

Abstract
Exome sequencing (ES) has revolutionized rare disease management, yet only ~25%–30% of patients receive a molecular diagnosis. A limiting factor is the quality of available phenotypic data. Here, we describe how deep clinicopathological phenotyping yielded a molecular diagnosis for a 19‐year‐old proband with muscular dystrophy and negative clinical ES. Deep phenotypic analysis identified two critical data points: (1) the absence of emerin protein in muscle biopsy and (2) clinical features consistent with Emery‐Dreifuss muscular dystrophy. Sequencing data analysis uncovered an ultra‐rare, intronic variant in EMD, the gene encoding emerin. The variant, NM_000117.3: c.188‐6A > G, is predicted to impact splicing by in silico tools. This case thus illustrates how better integration of clinicopathologic data into ES analysis can enhance diagnostic yield with implications for clinical practice.
Introduction
Molecular diagnostic rates have significantly improved since the widespread implementation of exome sequencing (ES). 1 , 2 At present, the molecular diagnostic rate of ES obtained in clinical diagnostic laboratories is approximately 25%–30%, although higher or lower rates may be seen in certain disease states and patient populations. 3 , 4 , 5 , 6 , 7 , 8 Advances in ES data analyses including copy number variation (CNV) assessment, 9 , 10 better detection of insertion/deletion (indel) variant alleles, 11 and homozygosity mapping using absence of heterozygosity (AOH) data as a surrogate measure of identity‐by‐descent (IBD) 10 have improved diagnostic rates. Nevertheless, the notion of a “diagnostic ceiling” has been proposed because of similar diagnostic rates observed in multiple disease cohorts 5 , 12 as well as known limitations of ES technology (e.g., poor coverage of noncoding regions, limited detection of structural variants and repeat expansions). 13
While both short read whole‐genome sequencing (SR‐WGS) and long‐read sequencing (LR‐WGS) technologies will likely improve molecular diagnostic rates by increasing coverage of noncoding regions, mounting evidence suggests the “molecular diagnostic gap” can be further narrowed by better integration of detailed phenotypic data into ES data analysis. 14 , 15 , 16 Here, we provide an illustrative example of how a detailed analysis of extant clinicopathologic data led to a molecular diagnosis in a patient with muscular dystrophy and negative clinical ES (cES) and reflect on its heuristic implications for clinical practice.
Methods
Participants
All participants in this study provided informed consent as part of the Baylor‐Hopkins Center for Mendelian Genomics (BHCMG) initiative, including consent to publish photographs. This study was approved through Baylor College of Medicine Institutional Review Board (IRB) protocol H‐29697.
Histology, immunofluorescence and western blot analysis of muscle samples
A vastus lateralis muscle biopsy was obtained during the proband’s clinical care. Hematoxylin and eosin (H&E) staining, immunostaining, and western blot analysis were performed by the Texas Children’s Hospital Neuropathology and Molecular Neuropathology Laboratory (Houston, TX) by board‐certified neuropathologists (CAM and AMA). For immunofluorescence, cryosections of skeletal muscle were stained using the nuclear stain 4′,6‐diamidino‐2‐phenylindole (DAPI) and antibodies against emerin or lamin A/C. Western blots were stained with antibodies for emerin and alpha‐sarcoglycan. Antibodies were obtained from Leica (emerin, cat. no. Emerin CE; alpha‐sarcoglycan, cat. no. A‐SARC‐L‐CE) or Abcam (lamin A/C, cat. no. AB5090).
Exome sequencing
Research trio ES of genomic DNA obtained from peripheral blood was performed in the Baylor College of Medicine Human Genome Sequencing Center (BCM‐HGSC). 1 , 17 Rare variant family‐based exome analysis was performed as previously described. 1 , 17 Identified variants after computational parsing and filtering were experimentally confirmed and segregated via orthogonal Sanger dideoxy sequencing.
Results
The proband is a 19‐year‐old male with muscular dystrophy. Since early childhood he had frequent falls, easy fatigability, joint stiffness, and motor difficulties. Weakness and stiffness gradually progressed with age. On last physical examination at 18 years of age he had bilateral ankle contractures, elbow contractures (right greater than left), and weakness of bilateral ankle dorsiflexion, biceps, and hand interossei (Fig. 1A and B). There was no family history of neuromuscular disease. His creatine kinase (CK) levels trended upward with time (588 U/L at 17 years old, normal <245 U/L), suggesting a dystrophic condition. Needle electromyography showed small amplitude, short duration, polyphasic motor unit action potentials consistent with a myopathic condition. Muscle biopsy at age 11 years demonstrated unremarkable hematoxylin and eosin staining but absent emerin staining by immunofluorescence and western blot (Fig. 1C–F). Trio cES was subsequently performed (Baylor Genetics, BG, Laboratories, Houston, TX) and failed to identify any variants in EMD or other myopathy genes.
Figure 1.
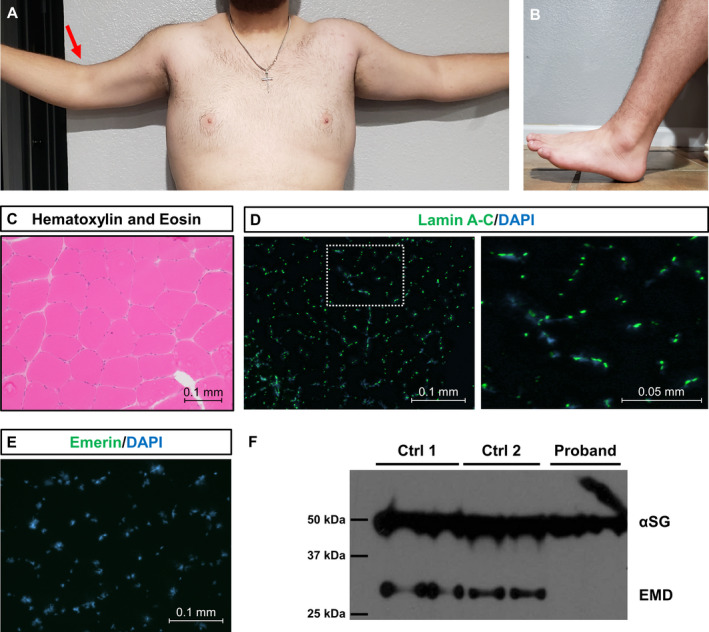
Clinicopathological features of proband with X‐linked Emery‐Dreifuss muscular dystrophy (A) Photograph of proband’s fully extended arms demonstrating limited active range of motion due to elbow contractures (right greater than left, red arrow). (B) Photograph of proband’s fully dorsiflexed foot demonstrating limited active range of motion due to ankle contractures. (C) Hematoxylin and eosin staining of left vastus lateralis muscle biopsy taken at 11 years old. (D) Immunofluorescence staining of lamin A‐C in the proband. Lamin A‐C is a nuclear protein involved in autosomal dominant Emery‐Dreifuss muscular dystrophy 1 (MIM #310300). Lamin A‐C (green) is widely expressed and co‐localizes with the nuclear stain DAPI (blue). (E) Immunofluorescence staining of emerin in the proband. Note the absence of emerin staining (green). Nuclei were counterstained with DAPI (blue). (F) Western blot of muscle biopsy protein lysates from proband and two control patients using antibodies for alpha‐sarcoglycan (αSG) and emerin (EMD). All samples were run in duplicate. Molecular weights are provided in kilodaltons (kDa). A 50 kDa band is seen in all samples corresponding to αSG. A 34 kDa band corresponding to EMD can be seen in the control samples but not in the proband.
The proband and his family were subsequently enrolled in a “molecularly undiagnosed” neuromuscular disease cohort in the BHCMG and underwent research ES. Analysis began with an extensive review of the proband’s medical history, laboratory findings, electrodiagnostic studies, and muscle biopsy pathology. Due to negative emerin staining of muscle biopsy as well as phenotypic features consistent with Emery‐Dreifuss muscular dystrophy, the proband’s BAM file, a data file containing aligned sequencing data in a format which facilitates visualization, was inspected, leading to the identification of a hemizygous variant in EMD intron 2 (Fig. 2A). The variant, EMD (NM_000117.3):c.188‐6A > G, has a high CADD‐v1.6 score (20.6) and is predicted to impact splicing by multiple in silico algorithms (SpliceAI acceptor gain score 0.99; MMSp acceptor score −2.996; Human Splice Finder, alteration of the wild‐type acceptor site) 18 , 19 , 20 , 21 (Fig. 2B). The variant is absent from gnomAD v2.1.1. 22 Segregation analysis within the family demonstrated that the variant was maternally inherited and absent from the proband’s unaffected brothers (Fig. 2C).
Figure 2.
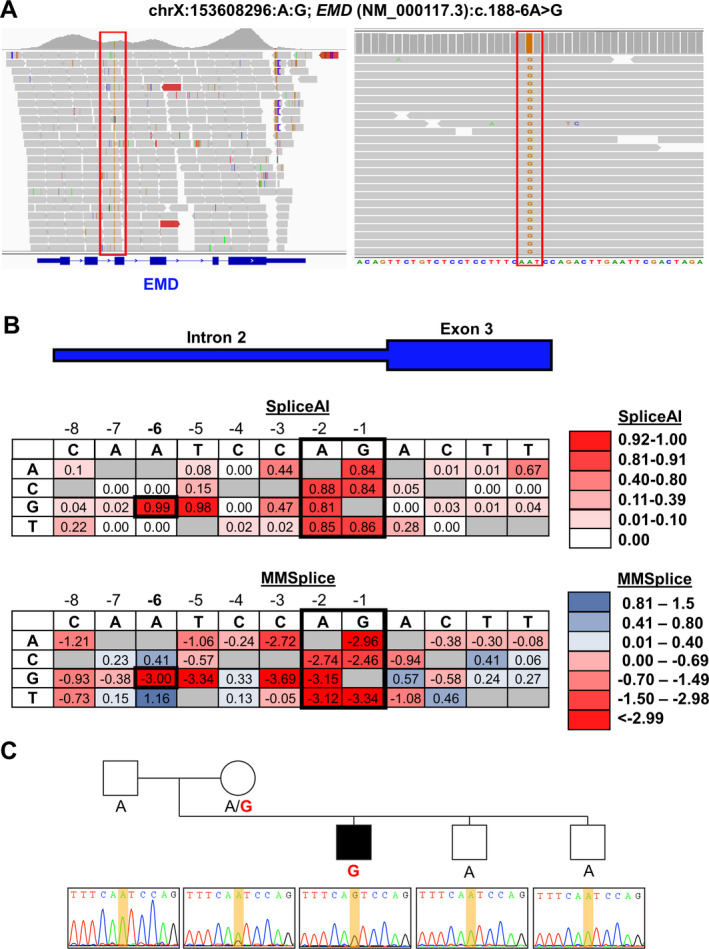
Research ES identifies a previously unrecognized EMD splice variant. (A) Screenshots of the proband’s BAM file data from Integrated Genomics Viewer (https://software.broadinstitute.org/software/igv/) showing a hemizygous intronic variant in intron 2 of EMD. (B) Heat map showing predicted impact on splicing from SpliceAI and MMSplice of all possible single‐nucleotide variants near c.188‐6A > G. Values in heat map were obtained from CADD v1.6 (https://cadd.gs.washington.edu/snv) and reflect the SpliceAI acceptor gain and MMSplice acceptor submodules, respectively. Darker shades of red indicate a greater predicted splicing impact. c.188‐6A > G and the consensus splice acceptor sites (−1, −2) are highlighted in black. (C) Sanger sequencing of the proband’s parents and unaffected brothers confirms the variant and demonstrates it is only found in the proband and his mother.
Discussion
While diagnostic rates have improved since the advent of ES, many patients with presumed Mendelian disorders still lack a definitive molecular diagnosis. This diagnostic gap is often attributed to: (1) yet unidentified “disease‐contributing genes” and variant alleles and (2) the limited ability of ES to detect non‐coding and structural variants. 13 By providing a complete sequence of all genic and intergenic regions, whole‐genome sequencing (WGS) has been regarded as a potential “panacea” and solution for the latter issue. However, WGS results to date have been underwhelming, with many additional diagnoses resulting from variants previously captured on ES and interim gene discoveries. 23 Future hopes for resolving the diagnostic gap include LR‐WGS and RNA‐seq. 24 , 25 , 26 , 27 Although these approaches are promising, they will remain inaccessible in the clinic for the foreseeable future due to unavailability, high cost, and/or lack of appropriate tissue specimens.
An alternative hypothesis to explain the diagnostic gap is the under‐utilization of extant ES data. For example, expansion from proband‐only to trio ES improves diagnostic yield by permitting detection of de novo mutations and phasing (i.e., cis or trans configuration). 1 , 28 Copy number analysis of ES data, a practice not routinely performed by clinical diagnostic laboratories, may identify large deletions or duplications (>100 Kb) or even smaller homozygous exonic deletions. 9 , 10 The absence of heterozygosity (AOH) analysis, as a surrogate measure of runs‐of‐homozygosity (ROH), recognizes genomic intervals of identity‐by‐descent in families with or without a known history of consanguinity which prompts a thorough investigation of those regions for a potentially causative homozygous variant. 1 , 28 , 29 Greater integration of clinicians and deep clinical phenotyping also improves diagnostic rates by enhancing variant prioritization and drawing increased scrutiny of extant single gene or gene families’ ES data. 14 , 15 , 16 Deep phenotyping, the process of comprehensively assessing and categorizing individual phenotypic features often through Human Phenotype Ontology (HPO) terms, is routinely performed by medical geneticists and neurologists, yet the requisition forms for clinical genetic and genomic testing often fail to capture the depth of phenotyping performed by clinicians. 14 , 30 Finally, large amounts of “off‐target” sequencing data, for example intronic and 3′/5′ untranslated regions, are generated by ES yet are often filtered by cES bioinformatic pipelines despite increasing evidence of their significance in Mendelian disorders and improved in silico tools for evaluating their pathogenicity. 19 , 21 , 31
Here we provide an illustrative example of how the incorporation of deep phenotyping into ES analysis improves molecular diagnostic yield. The proband carried a clinical diagnosis of muscular dystrophy with supporting laboratory and electrophysiologic data. The absence of emerin protein in his muscle biopsy strongly supported the clinical diagnosis of X‐linked Emery‐Dreifuss muscular dystrophy 1 (MIM #310300), and retrospective review of his clinical presentation identified compatible features including childhood‐onset joint contractures and slowly progressive muscle weakness. However, trio cES failed to identify pathogenic variants in EMD or other myopathy genes. Considering his clinical history and biopsy results, EMD sequencing data were reanalyzed, identifying a pathogenic hemizygous variant in EMD, c.188‐6A > G. The variant results in the substitution of a guanine for an adenine six nucleotides from the intron 2‐exon 3 boundary (Fig. 2B) which is predicted to create a new splice acceptor site by multiple in silico prediction tools. While the precise impact of the variant on splicing was not determined, the convergence of in silico algorithms predicting alteration of the splice acceptor site and the in vivo readout provided by western blot and immunofluorescence strongly suggests the mutant transcript either has a premature termination codon (PTC) resulting in nonsense‐mediated decay or encodes an unstable protein subject to rapid decay.
Identification of the variant had immediate clinical impact and management implications for the patient and his family. Gene therapy trials increasingly require a definitive genetic diagnosis for enrollment, and identification of a specific pathogenic intronic variant offers the opportunity for bespoke therapies like personalized antisense oligonucleotides (ASO). While personalized gene therapies may seem impractical, the recent story of milasen, an ASO customized and administered to a single patient with neuronal ceroid lipofuscinosis 7 (MIM # 610951), has demonstrated their feasibility and provide a pathway forward for rare disease. 32 Additionally, these studies identified the carrier status of the proband’s mother, a finding of considerable significance as female carriers can develop cardiac conduction defects and are at risk of sudden death. 33 Therefore, cardiology follow‐up and screening of the extended family was recommended.
The importance of intronic variants such as EMD c.188‐6A > G which impact cis‐acting elements in human disease is well‐recognized. 34 , 35 , 36 The proportion of human pathogenic variants disrupting cis‐acting elements has been estimated between 15% to 60%. 34 , 35 Prior to this report, only a single non‐consensus splice variant, EMD: c.449 + 23_450−35del, was recognized. 37 Located within intron 5, the variant was detected on a neuromuscular gene panel and would have been well‐covered in the BCM‐HGSC ES platform (Fig. 2A). Studies of EMD constructs with variably sized intron 5 deletions demonstrated the 23‐nucleotide deletion does not impact the major branchpoint c.450‐24A but rather causes splicing abnormalities due to excessive intronic shortening. 37 Such an intron size constraint mutational mechanism may disproportionately affect genes with small size introns and remains underappreciated despite the fact that it was described over a decade ago. 38 Additional pathogenic intronic EMD variants will undoubtedly be identified with increased implementation of WGS and closer scrutiny of extant ES data. Further identification and study of pathogenic intronic variants through ES/GS, mini‐gene assays, and RNA‐seq will clarify the mechanisms involved in splicing and in turn improve in silico predictive models.
In summary, this report illustrates how the integration of deep clinicopathological phenotypic data into ES analysis improves molecular diagnostic yield. Clinicians play a critical role in this process by providing accurate and detailed clinical data to clinical diagnostic laboratories and following up on all exome negative studies. Additionally, an active dialogue between clinicians and laboratories is essential to maximize diagnostic yield.
Conflict of Interest
J.R.L. has stock ownership in 23andMe, is a paid consultant for Regeneron Genetics Center, and is a co‐inventor on multiple United States and European patents related to molecular diagnostics for inherited neuropathies, eye diseases, and bacterial genomic fingerprinting. The Department of Molecular and Human Genetics at Baylor College of Medicine receives revenue from clinical genetic testing conducted at Baylor Genetics (BG) Laboratories; JRL is a member of the Scientific Advisory Board of BG. Other authors have no potential conflicts to report.
Data Sharing
All data supporting the findings of this study are available from authors DGC and JRL upon reasonable request.
Acknowledgments
This study was supported in part by the U.S. National Human Genome Research Institute (NHGRI) and National Heart Lung and Blood Institute (NHBLI) to the Baylor‐Hopkins Center for Mendelian Genomics (BHCMG, UM1 HG006542 to J.R.L); NHGRI grant to Baylor College of Medicine Human Genome Sequencing Center (U54HG003273 to R.A.G.); U.S. National Institute of Neurological Disorders and Stroke (NINDS) (R35NS105078 to J.R.L.) and Muscular Dystrophy Association (MDA) (512848 to J.R.L.). D.M. was supported by a Medical Genetics Research Fellowship Program through the United States National Institute of Health (T32 GM007526‐42). J.E.P. was supported by NHGRI K08 HG008986. D.P. is supported by a Clinical Research Training Scholarship in Neuromuscular Disease partnered by the American Academy of Neurology (AAN), American Brain Foundation (ABF), and Muscle Study Group (MSG), and International Rett Syndrome Foundation (IRSF grant #3701‐1). D.G.C. was supported by NIH ‐ Brain Disorders and Development Training Grant (T32 NS043124‐19) and MDA Development Grant (873841).
Funding Information
This study was supported in part by the U.S. National Human Genome Research Institute (NHGRI) and National Heart Lung and Blood Institute (NHBLI) to the Baylor‐Hopkins Center for Mendelian Genomics (BHCMG, UM1 HG006542 to J.R.L); NHGRI grant to Baylor College of Medicine Human Genome Sequencing Center (U54HG003273 to R.A.G.); U.S. National Institute of Neurological Disorders and Stroke (NINDS) (R35NS105078 to J.R.L.) and Muscular Dystrophy Association (MDA) (512848 to J.R.L.). D.M. was supported by a Medical Genetics Research Fellowship Program through the United States National Institute of Health (T32 GM007526‐42). J.E.P. was supported by NHGRI K08 HG008986. D.P. is supported by a Clinical Research Training Scholarship in Neuromuscular Disease partnered by the American Academy of Neurology (AAN), American Brain Foundation (ABF), and Muscle Study Group (MSG), and International Rett Syndrome Foundation (IRSF grant #3701‐1). D.G.C. was supported by NIH ‐ Brain Disorders and Development Training Grant (T32 NS043124‐19) and MDA Development Grant (873841).
Funding Statement
This work was funded by National Institute of Neurological Disorders and Stroke grant R35NS105078; Muscular Dystrophy Association grants 512848 and 873841; National Human Genome Research Institute grants K08 HG008986, U54HG003273, and UM1 HG006542; National Heart, Lung, and Blood Institute grant UM1 HG006542; National Institute of Health grant T32 GM007526‐42; American Academy of Neurology grant American Brain Foundation; Muscle Study Group; International Rett Syndrome Foundation grant 3701‐1; NIH ‐ Brain Disorders and Development grant T32 NS043124‐19.
References
- 1. Eldomery MK, Coban‐Akdemir Z, Harel T, et al. Lessons learned from additional research analyses of unsolved clinical exome cases. Genome Med. 2017;9:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Liu P, Meng L, Normand EA, et al. Reanalysis of clinical exome sequencing data. N Engl J Med. 2019;380:2478‐2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Helman G, Lajoie BR, Crawford J, et al. Genome sequencing in persistently unsolved white matter disorders. Ann Clin Transl Neurol. 2020;7:144‐152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Herman I, Lopez MA, Marafi D, et al. Clinical exome sequencing in the diagnosis of pediatric neuromuscular disease. Muscle Nerve. 2021;63:304‐310. [DOI] [PubMed] [Google Scholar]
- 5. Ngo KJ, Rexach JE, Lee H, et al. A diagnostic ceiling for exome sequencing in cerebellar ataxia and related neurological disorders. Hum Mutat. 2020;41:487‐501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yavarna T, Al‐Dewik N, Al‐Mureikhi M, et al. High diagnostic yield of clinical exome sequencing in Middle Eastern patients with Mendelian disorders. Hum Genet. 2015;134:967‐980. [DOI] [PubMed] [Google Scholar]
- 7. Bayram Y, Karaca E, Coban Akdemir Z, et al. Molecular etiology of arthrogryposis in multiple families of mostly Turkish origin. J Clin Invest. 2016;126:762‐778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Posey JE, Rosenfeld JA, James RA, et al. Molecular diagnostic experience of whole‐exome sequencing in adult patients. Genet Med. 2016;18:678‐685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fromer M, Moran J, Chambert K, et al. Discovery and statistical genotyping of copy‐number variation from whole‐exome sequencing depth. Am J Hum Genet. 2012;91:597‐607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gambin T, Akdemir ZC, Yuan B, et al. Homozygous and hemizygous CNV detection from exome sequencing data in a Mendelian disease cohort. Nucleic Acids Res. 2017;45:1633‐1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Farek J, Hughes D, Mansfield A, et al. xAtlas: scalable small variant calling across heterogeneous next‐generation sequencing experiments. bioRxiv, 295071. 10.1101/295071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Estrella EA, Kang PB. Hunting for the perfect test: neuromuscular diagnosis in the age of genomic bounty. Muscle Nerve. 2021;63:282‐284. [DOI] [PubMed] [Google Scholar]
- 13. Posey JE. Genome sequencing and implications for rare disorders. Orphanet J Rare Dis. 2019;14:153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Basel‐Salmon L, Orenstein N, Markus‐Bustani K, et al. Improved diagnostics by exome sequencing following raw data reevaluation by clinical geneticists involved in the medical care of the individuals tested. Genet Med. 2019;21:1443‐1451. [DOI] [PubMed] [Google Scholar]
- 15. Pena LDM, Jiang Y‐H, Schoch K, et al. Looking beyond the exome: a phenotype‐first approach to molecular diagnostic resolution in rare and undiagnosed diseases. Genet Med. 2018;20:464‐469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Aarabi M, Sniezek O, Jiang H, et al. Importance of complete phenotyping in prenatal whole exome sequencing. Hum Genet. 2018;137:175‐181. [DOI] [PubMed] [Google Scholar]
- 17. Karaca E, Harel T, Pehlivan D, et al. Genes that affect brain structure and function identified by rare variant analyses of Mendelian neurologic disease. Neuron. 2015;88:499‐513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rentzsch P, Schubach M, Shendure J, Kircher M. CADD‐Splice‐improving genome‐wide variant effect prediction using deep learning‐derived splice scores. Genome Med. 2021;13:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cheng J, Nguyen TYD, Cygan KJ, et al. MMSplice: modular modeling improves the predictions of genetic variant effects on splicing. Genome Biol. 2019;20:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Desmet FO, Hamroun D, Lalande M, Collod‐Beroud G, Claustres M, Beroud C. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009;37:e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jaganathan K, Kyriazopoulou Panagiotopoulou S, McRae JF, et al. Predicting splicing from primary sequence with deep learning. Cell. 2019;176(3):535‐548.e24. [DOI] [PubMed] [Google Scholar]
- 22. Karczewski KJ, Francioli LC, Tiao G, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434‐443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Alfares A, Aloraini T, Subaie LA, et al. Whole‐genome sequencing offers additional but limited clinical utility compared with reanalysis of whole‐exome sequencing. Genet Med. 2018;20:1328‐1333. [DOI] [PubMed] [Google Scholar]
- 24. Logsdon GA, Vollger MR, Eichler EE. Long‐read human genome sequencing and its applications. Nat Rev Genet. 2020;21:597‐614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Beck CR, Carvalho CMB, Akdemir ZC, et al. Megabase length hypermutation accompanies human structural variation at 17p11.2. Cell. 2019;176:1310‐1324 e1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Murdock DR, Dai H, Burrage LC. Transcriptome‐directed analysis for Mendelian disease diagnosis overcomes limitations of conventional genomic testing. J Clin Invest 2021;131(1): e141500. 10.1172/jci141500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cummings BB, Marshall JL, Tukiainen T, et al. Improving genetic diagnosis in Mendelian disease with transcriptome sequencing. Sci Transl Med. 2017;9(386):eaal5209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pehlivan D, Bayram Y, Gunes N, et al. The genomics of arthrogryposis, a complex trait: candidate genes and further evidence for oligogenic inheritance. Am J Hum Genet. 2019;105:132‐150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gonzaga‐Jauregui C, Yesil G, Nistala H, et al. Functional biology of the Steel syndrome founder allele and evidence for clan genomics derivation of COL27A1 pathogenic alleles worldwide. Eur J Hum Genet. 2020;28:1243‐1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Robinson PN, Kohler S, Bauer S, Seelow D, Horn D, Mundlos S. The Human Phenotype Ontology: a tool for annotating and analyzing human hereditary disease. Am J Hum Genet. 2008;83:610‐615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wright CF, Quaife NM, Ramos‐Hernández L, et al. Non‐coding region variants upstream of MEF2C cause severe developmental disorder through three distinct loss‐of‐function mechanisms. Am J Hum Genet. 2021;108:1083‐1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kim J, Hu C, Moufawad El Achkar C, et al. Patient‐customized oligonucleotide therapy for a rare genetic disease. N Engl J Med. 2019;381:1644‐1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Buckley AE, Dean J, Mahy IR. Cardiac involvement in Emery Dreifuss muscular dystrophy: a case series. Heart. 1999;82:105‐108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang GS, Cooper TA. Splicing in disease: disruption of the splicing code and the decoding machinery. Nat Rev Genet. 2007;8:749‐761. [DOI] [PubMed] [Google Scholar]
- 35. Park E, Pan Z, Zhang Z, Lin L, Xing Y. The expanding landscape of alternative splicing variation in human populations. Am J Hum Genet. 2018;102:11‐26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Calame DG, Fatih J, Herman I, et al. Biallelic pathogenic variants in TNNT3 associated with congenital myopathy. Neurol Genet. 2021;7:e589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bryen SJ, Joshi H, Evesson FJ, et al. Pathogenic abnormal splicing due to intronic deletions that induce biophysical space constraint for spliceosome assembly. Am J Hum Genet. 2019;105:573‐587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wang LL, Worley K, Gannavarapu A, Chintagumpala MM, Levy ML, Plon SE. Intron‐size constraint as a mutational mechanism in Rothmund‐Thomson syndrome. Am J Hum Genet. 2002;71:165‐167. [DOI] [PMC free article] [PubMed] [Google Scholar]