

Abstract
Rationale:
Therapeutic efforts to decrease atherosclerotic cardiovascular disease risk have focused largely on reducing atherogenic lipoproteins, yet lipid lowering therapies alone are insufficient to fully regress plaque burden. We postulate that arterial repair requires resolution of a maladaptive immune response, and that targeting factors that hinder inflammation resolution will facilitate plaque regression.
Objective:
The guidance molecule netrin-1 is secreted by macrophages in atherosclerotic plaques, where it sustains inflammation by enhancing macrophage survival and blocking macrophage emigration. We tested whether silencing netrin-1 in advanced atherosclerosis could resolve arterial inflammation and regress plaques.
Methods and Results:
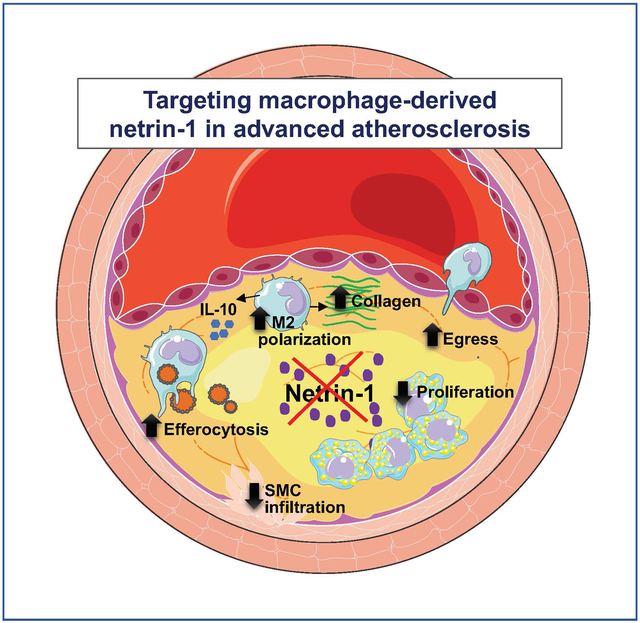
To temporally silence netrin-1 in myeloid cells, we generated genetically modified mice in which Ntn1 could be selectively deleted in monocytes and macrophages using a tamoxifen-induced CX3CR1-driven cre-recombinase (Ntn1fl/flCx3cr1creERT2+) and littermate control mice (Ntn1fl/flCx3cr1WT). Mice were fed western diet in the setting of hepatic PCSK9 overexpression to render them atherosclerotic, and then treated with tamoxifen to initiate deletion of myeloid netrin-1 (MøΔNtn1) or not in controls (MøWT). Morphometric analyses performed 4 weeks later showed that myeloid Ntn1 silencing reduced plaque burden in the aorta (–50%) and plaque complexity in the aortic root. Monocyte-macrophage tracing experiments revealed lower monocyte recruitment, macrophage retention, and proliferation in MøΔNtn1 compared to MøWT plaques, indicating a restructuring of monocyte-macrophage dynamics in the artery wall upon netrin-1 silencing. Single cell RNA-sequencing of aortic immune cells prior to and after netrin-1 silencing revealed upregulation of gene pathways involved in macrophage phagocytosis and migration, including the Ccr7 chemokine receptor signaling pathway required for macrophage emigration from plaques and atherosclerosis regression. Additionally, plaques from MøΔNtn1 mice showed hallmarks of inflammation resolution, including higher levels of pro-resolving macrophages, interleukin-10, and efferocytosis, as compared to plaques from MøWT mice.
Conclusion:
Our data show that targeting netrin-1 in advanced atherosclerosis ameliorates atherosclerotic inflammation and promotes plaque regression.
Subject Terms: Atherosclerosis, Inflammation, Vascular Biology, Vascular Disease
Graphical Abstract
INTRODUCTION
Cardiovascular disease (CVD) remains the number one cause of death in the U.S. and globally, and is responsible for 17.9 million deaths annually1. In the last decades, therapeutic efforts to decrease the risk of coronary artery disease (CAD) events have largely focused on reducing the levels of circulating atherogenic lipoproteins. Although these approaches have yielded drugs that have demonstrated substantial impacts on cardiovascular mortality and morbidity, a large burden of residual CVD risk remains and there is concern that we may be reaching a point of diminishing returns in targeting the lipoprotein axis. An orthogonal approach that has yielded recent success is targeting inflammation, a process critical to the development of atherosclerosis. The CANTOS trial of canakinumab, an inhibitor of the cytokine interleukin (IL)-1b, revealed that treating chronic inflammation in atherosclerosis decreases the occurrence of major cardiac events in CAD patients2. While this study highlighted the potential of targeting the immune response in CVD, inhibiting IL-1b led to an increase in fatal infections, raising concerns that such an approach could compromise the immune system’s ability to respond to pathogens. There is a need to better understand the pathways that inhibit the resolution of atherosclerotic inflammation in order to develop therapeutic strategies to target the maladaptive immune response that fuels plaque growth.
Inflammation is a critical factor in the initiation and clinical progression of atherosclerotic lesions. Insults to the vascular endothelial layer during hypercholesterolemia or hypertension allow subendothelial lipoprotein deposition, which is thought to trigger the initial inflammatory response3. Activated endothelial and smooth muscle cells (SMCs) release chemoattractants that elicit the recruitment of monocytes from the blood, and these monocyte-derived macrophages, as well as tissue-resident macrophages and SMCs, internalize the deposited lipoproteins, leading to the formation of lipid-laden foam cells – a hallmark of early atherosclerotic plaques4–7. The accruing macrophages are sources of pro-inflammatory mediators (e.g., IL-1b, CCL2/MCP-1, CCL5) that amplify the immune response by recruiting an arsenal of immune cells (e.g., monocytes, neutrophils, dendritic cells, T cells, B cells, natural killer cells and innate lymphoid cells)8. Despite these reinforcements, atherosclerotic lesions often fail to resolve, progressing instead to complex plaques of the artery wall8, 9 in which SMCs also contribute to lesion growth by undergoing transdifferentiation to macrophage-like and fibroblast-like states10–13. During the evolution of arterial lesions, macrophages and SMCs undergo cell death and contribute to the formation of the necrotic core, a lipid-rich, inflammatory milieu that characterizes advanced plaques14. Progressive extracellular lipid deposition and accumulation of macrophage-derived factors that contribute to fibrous cap weakening and a pro-coagulant state can render plaques vulnerable to rupture or erosion, leading to atherothrombotic events including myocardial infarction and stroke15–17.
While the factors and pathways that fuel inflammation in the artery wall have been intensively investigated4, the reasons why atherosclerotic inflammation fails to resolve are poorly understood. Acute inflammatory responses to injury or infection are typically self-limiting, and are followed by a reparative phase that allows for a return to homeostasis9. Studies have shown that inflammation resolution is an active process, requiring the initiation of pathways that counter-regulate pro-inflammatory responses and induce the production of pro-resolving mediators that terminate inflammation and promote tissue repair9. Atherosclerotic plaques exhibit features of defective inflammation resolution, including defective clearance of dead cells, necrosis, erosion of plaque stabilizing extracellular matrix, and deficit of specialized pro-resolving mediators (e.g., resolvins, maresins, lipoxins). These characteristics suggest that the transition from the pro-inflammatory to the pro-resolving phase of inflammation is impaired in plaques.
We previously identified the laminin-like protein netrin-1 as a regulator of macrophage persistence and chronic inflammation in atherosclerosis and obesity18–20. Although originally described as a guidance cue directing axonal migration during development21, 22, netrin-1 can also regulate the migration of immune cells, including monocytes, macrophages, and lymphocytes23, 24. Netrin-1 can play both protective and deleterious roles in inflammation, depending on its localization in the circulation or tissues, as well as the cell surface receptors that it engages20, 25–33. Recent studies in patients with coronary artery disease have shown that netrin-1 levels in the circulation, where it reduces monocyte adhesion to the endothelium, are inversely correlated with plaque burden34, 35, whereas expression levels of netrin-1 and its receptor Unc5b increase in plaque macrophages with disease progression19, 35, 36. Macrophage expression of netrin-1 is upregulated by oxidized forms of low density lipoprotein (LDL) and in response to hypoxia or activation of the transcription factor HIF-1a19, 37, 38. Netrin-1, acting via autocrine and paracrine activation of Unc5b, inhibits macrophage migration to chemotactic signals that direct emigration from tissues (e.g., CCL19-CCR7) and also fosters macrophage survival, leading to macrophage persistence18, 19, 39. Netrin-1 also induces SMC migration into the intima via the receptor neogenin (which is lowly expressed in macrophages), contributing to plaque progression19, 36. A rare variant in NTN1, which enhances netrin-1’s ability to block macrophage migration and increases it binding to neogenin, was identified in a family with premature atherosclerosis40. Furthermore, hematopoietic deficiency of netrin-1 in Ldlr–/– mice, attained by transplantation of Ntn1–/– bone marrow, reduced the development of atherosclerosis by 40%19. These studies support a role for macrophage-derived netrin-1 in sustaining the maladaptive immune response that fuels plaque progression. However, whether targeting netrin-1 in advanced atherosclerosis has therapeutic benefit remains to be explored.
To therapeutically silence myeloid netrin-1 expression in established atherosclerosis, we generated genetically modified mice in which expression of netrin-1 could be temporally deleted in monocytes and macrophages using a tamoxifen-inducible CX3CR1-driven cre recombinase (Ntn1fl/flCx3cr1CreER+) and littermate control mice (Ntn1fl/flCx3cr1WT). We found that in mice with complex atherosclerotic plaques, tamoxifen-induced deletion of Ntn1 promotes the regression of atherosclerosis as evidenced by a reduction of aortic plaque burden and plaque macrophage content. Single cell RNA sequencing (scRNA-seq) of plaque immune cells showed that netrin-1 silencing altered the gene expression profiles of plaque macrophages and reorganized the immune cell landscape in the artery wall. Using histological, flow cytometry and monocyte-macrophage tracking techniques, we show that netrin-1 silencing in macrophages reduced their retention, proliferation and survival in plaques, and promoted pro-resolving functions including macrophage emigration, polarization of tissue reparative M2-like macrophages and efferocytosis. These findings reveal the critical role of netrin-1 in sustaining chronic inflammation in the artery wall and support targeting netrin-1 in advanced atherosclerosis to resolve arterial inflammation and regress plaque.
METHODS
Data, Analytic Methods (Code), and Research Materials Transparency.
RNA-sequencing data have been deposited at the Gene Expression Omnibus (GEO) under accession number GSE168389. Code used to generate the tables have been deposited on a GitHub repository (https://github.com/EmilyJBrown/Schlegel_et_al_core_analysis). Other data, analytic methods, and study materials are available upon request from the corresponding author. Extended methods and Major Resources Table included in the Supplemental Materials.
Mouse studies.
Experimental procedures were in accordance with the US Department of Agriculture Animal Welfare Act and the US Public Health Service Policy on Humane Care and Use of Laboratory Animals, and approved by NYU’s Institutional Animal Care and Use Committee. Mice were housed in a specified pathogen-free (SPF) facility. Analyses of mouse experiments were blinded through numerical coding of samples. Netrin-1 floxed (Ntn1fl/fl) mice on a C57BL6 background were provided by H. Eltzschig (University of Texas Health Science Center at Houston) and crossed with C57BL6 Cx3cr1CreERT2+/– mice (The Jackson Laboratory, Stock# 020940). Ntn1fl/flCx3cr1CreERT2+/– mice were crossed with Ntn1fl/fl mice to generate experimental Ntn1fl/flCx3cr1CreERT2+/– (Ntn1fl/flCx3cr1CreER+) and Cre negative (Ntn1fl/flCx3cr1WT) littermate control mice. In the treatment cohort, administration of tamoxifen results in cre-mediated deletion of Ntn1 in monocytes and macrophages of Ntn1fl/flCx3cr1CreERT2+ mice (hereafter MøΔNtn1) and wild type levels of Ntn1 in monocytes and macrophages of Ntn1fl/flCx3cr1WTmice (hereafter MøWT). For in vitro studies, bone marrow derived macrophages (BMDM) were prepared from Ntn1fl/flUbcCre+ (Ntn1–/–) and Ntn1fl/flUbcCre– (Control) mice as described41.
Atherosclerosis analyses.
Atherosclerosis was induced in 8-week old male mice by administering AAV-mPCSK9 (5 × 1011; Penn Vector core, PA, USA) to reduce hepatic LDL-R levels and feeding western diet (40% fat kcal, 0.3% cholesterol; Dyets #101977GI) for 20 weeks. As AAV-mPCSK9 transduction of the liver is inconsistent in female mice42, only male mice were used. Atherosclerotic mice were randomly assigned to either the baseline or tamoxifen treatment groups. Mice in the treatment group were injected with tamoxifen (75 mg/kg; Sigma, cat# T5648) intraperitoneally (i.p.) for 5 days and fed a chow diet (13% fat kcal, 0% cholesterol; LabDiet cat# 5053) for 4 weeks. At the study endpoint, mice were euthanized with isoflurane, exsanguinated by cardiac puncture, and perfused with PBS. The heart was separated from the aorta at the root, embedded in optimal cutting temperature (OCT) compound and snap-frozen at −80°C. Hearts were sectioned (6 μm) through the aortic root and stained with hematoxylin and eosin. For morphometric analyses of plaque and necrotic area, 6 sections per mouse spanning the aortic root were assessed using ImageJ software (https://fiji.sc/). Descriptions of immunohistochemical staining, flow cytometric analyses and labeling and tracking of blood monocytes are in the extended methods section of the online supplement.
Single Cell RNA Sequencing.
Single cell suspensions of the aortic arch of 5 mice per group were combined and CD45+ immune cells were isolated by staining (anti-CD45 PerCp-Cy5.5, BioLegend) and flow sorting viable (fixable viability dye e780, eBioscience) single cells on a Facs Aria II cytometer (BD) equipped with a 100 μm nozzle. Aortic CD45+ cells were loaded into single cell gel beads (GEMs) and barcoded with a unique molecular identifier (UMI) using the Single Cell 3’ reagent kit (10X Genomics) according to the manufacturer’s protocol. Sequence libraries containing the full-length, barcoded cDNA were generated and sequenced on a NovaSeq 6000 (Illumina) in a dual paired-end sequencing. Data analyses are described in the extended methods section of the online supplement.
Statistics.
Statistical analyses were performed using GraphPad Prism 8. Experimental data are reported as mean ± standard error (SEM), and P ≤ 0.05 was considered as statistically significant. Gaussian distribution was determined using the Shapiro Wilk normality test. Data determined to be parametric were analyzed by unpaired 2-tailed Student’s t test to compare 2 independent groups, or for more than 2 groups, by 2-way ANOVA test followed by Tukey’s HSD post-hoc multiple comparison analysis. Data determined to be non-parametric were analyzed by Mann-Whitney U-test for 2 group comparison or a Kruskal-Wallis test with post-hoc Dunn’s test for more than 2 group comparison. Outliers were detected using the ROUT outlier test with an FDR of 1%. For single-cell RNA-seq analysis, unless otherwise noted, adjusted p-values are reported with correction for multiple testing using Benjamini-Hochberg. No experiment-wide multiple test correction was applied.
RESULTS
Macrophage expression of netrin-1 persists in plaques after cholesterol lowering.
In advanced atherosclerosis, the normalization of hypercholesteremia alone is insufficient to fully regress atherosclerotic plaques43. Hence, in addition to cholesterol lowering therapies, therapeutic strategies are needed to resolve atherosclerotic inflammation and promote tissue repair in the artery wall. To test if therapeutic targeting of netrin-1 in mice with advanced atherosclerosis could reverse macrophage retention and promote inflammation resolution, we generated Ntn1fl/flCx3cr1CreER+ mice which heterozygously express a tamoxifen-inducible CreERT2 fusion protein from the endogenous Cx3cr1 promoter (Cx3cr1CreER+), with normal levels of CX3CR1 expressed from the other allele (Figure 1a, Online Figure II). Upon tamoxifen treatment the netrin-1 gene (Ntn1) is inducibly deleted in cells of the myeloid lineage, including monocytes and macrophages (Figure 1a, Online Figure II), allowing temporal control of netrin-1 expression. To establish atherosclerosis in Ntn1fl/flCx3cr1CreER+ mice and control Ntn1fl/flCx3cr1WT mice, we used an adeno-associated virus (AAV) to induce sustained expression of PCSK9, which reduces hepatic LDLR expression and increases plasma cholesterol levels (Online Figure II). After 20 weeks of Western diet feeding, one group of mice was sacrificed for baseline plaque measurements, while the remaining mice were switched to chow diet to lower plasma cholesterol levels and treated with tamoxifen for 5 days to delete netrin-1 in macrophages (MøΔNtn1) or not (MøCtrl) (Figure 1a). Mice were sacrificed 4 weeks later to assess the effects of macrophage-specific netrin-1 deficiency on plaque area, cellular composition and inflammatory markers.
Figure 1. Myeloid-specific netrin-1 deletion in advanced atherosclerosis alters plaque composition and promotes plaque regression.
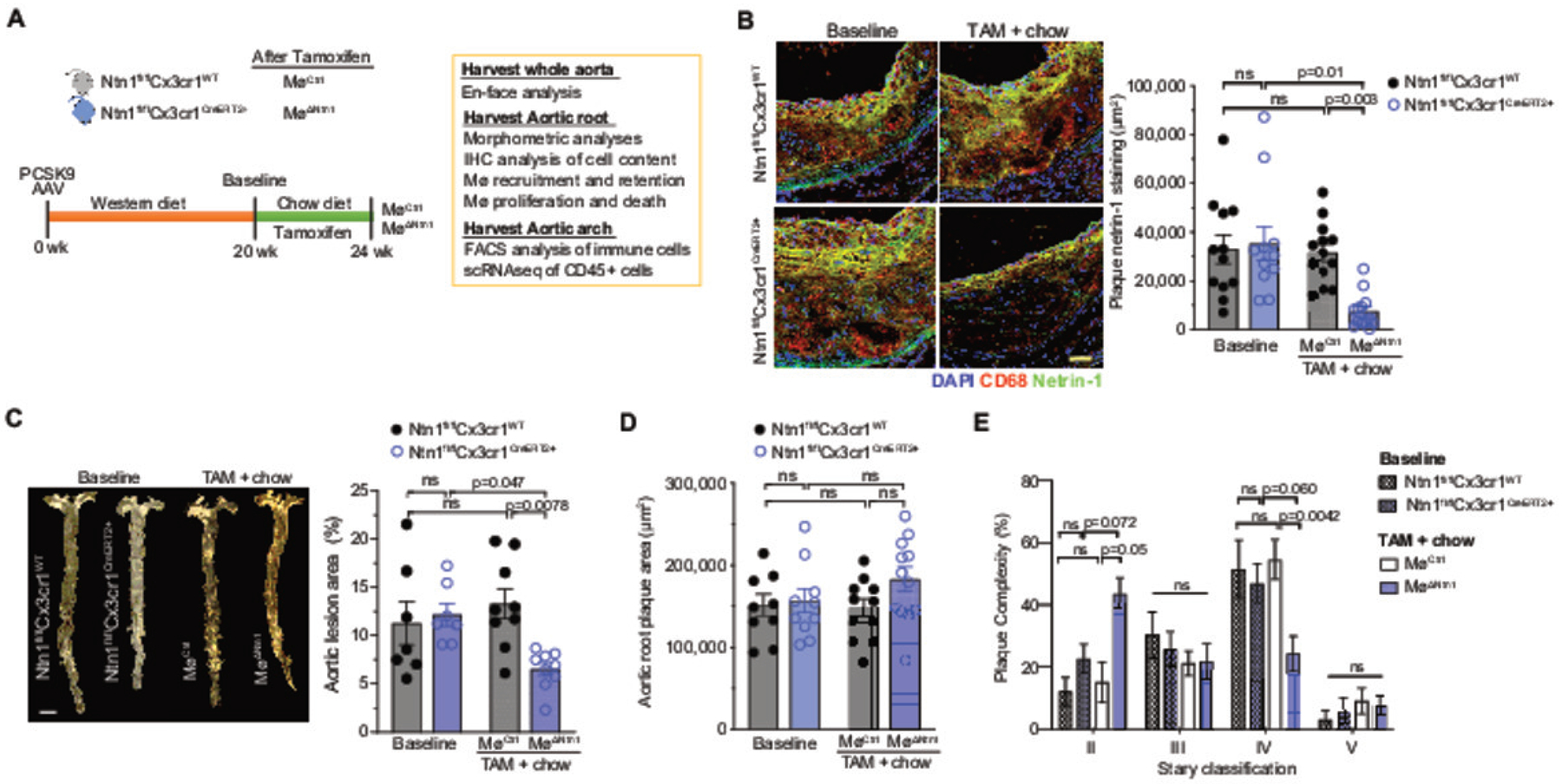
A) Experimental design: Ntn1fl/flCx3cr1WT and Ntn1fl/flCx3cr1CreER+ mice were injected with mPCSK9-AAV and fed Western diet for 20 weeks to establish advanced atherosclerosis. Mice were taken for baseline (BL) measurements or placed on chow diet for 4 weeks to halt atherosclerosis progression and injected with tamoxifen (TAM) to induce Ntn1 deletion (MøΔNtn1) or not (MøCtrl). B) Representative images (left) and quantification (right) of immunostaining for netrin-1 (green), CD68 (red) and DAPI (blue) in cross-sections of aortic root plaques. Scale bar = 50 μm. BL: Ntn1fl/flCx3cr1WT n=12, Ntn1fl/flCx3cr1CreER+ n=11; TAM: MøCtrl n=13, MøΔNtn1 n=13. C) Representative images and quantification of lesion area of the en face aorta of mice at baseline (n=7) and after TAM + chow diet (n=9). Scale bar = 2 mm. D) Quantification of plaque area in cross-sections of the aortic root at baseline (Ntn1fl/flCx3cr1WT n=9, Ntn1fl/flCx3cr1CreER+ n=10) and after TAM + chow diet (MøCtrl n=11, MøΔNtn1 n=12). E) Histological classification of aortic root plaques according to Stary: II, moderate lesions (foam cells, SMCs, intracellular lipid accumulation); III, pre-atheroma (foam cells, SMCs, pools of extracellular lipids); IV, atheroma (foam cells, SMCs, large pools of extracellular lipids, necrotic core); V, fibroatheroma (foam cells, SMCs, large pools of extracellular lipids, large irregular necrotic core). BL: Ntn1fl/flCx3cr1WT n=11, Ntn1fl/flCx3cr1CreER+ n=12; TAM: MøCtrl n=11, MøΔNtn1 n=15). Data are mean ± SEM. P values were determined by (B-D) one-way ANOVA with post-hoc Tukey’s test or (E) Kruskal-Wallis test with Dunn’s correction.
At baseline, aortic root plaques of Ntn1fl/flCx3cr1CreER+ mice and Ntn1fl/flCx3cr1WT mice showed accumulation of netrin-1 in macrophage-rich regions of the plaque (Figure 1b), as previously observed in Apoe–/– and Ldlr–/– mouse models of atherosclerosis19. Four weeks after switch to chow diet and tamoxifen treatment, MøWT mice had similar levels of plaque netrin-1 staining as baseline mice, despite an 80% reduction in total plasma cholesterol levels (Figure 1b, Online Figure II), indicating that macrophage expression of netrin-1 is maintained in plaques, even after normalization of hypercholesterolemia. By contrast, MøΔNtn1 showed an 85% reduction in plaque netrin-1 immunostaining (Figure 1b), and a 65% reduction in Ntn1 mRNA in peritoneal macrophages (Online Figure II), indicating efficient deletion of macrophage netrin-1 in our model and allowing us to test our hypothesis that targeting netrin-1 in advanced atherosclerosis would promote inflammation resolution. Importantly, we observed no differences in body weights, numbers of circulating leukocytes or bone marrow progenitors in MøCtrl and MøΔNtn1 mice (Online Figure III), indicating that myeloid deletion of netrin-1 does not alter hematopoiesis.
Deletion of myeloid-netrin-1 reduces aortic plaque burden and beneficially remodels aortic root plaques.
To evaluate the effects of macrophage netrin-1 silencing, we quantified plaque burden in the aorta en face and in cross-sections of the aortic root before and after tamoxifen treatment. As expected, we observed no difference in plaque area between Ntn1fl/flCx3cr1CreER+ mice and Ntn1fl/flCx3cr1WT mice at baseline (Figure 1c–d). Four weeks after Ntn1fl/flCx3cr1WT mice were switched to chow diet and treated with tamoxifen (MøCtrl), aortic lesion area remained similar to baseline, confirming that reversal of hypercholesterolemia alone does not induce regression of plaque area (Figure 1c–d). By contrast, MøΔNtn1 mice showed a 50% reduction in lesional area in the en face aorta compared to baseline mice (Figure 1d), including in the aortic arch (Online Figure II). In the aortic root, a site of more advanced atherosclerosis, plaque size was similar in MøΔNtn1 and MøCtrl mice (Figure 1d, Online Figure II), but histopathological classication using the Stary method44 revealed a marked decrease in plaque complexity after netrin-1 deletion (Figure 1e). While the majority of aortic root plaques in baseline and MøCtrl mice were advanced stage IV lesions, characterized by a lipid core, macrophage and SMC accumulation, and a thin fribrous cap, most plaques in MøΔNtn1 mice had regressed to stage II lesions (Figure 1e).
To understand how netrin-1 silencing induces plaque remodeling, we assessed the cellular composition of aortic root plaques. Consistent with previous studies showing that reversal of hypercholesterolemia induces a limited reduction in plaque macrophage content, plaque immunostaining for the macrophage marker CD68 was 25% lower in MøCtrl mice that had been switched to chow diet compared to baseline mice (Figure 2a). However, we observed a further decrease in plaque macrophage content in MøΔNtn1 mice, with a net loss of 50% of plaque CD68+ cells from baseline (Figure 2a). As CD68 can be expressed by some plaque SMCs after lipid loading12, we also assessed the macrophage marker F4/80 in the aortic arch by flow cytometry, which confirmed a 50% reduction in aortic macrophages in MøΔNtn1 mice (Online Figure IV). In addition, plasma levels of IL-1b, which is secreted by plaque macrophages, were lower in MøΔNtn1 mice compared to MøCtrl and baseline mice (Figure 2b). As macrophage-derived netrin-1 can also elicit the recruitment of vascular SMCs into the intima, we next quantified expression of alpha smooth muscle actin (ACTA2) as a marker SMCs. Immunostaining of aortic root plaques revealed a marked decrease in intimal SMCs in MøΔNtn1 mice, compared to MøCtrl and baseline mice, but no change in fibrous cap SMC area (Figure 2c, d). Staining of plaques with Movat’s pentachrome stain revealed an increase in minimal fibrous cap thickness and number of fibrous cap layers in plaques of MøΔNtn1 mice, indicating that netrin-1 deletion does not adversely affect the ACTA2+ SMC-rich fibrous cap (Online Figure IV). Finally, PicroSirius Red staining showed an increase in intralesional collagen content in MøΔNtn1 plaques, as compared to MøCtrl and baseline plaques (Figure 2e). Together, these data indicate that netrin-1 silencing in advanced atherosclerosis favorably remodels plaques by reducing macrophage content and enhancing markers of plaque stability, including collagen deposition and fibrous cap thickness.
Figure 2. Deletion of netrin-1 reduces plaque macrophage and smooth muscle burden.
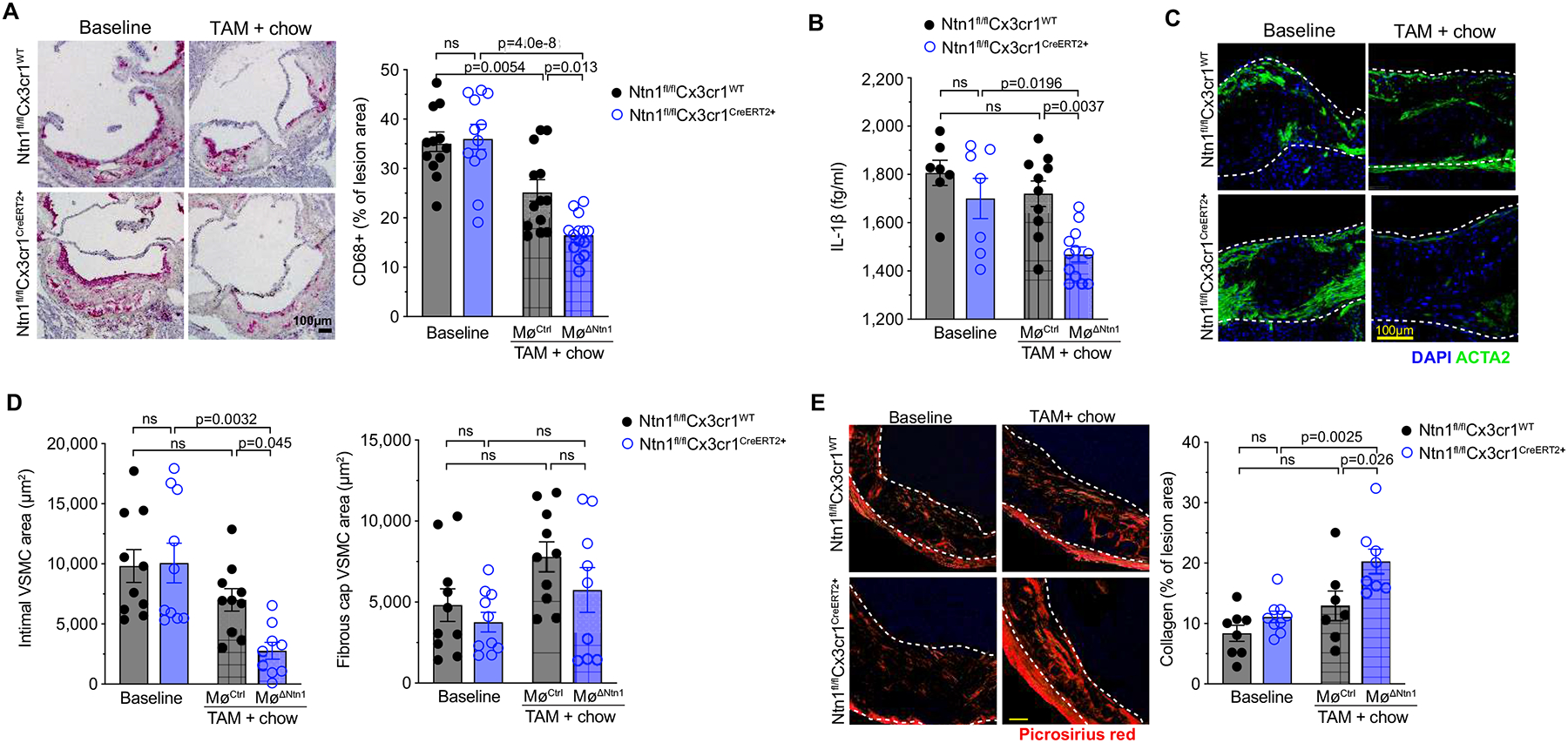
A) Representative images and quantification of CD68 staining in cross-sections of the aortic root (scale bar = 100 μm) of mice at baseline (Ntn1fl/flCx3cr1WT n=12, Ntn1fl/flCx3cr1CreER+ n=12) and 4 weeks after switch to chow diet and tamoxifen treatment (MøCtrl n=13, MøΔNtn1 n=14). B) Plasma levels of interleukin (IL)-1β (BL: n=7; TAM: MøCtrl n=10, MøΔNtn1 n=12). C) Representative images of alpha smooth muscle actin (ACTA2; green) and DAPI (blue) staining to detect putative vascular smooth muscle cells (VSMC) in aortic root plaques. Scale bar = 100 μm. D) Quantification of VSMC accumulation in the intima (left) and fibrous cap (right) of aortic root plaques (BL: Ntn1fl/flCx3cr1WT n=10, Ntn1fl/flCx3cr1CreER+ n=10; TAM: MøCtrl n=10, MøΔNtn1 n=9). E) Representative images and quantification of PicroSirius Red staining for collagen. Scale bar = 100 μm (BL: Ntn1fl/flCx3cr1WT n=8, Ntn1fl/flCx3cr1CreER+ n=9; TAM: MøCtrl n=7, MøΔNtn1 n=8). Data are mean ± SEM. P values were determined by [A, B, D (right panel), E] one-way ANOVA with post-hoc Tukey’s test, or [D (left panel)] Kruskal-Wallis test with Dunn’s correction.
Single cell RNA-seq reveals netrin-1-specific effects on plaque monocyte and macrophage gene expression.
To gain further insight into the mechanisms by which myeloid netrin-1 silencing promotes atherosclerosis regression, we performed scRNA-seq of CD45+ cells isolated from the aortic arches of mice using the 10X Genomics platform (Figure 3a). Using canonical correlation analysis (CCA), unsupervised Louvain clustering45 and t-SNE dimension reduction, we identified 13 distinct immune cell clusters (Figure 3b). Using SingleR, which leverages the ImmGen database to characterize cells by their closest match, in an unsupervised manner46, we identified one monocyte and two macrophage clusters, six T cell clusters, one natural killer (NK) cell clusters, one innate lymphoid cell (ILC) cluster and two dendritic cell (DC) clusters. We further characterized these immune cell subpopulations in the plaque by defining the genes with the highest differential expression for a given cluster as compared to all other cells in the dataset (Online Figure V), which showed concordance with recently published scRNA-seq of the immune cell repertoire of Apoe–/– and Ldlr–/– mouse atherosclerotic plaques47.
Figure 3. Single cell RNA-sequencing of aortic immune cells.
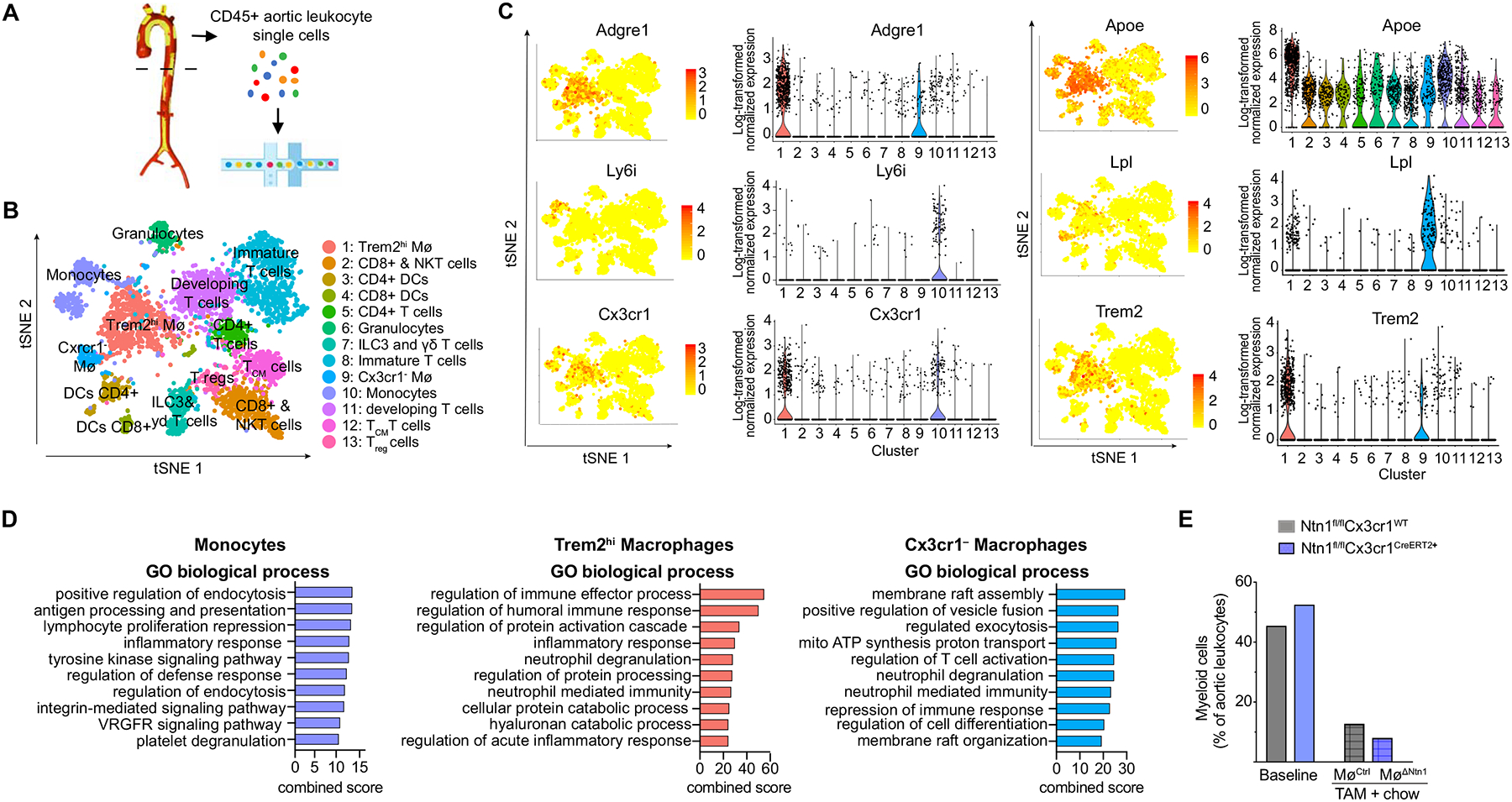
A) Experimental design of aortic leukocyte isolation and single cell transcriptome analysis. B) t-Stochastic neighbor embedding (t-SNE) visualization of aortic CD45+ cells showing 13 leukocyte clusters based on single cell transcriptomes of all groups merged. C) Gene expression of principal monocyte and macrophage markers projected onto t-SNE plots (left) and Violin plots (right) of log-transformed normalized gene expression in the clusters. D) Gene ontology analysis showing top biological processes of monocyte and macrophage clusters in the aortic arches of mice (combined score is multiplication of log p-value and the z-score). E) Combined frequency of myeloid cells in the aortic arch at baseline and after TAM + chow diet. Data are from n = 5 mice pooled per group; n = 4,350 cells (all groups merged).
All captured aortic leukocytes expressed Ptprc, the gene encoding CD45, and as expected, cells in the monocyte and macrophage clusters expressed high levels of macrophage markers such as Cd68, Csf1r, Fcgr1 (encoding CD64) and Itgam (encoding CD11b) (Online Figure VI). While monocytes (cluster 10) expressed high levels of monocyte-defining genes, Ly6i and Ly6c, the Trem2hi (cluster 1) and Cx3cr1– (cluster 9) macrophage populations were defined by high levels of Adrgre1 (encoding F4/80) (Figure 3c). Unlike some recently published studies, we did not detect a separate Cd11c expressing macrophage cluster in plaques (Online Figure VI). Notably, we observed high expression of Cx3cr1 in monocytes (cluster 10) and Trem2hi macrophages (cluster 1), which would drive Cre expression and Ntn1 deletion in these cell types (Figure 3c). However, expression of Cx3cr1 was absent in the second macrophage cluster (cluster 9), suggesting that Ntn1 deletion would be minimal or absent in this macrophage subset (Figure 3c). Cx3cr1– macrophages express high levels of lipid metabolism (Lpl, Pltp) and PPARγ signaling (Cd36, Dbi) genes (Figure 3c, Online Figure VI). The origin and fate of Cx3cr1– macrophages has been debated in the literature48, as mononuclear phagocytes and their progenitors in the bone marrow broadly express Cx3cr1, and we noted that Cx3cr1– macrophages had decreased expression of bone marrow derived macrophage genes (BRENDA Tissue and Enzyme Source Ontology, BTO_0004732) (Online Figure VI). To investigate whether Cx3cr1– macrophages might derive from a non-myeloid cell origin, we built a pseudotime trajectory of the monocyte and macrophages, which allowed us to track their differentiation based on biological progression. While monocytes continuously differentiated into Trem2hi macrophages (cluster 1), the Cx3cr1– macrophages (cluster 9) remained distinct (Online Figure VI). Cx3cr1– macrophages exhibited stemness genes, especially in G1 and G2M phases, and we found that they partially resembled embryonically seeded “arterial wall Mø”. However, our Cx3cr1– cluster did not match the previously described “resident-like Mø” signature47, 49, consistent with data that CX3CR1 is essential for the survival of embryonically-implanted tissue-resident macrophages Mø in the arterial wall50, 51. As vascular SMCs can acquire macrophage markers in atherosclerotic plaques12, 52, we also compared the transcriptome of our Cx3cr1– macrophages to that of arterial wall vascular SMCs. We found that the top 20 genes of the arterial wall vascular SMC (based on TISSUE Text-mining Tissue Protein Expression Evidence Scores, BTO_00045578) were enriched within Cx3cr1– macrophages, when compared to monocytes and Trem2hi macrophages (Online Figure VI). The Cx3cr1– macrophages expressed high levels of vascular SMC markers like Fn1 (encoding fibronectin 1), Tagln2 (encoding transgelin-2) and Lpl (encoding lipoprotein lipase), which has been shown to contribute to foam cell formation53. Other vascular SMC markers, such as Myh11, Acta2 or Smtn were only sparsely expressed in our single cell analyses. Gene set enrichment analysis of the transcriptome of Cx3cr1– macrophage revealed processes involved in membrane reorganization and cell differentiation (Figure 3d), suggesting that these cells might be undergoing macrophage to myofibroblast transition.11
scRNA-seq analysis confirmed that switching mice to chow diet reduced the frequency of myeloid cells in the aorta compared to baseline mice, and this decrease was more pronounced in MøΔNtn1 mice than in MøCtrl mice (Figure 3e). Analysis of genes differentially expressed in aortic monocytes [defined by expression of Ly6c2, Chil3, and Cebpb (Figure 4a, Online Figure V)] of MøΔNtn1 and MøCtrl mice showed that netrin-1 silencing was associated with upregulation of genes involved in limiting pro-inflammatory immune cell activation (e.g., Il4ra, Cd72, Tgfb1) and lipid metabolism (e.g., Lipin, Hacd4) (Figure 4b). Gene ontology analysis revealed that switching mice to chow diet upregulated monocyte pathways involved in cell motility and migration, which was further enhanced by silencing netrin-1 (Figure 4c). We also noted upregulation of phagocytosis and immune cell priming pathways in plaque monocytes of MøΔNtn1 mice compared to MøCtrl and baseline mice. Together, these findings show that silencing netrin-1 releases the brake on macrophage migration and innate immune pathways involved in tissue repair in the plaque.
Figure 4. Effects of Netrin-1 deletion on plaque monocyte and macrophage transcriptomic profiles.
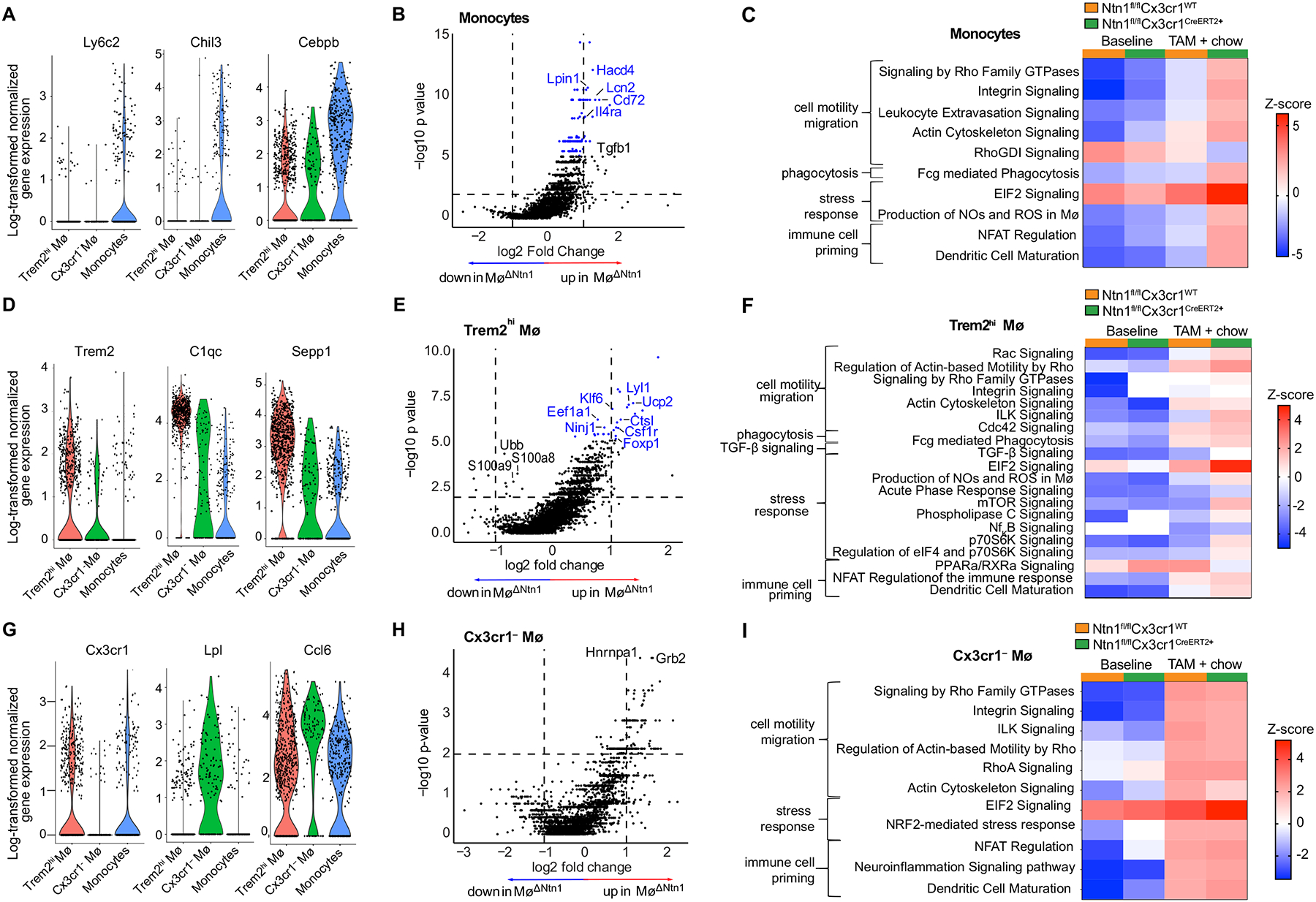
A, D, G) Violin plots of log-transformed normalized gene expression data showing cluster defining genes in (A) aortic monocytes, (D) Trem2hi macrophages and (G) Cx3cr1– macrophages. B, E, H) Volcano plots of the most differentially expressed genes in (B) monocytes, (E) Trem2hi macrophages, and (H) Cx3cr1– macrophages after Ntn1 deletion (Ntn1ΔMø). Horizontal line indicates an unadjusted p-value of 0.01, vertical lines indicate a logFC values of 1 and −1 (i.e. 2-fold change in either direction), and points in blue represent genes with an adjusted p-value (Benjamini-Hochberg) <0.1. C, F, I) Heatmaps of differentially regulated pathways in aortic (C) monocytes, (F) Trem2hi macrophages, and (I) Cx3cr1– macrophages of mice at baseline and after TAM + chow diet treatment (CtrlMø and Ntn1ΔMø). Significant pathways were identified based on a 5% false discovery rate using the Benjamini-Hochberg procedure within the Canonical Pathways annotations by Ingenuity Pathway Analysis (content version 62089861). The pathways shown were selected on the most differentially expressed z-scores of the sum of log-fold transformed gene expression between the groups. Data are from n = 5 mice pooled per group.
Tamoxifen-induced netrin-1 silencing also induced changes in Trem2hi macrophages (cluster 1), which have roles in lipid metabolism54 and are characterized by expression of genes involved in phagocytosis (C1qc, Fcgr, Dab2), monocyte to macrophage differentiation (Cd14, Csfr1), extracellular matrix remodeling cathepsins (Ctsc, Ctsb, Ctss), and the M2-like macrophage protein selenoprotein-1 (Sepp1) (Figure 4d, Online Figure V). Compared to MøCtrl mice, Trem2hi macrophages from MøΔNtn1 mice showed reduced expression of S100a8 and S100a9, which trigger pro-inflammatory responses via TLR4 and RAGE55, and increased expression of genes that protect from aortic inflammation and endoplasmic reticulum stress, including Klf656, 57, Ucp2, FoxP158, and Eef1a59 (Figure 4e). Gene ontology analysis revealed upregulation of cell motility and migration pathways in both MøΔNtn1 and MøCtrl Trem2hi macrophages compared to baseline, with higher induction in MøΔNtn1 mice (Figure 4f). Notably, MøΔNtn1 Trem2hi macrophages mice showed upregulation of Rac, Rho, and integrin-linked kinase (ILK) signaling (Figure 4f) – pathways that we previously showed were inhibited by netrin-1 in vitro19. Trem2hi macrophages from MøΔNtn1 mice also showed higher expression of pathways involved in phagocytosis, TGF-b signaling, mTOR signaling and dendritic cell maturation, as compared to Trem2hi macrophages from MøCtrl and baseline mice (Figure 4f). By contrast, we observed no difference in expression of these pathways in Cx3cr1– macrophages from MøΔNtn1 and MøCtrl mice, consistent with the lack of CX3CR1-driven CreERT2 expression required for Ntn1 deletion (Figure 4g–i). Collectively, these data indicate that netrin-1 silencing in plaque monocytes and Trem2hi macrophages upregulates gene pathways controlling cell motility and migration.
Targeting netrin-1 alters monocyte-macrophage dynamics in the plaque.
To dissect the mechanisms by which plaque macrophages are reduced upon netrin-1 silencing, we quantified monocyte-macrophage dynamics in plaques by immunohistochemistry, flow cytometry, and bead-pulse labeling. We first quantified cellular proliferation by Ki67 staining, as local macrophage proliferation contributes to macrophage burden in advanced plaques60. Compared to baseline hypercholesterolemic mice, we observed a 50% reduction in CD68+ Ki67+ cells in aortic root plaques of MøCtrl mice, indicating that just switching control mice to chow diet alone reduces macrophage proliferation (Figure 5a). Plaque macrophage proliferation was further reduced in MøΔNtn1 mice, with a net 75% decrease of CD68+ Ki67+ cells compared to baseline mice (Figure 5a). To assess the impact of netrin-1 silencing on macrophage death, we stained plaques for the apoptosis marker TUNEL. Although netrin-1 is a survival factor for macrophages, we did not observe an increase in TUNEL staining in MøΔNtn1 plaques (Figure 5b). Rather, MøΔNtn1 plaques contained 50% fewer apoptotic cells compared to MøCtrl and baseline mice. These data suggest that macrophage proliferation and apoptotic cell burden are both reduced after Ntn1 deletion in advanced plaques.
Figure 5. Deletion of netrin-1 alters monocyte – macrophage dynamics in the artery wall.
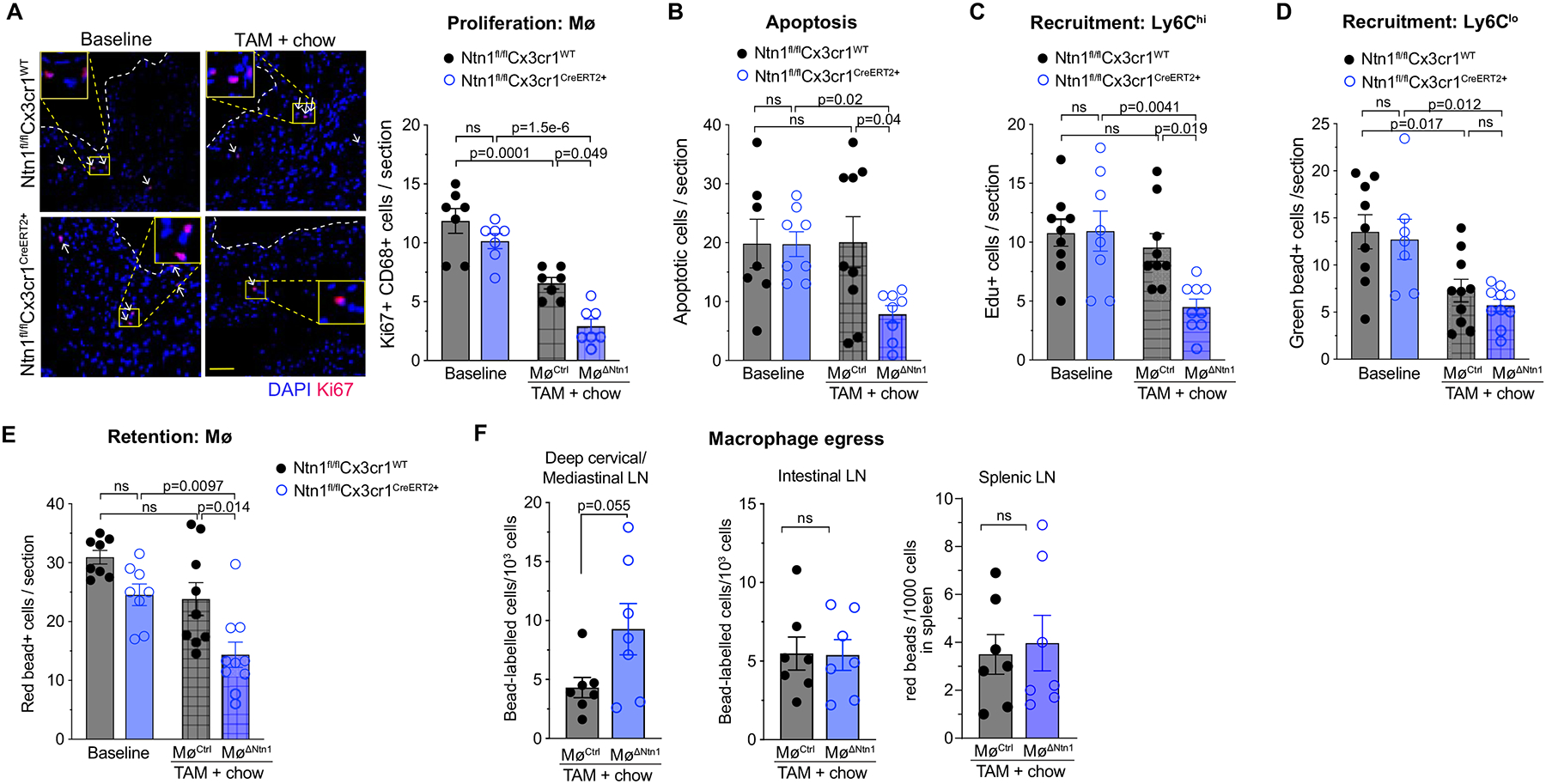
A) Representative images and quantification of immunostaining for the proliferation marker Ki67 (red) and DAPI (blue) in aortic root plaques of mice at baseline and after chow diet + tamoxifen (TAM) treatment (MøCtrl and MøΔNtn1). Scale bar = 100 μm. n = 7 mice/group. B) Quantification of TUNEL+ apoptotic cells in aortic root plaques of mice at baseline (Ntn1fl/flCx3cr1WT n=7, Ntn1fl/flCx3cr1CreER+ n=8) and after chow diet + TAM (MøCtrl n=9, MøΔNtn1 n=8). C) Quantification of EdU+ Ly6hi monocyte recruitment into aortic root plaques (BL: Ntn1fl/flCx3cr1WT n=9, Ntn1fl/flCx3cr1CreER+ n=8; TAM: MøCtrl n=9, MøΔNtn1 n=9). D) Quantification of green bead-labelled Ly6lo monocyte recruitment into aortic root plaques (BL: Ntn1fl/flCx3cr1WT n=9, Ntn1fl/flCx3cr1CreER+ n=7; TAM: MøCtrl n=10, MøΔNtn1 n=10). E) Retention of baseline-labelled macrophages in aortic root plaques of MøCtrl and MøΔNtn1 mice. (BL: Ntn1fl/flCx3cr1WT n=8, Ntn1fl/flCx3cr1CreER+ n=8; TAM: MøCtrl n=9, MøΔNtn1 n=10). F) Quantification of bead labeled macrophages in the mediastinal, intestinal, and splenic lymph nodes (LN) of MøCtrl and MøΔNtn1 (n=7 mice/group). A-F) Data are mean ± SEM. P values were determined by (A-E) one-way ANOVA with post-hoc Tukey’s test, or (F) two tailed Student’s t-test for mediastinal and intestinal LN; or Mann-Whitney test for Spleen LN.
Next, we assessed monocyte recruitment into and monocyte-derived macrophage egress from aortic root plaques after netrin-1 silencing. Monocyte migration into the plaque was measured 72 h prior to the end of the treatment period by pulse-labeling Ly6Chi and Ly6Clo monocytes using EdU61 or green fluorescent microspheres19, respectively. We observed a marked decrease in Ly6Chi monocyte recruitment into plaques of MøΔNtn1 mice compared to MøCtrl or baseline mice (Figure 5c), which was confirmed by flow cytometry of the aortic arch (Online Figure VII). By contrast, we observed no difference in recruitment of patrolling Ly6Clo monocytes into plaques of MøΔNtn1 and MøCtrl mice, although both groups showed lower Ly6Clo monocyte recruitment compared to baseline mice (Figure 5d). Next, to understand whether netrin-1 altered macrophage retention in plaques, we pulse-labeled Ly6Clo monocytes with red fluorescent microspheres at baseline immediately prior to the switch to chow diet + tamoxifen treatment, and measured the number of labelled monocyte-derived macrophages in plaques four weeks later. While we observed similar numbers of bead-labelled macrophages in baseline and MøCtrl plaques, significantly fewer bead-labelled macrophages were retained in MøΔNtn1 plaques (Figure 5e), suggesting macrophage eggress from plaques after netrin-1 silencing. Isolation of lymph nodes showed an increase in bead-labeled cells in the deep cervical and mediastinal lymph nodes draining the aorta of MøΔNtn1 compared to MøCtrl mice, but no difference in the splenic and intestinal lymph nodes (Figure 5f).
The chemokine receptor CCR7, which regulates cell migration to lymph nodes in response to CCL19 and CCL21, has been shown to be upregulated in CD68+ cells of regressing plaques and to be essential for regression62, 63. Pathway analysis of differentially expressed genes from the scRNA-Seq showed that the transendothelial migration pathway (KEGG pathway mmu6470), which includes CCR7, was upregulated in monocytes and Trem2hi macrophages (Figure 6a, Online Table I) from plaques of MøΔNtn1 mice compared to MøCtrl and baseline mice. Ccr7 transcript levels were higher in aortic myeloid cells of MøΔNtn1 compared to MøCtrl mice (Figure 6b), and immunostaining confirmed an increase in the number of CD68+ cells co-expressing CCR7 (Online Figure VII) and co-localization of CCR7 and CD68 staining (Figure 6c) in the intima of MøΔNtn1 compared to MøCtrl plaques. It should be noted that the CCR7 antibody used showed some non-specific staining in the medial layer of plaques from Ccr7–/–Apoe–/– mice, which were used as a control (Online Figure I). Pathway analysis of genes differentially expressed in the scRNA-Seq confirmed upregulation of the CCR7 signaling gene set in aortic monocytes and Trem2hi macrophages from MøΔNtn1 mice compared to MøCtrl and baseline mice (Figure 6d–e, Online Table I) that leads to increased cytoskeletal rearrangements and chemotaxis.
Figure 6. Netrin-1 alters expression of Ccr7 and downstream signaling genes.
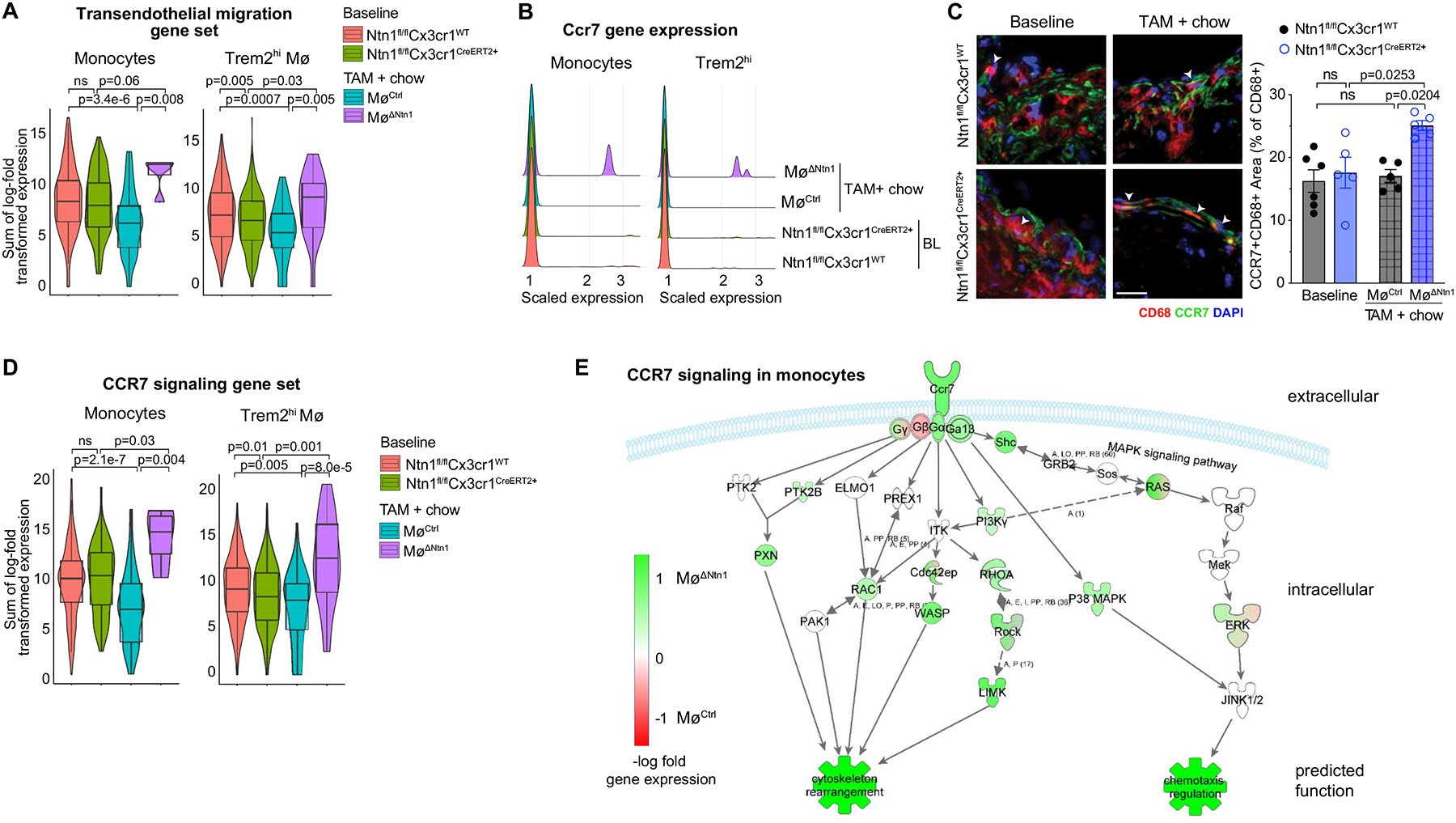
A) Violin plots of the sum of log-transformed normalized expression for transendothelial migration genes (KEGG mmu04670) at baseline and after chow diet + tamoxifen (TAM) treatment. B) Ridge plots of log-transformed normalized expression of Ccr7 in monocytes and Trem2hi macrophages. C) Immunostaining for CCR7 (green), CD68 (red) and DAPI (blue). Scale bar = 50 μm. BL: Ntn1fl/flCx3cr1WT n=6, Ntn1fl/flCx3cr1CreER+ n=5; TAM: MøCtrl n=5, MøΔNtn1 n=5. Quantification (right) shows percentage of CD68+ area that also stained positive for CCR7. D) Violin plots of the sum of log-transformed normalized expression for the 23 highest expressed genes involved in CCR7 signaling. E) Log-fold change of genes involved in the CCR7 signaling of monocytes in CtrlMø and Ntn1ΔMø mice (green upregulated, red downregulated in Ntn1ΔMø). (A-B, D-E) Data are from n = 5 mice pooled per group. (A, D) Box plots represent mean ± IQR and 95%-CI; P-values were calculated using a two-sided Wilcoxon rank-sum test. (C) Data are mean ± SEM. P values were determined by one-way ANOVA with post-hoc Tukey’s test.
Gene ontology analysis of monocytes and Trem2hi macrophages of MøΔNtn1 and MøCtrl mice also revealed an increase in stress response pathways upon netrin-1 silencing in plaques, including the production of reactive oxygen and nitrogen species, EIF2 signaling and mTOR signaling (Figure 4e, f). While netrin-1 has not previously been linked to regulation of stress responses, reactive oxygen generation and ER stress have been reported in the transition of plaque monocytes and macrophages to a tissue reparative state. The reactive oxygen burst promotes clearance of apoptotic/necrotic cells and actin polymerization needed for tissue repair64, 65, and ER stress favors macrophage differentiation to an M2-like tissue reparative state66. To test whether deletion of Ntn1 altered the accumulation of pro-resolving M2-like macrophages in plaques, we performed immunostaining for the M2-associated markers mannose receptor (MR1/CD206) and Arginase 1 (ARG1). Baseline and MøCtrl plaques showed similar MR1 and ARG1 expression levels (Figure 7a, Online Figure VI), consistent with findings that normalization of hypercholesterolemia is insufficient to induce a proresolving state in the plaque. However, upon netrin-1 silencing, we observed marked increases in MR1 and ARG1 staining (2.5-fold and 4-fold, respectively) in MøΔNtn1 plaques (Figure 7a, Online Figure VI). Furthermore, aortic Trem2hi macrophages from MøΔNtn1 mice showed an increase in TGF-b signaling compared to MøCtrl mice (Figure 4), which can promote M2 macrophage polarization67. While the macrophages clusters in our scRNA-Seq do not fall into classical M1 and M2 subsets, comparison of monocyte and Trem2hi macrophage gene signatures to a recently published data set describing gene profiles of mouse M2-like macrophages68 showed enrichment of M2 marker gene expression in MøΔNtn1 mice compared to MøCtrl and baseline mice (Figure 7b, Online Table I).
Figure 7. Deletion of Netrin-1 promotes a proresolving macrophage phenotype in the regressing plaque.
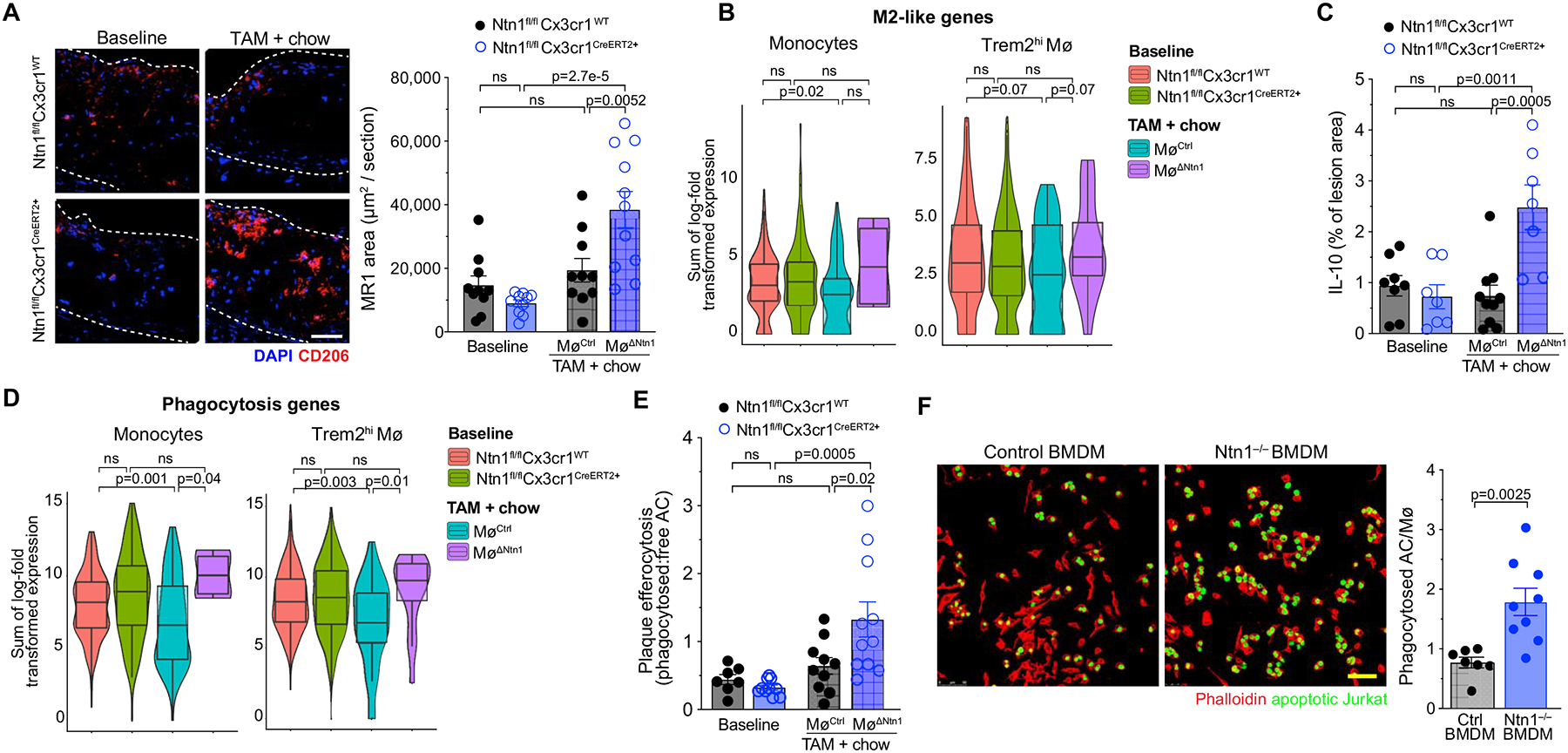
A) Representative immunostaining and quantification for mannose receptor (MR1; red) and DAPI (blue) in aortic root plaques of mice at baseline (n=10/group) and after chow diet + tamoxifen (TAM) treatment (MøCtrl n=10, MøΔNtn1 n=11). Scale bar = 100 μm. B) Violin plots of the sum of log-transformed normalized expression for the 21 most highly expressed genes of M2-like macrophages. C) Quantification of immunostaining for interleukin (IL)-10 in aortic root plaques of mice at baseline (Ntn1fl/flCx3cr1WT n=8, Ntn1fl/flCx3cr1CreER+ n=7) and after chow diet + TAM treatment (MøCtrl n=10, MøΔNtn1 n=7). D) Violin plots of the sum of log-transformed normalized expression for the most highly expressed phagocytosis genes (GO: 0006909). E) Quantification of efferocytosis of apoptotic cells in aortic root plaques of mice at baseline (Ntn1fl/flCx3cr1WT n=7, Ntn1fl/flCx3cr1CreER+ n=10) and after chow diet + TAM treatment (MøCtrl n=10, MøΔNtn1 n=10). F) Representative images and quantification of in vitro efferocytosis of apoptotic Jurkat cells (green) by control (n=7) and Ntn1–/– (n=9) bone marrow derived macrophages (BMDM). Scale bar = 50 μm. (B+D) Box plots represent mean ± IQR and 95%-CI; P-values were calculated using a two-sided Wilcoxon rank-sum test. (A,C,E-F) Data are mean ± SEM. P values were determined by one-way ANOVA with post-hoc Tukey’s test (A,C, E) or Student’s t-test (F).
Pro-resolving macrophages are sources of the cytokine IL-10, which dampens inflammation and promotes tissue reparative functions, including efferocytosis. Immunostaining showed a marked increase of IL-10 in plaques of MøΔNtn1 mice compared to MøCtrl and baseline mice (Figure 7c). Pathway analysis of differentially expressed genes from the scRNA-Seq showed that genes involved in phagocytosis (GO: 0006909) were upregulated in monocytes and Trem2hi macrophages from plaques of MøΔNtn1 mice compared to MøCtrl and baseline mice (Figure 7d, Online Table I). To assess phagocytosis of apoptotic cells (efferocytosis), we measured the ratio of Mac2-associated TUNEL+ cells to free TUNEL+ cells in aortic root plaques. We observed a 2-fold increase in macrophages associated with apoptotic cells in MøΔNtn1 plaques compared to MøCtrl and baseline plaques (Figure 7e). To understand whether the increase in efferocytosis upon netrin-1 silencing was due to cell-intrinsic changes in actin polymerization and phagocytosis or local increases in pro-resolving signals in the plaque such as IL-10, we performed an in vitro efferocytosis assay using BMDMs from mice with global cre-mediated deletion of Ntn1 (Ntn1fl/flUbccre+) or Ntn1-intact control (Ntn1fl/flUbcCre–) mice. Ntn1–/– BMDMs incubated with apoptotic Jurkat cells showed increased efferocytotic uptake of on a per cell basis than wild type macrophages (Figure 7f). These findings are consistent with an increase in actin polymerization upon netrin-1 silencing, which is required for phagocytosis of large cargo such as apoptotic cells.
DISCUSSION
In atherosclerosis, reversal of hypercholesteremia slows clinical disease progression and promotes plaque stabilization, resulting in a reduction of major adverse cardiovascular events. However, lipid lowering therapies alone have proven insufficient to fully regress advanced plaques, resulting in an enduring burden of atherosclerosis69. Studies in mouse models of atherosclerosis suggest that disease regression requires reshaping the plaque immune cell landscape to enable inflammation resolution and to enhance tissue repair61, 70. Macrophages play a central role in sustaining chronic inflammation in the plaque by secreting inflammatory mediators that act on other immune and vascular cells, and contribute to plaque instability. In advanced plaques, macrophages become dysfunctional and their inability to effectively phagocytose apoptotic cells or egress from the plaque hinders regression. Our data show that netrin-1 secretion by macrophages contributes to this maladaptive macrophage phenotype in atherosclerosis by impeding inflammation resolution19. We find that plasma cholesterol lowering after a diet change in mice with advanced atherosclerosis halts plaque progression in the aortic root, but fails to reduce plaque size or complexity, and this is associated with continued expression of netrin-1 in plaque macrophages. Induced deletion of netrin-1 expression in myeloid cells, coincident with lipid lowering, licensed plaque regression by reshaping the immune landscape at the transcriptional and phenotypic levels, and altering monocyte-macrophage kinetic processes to reduce macrophage burden. Four weeks after netrin-1 silencing, plaque burden in the aorta was decreased by 50% and advanced plaques in the aortic root, while not different in overall area, showed beneficial remodeling, including lower macrophage and intimal SMC content, and higher collagen content. This was accompanied by a shift in the phenotype of residual macrophages toward a pro-resolving state, as evidenced by increased M2-like marker expression, IL-10 expression and efferocytosis. Collectively, these data show that targeting netrin-1 in conjunction with lipid lowering releases the brake on inflammation resolution, allowing tissue repair and plaque contraction to proceed.
Since the 1980s, numerous studies have shown the importance of controlling the plaque’s macrophage content in order to reduce atherosclerosis and plaque vulnerability to rupture4, 71, 72. The plaque’s net macrophage content is derived from newly recruited blood monocytes4, 72, 73, proliferation of aortic resident and recruited macrophages60, 74, their unresolved cell death14, 75 and macrophage egress62, 76 from the artery wall. We performed a comprehensive analysis of these processes to understand how silencing netrin-1 reduces macrophage burden in the artery wall. Hypercholesteremia and atherosclerosis stimulate proliferation of hematopoietic precursors, expand monocyte reservoirs, and induce monocytosis77, 78, but this is reversable upon lipid lowering. Accordingly, we found that bone marrow precursor cells and monocytes returned to normal levels four weeks after switching atherosclerotic mice to chow diet, and levels were unaffected by myeloid netrin-1 silencing, implicating local changes at the arterial wall in driving regression. Using in vivo monocyte labeling to track monocyte recruitment to and macrophage retention in plaques, we found that netrin-1 silencing in myeloid cells selectively reduced the recruitment of pro-inflammatory Ly6Chi monocytes by 50%. This was complemented by decreased retention of monocyte-derived macrophages in plaques, suggesting increased egress of macrophages from the artery wall. The chemokine receptor CCR7 directs macrophage emigration from various inflamed tissues (e.g., intestine79, adipose tissue80 and atherosclerotic plaques62, 63, 81) to the lymph node. Consistent with this mechanism of macrophage egress, we observed upregulation of macrophage expression of Ccr7 and a defined set of genes involved in CCR7 signaling, as well as an increase in bead-labelled Mø in the aortic draining lymph nodes after netrin-1 silencing. In addition to the altered flux of monocyte-derived macrophages in plaques, we found that macrophage proliferation and apoptotic cell accumulation in plaques were quelled upon netrin-1 silencing. The decrease in apoptotic cells upon netrin-1 silencing was somewhat surprising as netrin-1 has been shown to be a survival signal for macrophages during oxidative stress, however, we observed a compensatory increase in efferocytosis which would reduce apoptotic cell burden in plaques. Notably, our studies suggest that both cell-intrinsic increases in actin polymerization and microenvironmental signals (i.e., IL-10) likely contribute to increasing efferocytosis in the plaque upon netrin-1 silencing.
The ability to sequence single cells in complex tissues has revolutionized the study of atherosclerosis in mice and humans, and revealed the heterogeneity of the immune cell populations in the artery wall. scRNA-Seq of CD45+ cells isolated from the aortic arches of Ntn1fl/flCx3cr1WT and Ntn1fl/flCx3cr1CreER+ mice identified 13 immune cell clusters, including one monocyte and two macrophage clusters. Analysis of the transcriptomic signature of plaque monocytes from atherosclerotic mice switched from Western to chow diet and treated with tamoxifen showed upregulation of genes involved in cell motility, phagocytosis, and immune cell activation after lipid lowering, with these changes more pronounced in monocytes of MøΔNtn1 mice compared to MøCtrl mice. Monocytes from MøΔNtn1 mice also showed higher expression of genes involved in limiting inflammation (e.g., Il4ra, Cd72, Tgfb1) and lipid metabolism (e.g., Lipin, Hacd4). Similarly, the Trem2hi macrophage cluster, which is prominent in metabolic disease47, 49, showed upregulation of pathways involved in cell motility and migration after netrin-1 silencing, including integrin-linked kinase (ILK) signaling, Rac signaling and actin cytoskeleton, some of which have previously been shown to be inhibited by netrin-1 in macrophages in vitro19. Trem2hi macrophages also showed higher expression of gene pathways involved in phagocytosis, and TGF-b signaling, and reduced expression of the pro-inflammatory mediators S100a8 and S100a9, which promote local and systemic inflammation55. Together, these data support the results of our monocyte-macrophage tracking experiments, which show that silencing netrin-1 reduces macrophage retention and inflammation, and stimulates the phagocytosis of apoptotic cells, all of which contribute to inflammation resolution.
Our scRNA-seq analyses also identified a plaque macrophage cluster that lacked expression of Cx3cr1, in which ERT2 would not be expressed. Consistent with this, we did not observe differences in the expression of migratory or phagocytic genes in Cx3cr1– macrophages from MøΔNtn1 and MøCtrl. Studies have shown that vascular SMCs can acquire macrophage markers in atherosclerotic plaques12, 52, raising the possibility that these Cx3cr1– macrophages may be of SMC origin. In support of this, Cx3cr1– macrophages expressed high levels of vascular SMC markers like Fn1 (encodes fibronectin 1), Tagln2 (encodes transgelin-2) and Lpl (encodes lipoprotein lipase, which contributes to foam cell formation53), but other vascular SMC markers such as Myh11, Acta2 or Smtn were sparsely expressed in our scRNA-seq and could not be used to elucidate cell origins. Notably, the aortic cells sequenced were all CD45+, and most studies have detected little to no CD45 expression in vascular SMC-derived macrophages6. It is also possible that these Cx3cr1– macrophages represent a population that has undergone macrophage to myofibroblast transitions (MMT), thereby expressing CD45 as well as mesenchymal markers,11 but further studies will be needed to understand the origins of aortic Cx3cr1– macrophages.
Clinically, the stabilization of plaques is a major goal in treating CAD patients to prevent adverse cardiovascular events, such as stroke and myocardial infarction82. Plaque stability is defined by indices including complexity, inflammatory status, and thickness of the fibrous cap, which protects the plaque from rupture. While aortic plaque burden regressed upon netrin-1 silencing, the more advanced plaques in the aortic root did not change in size. Nevertheless, we observed beneficial remodeling of aortic root plaques of MøΔNtn1 mice, including decreased plaque complexity, and increased intralesional collagen content and fibrous cap thickness. Netrin-1 induces chemoattraction of vascular SMCs via the neogenin receptor19, and while SMCs in the intimal space can promote collagen deposition that increase plaque stability, these cells can also transition into inflammatory phenotypes that are detrimental11. We found that myeloid netrin-1 silencing in advanced plaques reduced intimal vascular SMC infiltration, but importantly, ACTA2 expression and collagen deposition were maintained in the fibrous cap suggesting that targeting netrin-1 did not adversely affect plaque stabilization. M2-like macrophages, which were enriched in plaques upon netrin-1 silencing, may contribute to the synthesis of collagen and other extracellular matrices, as well as promote efferocytosis and clearance of debris to increase plaque stabilization. Notably, efficient efferocytosis of dying cells is essential for polarizing macrophages towards a pro-resolving phenotype83, which increases the production of anti-inflammatory (e.g., IL-10, TGF-b) and pro-resolving (e.g., resolvins) mediators84. Consistent with this, plaque monocytes and macrophages from MøΔNtn1 mice showed an increase in IL-10 and TGF-b signaling, and reduced expression of the alarmins S100A8/A9 and IL-1b. Together, these findings point to inflammation resolution and beneficial restructuring of the plaque upon netrin-1 silencing.
Collectively, our study shows that targeting netrin-1 in advanced atherosclerotic plaques induces therapeutically beneficial changes, including dampening of inflammation, tissue remodeling and plaque regression. Our data advances previous publications demonstrating detrimental effects of netrin-1 in chronic inflammatory conditions (e.g., arthritis25, abdominal aneurysm26, 27, diabetic retinopathy27, obesity20 and heart transplantation28), and highlights the benefit of netrin-1 silencing specifically in myeloid cells, where it contributes to prolonging inflammation. However, it should be noted that infusion of exogenous netrin-1 can also induce protective effects during acute inflammation by engaging the A2BAR and/or DCC receptors. In models of acute peritonitis29, liver-ischemia reperfusion30, hypoxic injury31 and myocardial infarction32, acute inflammation diminishes expression of netrin-1 in endothelial cells, which in turn facilitates the infiltration of immune cells into the inflamed site to fuel the proinflammatory response. Reconstitution of netrin-1 expression in these acute inflammatory settings can be protective by limiting the influx of leukocytes into tissues and reducing damaging effects of inflammation. Interestingly, although netrin-1 is thought to exert protective effects in the acute phase of myocardial infarction, a recent retrospective study in over 800 patients with acute coronary syndrome showed that elevated netrin-1 levels at admission positively correlated with increased 2-year mortality85. Thus, further study will be needed to understand the effects of netrin-1 during acute and chronic inflammation and the molecular pathways regulating expression of netrin-1 and its receptors in these contexts, as well as to develop cell type-specific therapeutic approaches to target netrin-1 or its receptors in disease settings. In addition, our study used AAV-mediated PCSK9 delivery in mice to induce hypercholesterolemia by reducing hepatic expression of the LDLR. While this has become a commonly used model of atherosclerosis, PCSK9 overexpression has been shown to have LDLR-independent effects86 and thus, it will be important to confirm the merits of targeting netrin-1 to achieve inflammation resolution and plaque regression in other mouse models (e.g., Apoe–/– or Ldlr–/– mice) of advanced atherosclerosis.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What Is known?
Atherosclerosis regression – the shrinkage and remodeling of arterial plaques – is an important clinical goal to reduce the burden of cardiovascular disease. Current therapies focus primarily on lowering atherogenic lipids, yet lipid lowering alone is insufficient to fully regress plaque burden.
Chronic inflammation is a critical factor in the clinical progression of atherosclerotic lesions, and thus targeting factors that sustain the maladaptive immune response in the artery wall holds promise for the treatment of atherosclerosis and regression of arterial plaques.
Netrin-1 is a protein secreted by macrophages in atherosclerotic plaques that fuels inflammation by enhancing the survival and accumulation of macrophages in the artery wall, and promoting the migration of smooth muscle cells into the intima.
Previous studies showed that hematopoietic deficiency of netrin-1 during the development of atherosclerosis reduces disease progression, but whether therapeutic targeting of netrin-1 in advanced atherosclerosis could resolve inflammation and induce arterial repair to regress plaque burden was not known.
What New Information Does This Article Contribute?
We used genetically modified mice in which tamoxifen treatment silenced the expression of the netrin-1 gene in myeloid cells (monocytes and macrophages) to test whether therapeutic targeting of netrin-1 in advanced atherosclerosis beneficially alters established disease.
Silencing of myeloid netrin-1 expression in mice with complex atherosclerotic plaques reduced macrophage accumulation in the artery wall and regressed plaque burden in the aorta.
Characterization of monocyte-macrophage dynamics in the artery wall showed that netrin-1 silencing reduced macrophage retention, proliferation and survival in plaques, and increased markers of inflammation resolution (e.g., tissue reparative macrophages, clearance of dying cells, and anti-inflammatory cytokines).
Single cell RNA sequencing of plaque immune cells showed that netrin-1 silencing reorganized the immune cell landscape in the artery wall to help promote inflammation resolution, and in particular, altered the gene expression profiles of plaque macrophages.
Our findings indicate that targeting netrin-1 in advanced atherosclerosis is beneficial in resolving arterial inflammation and regressing plaque burden.
Atherosclerotic cardiovascular disease is the leading cause of morbidity and mortality worldwide. Current therapies targeting atherogenic lipids reduce atherosclerosis progression, but are inadequate to fully reverse existing plaque burden. Chronic inflammation of the artery wall sustains atherosclerotic plaque progression and its complications, and thus therapies that induce inflammation resolution hold promise for the treatment of atherosclerosis. Our study shows that therapeutically targeting netrin-1, a factor that promotes macrophage and smooth muscle cell accumulation in the artery wall, induces beneficial changes in advanced atherosclerotic plaques, including dampening of inflammation, tissue remodeling and plaque contraction. Mechanistically, netrin-1 silencing increases macrophage migratory and phagocytic capacities, resulting in a net decrease in plaque macrophage content and an increase in apoptotic cell clearance (efferocytosis), which is critical for inflammation resolution and tissue repair. In addition, silencing of netrin-1 reduces the number of intimal smooth muscle cells, but does not compromise the fibrous cap or collagen content of lesions, which are important indices of plaque stability. Together these findings highlight the merits of targeting immune modulating factors such as netrin-1, in combination with lipid lowering therapies, to reshape the plaque immune landscape to achieve atherosclerosis regression.
SOURCES OF FUNDING
This work was supported by grants from the NIH [P01HL131481 to KJM, AMS and EAF; R01HL084312 to KJM, EAF, PL; R35HL135799 to KJM; T32HL098129 to CvS], the American Heart Association [19POST34380010 to MoS; 19CDA34630066 to CvS], German Research Foundation [Schl2173-2 to MS].
Nonstandard Abbreviations And Acronyms:
- AAV
adeno-associated virus
- apoB
apolipoprotein B
- ASO
anti-sense oligonucleotide
- BMDM
bone marrow derived macrophage
- CAD
coronary artery disease
- CCR
c-c motif chemokine receptor
- CVD
cardiovascular disease
- HDL
high density lipoprotein
- IL
interleukin
- ILC
innate lymphoid cell
- LDL
low density lipoprotein
- LDLR
low density lipoprotein receptor
- M1
classically activated
- M2
alternatively activated
- mAb
monoclonal antibody
- NK
natural killer
- PCSK9
proprotein convertase subtilisin/kexin type 9
- scRNA-seq
single cell RNA sequencing
- SMC
smooth muscle cell
- TGFb
transforming growth factor beta
- Th
T helper cell
- TNF
tumor necrosis factor
- Treg
regulatory T cell
- TUNEL
terminal deoxynucleotidyl transferase dUTP nick end labeling
- WD
western diet
Footnotes
Publisher's Disclaimer: This article is published in its accepted form. It has not been copyedited and has not appeared in an issue of the journal. Preparation for inclusion in an issue of Circulation Research involves copyediting, typesetting, proofreading, and author review, which may lead to differences between this accepted version of the manuscript and the final, published version.
DISCLOSURES
EAF and KJM hold a patent on the use of netrin-1 inhibitors to target inflammation.
REFERENCES
- 1.Benjamin EJ, Virani SS, Callaway CW, Chamberlain AM, Chang AR, Cheng S, Chiuve SE, Cushman M, Delling FN, Deo R, et al. Heart disease and stroke statistics-2018 update: A report from the american heart association. Circulation. 2018;137:e67–e492 [DOI] [PubMed] [Google Scholar]
- 2.Ridker PM, MacFadyen JG, Everett BM, Libby P, Thuren T, Glynn RJ, Group CT. Relationship of c-reactive protein reduction to cardiovascular event reduction following treatment with canakinumab: A secondary analysis from the cantos randomised controlled trial. Lancet. 2018;391:319–328 [DOI] [PubMed] [Google Scholar]
- 3.Moore KJ, Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell. 2011;145:341–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moore KJ, Sheedy FJ, Fisher EA. Macrophages in atherosclerosis: A dynamic balance. Nature reviews. Immunology 2013;13:709–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Allahverdian S, Chehroudi AC, McManus BM, Abraham T, Francis GA. Contribution of intimal smooth muscle cells to cholesterol accumulation and macrophage-like cells in human atherosclerosis. Circulation. 2014;129:1551–1559 [DOI] [PubMed] [Google Scholar]
- 6.Wang Y, Dubland JA, Allahverdian S, Asonye E, Sahin B, Jaw JE, Sin DD, Seidman MA, Leeper NJ, Francis GA. Smooth muscle cells contribute the majority of foam cells in apoe (apolipoprotein e)-deficient mouse atherosclerosis. Arterioscler Thromb Vasc Biol. 2019;39:876–887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Paulson KE, Zhu SN, Chen M, Nurmohamed S, Jongstra-Bilen J, Cybulsky MI. Resident intimal dendritic cells accumulate lipid and contribute to the initiation of atherosclerosis. Circ Res. 2010;106:383–390 [DOI] [PubMed] [Google Scholar]
- 8.Witztum JL, Lichtman AH. The influence of innate and adaptive immune responses on atherosclerosis. Annu Rev Pathol. 2014;9:73–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Back M, Yurdagul A Jr., Tabas I, Oorni K, Kovanen PT. Inflammation and its resolution in atherosclerosis: Mediators and therapeutic opportunities. Nat Rev Cardiol. 2019;16:389–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chappell J, Harman JL, Narasimhan VM, Yu H, Foote K, Simons BD, Bennett MR, Jorgensen HF. Extensive proliferation of a subset of differentiated, yet plastic, medial vascular smooth muscle cells contributes to neointimal formation in mouse injury and atherosclerosis models. Circ Res. 2016;119:1313–1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Newman AAC, Serbulea V, Baylis RA, Shankman LS, Bradley X, Alencar GF, Owsiany K, Deaton RA, Karnewar S, Shamsuzzaman S, et al. Multiple cell types contribute to the atherosclerotic lesion fibrous cap by pdgfrβ and bioenergetic mechanisms. Nature Metabolism. 2021;3:166–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rong JX, Shapiro M, Trogan E, Fisher EA. Transdifferentiation of mouse aortic smooth muscle cells to a macrophage-like state after cholesterol loading. Proc Natl Acad Sci U S A. 2003;100:13531–13536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shankman LS, Gomez D, Cherepanova OA, Salmon M, Alencar GF, Haskins RM, Swiatlowska P, Newman AA, Greene ES, Straub AC, et al. Klf4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat Med. 2015;21:628–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kavurma MM, Rayner KJ, Karunakaran D. The walking dead: Macrophage inflammation and death in atherosclerosis. Curr Opin Lipidol. 2017;28:91–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lendon CL, Davies MJ, Born GV, Richardson PD. Atherosclerotic plaque caps are locally weakened when macrophages density is increased. Atherosclerosis. 1991;87:87–90 [DOI] [PubMed] [Google Scholar]
- 16.Shah PK, Falk E, Badimon JJ, Fernandez-Ortiz A, Mailhac A, Villareal-Levy G, Fallon JT, Regnstrom J, Fuster V. Human monocyte-derived macrophages induce collagen breakdown in fibrous caps of atherosclerotic plaques. Potential role of matrix-degrading metalloproteinases and implications for plaque rupture. Circulation. 1995;92:1565–1569 [PubMed] [Google Scholar]
- 17.Moore KJ, Koplev S, Fisher EA, Tabas I, Bjorkegren JLM, Doran AC, Kovacic JC. Macrophage trafficking, inflammatory resolution, and genomics in atherosclerosis: Jacc macrophage in cvd series (part 2). J Am Coll Cardiol. 2018;72:2181–2197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ramkhelawon B, Hennessy EJ, Menager M, Ray TD, Sheedy FJ, Hutchison S, Wanschel A, Oldebeken S, Geoffrion M, Spiro W, et al. Netrin-1 promotes adipose tissue macrophage retention and insulin resistance in obesity. Nat Med. 2014;20:377–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van Gils JM, Derby MC, Fernandes LR, Ramkhelawon B, Ray TD, Rayner KJ, Parathath S, Distel E, Feig JL, Alvarez-Leite JI, et al. The neuroimmune guidance cue netrin-1 promotes atherosclerosis by inhibiting the emigration of macrophages from plaques. Nat Immunol. 2012;13:136–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sharma M, Schlegel M, Brown EJ, Sansbury BE, Weinstock A, Afonso MS, Corr EM, van Solingen C, Shanley LC, Peled D, et al. Netrin-1 alters adipose tissue macrophage fate and function in obesity. Immunometabolism. 2019;1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Keino-Masu K, Masu M, Hinck L, Leonardo ED, Chan SS, Culotti JG, Tessier-Lavigne M. Deleted in colorectal cancer (dcc) encodes a netrin receptor. Cell. 1996;87:175–185 [DOI] [PubMed] [Google Scholar]
- 22.Serafini T, Colamarino SA, Leonardo ED, Wang H, Beddington R, Skarnes WC, Tessier-Lavigne M. Netrin-1 is required for commissural axon guidance in the developing vertebrate nervous system. Cell. 1996;87:1001–1014 [DOI] [PubMed] [Google Scholar]
- 23.Ly NP, Komatsuzaki K, Fraser IP, Tseng AA, Prodhan P, Moore KJ, Kinane TB. Netrin-1 inhibits leukocyte migration in vitro and in vivo. Proc Natl Acad Sci U S A. 2005;102:14729–14734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Taylor L, Brodermann MH, McCaffary D, Iqbal AJ, Greaves DR. Netrin-1 reduces monocyte and macrophage chemotaxis towards the complement component c5a. PLoS One. 2016;11:e0160685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mediero A, Wilder T, Ramkhelawon B, Moore KJ, Cronstein BN. Netrin-1 and its receptor unc5b are novel targets for the treatment of inflammatory arthritis. FASEB J. 2016;30:3835–3844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hadi T, Boytard L, Silvestro M, Alebrahim D, Jacob S, Feinstein J, Barone K, Spiro W, Hutchison S, Simon R, et al. Macrophage-derived netrin-1 promotes abdominal aortic aneurysm formation by activating mmp3 in vascular smooth muscle cells. Nat Commun. 2018;9:5022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miloudi K, Binet F, Wilson A, Cerani A, Oubaha M, Menard C, Henriques S, Mawambo G, Dejda A, Nguyen PT, et al. Truncated netrin-1 contributes to pathological vascular permeability in diabetic retinopathy. J Clin Invest. 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mao X, Xing H, Mao A, Jiang H, Cheng L, Liu Y, Quan X, Li L. Netrin-1 attenuates cardiac ischemia reperfusion injury and generates alternatively activated macrophages. Inflammation. 2014;37:573–580 [DOI] [PubMed] [Google Scholar]
- 29.Mirakaj V, Dalli J, Granja T, Rosenberger P, Serhan CN. Vagus nerve controls resolution and pro-resolving mediators of inflammation. The Journal of experimental medicine. 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schlegel M, Kohler D, Korner A, Granja T, Straub A, Giera M, Mirakaj V. The neuroimmune guidance cue netrin-1 controls resolution programs and promotes liver regeneration. Hepatology. 2016;63:1689–1705 [DOI] [PubMed] [Google Scholar]
- 31.Rosenberger P, Schwab JM, Mirakaj V, Masekowsky E, Mager A, Morote-Garcia JC, Unertl K, Eltzschig HK. Hypoxia-inducible factor-dependent induction of netrin-1 dampens inflammation caused by hypoxia. Nat Immunol. 2009;10:195–202 [DOI] [PubMed] [Google Scholar]
- 32.Zhang J, Cai H. Netrin-1 prevents ischemia/reperfusion-induced myocardial infarction via a dcc/erk1/2/enos s1177/no/dcc feed-forward mechanism. J Mol Cell Cardiol. 2010;48:1060–1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van Gils JM, Ramkhelawon B, Fernandes L, Stewart MC, Guo L, Seibert T, Menezes GB, Cara DC, Chow C, Kinane TB, et al. Endothelial expression of guidance cues in vessel wall homeostasis dysregulation under proatherosclerotic conditions. Arterioscler Thromb Vasc Biol. 2013;33:911–919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bruikman CS, Vreeken D, Hoogeveen RM, Bom MJ, Danad I, Pinto-Sietsma SJ, van Zonneveld AJ, Knaapen P, Hovingh GK, Stroes ESG, et al. Netrin-1 and the grade of atherosclerosis are inversely correlated in humans. Arterioscler Thromb Vasc Biol. 2020;40:462–472 [DOI] [PubMed] [Google Scholar]
- 35.Fiorelli S, Cosentino N, Porro B, Fabbiocchi F, Niccoli G, Fracassi F, Capra N, Barbieri S, Crea F, Marenzi G, et al. Netrin-1 in atherosclerosis: Relationship between human macrophage intracellular levels and in vivo plaque morphology. Biomedicines. 2021;9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oksala N, Parssinen J, Seppala I, Raitoharju E, Kholova I, Hernesniemi J, Lyytikainen LP, Levula M, Makela KM, Sioris T, et al. Association of neuroimmune guidance cue netrin-1 and its chemorepulsive receptor unc5b with atherosclerotic plaque expression signatures and stability in human(s): Tampere vascular study (tvs). Circ Cardiovasc Genet. 2013;6:579–587 [DOI] [PubMed] [Google Scholar]
- 37.Ramkhelawon B, Yang Y, van Gils JM, Hewing B, Rayner KJ, Parathath S, Guo L, Oldebeken S, Feig JL, Fisher EA, et al. Hypoxia induces netrin-1 and unc5b in atherosclerotic plaques: Mechanism for macrophage retention and survival. Arterioscler Thromb Vasc Biol. 2013;33:1180–1188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sharma M, Boytard L, Hadi T, Koelwyn G, Simon R, Ouimet M, Seifert L, Spiro W, Yan B, Hutchison S, et al. Enhanced glycolysis and hif-1alpha activation in adipose tissue macrophages sustains local and systemic interleukin-1beta production in obesity. Sci Rep. 2020;10:5555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang X, Zhang J, Chen L, Yuan Z, Qin X, Wu Q, Shen D, He H, Yu C. The role of unc5b in ox-ldl inhibiting migration of raw264.7 macrophages and the involvement of ccr7. Biochem Biophys Res Commun. 2018;505:637–643 [DOI] [PubMed] [Google Scholar]
- 40.Bruikman CS, Vreeken D, Zhang H, van Gils MJ, Peter J, van Zonneveld AJ, Hovingh GK, van Gils JM. The identification and function of a netrin-1 mutation in a pedigree with premature atherosclerosis. Atherosclerosis. 2020;301:84–92 [DOI] [PubMed] [Google Scholar]
- 41.Ouimet M, Ediriweera HN, Gundra UM, Sheedy FJ, Ramkhelawon B, Hutchison SB, Rinehold K, van Solingen C, Fullerton MD, Cecchini K, et al. Microrna-33-dependent regulation of macrophage metabolism directs immune cell polarization in atherosclerosis. J Clin Invest. 2015;125:4334–4348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vozenilek AE, Blackburn CMR, Schilke RM, Chandran S, Castore R, Klein RL, Woolard MD. Aav8-mediated overexpression of mpcsk9 in liver differs between male and female mice. Atherosclerosis. 2018;278:66–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Koskinas KC, Windecker S, Raber L. Regression of coronary atherosclerosis: Current evidence and future perspectives. Trends Cardiovasc Med. 2016;26:150–161 [DOI] [PubMed] [Google Scholar]
- 44.Stary HC, Chandler AB, Dinsmore RE, Fuster V, Glagov S, Insull W Jr., Rosenfeld ME, Schwartz CJ, Wagner WD, Wissler RW. A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis. A report from the committee on vascular lesions of the council on arteriosclerosis, american heart association. Circulation. 1995;92:1355–1374 [DOI] [PubMed] [Google Scholar]
- 45.Butler A, Hoffman P, Smibert P, Papalexi E, Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol. 2018;36:411–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Aran D, Looney AP, Liu L, Wu E, Fong V, Hsu A, Chak S, Naikawadi RP, Wolters PJ, Abate AR, et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat Immunol. 2019;20:163–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cochain C, Vafadarnejad E, Arampatzi P, Pelisek J, Winkels H, Ley K, Wolf D, Saliba AE, Zernecke A. Single-cell rna-seq reveals the transcriptional landscape and heterogeneity of aortic macrophages in murine atherosclerosis. Circ Res. 2018;122:1661–1674 [DOI] [PubMed] [Google Scholar]
- 48.Swirski FK, Robbins CS, Nahrendorf M. Development and function of arterial and cardiac macrophages. Trends Immunol. 2016;37:32–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lin JD, Nishi H, Poles J, Niu X, McCauley C, Rahman K, Brown EJ, Yeung ST, Vozhilla N, Weinstock A, et al. Single-cell analysis of fate-mapped macrophages reveals heterogeneity, including stem-like properties, during atherosclerosis progression and regression. JCI Insight. 2019;4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Amit I, Winter DR, Jung S. The role of the local environment and epigenetics in shaping macrophage identity and their effect on tissue homeostasis. Nat Immunol. 2016;17:18–25 [DOI] [PubMed] [Google Scholar]
- 51.Ensan S, Li A, Besla R, Degousee N, Cosme J, Roufaiel M, Shikatani EA, El-Maklizi M, Williams JW, Robins L, et al. Self-renewing resident arterial macrophages arise from embryonic cx3cr1(+) precursors and circulating monocytes immediately after birth. Nat Immunol. 2016;17:159–168 [DOI] [PubMed] [Google Scholar]
- 52.Gomez D, Owens GK. Smooth muscle cell phenotypic switching in atherosclerosis. Cardiovasc Res. 2012;95:156–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Babaev VR, Fazio S, Gleaves LA, Carter KJ, Semenkovich CF, Linton MF. Macrophage lipoprotein lipase promotes foam cell formation and atherosclerosis in vivo. J Clin Invest. 1999;103:1697–1705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jaitin DA, Adlung L, Thaiss CA, Weiner A, Li B, Descamps H, Lundgren P, Bleriot C, Liu Z, Deczkowska A, et al. Lipid-associated macrophages control metabolic homeostasis in a trem2-dependent manner. Cell. 2019;178:686–698 e614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang S, Song R, Wang Z, Jing Z, Wang S, Ma J. S100a8/a9 in inflammation. Front Immunol. 2018;9:1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Son BK, Sawaki D, Tomida S, Fujita D, Aizawa K, Aoki H, Akishita M, Manabe I, Komuro I, Friedman SL, et al. Granulocyte macrophage colony-stimulating factor is required for aortic dissection/intramural haematoma. Nat Commun. 2015;6:6994. [DOI] [PubMed] [Google Scholar]
- 57.Blanc J, Alves-Guerra MC, Esposito B, Rousset S, Gourdy P, Ricquier D, Tedgui A, Miroux B, Mallat Z. Protective role of uncoupling protein 2 in atherosclerosis. Circulation. 2003;107:388–390 [DOI] [PubMed] [Google Scholar]
- 58.Zhuang T, Liu J, Chen X, Zhang L, Pi J, Sun H, Li L, Bauer R, Wang H, Yu Z, et al. Endothelial foxp1 suppresses atherosclerosis via modulation of nlrp3 inflammasome activation. Circ Res. 2019;125:590–605 [DOI] [PubMed] [Google Scholar]
- 59.Abbas W, Kumar A, Herbein G. The eef1a proteins: At the crossroads of oncogenesis, apoptosis, and viral infections. Front Oncol. 2015;5:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Robbins CS, Hilgendorf I, Weber GF, Theurl I, Iwamoto Y, Figueiredo JL, Gorbatov R, Sukhova GK, Gerhardt LM, Smyth D, et al. Local proliferation dominates lesional macrophage accumulation in atherosclerosis. Nat Med. 2013;19:1166–1172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rahman K, Vengrenyuk Y, Ramsey SA, Vila NR, Girgis NM, Liu J, Gusarova V, Gromada J, Weinstock A, Moore KJ, et al. Inflammatory ly6chi monocytes and their conversion to m2 macrophages drive atherosclerosis regression. J Clin Invest. 2017;127:2904–2915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Feig JE, Shang Y, Rotllan N, Vengrenyuk Y, Wu C, Shamir R, Torra IP, Fernandez-Hernando C, Fisher EA, Garabedian MJ. Statins promote the regression of atherosclerosis via activation of the ccr7-dependent emigration pathway in macrophages. PLoS One. 2011;6:e28534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Trogan E, Feig JE, Dogan S, Rothblat GH, Angeli V, Tacke F, Randolph GJ, Fisher EA. Gene expression changes in foam cells and the role of chemokine receptor ccr7 during atherosclerosis regression in apoe-deficient mice. Proc Natl Acad Sci U S A. 2006;103:3781–3786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hervera A, De Virgiliis F, Palmisano I, Zhou L, Tantardini E, Kong G, Hutson T, Danzi MC, Perry RB, Santos CXC, et al. Reactive oxygen species regulate axonal regeneration through the release of exosomal nadph oxidase 2 complexes into injured axons. Nat Cell Biol. 2018;20:307–319 [DOI] [PubMed] [Google Scholar]
- 65.Horn A, Van der Meulen JH, Defour A, Hogarth M, Sreetama SC, Reed A, Scheffer L, Chandel NS, Jaiswal JK. Mitochondrial redox signaling enables repair of injured skeletal muscle cells. Sci Signal. 2017;10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Oh J, Riek AE, Weng S, Petty M, Kim D, Colonna M, Cella M, Bernal-Mizrachi C. Endoplasmic reticulum stress controls m2 macrophage differentiation and foam cell formation. J Biol Chem. 2012;287:11629–11641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gong D, Shi W, Yi SJ, Chen H, Groffen J, Heisterkamp N. Tgfbeta signaling plays a critical role in promoting alternative macrophage activation. BMC Immunol. 2012;13:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Orecchioni M, Ghosheh Y, Pramod AB, Ley K. Macrophage polarization: Different gene signatures in m1(lps+) vs. Classically and m2(lps−) vs. Alternatively activated macrophages. Front Immunol. 2019;10:1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Goldberg IJ, Sharma G, Fisher EA. Atherosclerosis: Making a u turn. Annu Rev Med. 2020;71:191–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Peled M, Fisher EA. Dynamic aspects of macrophage polarization during atherosclerosis progression and regression. Front Immunol. 2014;5:579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gerrity RG. The role of the monocyte in atherogenesis: Ii. Migration of foam cells from atherosclerotic lesions. Am J Pathol. 1981;103:191–200 [PMC free article] [PubMed] [Google Scholar]
- 72.Potteaux S, Gautier EL, Hutchison SB, van Rooijen N, Rader DJ, Thomas MJ, Sorci-Thomas MG, Randolph GJ. Suppressed monocyte recruitment drives macrophage removal from atherosclerotic plaques of apoe−/− mice during disease regression. J Clin Invest. 2011;121:2025–2036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Swirski FK, Pittet MJ, Kircher MF, Aikawa E, Jaffer FA, Libby P, Weissleder R. Monocyte accumulation in mouse atherogenesis is progressive and proportional to extent of disease. Proc Natl Acad Sci U S A. 2006;103:10340–10345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tang J, Lobatto ME, Hassing L, van der Staay S, van Rijs SM, Calcagno C, Braza MS, Baxter S, Fay F, Sanchez-Gaytan BL, et al. Inhibiting macrophage proliferation suppresses atherosclerotic plaque inflammation. Sci Adv. 2015;1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tabas I. Consequences and therapeutic implications of macrophage apoptosis in atherosclerosis: The importance of lesion stage and phagocytic efficiency. Arterioscler Thromb Vasc Biol. 2005;25:2255–2264 [DOI] [PubMed] [Google Scholar]
- 76.Llodra J, Angeli V, Liu J, Trogan E, Fisher EA, Randolph GJ. Emigration of monocyte-derived cells from atherosclerotic lesions characterizes regressive, but not progressive, plaques. Proc Natl Acad Sci U S A. 2004;101:11779–11784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Swirski FK, Libby P, Aikawa E, Alcaide P, Luscinskas FW, Weissleder R, Pittet MJ. Ly-6chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J Clin Invest. 2007;117:195–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dutta P, Nahrendorf M. Regulation and consequences of monocytosis. Immunol Rev. 2014;262:167–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Diehl GE, Longman RS, Zhang JX, Breart B, Galan C, Cuesta A, Schwab SR, Littman DR. Microbiota restricts trafficking of bacteria to mesenteric lymph nodes by cx(3)cr1(hi) cells. Nature. 2013;494:116–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hellmann J, Sansbury BE, Holden CR, Tang Y, Wong B, Wysoczynski M, Rodriguez J, Bhatnagar A, Hill BG, Spite M. Ccr7 maintains nonresolving lymph node and adipose inflammation in obesity. Diabetes. 2016;65:2268–2281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mueller PA, Zhu L, Tavori H, Huynh K, Giunzioni I, Stafford JM, Linton MF, Fazio S. Deletion of macrophage low-density lipoprotein receptor-related protein 1 (lrp1) accelerates atherosclerosis regression and increases c-c chemokine receptor type 7 (ccr7) expression in plaque macrophages. Circulation. 2018;138:1850–1863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ahmadi A, Argulian E, Leipsic J, Newby DE, Narula J. From subclinical atherosclerosis to plaque progression and acute coronary events: Jacc state-of-the-art review. J Am Coll Cardiol. 2019;74:1608–1617 [DOI] [PubMed] [Google Scholar]
- 83.Yurdagul A Jr., Doran AC, Cai B, Fredman G, Tabas IA. Mechanisms and consequences of defective efferocytosis in atherosclerosis. Front Cardiovasc Med. 2017;4:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Watanabe S, Alexander M, Misharin AV, Budinger GRS. The role of macrophages in the resolution of inflammation. J Clin Invest. 2019;129:2619–2628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Leocadio P, Menta P, Dias M, Fraga J, Goulart A, Santos I, Lotufo P, Bensenor I, Alvarez-Leite J. High serum netrin-1 and il-1beta in elderly females with acs: Worse prognosis in 2-years follow-up. Arq Bras Cardiol. 2020;114:507–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Warden BA, Fazio S, Shapiro MD. The pcsk9 revolution: Current status, controversies, and future directions. Trends Cardiovasc Med. 2020;30:179–185 [DOI] [PubMed] [Google Scholar]
- 87.Tacke F, Alvarez D, Kaplan TJ, Jakubzick C, Spanbroek R, Llodra J, Garin A, Liu J, Mack M, van Rooijen N, et al. Monocyte subsets differentially employ ccr2, ccr5, and cx3cr1 to accumulate within atherosclerotic plaques. The Journal of clinical investigation. 2007;117:185–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.