

Abstract
Coronavirus disease 2019 (COVID-19), the third type of coronavirus pneumonia after severe acute respiratory syndrome (SARS) and Middle East respiratory syndrome (MERS), is spreading widely worldwide now. This pneumonia causes not only respiratory symptoms but also multiple organ dysfunction, including thrombotic diseases such as ischemic stroke. The purpose of this review is to explore whether COVID-19 is a risk factor for ischemic stroke and its related pathophysiological mechanisms. Based on the high thrombosis rate and frequent strokes of COVID-19 patients, combined with related laboratory indicators and pathological results, the discussion is mainly from two aspects: nerve invasion and endothelial dysfunction. SARS-CoV-2 can directly invade the CNS through blood-borne and neuronal retrograde pathways, causing cerebrovascular diseases. In addition, the endothelial dysfunction in COVID-19 is almost certain. Cytokine storm causes thromboinflammation, and downregulation of ACE2 leads to RAS imbalance, which eventually lead to ischemic stroke.
Keywords: COVID-19, Endothelial dysfunction, Ischemic stroke, SARS-CoV-2, Thrombosis
Coronavirus disease 2019 (COVID-19) is a severe acute respiratory infection caused by severe acute respiratory syndrome coronavirus 2(SARS-CoV-2). The pneumonia cases first appeared in Wuhan in December, 2019. Subsequently, the epidemic spread rapidly, with more than 100 million confirmed cases and more than one million deaths worldwide by March 4, 2021. At present, the world is still in the pandemic of COVID-19. In a systematic review of the literature on the neurological manifestations of patients with SARS-CoV-2, up to 21.3% of COVID-19 patients have neurological symptoms [1]. Headache, skeletal muscle injury, mental disorders, disturbance of consciousness, taste/olfactory dysfunction, acute cerebrovascular events, and dizziness are the most commonly reported neurological manifestations [1]. 1.3% of COVID-19 patients have ischemic stroke, and the incidence of hemorrhagic stroke is 0.15% [1]. Other studies have also reported that patients with COVID-19 have similar neurological symptoms [2, 3]. It is worth noting that cerebrovascular events occur more frequently than other neurological symptoms. The ischemic stroke that occurred in COVID-19 patients do not seem to be accidental. Therefore, we collected and compiled a number of articles about COVID-19-associated ischemic stroke, aiming to explore whether COVID-19 is a risk factor for ischemic stroke and its related mechanisms, and to provide clinical workers and researchers with inspirations for COVID-19 associated treatments and researches.
Thrombotic events are the main complications of COVID-19
The rates of ischemic stroke
The rate of thrombosis in COVID-19 patients is generally high, ischemic stroke accounts for most of them. Lodigian studied the rates of venous and arterial thromboembolic complications in 388 COVID-19 patients, of which 21% had thromboembolic events, and the rates of ischemic stroke and acute coronary syndrome (ACS)/myocardial infarction (MI) were 2.5% and 1.1%, respectively [4]. The rate of thrombotic complications in ICU patients infected with COVID-19 was as high as 31%. [5] Similarly, among 73 patients with COVID-19, 43 patients had pathological MRI findings (58.9%) within 2 to 4 weeks after the onset of symptoms (58.9%), including 17 cases of acute ischemic infarction (23.3%), one case of deep vein thrombosis (1.4%). [6] Additionally, Merkler compared the rate of ischemic stroke between patients with COVID-19 and patients with influenza and found that approximately 1.6% of adults with COVID-19 who visited the emergency department or were hospitalized experienced ischemic stroke, a higher rate of stroke compared with a cohort of patients with influenza (0.2%). [7] Furthermore, a retrospective study demonstrated that COVID-19 is an independent risk factor for stroke in hospitalized patients and mortality, in that ischemic stroke (83.7%) and nonfocal neurological presentations (67.4%) predominated, commonly involving multivascular distributions (45.8%) with associated hemorrhage (20.8%). [8] Interestingly, stroke is also uncommon among young patients with COVID-19. It was reported that five patients under the age of 50 who were diagnosed with SARS-CoV-2 had large-vessel stroke, and the youngest two patients among them who had not medical history were 33 and 37 years old. [9] Aortic occlusion in COVID-19 may be mainly due to cardiogenic embolism or paradoxical embolism, and less frequently due to aortic atherosclerosis and plaque rupture, which explains the occurrence of stroke in young people without vascular risk factors. [10] All of the above support the fact of ischemic stroke in COVID-19, indicating that COVID-19 patients have coagulation dysfunction.
Related laboratory indicators and pathological findings
The markers of coagulation and inflammation of COVID-19 patients are usually high, especially in critical patients. d-dimer, fibrinogen, and interleukin-6(IL-6) are generally elevated. [11, 12] Similarly, other studies also found that coagulation and inflammation were common in COVID-19 patients. [13–16] In some cases of COVID-19-associated ischemic stroke, inflammation and coagulation indicators are frequently elevated. [17] It was reported that 6 patients with ischemic stroke had great vessel occlusion, with significantly increased levels of d-dimer (≥ 1000 μg/L), decreased albumin, and increased LDH. [18] Similarly, among 4 cases of COVID-19 patients with ischemic stroke, 2 patients were considered to have high causality: they had cortical infarction, not obvious cardiac embolism or arterial disease, and systemic inflammation and hypercoagulability parameters; the other 2 patients were elderly patients, and ischemic stroke considered to be heart embolism may be related to COVID-19. [19] Four patients had high levels of LDH and CRP, and two patients had high levels of serum ferritin, d-dimer and fibrinogen. [19] In addition to coagulation and inflammation indicators, the pathological study of COVID-19 also found evidences of endothelial damage and cytokine storms.
A pathological study described the pathological characteristics of the lungs of patients who died of COVID-19, with extensive thrombosis with microvascular disease. [20] Compared with patients who died of influenza, the lungs from COVID-19 patients showed unique vascular characteristics, including severe endothelial damage related to the presence of intracellular viruses and cell membrane rupture. [20] Another pathological study indicated the presence of viral elements within endothelial cells and an accumulation of inflammatory cells, with evidence of endothelial and inflammatory cell death. [21] These findings suggest that SARS-CoV-2 infection facilitates the induction of endotheliitis in several organs as a direct consequence of viral involvement (as noted with presence of viral bodies) and of the host inflammatory response. [21] In addition to the pathological evidences of patients, the mouse models of COVID-19 are also under continuous development and provide many findings. Jiang successfully developed a model of SARS-CoV-2 infection in hACE2 transgenic mice, and their results provided strong evidences that SARS-CoV-2 could cause typical interstitial pneumonia in hACE2 mice, and progeny viruses could be re-isolated from diseased animal tissues. [22] Furthermore, other researchers also provided similar results that interstitial pneumonia and increased cytokines were found in hACE2 mice infected SARS-CoV-2. [23, 24] Therefore, combined laboratory indicators with pathological results, endothelial dysfunction is likely to be the main cause of thrombosis caused by COVID-19. Considering that the most common stroke mechanism of acute ischemic stroke is cryptogenic, followed by cardiogenic embolism and large vessel atherosclerosis. [10] Studies shown that the radiological characteristics of ischemic stroke in COVID-19 are large artery occlusion and multi-arterial area involvement. [10] Therefore, in situ thrombus formed in other organs is likely to enter the cerebrovascular through blood circulation and cause embolism.
Does SARS-CoV-2 invade the central nervous system?
Whether SARS-CoV-2 is present in the cerebrospinal fluid (CSF) of COVID-19 patients with neurological symptoms is always a controversial topic. SARS-CoV-2 was not found in the CSF of two COVID-19 patients with severe neurological symptoms. [25] CSF analysis of all COVID-19 patients with acute neurological symptoms who were tested in a study was negative for SARS-CoV-2 [6]. Moriguchi reported a case of meningitis associated with SARS-CoV-2. [26] Specific SARS-CoV-2 RNA was not detected in nasopharyngeal swabs, but was detected in the CSF. [26] In a case of COVID-19-related acute necrotizing encephalopathy, SARS-CoV-2 RNA was found in the CSF. [27] If SARS-CoV-2 is found to bind to neurons or cerebral vascular endothelial cells, it will directly indicate that COVID-19 can affect the nervous system and cause encephalitis, cerebral thrombosis, and other diseases. Two main pathways for virus to enter the central nervous system (CNS) were proposed, namely, blood-borne and neuronal retrograde pathways. [28–38] In the neuronal retrograde pathway, the virus undergoes retrograde axon transport to reach the neuronal cell bodies of the surrounding and/or central nervous system. In the blood-borne pathway, the virus can enter the CNS by infecting endothelial cells of the blood–brain barrier (BBB), epithelial cells of the blood-cerebrospinal fluid barrier in the choroid plexus or using inflammatory cells for concealment. [39] According to the existing relevant literature research, these two approaches are possible.
Wrapp’s research provided biophysical and structural evidences that the S protein of SARS-CoV-2 binds angiotensin converting enzyme 2(ACE2) with a higher affinity than SARS-CoV. [40] Human angiotensin converting enzyme 2 (hACE2) is a transmembrane carboxypeptidase that contains a highly glycosylated N-terminal extracellular domain, which contains the active site of the enzyme, a hydrophobic transmembrane domain and a short intracellular C-terminal tail.41 ACE2 receptors are ubiquitously and widely expressed in the heart, blood vessels, intestines, lungs (especially type 2 alveolar epithelial cells and macrophages), kidneys, testes, and brain. [42] The extensive expression of ACE2 on endothelial cells determines that the endothelium may be the main target organ of SARS-CoV-2. Related pathological studies also found inclusion bodies of SARS-CoV-2 in endothelial cells. [21] Importantly, ACE2 may play a key role in the neural invasion of SARS-CoV-2 because it is expressed on neurons and glial cells in the brain, especially in the brain stem and cardiovascular regulatory areas. [43] In addition, ACE2 is expressed in the endothelium of cerebral blood vessels, which may cause endothelial damage and subsequently cause the virus to enter the brain. [43] A pathological study reported that the virus was detected in nerve cells and capillary endothelial cells in the frontal lobe tissues of patients with SARS-CoV-2. [39] Ultrastructural analysis of the tissue in this case showed the presence of virus-like particles in the brain and capillary endothelium, which was further confirmed by molecular detection of SARS-CoV-2. [39] In addition, the study recorded the exudation of virus-like particles into/out of the endothelial wall, indicating that active pathogens entered the neural environment by entering-transporting(cross-cell penetration) brain microvascular endothelial cells. [39] This seems to better explain the pathological mechanism of cerebrovascular disease caused by COVID-19: the endothelial bed and blood-borne pathway are likely to be the pathways for SARS-CoV-2 to reach the brain. In another pathology study, SARS-CoV-2 was detected at low levels in six brain slices obtained from five COVID-19 patients. [44] Furthermore, in three studies of hACE2 mice infected with SARS-CoV-2, brain infections were observed in mouse brain tissues. [22–24] One of them only observed brain infections in dead mice, indicating that neural invasion is sporadic in this model and likely represents the cause of death. [22] Another immunostaining of brain sections of infected hACE2 mice showed that strong expression of viral S protein was detected in neurons, astrocytes and microglia. [23] Interestingly, a study used SARS-CoV-2 spike pseudovirus and live virus to challenge brain organoids, but almost no neurons or glial cells were infected; the infected cells were the apolipoproteins of the choroid plexus epithelial barrier and ACE2 expressing cells. [45] It indicated that infection with SARS-CoV-2 can damage the choroid plexus epithelium, leading to the leakage of this important barrier, allowing pathogens, immune cells, and cytokines to enter the CSF and brain. [45] In fact, the rapid release of pro-inflammatory cytokines during the cytokine storm may cause an increase of the inflammatory response and the destruction of the BBB. [28] Studies showed that the surge of pro-inflammatory cytokines can cause focal damage to the BBB, followed by edema and necrosis. [27] The presence of SARS-CoV-2 in the general circulation can make it enter the cerebral circulation. Because ACE2 is expressed on neurons and cerebral vascular endothelial cells, SARS-CoV-2 may invade neuronal cell membranes through ACE2 to cause brain damage and neurological symptoms, and attack cerebral vascular endothelial cells to destroy the BBB. [38] The damaged BBB also promote its invasion of brain tissue and neurons.
In addition to the aforementioned pathways, SARS-CoV-2 may also spread from the olfactory epithelium along the olfactory nerve to the olfactory bulb in the CNS through the transcription pathway, or use the mechanisms of endocytosis or exocytosis and the rapid axonal transport mechanism of vesicle transport, which propagates retrogradely through the transsynaptic transfer along the microtubules back to the neuron cell body. [34] The dysfunction of smell and taste in COVID-19 patients is a direct evidence. [1, 3, 46–48] A study showed that half of COVID-19 patients in Europe had anosmia, [46] and may not be related to nasal congestion or inflammation. [48] It might mean nerve invasion through the olfactory bulb, because the retrograde transport of HCoV from the nasal epithelium to the olfactory nerve and central nervous system has been confirmed in mouse models. [49] Sequencing studies found that mice, non-human primates, and human olfactory mucosa express two key genes involved in the entry of SARS-CoV-2, ACE2 and transmembrane protease serine 2 (TMPRSS2). [50] However, the study revealed the expression of ACE2 protein in mouse dorsal olfactory epithelial sertoli cells and olfactory bulb pericytes, but not in neurons, and immunostaining confirmed these results. [50] These findings indicated that SARS-CoV-2 infection of non-neuronal cell types causes loss of smell and related odor perception disorders in COVID-19 patients. Someone summarized the researches related to other coronaviruses invading the CNS. In transgenic mice, SARS-CoV and MERS-CoV entered the brain through the olfactory nerve and quickly spread to the brainstem and thalamus. [37, 38, 51] Perhaps, we can be inspired by similar studies on other coronaviruses. More future researches need to understand the pathophysiological mechanisms of the loss of smell and taste in COVID-19, including potential virus transmission through olfactory neuroepithelial cells and invasion of the olfactory bulb and CNS.
Endothelial dysfunction: a collision of thromboinflammation, cytokine storm, and ACE2 downregulation
The mechanism of thromboinflammation
The concept of thromboinflammation
More and more evidences indicate that the cause of COVID-19 pneumonia is both coagulative and inflammatory, because the two pathways are closely related, which are characterized by high levels of procoagulant factors including d-dimer and inflammatory factors including IL-6. In an in vitro study, the addition of fibrin markedly increased the secretion of cytokines and chemokines by human peripheral mononuclear blood cells (PBMCs) in a dose-dependent manner. [52] It confirmed the link between coagulation and inflammatory response of the immune system. Ranucci’s study showed that IL-6 levels of all COVID-19 patients were elevated, and proved that there was a clear correlation between IL-6 and fibrinogen. [53] Furthermore, the coagulation parameters of COVID-19 patients are extensively and closely related to liver function and inflammation indicators in a retrospective study, demonstrating that the liver injury and inflammatory storm may cause coagulation dysfunction of these patients. [14] Regarding the link between inflammation and coagulation, someone proposed the concept of thromboinflammation that platelets, coagulation factors, and innate immune effector systems (such as macrophages, neutrophils, and the complement system) interact to form a clot during the infection process. [54] The center of thromboinflammation is the loss of the normal antithrombotic and anti-inflammatory functions of endothelial cells, which leads to the disorders of coagulation, complement, platelet activation, and leukocyte recruitment in microvessels. [55] The endothelium, a key regulator of thrombotic inflammation, covers the entire internal cavity of the circulatory system, from the heart chamber to the microcapillary bed. [55] When some factors cause endothelial dysfunction, microcirculation disorders are prone to occur in the body. In addition to the pathological tissues of the lungs from COVID-19 patients mentioned above that provided the evidences of endothelial cell infection and endotheliitis, it was also found that COVID-19 patients had elevated markers of endothelial damage in several studies. In a retrospective cohort study, the plasma levels of soluble fumes-like tyrosine kinase 1 (sFlt-1), which has been demonstrated to promote endothelial dysfunction, correlated with respiratory symptoms severity, expression of endothelial dysfunction biomarker and incidence of organ failure in COVID-19 patients. [56] Besides, endothelial disease appeared during SARS-CoV-2 infection, as evidenced by the increased level of circulating endothelial cells (CEC) considered to be related markers of endothelial disease or dysfunction. [57] Therefore, it is reasonable to speculate that thromboinflammation promotes endothelial damage. The specific mechanism of thromboinflammation needs further discussion.
The cytokine storm in COVID-19
After SARS-CoV-2 infects endothelial cells, it produces a high-inflammatory state that promotes endothelial cell activation and endothelial dysfunction, which may induce a prethrombotic state. Recently, the cytokine storm receives frequent attentions in COVID-19. SARS-CoV-2 can activate mast cells and other innate immune cells, and extensive mast cell activation leads to an increase in the release of inflammatory cytokines and chemokines, which further aggravates inflammation and increases the severity of the disease. [58] IL-6 is one of the frequently elevated inflammatory indicators in COVID-19 patients and is associated with a poor prognosis. Interestingly, the key role of IL-6 in the cytokine storm and endothelial cell damage was mentioned in some reviews. [41, 59–61] IL-6 is a major highly inducible pro-inflammatory cytokine, secreted by several different types of cells, including monocytes, lymphocytes, fibroblasts, and ECs. IL-6 can promote macrophage activation syndrome (MAS) and cause harmful endothelial cell activation, trigger the mass production of pro-inflammatory cytokines, and induce the migration of neutrophils and fibroblasts to the lung epithelium. [41, 59–61] These increase the deposition of collagen and fibrin, resulting in damage to the lower lung tissue. The IL-6 receptor antibody, tocilizumab, is also used clinically to treat the cytokine storm of COVID-19. Therefore, inflammatory cytokines, mainly IL-6, participate in the formation of cytokine storm in COVID-19. Furthermore, Wang confirmed that SARS-CoV-2 infected macrophages and determined the expression of ACE2 which is the receptor of SARS-CoV-2 on the surface of macrophages. [62] Their findings emphasize the role of macrophages as the direct host cells of SARS-CoV-2 and the potential driving factors of “cytokine storm syndrome” in COVID-19, and support the effects of anti-IL-6/IL-6R antibody that activate alveolar macrophages and inhibit inflammatory damage in patients with COVID-19. [62] In the context that the inflammatory response of the lungs of K18-hACE2 mice to SARS-CoV-2, many cytokines and chemokines were induced, some of which continued to express, while others displayed a fast up and down adjustment mode. [24] Harky proposed that the downstream effects of inflammatory cytokines can be roughly divided into two sequelae. [63] First, the production of procoagulant factors, and second, the damage to the capillary endothelium leads to the imbalance of its antithrombotic properties. [63] Both of them can lead to microvascular thrombosis, which may result in systemic embolism.
Mechanism of triggering tissue factor coagulation pathway
Endothelial cell damage can strongly activate the coagulation system by exposing tissue factor (TF) and other means. [64] TF activates the tissue factor (“exogenous”) pathway and is amplified by the previously known “intrinsic” pathway (now called “thrombin burst”), and finally converges to activate the “common” pathway. [54] TF triggers coagulation and becomes a cofactor for coagulation factor VIIa (FVIIa) to mediate the production of FXa. [65] Factor Va (FVa) and factor Xa (FXa) convert prothrombin into thrombin, and then thrombin converts fibrinogen into fibrin and stabilizes it in the blood clot through factor XIIIa (FXIIIa) activity. [54] TF can also function from the nascent TF/FVIIa/FXa complex, thereby triggering profound changes in many types of cells (including vegetative cells of SARS-CoV-2) through protease-activated receptors (PARs). [65] Therefore, the upregulation of TF is coupled with FVII (TF-Factor Vila complex) activated by the external coagulation pathway, and is related to thrombin generation and fibrin deposition in various organs including the bronchoalveolar system. [66] On the other hand, overactive fibrinolysis leads to increased plasma concentrations of plasminogen and plasmin. [66] The increased activity of plasmin cleavage can explain the extreme increase of d-dimer, which has a linear relationship with the severity of the disease. [66] These mechanisms can explain the pathological findings of lung tissues. In addition, some studies show that TF-dependent coagulation activation is a part of the innate immune response to viral infections, which helps prevent intrapulmonary hemorrhage. [67] However, the complication of this reaction is thrombosis. In general, the activation of tissue factor pathway through the action of multiple inflammatory mediators promotes the occurrence of coagulation and thrombosis, and TF is at the central link of inflammation and coagulation.
The activation of complement pathway
In addition to activation of the tissue factor coagulation pathway, there seems to be some new evidences for the activation of complement pathway. Magro found that the lungs of patients with severe COVID-19 were accompanied by significant deposits of terminal complement components C5b-9 (membrane attack complex), C4d, and mannose binding lectin (MBL)-associated serine protease (MASP)2, in the microvasculature, consistent with sustained, systemic activation of the alternative and lectin-based complement pathways. [68] The complement pathway is the basis of innate host immunity and plays a key role in the defense against pathogens. After infection, C5a stimulates inflammation, microvascular thrombosis, fibrinolysis, and innate immune response. [69] C5a activates neutrophils and mast cells to release pro-inflammatory cytokines, such as IL-12 and TNF, and also activates macrophages and endothelial cells, thereby promoting vascular leakage and the release of enzymes and oxidants. [69] Besides, studies reported that von Willebrand factor (vWF) was elevated in COVID-19 patients. [13, 70] It is a multimeric glycoprotein that exists in plasma and subendothelial matrix, and is believed to have a mechanism that promotes the connection of red blood cells. [71] In fact, vWF can be regarded as a marker of endothelial injury, because it is usually stored in Weibel-Palade of endothelial cells. The accumulation of platelets and vWF in microvessels is a unified step of endothelial cell activation, impaired vascular integrity, leukocyte recruitment, transendothelial migration, tissue infiltration, and target organ damage. [72]
The mechanism of inflammation triggering coagulation is complex and mediated by multiple pathways. The endothelium is the hub of inflammation and coagulation, and it plays an important role in regulating inflammatory mediators and coagulation factors. SARS-CoV-2 triggers cytokine storm in the body after infection, and the endothelium located in the microcirculation becomes dysfunctional under stimulation, which activates a variety of coagulation mechanisms including tissue factor pathways, and promotes microcirculation thrombosis. When thrombosis occurs in the cerebral circulation, it can cause ischemic stroke. However, more evidences are needed to prove that the thromboinflammation in COVID-19 causes ischemic stroke.
The immune mechanisms of thrombocytopenia and vaccine-induced thrombocytopenia
Under inflammatory conditions, the crosstalk between platelets, coagulation system, and endothelium can be disturbed. Inflammation and/or procoagulant mediators in COVID-19 patients may contribute to platelet activation. [32] Interestingly, immune thrombocytopenia, commonly referred to as immune thrombocytopenic purpura (ITP), has become an important complication of COVID-19. [73–79] Low platelet count was associated with an increased risk of serious illness and death in COVID-19 patients. [74, 78] Another study showed that platelet count was an independent risk factor related to in-hospital mortality and was dose-dependent. [79] The causal relationship between SARS-CoV-2 infection and ITP has been determined. [77] The mechanisms of thrombocytopenia patients with COVID-19 may be multifactorial. The virus directly infects bone marrow cells and inhibits platelet synthesis or the cytokine storm after virus infection destroys bone marrow progenitor cells and leads to reduced platelet production. [80–83] COVID-19 may increase the levels of autoantibodies and immune complexes, causing the immune system to specifically destroy platelets. [81, 83] There was a case of immune thrombocytopenic purpura that indicated that COVID-19 patients with thrombocytopenia should consider immune causes. [84] In addition, antibodies produced during viral infection may specifically bind to antigens on platelets through molecular simulations, leading to increased platelet destruction. [81, 82] Extensive alveolar damage in COVID-19 can reduce the availability of effective capillary beds in the lungs, cause megakaryocytes to rupture and block platelet release, affect the release of platelets to the pulmonary circulation, and indirectly lead to a decrease in platelet synthesis in the systemic circulation. [81–83] Damaged tissue and endothelial cells may activate platelets in the lungs, leading to the formation of microthrombi aggregation, thereby increasing platelet consumption. [80–83] Antiviral drugs can also cause liver damage and affect the production of thrombopoietin (TPO), which indirectly causes thrombocytopenia. [82] Combined with the above mechanisms, thrombocytopenia may be the result of cytokine storm, bone marrow suppression, and platelet destruction caused by the uncontrolled immune response of COVID-19 patients.
Recently, sporadic cases of rare thrombotic syndrome related to thrombocytopenia have been observed in individuals receiving ChAdOx1 nCov-19 vaccine. [85–89] Thrombosis and thrombocytopenia syndrome (TTS) after vaccination is now known as vaccine-induced immune thrombotic thrombocytopenia (VITT) or vaccine-induced prethrombotic immune thrombocytopenia (VIPIT). [85] VITT is characterized by low platelet counts, thrombosis, and platelet factor 4 (PF4) antibodies. [85–89] Another distinguishing feature of this syndrome is the unusual location of thrombosis, which mainly occurs in cerebral venous sinus thrombosis or adrenal vein thrombosis. [90] In addition, there are also deep vein thrombosis and/or pulmonary embolism, or arterial thrombosis, including ischemic stroke and/or peripheral arterial occlusion. [90] It is often found that high levels of d-dimer and low levels of fibrinogen in VITT indicate systemic activation of coagulation. [87, 90, 91] The production and binding of anti-PF4 antibodies in VITT occurs without heparin [85, 87, 89, 92]. This serological feature is very similar to severe heparin-induced thrombocytopenia (HIT) [85–89]. PF4 is one of the most abundant chemokines released when platelets are activated. [93] It can induce a strong immune response to produce antibodies, activate and aggregate platelets, ultimately leading to clot formation and decreased platelet levels. [93] The pathogenesis of VITT is still uncertain. It may be that vaccination induces a strong inflammatory response, which in turn leads to the production of autoantibodies against PF4 or PF4-polyanion complex, respectively. [90, 93] Several components of the vaccine such as the adenovirus itself, the spike protein cassette, or other components of the vaccine (polysorbate 80) may cause the production of anti-PF4 antibodies. [90, 93]
Downregulation of ACE2 causes imbalance of renin-angiotensin system
The renin-angiotensin system (RAS) is a potential therapeutic target for ischemic stroke, because the excessive activation of angiotensin converting enzyme/angiotensin II/angiotensin type 1 receptor(ACE/Ang II /AT1R) in RAS is highly involved in the process of inducing brain injury after ischemia. [94] Ang II activated by AT1R plays a harmful role in stroke and other cardiovascular diseases, but more and more evidences demonstrate that the angiotensin converting enzyme 2/angiotensin 1–7/Mas receptor (ACE2/Ang(1–7)/Mas R) axis plays a beneficial role in several cardiovascular diseases. [94] A mouse model proved that ACE2 protected the brain from ischemic damage by regulating the Ang(1–7)/Ang II ratio and the NADPH oxidase/Endothelial Nitric Oxide Synthases (eNOS) pathway. [95] Therefore, ACE2 plays an important role in maintaining cardiovascular and cerebrovascular homeostasis.
SARS-CoV-2 enters the cell through membrane fusion, leading to a significant downregulation of the ACE2 receptor and the loss of the catalytic effect of the ACE2 receptor on the outside of the membrane. [42] The mechanism is similar to SARS-CoV. The entry of SARS-CoV-2 into cells is relevant to the internalization of ACE2 and the reduction of viral particles and ACE2 on the cell. [41] Because the enzymatic activity of ACE2 is located in the N-terminal extracellular domain exposed to circulating vasoactive peptides, ACE2 internalization is relevant to the increased balance of cell-related vasoactive mediators to Ang II, which triggers vasoconstriction, inflammation and release of Ang II. [41] After internalization, ACE2 will undergo Ang II-mediated ubiquitination and degradation. [41] In a mouse model, the level of hACE2 in the lung decreased during infection, indicating that the receptor was downregulated after SARS-CoV-2 infection, hACE2 fell off, or cells expressing hACE2 died, which further confirmed the mechanism of ACE downregulation. [24] ACE2 can play a protective role in the brain by balancing the ACE/Ang II/AT1R axis. Disturbance of balance may change the level of Ang II and lead to the development of hypertension and other cardiovascular diseases. [96] Therefore, when SARS-CoV-2 infection causes the downregulation of ACE2 expression, it will lead to an increase in Ang II. It was reflected in recent studies. The plasma level of Ang II of COVID-19 pneumonia patients was significantly higher than that of healthy controls, and the Ang II level of COVID-19 patients was related to viral load and lung injury. [97] In a prospective study, soluble E-selectin and Ang II significantly increased in 40 patients with COVID-19. [98] There is a strong relationship between Ang II and CRP, creatinine and d-dimer in plasma, and it is determined that Ang II is a relevant predictor of direct ICU admission for COVID-19 patients. [98] The two studies confirmed the hypothesis that endothelial activation enhances the microvascular dysfunction associated with COVID-19, and the increase in Ang II seems to be connected with the downregulation of ACE2. Additionally, Giardini’ s data offer a link between ACE2 downregulation and an Ang II/s-Flt-1 mediated ED, a model that strictly resembles preeclampsia. [99]
In different experimental and clinical models, Ang II triggers a variety of important adverse reactions, including cardiac hypertrophy and dysfunction, interstitial fibrosis, endothelial dysfunction, increased inflammation, obesity-related hypertension, oxidative stress, and increased coagulation. [42] In the current SARS-CoV-2 pandemic and related lung inflammation, Ang II also interferes with adaptive immunity by activating macrophages and other immune cells, thereby increasing the production of IL-6, TNF-α, and other inflammatory cytokines. [42] It is worth noting that Ang II can increase the permeability of capillaries, induce the transcription and expression of tissue factor on endothelial cells, and activate platelets. [54, 100, 101] In addition, Ang II can also trigger endothelial cells to release certain components of the complement system, further confirming the key role of endothelium in the pathogenesis of venous and arterial thrombosis in patients with COVID-19. [54, 100, 101] Ang (1–7) acts by binding to Mas receptors to counteract the pro-inflammatory, procoagulant and vasoconstrictive effects of Ang II. Sumners pointed out that the brain protection of Ang (1–7) may have anti-inflammatory effects, which may be caused by interrupting the excessive activation of microglia that occurs during stroke. [94]
In summary, ACE converts angiotensin I into angiotensin II, which binds to angiotensin II type 1 receptor (AT1R) to induce vasoconstriction and increase blood pressure. ACE2 indirectly and directly catalyzes the conversion of angiotensin I and angiotensin II to angiotensin 1–7; the latter interacts with the G protein-coupled receptor Mas to antagonize angiotensin II and weaken the effects of angiotensin II, such as vasoconstriction, pro-inflammatory, pro-apoptosis, pro-thrombosis, mitogenic, metabolic, and other vascular effects. The high-affinity combination of SARS-CoV-2 and ACE2 reduces the expression of ACE2 on the cell surface, which in turn affects the balance of ACE2/Ang(1–7)/Mas R axis in RAS, which may eventually lead to cerebrovascular diseases, such as ischemic stroke (Fig. 1). The downregulation of ACE2 and the overexpression of tissue factor in thromboinflammation are both mechanisms that lead to endothelial dysfunction (Fig. 2). Eventually, it develops into different outcomes due to the severity of the disease and physical differences, and ischemic stroke is one of them.
Fig. 1.

The balance mechanism of the two axes in RAS. ① is ACE/Ang II/AT1R axis; ② is ACE2/Ang (1–7)/Mas R axis. The combination of SARS-CoV-2 and ACE2 downregulates ACE2, which leads to the balance in RAS that tends to ACE/Ang II/AT1R axis, and develops toward the direction of cardiovascular and cerebrovascular damage
Fig. 2.
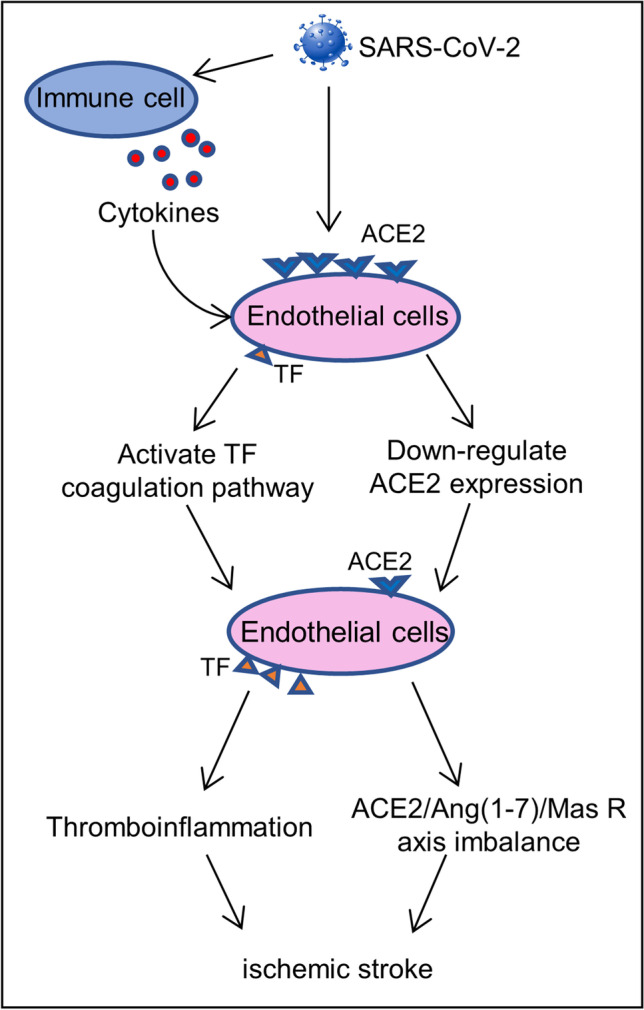
Main mechanisms of endothelial dysfunction. After SARS-CoV-2 infection, it causes the cytokine storm that produces a large number of inflammatory factors to promote the overexpression of TF on endothelial cells. The combination of SARS-CoV-2 and ACE2 leads to downregulation of ACE2 that increases the level of Ang II, and its effect contributes to the pathophysiology of ischemic stroke. The downregulation of ACE2 and the overexpression of TF on endothelial cells collectively cause endothelial dysfunction
Influences of other factors (age, gender, underlying diseases, and hypoxia)
COVID-19 patients with ischemic stroke are usually older, and some have underlying diseases. Thromboinflammation and ACE downregulation are the main mechanisms leading to endothelial dysfunction, but external factors also determine the severity and final outcome of the disease.
COVID-19 patients complicated by ischemic stroke are mostly male, and most of them are over 50 years old, up to 80 years old. [18, 19] A retrospective cohort study identified several risk factors for death in Wuhan adults hospitalized with COVID-19, and advanced age was one of them. [11] Among the 219 COVID-19 patients, COVID-19 patients with cerebrovascular disease are significantly older, are more likely to develop into severe COVID-19 and may have cardiovascular risk factors, including a history of hypertension and diabetes. [102] In a case study of SARS-CoV-2 infection, there were more male patients than females. In addition, about half of infected patients suffered from chronic underlying diseases, mainly cardiovascular and cerebrovascular diseases and diabetes. [103] Diabetic patients with severe COVID-19 are older, have a higher mortality rate, and usually show severe inflammation. [104] Therefore, age, gender, and underlying diseases are also important factors affecting the complications and prognosis of COVID-19. It is reasonable to speculate that advanced age, men, and underlying diseases are also involved in the pathological mechanism of COVID-19. This review discusses that endothelial dysfunction is the main pathological mechanism of COVID-19. Correspondingly, the difference in the expression of ACE2 on the endothelium due to age, gender, and underlying diseases may be the main reason for the poor prognosis of aged and male patients with COVID-19. The expression of ACE2 in the lungs and the viral load of SARS-CoV-2 are thought to increase with age, which may explain the higher severity of the disease in elderly patients with COVID-19. [43] Similar to influenza and other respiratory viral infections, it is expected that the gradual decline in innate and adaptive immune responses may play an important role in the increase of age-related susceptibility. [43] Aging is accompanied by structural and functional modifications of the vasculature, which ultimately results in dysfunction of endothelial and smooth muscle cells. [105] Studies shown that the expression of ACE2 in men is nearly three times that of women. In addition, the reduced sensitivity of women to viral infections may be attributed to the protection of X chromosomes and sex hormones. [103] Relevant mouse experiments showed more evidences. In the mouse experimental model of COVID-19, it was found that the mortality rate of male mice was higher than that of females, [22] and the disease of elderly mice was more serious. [106] More severe pathological changes and increased cytokine responses were also observed in aged hACE2 mice. [23] In addition, an experimental study in mice indicated that ACE2 expression increased in the ischemic brain and blood vessels exposed to diabetes or smoking, making it susceptible to COVID-19. [107]
Histopathological examination of brain specimens from patients who died of COVID-19 showed hypoxic changes. [44] Some reviews pointed out that hypoxic blood diseases and abnormal iron metabolism should also be considered in COVID-19, in addition to the classic lung immune inflammation. [108] Li explained a case in which a right limb weakness was diagnosed as COVID-19. [109] It is hypothesized that SARS-CoV-2 infection will cause hypoxemia and excessive secretion of inflammatory cytokines, thereby inducing acute ischemic stroke. [109] Hypoxemia triggers increased expression of hypoxia-inducible factor (HIF), which may promote further inflammation, including inflammatory cell infiltration and cytokine release. [54, 70] This leads to further tissue ischemia, increased blood viscosity, and worsening hypercoagulability.
In conclusion, advanced age, male, underlying diseases, and hypoxia can all exacerbate endothelial dysfunction in COVID-19. Therefore, they are also potential factors for COVID-19 to cause ischemic stroke.
Cerebral hemorrhage in COVID-19 is another outcome of endotheliitis and coagulopathy
Compared with ischemic stroke, there were fewer hemorrhagic strokes associated with COVID-19. [10, 17, 102, 110–116] Although the pathological mechanism of ischemic stroke in COVID-19 is the focus of this review, cerebral hemorrhage events cannot be ignored. COVID-19 patients with hemorrhagic stroke had higher levels of ferritin, and their characteristic radiological patterns were subarachnoid hemorrhage, parietooccipital leukoencephalopathy, microhemorrhage, and single or multiple focal hematomas. [117] The brain biopsy performed showed signs of thrombotic microangiopathy and endothelial damage, and there was no evidence of vasculitis or necrotizing encephalitis. [117] In the 5 cases of cerebral hemorrhage mainly related to COVID-19, the author proposed that endothelial injury is the main mechanism of cerebral hemorrhage. Microvascular thrombosis is caused by direct invasion of endothelial cells and a combination of systemic factors including the production of pro-thrombotic factors and inflammatory cytokines, activation of the coagulation cascade, and complement-mediated. [118] In addition, diffuse intravascular coagulation (DIC) associated with fibrinogen depletion may also increase the risk of cerebral hemorrhage. [10] Accompanying the immune response to SARS-CoV-2 infection, the massive release of cytokines and proteases may damage the integrity of the BBB. [10, 119] In addition to cerebral hemorrhage, the breakdown of the BBB can also explain the hemorrhagic transformation of ischemic stroke. [10] It is worth noting that some patients with thrombocytopenia have intracranial hemorrhage. [80] Thrombocytopenia is also a potential risk of cerebral hemorrhage.
Moreover, the affinity of SARS-CoV-2 to the ACE2 receptor may cause the virus to directly damage the intracranial arteries and cause the vascular wall to rupture. The mechanism of downregulation of RAS has been introduced above. It may also increase blood pressure and increase the risk of hemorrhagic stroke in patients who have been diagnosed with high blood pressure. [10] Older people affected by age-related ACE2 deficiency may be particularly vulnerable to hemorrhagic brain injury in this situation. [10]
Necessary preventive and therapeutic measures
Early use of drugs to curb the development of inflammation in COVID-19 is necessary. A retrospective study shown that the implementation of early home treatment algorithms failed to accelerate the recovery of the main symptoms of COVID-19, but reduced the risk of hospitalization and related treatment costs. [120] There are increasing evidences that the excessive inflammatory response in SARS-CoV-2 infection, rather than the virus itself, is the basis for the development of severe COVID-19 cases. Therefore, it is recommended to start using non-steroidal anti-inflammatory drugs to treat early COVID-19 symptoms. [120, 121] Nimesulide or celecoxib is safe and well tolerated. When the signs of toxicity or contraindications of these drugs are brought to the attention of family doctors, aspirin should be used as an alternative therapy to nimesulide and celecoxib. [120] The use of corticosteroids in COVID-19 patients is controversial. Although corticosteroids can be quickly used to target cytokine storms, their use in respiratory viral infections is associated with increased mortality, increased risk of secondary bacterial or fungal infections, and longer stay in the ICU. [121] Therefore, although it is theoretically possible to reduce the risk of cytokine storm, corticosteroids are not recommended for routine treatment of COVID-19 patients. [76] IL-6 plays a key role in the cytokine storm, and blocking IL-6 is another potential treatment strategy. Tocilizumab is an anti-IL-6 receptor monoclonal antibody, which can effectively improve the clinical symptoms of COVID-19 patients. [121] Importantly, therapies aimed at preventing platelet and endothelial cell dysfunction should also be considered. The two main natural medicines that are beneficial to both endothelium and platelets are endothelial-derived NO and prostacyclin. [122] Prostacyclin can resist thrombosis, is an effective inhibitor of platelet activation, and has anti-inflammatory effects. NO can prevent vascular diseases by promoting vasodilation, inhibiting platelet activation, and reducing white blood cell adhesion and inflammation. [122] Given that prostacyclin has a beneficial effect on platelets and endothelial cells by increasing cAMP levels, other oral drugs that increase cAMP levels in these cell types may also be potential treatment strategies for COVID-19. These include phosphodiesterase 3 inhibitors such as dipyridamole and cilostazil, which are currently used to prevent stroke and peripheral vascular disease. [122]
Conclusion
In this review, first based on the reports of high thrombosis rate and frequent ischemic stroke in COVID-19, combined with relevant laboratory indicators and pathological results, the pathophysiological mechanisms of ischemic stroke in COVID-19 are mainly discussed. SARS-CoV-2 can directly invade the CNS through blood-borne and neuronal retrograde pathways, causing cerebrovascular diseases. In addition, the endothelial dysfunction in COVID-19 is almost certain. Cytokine storm causes thromboinflammation, and downregulation of ACE2 leads to RAS imbalance, which eventually lead to systemic thrombotic microangiopathies and also cause ischemic stroke.
Author contribution
Xuhuan Tang retrieved, organized, and analyzed references; framed the content; writing—original draft preparation; visualization. Fang Zheng proposed the idea; review and revision of the first draft.
Funding
This work was supported by the grant awarded by the National Natural Science Foundation of China (Grant No. 82171761) to Fang Zheng.
Declarations
Competing interests
The authors declare no competing interests.
Footnotes
Xuhuan Tang is the first author
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Cagnazzo F, et al. Neurological manifestations of patients infected with the SARS-CoV-2: a systematic review of the literature. J Neurol. 2021;268:2656–2665. doi: 10.1007/s00415-020-10285-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Collantes MEV, Espiritu AI, Sy MCC, Anlacan VMM, Jamora RDG. Neurological manifestations in COVID-19 infection: a systematic review and meta-analysis. Can J Neurol Sci. 2021;48:66–76. doi: 10.1017/cjn.2020.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Orru G, et al. Neurological complications of COVID-19 and possible neuroinvasion pathways: a systematic review. Int J Environ Res Public Health. 2020;17:18 6688. doi: 10.3390/ijerph17186688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lodigiani C, et al. Venous and arterial thromboembolic complications in COVID-19 patients admitted to an academic hospital in Milan. Italy Thromb Res. 2020;191:9–14. doi: 10.1016/j.thromres.2020.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Klok FA, et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb Res. 2020;191:145–147. doi: 10.1016/j.thromres.2020.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chougar L, et al. Retrospective observational study of brain magnetic resonance imaging findings in patients with acute SARS-CoV-2 infection and neurological manifestations. Radiology. 2020;297(3):E313–E323. doi: 10.1148/radiol.2020202422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Merkler, A. E. et al. 2020 Risk of ischemic stroke in patients with coronavirus disease 2019 (COVID-19) vs patients with influenza. JAMA Neurol, doi:10.1001/jamaneurol.2020.273010.1001/jamaneurol.2020.2730 [DOI] [PMC free article] [PubMed]
- 8.Katz JM, et al. Cerebrovascular complications of COVID-19. Stroke. 2020;51:e227–e231. doi: 10.1161/STROKEAHA.120.031265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oxley TJ, et al. Large-vessel stroke as a presenting feature of Covid-19 in the young. N Engl J Med 382 20 e60. 2020 doi: 10.1056/NEJMc2009787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nannoni S, de Groot R, Bell S, Markus HS. Stroke in COVID-19: a systematic review and meta-analysis. Int J Stroke. 2021;16:137–149. doi: 10.1177/1747493020972922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou F, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. The Lancet. 2020;395:1054–1062. doi: 10.1016/s0140-6736(20)30566-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tang N, Li D, Wang X, Sun Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J Thromb Haemost. 2020;18:844–847. doi: 10.1111/jth.14768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Panigada M, et al. Hypercoagulability of COVID-19 patients in intensive care unit: a report of thromboelastography findings and other parameters of hemostasis. J Thromb Haemost. 2020;18:1738–1742. doi: 10.1111/jth.14850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang Y, et al. Manifestations of blood coagulation and its relation to clinical outcomes in severe COVID-19 patients: Retrospective analysis. Int J Lab Hematol. 2020 doi: 10.1111/ijlh.13273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang C, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan. China The Lancet. 2020;395:497–506. doi: 10.1016/s0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Han H, et al. Prominent changes in blood coagulation of patients with SARS-CoV-2 infection. Clinical Chemistry and Laboratory Medicine (CCLM) 2020;58:1116–1120. doi: 10.1515/cclm-2020-0188. [DOI] [PubMed] [Google Scholar]
- 17.Morassi M, et al. Stroke in patients with SARS-CoV-2 infection: case series. J Neurol. 2020;267:2185–2192. doi: 10.1007/s00415-020-09885-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beyrouti R, et al. Characteristics of ischaemic stroke associated with COVID-19. J Neurol Neurosurg Psychiatry. 2020;382(20):889–891. doi: 10.1136/jnnp-2020-323586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barrios-Lopez JM, et al. Ischaemic stroke and SARS-CoV-2 infection: a causal or incidental association? Neurologia. 2020;35:295–302. doi: 10.1016/j.nrl.2020.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ackermann M, et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid-19. N Engl J Med. 2020;383:120–128. doi: 10.1056/NEJMoa2015432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Varga Z, et al. Endothelial cell infection and endotheliitis in COVID-19. The Lancet. 2020;395:1417–1418. doi: 10.1016/s0140-6736(20)30937-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jiang RD, et al. Pathogenesis of SARS-CoV-2 in transgenic mice expressing human angiotensin-converting enzyme 2. Cell. 2020;182(50):58 e58. doi: 10.1016/j.cell.2020.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sun SH, et al. A mouse model of SARS-CoV-2 infection and pathogenesis. Cell Host Microbe. 2020;28(124):133 e124. doi: 10.1016/j.chom.2020.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Winkler, E. S. et al. 2020 SARS-CoV-2 infection in the lungs of human ACE2 transgenic mice causes severe 2 inflammation, immune cell infiltration, and compromised respiratory function. bioRxivJul 10;2020.07.09.196188., doi:doi 10.1101/2020.07.09.196188
- 25.Al Saiegh F, et al. Status of SARS-CoV-2 in cerebrospinal fluid of patients with COVID-19 and stroke. J Neurol Neurosurg Psychiatry. 2020;91:846–848. doi: 10.1136/jnnp-2020-323522. [DOI] [PubMed] [Google Scholar]
- 26.Moriguchi T, et al. A first case of meningitis/encephalitis associated with SARS-Coronavirus-2. Int J Infect Dis. 2020;94:55–58. doi: 10.1016/j.ijid.2020.03.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Virhammar J, et al. Acute necrotizing encephalopathy with SARS-CoV-2 RNA confirmed in cerebrospinal fluid. Neurology. 2020 doi: 10.1212/WNL.0000000000010250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Achar A, Ghosh C. COVID-19-associated neurological disorders: the potential route of cns invasion and blood-brain relevance. Cells. 2020;9(11):2360. doi: 10.3390/cells9112360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gasmi A, et al. Neurological involvements of SARS-CoV2 infection. Mol Neurobiol. 2021;58:944–949. doi: 10.1007/s12035-020-02070-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bougakov D, Podell K, Goldberg E. Multiple neuroinvasive pathways in COVID-19. Mol Neurobiol. 2021;58:564–575. doi: 10.1007/s12035-020-02152-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Najjar S, et al. Central nervous system complications associated with SARS-CoV-2 infection: integrative concepts of pathophysiology and case reports. J Neuroinflammation. 2020;17:231. doi: 10.1186/s12974-020-01896-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hottz ED, et al. Platelet activation and platelet-monocyte aggregate formation trigger tissue factor expression in patients with severe COVID-19. Blood Sep. 2020;10:1330–1341. doi: 10.1182/blood.2020007252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mishra R, Banerjea AC. Neurological damage by coronaviruses: a catastrophe in the queue! Front Immunol. 2020;11:565521. doi: 10.3389/fimmu.2020.565521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pennisi M, et al. SARS-CoV-2 and the nervous system: from clinical features to molecular mechanisms. Int J Mol Sci. 2020;21:15–5475. doi: 10.3390/ijms21155475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kumar A, et al. Possible routes of SARS-CoV-2 invasion in brain: in context of neurological symptoms in COVID-19 patients. J Neurosci Res. 2020;98:2376–2383. doi: 10.1002/jnr.24717. [DOI] [PubMed] [Google Scholar]
- 36.Alam SB, Willows S, Kulka M, Sandhu JK. Severe acute respiratory syndrome coronavirus 2 may be an underappreciated pathogen of the central nervous system. Eur J Neurol. 2020;27:2348–2360. doi: 10.1111/ene.14442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Finsterer J, Stollberger C. Update on the neurology of COVID-19. J Med Virol. 2020 doi: 10.1002/jmv.26000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li H, Xue Q, Xu X. Involvement of the nervous system in SARS-CoV-2 infection. Neurotox Res. 2020;38:1–7. doi: 10.1007/s12640-020-00219-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Paniz-Mondolfi A, et al. Central nervous system involvement by severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) J Med Virol. 2020;92:699–702. doi: 10.1002/jmv.25915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wrapp D, et al. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020;367(6483):1260–1263. doi: 10.1126/science.abb2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Labo N, Ohnuki H, Tosato G. Vasculopathy and Coagulopathy Associated with SARS-CoV-2 Infection. Cells. 2020;9(7):1583. doi: 10.3390/cells9071583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Verdecchia P, Cavallini C, Spanevello A, Angeli F. The pivotal link between ACE2 deficiency and SARS-CoV-2 infection. Eur J Intern Med. 2020;76:14–20. doi: 10.1016/j.ejim.2020.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bourgonje, A. R. et al. 2020 Angiotensin-converting enzyme 2 (ACE2), SARS-CoV-2 and the pathophysiology of coronavirus disease 2019 (COVID-19). J Pathol, doi:10.1002/path.547110.1002/path.5471 [DOI] [PMC free article] [PubMed]
- 44.Solomon IH, et al. Neuropathological Features of Covid-19. N Engl J Med. 2020;383(10):989–992. doi: 10.1056/NEJMc2019373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pellegrini L, et al. SARS-CoV-2 Infects the brain choroid plexus and disrupts the blood-CSF barrier in human brain organoids. Cell Stem Cell. 2020;27:951–961 e955. doi: 10.1016/j.stem.2020.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Klopfenstein T, et al. Features of anosmia in COVID-19. Med Mal Infect. 2020;50:436–439. doi: 10.1016/j.medmal.2020.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Politi LS, Salsano E, Grimaldi M. Magnetic resonance imaging alteration of the brain in a patient with coronavirus disease 2019 (COVID-19) and anosmia. JAMA Neurol. 2020;77:1028–1029. doi: 10.1001/jamaneurol.2020.2125. [DOI] [PubMed] [Google Scholar]
- 48.Lechien JR, et al. Loss of smell and taste in 2013 European patients with mild to moderate COVID-19. Ann Intern Med. 2020;173(8):672–675. doi: 10.7326/M20-2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Aghagoli G, et al. Neurological involvement in COVID-19 and potential mechanisms: a review. Neurocrit Care. 2021;34:1062–1071. doi: 10.1007/s12028-020-01049-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brann DH, et al. Non-neuronal expression of SARS-CoV-2 entry genes in the olfactory system suggests mechanisms underlying COVID-19–associated anosmia. Sci Adv. 2020;31(6):eabc5801. doi: 10.1126/sciadv.abc5801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhou Z, Kang H, Li S, Zhao X. Understanding the neurotropic characteristics of SARS-CoV-2: from neurological manifestations of COVID-19 to potential neurotropic mechanisms. J Neurol. 2020;267:2179–2184. doi: 10.1007/s00415-020-09929-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Paar V, et al. Anti-coagulation for COVID-19 treatment: both anti-thrombotic and anti-inflammatory? J Thromb Thrombolysis. 2020 doi: 10.1007/s11239-020-02212-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ranucci M, et al. The procoagulant pattern of patients with COVID-19 acute respiratory distress syndrome. J Thromb Haemost. 2020;18:1747–1751. doi: 10.1111/jth.14854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Henry BM, Vikse J, Benoit S, Favaloro EJ, Lippi G. Hyperinflammation and derangement of renin-angiotensin-aldosterone system in COVID-19: a novel hypothesis for clinically suspected hypercoagulopathy and microvascular immunothrombosis. Clin Chim Acta. 2020;507:167–173. doi: 10.1016/j.cca.2020.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jackson SP, et al. Thromboinflammation: challenges of therapeutically targeting coagulation and other host defense mechanisms. Blood. 2020;133(9):906–918. doi: 10.1182/blood-2018-11-882993. [DOI] [PubMed] [Google Scholar]
- 56.Dupont V, et al. Excess soluble fms-like tyrosine kinase 1 correlates with endothelial dysfunction and organ failure in critically ill COVID-19 patients. Clin Infect Dis. 2020 doi: 10.1093/cid/ciaa1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Khider L, et al. Curative anticoagulation prevents endothelial lesion in COVID-19 patients. J Thromb Haemost. 2020 doi: 10.1111/jth.14968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kempuraj D, et al. COVID-19, Mast Cells, Cytokine Storm, Psychological Stress, and Neuroinflammation. Neuroscientist. 2020;26(5-6):402–414. doi: 10.1177/1073858420941476. [DOI] [PubMed] [Google Scholar]
- 59.Pearce L, Davidson SM, Yellon DM. The cytokine storm of COVID-19: a spotlight on prevention and protection. Expert Opin Ther Targets. 2020;24(8):723–730. doi: 10.1080/14728222.2020.1783243. [DOI] [PubMed] [Google Scholar]
- 60.Pelaia C, Tinello C, Vatrella A, De Sarro G, Pelaia G. Lung under attack by COVID-19-induced cytokine storm: pathogenic mechanisms and therapeutic implications. Ther Adv Respir Dis. 2020;14:1753466620933508. doi: 10.1177/1753466620933508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pons S, Fodil S, Azoulay E, Zafrani L. The vascular endothelium: the cornerstone of organ dysfunction in severe SARS-CoV-2 infection. Critical Care. 2020;24:353. doi: 10.1186/s13054-020-03062-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang, C. et al. 2020Aveolar macrophage activation and cytokine storm in the pathogenesis of severe COVID-19. Research Square 25 Mar, doi:10.21203/rs.3.rs-19346/v1
- 63.Whittaker A, Anson M, Harky A. Neurological manifestations of COVID-19: a systematic review and current update. Acta Neurol Scand. 2020;142:14–22. doi: 10.1111/ane.13266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jung F, Kruger-Genge A, Franke RP, Hufert F, Kupper JH. COVID-19 and the endothelium. Clin Hemorheol Microcirc. 2020;75:7–11. doi: 10.3233/CH-209007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pryzdial ELG, Sutherland MR, Lin BH, Horwitz M. Antiviral anticoagulation. Res Pract. Thromb Haemost. 2020;4:774–788. doi: 10.1002/rth2.12406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kasinathan G, Sathar J. Haematological manifestations, mechanisms of thrombosis and anti-coagulation in COVID-19 disease: a review. Ann Med Surg (Lond) 2020;56:173–177. doi: 10.1016/j.amsu.2020.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mackman N, Antoniak S, Wolberg AS, Kasthuri R, Key NS. Coagulation abnormalities and thrombosis in patients infected With SARS-CoV-2 and other pandemic viruses. Arterioscler Thromb Vasc Biol. 2020;40(9):2033–2044. doi: 10.1161/ATVBAHA.120.314514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Magro C, et al. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: a report of five cases. Transl Res. 2020;220:1–13. doi: 10.1016/j.trsl.2020.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chauhan AJ, Wiffen LJ, Brown TP. COVID-19: a collision of complement, coagulation and inflammatory pathways. J Thromb Haemost. 2020 doi: 10.1111/jth.14981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Helms J, et al. High risk of thrombosis in patients with severe SARS-CoV-2 infection: a multicenter prospective cohort study. Intensive Care Med. 2020;46:1089–1098. doi: 10.1007/s00134-020-06062-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Grobler C, et al. Covid-19: The Rollercoaster of Fibrin(Ogen), D-Dimer, Von Willebrand Factor, P-selectin and their interactions with endothelial cells, platelets and erythrocytes. Int J Mol Sci. 2020;21(14):5168. doi: 10.3390/ijms21145168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Becker RC. COVID-19 update: Covid-19-associated coagulopathy. J Thromb Thrombolysis. 2020;50:54–67. doi: 10.1007/s11239-020-02134-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bomhof G, et al. COVID-19-associated immune thrombocytopenia. Br J Haematol. 2020;190:e61–e64. doi: 10.1111/bjh.16850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lippi G, Plebani M, Henry BM. Thrombocytopenia is associated with severe coronavirus disease 2019 (COVID-19) infections: a meta-analysis. Clin Chim Acta. 2020;506:145–148. doi: 10.1016/j.cca.2020.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Murt A, Eskazan AE, Yilmaz U, Ozkan T, Ar MC. COVID-19 presenting with immune thrombocytopenia: a case report and review of the literature. J Med Virol. 2021;93:43–45. doi: 10.1002/jmv.26138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sahu KK, Borogovac A, Cerny J. COVID-19 related immune hemolysis and thrombocytopenia. J Med Virol. 2021;93:1164–1170. doi: 10.1002/jmv.26402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mahevas M, et al. Clinical characteristics, management and outcome of COVID-19-associated immune thrombocytopenia: a French multicentre series. Br J Haematol. 2020;190:e224–e229. doi: 10.1111/bjh.17024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Maquet J, Lafaurie M, Sommet A, Moulis G. Thrombocytopenia is independently associated with poor outcome in patients hospitalized for COVID-19. Br J Haematol. 2020;190:e276–e279. doi: 10.1111/bjh.16950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Liu Y, et al. Association between platelet parameters and mortality in coronavirus disease 2019: Retrospective cohort study. Platelets. 2020;31:490–496. doi: 10.1080/09537104.2020.1754383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bhattacharjee S, Banerjee M. Immune thrombocytopenia secondary to COVID-19: a systematic review. SN Compr Clin Med. 2020;19(1):11. doi: 10.1007/s42399-020-00521-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Xu P, Zhou Q, Xu J. Mechanism of thrombocytopenia in COVID-19 patients. Ann Hematol. 2020;99:1205–1208. doi: 10.1007/s00277-020-04019-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mei H, Luo L, Hu Y. Thrombocytopenia and thrombosis in hospitalized patients with COVID-19. J Hematol Oncol. 2020;13:161. doi: 10.1186/s13045-020-01003-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhang Y, et al. Mechanisms involved in the development of thrombocytopenia in patients with COVID-19. Thromb Res. 2020;193:110–115. doi: 10.1016/j.thromres.2020.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Deruelle E, et al. Immune thrombocytopenia in a patient with COVID-19. Int J Hematol. 2020;112:883–888. doi: 10.1007/s12185-020-02943-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Porres-Aguilar M, Lazo-Langner A, Panduro A, Uribe M. COVID-19 vaccine-induced immune thrombotic thrombocytopenia: an emerging cause of splanchnic vein thrombosis. Ann Hepatol. 2021;23:100356. doi: 10.1016/j.aohep.2021.100356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bjørnstad-Tuveng, T. H. et al. 2021 Fatal cerebral haemorrhage after COVID-19 vaccine. Tidsskr Nor LaegeforenApr 29, doi:10.4045/tidsskr.21.0312. Print 2021 Apr 30 [DOI] [PubMed]
- 87.Dotan A, Shoenfeld Y. Perspectives on vaccine induced thrombotic thrombocytopenia. J Autoimmun. 2021;121:102663. doi: 10.1016/j.jaut.2021.102663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Douglas C, James B. SARS-CoV-2 vaccine–induced immune thrombotic thrombocytopenia. N Engl J Med. 2021;384(23):2254–2256. doi: 10.1056/NEJMe2106315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tiede A, et al. Prothrombotic immune thrombocytopenia after COVID-19 vaccination. Blood. 2021;138(4):350–353. doi: 10.1182/blood.2021011958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Scharf RE, Alberio L. COVID-19: SARS-CoV-2 vaccine-induced immune thrombotic thrombocytopenia. Hamostaseologie. 2021;41:179–182. doi: 10.1055/a-1369-3488. [DOI] [PubMed] [Google Scholar]
- 91.Favaloro EJ. Laboratory testing for suspected COVID-19 vaccine-induced (immune) thrombotic thrombocytopenia. Int J Lab Hematol. 2021;43:559–570. doi: 10.1111/ijlh.13629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cines DB. SARS-CoV-2 vaccine–induced immune thrombotic thrombocytopenia. N Engl J Med. 2021;384(23):2254–2256. doi: 10.1056/NEJMe2106315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chen PW, et al. Addressing vaccine-induced immune thromboticthrombocytopenia (VITT) following COVID-19 vaccination: a mini-review of practical strategies. Acta Cardiol Sin. 2021;37(4):355–364. doi: 10.6515/ACS.202107_37(4).20210628A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Regenhardt RW, et al. Anti-inflammatory effects of angiotensin-(1–7) in ischemic stroke. Neuropharmacology. 2013;71:154–163. doi: 10.1016/j.neuropharm.2013.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chen J, et al. Neuronal over-expression of ACE2 protects brain from ischemia-induced damage. Neuropharmacology. 2014;79:550–558. doi: 10.1016/j.neuropharm.2014.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Xia H, Lazartigues E. Angiotensin-converting enzyme 2 in the brain: properties and future directions. J Neurochem. 2008;107:1482–1494. doi: 10.1111/j.1471-4159.2008.05723.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Liu Y, et al. Clinical and biochemical indexes from 2019-nCoV infected patients linked to viral loads and lung injury. Sci China Life Sci. 2020;63:364–374. doi: 10.1007/s11427-020-1643-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Smadja DM, et al. Angiopoietin-2 as a marker of endothelial activation is a good predictor factor for intensive care unit admission of COVID-19 patients. Angiogenesis. 2020 doi: 10.1007/s10456-020-09730-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Giardini V, et al. Increased sFLT-1/PlGF ratio in COVID-19: a novel link to angiotensin II-mediated endothelial dysfunction. Am J Hematol. 2020;95:E188–E191. doi: 10.1002/ajh.25882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sardu C, et al. Hypertension, thrombosis, kidney failure, and diabetes: is COVID-19 an endothelial disease? A comprehensive evaluation of clinical and basic evidence. J Clin Med. 2020;9(5):1417. doi: 10.3390/jcm9051417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bautista-Vargas M, Bonilla-Abadia F, Canas CA. Potential role for tissue factor in the pathogenesis of hypercoagulability associated with in COVID-19. J Thromb Thrombolysis. 2020 doi: 10.1007/s11239-020-02172-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Li Y, et al. Acute cerebrovascular disease following COVID-19: a single center, retrospective, observational study. Stroke and Vascular Neurology. 2020 doi: 10.1136/svn-2020-000431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chen N, et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. The Lancet. 2020;395:507–513. doi: 10.1016/s0140-6736(20)30211-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yan, Y. et al. Clinical characteristics and outcomes of patients with severe covid-19 with diabetes. BMJ Open Diabetes Res Care8, doi:10.1136/bmjdrc-2020-001343 (2020). [DOI] [PMC free article] [PubMed]
- 105.Amraei R, Rahimi N. COVID-19, Renin-angiotensin system and endothelial dysfunction. Cells. 2020;9(7):165. doi: 10.3390/cells9071652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.KH Dinnon,. et al. 2020 A mouse-adapted SARS-CoV-2 model for the evaluation of COVID-19 medical countermeasures. bioRxiv, doi:10.1101/2020.05.06.081497
- 107.Choi JY, et al. Altered COVID-19 receptor ACE2 expression in a higher risk group for cerebrovascular disease and ischemic stroke. Biochem Biophys Res Commun. 2020;528:413–419. doi: 10.1016/j.bbrc.2020.05.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Cavezzi A, Troiani E, Corrao S. COVID-19: hemoglobin, iron, and hypoxia beyond inflammation. A narrative review Clin Pract. 2020;10(2):1271. doi: 10.4081/cp.2020.1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zhai P, Ding Y, Li Y. The impact of COVID-19 on ischemic stroke. Diagn Pathol. 2020;15:78. doi: 10.1186/s13000-020-00994-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Vogrig A, Gigli GL, Bna C, Morassi M. Stroke in patients with COVID-19: clinical and neuroimaging characteristics. Neurosci Lett. 2021;743:135564. doi: 10.1016/j.neulet.2020.135564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Varatharaj A, et al. Neurological and neuropsychiatric complications of COVID-19 in 153 patients: a UK-wide surveillance study. The Lancet Psychiatry. 2020;7:875–882. doi: 10.1016/s2215-0366(20)30287-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hernández-Fernández F, et al. Cerebrovascular disease in patients with COVID-19: neuroimaging, histological and clinical description. Brain. 2020;143(10):3089 3103. doi: 10.1093/brain/awaa239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sweid A, et al. Cerebral ischemic and hemorrhagic complications of coronavirus disease 2019. Int J Stroke. 2020;15:733–742. doi: 10.1177/1747493020937189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Fatima N, Saqqur M, Qamar F, Shaukat S, Shuaib A. Impact of COVID-19 on neurological manifestations: an overview of stroke presentation in pandemic. Neurol Sci. 2020;41:2675–2679. doi: 10.1007/s10072-020-04637-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Katsanos AH, et al. The impact of SARS-CoV-2 on stroke epidemiology and care: a meta-analysis. Ann Neurol. 2021;89:380–388. doi: 10.1002/ana.25967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Al-Samkari H, et al. COVID-19 and coagulation: bleeding and thrombotic manifestations of SARS-CoV-2 infection. Blood. 2020;136(4):489–500. doi: 10.1182/blood.2020006520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hernández-Fernández F. Cerebrovascular disease in patients with COVID-19: neuroimaging, histological and clinical description. Brain. 2021;143(10):3089–3103. doi: 10.1093/brain/awaa239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Benger M, Williams O, Siddiqui J, Sztriha L. Intracerebral haemorrhage and COVID-19: clinical characteristics from a case series. Brain Behav Immun. 2020;88:940–944. doi: 10.1016/j.bbi.2020.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Huang X, Hussain B, Chang J. Peripheral inflammation and blood-brain barrier disruption: effects and mechanisms. CNS Neurosci Ther. 2021;27:36–47. doi: 10.1111/cns.13569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Suter F, et al. A simple, home-therapy algorithm to prevent hospitalisation for COVID-19 patients: A retrospective observational matched-cohort study. EClinicalMedicine. 2021;37:100941. doi: 10.1016/j.eclinm.2021.100941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Bhaskar S, et al. Cytokine storm in COVID-19-immunopathological mechanisms, clinical considerations, and therapeutic approaches: the REPROGRAM consortium position paper. Front Immunol. 2020;11:1648. doi: 10.3389/fimmu.2020.01648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Gu SX, et al. Thrombocytopathy and endotheliopathy: crucial contributors to COVID-19 thromboinflammation. Nat Rev Cardiol. 2021;18:194–209. doi: 10.1038/s41569-020-00469-1. [DOI] [PMC free article] [PubMed] [Google Scholar]