

Abstract
Originally described as a risk factor for autism, CHD8 loss‐of‐function variants have recently been associated with a wider spectrum of neurodevelopmental abnormalities. We further expand the CHD8‐related phenotype with the description of two unrelated patients who presented with childhood‐onset progressive dystonia. Whole‐exome sequencing conducted in two independent laboratories revealed a CHD8 nonsense variant in one patient and a frameshift variant in the second. The patients had strongly overlapping phenotypes characterized by generalized dystonia with mild‐to‐moderate neurodevelopmental comorbidity. Deep brain stimulation led to clinical improvement in both cases. We suggest that CHD8 should be added to the growing list of neurodevelopmental disorder‐associated genes whose mutations can also result in dystonia‐dominant phenotypes.
Introduction
Heterozygous loss‐of‐function variants in CHD8 (MIM:610528) were first described as a highly penetrant risk factor for autism spectrum disorder. 1 CHD8 encodes a member of the Chromodomain‐Helicase‐DNA binding family of chromatin remodelers, a class of proteins involved in various biological processes including development, cognition, and motor functioning. 1 Over the past few years, a few dozens of pathogenic CHD8 variants have been reported, mostly comprising nonsense and frameshifting alleles. 1 , 2 , 3 , 4 Although still referred to as „susceptibility to autism‐18“ in the OMIM database (MIM:615032), the phenotype associated with truncating variants in CHD8 is known to extend beyond autistic traits. 2 , 3 , 4 CHD8 variants have been found in cohorts of patients with developmental delay, overgrowth and intellectual disability, behavioral disorders, and cerebellar malformations. 2 , 3 , 4 Epilepsy, cerebral palsy, and ataxic movements have been documented in some affected individuals. 2 , 3 As for most rare neurodevelopmental diseases, the full spectrum of phenotypes presented by patients with CHD8 mutations is not well understood.
We report two unrelated female patients with childhood‐onset progressive dystonia, connected through personal communication between independent research groups, who were identified to have CHD8 pathogenic variants.
Methods
The index patients (patient 1 from family 1; patient 2 from family 2) underwent in‐depth phenotyping and whole‐exome sequencing using published standard protocols and pipelines. 5 , 6 Written informed consent was obtained from all subjects participating in this case study. The patient shown in the video (patient 1) provided explicit consent to the publication including online publication of the video material. Consent was obtained for publication of facial photographs from patient 2.
Results
Molecular analysis
In patient 1, we uncovered a heterozygous CHD8 nonsense substitution, c.6649C>T (p. Arg2217*). This variant was unobserved in gnomAD and 20,000 in‐house control exomes and has been identified previously in individuals with CHD8‐related neurodevelopmental disorder (ClinVar:VCV000588945.3; HGMD:CM1914505). Sanger sequencing‐based segregation analysis revealed that the variant was absent in patient 1ˋs healthy mother and daughter but present in the DNA from her two phenotypically abnormal sons (Fig. 1A). Patient 1ˋs CHD8 variant finding has been mentioned previously but without detailed clinical information. 5 A novel heterozygous CHD8 2‐bp deletion, c.4800_4801delAG (p. Gly1602Cysfs*5), was detected in patient 2. The variant was absent from the abovementioned control databases. Sanger sequencing showed that the variant was not carried by patient 2ˋs healthy family members (Fig. 1A). Each CHD8 variant was classified as pathogenic, based on the ACMG guidelines. 7 No alternative genetic causes for the observed disease manifestations were identified in either of the two families.
Figure 1.
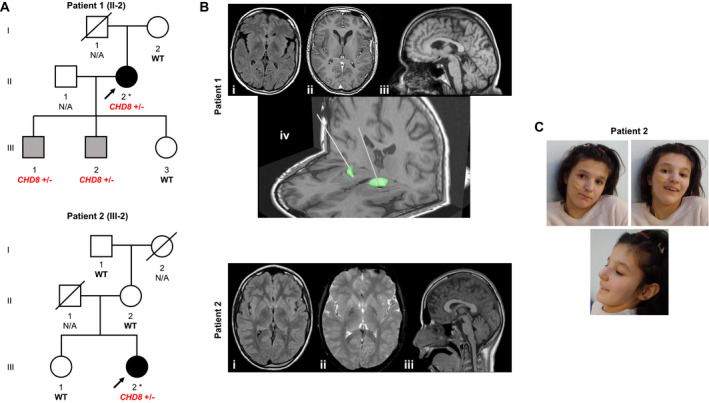
(A) Families with dystonia affected by CHD8 variants. Index patients 1 and 2 underwent whole‐exome sequencing, as indicated by asterisks. Open symbols are unaffected individuals, black symbols are individuals with generalized dystonia, and gray symbols are individuals with neurodevelopmental issues but no movement disorder. CHD8 variant status is depicted for all family members for whom DNA was available; WT, CHD8 homozygous wild‐type allele; N/A, no DNA available. (B) MRI data of patients 1 and 2; patient 1: axial FLAIR (i), axial T1 (ii), sagittal T1 (iii), DBS lead implantation into the globus pallidus internus (iv); patient 2: axial FLAIR (i), axial T2*WI (ii), sagittal T1 (iii). (C) Clinical photographs of patient 2 showing subtle facial stigmata including high forehead and supraorbital ridge.
Clinical vignettes
Patient 1, a 53‐year‐old woman of Austrian descent, was born following a normal pregnancy and delivery. The postnatal period was uneventful. Although some mild developmental delay concerns were documented, there were no gross abnormalities in infancy and early childhood. By age 10 years, she began to manifest action‐dependent involuntary cramps in her right hand leading to writing difficulties. In the next few years, abnormal muscle contractions spread progressively involving the neck, trunk, and both upper limbs, with generalization of symptoms by the age of 15 years. She was diagnosed with limb‐onset childhood dystonia with no identifiable cause on routine diagnostic investigations. She first presented to us at the age of 48 years with complaints of increasingly painful generalized muscle spasms and articulation deficits. On examination, she displayed generalized dystonia including oromandibular dystonia with dysarthric speech, cervical dystonia with left‐sided torticollis and fluctuating ante‐ and retrocollis, upper‐limb dystonic movements, both‐sided writer`s cramp, truncal dystonia, and dystonic foot posturing. She scored 42/120 points in the Burke‐Fahn‐Marsden Dystonia Rating Scale (BFMDRS). On assessment of cerebellar signs, dysdiadochokinesia and difficulties in tandem walk were observed but no dysmetria, impairments in smooth pursuit and saccades, or tremor; electro‐nystagmic recordings showed intermittent spontaneous downbeat nystagmus. The remainder of the neurological examination was normal. Brain magnetic resonance imaging (MRI) showed flattened caudate nuclei and cerebellar vermis atrophy (Fig. 1B). A neuropsychological examination disclosed mild intellectual impairment. Deficits in verbal fluency and visual working memory were also observed. Dystonic symptoms did not respond to levodopa, trihexyphenidyl, and tetrabenazine. At the age of 50 years, deep brain stimulation (DBS) surgery was performed (Fig. 1B). DBS settings were programmed in bipolar stimulation mode with three active contacts per side: C0 = negative, C1 and C2 = positive; the pulse width was set to 120 μs and the frequency to 130 Hz on both sides; the amplitude was self‐adjusted by the patient at around 5.0 V. Particular improvements were seen for neck and truncal dystonia, with an overall improvement of dystonic features of ~30%. During a 3‐year follow‐up period, DBS has remained effective, and the patient experienced significant worsening of dystonia when turning off the stimulation (Video S1). Patient 1’s family history was unremarkable with regard to movement disorders. Her parents were neurologically normal and she had a healthy daughter. Her two sons, aged 27 and 29 years, had a history of delayed psychomotor development and were reported to manifest gait instability and poor fine motor task performance. On examinations by movement‐disorder neurologists, none of the sons had signs of dystonia or other extrapyramidal features; they had slightly ataxic gait but no other cerebellar motor deficits.
Patient 2, a 17‐year‐old girl, was the second child of non‐consanguineous parents of Polish‐French descent. Family history was unremarkable. She was born at term after an uneventful pregnancy and labor via scheduled C‐section. Birth weight was 3450 g (50th‐75th percentile), length 52 cm (90th percentile), and head circumference 39 cm (>98th percentile). During her first month of life, feeding difficulties were observed, followed by appearance of axial hypotonia (3 months). Milestone acquisition was delayed with autonomous walking by age 2 years and 7 months. She had impairments in social interactions, speech, and nonverbal communication with stereotypic behaviors. Psychometric tests showed normal intelligence (7 years old). Assessment in a French autism reference center suggested a diagnosis of autism spectrum disorder. At the age of 9 years, neck and upper‐limb dystonia with writing difficulties appeared, followed by the manifestation of gait problems with marked fatigue. She became wheelchair‐dependent by the age of 12 years because of bilateral progressive lower‐limb dystonia. Axial dystonia and scoliosis were noted since the age of 15 years. Abnormal muscle spasms progressed over the next two years, involving cranial muscles with severe dysarthria and dysphagia. Additional features were present including bilateral ankle clonus and facial dysmorphia (high forehead, supraorbital ridge, and pointed chin; Fig. 1C). Autoimmune diabetes mellitus manifested by the age of 11 years. Brain MRI at the age of 12 years showed slight cerebellar vermis atrophy and bilateral hypointense signal alterations of globus pallidus in T2*‐weighted imaging (Fig. 1B); no signal alterations were detected on T1, T2, and FLAIR sequences, while DWI and ADC sequences were not obtained. A working diagnosis of a neurodegeneration with brain iron accumulation disorder was proposed and conservative iron chelation was initiated with deferiprone (30 mg/kg/d). Under deferiprone, there was a tendency of slowed progression of disease but no sustained effect. Dystonic features in the neck, trunk, and limbs further worsened and were unresponsive to levodopa and trihexyphenidyl. At the time of her referral for DBS (17 years old), she demonstrated permanent generalized dystonia, with left tilt of the trunk, hip rotation, dystonic leg postures with bilateral hip and knee flexion, bilateral phasic upper‐limb dystonia, left laterocaput, torticaput, antecollis, and horizontal cephalic oscillations. Jaw‐closing dystonia and dysarthria were also seen. She scored 88/120 points in the motor section of the BFMDRS and 26/30 points in the disability section. There were no motor symptoms in addition to dystonia and examination of cranial nerves, sensation, and cerebellar function was normal. DBS settings were programmed in monopolar stimulation mode with two active contacts per side: right GPI: C1 and C2 = negative, left GPI: C0 and C1 = negative; the pulse width was set to 90μs and the frequency to 130Hz on both sides; the amplitude was 2.4V on both sides. At 6‐month follow‐up after DBS implantation, improvements in the phasic component and the dystonic postures of both upper limbs, her cranial dystonia, and cephalic oscillations were observed; however, DBS response was only partially reflected in a change of BFMDRS parameters (82/120 and 22/30 in motor and disability scores, respectively).
Discussion
The adoption of unbiased genomic sequencing as a routine biomedical tool has enabled the characterization of many phenotype‐gene relationships not previously described. CHD8 pathogenic variants have originally been linked to a specific subtype of autism, 1 but the ascertainment of additional patients has revealed a more complex clinical constellation encompassing neurological, neuropsychiatric, and systemic abnormalities. 2 , 3 , 4
In the present case study, we further expand the neurological phenotype associated with CHD8 variants to include childhood‐onset progressive dystonia. Although our independent index patients harboring CHD8 heterozygous loss‐of‐function variants displayed previously reported clinical features of CHD8‐associated disease (e.g., mild intellectual impairment and autistic traits), their chief complaints were neck/limb‐onset dystonic symptoms with evolution towards generalized dystonia.
Childhood dystonia is a clinically and genetically heterogeneous condition. 5 Mounting evidence suggests that mutational defects in neurodevelopmental disorder‐associated genes play an important role in the etiopathogenesis of dystonia manifesting early in life. 5 For example, NUS1 (MIM:610463) and YY1 (MIM:600013) have been associated with severe neurodevelopmental syndromes (MIM:617831 and MIM:617557, respectively), while at the same time mutations in these genes were also shown to result in generalized dystonia with only minimal neurodevelopmental comorbidity. 8 , 9 CHD8 is widely expressed in the central nervous system and involved in the regulation of hundreds of developmentally important target genes. 1 One of the best characterized CHD8 targets is beta‐catenin, 10 encoded by CTNNB1 (MIM:116806), in which mutations can also cause a neurodevelopmental disorder with dystonia (MIM:615075). 11 As such, an association between CHD8 pathogenic variants, as carried by our patients, and an expression of dystonia might well be conceivable.
The index patients presented in this study exhibited a combination of limb, truncal, and cranio‐cervical dystonia. The presentations resembled dystonic manifestations seen in THAP1‐, KMT2B‐, and VPS16‐related diseases. 5 , 6 Patient 2 also had signal hypointensities in basal ganglia on MRI similar to those observed in some patients with KMT2B‐ and VPS16‐related dystonia. 5 , 6 Our findings thus extend the differential diagnosis of early‐onset progressive dystonia and suggest that CHD8 should be considered in genetic screening panels for this indication. Notably, our two patients had undergone pallidal DBS with partial clinical response, confirming the previously reported trend that neurostimulation appears to represent an effective treatment option for many genetically determined forms of generalized dystonia. 5 , 6
To conclude, we have clinically characterized two novel families with CHD8‐related neurodevelopmental disorder and expanded the phenotypic spectrum linked to CHD8 heterozygous loss‐of‐function variation. The identification of additional cases of the CHD8‐related dystonic condition will be important to confirm our observations and provide us with the opportunity to elucidate the factors contributing to clinical variability in the context of CHD8 dysfunction.
Author Contributions
D.D. and M.T. contributed to data acquisition and analysis and drafted a significant portion of the manuscript. L.Q., D.D., J.G., C.R., G.K., M.K., D.D., L.C., J.‐B.D., and F.Z. contributed to data acquisition and analysis. D.D., L.B., and M.Z. contributed with design and conception of the study, data acquisition and analysis, and supervised the study. M.Z. drafted a significant portion of the manuscript. All authors performed a critical review of the manuscript.
Conflict of Interest
The authors declare that they have no conflict of interest.
Supporting information
Video S1. Patient 1 at the age of 53 years showing generalized dystonia with involvement of the neck (prominent retrocollis component), trunk, oromandibular region, and all four limbs.
Acknowledgment
We are grateful to the families who participated in this study.
Funding Information
MZ receives research support from the German Research Foundation (DFG 458949627; ZE 1213/2‐1).
References
- 1. Bernier R, Golzio C, Xiong B, et al. Disruptive CHD8 mutations define a subtype of autism early in development. Cell 2014;158(2):263–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Douzgou S, Liang HW, Metcalfe K, et al. The clinical presentation caused by truncating CHD8 variants. Clin Genet 2019;96(1):72–84. [DOI] [PubMed] [Google Scholar]
- 3. Ostrowski PJ, Zachariou A, Loveday C, et al. The CHD8 overgrowth syndrome: a detailed evaluation of an emerging overgrowth phenotype in 27 patients. Am J Med Genet C Semin Med Genet 2019;181(4):557–564. [DOI] [PubMed] [Google Scholar]
- 4. Sadler B, Wilborn J, Antunes L, et al. Rare and de novo coding variants in chromodomain genes in Chiari I malformation. Am J Hum Genet 2021;108(3):530–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zech M, Jech R, Boesch S, et al. Monogenic variants in dystonia: an exome‐wide sequencing study. Lancet Neurol 2020;19(11):908–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cif L, Demailly D, Lin JP, et al. KMT2B‐related disorders: expansion of the phenotypic spectrum and long‐term efficacy of deep brain stimulation. Brain 2020;143(11):3242–3261. 10.1093/brain/awaa304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17(5):405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gunzler SA, DeBrosse SD. Generalized dystonia as a prominent feature in a case of NUS1 gene mutation. Can J Neurol Sci 2021;48(3):433–434. [DOI] [PubMed] [Google Scholar]
- 9. Zorzi G, Keller Sarmiento IJ, Danti FR, et al. YY1‐related dystonia: clinical aspects and long‐term response to deep brain stimulation. Mov Disord 2021;36(6):1461–1462. 10.1002/mds.28547. [DOI] [PubMed] [Google Scholar]
- 10. Thompson BA, Tremblay V, Lin G, Bochar DA. CHD8 is an ATP‐dependent chromatin remodeling factor that regulates beta‐catenin target genes. Mol Cell Biol 2008;28(12):3894–3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pipo‐Deveza J, Fehlings D, Chitayat D, Yoon G, Sroka H, Tein I. Rationale for dopa‐responsive CTNNB1/ss‐catenin deficient dystonia. Mov Disord 2018;33(4):656–657. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1. Patient 1 at the age of 53 years showing generalized dystonia with involvement of the neck (prominent retrocollis component), trunk, oromandibular region, and all four limbs.