lagorce D. Miteva M. A. Villoutreix B. O. Sperandio O. Nucleic Acids Res. 2016;44:D542–D547. doi: 10.1093/nar/gkv982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torchet R. Druart K. Ruano L. C. Moine-Franel A. Borges H. Doppelt-Azeroual O. Brancotte B. Mareuil F. Nilges M. Menager H. Sperandio O. Bioinformatics. 2021;37(1):89–96. doi: 10.1093/bioinformatics/btaa1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nero T. L. Morton C. J. Holien J. K. Wielens J. Parker M. W. Nat. Rev. Cancer. 2014;14:248–262. doi: 10.1038/nrc3690. [DOI] [PubMed] [Google Scholar]
- Shin W. H. Christoffer C. W. Kihara D. Methods. 2017;131:22–32. doi: 10.1016/j.ymeth.2017.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin W.-H. Kumazawa K. Imai K. Hirokawa T. Kihara D. Adv. Appl. Bioinf. Chem. 2020;13:11–25. doi: 10.2147/AABC.S235542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaro R. E. Baudry J. Chodera J. Demir Ö. Mccammon J. A. Miao Y. Smith J. C. Biophys. J. 2018;114:2271–2278. doi: 10.1016/j.bpj.2018.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H. Zhou Q. He J. Jiang Z. Peng C. Tong R. Shi J. Signal Transduction Targeted Ther. 2020;5:213–235. doi: 10.1038/s41392-020-00315-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlieghe P. Lisowski V. Martinez J. Khrestchatisky M. Drug Discovery Today. 2010;15:40–56. doi: 10.1016/j.drudis.2009.10.009. [DOI] [PubMed] [Google Scholar]
- Cheng S.-S. Yang G.-J. Wang W. Leung C.-H. Ma D.-L. J. Hematol. Oncol. 2020;13:26–39. doi: 10.1186/s13045-020-00850-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh J. Petter R. C. Baillie T. A. Whitty A. Nat. Rev. Drug Discovery. 2011;10:307–317. doi: 10.1038/nrd3410. [DOI] [PubMed] [Google Scholar]
- Robertson N. Spring D. Molecules. 2018;23:959. doi: 10.3390/molecules23040959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith M. C. Gestwicki J. E. Expert Rev. Mol. Med. 2012;14:e16. doi: 10.1017/erm.2012.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski C. A. Lombardo F. Dominy B. W. Feeney P. J. Adv. Drug Delivery Rev. 2001;46:3–26. doi: 10.1016/S0169-409X(00)00129-0. [DOI] [PubMed] [Google Scholar]
- Moreira I. S. Fernandes P. A. Ramos M. J. Proteins: Struct., Funct., Bioinf. 2007;68:803–812. doi: 10.1002/prot.21396. [DOI] [PubMed] [Google Scholar]
- Higueruelo A. P. Schreyer A. Bickerton G. R. J. Pitt W. R. Groom C. R. Blundell T. L. Chem. Biol. Drug Des. 2009;74:457–467. doi: 10.1111/j.1747-0285.2009.00889.x. [DOI] [PubMed] [Google Scholar]
- Egbert M. Whitty A. Keserű G. M. Vajda S. J. Med. Chem. 2019;62:10005–10025. doi: 10.1021/acs.jmedchem.8b01732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choy Y. B. Prausnitz M. R. Pharm. Res. 2010;28:943–948. doi: 10.1007/s11095-010-0292-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski C. A. Adv. Drug Delivery Rev. 2016;101:34–41. doi: 10.1016/j.addr.2016.04.029. [DOI] [PubMed] [Google Scholar]
- Morelli X. Bourgeas R. Roche P. Curr. Opin. Chem. Biol. 2011;15:475–481. doi: 10.1016/j.cbpa.2011.05.024. [DOI] [PubMed] [Google Scholar]
- Wishart D. S. Feunang Y. D. Guo A. C. Lo E. J. Marcu A. Grant J. R. Sajed T. Johnson D. Li C. Sayeeda Z. Assempour N. Iynkkaran I. Liu Y. Maciejewski A. Gale N. Wilson A. Chin L. Cummings R. Le D. Pon A. Knox C. Wilson M. Nucleic Acids Res. 2018;46:D1074–D1082. doi: 10.1093/nar/gkx1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperandio O. Reynès C. H. Camproux A.-C. Villoutreix B. O. Drug Discovery Today. 2010;15:220–229. doi: 10.1016/j.drudis.2009.11.007. [DOI] [PubMed] [Google Scholar]
- Lagorce D. Douguet D. Miteva M. A. Villoutreix B. O. Sci. Rep. 2017;7:46277. doi: 10.1038/srep46277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veber D. F. Johnson S. R. Cheng H.-Y. Smith B. R. Ward K. W. Kopple K. D. J. Med. Chem. 2002;45:2615–2623. doi: 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]
- Clark D. E. J. Pharm. Sci. 1999;88:807–814. doi: 10.1021/js9804011. [DOI] [PubMed] [Google Scholar]
- Clark D. E. J. Pharm. Sci. 1999;88:815–821. doi: 10.1021/js980402t. [DOI] [PubMed] [Google Scholar]
- Jubb H. Blundell T. L. Ascher D. B. Prog. Biophys. Mol. Biol. 2015;119:2–9. doi: 10.1016/j.pbiomolbio.2015.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arkin M. R. Tang Y. Wells J. A. Chem. Biol. 2014;21:1102–1114. doi: 10.1016/j.chembiol.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y. Coulombe R. Cameron D. R. Thauvette L. Massariol M.-J. Amon L. M. Fink D. Titolo S. Welchner E. Yoakim C. Archambault J. White P. W. J. Biol. Chem. 2004;279:6976–6985. doi: 10.1074/jbc.M311376200. [DOI] [PubMed] [Google Scholar]
- Wilson C. G. M. and Arkin M. R., in Small-Molecule Inhibitors of Protein-Protein Interactions, ed. L. Vassilev and D. Fry, Springer Berlin Heidelberg, Berlin, Heidelberg, 2011, pp. 25–59, 10.1007/82_2010_93 [DOI] [Google Scholar]
- Cai Q. Sun H. Peng Y. Lu J. Nikolovska-Coleska Z. McEachern D. Liu L. Qiu S. Yang C. Y. Miller R. Yi H. Zhang T. Sun D. Kang S. Guo M. Leopold L. Yang D. Wang S. J. Med. Chem. 2011;54:2714–2726. doi: 10.1021/jm101505d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H. Nikolovska-Coleska Z. Yang C.-Y. Xu L. Liu M. Tomita Y. Pan H. Yoshioka Y. Krajewski K. Roller P. P. Wang S. J. Am. Chem. Soc. 2004;126:16686–16687. doi: 10.1021/ja047438+. [DOI] [PubMed] [Google Scholar]
- Leeson P. D. Springthorpe B. Nat. Rev. Drug Discovery. 2007;6:881–890. doi: 10.1038/nrd2445. [DOI] [PubMed] [Google Scholar]
- Jamieson C. Moir E. M. Rankovic Z. Wishart G. J. Med. Chem. 2006;49:5029–5046. doi: 10.1021/jm060379l. [DOI] [PubMed] [Google Scholar]
- Shamovsky I. Connolly S. David L. Ivanova S. Norden B. Springthorpe B. Urbahns K. J. Med. Chem. 2008;51:1162–1178. doi: 10.1021/jm070543k. [DOI] [PubMed] [Google Scholar]
- Freise K. J. Shebley M. Salem A. H. J. Clin. Pharmacol. 2017;57:796–804. doi: 10.1002/jcph.858. [DOI] [PubMed] [Google Scholar]
- FDA, Venclexta (Venetoclax) Clinical Pharmacology and Biopharmaceutics Review(s) Application Number: 208573, https://www.accessdata.fda.gov/drugsatfda_docs/nda/2016/208573Orig1s000ClinPharmR.pdf, (accessed 14th April, 2021)
- Koehl N. J. Henze L. J. Kuentz M. Holm R. Griffin B. T. Pharmaceutics. 2020;12:564–584. doi: 10.3390/pharmaceutics12060564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EMEA, Assessment Report Venclyxto, https://www.ema.europa.eu/en/documents/assessment-report/venclyxto-epar-public-assessment-report_en.pdf, (accessed 14th April, 2021)
- Tse C. Shoemaker A. R. Adickes J. Anderson M. G. Chen J. Jin S. Johnson E. F. Marsh K. C. Mitten M. J. Nimmer P. Roberts L. Tahir S. K. Xiao Y. Yang X. Zhang H. Fesik S. Rosenberg S. H. Elmore S. W. Cancer Res. 2008;68:3421–3428. doi: 10.1158/0008-5472.CAN-07-5836. [DOI] [PubMed] [Google Scholar]
- Mohamad Anuar N. N. Nor Hisam N. S. Liew S. L. Ugusman A. Front. Pharmacol. 2020;11:564108. doi: 10.3389/fphar.2020.564108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuenemann M. A. Bourbon L. M. L. Labbé C. M. Villoutreix B. O. Sperandio O. J. Chem. Inf. Model. 2014;54:3067–3079. doi: 10.1021/ci500487q. [DOI] [PubMed] [Google Scholar]
- Ran X. Gestwicki J. E. Curr. Opin. Chem. Biol. 2018;44:75–86. doi: 10.1016/j.cbpa.2018.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leeson P. D. Adv. Drug Delivery Rev. 2016;101:22–33. doi: 10.1016/j.addr.2016.01.018. [DOI] [PubMed] [Google Scholar]
- Hann M. M. MedChemComm. 2011;2:349–355. doi: 10.1039/C1MD00017A. [DOI] [Google Scholar]
- Wenlock M. C. Austin R. P. Barton P. Davis A. M. Leeson P. D. J. Med. Chem. 2003;46:1250–1256. doi: 10.1021/jm021053p. [DOI] [PubMed] [Google Scholar]
- Hamon V. Brunel J. M. Combes S. Basse M. J. Roche P. Morelli X. MedChemComm. 2013;4:797–809. doi: 10.1039/C3MD00018D. [DOI] [Google Scholar]
- Johnson T. W. Dress K. R. Edwards M. Bioorg. Med. Chem. Lett. 2009;19:5560–5564. doi: 10.1016/j.bmcl.2009.08.045. [DOI] [PubMed] [Google Scholar]
- Zhang X. Betzi S. Morelli X. Roche P. Future Med. Chem. 2014;6:1291–1307. doi: 10.4155/fmc.14.57. [DOI] [PubMed] [Google Scholar]
- Hann M. M. Keserü G. M. Nat. Rev. Drug Discovery. 2012;11:355–365. doi: 10.1038/nrd3701. [DOI] [PubMed] [Google Scholar]
- Bogan A. A. Thorn K. S. J. Mol. Biol. 1998;280:1–9. doi: 10.1006/jmbi.1998.1843. [DOI] [PubMed] [Google Scholar]
- Clackson T. Wells J. A. Science. 1995;267:383–386. doi: 10.1126/science.7529940. [DOI] [PubMed] [Google Scholar]
- Xu D. Si Y. Meroueh S. O. J. Chem. Inf. Model. 2017;57:2250–2272. doi: 10.1021/acs.jcim.7b00181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogan A. A. Thorn K. S. J. Mol. Biol. 1998;280:1–9. doi: 10.1006/jmbi.1998.1843. [DOI] [PubMed] [Google Scholar]
- Kozakov D. Hall D. R. Chuang G.-Y. Cencic R. Brenke R. Grove L. E. Beglov D. Pelletier J. Whitty A. Vajda S. Proc. Natl. Acad. Sci. U. S. A. 2011;108:13528–13533. doi: 10.1073/pnas.1101835108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morphy R. J. Med. Chem. 2006;49:2969–2978. doi: 10.1021/jm0512185. [DOI] [PubMed] [Google Scholar]
- Laraia L. McKenzie G. Spring D. R. Venkitaraman A. R. Huggins D. J. Chem. Biol. 2015;22:689–703. doi: 10.1016/j.chembiol.2015.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White P. W., Faucher A.-M. and Goudreau N., in Small-Molecule Inhibitors of Protein-Protein Interactions, ed. L. Vassilev and D. Fry, Springer Berlin Heidelberg, Berlin, Heidelberg, 2011, pp. 61–88, 10.1007/82_2010_92 [DOI] [Google Scholar]
- Toogood P. L. J. Med. Chem. 2002;45:1543–1558. doi: 10.1021/jm010468s. [DOI] [PubMed] [Google Scholar]
- Asinex, Protein-Protein Interactions - Asinex.com, https://www.asinex.com/ppi/, (accessed 4th April 2021)
- ChemDiv, PPI Library - 100% quality control. Custom Design. Worldwide delivery, https://www.chemdiv.com/protein-protein-interaction-ppi-library, (accessed 4th April, 2021)
- ChemDiv, Eccentric PPI Library, https://www.chemdiv.com/eccentric-ppi-library, (accessed 4th April, 2021)
- Bosc N. Muller C. Hoffer L. Lagorce D. Bourg S. Derviaux C. Gourdel M.-E. Rain J.-C. Miller T. W. Villoutreix B. O. Miteva M. A. Bonnet P. Morelli X. Sperandio O. Roche P. ACS Chem. Biol. 2020;15:1566–1574. doi: 10.1021/acschembio.0c00179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Life Chemicals, PPI Focused Libraries by Ligand-based Approach, https://lifechemicals.com/screening-libraries/targeted-and-focused-screening-libraries/ppi-libraries/ppi-focused-library, (accessed 7th April, 2021)
- Garland S. L. Dean P. M. J. Comput.-Aided Mol. Des. 1999;13:485–498. doi: 10.1023/A:1008014620568. [DOI] [PubMed] [Google Scholar]
- Garland S. L. Dean P. M. J. Comput.-Aided Mol. Des. 1999;13:469–483. doi: 10.1023/A:1008045403729. [DOI] [PubMed] [Google Scholar]
- Otava Chemicals, iPPI Focused Libraries, https://www.otavachemicals.com/products/targeted-libraries-and-focused-libraries/protein-protein-interaction, (accessed 8th April, 2021)
- Otava Chemicals, Peptidomimetic Libraries, https://www.otavachemicals.com/products/targeted-libraries-and-focused-libraries/peptidomimetics, (accessed 8th April, 2021)
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.