

Abstract
Introduction:
Neurophysiological manifestations selectively associated with amyloid beta and tau depositions in Alzheimer’s disease (AD) are useful network biomarkers to identify peptide specific pathological processes. The objective of this study was to validate the associations between reduced neuronal synchrony within alpha oscillations and neurofibrillary tangle (NFT) density in autopsy examination, in patients with AD.
Methods:
In a well-characterized clinicopathological cohort of AD patients (n = 13), we quantified neuronal synchrony within alpha (8–12 Hz) and delta-theta (2–8 Hz) oscillations, using magnetoencephalography during the disease course, within six selected neocortical and hippocampal regions, including angular gyrus, superior temporal gurus, middle frontal gyrus, primary motor cortex, CA1, and subiculum, and correlated these with regional NFT density quantified at histopathological examination.
Results:
Abnormal synchrony in alpha, but not in delta-theta, significantly predicted the NFT density at post mortem neuropathological examination.
Discussion:
Reduced alpha synchrony is a sensitive neurophysiological index associated with pathological tau, and a potential network biomarker for clinical trials, to gauge the extent of network dysfunction and the degree of rescue in treatments targeting tau pathways in AD.
Keywords: alpha oscillations, Alzheimer’s disease, magnetoencephalography, neurofibrillary tangle density, neuropathology, neurophysiology
1|. INTRODUCTION
The pathological hallmarks of Alzheimer’s disease (AD) consist of extracellular amyloid beta (Aβ) plaques, and intracellular neurofibrillary tangles (NFT) of abnormally phosphorylated tau proteins.1,2 Cognitive decline in AD, however, correlates strongly with the NFT burden, but only poorly with the Aβ plaque burden.3 Studies using positron emission tomography (PET) with tau binding tracers and quantitative post mortem studies, in patients with AD, have also demonstrated that regional density of aggregated tau is strongly correlated with the patterns of cortical atrophy and corresponding domain-specific cognitive impairments.4–7 While this evidence strongly indicates that tau pathology is a major driver of local neurodegeneration, the mechanisms of neuronal damage in the milieu of abnormal tau accumulations remains under intense investigation.
Preclinical research in transgenic mice overexpressing pathological variants of tau has shown disruption of neuronal firing patterns and neurodegeneration.8–10 Although these studies suggest that tau may play a role in both pro-excitable and hypo-excitable pathophysiological mechanisms, more recent in vivo studies have clearly demonstrated tau-associated neuronal hypoactivity in neocortical networks. For example, transgenic mice overexpressing mutant tau variants or those that accumulate NFT have suppressed neocortical and hippocampal neuronal activity,11,12 reduced functional connectivity,13 impaired synaptic plasticity,14 and downregulation of neuronal and synaptic genes.15 In line with these circuit-level dysfunctions, human neuroimaging studies examining the electrical field potentials generated by pyramidal neurons using electroencephalography (EEG) and magnetoencephalography (MEG) in patients with AD clinical syndrome have reported network-level dysfunctions as impaired synchrony and altered functional connectivity.16–18 Understanding the relationships across the levels of organization–from cellular physiology to neural networks, is a fundamental prerequisite for successful translation between preclinical mouse models and patients with AD. As such, delineating the specific associations between tau and network level dysfunctions in patients with AD is a crucial translational link.
In a previous multimodal imaging study using magnetoencephalo-graphic imaging (MEGI) alongside tau- and Aβ-PET, we demonstrated that abnormal neuronal synchrony within alpha (8–12 Hz) and delta-theta (2–8 Hz) frequency oscillations in patients with AD are differentially associated with tau and Aβ accumulations.16 Consistent with the hypoactivity phenotype reported in preclinical animal models we found that higher tau-tracer uptake was uniquely associated with the degree and distribution of the reduced synchrony within the alpha band. We also found that focal alpha synchronization deficits, but not delta-theta, significantly predicted the impairments in Mini-Mental State Examination (MMSE) performance in AD patients. Other studies have shown that higher levels of phosphorylated tau and total tau in cerebrospinal fluid (CSF) assays are correlated with reduced oscillatory power and synchrony in EEG spectral signatures, in patients with AD dementia as well as with mild cognitive impairment (MCI) due to AD.19–21 As such, abnormal indices of alpha synchronizations could potentially aid in clinical trials targeting tau pathways to determine the degree of network deficits at screening and rescue of network function after the putative disease-modifying therapies.
An important and an essential step in identifying a disease marker is to demonstrate its direct relationship with the gold-standard indices. The current study addresses this important link in validating the associations between neurophysiological markers and histopathological hallmarks of AD. Here, we examine the relationships between MEGI-derived neuronal synchrony abnormalities within the alpha and delta-theta bands and NFT density determined in a subsequent autopsy assessment, in select brain regions, in a cohort of patients with pathologically proven AD. We predicted that the functional impact of tau on neural networks during life will be correlated with the degree of tau accumulation indexed in the post mortem neuropathological examination. Specifically, we tested the hypothesis that focal abnormalities of neuronal synchrony within the alpha band, but not within delta-theta, will be correlated with regional NFT density.
2|. METHODS
2.1|. Participants
A cohort of 13 individuals with pathologically proven AD and free of other contributing neuropathological changes (to avoid confounders) were included in the study (Table S1 in supporting information). All patients were evaluated with MEGI during their disease course and were enrolled in the Neurodegenerative Disease Brain Bank (NDBB) which is part of the University of California San Francisco, Memory and Aging Center (UCSF-MAC). At the NDBB, an extensive dementia-oriented post mortem assessment covering the dementia-related regions of interest on one of the hemispheres, selected a priori based on higher degree of atrophy in gross inspection was performed following the currently accepted guidelines for neuropathological diagnosis of AD and other age-associated neuropathological changes.22,23
All patients underwent a complete clinical history, neurological examination, neuropsychological evaluation, and functional assessment using Clinical Dementia Rating (CDR and CDR Sum of Boxes [CDR-SOB).24 Each participant underwent a resting MEG scan and a brain structural magnetic resonance imaging (MRI). Eleven out of 13 patients were also assessed for AD biomarker status around the time of MEG evaluation, using CSF protein assays or using PET with 11C-PIB (amyloid-PET) and FDG-PET (Table S2 in supporting information). At the time of MEGI evaluation all patients underwent a complete bedside cognitive evaluation (duration between MEGI and bedside assessments: 90.62 ± 70.43 days; Methods and Table S3 in supporting information). All patients fulfilled the diagnostic criteria for probable AD1 or MCI due to AD25 at the time of MEGI assessment. Each participant also underwent genetic analysis for familial AD mutations and apolipoprotein E (APOE) allele status. Familial AD mutations, namely, APP, PSEN1, and PSEN2 were analyzed by direct sequencing of amplified DNA. For genetic analysis of APOE, genomic DNA was purified from blood samples and amplified by polymerase chain reaction using primers that straddle the polymorphism encoding the ε2, ε3, or ε4 genotypes. One out of 13 patients in our cohort was positive for familial APP mutation and none were positive for PSEN1 or PSEN2 mutations. Each participant’s APOE allele status is shown in Table S1. To determine the normalized values (z-scores) of regional neurophysiological indices, we also recruited an age-matched control cohort (n = 23; Table S4 in supporting information) from the research cohorts of UCSF-MAC. Informed consent was obtained from all participants or their assigned surrogate decision makers. The study was approved by the Institutional Review Board (IRB) at UCSF.
2.2|. General and project-specific neuropathological assessment
The brains were obtained post mortem, processed, and analyzed according to standard protocols, as previously described.26 Primary pathological diagnosis was herein defined as the most developed neuropathological entity and which severity and regional distribution are thought to explain the majority of the patient’s clinical cognitive and behavioral phenotype.22,27 All cases were classified according to the Alzheimer’s Disease Neuropathological Change (ADNC) severity (i.e., ABC score), including Thal phase, Braak Stage, and Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) neuritic plaque frequency stage22 (Table 1 and Table S1). Using thioflavin-S fluorescent microscopy, we quantified NFT burden, manually, from microphotographs obtained with a Zeiss Axio Scan.Z1 fluorescent slide scanner microscope at the Molecular Imaging Center at University of California Berkeley, in the most affected hemisphere (left hemisphere in nine and right hemisphere in four individuals).6 We examined four neocortical regions of interest (ROI), middle frontal gyrus (MFG), superior temporal gyrus (STG), primary motor cortex (PMC), and angular gyrus (AG), and two hippocampal ROIs, the CA1 sector and subiculum (SUBI). NFT counts included intracellular and extracellular NFTs (Methods in supporting information).These regions were chosen as representative anatomic regions of the functional domains affected in AD and their classical vulnerability to AD pathology6 and there were no statistically significant regional differences in NFT density measures across the six regions (one-way analysis of variance, F = 1.59, P = 0 .1782).
TABLE 1.
Demographic characteristics of patients with AD (n = 13)
| Age at MEG – y | 62.88 ± 7.47 |
| Age of disease onset –y | 56.77 ± 8.07 |
| Age at death – y | 67.15 ± 6.82 |
| Sex (female) – no. (%) | 7 (53.85) |
| Race (White) – no. (%) | 13 (100.00) |
| Education – y | 15.92 ± 3.38 |
| CDR at MEG | 0.96 ± 0.52 |
| CDR-SOB at MEG | 5.46 ± 2.95 |
| MMSE at MEG | 20.62 ± 4.81 |
| CDR-SOB at death | 14.58 ± 4.14 |
| CDR at death | 2.46 ± 0.88 |
| Braak stage | 5.92 ± 0.28 |
| Thal stage | 4.92 ± 0.28 |
| Duration between MEG and death – y | 4.27 ± 2.29 |
Abbreviations: AD, Alzheimer’s disease; CDR, Clinical Dementia Rating; CDR-SOB, CDR Sum of Boxes; MEG, magnetoencephalography; MMSE, Mini-Mental State Examination.
2.3|. Resting state MEG data acquisition and analysis
Each subject underwent an MEG scan (average duration between MEG and death for patients with AD, 4.27 ± 2.29 years), on a whole-head biomagnetometer system consisting of 275 axial gradiometers (CTF). A minimum of 10 minutes of continuous data was collected from each subject, lying supine, awake, and eyes-closed (sampling-rate: 600 Hz). Three fiducial coils including nasion, left, and right pre-auricular points were placed to localize the position of head relative to sensor array, and later coregistered to each individual’s MRI to generate an individualized head shape model. Data collection was optimized to minimize within-session head movements (< 0.5 cm).
Data were then pre-processed using the Fieldtrip toolbox, and source space reconstruction was performed using custom-built MATLAB software tools (Methods in supporting information). We computed the average imaginary coherence per ROI, for each patient and control, within the alpha (8–12 Hz) and delta-theta (2–8 Hz) bands. Imaginary coherence captures only the coherence that cannot be explained by volume spread,28 and is a reliable metric of resting state functional connectivity.29,30 For each ROI, we identified the best representative anatomic region from the Brainnetome atlas in both hemispheres (Table S5 in supporting information), and computed the average normalized imaginary coherence (z-score) per subject (Methods in supporting information).
2.4|. Statistical analyses
We used a linear mixed model analysis to quantify the relationship between neurophysiological indices and NFT densities. Mixed models using random coefficients are robust for analysis of data with repeated observations per subject accounting for both within- and between-subject factors to provide a more accurate estimate of error. Specifically, we used SAS MIXED procedure with a general covariance structure approach that is better suited to analyze data with repeated observations per subject, compared to traditional univariate or multivariate approaches. We used two separate mixed models to examine the associations of alpha and delta-theta synchrony. The models included a repeated measures design with unstructured covariance type to estimate the within-subject covariance matrix incorporating data from six ROIs per subject. The dependent variable in each model included regional NFT density, from the six anatomical regions, per subject. The predictor variable of the models included the neuronal synchrony (i.e., alpha or delta-theta) from each of the six anatomical regions, per each subject. Subject identity and ROI identity were included into the models as categorical variables. To account for the clinical decline between the MEG scan and death in the models, we included the difference in CDR-SOB. We chose CDR-SOB to indicate the difference in clinical severity because it is a more dimensional score than CDR and hence able to better quantify a longitudinal change. Duration between MEG scan and death and CDR at the closest evaluation to death were also included in the models to account for the time lag between MEG scan and autopsy exam and the clinical severity at autopsy exam, respectively.
3|. RESULTS
3.1|. Demographics
Our cohort predominantly included patients with early-onset AD in which all but one patient had their disease onset before age 65 (average disease onset, 56.8 ± 8.1 years; 54% female and 46% male). At the time of MEG evaluation, the participants were mild-to-moderately impaired with an average MMSE of 20.62 ± 4.81 and an average CDR of 0.96 ± 0.52 (Table 1 and Table S3). The average disease duration at death was 10.4 ± 2.4 years and the average age-at-death was 67.15 ± 6.82 years. All participants were assigned Braak stage V or VI for neurofibrillary changes31 and had “frequent” neuritic plaque pathology by CERAD criteria (Table 1 and Table S1).
3.2|. Regional patterns of NFT density and frequency-specific neuronal synchrony deficits
Regional distribution of NFT burden, quantified using thioflavin-S fluorescent microscopy (Figure 1A), showed more NFT in the neocortex than the hippocampus (CA1 and subiculum) in our cohort (Figure 1B and C). For example, the angular gyrus and superior temporal gyrus showed the highest average NFT burden values (126.2 ± 64.7, 126.1 ± 51.3 per mm2, for angular gyrus and superior temporal gyrus, respectively), while CA1 showed the lowest average NFT burden (82.5 ± 49.5 per mm2). This is consistent with our cohort having a high percentage (92%) of patients with early-onset and atypical AD who selectively have high tau density in the neocortex.6
FIGURE 1.
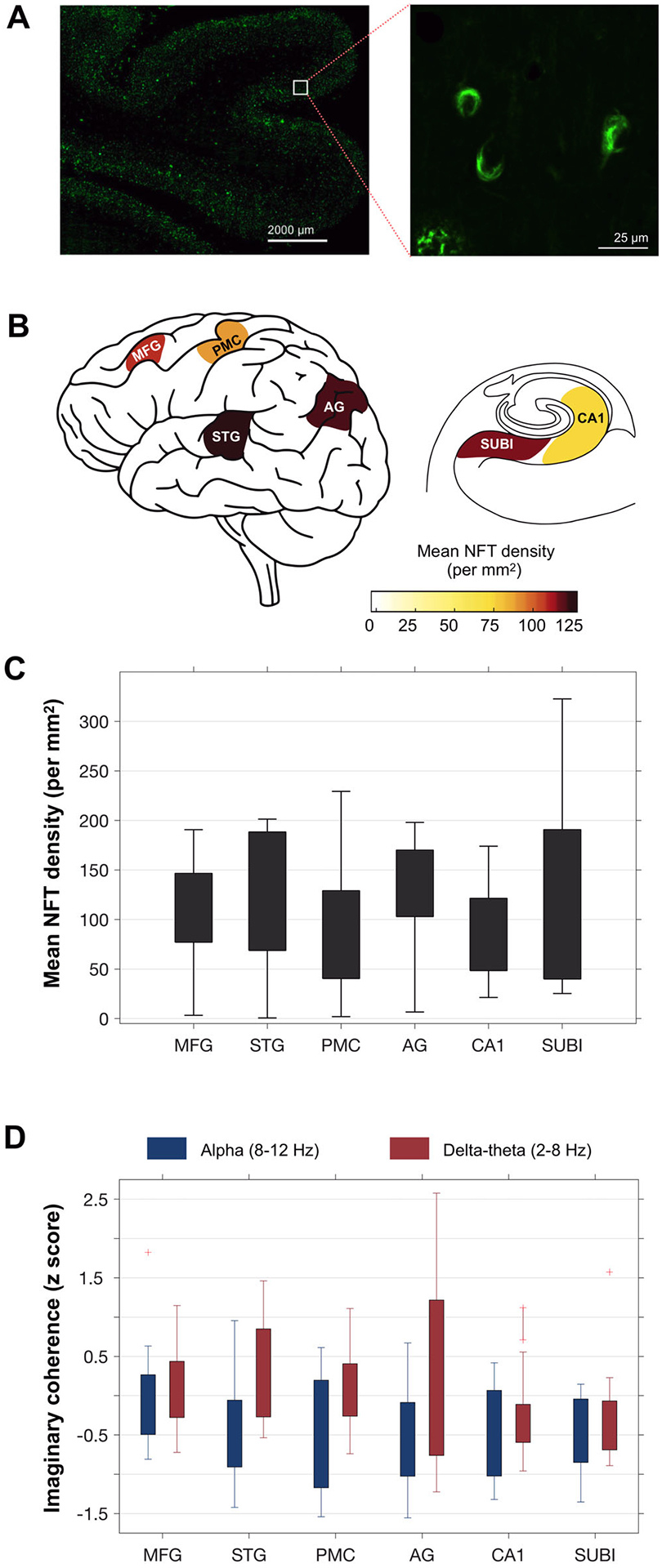
Estimation of regional neurofibrillary tangle (NFT) densities and frequency-specific neuronal synchrony deficits: Regional tau NFT densities were quantified via thioflavin-S fluorescent microscopy (A), where NFT pathology is distinguished by flame-shaped or globose morphology of fibrous aggregates (A-inset). Mean NFT densities per mm2 were estimated for six regions, including angular gyrus (AG), primary motor cortex (PMC), superior temporal gyrus (STG), middle frontal gyrus (MFG), hippocampus-CA1 (CA1), and subiculum (SUBI), in patients with Alzheimer’s disease (AD; B-C). For each region, the degree of neuronal synchrony was quantified within alpha (8–12 Hz) and delta-theta (2–8 Hz) oscillatory bands (D). Neuronal synchrony is depicted as the average imaginary coherence estimate per each region, normalized based on an age-matched healthy control population. (n = 13, patients with AD; n = 23 age-matched controls)
Patients with AD consistently showed reduced neuronal synchrony within the alpha band and increased neuronal synchrony within the delta-theta band in most ROIs, compared to age-matched controls (Figure S1 in supporting information). Across the six ROIs, the angular gyrus showed the largest reductions in alpha synchrony as well as the highest increases in delta-theta synchrony (Figure 1D; normalized imaginary coherence, −0.57 ± 0.71, 0.42 ± 1.21, for alpha and delta-theta, respectively). At each ROI, the z-score distribution of delta-theta range was higher than that of the alpha range (Figure 1D; range, −1.6–1.8, and −1.2–2.6, for alpha and delta-theta, respectively). Furthermore, reduced alpha synchrony, but not increased delta-theta synchrony, showed a significant negative correlation with the CDR-SOB measured around the time of the MEG (Figure S2A–B in supporting information). The pattern of alpha hyposynchrony and delta-theta hypersynchrony as well as their distinct associations with clinical deficits are consistent with previous reports in patients with AD, from our group as well as from others.16,19,32
3.3|. Associations of regional NFT density and frequency-specific neuronal synchrony deficits
Next, we examined the associations between the regional NFT density estimates and the regional deficits of neuronal synchrony within alpha and delta-theta frequency bands (Figure 2). A linear mixed model predicting the degree of NFT density from frequency-specific neuronal synchrony deficits adjusted for the clinical decline and time lag between MEG scan and death, showed significant associations with alpha deficits but not with delta-theta deficits. For example, lower regional z-scores of alpha (more hyposynchronous alpha) predicted higher NFT burden (Figure 2A; F = 10.17, P = 0 .01). In contrast, delta-theta synchrony deficits did not show significant associations with the regional NFT burden (Figure 2B; F = 0.07, P = 0.80). Figure 2A–B depict the model fits computed at group average values of duration between MEG and death, CDR-SOB difference between MEG and death, and CDR at death, and Figure 2C–D depict raw data.
FIGURE 2.
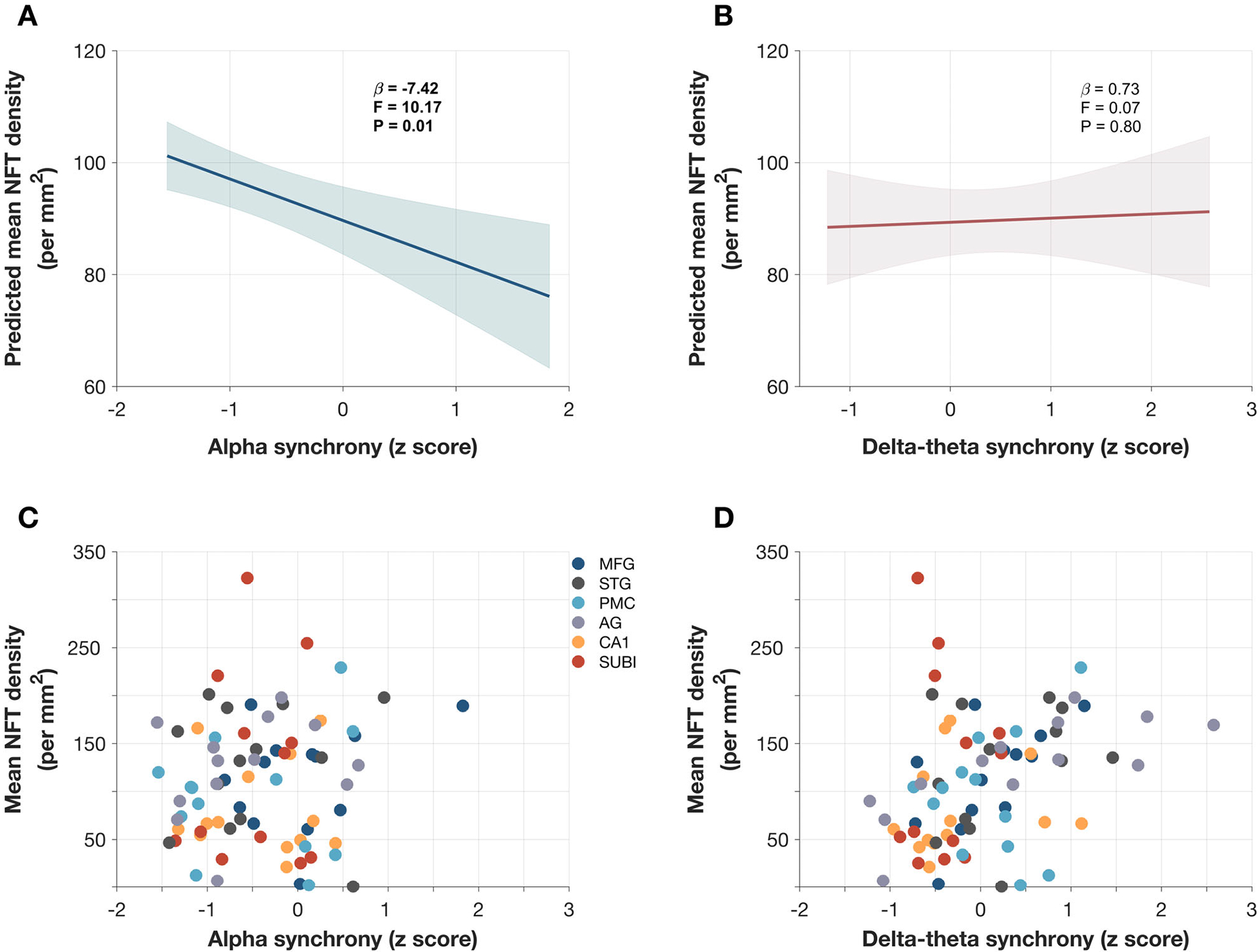
Predictions of neurofibrillary tangle (NFT) density by frequency-specific neuronal synchrony deficits in patients with Alzheimer’s disease (AD): Estimates from linear mixed-effects models predicting the NFT density by alpha (8–12) synchrony deficits and delta-theta (2–8 Hz) synchrony deficits, in patients with AD showed that greater alpha deficits significantly predicts with higher NFT burden (A), while delta-theta does not predict NFT burden (B). Each mixed-effects model included a repeated measured design to include six regions per subject and additional variables of CDR at death, CDR-SOB difference from MEG to death and time duration from MEG to death. The model fits depicted in (A-B) are computed at group averages for other additional variables (2.45, 9.07, and 4.74, for CDR, CDR-SOB difference, and time difference, respectively). Raw data points depicting the regional values of mean NFT densities and mean imaginary coherence in alpha (8–12 Hz) band (C), and delta-theta (2–8 Hz) band (D), in patients with AD. AG, angular gyrus; CA1, hippocampus-CA1; CDR, Clinical Dementia Rating; CDR-SOB, Clinical Dementia Rating Sum of Boxes; MEG, magnetoencephalography; MFG, middle frontal gyrus; PMC, primary motor cortex; STG, superior temporal gyrus; SUBI, subiculum
The random intercepts at different levels of CDR at death illustrated the associations of the alpha band synchrony deficits and NFT density (Figure 3, CDR = 0–3). For example, at CDR = 0, and at an unimpaired level of alpha hyposynchrony (i.e., z-score = 0), which may essentially represent a cognitively normal individual with intact functional connectivity, the model predictions of cortical NFT burden was close to zero (Figure 3A, CDR = 0). At higher values of CDR scores, the model predicted higher levels of NFT density for a given z-score measure of neuronal synchrony within alpha band (Figure 3A, CDR = 1–3). Delta-theta synchrony deficits (Figure 3B) were not significant predictors of NFT density.
FIGURE 3.
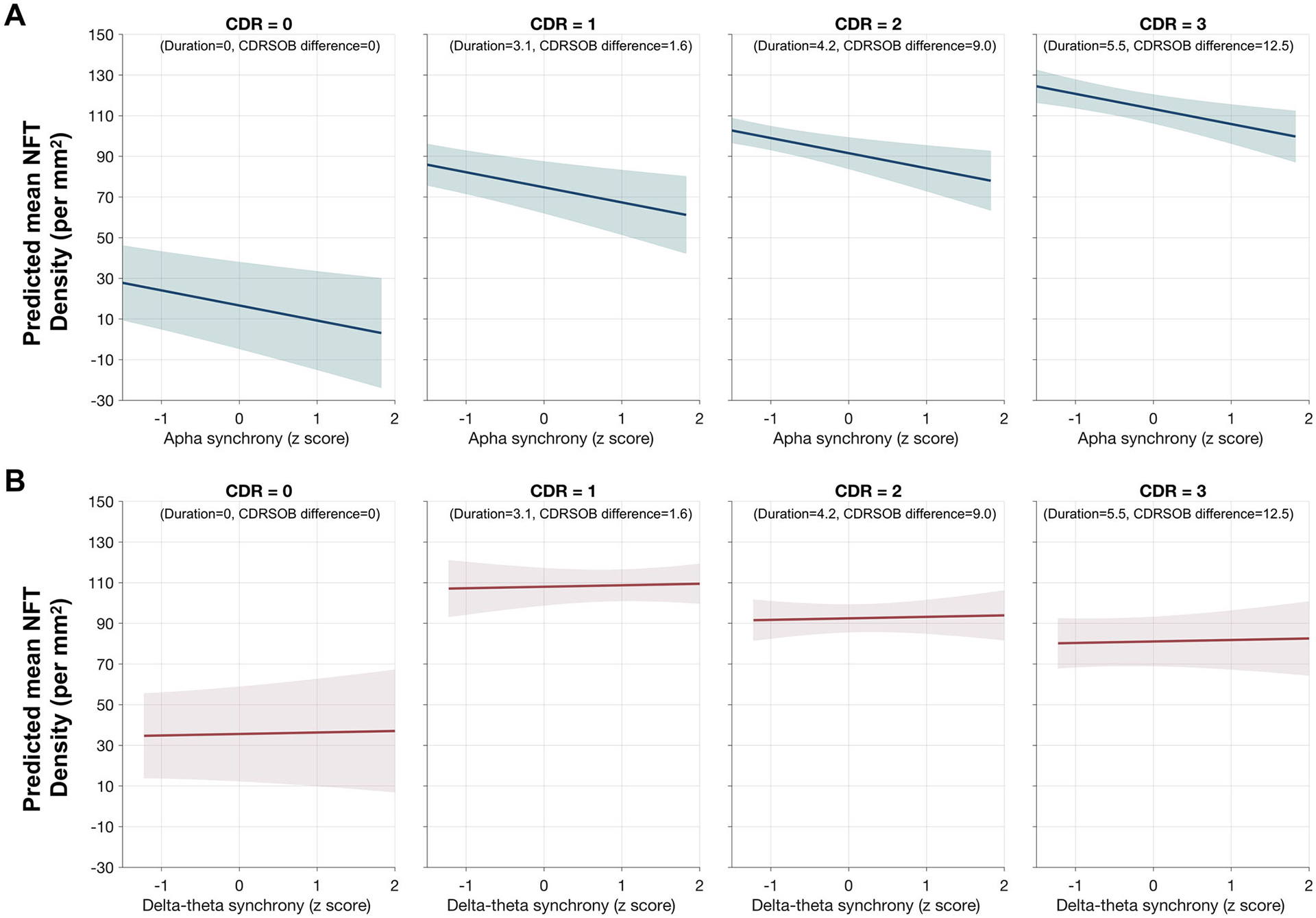
Predictions of neurofibrillary tangle (NFT) density by frequency-specific neuronal synchrony deficits at different degrees of functional severity at death in patients with Alzheimer’s disease (AD): Estimates from linear mixed-effects models predicting the NFT burden by alpha (8–12) synchrony deficits (A) and delta-theta (2–8 Hz) synchrony deficits (B), in patients with AD. From left to right each subplot depicts model fits computed at varying combination of Clinical Dementia Rating (CDR) at death and Clinical Dementia Rating Sum of Boxes (CDR-SOB) difference as such to represent increasing functional deficits. For example, the left most subplot is computed at the CDR = 0 and CDR-SOB difference = 0, while the next three subplots from left to right are computed at first quartile (Q1), median and the third quartile (Q3) values for CDR-SOB difference from magnetoencephalography (MEG) to death and duration from MEG to death
MEG source localization estimates have a relatively higher reliability in superficial cortical regions than deep brain regions such as medial temporal cortex. To examine the associations between neuronal synchrony deficits and NFT densities exclusively within the neocortex, we repeated the mixed model analyses using only the four neocortical ROIs (angular gyrus, middle frontal gyrus, superior temporal gyrus, and primary mortor cortex). Consistent with our previous result with six ROIs, we found robust correlations between the NFT burden and frequency-specific neuronal synchrony deficits (Figure S3 in supporting information). Specifically, alpha deficits showed a significant negative association with the NFT density, where greater reductions in alpha synchronizations predicted higher NFT burden (Figure S3A,C). In contrast, delta-theta synchrony deficits did not show significant associations with neocortical NFT density (Figure S3B,D).
4|. DISCUSSION
In this study, we demonstrated the neuronal synchrony abnormalities within alpha and delta-theta oscillatory bands and their associations with the NFT burden, in a well-characterized clinicopathological cohort of patients with AD. To the best of our knowledge, this is the first study to examine the associations between neurophysiological signatures of network level dysfunction and histopathologically quantified NFT density–the gold-standard method to measure tau burden, in patients with AD. Our results indicate that reductions within alpha band neuronal synchrony are sensitive indices of neurodegenerative effects associated with abnormally phosphorylated, aggregated tau, in AD. The current results are consistent with, and extend the previous findings from tau-PET imaging in which the degree and distribution of tau-tracer uptake was significantly correlated with the degree and distribution of alpha hyposynchrony,16 and from CSF tau assays in which higher CSF phospho-tau and total-tau were associated with reduced electrophysiological signatures of alpha oscillations, in patients with AD.19,21,33 These findings indicate the specific vulnerabilities of cellular and molecular pathways underlying alpha oscillatory dynamics to tau-associated pathomechanisms in AD.
4.1|. Disrupted alpha oscillations may provide a biomarker beyond NFT in AD pathophysiology
Previous neurophysiological studies using EEG and MEG have demonstrated a dynamic pattern of abnormalities within alpha band oscillatory activity during the disease course in patients with AD neuropathological spectrum. In preclinical and prodromal stages of AD there is a frontal predominant increase in alpha power and synchrony where the increased alpha oscillatory signal has been shown to discriminate patients with MCI and individuals with subjective cognitive decline from healthy controls,34,35 associated with increased CSF phosphotau20 and to be of higher predictive value Proof.aspxof conversion from MCI to AD.36–38
With advanced stages of AD dementia, alpha power and synchrony become significantly reduced in frontal as well as posterior temporal and parietal cortices in the brain, compared to healthy older adults.16,39–41 The current study represents the latter associations–reduced alpha oscillatory activity in patients with AD dementia stage–and further establishes the specific associations of reduced alpha signatures with underlying tau pathology in AD. Although the specific relationships between dynamic patterns of alpha oscillations and progressive changes in tau and Aβ accumulations are yet to be demonstrated, current evidence supports the hypothesis that alpha deficits may have distinct temporal relationships with tau and Aβ and may provide reliable electrophysiological indices of disease progression. For example, the early increase of alpha power and synchrony may be driven by the neuronal hyperactivity that is strongly associated with Aβ in animal models of AD. This idea is supported by MEG and EEG evidence of alpha power augmentation significantly associated with Aβ tracer uptake in Aβ-positive MCI patients.18,42 In the backdrop of novel peptide modulatory therapies in the lineup for clinical trials the demand for precise deciphering of neuronal circuit dysfunction and measures to quantify therapeutic efficacy is felt more than ever and alpha oscillatory indices provide promising tools for such requirement.
The strong and distinct association between alpha hyposynchrony and histopathologically quantified NFT density, however, does not limit alpha hyposynchrony as a neurophysiological measure solely representing NFT. In a previous study, using 18F-flortaucipir we showed that regional patterns of alpha hyposynchrony in AD patients were spatially colocalized with tau tracer uptake, and that greater degree of hyposynchrony is associated with higher amount of tracer uptake.16 Although there are important distinctions including the apriori selected regional analysis in the current study as opposed to the voxelwise whole-brain analysis in the previous multimodal imaging investigation, we can identify some important parallels. First, STG and AG showed greater degree of alpha synchrony reductions compared to other neocortical regions in both investigations. Second, the regional patterns of colocalized reduced-alpha synchrony and increased flortaucipir uptake overlapped with the neocortical regions of AG, STG, and MFG. Of importance is the finding that alpha-hyposynchrony is correlated with both NFT density at histopathological assessment and with the 18F-flortaucipir uptake, suggesting that it is a biomarker sensitive to the effects of tau measured by both these modalities. Because 18F-flortaucipir binds to tau filaments in neurites (neuropil threads) as well as NFT,43 tau-PET may represent a different load of pathological tau than what is quantified as NFT density in histopathological assessment. As such, alpha hyposynchrony may be a sensitive index of multiple and heterogeneous effects of tau-associated toxicity in AD pathophysiology.
4.2|. Alpha hyposynchrony provides a common translational thread between human neuroimaging and AD transgenic mice
Our finding that reduced alpha synchrony, but not the increased delta-theta synchrony, is strongly correlated with the NFT density within select brain regions is consistent with in-vivo experiments in AD transgenic mice showing tau-dependent suppression of neurons. In transgenic mice, pathological tau was found to reduce the activity within the neocortical networks via suppression and disruption of neuronal firing patterns.11,12,44 In addition, the toxic effect of tau appears to preferentially target excitatory neurons and leads to downregulation of the genes involved in glutamate receptor signaling including several AMPA and NMDA receptor subunits.12,45 Although several fundamental questions remain to be answered in these preclinical models, including whether tau is pathogenic in its oligomeric or fibrillar form and whether the toxic effects of tau are intracellular or extracellular, the results from the current study provide a consistent link between cellular abnormalities found in preclinical mouse models and network-level dysfunction in patients with AD. Alpha hyposynchrony thus poses an index of high translational value in AD research, which needs to be pursued in future studies researching the physiological and pathological bases of this rhythm.
4.3|. Tau is related to abnormal cortical neural synchronization mechanisms
Recent studies have also reported close associations between disrupted sleep-wake cycles and tau, both in humans and in rodent models.46,47 Neuropathological studies in human patients, have shown that wake-promoting neurons in the brain are extremely vulnerable to tau depositions in AD.48 Given that alpha oscillations are tightly related to the awake brain states, these relationships posit an intriguing connection between the selective vulnerabilities of neurons to tau pathology in AD and deficits in alpha oscillatory dynamics.49 The demonstration of strong overlap of alpha hyposynchrony with the brain regions that ultimately develop pathological hallmarks of tau–the key emphasis delivered in the current results–suggests that the disrupted alpha rhythms may be a harbinger of pathological processes targeting “awake” states of neural circuits and may even contribute to aberrant neuroplasticity.
A growing literature illustrates that neural oscillations are a fundamental mechanism of information processing in neural networks. Modulation of oscillatory power in different frequency bands is thought to reflect changes in the synchronization of local and long-range neuronal assemblies. The prominent view in the current literature is that alpha oscillations exert an inhibitory modulation of irrelevant neuronal activity thus reducing the neural noise, and plays an important role in gating the information in neural networks.50 As such, reduced alpha oscillations in patients with AD is consistent with network dysfunction associated with AD pathophysiology. Previous EEG and MEG studies in patients with AD have demonstrated that the reduced alpha oscillatory activity is significantly correlated with global cognitive deficits such as reduced MMSE scores or increased CDR-SOB ratings as well as some domain-specific cognitive deficits such as executive and working memory.16,51–53 As the current study establishes an important link between alpha hyposynchrony and tau accumulation in patients with AD, it highlights the importance of including neurophysiological indices together with cognitive measures in the models predicting tau accumulation and neurodegeneration.
4.4|. Delta-theta oscillatory deficits have a variable association with tau and Aβ during the disease course in AD
In our previous multimodal imaging investigation combining MEG and Aβ- and tau-PET we found that Aβ tracer uptake only showed significant relationships with the degree and distribution of delta-theta hypersynchrony, whereas tau tracer uptake showed spatial as well as functional associations with both alpha hyposynchrony and delta-theta hypersynchrony, in patients with AD.16 In this context, the current finding of delta-theta hypersynchrony showing no relationship to NFT density is seemingly inconsistent. However, the time and space dynamics of Aβ and tau in AD pathophysiology may explain these diverse relationships. Neuronal hyperactivity and hypersynchrony has been identified as an early-stage abnormality along the AD continuum in both preclinical mouse models as well as in human neuroimaging studies.9,18,35,42,54–56 It has been suggested that this early hyperactivity is driven by the pathogenic effects of Aβ on neuronal circuits, which later become hypoactive with the emergence of a predominant tau-associated pathogenic effect.10 It is therefore reasonable to expect that delta-theta hypersynchrony may only covary with protein depositions in the brain regions that have a predominance of ongoing Aβ-associated toxic effect. Our results indicate that the select ROIs considered in the current autopsy analysis do not represent these regional combinatorial effects of Aβ and tau. On the other hand, in our previous investigation of voxel-level associations between delta-theta hypersynchrony and the protein-specific tracer uptake in PET imaging during the disease course in AD patients we demonstrated the main effects as well as interactive effects of tau and Aβ on delta-theta hypersynchrony.
4.5|. Limitations
Our study is not without limitations. First, we chose to focus on six specific brain regions because of their relevance across a range of functional domains and classical vulnerability to AD pathology. Future work to further elucidate the relationship between regionally specific tau accumulation and neurophysiological abnormalities would benefit from assessing additional brain regions, as well as intra-regional differences such as NFT pathology in layers III and V. The latter may provide additional insights about cellular-level vulnerabilities. Second, except for one patient our cohort exclusively consisted of the early-onset AD phenotype. Although this selection allowed us to chose a clinical cohort of AD excluding other co-pathologies, it also limited the ability to generalize our findings to late-onset AD phenotype. Third, despite the benefit of close clinical follow-up over many years, our sample size is relatively small. Future studies with more patients and representing all phenotypic presentations of AD will be able to examine the sensitivity and specificity of alpha hyposynchrony as a neurophysiological index of NFT density. Notwithstanding the constraints imposed by small sample size, the robust associations between neurophysiological and histopathological indices of AD from the current study provide a solid rationale for the design and analyses of large-scale cohort studies in future.
4.6|. Summary and future directions
During the past decades, intense research into the mechanisms of neurodegenerative diseases have discovered novel genetic and molecular features as well as exposed the complexity and heterogeneity of tau proteinopathies.57 Characterizing the abnormal neurophysiological indices associated with tau in AD patients not only will help address fundamental questions about the mechanisms of AD pathophysiology, but also will deliver translational biomarkers to facilitate the preclinical-to-clinical discovery of new treatments. The important link established by the current study, between neurophysiological manifestations and the histopathological indices of AD pathophysiology, marks a significant step toward a long-term goal of developing validated electrophysiological biomarkers. Key topics for future research include the study of cross-sectional and longitudinal relationships between neurophysiological manifestations and tau accumulation and neurodegeneration, and integration of these findings into structure-function modeling that can inform clinical trials and new therapeutic options.
Supplementary Material
RESEARCH IN CONTEXT.
Systematic review: The authors reviewed the literature using traditional (e.g., PubMed) sources and conference proceedings. The relevant publications describing the neurophysiological manifestations and neuropathological characteristics of Alzheimer’s disease (AD) are appropriately cited.
Interpretation: Associations between alpha oscillatory (8–12 Hz) deficits during the disease course and tau burden at the post mortem examination, in patients with AD, offer neuropathological validation of frequency-specific neurophysiological signatures of tau-mediated pathophysiological mechanisms in AD.
Future directions: The article emphasizes that characterization of abnormal neurophysiological indices associated with tau in AD patients will not only help address fundamental questions about the mechanisms of AD pathophysiology, but also facilitate the translation between preclinical animal models and human condition. Key topics for future research include the study of cross-sectional and longitudinal relationships between neurophysiological manifestations and tau accumulation and integration of these findings into structure-function modeling that can inform clinical trials and new therapeutic options.
HIGHLIGHTS.
An observational cohort study combining magnetoencephalography and neuropathology.
Abnormal alpha but not delta-theta oscillations predicted post mortem tau density.
Frequency-specific neurophysiological indices are promising network biomarkers in Alzheimer’s disease.
ACKNOWLEDGMENTS
We would like to thank all of the study participants and their families for their generous support to our research. This study was supported by the National Institutes of Health grants: K08AG058749 (KGR), F32AG050434-01A1 (KGR), K23 AG038357 (KAV); P50 AG023501, P01 AG19724 (BLM), R01 NS100440 (SSN), DC013979 (SSN), AG062196 (SSN), EB022717 (SSN), UCOP MRIP MRP-17-454755 (SSN), K24AG053435 (LTG), R01 AG064314 (LTG), R01 AG060477 (LTG), U01 AG057195 (LTG); a grant from Ricoh MEG Inc. (SSN); a grant from John Douglas French Alzheimer’s Foundation (KAV); grants from Larry L. Hillblom Foundation: 2015-A-034-FEL (KGR); and 2019-A-013-SUP (KGR); a grant from the Alzheimer’s Association: (PCTRB-13-288476) (KAV), and made possible by Part the CloudTM, (ETAC-09-133596).
Funding information
National Institutes of Health, Grant/Award Numbers: K08AG058749 (KGR), F32AG050434-01A1 (KGR), K23 AG038357 (KAV), P50 AG023501, P01 AG19724 (BLM), R01 NS100440 (SSN), DC013979 (SSN), AG062196 (SSN), EB022717 (SSN), UCOP MRIP MRP-17-454755 (SSN), K24AG053435 (LTG), R01 AG064314 (LTG), R01 AG060477 (LTG), U01 AG057195 (LTG); Ricoh MEG Inc. (SSN); John Douglas French Alzheimer’s Foundation (KAV); Larry L. Hillblom Foundation, Grant/Award Numbers: 2015-A-034-FEL (KGR), 2019-A-013-SUP (KGR); Alzheimer’s Association, Grant/Award Number: PCTRB-13-288476) (KAV); CloudTM, Grant/Award Number: ETAC-09-133596
Footnotes
CONFLICTS OF INTEREST
The authors report no conflicts of interest.
REFERENCES
- 1.McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jack CR Jr, Bennett DA, Blennow K, et al. NIA-AA Research Framework: toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018;14:535–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Giannakopoulos P, Herrmann FR, Bussiere T, et al. Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer’s disease. Neurology. 2003;60:1495–1500. [DOI] [PubMed] [Google Scholar]
- 4.Ossenkoppele R, Schonhaut DR, Scholl M, et al. Tau PET patterns mirror clinical and neuroanatomical variability in Alzheimer’s disease. Brain. 2016;139:1551–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bejanin A, Schonhaut DR, La Joie R, et al. Tau pathology and neurodegeneration contribute to cognitive impairment in Alzheimer’s disease. Brain. 2017;140:3286–3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Petersen C, Nolan AL, de Paula Franca Resende E, et al. Alzheimer’s disease clinical variants show distinct regional patterns of neurofibrillary tangle accumulation. Acta Neuropathol. 2019;138:597–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.La Joie R, Visani AV, Baker SL, et al. Prospective longitudinal atrophy in Alzheimer’s disease correlates with the intensity and topography of baseline tau-PET. Sci Transl Med. 2020;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Palop JJ, Mucke L. Network abnormalities and interneuron dysfunction in Alzheimer disease. Nat Rev Neurosci. 2016;17:777–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zott B, Busche MA, Sperling RA, Konnerth A. What happens with the circuit in Alzheimer’s disease in mice and humans?. Annu Rev Neurosci. 2018;41:277–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harris SS, Wolf F, De Strooper B, Busche MA. Tipping the scales: peptide-dependent dysregulation of neural circuit dynamics in Alzheimer’s disease. Neuron. 2020;107(3):417–435. [DOI] [PubMed] [Google Scholar]
- 11.Busche MA, Wegmann S, Dujardin S, et al. Tau impairs neural circuits, dominating amyloid-beta effects, in Alzheimer models in vivo. Nat Neurosci. 2019;22:57–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fu H, Rodriguez GA, Herman M, et al. Tau pathology induces excitatory neuron loss, grid cell dysfunction, and spatial memory deficits reminiscent of early Alzheimer’s disease. Neuron. 2017;93:533–541.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ahnaou A, Moechars D, Raeymaekers L, et al. Emergence of early alterations in network oscillations and functional connectivity in a tau seeding mouse model of Alzheimer’s disease pathology. Sci Rep. 2017;7:14189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Muller-Thomsen L, Borgmann D, Morcinek K, et al. Consequences of hyperphosphorylated tau on the morphology and excitability of hippocampal neurons in aged tau transgenic mice. Neurobiol Aging. 2020;93:109–123. [DOI] [PubMed] [Google Scholar]
- 15.Castanho I, Murray TK, Hannon E, et al. Transcriptional signatures of tau and amyloid neuropathology. Cell Rep. 2020;30:2040–2054.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ranasinghe KG, Cha J, Iaccarino L, et al. Neurophysiological signatures in Alzheimer’s disease are distinctly associated with TAU, amyloid-beta accumulation, and cognitive decline. Sci Transl Med. 2020;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Babiloni C, Del Percio C, Lizio R, et al. Abnormalities of cortical neural synchronization mechanisms in patients with dementia due to Alzheimer’s and Lewy body diseases: an EEG study. Neurobiol Aging. 2017;55:143–158. [DOI] [PubMed] [Google Scholar]
- 18.Nakamura A, Cuesta P, Fernandez A, et al. Electromagnetic signatures of the preclinical and prodromal stages of Alzheimer’s disease. Brain. 2018;141:1470–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smailovic U, Koenig T, Kareholt I, et al. Quantitative EEG power and synchronization correlate with Alzheimer’s disease CSF biomarkers. Neurobiol Aging. 2018;63:88–95. [DOI] [PubMed] [Google Scholar]
- 20.Canuet L, Pusil S, Lopez ME, et al. Network disruption and cerebrospinal fluid amyloid-beta and phospho-tau levels in mild cognitive impairment. J Neurosci. 2015;35:10325–10330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kramberger MG, Kareholt I, Andersson T, Winblad B, Eriksdotter M, Jelic V. Association between EEG abnormalities and CSF biomarkers in a memory clinic cohort. Dement Geriatr Cogn Disord. 2013;36:319–328. [DOI] [PubMed] [Google Scholar]
- 22.Montine TJ, Phelps CH, Beach TG, et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol. 2012;123:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grinberg LT, Rueb U, Heinsen H. Brainstem: neglected locus in neurodegenerative diseases. Front Neurol. 2011;2:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology. 1993;43:2412–2414. [DOI] [PubMed] [Google Scholar]
- 25.Albert MS, DeKosky ST, Dickson D, et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:270–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Spina S, Brown JA, Deng J, et al. Neuropathological correlates of structural and functional imaging biomarkers in 4-repeat tauopathies. Brain. 2019;142:2068–2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Banerjee G, Carare R, Cordonnier C, et al. The increasing impact of cerebral amyloid angiopathy: essential new insights for clinical practice. J Neurol Neurosurg Psychiatry. 2017;88:982–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nolte G, Bai O, Wheaton L, Mari Z, Vorbach S, Hallett M. Identifying true brain interaction from EEG data using the imaginary part of coherency. Clin Neurophysiol. 2004;115:2292–2307. [DOI] [PubMed] [Google Scholar]
- 29.Guggisberg AG, Honma SM, Findlay AM, et al. Mapping functional connectivity in patients with brain lesions. Ann Neurol. 2008;63:193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hinkley LB, Vinogradov S, Guggisberg AG, Fisher M, Findlay AM, Nagarajan SS. Clinical symptoms and alpha band resting-state functional connectivity imaging in patients with schizophrenia: implications for novel approaches to treatment. Biol Psychiatry. 2011;70:1134–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. [DOI] [PubMed] [Google Scholar]
- 32.Sami S, Williams N, Hughes LE, et al. Neurophysiological signatures of Alzheimer’s disease and frontotemporal lobar degeneration: pathology versus phenotype. Brain. 2018;141:2500–2510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cuesta P, Garces P, Castellanos NP, et al. Influence of the APOE epsilon4 allele and mild cognitive impairment diagnosis in the disruption of the MEG resting state functional connectivity in sources space. J Alzheimers Dis. 2015;44:493–505. [DOI] [PubMed] [Google Scholar]
- 34.Bajo R, Maestu F, Nevado A, et al. Functional connectivity in mild cognitive impairment during a memory task: implications for the disconnection hypothesis. J Alzheimers Dis. 2010;22:183–193. [DOI] [PubMed] [Google Scholar]
- 35.Lopez-Sanz D, Bruna R, Garces P, et al. Functional connectivity disruption in subjective cognitive decline and mild cognitive impairment: a common pattern of alterations. Front Aging Neurosci. 2017;9: 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lopez ME, Bruna R, Aurtenetxe S, et al. Alpha-band hypersynchronization in progressive mild cognitive impairment: a magnetoencephalography study. J Neurosci. 2014;34:14551–14559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jelic V, Johansson SE, Almkvist O, et al. Quantitative electroencephalography in mild cognitive impairment: longitudinal changes and possible prediction of Alzheimer’s disease. Neurobiol Aging. 2000;21:533–540. [DOI] [PubMed] [Google Scholar]
- 38.Rossini PM, Del Percio C, Pasqualetti P, et al. Conversion from mild cognitive impairment to Alzheimer’s disease is predicted by sources and coherence of brain electroencephalography rhythms. Neuroscience. 2006;143:793–803. [DOI] [PubMed] [Google Scholar]
- 39.Babiloni C, Ferri R, Moretti DV, et al. Abnormal fronto-parietal coupling of brain rhythms in mild Alzheimer’s disease: a multicentric EEG study. Eur J Neurosci. 2004;19:2583–2590. [DOI] [PubMed] [Google Scholar]
- 40.Babiloni C, Ferri R, Binetti G, et al. Directionality of EEG synchronization in Alzheimer’s disease subjects. Neurobiol Aging. 2009;30:93–102. [DOI] [PubMed] [Google Scholar]
- 41.Briels CT, Schoonhoven DN, Stam CJ, de Waal H, Scheltens P, Gouw AA. Reproducibility of EEG functional connectivity in Alzheimer’s disease. Alzheimers Res Ther. 2020;12:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gaubert S, Raimondo F, Houot M, et al. EEG evidence of compensatory mechanisms in preclinical Alzheimer’s disease. Brain. 2019;142:2096–2112. [DOI] [PubMed] [Google Scholar]
- 43.Smith R, Wibom M, Pawlik D, Englund E, Hansson O. Correlation of in vivo [18F]flortaucipir with postmortem Alzheimer disease tau pathology. JAMA Neurol. 2019;76:310–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Menkes-Caspi N, Yamin HG, Kellner V, Spires-Jones TL, Cohen D, Stern EA. Pathological tau disrupts ongoing network activity. Neuron. 2015;85:959–966. [DOI] [PubMed] [Google Scholar]
- 45.Pickett EK, Herrmann AG, McQueen J, et al. Amyloid beta and tau cooperate to cause reversible behavioral and transcriptional deficits in a model of Alzheimer’s disease. Cell Rep. 2019;29:3592–3604.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang C, Holtzman DM. Bidirectional relationship between sleep and Alzheimer’s disease: role of amyloid, tau, and other factors. Neuropsychopharmacology. 2020;45:104–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Winer JR, Mander BA, Helfrich RF, et al. Sleep as a potential biomarker of tau and beta-amyloid burden in the human brain. J Neurosci. 2019;39:6315–6324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Oh J, Eser RA, Ehrenberg AJ, et al. Profound degeneration of wake-promoting neurons in Alzheimer’s disease. Alzheimers Dement. 2019;15:1253–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Babiloni C, Pascarelli MT, Lizio R, et al. Abnormal cortical neural synchronization mechanisms in quiet wakefulness are related to motor deficits, cognitive symptoms, and visual hallucinations in Parkinson’s disease patients: an electroencephalographic study. Neurobiol Aging. 2020;91:88–111. [DOI] [PubMed] [Google Scholar]
- 50.Klimesch W, Sauseng P, Hanslmayr S. EEG alpha oscillations: the inhibition-timing hypothesis. Brain Res Rev. 2007;53:63–88. [DOI] [PubMed] [Google Scholar]
- 51.Ranasinghe KG, Hinkley LB, Beagle AJ, et al. Regional functional connectivity predicts distinct cognitive impairments in Alzheimer’s disease spectrum. NeuroImage Clin. 2014;5:385–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.de Haan W, Stam CJ, Jones BF, Zuiderwijk IM, van Dijk BW, Scheltens P. Resting-state oscillatory brain dynamics in Alzheimer disease. J Clin Neurophysiol. 2008;25:187–193. [DOI] [PubMed] [Google Scholar]
- 53.Engels MM, Hillebrand A, van der Flier WM, Stam CJ, Scheltens P, van Straaten EC. Slowing of hippocampal activity correlates with cognitive decline in early onset alzheimer’s disease. An MEG study with virtual electrodes. Front Hum Neurosci. 2016;10:238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schultz AP, Chhatwal JP, Hedden T, et al. Phases of hyperconnectivity and hypoconnectivity in the default mode and salience networks track with amyloid and tau in clinically normal individuals. J Neurosci. 2017;37:4323–4331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sepulcre J, Sabuncu MR, Li Q, El Fakhri G, Sperling R, Johnson KA. Tau and amyloid beta proteins distinctively associate to functional network changes in the aging brain. Alzheimers Dement. 2017;13:1261–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pusil S, Lopez ME, Cuesta P, Bruna R, Pereda E, Maestu F. Hyper-synchronization in mild cognitive impairment: the ‘X’ model. Brain. 2019;142:3936–3950. [DOI] [PubMed] [Google Scholar]
- 57.Guo T, Noble W, Hanger DP. Roles of tau protein in health and disease. Acta Neuropathol. 2017;133:665–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.