

Abstract
Objective
Cellular prion protein (PrPC), the primary form of prion diseases pathogen, has received increasing attention for its protective effect against ischaemic stroke. Little is known about its role in peripheral immune responses after cerebral ischaemia/reperfusion (I/R) injury. This study is to detect the variation of splenic CD4+ T lymphocytes differentiation and the concentration of inflammatory cytokines after murine cerebral I/R injury in the context of PRNP expression as well as its influence on the ischaemic neuronal apoptosis.
Methods
We established the cerebral ischaemic murine model of different PRNP genotypes. We detected the percentage of splenic CD4+PrPC+ T cells of PRNP wild‐type mice and the ratio of splenic Th1/2/17 lymphocytes of mice of different PRNP expression. The relevant inflammatory cytokines were then measured. Oxygen glucose deprivation/reperfusion (OGD/R) HT22 mouse hippocampal neurons were co‐cultured with the T‐cell‐conditioned medium harvested from the spleen of modelled mice and then the neuronal apoptosis was detected.
Results
CD4+ PrPC+ T lymphocytes in wild‐type mice elevated after MCAO/R. PRNP expression deficiency led to an elevation of Th1/17 phenotypes and the promotion of pro‐inflammatory cytokines, while PRNP overexpression led to the elevation of Th2 phenotype and upregulation of anti‐inflammatory cytokines. In addition, PrPC‐overexpressed CD4+T cells weakened the apoptosis of OGD/R HT‐22 murine hippocampal neurons caused by MCAO/R CD4+ T‐cell‐conditioned medium, while PrPC deficiency enhanced apoptosis.
Interpretation
PrPC works as a neuron protector in the CNS when I/R injury occurs and affects the peripheral immune responses and defends against stroke‐induced neuronal apoptosis.
Introduction
Ischaemic stroke is one of the major causes of high disability and mortality rates worldwide. The key to its treatment is restoring blood flow to the offending vessels and salvaging ischaemic penumbra in order to gradually recover mental functions. However, after reperfusion, a series of post‐stroke inflammatory responses that may not be the most important pathological responses are triggered, relating to both the central nervous system (CNS) and peripheral immune system. These responses lead to further neuronal impairment and cell death. 1 , 2 , 3
During the acute phase of ischaemic stroke, microglial activation and damage to the blood–brain barrier (BBB) exaggerate the release of inflammatory cytokines, chemokine/chemokine receptors, or CNS‐relevant antigens, leading to chemotaxis and infiltration of peripheral immunocytes. 4 The spleen is considered to play an important role in post‐stroke peripheral immunoreaction. 5 The activated splenocytes are attracted and migrate to the infarction area through the damaged BBB, thereby triggering intracerebral immune reactions. 6
T cells are regarded as injurious in the early phase of post‐stroke injury; they promote platelet and leucocyte adhesion to the vascular endothelium and initiate thrombotic inflammation, which promotes pro‐inflammatory responses. 7 Severe combined immune deficiency (SCID) mice with T‐cell defects (Rag−/−, etc.) have been shown to have smaller infarction volume and milder neurological impairment in the acute stage of cerebral ischaemic stroke (CIS) than wild‐type (WT) mice. 8 However, recent studies have shown that T‐cell phenotypes may have dual effects on stroke. Activated CD4+ T cells can differentiate into pro‐inflammatory phenotypes such as Th1 and Th17 and anti‐inflammatory phenotypes such as Th2, thereby generating pro‐ and anti‐inflammatory cytokines and polarising macrophages into different phenotypes to harm or protect the brain. 9 , 10
Cellular prion protein (PrPC) is well‐known as the primary structure of scrapie prion protein (PrPSc), the pathogen of transmissible spongiform encephalopathies (TSEs) and is a highly conserved membrane‐bound glycoprotein anchored by glycosylphosphatidylinositol (GPI). PrPC has also been found to be neuroprotective in post‐stroke injury. Nicholas et al. detected a lesion‐related increase in PrPC expression in the plasma of stroke patients and the periinfarct area of middle cerebral artery occlusion (MCAO)‐operated mice. 11 , 12 In addition, PrPC exempts neurons from Bax‐mediated‐cell apoptosis and reduces the infarct volume in mice. 13
Many studies have shown that PrPC expression varies in different immunocytes and phases in both the innate and adaptive immune systems, participating in intercellular communication, phagocytosis, differentiation and immunity homeostasis. 14 More PrPC mRNA was detected in T cells activated by mitogens (such as ConA and PHA), while T‐cell mitosis decreased along with PrPC expression inhibition. PrPC balances pro‐ and anti‐inflammatory cytokines during the acute and chronic stages of bacterial infection. 15 In studies on experimental autoimmune encephalomyelitis (EAE), the cerebral inflammatory responses have been found to occur earlier and be more severe and Th1/17 phenotypes were enhanced at the same time when PRNP was knocked out. 16 , 17
In summary, PrPC is known to play an important role in various inflammatory reactions. Therefore, we hypothesise that PrPC may be a regulator of peripheral immune responses during the acute phase of cerebral I/R injury. We explored the alteration of PrPC expression in splenic CD4+ T helper (Th) cells of transient middle cerebral artery occlusion/reperfusion (tMCAO/R) mice. We also detected the differentiation of Th1/2/17 lymphocytes and changes in peripheral blood and splenic supernatant inflammatory cytokines after the first 24 h of cerebral I/R injury. Furthermore, we investigated the effect of PrPC‐regulated Th cells on the survival of neuronal death in vitro. The aforementioned methods may help us explore new therapeutic targets for ischaemic stroke.
Materials and methods
Ethic statement
The experimental protocols were approved by the Experimental Animal Research Ethics Committee of Jilin University. All efforts were made to minimise suffering and to reduce the number of animals used.
Animals and surgery
Male FVB/N WT mice (Vital River Laboratory Animal Technology Co. Ltd., Beijing, China) with PRNP knockout (KO) and PRNP‐overexpressed 6‐ to 8‐week‐old mice (Tga20, Chinese Center for Disease Control and Prevention) weighing 20–25 g were used in this study. A total of 72 mice were used in this study.
tMCAO/R was performed in WT mice to detect PrPC expression in splenic CD4+ T lymphocytes after cerebral I/R (n = 8). Mice were anaesthetised with isoflurane (RWD Life Science, Shenzhen, China, 3% induced concentration, and 1.5% sustained concentration). Following midline skin incision of the neck, the left common carotid artery (CCA), internal carotid artery (ICA) and external carotid artery (ECA) were exposed. The ECA was transiently clipped, and the proximal part of the CCA was ligated. A silicon‐coated monofilament (diameter 0.21 mm, Beijing Cinotech Co. Ltd, Beijing, China) was then inserted into the ICA from the lumen of the distal CCA. During ligation and insertion, the vagal nerve was carefully preserved. The appropriate placement of the filament was monitored by measuring the regional cerebral blood flow (CBF) using laser Doppler flowmetry (PeriFlux 5000, Perimed AB, Järfälla, Sweden) both ipsilaterally and contralaterally. The baseline CBF before the insertion of the filament into the ICA was monitored, as well as the blood flow during the 90 min occlusion until the filament was removed. Animals that showed a CBF drop of over 50% after the filament insertion and recovered to almost normal levels were included in the study. Sham‐operated animals (n = 6) were treated with the same anaesthesia and surgical procedures, except that the MCA occlusion and control animals (n = 6) were handled without any processing. During the operation, body temperature was maintained at 37.0 ± 0.5°C. After tMCAO/R, animals were allowed to recover for 24 h for flow cytometry. To clarify the ratio of splenic CD4+ T cell Th1/2/17 phenotypes and the expression of inflammatory cytokines in different genotypes of supernatant and peripheral blood, tMCAO/R was performed with WT, KO and Tga20 mice following the same procedure as described above. The numbers of animals in the experimental groups were as follows: control (n = 26), sham (n = 26), WT tMCAO/R (n = 30), KO tMCAO/R (n = 35) and Tga20 tMCAO/R (n = 29).
Behavioural evaluation
The tMCAO/R mice were evaluated by Longa Score, a five‐point scale: a score of 0 indicated no neurologic deficit, a score of 1 (failure to extend left forepaw fully) a mild focal neurologic deficit, a score of 2 (circling to the left) a moderate focal neurologic deficit, and a score of 3 (falling to the left) a severe focal deficit; rats with a score of 4 did not walk spontaneously and had a depressed level of consciousness. 18
Collection of mice orbital venous serum
Mice were deeply anaesthetised and 0.5–1 mL peripheral blood was collected from the orbital vein in a non‐heparinised tube. The blood collected 24 h post‐tMCAO/R was preserved in a 4°C refrigerator for 2 h and centrifuged at 3000 rpm. The supernatant was then collected. The numbers of animals for each genotype were as follows: control (n = 6), sham (n = 6), tMCAO/R (n = 6).
TTC staining
Mice were subjected to transcardial perfusion with 0.9% NaCl after decapitation. Cerebrums were immediately removed and sliced into five consecutive coronal sections, which were then stained in 2% 2,3,5‐triphenyltetrazolium chloride (TTC, Solarbio, Beijing, China) solution (30 min, 20°C). The slices were arranged in the order of frontal to occipital and photographed using a digital camera. The unstained areas indicate the infarction, and they were measured using the morphometric software ImageJ (NIH image, version 1.53c, NIH, Bethesda, MD, USA). Total infarct volumes were calculated by adding the mean area of each section multiplied by the thickness of the sections. (WT control n = 6, sham n = 6, tMCAO/R n = 6).
Immunofluorescence analysis
Brains of different genotypes after 24h of tMCAO/R were dissected and fixed with 4% PFA before paraffin embedding (WT tMCAO/R (n = 20), KO tMCAO/R (n = 20), and Tga20 tMCAO/R (n = 20)). Neuron survival was evaluated by immunofluorescence in 4‐μm‐thick paraffin section. Three sections per animal were used and incubated with monoclonal mouse anti‐NeuN antibodies (1:100; Cat # 24307 S, Cell Signaling Technology, Danvers, MA) and then with appropriate secondary antibodies (Goat anti‐Mouse IgG (H&L)‐ Alexa Fluor 488, Cat #CSA3208, Cohesionbio, China). All sections were mounted with DAPI mounting medium (Cat #S2110, Solarbio, Beijing, China). Cell density in four regions of interest (ROI) in the ischaemic striatum was assessed under a fluorescence microscope as described before 19 and counted using the morphometric software ImageJ (NIH image, version 1.53c, NIH, Bethesda, MD, USA).
Isolation of mice lymphocytes from the spleen
Mouse spleens were harvested in tissue culture dishes containing 20 mL cold RPMI1640. To isolated the lymphocytes, spleens were chopped into small pieces, homogenised by the plunger tip of a 5 mL syringe, filtered through a 40 μm strainer and centrifuged at 4°C and 1500 rpm for 5 min. After erythrocytes were lysed using ACK lysis buffer, the cells were washed twice, resuspended in 5 mL RPMI1640 medium, and adjusted the cell concentration to 1 × 107/ mL.
FACS analysis for immune cell lymphocyte population
For the WT tMCAO/R mice, the lymphocytes were labelled with monoclonal antibodies (mAbs) for membrane expression of CD4 (APC‐H7 Rat anti‐mouse CD4, BD Pharmingen™, San Diego, CA, USA, Cat. #560181) and PrPC (CD230 (PrP) Recombinant Rabbit Monoclonal Antibody, Thermo Fisher Scientific, Waltham, MA, USA, Cat. #MA5‐32202) (control n = 6, sham n = 6, tMCAO/R n = 6). Before labelling, the lymphocytes were incubated in cold 10% goat serum for 30 min to seal off the non‐specific adsorption sites on the membrane. Cells were washed once with 2 mL cold FACS buffer (PBS with 1% FBS) and labelled with the primary antibodies. The labelled cells were then washed for a second time in 2 mL cold FACS buffer and incubated in cold 5 μg/mL secondary antibody (Goat anti‐Rabbit IgG (H + L) Highly Cross‐Adsorbed Secondary Antibody, Alexa Fluor 488, Thermo Fisher Scientific, Waltham, MA, USA, Cat. #A‐11034) for 30 min. The cells were then washed with 2 mL FACS buffer.
Cytokines of three different phenotypes were intracellularly stained to determine the percentage of Th1/2/17 cell populations. The harvested lymphocyte solution was diluted 1:1 with RPMI‐1640 culture medium and stimulated with 25 ng/mL Phorbol 12‐Myristate 13‐Acetate (PMA) and 1 μM Ionomycin in the presence of protein transport inhibitor (containing monensin, BD GolgiStop™, San Diego, CA, USA, Cat. #560758) in a total volume of 1 mL. Unstimulated samples were prepared in parallel with the addition of a protein transport inhibitor. After 5 h of culture, the sample was fixed and permeabilised with cytofix and cytoperm buffer and incubated with the mixed mAbs against membrane CD4 and relevant cytokines: mouse anti‐CD4‐PerCP‐Cy5.5 (clone: RM4‐5), anti‐IL‐17A‐PE (clone: TC11‐18H10.1), anti‐IFN‐GMA‐FITC (clone: XMG1.2) and anti‐IL‐4‐APC (clone: 11B11) (BD Pharmingen™, San Diego, CA, USA, Cat. #560758). Cells were then analysed and counted using a FACS Calibur. Experiments were carried out in biologically triplicate. Data were analysed using FlowJo software.
Preparation of conditioned medium
The harvested lymphocyte solution was diluted 1:1 with RPMI‐1640 culture medium and stimulated with 25 ng/mL PMA and 1 µM ionomycin without protein transport inhibitor in a total volume of 1 mL. Then, the sample was cultured for 5 h, and the supernatant was preserved as the T‐cell‐conditioned medium (TCCM). The sample was used for cytokine evaluation by cytometric bead array (CBA) and the following cell apoptosis research.
CBA
Cytokines in PMA/iono stimulated lymphocyte supernatants harvested from the lymphocytes of MCAO/R mice of different genotypes without protein transport inhibitors as well as serum were measured using a CBA mouse Th1/Th2/Th17 Cytokine Kit (BD Biosciences, San Jose, CA, USA, Cat. #560485), and IL‐2, IL‐4, IL‐6, IL‐10, IL‐17A and IFN‐γ were selected as the representative cytokines for Th1, Th2 and Th17 lymphocytes. Experiments were carried out in biologically triplicate.
Apoptosis
HT‐22 murine hippocampal neuronal cells were resuscitated from cryopreservation. HT‐22 neurons were maintained in DMEM, which was supplemented with 10% FBS, 2 mM glutamine and penicillin/streptomycin. Neurons were kept at 37°C and 5% CO2 and passed twice a week with 0.125% trypsin. Cell apoptosis was induced by OGD for 10 h, and cells were reperfused with TCCM of each genotype for 24 h. HT‐22 neurons were subjected to oxygen glucose deprivation (OGD) for 10 h, reperfused with TCCM, and divided into four groups: the vehicle group (n = 6), oxygen glucose deprivation /reperfusion (OGD/R) + control TCCM group (n = 6), OGD/R+ sham TCCM group (n = 6) and OGD/R+ MCAO/R TCCM group (n = 6). These groups were then cultured for 24 h. Neuronal apoptosis was detected using the FITC Annexin V Apoptosis Detection Kit (BD Biosciences, San Jose, CA, USA, Cat. #556570) according to the manufacturer’s instructions and analyzed by flow cytometry. Experiments were carried out in biologically triplicate.
Statistical analysis
Statistical analysis was performed using SPSS version 24.0 software. Analysis of variance (ANOVA), and post hoc Duncan’s test and Dunnett’s test were used to assess differences among multiple groups. P < 0.05 was considered statistically significant. Data are displayed as mean ± standard deviation (SD). The relationship between Th1/2/17 population and survival neurons was evaluated using multiple linear regression (MLR) and the variables were considered significant at P < 0.05 (WT tMCAO/R n = 20, KO tMCAO/R n = 20, Tga20 tMCAO/R n = 20).
Results
The discrepancy of infarction severity in MCAO/R mice of different PRNP genotypes
We conducted MCAO/R injury in WT, KO and Tga20 genotypes. A total amount of 15 mice were excluded due to death before the expected time of euthanasia. Mortality was as follows: 15.8% (6/38) in WT MCAO/R group, 25.7% (9/35) in KO MCAO/R group and 10.3% (3/29) in Tga20 MCAO/R group. There were no mortalities among the control and sham group. The Longa score and TTC staining were applied to measure the ischaemic severity of each group (Fig. 1). There was no significant difference in the Longa score and infarction volume between the control group and sham group for each genotype (data not shown). We then measured the infarction severity of WT mice after MCAO/R injury and found that the neurobehavioural scores of the MCAO/R group significantly increased compared with the sham group (2.5 ± 0.3416 vs. 0, P < 0.001). The infarction area was prominently detected by TTC staining (0.4755 ± 0.0130), which suggested that the MCAO/R models were successfully established. Compared with WT MCAO/R mice, the Longa score of KO MCAO/R mice was significantly higher (3.5 ± 0.2236 vs. 2.5 ± 0.3416, P < 0.05), while the score of Tga20 MCAO/R mice was significantly lower (1.167 ± 0.3073, P < 0.01). We discovered a similar trend for the infarction volume of each group. The KO MCAO/R group exhibited a larger ischaemic lesion than the WT MCAO/R group (0.5629 ± 0.0296 vs. 0.4755 ± 0.0130, P < 0.05), while the Tga20 MCAO/R group relieved the impairment (0.3467 ± 0.0227 vs. 0.4755 ± 0.0130, P < 0.01) (Fig. 1A and B).
Figure 1.
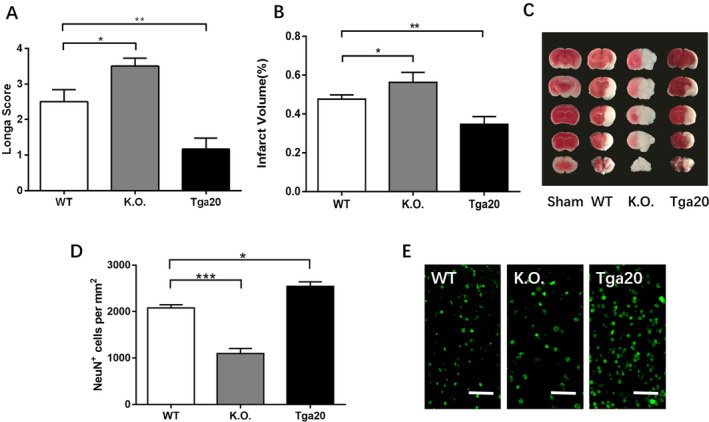
The comparison of lesion severity of experimental animals (neurological score, infarction volume and NeuN+ cell density) of different genotypes after 90 min of ischaemia and 24h of recovering. The percentage of infarction volume was calculated by TTC staining (WT control n = 6, sham n = 6, tMCAO/R n = 6). (A). The Longa score of each genotype after MCAO/R injury. (B). The infarction volume of each genotype. (C) The example of the TTC staining of each group. (D) The neuronal density of each genotype determined by NeuN immunohistochemistry in the striatum. (E) The representative photographs of neuronal density of each genotype. Scale bars: 50 μm. Analysis of variance (ANOVA), and post hoc Duncan’s test was used to assess differences among multiple groups and data are displayed as mean ± standard deviation (SD), *P < 0.05, **P < 0.01, ***P < 0.001.
We also analysed the severity of ischaemia brain injury by measuring the cell density of neurons after MCAO/R and compared the amount of NeuN+ cells of mice of different genotypes and found similar results. KO MCAO/R group showed significantly reduced survived neurons in ROIs than WT MCAO/R group (P < 0.05), while Tga20 MCAO/R group showed less severe brain injury with larger neuronal density than WT MCAO/R group (P < 0.001) (Fig. 1D and E).
Alternations of WT mice CD4+ and CD4+PrPC+ T splenocyte expression after MCAO/R injury
We extracted splenic T cells from WT mice and measured the CD4+ and CD4+PrPC+ T cells separately by flow cytometry. Neither CD4+ nor CD4+PrPC+ T splenocyte levels showed significant differences between the control and sham groups (28.87 ± 0.9191 vs. 27.88 ± 1.315, 4.003 ± 0.1014 vs. 3.778 ± 0.1638, both P > 0.05). After MCAO/R, the number of CD4+ T cells was significantly lower than in the sham group (18.9 ± 0.3755 vs. 27.88 ± 1.305, P < 0.001), while the number of CD4+PrPC+ T cells was significantly higher (8.503 ± 0.4481 vs. 3.778 ± 0.1638, P < 0.001) (Fig. 2B and D).
Figure 2.
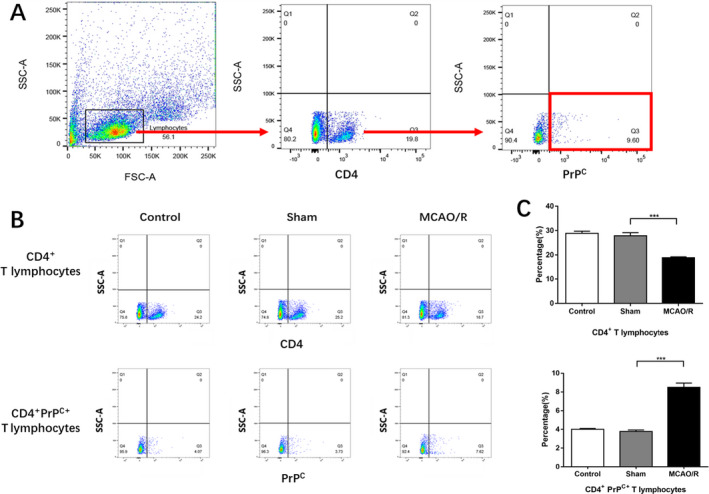
The post‐stroke expression of CD4+ and CD4+PrPC+ T cells in WT group (control n = 6, sham n = 6, tMCAO/R n = 6). (A) An example showing the gating strategy used to identify CD4+ T cells and CD4+PrPC+ T cells after MCAO/R injury of each genotype in the presence of the anti‐ CD4 anti‐ CD230 (PrP) mAbs. The first gate was set on physical parameters, then on CD4+ events and then on CD230+ events. (B) The example of the analysis of the CD4+ and CD4+PrPC+ T cell percentage through flow cytometry. The Q3 quadrant represented the CD4+ T cells (upper line) and the CD4+PrPC+ T cells (lower line). (C) The proportion of CD4+ T cells and CD4+PrPC+ T cells Experiments were carried out in biologically triplicate. Analysis of variance (ANOVA), and post hoc Duncan’s test was used to assess differences among multiple groups and data are displayed as mean ± standard deviation (SD), *** P < 0.001.
The expression of CD4+ T splenocyte phenotypes in MCAO/R mice of different PRNP genotypes
Since PrPC expression in CD4+ T cells was verified to increase after MCAO/R injury, we explored whether and how it would influence the differentiation of CD4+ T cells. We extracted the T splenic lymphocytes from the mice of each genotype and used flow cytometry to measure the expression of IFN‐γ, IL‐4 and IL‐17, which are markers of Th1, Th2 and Th17 phenotypes respectively. No significant differences in cytokine production were detected between the control and sham groups in each phenotype, while the cytokine production in the MCAO/R group was higher than that in the sham group (P < 0.05). Moreover, we found that the KO MCAO/R group exhibited an elevated percentage of Th1 and Th17 lymphocytes (P < 0.05) and a downward trend in the proportion of Th2 lymphocytes, although this change was not statistically significant in comparison with the WT MCAO/R group. In contrast, we detected an ascending percentage of Th2 lymphocytes (P < 0.05) and a descending percentage of Th1 lymphocytes (P < 0.01) in the Tga20 group. In addition, the expression of Th17 cells exhibited a decreasing trend, but this was without significant variation (Fig. 3).
Figure 3.
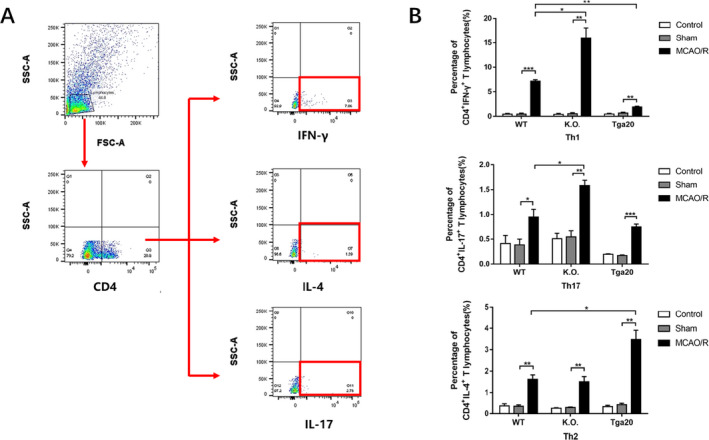
The percentage of splenic Th1/2/17 phenotypes of each genotype. (WT n = 6, KO n = 6, Tga20 n = 6) after stimulated by PMA/iono and protein transport inhibitor for 5h. (A) An example showing the gating strategy used to identify Th1/2/17 phenotypes respectively. The first gate was set on physical parameters and then on CD4+ events. Then IFN‐γ+, IL‐4+ and IL‐17+ events were separately detected and showed in the Q3 quadrant. (B) The ratio of Th1/2/17 lymphocytes respectively. Experiments were carried out in biologically triplicate. Analysis of variance (ANOVA), and post hoc Duncan’s test and Dunnett’s test were used to assess differences among multiple groups and data are displayed as mean ± standard deviation (SD), *P < 0.05, **P < 0.01, ***P < 0.001.
Correlation between cytometry of Th1/2/17 splenocytes and survived neurons in MCAO/R mice of different genotypes
In order to verify whether the variation of Th1/2/17 population affected the survival of neurons after MCAO/R injury, we analysed the two factors through MLR and referred the percentage of each phenotype and the number of NeuN+ cells per mm2 as the independent and dependent variables respectively. The relative parameters are listed in Table 1. The percentage of Th1 and Th17 lymphocytes showed negative correlation and the percentage of Th2 lymphocytes showed a positive correlation with the number of NeuN+ cells per mm2. The R2 was 0.900 and the adjusted R2 was 0.894. The model was NeuN+ cells (/mm2) = 2736.57‐0.596×Th1 (%) + 0.150 × Th2 (%) −0.269 × Th17 (%) and was statistically significant (P < 0.05).
Table 1.
Multiple linear regression of Th1 (%), Th2 (%) and Th17 (%) and NeuN+ cells (/mm2) after MCAO/R injury (WT tMCAO/R n = 20, KO tMCAO/R n = 20, Tga20 tMCAO/R n = 20).
| Independent variable | Unstandardised coefficient | Standardised coefficient β | t value | P value | |
|---|---|---|---|---|---|
| β | MSE | ||||
| Constant | 2736.568 | 159.599 | 17.147 | <0.001 | |
| Th1 (%) | −82.883 | 16.161 | −0.596 | −5.129 | <0.001 |
| Th2 (%) | 124.976 | 53.188 | 0.150 | 2.350 | 0.022 |
| Th17 (%) | −288.800 | 102.322 | −0.269 | −2.822 | 0.007 |
The model verified the significant positive correlation of Th2 percentage and negative correlation of Th1 and Th17 percentage with survived neurons and P < 0.05.
Effects of PrPC on CD4+ T splenocyte cytokine production after MCAO/R injury
Under resting conditions, cytokines produced by CD4+ T cells remain at fairly low levels. Meanwhile, after ischaemia impairment, the activated T lymphocytes can differentiate into either Th1/17‐pro‐inflammatory phenotype or Th2‐anti‐inflammatory phenotype. These cells secrete corresponding cytokines into the extracellular domain and then travel into the bloodstream, which regulates the downstream immunoinflammatory responses. We first separated and stimulated CD4+ T lymphocytes to detect pro‐ and anti‐inflammatory cytokines after MCAO/R injury. We used flow cytometry with a CBA kit to measure concentrations of Th1 phenotype‐related pro‐inflammatory cytokines (IFN‐γ, IL‐2), the Th17 phenotype‐related pro‐inflammatory cytokine (IL‐17 and IL‐6) and Th2 phenotype‐related anti‐inflammatory cytokines (IL‐4 and IL‐10) in stimulated cell supernatants (Fig. 4). The MCAO/R group of the KO genotype had significantly increased pro‐inflammatory cytokine concentrations (P < 0.05), except for IL‐2, which increased in a statistically significant manner in the Tga20 group instead of the KO group (P < 0.05). Nevertheless, the MCAO/R group of the Tga20 group exhibited higher anti‐inflammatory cytokine concentrations than the WT group (P < 0.05).
Figure 4.
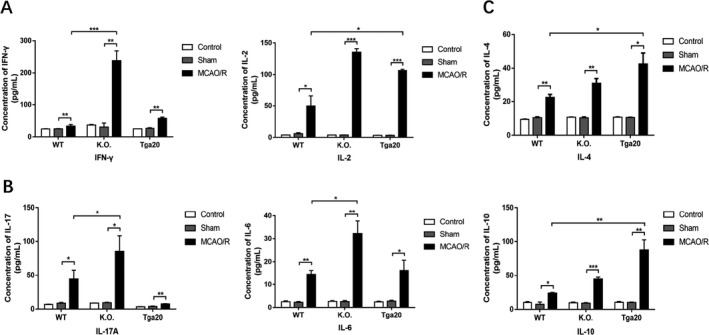
Flow cytometry‐based evaluation of concentrations of Th1/17‐related pro‐inflammatory and Th2‐related anti‐inflammatory cytokines in cultured T lymphocyte supernatants stimulated by PMA/iono for 5h. (WT n = 6, KO n = 6, Tga20 n = 6). (A) The expression of Th1‐related pro‐inflammatory cytokines. (B) The expression of Th17‐related pro‐inflammatory cytokines. (C) The expression of Th2‐related anti‐inflammatory cytokines. Experiments were carried out in biologically triplicate. Analysis of variance (ANOVA), and post hoc Duncan’s test and Dunnett’s test were used to assess differences among multiple groups and data are displayed as mean ± standard deviation (SD), *P < 0.05, **P < 0.01, ***P < 0.001.
We then collected the peripheral blood serum and measured concentrations of the same kinds of cytokines. Most of the detected cytokines showed increased concentrations after MCAO/R injury compared to the sham group (Fig. 5). We also detected near‐significant elevation of IFN‐γ in the Tga20 group, IL‐4 and IL‐17 elevation in the WT group and IL‐4 elevation in the KO group, although these changes were not statistically significant. In addition, the IL‐17 concentration in the Tga20 group exhibited a decline compared to the sham group (Fig. 5A and B). In addition, all the pro‐inflammatory cytokine concentrations showed a significant increase in the KO group compared to that in the WT group (P < 0.05), while the IL‐2 concentration increased in the Tga20 group as well (P < 0.01) (Fig. 5A). Both anti‐inflammatory cytokine concentrations showed significant increases in the MCAO/R group of the Tga20 genotype (P < 0.05) (Fig. 5C).
Figure 5.
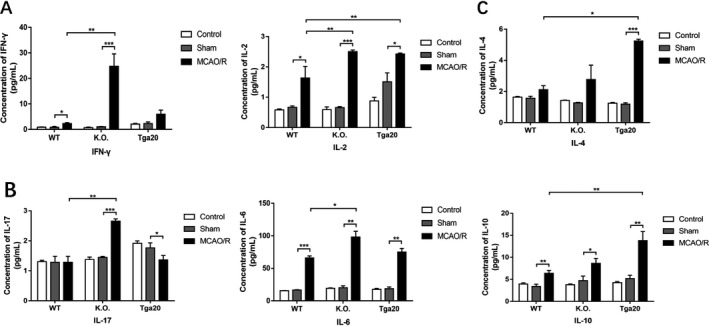
Flow cytometry‐based evaluation of concentrations of Th1/17‐related pro‐inflammatory and Th2‐related anti‐inflammatory cytokines in serum (WT n = 6, KO n = 6, Tga20 n = 6). (A) The expression of Th1‐related pro‐inflammatory cytokines. (B) The expression of Th17‐related pro‐inflammatory cytokines. (C) The expression of Th2‐related anti‐inflammatory cytokines. Experiments were carried out in biologically triplicate and data are displayed as mean ± standard deviation (SD), *P < 0.05, **P < 0.01, ***P < 0.001.
Effects of PrPC of CD4+ T lymphocytes on HT‐22 neuronal cell lines after OGD/R injury
To explore the effects of PrPC on neuronal survival, HT‐22 murine hippocampal neurons were cultured and subjected to OGD for 10 h. The cells were then reperfused with TCCM, and cell viability was examined after a 24 h incubation period. We used flow cytometry analysis with an Annexin V‐FITC/PI Apoptosis Detection Kit (Fig. 6) and found that OGD/R‐ MCAO/R TCCM‐treated neurons of WT and KO genotypes showed a higher apoptosis rate than the OGD/R‐sham TCCM‐treated neurons (P < 0.01), whereas the OGD/R‐MCAO/R TCCM‐treated neurons of the Tga20 group had reduced apoptosis compared to the sham group (Fig. 6C). When comparing the MCAO/R group of three genotypes, we found that the apoptosis of OGD/R‐KO MCAO/R TCCM‐treated neurons was much higher than that of OGD/R‐ WT MCAO/R TCCM‐treated neurons (P < 0.001), while OGD/R‐Tga20 MCAO/R TCCM‐treated neurons showed a sharp decrease in apoptosis compared to the WT group (P < 0.001) (Fig. 6D).
Figure 6.
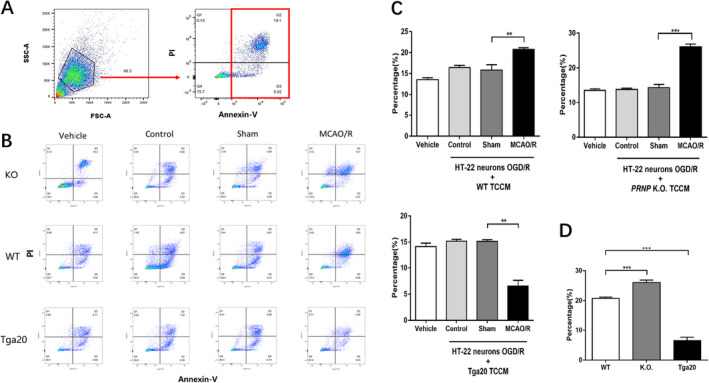
Effects of PrPC‐affected TCCM of each genotype on HT‐22 neuronal cell lines subjected to OGD/R injury. The vehicle group (n = 6), OGD/R+ control TCCM group (n = 6), OGD/R+ sham TCCM group (n = 6) and OGD/R+ MCAO/R TCCM group (n = 6). (A) An example showing the gating strategy to identify the apoptosis of HT‐22 murine hippocampal after co‐culture with TCCM in the presence of annexin V. The apoptotic neurons were showed in Q2 and Q3 quadrant. (B) Examples of cell apoptosis rates in HT‐22 murine hippocampal cells in each group of different genotypes. (C) The percentage of apoptotic OGD/R HT‐22 neurons after co‐cultured with TCCM of different genotype. (D) The comparison of OGD/R HT‐22 neurons apoptosis with MCAO/R TCCM of different genotype. Experiments were carried out in biologically triplicate and data are displayed as mean ± standard deviation (SD), **P < 0.01, ***P < 0.001.
Discussion
PrPC participated in the peripheral CD4+ cells inflammation responses of cerebral ischaemic/reperfusion injury
In this study, we measured the post‐ischaemic proportion of CD4+PrPC+ splenic T cells. Some studies have suggested that PrPC reflects the level of neuronal damage caused by ischaemia and regulates induced neuronal death in vivo. 20 However, there has been no definitive evidence regarding its role in peripheral immune inflammation. We detected a significant decrease in the proportion of splenic CD4+ T cells, which previous research has demonstrated to be stroke‐induced immunosuppression caused by the activation of the neuroendocrine axis and sympathetic/parasympathetic nervous system. 9 However, the percentage of CD4+PrPC+ T cells inversely increased, which suggested that PrPC may participate in the acute phase of post‐ischaemic splenic CD4+T cell proliferation; thus, we used mice with different levels of PrPC expression to detect its impact on inflammation.
PrPC regulated splenic CD4+ T cell differentiation after cerebral ischaemic/reperfusion injury
PrPC deficiency has been shown to lead to a two‐fold increase in cerebral infarction volume. 21 Meanwhile, we found that both the lesion and neurobehavioural score of the Tga20 group were attenuated when compared with that of the WT group, which emphasised its protective effect.
We measured the proportion of Th1/2/17 phenotypes in splenic lymphocytes of each genotype before and after ischaemia, of which IFN‐γ, IL‐4 and IL‐17 are the symbolic cytokines. The results showed that the percentage of Th1, Th2 and Th17 phenotypes of WT mice significantly increased after MCAO/R, indicating peripheral T‐cell activation by cerebral I/R. PrPC deficiency promoted pro‐inflammatory phenotypes (Th1 and Th17), followed by a shift in the balance of Th1/Th2 towards Th1. However, PrPC overexpression significantly facilitated anti‐inflammatory lymphocyte (Th2) expression and weakened the Th1 phenotype, which demonstrated a shift in the Th1/Th2 balance in the other direction. Previous studies have confirmed that Th1 deficiency leads to decreased ischaemic lesions, while Th2 deficiency has the opposite effect in murine MCAO/R injury. 22 These findings are in agreement with our experimental results. What’s more, an MLR model was constructed to verify the direct influence between Th percentage and the NeuN+ cells. We found Th2 committed a positive correlation while Th1 and Th17 committed a negative correlation with survived neurons, which emphasised the direct impact of CD4+ T cell differentiation on the ischaemia brain. The model also suggested that Th1 percentage was the most influential factor among the three phenotypes. In conclusion, our research indicates that the level of PrPC influences the percentage of pro/anti‐inflammatory Th phenotypes, which may regulate the severity of murine cerebral I/R injury.
Effects of PrPC expression on secretion of CD4+T cell‐related inflammatory cytokines
After cerebral I/R injury, T cells regulate the balance of the inflammatory responses by secreting and being affected by pro‐and anti‐inflammatory cytokines, thereby regulating the development of the injury. They also infiltrate around the lesion area through the impaired BBB, 23 promoting the polarisation of either classical‐activated macrophages (M1) or alternatively activated macrophages (M2). 10 We measured the CD4+ T cell‐related inflammatory cytokines in both the serum and cellular supernatant of all groups after ischaemic injury. The increase in supernatant cytokines in the WT group showed that T cells were activated after ischaemic injury, accompanied by the enhancement of proliferation and differentiation. Offner et al. found that IFN‐γ, IL‐6 and IL‐2 released by activated lymphocytes were significantly increased both 6 and 22 h after I/R, while IL‐10 only increased 22 h after modelling. 5 These results are consistent with our findings.
The cytokines detected in the supernatant represented the function of differentiated CD4+ T cells. We found that the levels of pro‐inflammatory cytokines such as IFN‐γ, IL‐6 and IL‐17 increased in the KO group, while the anti‐inflammatory cytokines IL‐4 and IL‐10 were significantly elevated in the Tga20 group. This trend was consistent with the differentiation and proportion of the CD4+ T cell phenotype, as detected above. These results suggest that PrPC may affect CD4+ T cell immune responses by cytokine production. The only exception was IL‐2, which showed a near‐significant increase in the KO group and an apparent rise in the Tga20 group. IL‐2 acts as an activator of T cells and is also secreted by NK cells and B cells; therefore, its concentration may increase in both groups. In future, the number of experimental subjects should be expanded for further exploration.
Cytokine concentrations in serum reflect the integrated result of CD4+ T cell inflammatory responses, demonstrating a variation trend that is in accordance with Th cell differentiation. The consistency between inflammatory cytokines and the variation in the ratio of Th1/2/17 phenotypes demonstrates that PrPC regulates CD4+ T cell differentiation and adjusts the secretion of relevant inflammatory cytokines. However, the variation in certain cytokines was not consistent with the expected result. IL‐2 levels were also increased in the serum of the Tga20 group. We speculate that although IL‐2 was secreted at much lower levels than CD4+ T cells, it is also released by CD8+ T cells, NK T cells and dendritic cells. 24 , 25 The above variations also indicate that the cytokine level is not simply affected by Th cells, and that PrPC may play an unknown role in peripheral inflammatory cytokine secretion in other immunocytes.
Effects of PrPC‐effected CD4+T cells on neuronal apoptosis after OGD/R injury
Previous studies have focused on the effects of T cells infiltrating the surrounding cerebral lesion, while our research explored the interaction between peripheral immunocytes and impaired neurons. The apparent increase in apoptosis of the OGD/R neurons after co‐cultivation in WT OGD/R TCCM illustrates that splenic lymphocytic cytokine may aggravate neuronal death; nevertheless, increased PrPC expression leads to a relative decline in apoptosis. The change in apoptosis also matched the lower neurobehavioural score and less severe infarct lesion in the Tga20 group in the above studies. In the PRNP KO group, neuronal apoptosis did not show a significant difference when compared with the WT group. Gu et al. co‐cultivated splenocytes and MCAO murine primary neurons and found that compared with the control group, there was no significant difference in neuronal death of splenocytes of CD4+T cell‐deficient mice; however, apoptosis may be inhibited by Th1‐deficient mice. 10 The difference between our studies was speculated to be because of the following factors: the cells we applied were neuronal cell lines instead of primary neurons; and the neurons were co‐cultivated in the conditioned medium rather than with the splenocytes, thus direct interaction between cells in the integrated system was not considered.
Study limitations
There are some limitations to this study. As only CD4+ T cells were analysed, the relationship between PrPC and other T‐cell phenotypes was not discussed. Moreover, the role of PrPC was explained only by the change in cell proportion and the fluctuation of inflammatory cytokines, while the concrete interaction sites and mechanism between PrPC and T cells were not provided. In addition, in vivo studies may also be necessitated to elucidate these interaction sites and mechanisms.
Conclusion
In this study, we showed that PrPC expression was altered in splenic CD4+ T cells after the cerebral I/R injury. From these results, we speculated that PrPC may participate in the stroke‐associated peripheral T‐cell responses. Furthermore, different levels of PrPC expression regulate the proportion of splenic Th1/2/17 phenotypes and the secretion of relevant pro‐ and anti‐inflammatory cytokines. Lack of PrPC expression led to an increase in the percentage of Th1/17 cells and the quantity of pro‐inflammatory cytokines, which deteriorated I/R injury. Conversely, PrPc overexpression induced an increase in Th2 cells and anti‐inflammatory cytokine expression, which alleviated the illness. We found that by varying the balance of Th cell phenotypes, PrPC deficiency aggravated the apoptosis of OGD/R HT‐22 neurons, while overexpression protected the neurons. In conclusion, PrPC works as a neuron protector in the CNS when I/R injury occurs. Furthermore, PrPc affects the peripheral immune responses and defends against stroke‐induced apoptosis.
Conflict of Interest
None.
Authors’ Contributions
Baizhuo Zhang contributed to execute the animal surgery, TTC staining, flow cytometry quantification and analysis; and prepared the draft of the manuscript. Xiang Yin contributed to the study design, data analysis and revised the manuscript. Yue Lang interpreted the data and contributed to the writing of the manuscript. Xiaoou Han mainly isolated and cultured T lymphocytes executed the flow cytometry quantification. Jie Shao was responsible for the data interpretation and the cell culturation. Rongrong Bai contributed to the data interpretation and manuscript writing. Li Cui was responsible for the study design and funding as well as supervising and approving the final version of the manuscript. All authors have read and approved the final version of the manuscript.
Funding Information
This work was supported by a grant from the Chinese National Natural Science Foundation (82071351).
Funding Statement
This work was funded by National Natural Science Foundation of China grant 82071351.
References
- 1. Liu T, Clark RK, McDonnell PC, et al. Tumor necrosis factor‐alpha expression in ischemic neurons. Stroke. 1994;25(7):1481‐1488. doi: 10.1161/01.STR.25.7.1481 [DOI] [PubMed] [Google Scholar]
- 2. Wang X, Yue T‐L, Young PR, et al. Expression of interleukin‐6, c‐Fos, and zif268 mRNAs in rat ischemic cortex. J Cereb Blood Flow Metab. 1995;15(1):166‐171. doi: 10.1038/jcbfm.1995.18 [DOI] [PubMed] [Google Scholar]
- 3. Wang X, Barone FC, Aiyar NV, Feuerstein GZ. Interleukin‐1 receptor and receptor antagonist gene expression after focal stroke in rats. Stroke. 1997;28(1):155‐162. doi: 10.1161/01.STR.28.1.155 [DOI] [PubMed] [Google Scholar]
- 4. Yoshimura A. Post‐ischemic inflammation regulates neural damage and protection. Front Cell. 2014;8:1‐8. doi: 10.3389/fncel.2014.00319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Offner H, Subramanian S, Parker SM, et al. Experimental stroke induces massive, rapid activation of the peripheral immune system. J. Cereb Blood Flow Metab. 2006;26:654‐665. doi: 10.1038/sj.jcbfm.9600217 [DOI] [PubMed] [Google Scholar]
- 6. Vahidy FS, Parsha KN, Rahbar H, et al. Acute splenic responses in patients with ischemic stroke and intracerebral hemorrhage. J Cereb Blood Flow Metab. 2016;36(6):1012‐1021. doi: 10.1177/0271678X15607880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jayaraj RL, Azimullah S, Beiram R, et al. Neuroinflammation: friend and foe for ischemic stroke. J Neuroinflammation 2019;16(1):142. Available from: https://pubmed.ncbi.nlm.nih.gov/31291966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Selvaraj UM, Stowe AM. Long‐term T cell responses in the brain after an ischemic stroke. Discov Med. 2017;24(134):323‐333. [PMC free article] [PubMed] [Google Scholar]
- 9. Gu L, Jian Z, Stary C, Xiong X. T Cells and cerebral ischemic stroke. Neurochem Res. 2015;40(9):1786‐1791. [DOI] [PubMed] [Google Scholar]
- 10. Manuscript A. Distinctive effects of T cell subsets in neuronal injury induced by co‐cultured splenocytes in vitro and by in vivo stroke in mice. Stroke. 2012;43(7):1941‐1946. doi: 10.1161/STROKEAHA.112.656611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mitsios N, Saka M, Krupinski J, et al. Cellular prion protein is increased in the plasma and peri‐infarcted brain tissue after acute stroke. J Neurosci Res. 2007;85(3):602‐611. Available from https://www.ncbi.nlm.nih.gov/pubmed/17149767 [DOI] [PubMed] [Google Scholar]
- 12. Lo RY‐Y, Shyu W‐C, Lin S‐Z, et al. New molecular insights into cellular survival and stress responses: neuroprotective role of cellular prion protein (PrPC). Mol Neurobiol. 2007;35(3):236‐244. doi: 10.1007/s12035-007-8003-y [DOI] [PubMed] [Google Scholar]
- 13. Bakkebø MK, Mouillet‐Richard S, Espenes A, et al. The cellular prion protein: a player in immunological quiescence. Front Immunol. 2015;6:450. Available from https://www.ncbi.nlm.nih.gov/pubmed/26388873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mabbott NA. Immunology of prion protein and prions. Prog Mol Biol Transl Sci. 2017;150:203‐240. Available from https://www.ncbi.nlm.nih.gov/pubmed/28838662 [DOI] [PubMed] [Google Scholar]
- 15. Ezpeleta J, Boudet‐Devaud F, Pietri M, et al. Protective role of cellular prion protein against TNFα‐mediated inflammation through TACE α‐secretase. Sci Rep. 2017;7(1):7671. Available from: https://www.ncbi.nlm.nih.gov/pubmed/28794434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Onodera T, Sakudo A, Tsubone H, Itohara S. Review of studies that have used knockout mice to assess normal function of prion protein under immunological or pathophysiological stress. Microbiol Immunol. 2014;58(7):361‐374. Available from https://www.ncbi.nlm.nih.gov/pubmed/24866463 [DOI] [PubMed] [Google Scholar]
- 17. Ingram RJ, Isaacs JD, Kaur G, et al. A role of cellular prion protein in programming T‐cell cytokine responses in disease. FASEB J. 2009;23(6):1672‐1684. doi: 10.1096/fj.08-116087 [DOI] [PubMed] [Google Scholar]
- 18. Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20(1):84‐91. doi: 10.1161/01.STR.20.1.84 [DOI] [PubMed] [Google Scholar]
- 19. Doeppner TR, Kaltwasser B, Bähr M, Hermann DM. Effects of neural progenitor cells on post‐stroke neurological impairment—a detailed and comprehensive analysis of behavioral tests. Front Cell Neurosci. 2014;8:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Weise J, Crome O, Sandau R, et al. Upregulation of cellular prion protein (PrP c) after focal cerebral ischemia and influence of lesion severity. Neurosci Lett. 2004;372(1–2):146‐150. doi: 10.1016/j.neulet.2004.09.030 [DOI] [PubMed] [Google Scholar]
- 21. Jens W, Raoul S, Sönke S, et al. Deletion of cellular prion protein results in reduced akt activation, enhanced postischemic caspase‐3 activation, and exacerbation of ischemic brain injury. Stroke. 2006;37(5):1296‐1300. doi: 10.1161/01.STR.0000217262.03192.d4 [DOI] [PubMed] [Google Scholar]
- 22. Huang Y, Rabb H, Womer KL. Ischemia‐reperfusion and immediate T cell responses. Cell Immunol. 2007;248(1):4‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gelderblom M, Leypoldt F, Steinbach K, et al. Temporal and spatial dynamics of cerebral immune cell accumulation in stroke. Stroke. 2009;40(5):1849‐1857. doi: 10.1161/STROKEAHA.108.534503 [DOI] [PubMed] [Google Scholar]
- 24. Granucci F, Vizzardelli C, Pavelka N, et al. Inducible IL‐2 production by dendritic cells revealed by global gene expression analysis. Nat Immunol. 2001;2(9):882‐888. doi: 10.1038/ni0901-882 [DOI] [PubMed] [Google Scholar]
- 25. Mitra S, Leonard WJ. Biology of IL‐2 and its therapeutic modulation: mechanisms and strategies. J Leukoc Biol. 2018;103(4):643‐655. doi: 10.1002/JLB.2RI0717-278R [DOI] [PubMed] [Google Scholar]