

Abstract
Defining and identifying causal intervention effects for transmissible infectious disease outcomes is challenging because a treatment – such as a vaccine – given to one individual may affect the infection outcomes of others. Epidemiologists have proposed causal estimands to quantify effects of interventions under contagion using a two-person partnership model. These simple conceptual models have helped researchers develop causal estimands relevant to clinical evaluation of vaccine effects. However, many of these partnership models are formulated under structural assumptions that preclude realistic infectious disease transmission dynamics, limiting their conceptual usefulness in defining and identifying causal treatment effects in empirical intervention trials. In this paper, we propose causal intervention effects in two-person partnerships under arbitrary infectious disease transmission dynamics, and give nonparametric identification results showing how effects can be estimated in empirical trials using time-to-infection or binary outcome data. The key insight is that contagion is a causal phenomenon that induces conditional independencies on infection outcomes that can be exploited for the identification of clinically meaningful causal estimands. These new estimands are compared to existing quantities, and results are illustrated using a realistic simulation of an HIV vaccine trial.
Keywords: infectiousness, interference, mediation, susceptibility, transmission, vaccine
1. Introduction
Estimating the causal effect of an intervention can be challenging when the outcome of interest is contagious [41]. For example, a vaccine intended to prevent infection by a transmissible disease may reduce the risk of infection in individuals who receive it, and may reduce transmissibility if a vaccinated individual becomes infected. When study subjects are embedded in interacting groups among whom the disease may be transmitted, it can be difficult to separate the effect of one subject’s vaccination on themselves from its effect on other individuals and the group as a whole. Usually, the estimand of greatest clinical interest is the effect of an intervention on individual risks of infection, holding all else constant.
The pursuit of empirically meaningful definitions of population-level causal vaccine effects has a long history. Greenwood and Yule [19] first described informally the conditions under which vaccine effects can be estimated. Halloran et al. [24] established some of the first theory and definitions for clinically meaningful vaccine effects, and subsequent work by Halloran and colleagues [22, 26, 27] described epidemiological study designs for identifying these quantities. Halloran and Struchiner [23] gave the first formal definitions of causal vaccine estimands using notation and assumptions of a modern counterfactual-based causal inference framework [54]. Hudgens and Halloran [31] and Tchetgen Tchetgen and VanderWeele [60] showed how this formalism could be applied in empirical randomized trials of clustered individuals [21, 29]. More recently, researchers have shown that randomized trials may not measure clinically meaningful intervention effects when infection can be transmitted within groups [15, 40, 59].
Researchers have described two-person partnership models of infectious disease transmission for defining more granular, or individual, causal intervention effects. VanderWeele and Tchetgen Tchetgen [64] introduced a partnership model consisting of two interacting individuals who may be vaccinated and can transmit the infection to each other. By limiting the extent of potential disease transmission to two individuals, effects can be more easily defined in terms of potential outcomes indexed by treatments of both individuals and the outcome of their partner. The partnership model can accommodate many types of epidemiological relationship where infectious disease transmission may occur between indidivudals. The partnership model can accommodate, for example, parent-child relationships, sibling relationships, needle-sharing partnerships among injection drug users, or sexual partnerships. While nearly all real-world partnerships occur in the context of a broader network of epidemiological relationships with others, partnership models may be useful when pairs are drawn nearly independently from disparate networks, so that pairs experience independent exposure to infection from outside the partnership. For example, a study of disease transmission among cohabitating couples chosen from different cities could plausbly claim that the pairs experienced independent exposure to infection from outside the relationship.
Using a principal stratification approach, the partnership model permits computations of bounds for the infectiousness effect [10, 20, 64]. VanderWeele et al. [66] presented a special case of the partnership model in which one individual is home-bound, and can only be infected via transmission from the other. The assumed asymmetry in the disease transmission structure – the home-bound partner cannot be infected from a source external to the partnership and cannot infect the other partner – makes this model tractable for point identification of contagion and infectiousness effects by ensuring that interference only happens in one direction. Interference arises when an individual’s potential outcomes depend on the treatment status of others [13]. To allow for mutual dependence of individuals’ potential outcomes on others’ treatments, Ogburn and VanderWeele [43] extend this approach to allow both individuals to be treated, with transmission occurring only from one specified individual to the other. However, Ogburn and VanderWeele [42] show using causal diagrams that transmission complicates application of existing mediation techniques, requiring additional structural assumptions about the nature of dependence among outcomes under different forms of interference [6, 44, 55, 57]. Shpitser et al. [57] proposed extensions of mediation analysis to symmetric mediation settings, using statistical chain graph models that do not require a priori fixing the individual whose outcome plays the role of the mediator within the partnership.
When the outcomes are time-dependent processes – as is often in infectious disease transmission dynamics – binary outcome indicators and specified time windows may be used to define outcomes so that the mediation-based approaches may be applied. But these definitions can complicate identification of causal effects because (i) a repeatedly measured outcome over time may introduce multiple mediators, and (ii) absence of the outcome at prior time points as a prerequisite for later measurements induces time-varying confounding. Existing methods for longitudinal mediation analysis have therefore either focused on defining “interventional” indirect effects in terms of combined path-specific effects that can be non-parametrically identified [36, 56, 65, 67, 69], or adopted approaches that avoid defining nested counterfactuals for time-to-event outcomes [1, 14]. These approaches to longitudinal mediation share the common prerequisite that the roles of the outcomes within each partnership are asymmetric.
Statisticians and epidemiologists have developed parallel literature devoted to mathematical modeling of infectious disease transmission dynamics. This work treats infectious disease transmission as a dynamic temporal phenomenon: the risk of infection in a given subject may change over time, as a function of the infection status of their contacts, and covariates. For example, Rhodes et al. [51] present hazard models of infectious disease transmission in groups that accommodate individual-level (e.g. treatment) variables with possibly different effects on susceptibility and infectiousness. Kenah [33, 34] extends these ideas to develop nonparametric and semi-parametric statistical models for estimating covariate effects under contagion. Structural transmission modeling has gained wide use in clinical studies of infectious disease dynamics because it combines mechanistic assumptions about infectious disease transmission with regression-style covariate adjustment [3–5, 7–9, 46, 61, 62, 68].
In this paper, we take a different approach to define and identify intervention effects in symmetric two-person partnerships under contagion. We seek to combine approaches from causal mediation analysis and mathematical modeling of transmission to develop a nonparametric framework that formalizes the role of time in infectious disease transmission from a causal perspective. In our construction, either individual can be vaccinated, can be infected from outside, and can infect the other if infected themselves. An individual’s treatment (or vaccine status) and covariates may affect both susceptibility to, and infectiousness of, their infection outcome. We first introduce a generic causal model and straightforward assumptions that permit non-parametric identification of “exposure-controlled” and natural “exposure-marginalized” contagion, susceptibility, and infectiousness effects. Briefly, the contagion effect captures how transmissible the infection is from an infected individual to an uninfected individual. The susceptibility effect summarizes the effect of treatment on the infection outcome of individual who receives it. The infectiousness effect indicates the effect of an individual’s treatment on others’ outcomes, when that individual is infected. We propose a framework that is non-parametric and imposes no restrictions on the joint distribution of infection times in a partnership. Before any infections have occurred in a partnership, the potential first infection times are conditionally independent, because neither partner can yet transmit the infection to the other. After the first infection, the time to infection of the remaining susceptible partner is now a function their partner’s, as well as their own, treatment and covariates. Because the resulting causal model incorporates this temporally changing structure, it is more complex than settings considered in other proposals. In particular, the causal effects defined in this paper differ from the “direct” and “indirect” effects defined using the interference framework developed by [31] in ways we describe formally. On the other hand, this added complexity yields straightforward point identification results that cannot be obtained by treating the infection outcomes of both individuals as simultaneous mediating variables [10, 20, 64]. Lastly, we discuss nonparametric identification under randomization and in observational settings, compare these estimands to existing quantities proposed by other authors, and conduct a simulation analysis of a hypothetical HIV vaccine trial to illustrate the estimands.
2. Setting
Consider a population consisting of pairs of individuals, henceforth referred to as partnerships. Within a partnership, either individual can be infected from an external source (exogenous to the partnership), and once infected, an individual may internally (endogenous to the partnership) transmit the infection to their uninfected partner. Label the individuals in the partnership 1 and 2. In a given partnership, let Ti be the infection time of person i, and let be the indicator of prior infection at time t. Let Xi be the binary treatment status of i, and let X = (X1, X2) be the joint binary treatment vector for the partnership. Let L = (L1, L2) be measured baseline covariates for the two individuals, including shared covariates for the partnership as a whole. In each partnership, we observe (T1, T2, X1, X2, L1, L2). In a symmetric partnership, the labels for individuals 1 and 2 may carry meaning (e.g. in mother-child pairs), or may be arbitrary and interchangeable. We will use the index i to refer generically to one individual, either 1 or 2, and j to refer to the partner of i.
To describe the causal structure of infectious disease transmission within a partnership, we consider a decomposition of the infection time Ti that will help us define counterfactual infection times under different circumstances. Recall that both individuals are uninfected at baseline, and let Wi be the time to initial infection of i from a source of infection external to the partnership. If i is the first in their partnership to become infected, then we observe Wi. If their partner j is infected first, we observe Wj = wj and Wi is censored at time wj. When Wi is censored by earlier infection of j, let Zi be the additional time to infection of i, beyond the infection time wj of their partner. Formally, we decompose Ti as follows.
| (1) |
We emphasize that the decomposition (1) is purely notational, and places no a priori restrictions on the joint distribution of infection times (Ti, Tj). Instead, (1) shows how observation of (Ti, Tj) reveals information about these infection waiting times: if Ti < Tj, then we can determine Wi = Ti, Wj > Ti, and Zj = Tj − Ti. Figure 1 illustrates this decomposition and motivates the contagion effect presented in Definition 1 below: the disease is said to be “contagious” if the distribution of Ti is different from that of Wi, or equivalently, if prior infection of j (Wj < Wi) changes the conditional distribution of the remaining time to infection of i (Zi). The definition (1) permits specification of causal assumptions, outlined below, to capture the way treatments to both i and j may affect different parts of the waiting times to infection.
Figure 1:
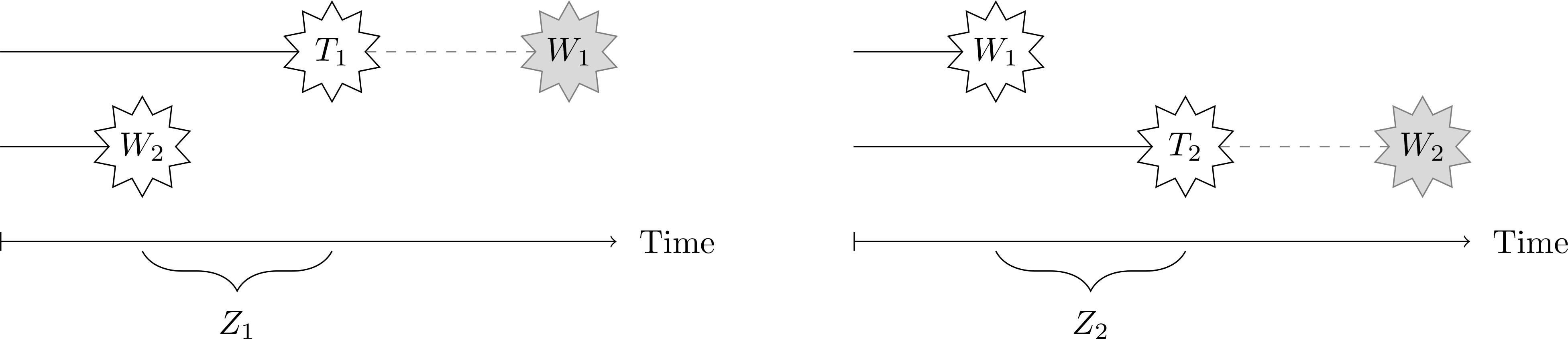
Illustration of contagion in a two-person partnership. At left, when subject 2 becomes infected first (W2 < W1), then W1 is censored, and Z1 is the remaining time to infection of subject 1. At right, when subject 1 is infected first (W1 < W2), then W2 is censored, and Z2 is the remaining time to infection of subject 2. Informally, the outcome is said to be “contagious” when the distribution of Ti is different from that of Wi.
In line with existing partnership models, it is assumed throughout that the partnerships are independent, thereby ruling out transmission between partnerships [31, 53]. Though partnerships are assumed to be independent, the waiting times Wi and Wj, or Zi and Zj, need not be identically distributed. The potential for transmission between partners is assumed to be symmetric – that is, either can infect the other – but the framework accommodates asymmetries in transmission if the distributions of Wi and Wj, or Zi and Zj, differ.
2.1. Assumptions
In this section, we describe assumptions that are sufficient to identify the causal effects defined in Section 3 from observable infection time data for each partnership. We state assumptions for a generic individual i and their partner j. To define potential, or counterfactual, infection times for individual i, let Wi(x) be the potential value of Wi under the joint treatment allocation x = (x1, x2). Let Zi(wj, x) be the additional potential time to infection of i, following the infection of j at time Wj(x) = wj, under joint treatment allocation x.
Assumption 1 (Exclusion restriction and independence of the initial infection).
Assumption 1 states that individual i’s initial infection time Wi(x) is invariant to the partner’s treatment status xj. Hence it may be viewed as a “no-interference” assumption on Wi, because Wi is the initial infection time from an external source, which can only be realized when Wi precedes Wj. Further, Wi(xi) is independent of Wj(xj) given (observed) covariates L. Assumption 1 respects a unique property of infectious disease: neither transmission nor treatment interference can occur without prior infection.
Assumption 2 (Initial infection exchangeability).
Assumption 2 states that there is sufficient covariate information in L so that the potential further time to infection Zi(wj, x) when j is infected at wj is conditionally independent of the potential initial infection time Wj(xj) of j. While this assumption bears similarity to the assumption of no unobserved confounding between the counterfactual mediator and nested potential outcome (for the same individual) under the single mediator setting [49, 52], we note that this assumption relates to counterfactual outcomes for different individuals.
Assumption 3 (Treatment exchangeability).
Assumption 3 means that the potential waiting times Wi(xi) and Zi(wj, x) are independent of the assigned treatment X within levels of the (observed) covariates L. This assumption prima facie resembles the conventional unconfoundedness assumptions for the (individual) exposure-mediator and exposure-outcome relations in mediation analysis. But in this context, Assumption 3 states that there is no unmeasured confounding between an individual’s infection times and the joint treatments for both individuals in the same partnership.
Two additional assumptions, commonly made in the literature when identifying causal estimands, ensure identifiability of potential infection outcomes from observational data.
Assumption 4 (Consistency).
Wi = Wi(xi), and Zi = Zi(wj, x) under the observed treatment X = x and infection time Wj = wj, for all x, wj > 0.
Assumption 5 (Positivity).
0 < Pr(Wj < w|Xi = xi, Li = li) < 1 for all w > 0, xi, and li; 0 < Pr(Zj < z|X = x, L = l) < 1 for all z > 0, x and l; and 0 < Pr(X = x|L = l) < 1 for all l.
A final assumption permits identification of certain “cross-world” potential infection outcomes.
Assumption 6 (Cross-world initial infection exchangeability).
.
Assumption 6 states that within levels of the observed covariates L, the potential waiting time of i to infection, after j is infected at wj under treatment xj, is independent of the potential infection time Wj under a different treatment . Informally, when Assumption 6 holds, after j becomes infected at (some fixed time) wj, the waiting time until i becomes infected under treatments x = (xi, xj) is independent of the time it would have taken j to be infected under a different treatment . We call this assumption a “cross-world” assumption because it makes explicit a probabilistic relationship between variables that cannot co-exist in the same realization of the process, namely Zi(wj, x) or .
Finally, let Ti(Wj(xj), x) be the potential outcome for the infection time of subject i, when j is infected at time Wj(xj) and the assigned treatments are x = (xi, xj). Following the decomposition (1) and by Assumptions 1–3, we can construct the potential infection time Ti(Wj(xj), x) as follows:
| (2) |
The potential infection time with Wj = wj fixed is denoted as Ti(wj, x).
For convenience, define the binary potential infection outcome evaluated at time t, . We refer to the potential infection time Ti(wj, x) and infection outcome Yi(t; wj, x) as exposure-controlled potential outcomes because they hold the partner’s infection time Wj = wj constant, thereby controlling the exposure to infection experienced by i. Similarly, we define , and refer to and as natural potential outcomes because they do not control the exact infection time wj of the partner, and instead rely on the natural distribution of Wj under the treatment xj.
The potential infection time decomposition (2) formalizes intuition about the structure of interference under contagion: there can be no interference without prior infection. When neither i nor j is infected, the time to infection of i is solely a function of the treatment xi, and there is no interference within the partnership. This is because the treatment xj of j can only affect i after j becomes infected. When j is the first to be infected, the remaining time to infection of i is now a function of both xi and xj, because j has now gained the ability to transmit to i. This apparent complexity simplifies identification of causal effects, as we show below.
3. Causal estimands
Contrasts of potential infection outcomes under different treatments x and infection times wj can yield epidemiologically meaningful causal estimands. In this paper, we express causal contrasts as differences of average potential infection outcomes. Effect measures on the hazard ratio, risk ratio, or odds ratio scales may be defined similarly [e.g. 24, 45].
First, the contagion effect captures the change in infection risk in one individual due to a change in the infection time of their partner.
Definition 1 (Contagion effect).
For and treatment x = (xi, xj), the controlled contagion effect is and the natural contagion effect is .
We say that the infection outcome (absent treatment) is “positively contagious” if for all infection times with wj < t, the controlled contagion effect under no treatment is . In this way, we interpret contagion, or outcome transmissibility, as a causal phenomenon that need not depend on treatments: under positive contagion, earlier infection of one’s partner causes one to become infected earlier, on average. On the other hand, the natural contagion effect CE(t, x) incorporates features of the treatment effect: it replaces fixed values of wj and with their counterfactual distributions Wj(0) and Wj(1) when j is treated versus untreated, similar to the effect proposed by VanderWeele et al. [66] for an asymmetric partnership. The natural contagion effect is a “cross-world” estimand because it integrates the average potential infection outcome with respect to the distribution of under a treatment that cannot arise in the same realization as Xj = xj. Figure 1 can be reinterpreted in light of Definition 1: positive contagion means that earlier infection of j causes i to become infected earlier, compared to the infection time of i that would have occurred, had Wj happened later.
The susceptibility effect is of interest in vaccine trials because it summarizes the clinical effect of an intervention on the individual who receives it, holding the treatment status and infection time of their partner constant [18, 23, 26]. The susceptibility effect is sometimes called the “per-exposure effect” because it holds the distribution of exposure to infectiousness constant [45].
Definition 2 (Susceptibility effect).
For wj > 0 and Xj = xj, the controlled susceptibility effect is and the natural susceptibility effect is .
If the controlled susceptibility effect is negative for every wj and xj, this means that the treatment is beneficial to the individual who receives it. Note that the natural susceptibility effect is not a cross-world estimand: it averages potential infection outcomes with respect to the distribution of Wj(xj), where xj is the treatment under which the infection outcome of i is realized.
The infectiousness effect summarizes the effect of changing the treatment to j on the infection risk of i, while holding the treatment to i and the infection time of j unchanged.
Definition 3 (Infectiousness effect).
For wj > 0 and Xi = xi, the controlled infectiousness effect is and the natural infectiousness effect is .
The natural infectiousness effect is a cross-world estimand because the first term in the contrast specifies that the infection time of j is realized under xj = 0, but the infectiousness of j subsequently is realized under xj = 1. Several authors have described the natural infectiousness effect as unidentified even under randomization when only binary infection outcomes are recorded at follow-up [10, 10–12, 20, 64, 66].
4. Identification of potential infection outcomes
We wish to non-parametrically identify the average potential infection outcome using observations of pairwise infection times, treatments, and covariates (Ti, Tj, Xi, Xj, Li, Lj). A preliminary result identifies the distribution of Wi(xi) in Lemma 1 using information about infection times. The proof is given in the Appendix.
Lemma 1.
Suppose Assumptions 1, 3–5 hold. Then the distribution function of Wi(xi) given Li = li is identified by
for any fixed values of xj, and lj, where p(Ti = u, Tj > u|X = (xi, xj),L = (li, lj)) is the joint probability density of Ti and survivor function of Tj.
Lemma 1 is a standard distributional identification result in competing risks [2]. Here, Wi and Wj are competing event times within the same partnership. The distribution of Wi or Wj is identified utilizing both waiting times in the partnerships, even when the waiting times are censored due to lost to follow-up or administrative censoring for some partnerships. The identified distribution function Fi(w|xi, li) is a function of xi and li only, and is invariant to values of xj and lj. However, in order to identify this function in the presence of the competing event Wj, particular values of xj and lj must be held constant.
The main result shows that average exposure-controlled potential infection outcomes given L = l are identified. Proofs are given in the Appendix.
Theorem 1 (Identification of the average exposure-controlled potential infection outcome).
Suppose Assumptions 1–5 hold and x = (x1, x2). For fixed values of wj and t, if wj < t,
| (3) |
otherwise, if t ≤ wj, .
In Theorem 1, Fi(wj|xi, li) is identified by Lemma 1 using all infection times (including censored infection times), and is estimated by the average outcome Yi(t) among observations when Tj = wj, Ti > Tj under X = x and L = l.
The structure of (3) shows that the average exposure-controlled potential infection outcome is identified by two types of observable events: when i is infected before their partner, and when i is infected after their partner. In contrast to most work studying causal effect of vaccine using binary infection outcomes by the end of observation, the causal identification in (3) is built on observation of infection time, which provides sufficient control for exposure to infection. Figure 2 shows a causal diagram [48] that captures the causal structure among the variables in the system outlined by Assumptions 1–5. This causal diagram does not necessarily represent a causal non-parametric structural equation model (NPSEM). The approach proposed in this paper is not contingent on having a well-defined joint (probabilistic) density of the counterfactuals under every possible intervention, whereas, Shpitser et al. [57] build on NPSEMs that are represented using such causal diagrams.
Figure 2:
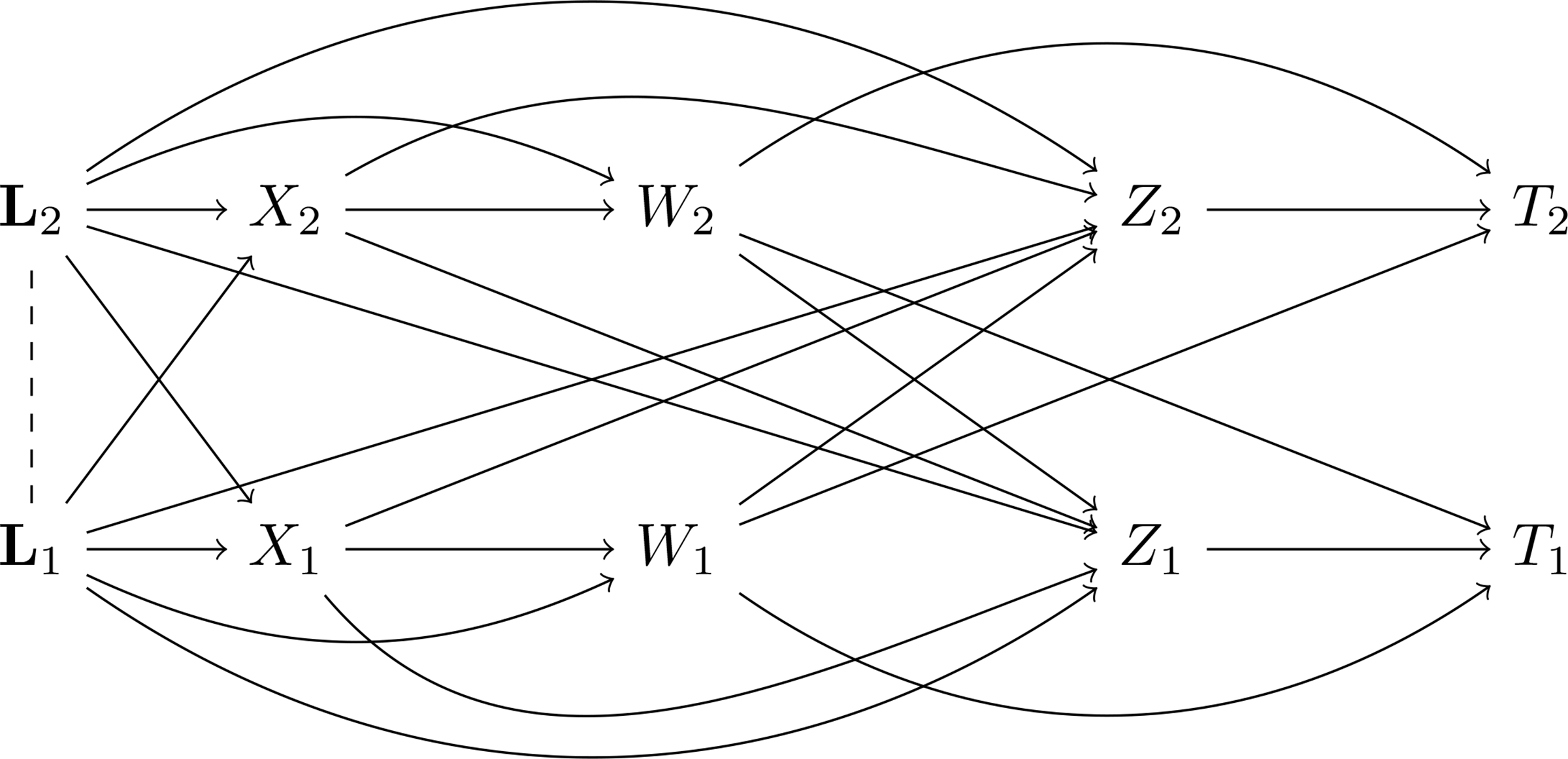
Causal graphical model for infection outcomes in a two-person partnership, under Assumptions 1–5. Covariates L1 and L2 may be dependent within partnerships, and covariates of both subjects may affect the joint treatments (X1, X2). The initial infection times W1 and W2 are functions of individual covariates and treatments alone by Assumption 1, and thus no arrows exist from Xj to Wi or from Lj to Wi. Subsequent waiting times Z1 and Z2 are functions of treatments and covariates of both subjects, and the infection time of the first infected subject. From the decomposition of the infection time (1), the latent additional infection time Zi and the (possibly latent) time Wi are relevant to exclusive cases of realization of Ti, so they are no arrows between them. The overall infection time Ti is determined by Wi, Wj and Zi, as specified in (1).
If we do not fix the infection time Wj = wj, and instead allow it to take its “natural” value under a particular treatment to j, we obtain the marginal average potential infection outcome when L = l as follows.
Corollary 1 (Identification of average natural/exposure-marginalized potential infection outcome).
Suppose Assumptions 1–5 hold. Then for x = (xi, xj), . If in addition and Assumption 6 holds,
where is given by Lemma 1 and by Theorem 1.
Definition 3 and Corollary 1 together show why the natural infectiousness effect is not identified even under randomization when only binary infection outcomes are recorded at follow-up [10–12, 20, 64, 66]. The correct marginalization over infection times cannot be computed unless the distribution of is identified as in Lemma 1. The controlled and natural infectiousness effects are similar to those proposed by Chiba and Taguri [12], but here the marginalization is over the infection time of j, not their binary infection outcome.
Finally, by standardization of the potential infection outcome across the distribution of covariates L, we can identify the average potential infection outcome. Let G(l) be the distribution function of the joint covariate vector L = l in the population of partnerships. Then
| (4) |
and
| (5) |
where and are given by Theorem 1 and Corollary 1 respectively. Because this paper is focused on nonparametric identification, we leave discussion of non-parametric statistical estimation of both controlled and natural causal estimands to the Appendix.
5. Comparison to other infectious disease intervention effects
Statisticians and epidemiologists have proposed a wide variety of estimands summarizing the effect of interventions for contagious outcomes, often in the two-person partnership setting. In this section, we evaluate the meaning of alternative definitions of vaccine effect estimands in the context of the causal effects defined above. We take the controlled contagion, susceptibility, and infectiousness effects defined above as fundamental characteristics of the disease transmission process and intervention under study. Whenever possible, we characterize the sign, or direction, of alternative effects, as a function of these primitives. In some cases, where the relationship is complex, we evaluate the alternative estimands under a null hypothesis, for example when the controlled susceptibility or infectiousness effect is zero, so that explicit results can be analytically proven. For simplicity, we omit the role of covariates L in the comparison of estimands.
The “attack rate” of an infectious disease is defined for individuals with treatment x as . The ratio of attack rates, sometimes called “relative cumulative incidence”, is a traditional measurement for the vaccine effect on susceptibility [12, 16, 17, 19, 24–27, 29, 30, 38, 47], defined as VEAR(t) = 1 − AR1(t)/AR0(t). A related estimand, called the “direct effect”, is a contrast on the difference scale, DE(t) = AR1(t) − AR0(t) when treatment is randomized within groups [31]. In the symmetric partnership setting, attack rates ARx(t) that condition only on the treatment to i implicitly marginalize over treatment to j.
Theorem 2.
Suppose SE(t, wj, xj) = 0 and IE(t, wj, xj) < 0 for all xj and wj > 0. If X = (Xi, Xj) is positively dependent, then DE(t) < 0 and VEAR(t) > 0; if X is negatively dependent then DE(t) > 0 and VEAR(t) < 0; and if Xi ⫫ Xj then DE(t) = VEAR(t) = 0. If there is no treatment effect whatsoever, SE(t, wj, x) = IE(t, wj, x) = 0 for all x and wj > 0, then DE(t) = VEAR(t) = 0 for any joint distribution of X.
In other words, VEAR(t) and DE(t) may or may not recover the sign, or direction, of the susceptibility effect, depending on the susceptibility and infectiousness effects, and the joint distribution of X within clusters. Morozova et al. [40] and Eck et al. [15] proved similar results in a parametric setting under Bernoulli, block, and cluster randomization for the joint treatment X in clusters or partnerships. Longini et al. [38], Halloran et al. [24], Halloran et al. [25], Halloran et al. [26] and Rhodes et al. [51] warned that VEAR(t) may be a biased approximation to the susceptibility effect due to differential exposure to infection between treated and untreated individuals in clusters. We show simulation examples that result in biased DE(t) under block randomization in Table 1 and Figure 4(d) below.
Table 1:
Simulation results showing true values of the natural contagion, susceptibility, infectiousness effects, and alternative estimands defined by Hudgens and Halloran [31], Halloran and Hudgens [20], and VanderWeele et al. [66]. Estimands are evaluated under six different scenarios - (i) constant hazards with α = 0.2 and γ = 10 in (8), (ii) constant hazards without contagion with α = 0.2, γ = 0 in (8), (iii) time-varying hazards with a = 0.4, b = 25 and w = 0.5 in (9), (iv) time-varying external hazard without contagion with a = 0.4, b = 0 and w = 0.5 in (9), (v) time-varying hazards with a = 0.2, b = 40, k = 1.5 and θ = 3 in (10), and (vi) time-varying hazard without contagion with a = 0.2, b = 0, k = 1.5 and θ = 3 in (10), respectively. The effect of vaccination is the same across all scenarios with and eσ = 0.01. The individual covariates (li, lj) are correlated with ρ = 0.1 and coefficients of .
| Treatment | CE(t, 0) | SE(t, 0) | IE(t, 0) | DE(t) | IDE(t) | ||
|---|---|---|---|---|---|---|---|
|
| |||||||
| Constant hazards | |||||||
| Observational | 0.12 | −0.14 | −0.19 | −0.16 | −0.20 | −0.70 | - |
| Bernoulli | 0.12 | −0.14 | −0.19 | −0.16 | −0.20 | −0.70 | (−0.73, −0.66) |
| Block | - | - | - | 0.06 | - | - | - |
| Cluster | - | - | - | −0.39 | - | - | - |
| Constant hazards without contagion | |||||||
| Observational | 0.00 | −0.18 | 0.00 | −0.18 | 0.00 | −0.01 | - |
| Bernoulli | 0.00 | −0.18 | 0.00 | −0.18 | 0.00 | −0.01 | (−0.25, 0.19) |
| Block | - | - | - | −0.18 | - | - | - |
| Cluster | - | - | - | −0.18 | - | - | - |
| Time-varying external and decreasing internal hazards | |||||||
| Observational | 0.12 | −0.14 | −0.20 | −0.21 | −0.22 | −0.51 | - |
| Bernoulli | 0.12 | −0.14 | −0.20 | −0.21 | −0.22 | −0.51 | (−0.53, −0.50) |
| Block | - | - | - | 0.08 | - | - | - |
| Cluster | - | - | - | −0.50 | - | - | - |
| Time-varying external and decreasing internal hazards without contagion | |||||||
| Observational | 0.00 | −0.28 | 0.00 | −0.28 | 0.00 | −0.02 | - |
| Bernoulli | 0.00 | −0.28 | 0.00 | −0.28 | 0.00 | −0.02 | (−0.43, 0.36) |
| Block | - | - | - | −0.28 | - | - | - |
| Cluster | - | - | - | −0.28 | - | - | - |
| Time-varying external and increasing-then-decreasing internal hazards | |||||||
| Observational | 0.10 | −0.16 | −0.17 | −0.17 | −0.18 | −0.64 | - |
| Bernoulli | 0.10 | −0.16 | −0.17 | −0.17 | −0.18 | −0.64 | (−0.62, −0.39) |
| Block | - | - | - | 0.02 | - | - | - |
| Cluster | - | - | - | −0.37 | - | - | - |
| Time-varying external and increasing-then-decreasing internal hazards without contagion | |||||||
| Observational | 0.00 | −0.18 | 0.00 | −0.18 | 0.00 | −0.01 | - |
| Bernoulli | 0.00 | −0.18 | 0.00 | −0.18 | 0.00 | −0.01 | (−0.43, 0.36) |
| Block | - | - | - | −0.18 | - | - | - |
| Cluster | - | - | - | −0.18 | - | - | - |
Figure 4:
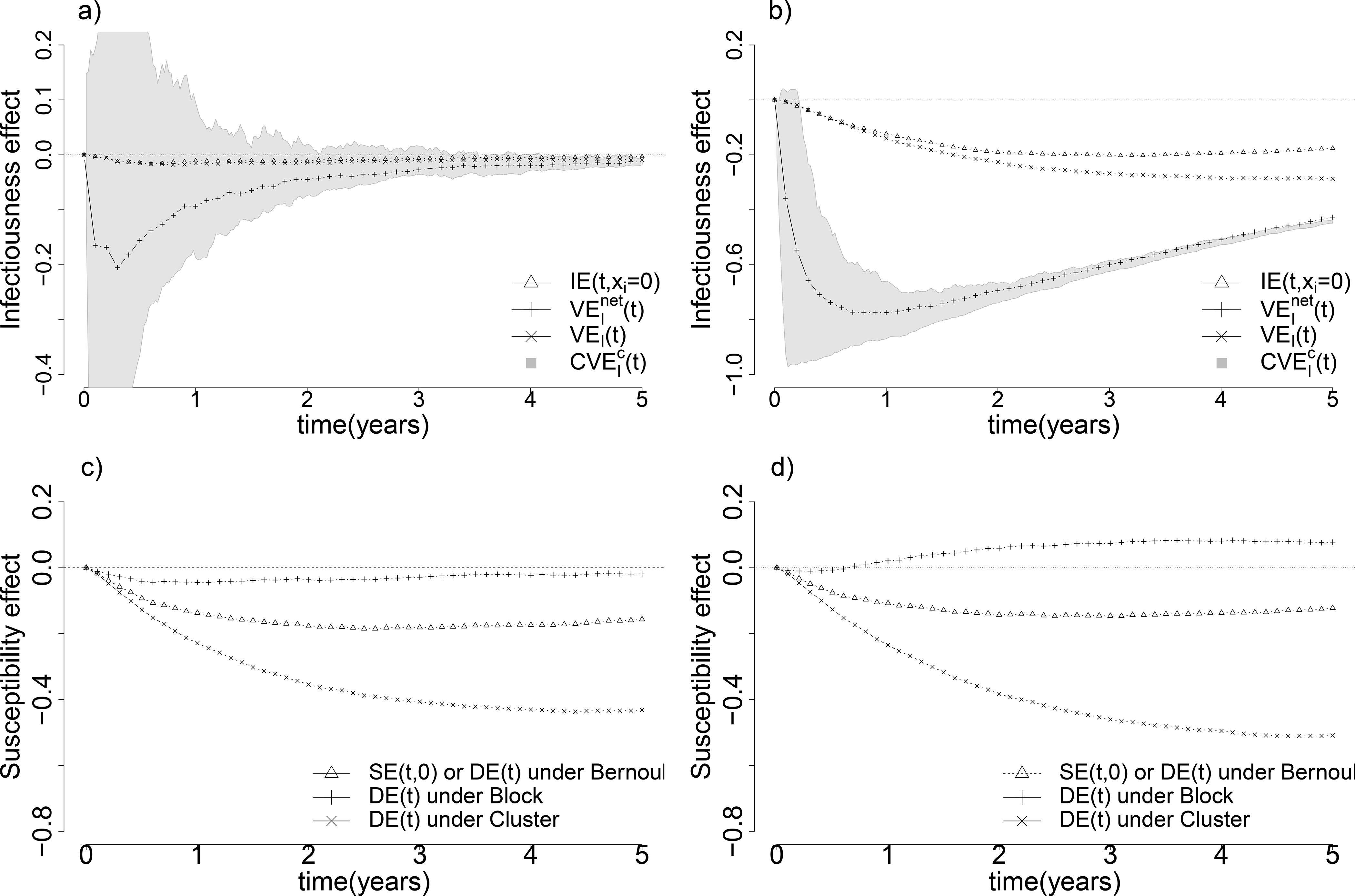
Comparison of different natural infectiousness and susceptibility effects. Figure a) compares different natural infectiousness effects – natural infectiousness effect IE(t, xi = 0), crude infectiousness effect , the infectiousness defined in mediation analysis VEI(t) and bounds identified by principal stratification – when both true susceptibility effect and true infectiousness effect are beneficial (eβ = 0.3, eσ = 0.5). Similarly, Figure b) shows the same comparison of multiple natural infectiousness effects as in Figure a) when the true infectiousness effect is much stronger than the true susceptibility effect (eβ = 0.4, eσ = 0.01). Figure c) shows the comparison of different types of natural susceptibility effect – the natural susceptibility effect SE(t, 0), the crude susceptibility effect DE(t) under Bernoulli, Complete, and Cluster randomization – when both true susceptibility effect and true infectiousness effect are beneficial (eβ = 0.3, eσ = 0.5) as in Figure a). Likewise, Figure d) shows the same comparison of multiple natural susceptibility effects when the true infectiousness effect is much stronger than the true susceptibility effect (eβ = 0.4, eσ = 0.01). All four graphs are under constant baseline hazards α(t) = 0.2 and γ(t) = 10.
Related definitions of the attack rate condition on the treatments to both individuals in the partnership. The attack rate among individuals with treatment x whose partner has treatment x′ is . The indirect effect is defined as IDE(t) = AR01(t) − AR00(t) [12, 31], and is equivalent to the difference of the natural infectiousness and contagion effects defined above:
The secondary attack rate is the proportion in a cluster infected after being exposed to an earlier infected individual, formally defined as . The SAR is the average infection status of i when j is infected during the study before i, under treatments x and x′ to i and j respectively. Based on the potential pitfalls of SAR, researchers proposed as “secondary attack rate for infectiousness” Halloran and Hudgens [20], Halloran et al. [24, 26, 27, 28, 29], Orenstein et al. [47]. We analyze under the null hypothesis of no infectiousness effect, and show that when the infection is contagious and there is a susceptibility effect, may nevertheless be nonzero. Let h0(u|0) be the hazard of the potential infection time Wi(0), and let h0(u|1) be the hazard of Wi(1).
Theorem 3.
Suppose IE(t, wj, 0) = 0, for all , and h0(u|1) = εh0(u|0) with ε ∈ [0,1), then . If SE(t, wj, xj) = 0 for all wj and xj, then preserves the same sign as IE(t, wj, 0). Suppose for all and h0(u|1) = εh0(u|0) with ε ∈ [0,1), then .
In other words, when the true infectiousness effect is null, the infection outcome is positively contagious, and the vaccine has a favorable susceptibility effect prior to the first infection, can nevertheless be nonzero. In a more extreme case, when the true contagion effect is null, the disease is not transmissible so that the true infectiousness is null; if the vaccine has a favorable susceptibility effect prior to the first infection, then is still nonzero. Simulation examples show biased under a null contagion effect in Tables 1 and 2 below.
Table 2:
Simulation results showing true values of the natural contagion, susceptibility, infectiousness effects, and alternative estimands defined by Hudgens and Halloran [31], Halloran and Hudgens [20], and VanderWeele et al. [66]. Estimands are evaluated under six different scenarios - (i) constant hazards with α = 0.2 and γ = 10 in (8), (ii) constant hazards without contagion with α = 0.2, γ = 0 in (8), (iii) time-varying hazards with a = 0.4, b = 25 and w = 0.5 in (9), (iv) time-varying external hazard without contagion with a = 0.4, b = 0 and w = 0.5 in (9), (v) time-varying hazards with a = 0.2, b = 40, k = 1.5 and θ = 3 in (10), and (vi) time-varying hazard without contagion with a = 0.2, b = 0, k = 1.5 and θ = 3 in (10), respectively. The effect of vaccination is the same across all scenarios with and eσ = 0.5. The individual covariates (li, lj) are correlated with ρ = 0.1 and coefficients of .
| Treatment | CE(t, 0) | SE(t, 0) | IE(t, 0) | DE(t) | IDE(t) | ||
|---|---|---|---|---|---|---|---|
|
| |||||||
| Constant hazards | |||||||
| Observational | 0.14 | −0.18 | −0.01 | −0.20 | −0.14 | −0.04 | - |
| Bernoulli | 0.14 | −0.18 | −0.01 | −0.20 | −0.14 | −0.04 | (−0.08, 0.02) |
| Block | - | - | - | −0.04 | - | - | - |
| Cluster | - | - | - | −0.36 | - | - | - |
| Constant hazards without contagion | |||||||
| Observational | 0.00 | −0.22 | 0.00 | −0.22 | 0.00 | −0.01 | - |
| Bernoulli | 0.00 | −0.22 | 0.00 | −0.22 | 0.00 | −0.01 | (−0.39, 0.19) |
| Block | - | - | - | −0.22 | - | - | - |
| Cluster | - | - | - | −0.22 | - | - | - |
| Time-varying external and decreasing internal hazards | |||||||
| Observational | 0.15 | −0.18 | −0.01 | −0.23 | −0.15 | −0.03 | - |
| Bernoulli | 0.15 | −0.18 | −0.01 | −0.23 | −0.15 | −0.03 | (−0.04, 0.00) |
| Block | - | - | - | −0.03 | - | - | - |
| Cluster | - | - | - | −0.44 | - | - | - |
| Time-varying external and increasing-then-decreasing internal hazards without contagion | |||||||
| Observational | 0.00 | −0.34 | 0.00 | −0.34 | 0.00 | −0.02 | - |
| Bernoulli | 0.00 | −0.34 | 0.00 | −0.34 | 0.00 | −0.02 | (−0.64, 0.36) |
| Block | - | - | - | −0.34 | - | - | - |
| Cluster | - | - | - | −0.34 | - | - | - |
| Time-varying external and increasing-then-decreasing internal hazards | |||||||
| Observational | 0.12 | −0.21 | −0.02 | −0.22 | −0.13 | −0.08 | - |
| Bernoulli | 0.12 | −0.21 | −0.02 | −0.22 | −0.13 | −0.08 | (−0.21, 0.07) |
| Block | - | - | - | −0.08 | - | - | - |
| Cluster | - | - | - | −0.36 | - | - | - |
| Time-varying external and increasing-then-decreasing internal hazards without contagion | |||||||
| Observational | 0.00 | −0.22 | 0.00 | −0.22 | 0.00 | −0.01 | - |
| Bernoulli | 0.00 | −0.22 | 0.00 | −0.22 | 0.00 | −0.01 | (−0.64, 0.36) |
| Block | - | - | - | −0.22 | - | - | - |
| Cluster | - | - | - | −0.22 | - | - | - |
A simple explanation shows why can behave in unexpected ways: it is not solely a function of the infectiousness effect. Instead, also incorporates reduced exposure to infection from delaying the infection of partner j due to vaccination, which in fact is the susceptibility effect on the partner j before the first infection occurs. Therefore, when the true susceptibility effect is null, is only a function of the infectiousness effect and thus recovers the correct sign of infectiousness effect. From a sightly different perspective, several authors have also pointed out that may suffer from selection bias because it conditions on post-randomization variables – the infection status of both partners [20, 24–26, 51]. Specifically, relies on the eventual infection outcome of partner j, rather than the infection time of partner j. Halloran and Hudgens [20] use tools from principal stratification to derive bounds for the infectiousness effect to correct this selection bias, and propose a bound estimator for under Bernoulli randomization. We analyze these bounds by simulation below.
Several authors have recognized that simple comparison of outcomes in treated versus untreated individuals may not suffice to identify meaningful causal effects for infectious disease interventions, even under randomization. For example, VanderWeele [63], VanderWeele et al. [66], Ogburn and VanderWeele [42], and Ogburn and VanderWeele [43] apply tools from mediation analysis to a simplified partnership model to identify contagion and infectiousness effects similar to those we have defined above. This “asymmetric partnership” model focuses on pairs of individuals i and j when i is restricted to be home-bound, unvaccinated, and may only be infected by their (possibly vaccinated) partner j. Partner j is randomized to receive treatment or placebo, and may be infected by a source of infection outside the partnership. In other words, the relative role of the two subjects cannot be swapped. For example, in a HIV trial of zidovudine, the study units are mother-child pairs, and only mothers are vaccinated and may transmit HIV to the children, not vice versa [39]. This is different from the symmetric partnership setting we considered, when both i and j can be treated and infected by the outside or each other.
To represent this structural assumption in the framework outlined here, we force the infection time of the home-bound partner, in the absence of infection in their partner, to be infinite. To this end, let hazard of Wi(0) be , so that infection of i from an external source can never occur. These authors define the infectiousness effect as , which contrasts the infection outcomes of i when j is treated versus untreated, with j’s infection status Yj(xj) set to the value it would take if j were treated.
Theorem 4.
Suppose for all t > 0. Then VEI(t) = IE(t, 0).
In other words, under the asymmetric setting where i is unvaccinated and cannot be infected from outside the partnership, VEI(t) is equivalent to the natural infectiousness effect in Definition 3.
A contagion effect is defined by VanderWeele et al. [66] as
contrasting the infection outcome of i when the infection status of j is set to the value it would obtain if j were treated versus untreated. Note that this quantity reverses the difference in the natural contagion effect in Definition 1, as VEC(t) = −CE(t, x). We provide sufficient conditions for the controlled contagion effect CE(t, u, u′, 0) and VEC(t) (or equivalently, −CE(t, x)) to behave similarly, that is, to have opposite sign.
Theorem 5.
Suppose for all t > 0 and SE(t, wj, 0) > 0. Then VEC(t) has opposite sign as for . Suppose , SE(t, wj, 0) = 0 and , then VEC(t) = 0.
In other words, in the asymmetric partnership setting, −VEC(t) recovers the sign of the true contagion effect, when the vaccine has a favorable susceptibility effect prior to the first infection. However, if the true susceptibility effect is null, VEC(t) = 0 regardless of the true contagion effect.
6. Application: a hypothetical vaccine trial
We simulate observational and randomized trials of a hypothetical HIV vaccine in a large population of sexual partnerships [25]. We assume individuals are not infected at baseline, but that either individual may become infected from outside the partnership, and transmission within partnerships may occur. To parameterize the infection transmission process, we specify hazard models for the infection times Wi(xi) and Zi(wj, x). This approach has been employed in extensive prior work on statistical models for time-to-infection data [25, 32–35, 51]. For a time t > 0, Let the hazard of Wi(xi) given covariates Li = li be given by
| (6) |
In words, the hazard of infection in an individual whose partner is not infected, is given by a Cox model with baseline hazard α(t). Following infection of j at time Wj = wj, the remaining potential infection time Zi(wj, x) given L = l = (li, lj) has hazard
| (7) |
for t > wj. The coefficients β0 and β1 represent the change in infection risk due to vaccination of i, and σ represents the change in transmission risk due to vaccination in j when j is infected. Covariate effects are represented by θ0, θ1, and θ2, and α(t) and γ(t−wj) are baseline transmission hazards for the external and internal forces of infection respectively. This specification implies that the external force of infection and transmissibility are competing risks for infection of i [37, 38, 50]. That is, a susceptible individual can be infected by a source of infectiousness outside their partnership, or from an infected partner. We consider three specifications of the baseline transmission hazards for the external and internal forces of infection: (i) both are time-invariant as in (8)
| (8) |
, (ii) the external baseline hazard varies seasonally and the internal baseline hazard decays over time as in (9)
| (9) |
, to (iii) when the external baseline hazard varies over seasons and the internal baseline hazard increases first then decreases over time as in (10).
| (10) |
When the baseline hazards α(t) and γ(t − wj) are time-invariant as specified in (8), the model reduces to a Markov susceptible-infective process with an external force of infection [e.g 15, 40]. For any functional forms of the baseline hazards α(t) and γ(t − wj), the hazard specifications (6) and (7) imply distributions for Wi(xi) and Zi(wj, x), and hence Ti(wj, x), that obey the required identification Assumptions 1–6.
Subjects in partnerships are endowed with individual characteristics L = (Li, Lj) that may be correlated. In the randomized trial simulation, the vaccine is randomized in accordance with a specified distribution – Bernoulli, block, or cluster randomization – without regard to these traits. Under each randomization design, the marginal treatment probability Pr(Xi = xi) is 1/2. For Bernoulli randomization, Pr(X = x) = 1/4, for block randomization, , and for cluster randomization, Pr(X = (1, 1)) = 1/2 and Pr(X = (0, 0)) = 1/2. In the observational study simulation, we consider a univariate individual covariate for illustration, and the traits L = (Li, Lj) together determine the joint distribution of vaccine in the partnership as
where
with v > 0. Non-parametric estimation of both controlled and natural causal estimands is described in detail in the Appendix.
Figure 3 illustrates controlled infection outcomes over time for different choices of wj and x under the time-invariant hazard scenario, estimated non-parametrically with sufficiently large numbers of pairs (N = 100, 000) so as to represent their true values in the simulation. Estimated controlled infection outcomes area aligned with their true values in Figure 3. Contrasts of these potential infection outcomes give the controlled contagion, susceptibility and infectiousness effects, shown in the lower-right corner of Figure 3.
Figure 3:
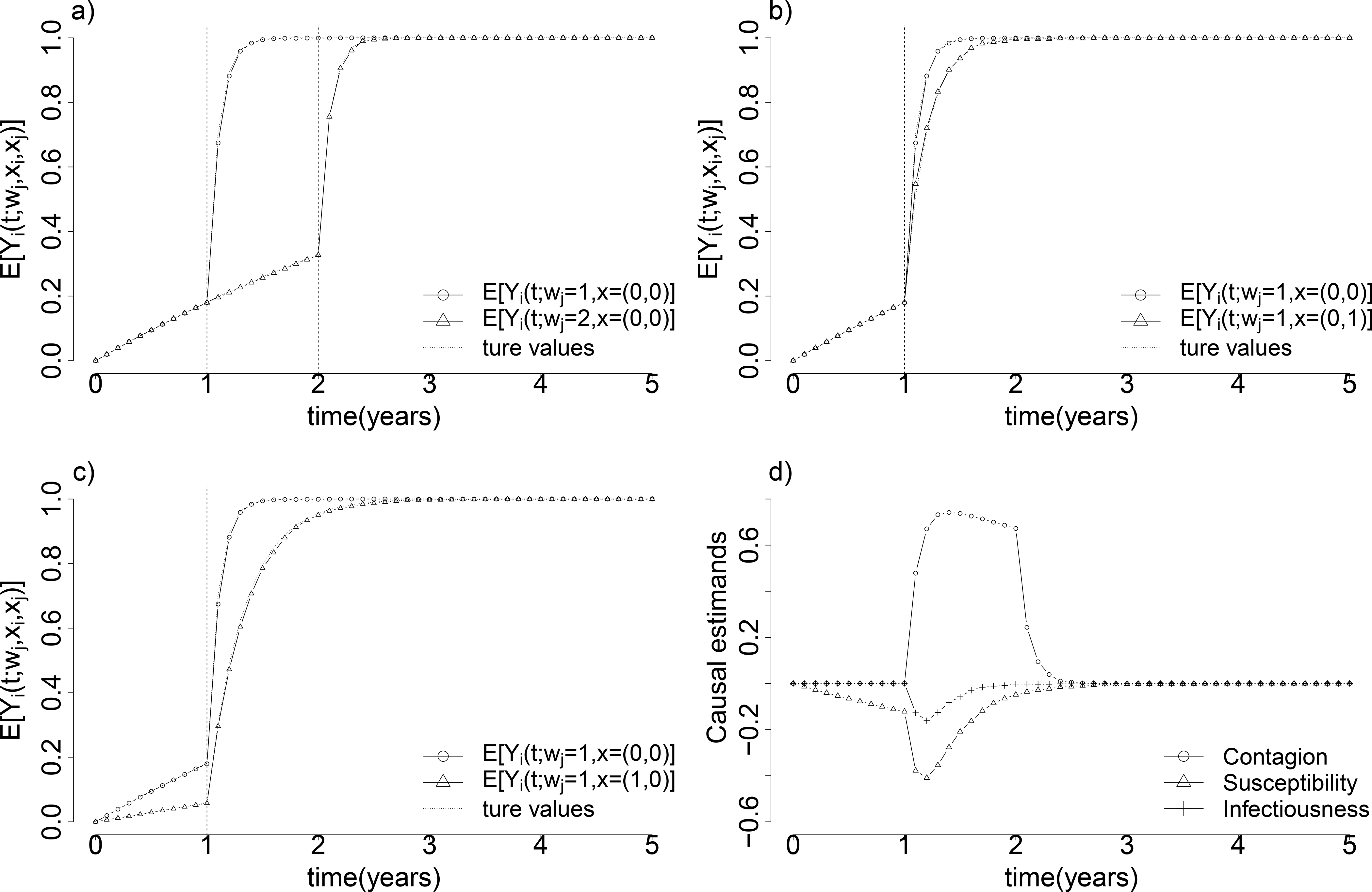
Illustration of average controlled potential infection outcomes under different values of the infection time wj and joint treatment x, under time-invariant baseline hazards α(t) = 0.2 and γ(t − wj) = 10 and coefficients and eσ = 0.5. Contrasts of potential outcomes in (a), (b) and (c) show the controlled contagion effect, the infectiousness effect, and the susceptibility effect evaluated at different times, shown together in (d).
Tables 1 and 2 show the true values of the natural contagion, susceptibility and infectiousness effects, and compare these values to the true values of alternative estimands proposed by other authors, including the direct effect DE(t), the indirect effect IDE(t), the secondary attack rate infectiousness effect , and bounds introduced by [20]. All natural or marginal estimands are evaluated at time t = 2 years under each design and under both time-invariant and time-varying baseline hazards. Estimands that are not identified under a given design are not evaluated. In Table 1, when the true infectiousness effect much stronger than the true susceptibility effect, the direct effect DE(t) is positive (0.06 and 0.08) under block randomization when the disease is contagious, even though the true susceptibility effect is negative, or beneficial [see, e.g. 15, 40]. Table 2 shows another simulation setting where DE(t) achieves the same sign as the susceptibility effect when the true infectiousness effect is on the same scale of the true susceptibility effect. In the three scenarios without contagion, the disease is not contagious and infection outcomes are realized independently. Therefore, all “indirect” and “infectiousness” effects should be null. However, is negative (−0.01 and −0.02 in both Table 1 and 2), conflicting with the fact that the disease is not transmissible (as proved in Theorem 3). The identification interval has nonzero width, but covers zero.
Figure 4 compares different types of natural susceptibility and infectiousness effects over time, when both effects are beneficial (negative). In the bottom-right panel of Figure 4, we show that DE(t) under block randomization can suffer from directional bias.
7. Discussion
We have described a nonparametric framework for identifying causal intervention effects under contagion in general two-person partnerships. The estimands and identification results generalize those given in prior work [20, 43, 64, 66], and establish that point identification of clinically meaningful causal estimands under contagion is possible even when relationships are symmetric and either individual can be treated. We take a nonparametric approach that does not ascribe infections to particular sources. Instead, the approach focuses on the effect of changing treatments or exposure to infections on the expectations of potential outcomes without information about “who-infected-whom.” We have made no assumptions about the functional form of infection risks (beyond the independencies and exclusion restrictions implied by Assumptions 1–6), how the risk of infection to a susceptible individual changes when their partner becomes infected, or how the vaccine changes susceptibility or infectiousness over time. The framework respects the logic of infectious disease transmission: if the outcome is not transmissible, the contagion and infectiousness effects are zero.
By studying the role of a partner’s infection time in the identification of controlled causal effects, we can identify causal estimands that are both more fundamental and more directly linked to the biological effect of a vaccine on infection risk than simple contrasts of infection rates. Our results also show that while some crude contrasts can recover causal effects in restricted settings (e.g. the infectious effect VEI(t) in the asymmetric partnership setting) or under a particular randomization design (e.g. the direct effect DE(t) under independent Bernoulli randomization), they may not deliver useful summaries of vaccine effects in more general situations. Finally, the framework developed in this paper may be useful in settings beyond infectious disease epidemiology, where symmetric mediated effects are of interest [e.g. 55, 58].
One important limitation of our identification approach is that the controlled estimands and cross-world natural estimands require observation of infection times, and not just binary infection indicators at a follow-up time t. In real-world vaccine trials, it may be unreasonable to require investigators to measure infection times Ti with precision, as is required by Lemma 1 and Theorem 1. Instead, cross-sectional infection assessment, follow-up surveys, or tests for biomarkers of prior infection are commonly used as the primary outcome. Corollary 1 shows exactly how controlled effects that rely on infection times relate to natural effects that do not. Attempts to disentangle individual effects from the mediating effects of treatment to partners using only binary infection outcomes may fail to recover useful controlled or marginal effects [see, e.g. , analyzed by 20]. One exception is the natural susceptibility effect, which can be estimated by binary outcomes under Bernoulli randomizations, as shown by Corollary 1.
Finally, while the symmetric partnership setting is useful for conceptualizing, defining, and identifying causal estimands, real-world vaccine trials usually happen in clusters of varying sizes. Adapting the setting outlined here to larger clusters results in rapid expansion of the number of potential outcomes, corresponding to every possible ordering of infections, necessitating simplifying structural assumptions to reduce the dimensionality of the problem. One promising avenue for dramatically reducing the number of potential outcomes without imposing a parametric structure was proposed by Kenah [33, 34]. The idea is that contagion works by competing risks, where hazards of infection from different sources are additive. This approach imposes no additional structure on the distribution of the initial time to infection, but assumes that new infected cluster members always add a competing risk of infection to the already existing risks of infection for susceptibles.
Acknowledgements
WWL was supported by NIH grant R01 AI085073 and by a Gillings Innovation Laboratory award from the UNC Gillings School of Global Public Health. FWC was supported by NIH grants DP2 OD022614, R01 AI112438, and R01 AI112970. We are grateful to Peter M. Aronow, Soheil Eshghi, Eben Kenah, Olga Morozova, Virginia E. Pitzer, and Li Zeng for helpful comments and discussion.
A. Proofs
Proof of Lemma 1.
Let fi(w|xi, li) be the density of Wi(xi) when Li = li and let Fi(w|xi, li) be the corresponding cumulative distribution function. By Assumption 5, 0 < Fi(w|xi, li) < 1 for all w > 0, xi, and li, so we can write
Then rearranging, we have
by Assumption 1
by Assumption 3
by Assumption 4
where xj is any fixed value of Xj and lj is any fixed value of Lj. ⎕
Lemma 2.
Under Assumptions 1–3, Yi(t;wj; x) ⫫ Wj(xj) | L and Yi(t;wj; x) X | L.
Proof of Lemma 2.
Fix a value wj > 0 and let x = (xi, xj). If Wi(xi) < wj, then Ti(wj, x) = Wi(xi) and by Assumption 1, Wi(xi) ⫫ Wj(xj) | L, so Ti(wj, x) ⫫ Wj(xj) | L. If Wi(xi) > wj then Ti(wj, x) = wj + Zi(wj, x) and by Assumption 2 Zi(wj, x) ⫫ Wj | L, so Ti(wj, x) ⫫ Wj(xj) | L. Therefore, since , it follows that Yi(t;wj; x) ⫫ Wj(xj) | L.
By the same reasoning, if Wi(xi) < wj, then Ti(wj, x) = Wi(xi) and by Assumption 3, Wi(xi) ⫫ X | L. If Wi(xi) > wj then Ti(wj, x) = wj + Zi(wj, x) and by Assumption 3, Zi(wj, x) ⫫ X | L. Therefore, since , it follows that Yi(t;wj; x) ⫫ X | L. ⎕
Lemma 3.
Under Assumptions 1–4, .
Proof of Lemma 3.
Fix a value wj > 0 and x = (xi, xj). If Wi(xi) ≥ wj then
If Wi(xi) < wj then
⎕
Proof of Theorem 1.
The average potential infection outcome when L = l is given by by Lemma 2
by the definition of Yi(t; wj, x)
by the definition of Ti(wj, x)
by Assumption 1
by Assumption 4 and Lemma 3
When t ≥ wj, then
Likewise, when t < wj, then
since when t < wj
Proof of Corollary 1.
Likewise, when x = (xi, xj) and ,
⎕
Lemma 4.
When SE(t, wj, xj) = 0, then Fj(t|xj) = Fj(t|1 − xj) and , for all xj ∈ {0,1} and t ≥ 0.
When SE(t, wj, xj) = IE(t, wj, xi) = 0, then .
When SE(t, wj, xj) = 0 and IE(t, wj, xi) < 0, then .
Proof of Lemma 4.
First we prove Fj(t|xj) = Fj(t|1−xj), for all xj ∈ {0, 1} when SE(t, wj, xj) = 0.
| (11) |
Second, we prove for all xj ∈ {0,1}, if SE(t, wj, xj) = 0.
| (12) |
Third, by (11), we prove , if SE(t, wj, xj) = IE(t, wj, xi) = 0.
| (13) |
Fourth, by (11), we prove , if SE(t, wj, xj) = 0 and IE(t, wj, xi) < 0.
| (14) |
⎕
Proof of Theorem 2.
Given the conclusions from (12) and (14), we have
| (15) |
Note by (14) in Lemma 4, we have the first term at the last line of (15) being negative. The sign of DE(t) then depends only on the treatment assignment mechanism, which leads to the following conclusions for DE(t).
- If the treatment assignment is positively correlated (Pr(Xi = c, Xj = c) > Pr(Xi = c) Pr(Xj = c) for c ∈ {0, 1}), we have:
Thus, DE(t) < 0.(16) If the treatment assignment is independent (Pr(Xi = c, Xj = c) = Pr(Xi = c) Pr(Xj = c) for c ∈ {0, 1}), then by similar arguments of (16), we have Pr(Xj = 1|Xi = 1)−Pr(Xj = 1|Xi = 0) = 0. Thus, DE(t) = 0.
If the treatment assignment is negatively correlated (Pr(Xi = c, Xj = c) < Pr(Xi = c) Pr(Xj = c) for c ∈ {0, 1}), then by similar arguments of (16), we have Pr(Xj = 1|Xi = 1)−Pr(Xj = 1|Xi = 0) < 0. Thus, DE(t) > 0.
When IE(t, wj, xi) = 0, following (13) and (15) in Lemma 4, we have and thus DE(t) = 0.
Similar arguments apply for VEAR(t). ⎕
Proof of Theorem 3.
We evaluate the sign of by analyzing SAR00(t) − SAR10(t).
First, we analyze the sign of under a null true infectiousness effect, when the infection outcome is positively contagious and vaccine has a favorable effect prior to first infection through h0(u|1) = εh0(u|0), for ε ∈ [0,1).
by applying the law of total probability
| (17) |
By IE(t, wj, 0) = 0 and Lemma 3 To ease the notation in Equation (17), we denote . Denote and , and and . Then by integration by parts, (17) can be re-written as follows:
By their definitions, we have G(0|1)−G(0|0) = 0 and G(t|1)−G(t|0) = 0, and thus . In other words, the sign of SAR10(t) − SAR00(t) only depends on the sign of G(u|1) − G(u|0) and dk(u) for all u > 0. First, we can show that dk(u) < 0 for 0 ≤ u < t. For 0 ≤ u < u′ < t, we have
| (18) |
Next, we analyze the property of G(u|1) − G(u|0) for ∀u > 0. Denote . Given h0(u|1) = εh0(u|0) with ε ∈ [0,1), we can write out G(u|0) and G(u|1) in terms of h0(u|0) as follows.
| (19) |
From (19), we observe that G(s|1) and G(s|0) only differ by the terms in front of H0. Treat G(s|1) and G(s|0) as functions of ε, and we can re-express them as and , given ε < 1. Then, if G(ε) is a decreasing function of ε, we have G(u|1) − G(u|0) ≤ 0.
| (20) |
Divide the numerator of (20) by a positive constant . We then have if for u < t, then G(u|1) − G(u|0) ≤ 0. Treat as a function of u, given 0 ≤ u < t. We have,
| (21) |
Combining (20) and (21), we have G(u|1) − G(u|0) ≤ 0.
In summary, we can see that
Thus, .
Next, we analyze the sign of under a null true susceptibility effect.
by (17)
by SE(t, wj, xj) = 0 and (11)
by Assumption 1
Thus, has the same sign as the true infectiousness effect, when the true susceptibility effect is null.
Third, we analyze the sign of in the case of no contagion, when the true susceptibility effect is beneficial. First, for all implies IE(t, wj, 0) = 0.
Following the same proof for the first case except replacing the second line of (18) by an equal sign, we know . ⎕
Proof of Theorem 4.
Given , we have for Wi(0).
by Fi(s|xi) = 0 for ∀s > 0
given and when Wi(0) = ∞
| (22) |
Thus, by the definition of VEI(t) and IE(t, xi), we have:
Thus, VEI is equivalent to the natural infectiousness effect under the asymmetric partnership. ⎕
Proof of Theorem 5.
Given , we have .
by Corollary 1
by Theorem 1
by Fi(t|xi) = 0 for ∀t > 0
since for wj > t
by the definition of k(u) in the proof of Theorem 3
by integration by parts
By the definition of k(u) and Fj(u|xj), we know k(t) = 0 and Fj(0|1) − Fj(0|0) = 0, and thus If SE(t, wj, x) > 0, we have Fj(wj|1) − Fj(wj|0) > 0. If CE(t, u, u′, (0, 0)) > 0 for 0 ≤ u < u′< t, dk(u) < 0 as shown in the the proof of Theorem 3. Thus, we have the following conclusions.
When SE(t, wj, x) > 0, VEC(t) has the opposite sign as CE(t, u′, u, (0, 0)).
If SE(t, wj, x) = 0 and CE(t, u, u′, (0, 0)) > 0, we have VEC(t) = 0. ⎕
Figure 5:
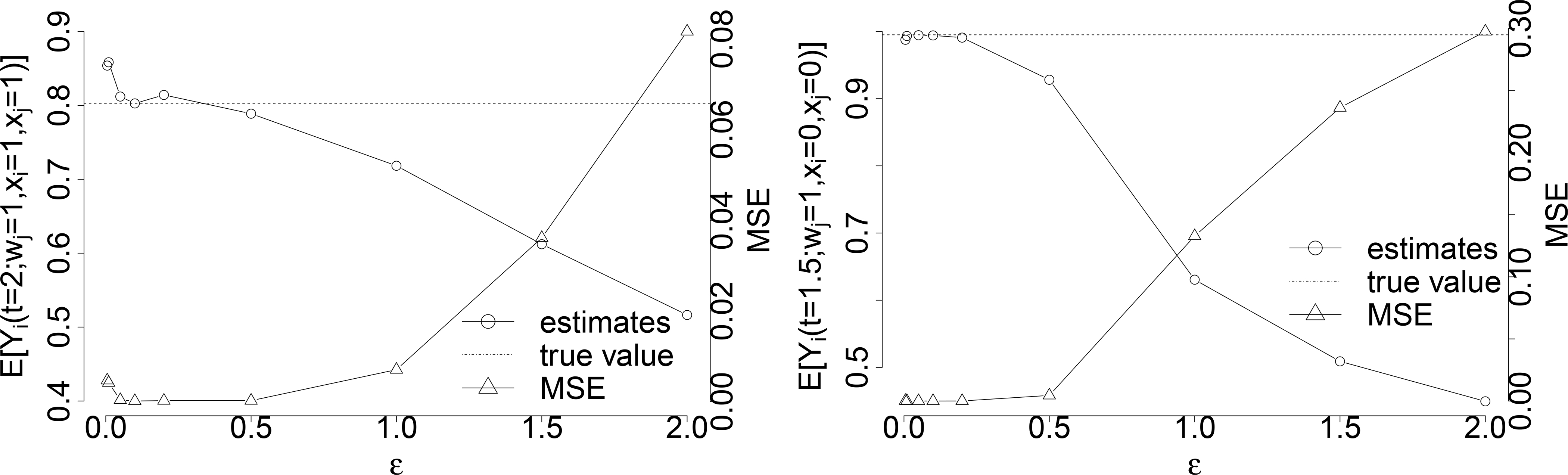
The choice of ε in the estimation of with sample size n = 100, 000 under the constant hazards α(t) = 0.2, γ(t) = 10 and coefficients and eσ = 0.5. Figure on the left shows the estimation of and its corresponding MSE under different choices of ϵ, and Figure on the right shows the estimation of and its corresponding MSE under difference choices of ϵ.
B. Statistical estimation
B.1. Statistical estimation for the controlled potential outcomes in Theorem 1
In Theorem 1, for t < wj, the estimation of is achieved by the estimation of Fi(wj|xi, li) by Lemma 1, which follows the standard technique of estimating distribution of time-to-event data in competing risks. For t ≥ wj, the estimation of is achieved by the estimation of Fi(wj|xi, li) by Lemma 1 and the estimation of . Let ϵ be a small positive number, then
Therefore, we estimate by averaging Yi(t) among observations when Tj falls into a narrow region around wj under X = x and L = l. With finite samples of observations, if ϵ is chosen too small, sample size for the estimation becomes smaller and variance gets bigger; if the ϵ is chosen too big, the selected observations no longer approximate Tj = wj well enough so that the estimation is more biased. The ϵ should be chosen to minimize the MSE of the estimation.
We choose ϵ = 0.1 in the estimations of controlled potential outcomes in Figure 3 when t ≥ wj with sample size N = 100, 000 (under the constant hazard scenario α(t) = 0.2 and γ(t) = 10 with beneficial susceptibility effect β1 = 0.3 and infectiousness effect β2 = 0.5), as it gives the smallest (or almost smallest) MSE for most observational times under different treatments and partner’s infection time. Figure 5 illustrates the estimations of and as well as their MSEs for the choice of ϵ among ϵ ∈ {0.005, 0.01, 0.05, 0.1, 0.2, 0.5, 1, 1.5, 2}, and ϵ = 0.1 gives the smallest MSE for the estimation.
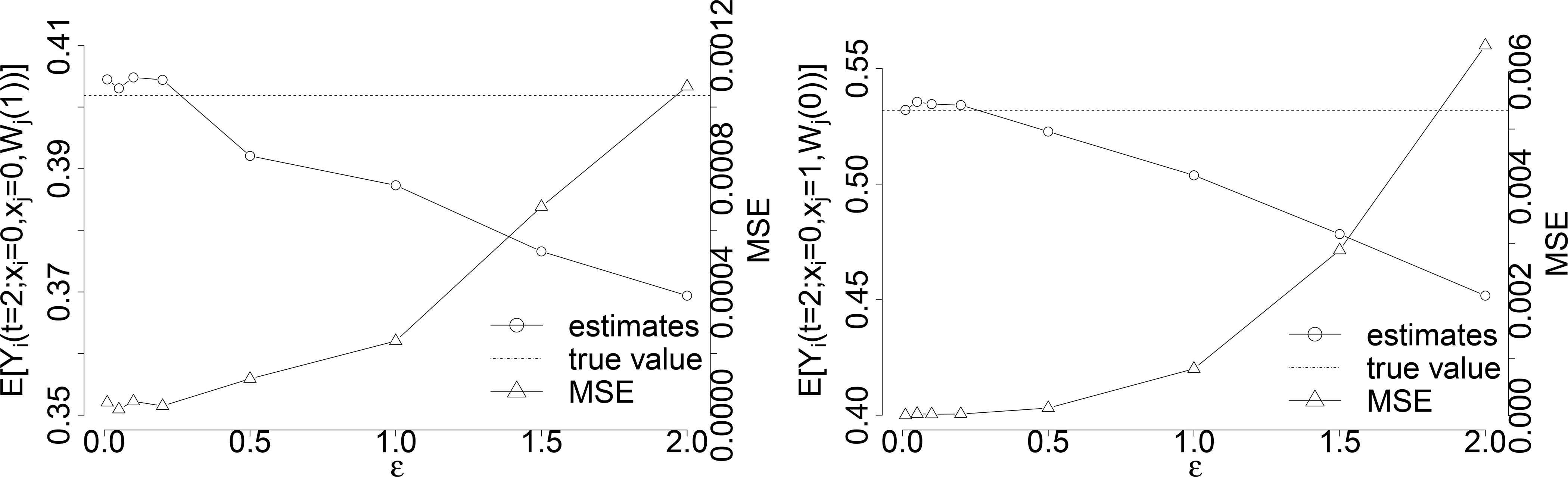
Figure 6:
The choice of ε in the estimation of with sample size n = 100, 000 under the constant hazards α(t) = 0.2, γ(t) = 10 and coefficients and eσ = 0.5. Figure on the left shows the estimation of and its corresponding MSE under different choices of ε, and Figure on the right shows the estimation of and its corresponding MSE under difference choices of ϵ.
B.2. Statistical estimation for the natural potential outcomes in Corollary 1
From Corollary 1, can be estimated by the average of Yi(t) when X = x.
For the identification of cross-world natural potential outcomes when , is estimated with the help of the estimation of by Lemma 1 and the estimation of in Theorem 1, which requires a proper choosing of ε again.
We illustrate examples of estimating cross-world natural potential outcomes of and with sample size N = 1, 000, 000 under the constant hazard scenario (α(t) = 0.2 and γ(t) = 10) with beneficial susceptibility effect (β1 = 0.3) and infectiousness effect (β2 = 0.5). We show their estimations as well as the MSEs under the choice among ϵ ∈ {0.005, 0.01, 0.05, 0.1, 0.2, 0.5, 1, 1.5, 2} in Figure 6, and ϵ = 0.1 gives the smallest MSE for the estimations.
B.3. Covariate adjustment for controlled and natural potential infection outcomes in Equations (4)–(5)
For the adjustment of covariates in Equations (4)–(5), the estimation is achieved by estimating (controlled or natural) potential outcomes by Theorem 1 and Corollary 1, and then integrate it over the estimated empirical distribution of the covariates.
We approximate the joint distribution of covariates G(l) empirically by dividing the space of L into small bins of size Δ × Δ. The probability of L in one bin centered around (ci, cj) is estimated by . The size of Δ should be chosen to minimize the MSE of the estimations. Within each bin centered, for example the one around (ci, cj), we estimate and by Theorem 1 and in Corollary 1, respectively. Finally, we integrate and over the estimated empirical distribution of G(l) by:
We illustrate the estimation of and with one covariate for each individual, so L = (Li, Lj), with sample size n = 1, 000, 000 under the constant hazards α(t) = 0.2, γ(t) = 10 and coefficients and eσ = 0.5. In our simulation, the covariates are generated by
so that the majority of them fall into (−4,4). Therefore, we separate the covariates space into bins from −4 to 4 by Δ as well as the 4 left regions at the corners. Specifically, the space of (Li, Lj) are separated into bins of , where , as well as (−∞, −4] × (−∞, −4], (−∞, −4] × (4, ∞), (4, ∞) × (−∞, −4], and (4, ∞) × (4, ∞) at the corners.
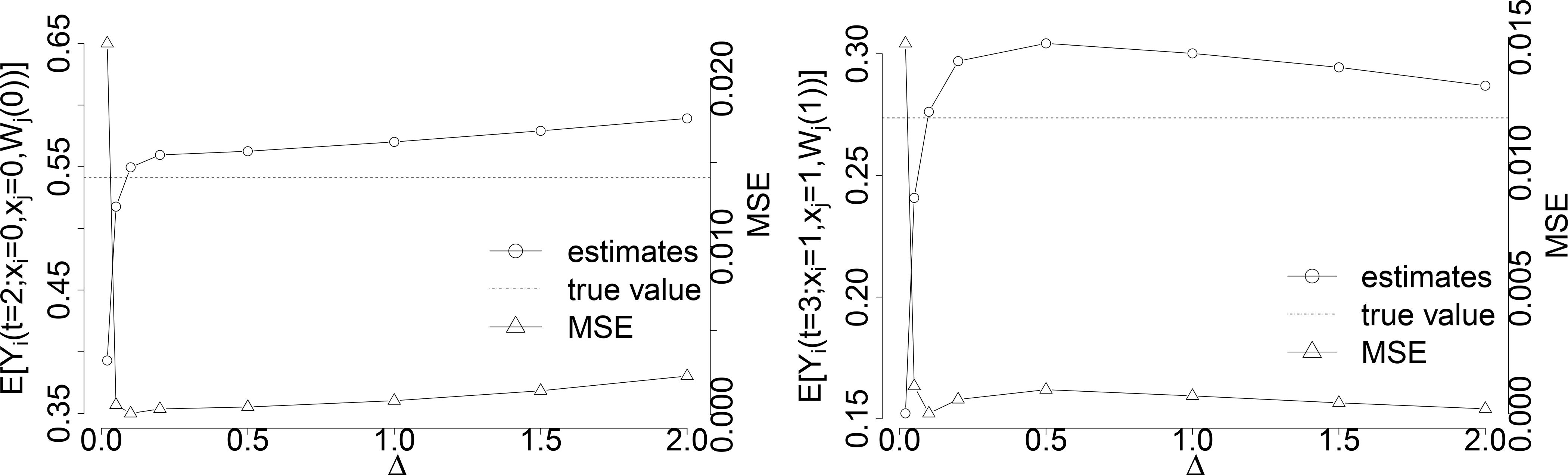
We show the estimations of and as well as MSE under the choice among Δ ∈ {0.005, 0.01, 0.05, 0.1, 0.2, 0.5, 1, 1.5, 2} in Figure 7, and Δ = 0.1 gives the smallest MSE for the estimations.
Figure 7:
The choice of Δ in the estimation of natural potential outcomes with sample size n = 1, 000, 000 under the constant hazards α(t) = 0.2, γ(t) = 10 and coefficients and eσ = 0.5. Figure on the left shows the estimation of and its corresponding MSE under different choices of Δ among Δ ∈ {0.02, 0.05, 0.1, 0.2, 0.5, 1, 1.5, 2}, and Figure on the right shows the estimation of and its corresponding MSE under difference choices of Δ among Δ ∈ {0.02, 0.05, 0.1, 0.2, 0.5, 1, 1.5, 2}.
References
- [1].Aalen OO, Stensrud MJ, Didelez V, Daniel R, Røysland K, and Strohmaier S. Time-dependent mediators in survival analysis: Modeling direct and indirect effects with the additive hazards model. Biometrical Journal, 62(3):532–549, 2020. [DOI] [PubMed] [Google Scholar]
- [2].Akritas MG. Nonparametric survival analysis. Statistical Science, 19(4):615–623, 2004. [Google Scholar]
- [3].Auranen K, Arjas E, Leino T, and Takala AK. Transmission of pneumococcal carriage in families: A latent Markov process model for binary longitudinal data. Journal of the American Statistical Association, 95(452):1044–1053, 2000. [Google Scholar]
- [4].Becker NG, Britton T, and O’Neill PD. Estimating vaccine effects on transmission of infection from household outbreak data. Biometrics, 59(3):467–475, 2003. [DOI] [PubMed] [Google Scholar]
- [5].Becker NG, Britton T, and O’Neill PD. Estimating vaccine effects from studies of outbreaks in household pairs. Statistics in Medicine, 25(6):1079–1093, 2006. [DOI] [PubMed] [Google Scholar]
- [6].Bhattacharya R, Malinsky D, and Shpitser I. Causal inference under interference and network uncertainty. Uncertainty in Artificial Intelligence, 2019, 2019. [PMC free article] [PubMed] [Google Scholar]
- [7].Cauchemez S, Carrat F, Viboud C, Valleron AJ, and Boëlle P. A Bayesian MCMC approach to study transmission of influenza: application to household longitudinal data. Statistics in Medicine, 23(22):3469–3487, 2004. [DOI] [PubMed] [Google Scholar]
- [8].Cauchemez S, Temime L, Guillemot D, Varon E, Valleron A-J, Thomas G, and Bo P-Y ëlle. Investigating heterogeneity in pneumococcal transmission: a Bayesian MCMC approach applied to a follow-up of schools. Journal of the American Statistical Association, 101 (475):946–958, 2006. [Google Scholar]
- [9].Cauchemez S, Donnelly CA, Reed C, Ghani AC, Fraser C, Kent CK, Finelli L, and Ferguson NM. Household transmission of 2009 pandemic influenza A (H1N1) virus in the United States. New England Journal of Medicine, 361(27):2619–2627, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Chiba Y. A note on bounds for the causal infectiousness effect in vaccine trials. Statistics & Probability Letters, 82(7):1422–1429, 2012. [Google Scholar]
- [11].Chiba Y. A simple method of measuring vaccine effects on infectiousness and contagion. Open Journal of Statistics, 3(4A):7–15, 2013. [Google Scholar]
- [12].Chiba Y and Taguri M. Conditional and unconditional infectiousness effects in vaccine trials. Epidemiology, 24(2):336–337, 2013. [DOI] [PubMed] [Google Scholar]
- [13].Cox D. Planning of Experiments. John Wiley & Sons, New York, 1958. [Google Scholar]
- [14].Didelez V. Defining causal mediation with a longitudinal mediator and a survival outcome. Lifetime Data Analysis, 25(4):593–610, 2019. [DOI] [PubMed] [Google Scholar]
- [15].Eck DJ, Morozova O, and Crawford FW. Randomization for the susceptibility effect of an infectious disease intervention in a clustered study population. arXiv preprint arXiv:1808.05593, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Fine PEM, Clarkson JA, and Miller E. The Efficacy of Pertussis Vaccines under Conditions of Household Exposure: Further Analysis of the 1978–80 PHLS/ERL Study in 21 Area Health Authorities in England. International Journal of Epidemiology, 17(3):635–642, September 1988. [DOI] [PubMed] [Google Scholar]
- [17].Francis T Jr. Evaluation of the 1954 poliomyelitis vaccine field trial: Further studies of results determining the effectiveness of poliomyelitis vaccine (Salk) in preventing paralytic poliomyelitis. Journal of the American Medical Association, 158(14):1266–1270, 1955. [DOI] [PubMed] [Google Scholar]
- [18].Golm GT, Elizabeth Halloran M, and Longini IM. Semiparametric methods for multiple exposure mismeasurement and a bivariate outcome in HIV vaccine trials. Biometrics, 55(1):94–101, 1999. [DOI] [PubMed] [Google Scholar]
- [19].Greenwood M and Yule GU. The statistics of anti-typhoid and anti-cholera inoculations, and the interpretation of such statistics in general. Proceedings of the Royal Society of Medicine, 8(Sect Epidemiol State Med):113, 1915. [PMC free article] [PubMed] [Google Scholar]
- [20].Halloran ME and Hudgens MG. Causal inference for vaccine effects on infectiousness. The International Journal of Biostatistics, 8(2), 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Halloran ME and Hudgens MG. Dependent happenings: a recent methodological review. Current Epidemiology Reports, 3:297–305, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Halloran ME and Struchiner CJ. Study designs for dependent happenings. Epidemiology, 2(5):331–338, 1991. [DOI] [PubMed] [Google Scholar]
- [23].Halloran ME and Struchiner CJ. Causal inference in infectious diseases. Epidemiology, 6 (2):142–151, 1995. [DOI] [PubMed] [Google Scholar]
- [24].Halloran ME, Haber M, Longini IM, and Struchiner CJ. Direct and indirect effects in vaccine efficacy and effectiveness. American Journal of Epidemiology, 133(4):323–331, 1991. [DOI] [PubMed] [Google Scholar]
- [25].Halloran ME, Longini IM, Haber MJ, Struchiner CJ, and Brunet RC. Exposure efficacy and change in contact rates in evaluating prophylactic HIV vaccines in the field. Statistics in Medicine, 13(4):357–377, 1994. [DOI] [PubMed] [Google Scholar]
- [26].Halloran ME, Struchiner CJ, and Longini IM. Study designs for evaluating different efficacy and effectiveness aspects of vaccines. American Journal of Epidemiology, 146(10):789–803, 1997. [DOI] [PubMed] [Google Scholar]
- [27].Halloran ME, Longini IM, and Struchiner CJ. Design and interpretation of vaccine field studies. Epidemiological Reviews, 21(1):73–88, 1999. [DOI] [PubMed] [Google Scholar]
- [28].Halloran ME, Préziosi MP, and Chu H. Estimating vaccine efficacy from secondary attack rates. Journal of the American Statistical Association, 98(461):38–46, 2003. [Google Scholar]
- [29].Halloran ME, Longini IM, and Struchiner CJ. Design and Analysis of Vaccine Studies. Springer, 2010. [Google Scholar]
- [30].Hudgens MG and Halloran ME. Causal vaccine effects on binary postinfection outcomes. Journal of the American Statistical Association, 101(473):51–64, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Hudgens MG and Halloran ME. Toward causal inference with interference. Journal of the American Statistical Association, 103(482):832–842, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Kenah E. Contact intervals, survival analysis of epidemic data, and estimation of R0. Biostatistics, 12(3):548–566, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Kenah E. Non-parametric survival analysis of infectious disease data. Journal of the Royal Statistical Society: Series B (Statistical Methodology), 75(2):277–303, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kenah E. Semiparametric relative-risk regression for infectious disease transmission data. Journal of the American Statistical Association, 110(509):313–325, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Kenah E, Lipsitch M, and Robins JM. Generation interval contraction and epidemic data analysis. Mathematical Biosciences, 213(1):71–79, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Lin S-H, Young JG, Logan R, and VanderWeele TJ. Mediation analysis for a survival outcome with time-varying exposures, mediators, and confounders. Statistics in Medicine, 36 (26):4153–4166, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Longini IM, Koopman JS, Monto AS, and Fox JP. Estimating household and community transmission parameters for influenza. American Journal of Epidemiology, 115(5):736–751, 1982. [DOI] [PubMed] [Google Scholar]
- [38].Longini IM, Koopman JS, Haber M, and Cotsonis GA. Statistical inference for infectious diseases: risk-specific household and community transmission parameters. American Journal of Epidemiology, 128(4):845–859, 1988. [DOI] [PubMed] [Google Scholar]
- [39].McSherry GD, Shapiro DE, Coombs RW, McGrath N, Frenkel LM, Britto P, Culnane M, and Sperling RS. The effects of zidovudine in the subset of infants infected with human immunodeficiency virus type-1 (Pediatric AIDS Clinical Trials Group Protocol 076). The Journal of Pediatrics, 134(6):717–724, 1999. [DOI] [PubMed] [Google Scholar]
- [40].Morozova O, Cohen T, and Crawford FW. Risk ratios for contagious outcomes. Journal of The Royal Society Interface, 15:1–12, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Ogburn EL. Challenges to estimating contagion effects from observational data. In Complex Spreading Phenomena in Social Systems, pages 47–64. Springer, 2018. [Google Scholar]
- [42].Ogburn EL and VanderWeele TJ. Causal diagrams for interference. Statistical science, 29 (4):559–578, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Ogburn EL and VanderWeele TJ. Vaccines, contagion, and social networks. The Annals of Applied Statistics, 11(2):919–948, 2017. [Google Scholar]
- [44].Ogburn EL, Shpitser I, and Lee Y. Causal inference, social networks, and chain graphs. arXiv preprint arXiv:1812.04990, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].O’Hagan JJ, Lipsitch M, and Hernán MA. Estimating the per-exposure effect of infectious disease interventions. Epidemiology, 25(1):134, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].O’Neill PD, Balding DJ, Becker NG, Eerola M, and Mollison D. Analyses of infectious disease data from household outbreaks by Markov chain Monte Carlo methods. Journal of the Royal Statistical Society: Series C (Applied Statistics), 49(4):517–542, 2000. [Google Scholar]
- [47].Orenstein WA, Bernier RH, and Hinman AR. Assessing vaccine efficacy in the field: Further observations. Epidemiologic Reviews, 10(1):212–241, March 1988. [DOI] [PubMed] [Google Scholar]
- [48].Pearl J. Causality: models, reasoning and inference. Cambridge University Press, 2000. [Google Scholar]
- [49].Pearl J. Direct and indirect effects. In Proceedings of the 17th Conference in Uncertainty in Artificial Intelligence, pages 411–420, San Francisco, CA, USA, 2001. Morgan Kaufmann Publishers Inc. [Google Scholar]
- [50].Rampey AH Jr, Longini IM, Haber M, and Monto AS. A discrete-time model for the statistical analysis of infectious disease incidence data. Biometrics, 48(1):117–128, 1992. [PubMed] [Google Scholar]
- [51].Rhodes PH, Halloran ME, and Longini IM. Counting process models for infectious disease data: Distinguishing exposure to infection from susceptibility. Journal of the Royal Statistical Society: Series B (Methodological), 58(4):751–762, 1996. [Google Scholar]
- [52].Robins JM and Greenland S. Identifiability and exchangeability for direct and indirect effects. Epidemiology, 3(2):143–155, 1992. [DOI] [PubMed] [Google Scholar]
- [53].Rosenbaum PR. Interference between units in randomized experiments. Journal of the American Statistical Association, 102(477):191–200, 2007. [Google Scholar]
- [54].Rubin DB. Causal inference using potential outcomes: Design, modeling, decisions. Journal of the American Statistical Association, 100(469):322–331, 2005. [Google Scholar]
- [55].Sherman E and Shpitser I. Identification and estimation of causal effects from dependent data. In Advances in Neural Information Processing Systems 31, pages 9424–9435. Curran Associates, Inc., 2018. [PMC free article] [PubMed] [Google Scholar]
- [56].Shpitser I. Counterfactual graphical models for longitudinal mediation analysis with unobserved confounding. Cognitive Science, 37(6):1011–1035, 2013. [DOI] [PubMed] [Google Scholar]
- [57].Shpitser I, Tchetgen Tchetgen E, and Andrews R. Modeling interference via symmetric treatment decomposition. arXiv preprint arXiv:1709.01050, 2017. [Google Scholar]
- [58].Sjölander A, Frisell T, Kuja-Halkola R, Öberg S, and Zetterqvist J. Carryover effects in sibling comparison designs. Epidemiology, 27(6):852–858, 2016. [DOI] [PubMed] [Google Scholar]
- [59].Struchiner C and Halloran M. Randomization and baseline transmission in vaccine field trials. Epidemiology & Infection, 135(2):181–194, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Tchetgen Tchetgen EJ and VanderWeele TJ. On causal inference in the presence of interference. Statistical Methods in Medical Research, 21(1):55–75, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Tsang TK, Cowling BJ, Fang VJ, Chan K-H, Ip DKM, Leung GM, Peiris JSM, and Cauchemez S. Influenza A virus shedding and infectivity in households. The Journal of Infectious Diseases, 212(9):1420–1428, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Tsang TK, Fang VJ, Chan K-H, Ip DKM, Leung GM, Peiris JSM, Cowling BJ, and Cauchemez S. Individual correlates of infectivity of influenza A virus infections in households. PLOS ONE, 11(5):1–11, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].VanderWeele TJ. Direct and indirect effects for neighborhood-based clustered and longitudinal data. Sociological Methods & Research, 38(4):515–544, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].VanderWeele TJ and Tchetgen Tchetgen EJ. Bounding the infectiousness effect in vaccine trials. Epidemiology, 22(5):686–693, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].VanderWeele TJ and Tchetgen Tchetgen EJ. Mediation analysis with time varying exposures and mediators. Journal of the Royal Statistical Society: Series B (Statistical Methodology), 79(3):917–938, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].VanderWeele TJ, Tchetgen Tchetgen EJ, and Halloran ME. Components of the indirect effect in vaccine trials: identification of contagion and infectiousness effects. Epidemiology, 23 (5):751–761, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Vansteelandt S, Linder M, Vandenberghe S, Steen J, and Madsen J. Mediation analysis of time-to-event endpoints accounting for repeatedly measured mediators subject to time-varying confounding. Statistics in medicine, 38(24):4828–4840, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Yang Y, Longini IM, and Halloran ME. Design and evaluation of prophylactic interventions using infectious disease incidence data from close contact groups. Journal of the Royal Statistical Society: Series C (Applied Statistics), 55(3):317–330, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Zheng W and van der Laan M. Longitudinal mediation analysis with time-varying mediators and exposures, with application to survival outcomes. Journal of Causal Inference, 5(2):20160006, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]