

Summary
Delivering peptides into cells could open up possibilities for targeting intracellular proteins. Although fatty acylation of peptide therapeutics improves their systemic half-life, it remains unclear how it influences their cellular uptake. Here, we demonstrate that a fatty acylated peptide exhibits enhanced cellular internalization and cytosolic distribution compared to the un-acylated version. By using a cystine-knot peptide as a model system, we report an efficient strategy for site-specific conjugation of fatty acids. Peptides modified with fatty acids of different chain lengths entered cells through clathrin-mediated and macropinocytosis pathways. The cellular uptake was mediated by the length of the hydrocarbon chain, with myristic acid conjugates displaying the highest distribution across the cytoplasm including the cytosol, and endomembranes of the ER, Golgi and mitochondria. Our studies demonstrate how fatty acylation improves the cellular uptake of peptides, and lay the groundwork for future development of bioactive peptides with enhanced intracellular distribution.
Subject areas: Biological sciences, Biochemistry, Biochemical engineering, Biotechnology
Graphical abstract
Highlights
-
•
A synthetic strategy comprises site-specific conjugation of fatty acids to peptides
-
•
Fatty acylation of a peptide enhances its cellular uptake and cytosolic distribution
-
•
Myristoylated peptides display a high cytoplasmic distribution
-
•
Fatty acylated peptides are internalized via multiple endocytic routes
Biological sciences; Biochemistry; Biochemical engineering; Biotechnology
Introduction
Fatty acylation, a post-translational modification that comprises the addition of fatty acids onto proteins, regulates protein function, stability, and membrane association (Chamberlain and Shipston, 2015; Gao and Hannoush, 2018; Hannoush and Sun, 2010; Resh, 2004; Smotrys and Linder, 2004). Myristoylation and palmitoylation, two distinct forms of fatty acylation of proteins, have been shown to promote membrane targeting and binding of proteins. They also could influence protein structure, as well as direct protein interactions. Palmitoylation has also been observed to regulate other posttranslational modifications including phosphorylation or ubiquitination at nearby sites (Blaskovic et al., 2013; Shipston, 2011). Many fatty acylated proteins play a key role in physiology and disease. Examples include the Ras family of proteins (Rocks et al., 2005), c-Abl kinase (Hantschel et al., 2003), Src family kinases (Resh, 1994), secreted morphogens such as Hedgehog and Wnt (Gao and Hannoush, 2014a; Pepinsky et al., 1998; Willert et al., 2003), and the peptide hormone ghrelin (Kojima and Kangawa, 2005). N-palmitoylation of hedgehog proteins is essential for their production, secretion and signaling activity (Chen et al., 2004; Goetz et al., 2006). Wnt proteins are palmitoleoylated and the lipid modification is required for their trafficking, secretion, and binding to their cognate frizzled receptors to initiate signaling (Gao and Hannoush, 2014b; Janda et al., 2012; Nile and Hannoush, 2016). Octanoate covalently conjugated to the N-terminal region of ghrelin mediates its binding to the growth hormone secretagogue type 1a receptor, thereby controlling physiologic outputs such as appetite stimulation and growth hormone release (Darling et al., 2015; Kojima and Kangawa, 2005).
A large body of literature details how fatty acylation regulates protein activity by modulating binding to different effectors both inside and outside the cell (Chamberlain and Shipston, 2015; Resh, 2016). Yet, how fatty acylation could influence the behavior of smaller bioactive peptide ligands, in particular their delivery across cell membranes and their mechanism of cellular uptake, remains poorly understood. This is of particular importance as delivering small peptides from the extracellular milieu into cells would open up possibilities for intracellular targeting of proteins. Despite earlier reports demonstrating that myristoylation could increase the cellular uptake of short peptides (Enseñat-Waser et al., 2002; Nelson et al., 2007), the understanding of myristate-mediated cellular import route and its subcellular distribution has been limited. Fatty acylation has drawn considerable interest in the field of peptide drug development due to its impact on the pharmacokinetics of biotherapeutics, in particular extending the systemic half-life of drugs via promoting their binding to serum albumin (Kowalczyk et al., 2017; Myers et al., 1997). Examples of marketed fatty acylated peptide therapeutics used in diabetic patients include long-acting insulin detemir (Levemir) (Home and Kurtzhals, 2006; Le Floch, 2010), which is myristoylated, and glucagon-like peptide-1(GLP-1) receptor agonist liraglutide (Victoza) (Jackson et al., 2010; Knudsen et al., 2000), which is N-palmitoylated. Although the effects of fatty acylation on the pharmacokinetic behavior and serum binding of peptide drugs have been extensively studied, their impact on delivery to the cytosol remains elusive.
The intracellular delivery of peptides into mammalian cells has enormous potential in the development of therapeutics, as it enables peptides to interact with their intended intracellular targets. We sought to explore the impact of fatty acylation on permeability and subcellular distribution of bioactive peptides. One particular class of emerging peptides with drug-like properties is the cystine-knot peptide (CKP) family. Naturally occurring CKPs exhibit diverse pharmacological activities such as antiviral, anti-microbial, uterotonic, and protease inhibitory activity (Čemažar et al., 2006; Craik and Du, 2017). In our laboratory, we used EETI-II (Ecballium elaterium trypsin inhibitor-II), a potent trypsin inhibitor found in squirting cucumbers, as a model system to study the cellular uptake of CKPs. Fluorescence and electron microscopy studies demonstrated that EETI-II was internalized into mammalian cells via macropinocytosis and clathrin-mediated endocytosis, and it remained trapped in the lumen of lysosomes with no cytosolic distribution observed (Gao et al., 2016, Gao et al., 2019).
Here we systematically examined the impact of site-specific fatty acyl modification on the uptake and intracellular delivery of EETI-II. In particular, we designed and synthesized fluorescently-labeled EETI-II that is conjugated to lauroyl (C12:0), myristoyl (C14:0), palmitoyl (C16:0), or stearoyl (C18:0) fatty acyl moieties. The introduction of these moieties to the N-terminal region of EETI-II did not alter its native trypsin inhibitory activity. Using fluorescence microscopy, we demonstrated that fatty acylation substantially increased the cellular uptake efficiency of EETI-II in mammalian cells. The extent of cellular uptake was dependent on the hydrocarbon chain length of the fatty acid, with C14 being the most favorable followed by C12 and C16. All fatty acylated EETI-II peptides studied shared common internalization mechanisms with that of the unmodified parent CKP, primarily through macropinocytosis and clathrin-mediated endocytosis. Despite the majority of fatty acylated molecules accumulating in endosomal and lysosomal compartments (similar to EETI-II), a pool of acylated peptides was found in the cytosol and on endomembranes as observed by immunoelectron microscopy (immunoEM). The internalized fatty acylated peptides also demonstrated enhanced resistance to detergent extraction compared to their unmodified peptide counterparts. Our studies demonstrate that fatty acylation could improve the intracellular uptake and subcellular distribution of peptides and lay the groundwork for future development of bioactive peptides with enhanced subcellular distribution. They also provide the first examples of fatty acylated bifunctional CKPs with both trypsin and albumin binding properties.
Results
Design and synthesis of fatty acylated cystine-knot peptides
We used the cystine-knot peptide EETI-II as a model system to systematically investigate the effect of fatty acylation on cellular uptake of peptides. Based on structural observations, we predicted that N-terminal modifications of EETI-II should be least disruptive to its trypsin inhibitory activity. Accordingly, we designed fatty acids of different chain lengths (C12, C14, C16, and C18) to be incorporated at the N terminus of EETI-II or through a lysine residue that was engineered at the N terminus (Figures 1A and 1B). The strategy to modify the lysine and introduce gamma-L-glutamyl moiety as a linker in the conjugation of palmitic and stearic acid to the peptide was chosen in order to mimic the spacer architecture present in the approved drugs detemir and liraglutide (Home and Kurtzhals, 2006; Knudsen et al., 2000). For the stearoyl peptide conjugate, octadecanedioic acid was incorporated via a gamma-L-glutamyl linker, resulting in a free carboxylic acid head group which was postulated to improve the solubility of the long-chain fatty acyl peptide conjugate. All the other chosen lipid modifications comprised a free ω-methyl group. The minor difference in the linker region of these model peptides should minimally impact their functions since the lipid tags rather than the linker region are the functional groups responsible for mediating membrane interactions. We elected to introduce the fatty acyl modifications during solid-phase peptide synthesis, leading to generation of linear fatty acylated peptide precursors. They were then screened in different buffers to identify the optimal folding conditions by using analytical RP-HPLC and MS. This synthetic strategy provided an efficient workflow (Figure 1C) for site-specific conjugation of the fatty acids to the peptides as it minimized the number of purification steps. Subsequently, we scaled up the folding reactions and purified the fatty acylated products to near homogeneity (Figure S1) in multi milligram quantities. All the conjugates, except C18-EETI-II, were more lipophilic than the unconjugated parent EETI as determined by their slower HPLC retention times. C18-EETI-II displayed a similar retention time as EETI-Il, likely because of the presence of the carboxylic acid group. The folded lipid-modified peptides were then labeled with Alexa 488 NHS using N-hydroxysuccinimidyl ester chemistry(Gao et al., 2016), desalted on a C18 column and then purified by RP-HPLC. The identity and purities of all folded peptides were confirmed by LC-MS (Figures S1 and S2). In certain cases, two fluorophores were observed to be conjugated to the peptide because of the presence of two reactive amino groups, one at the free N terminus and the other at the lysine residue located in loop 2 of EETI-II (Figure S2). Conjugation at this lysine residue is not predicted to affect the inhibitory activity of EETI-II based on structural analysis.
Figure 1.

Fatty acylated EETI-II molecules are internalized into mammalian cells more efficiently than WT EETI-II
(A) Sequence and disulfide bond connectivity of EETI-II. Each colored region represents a loop region that is flanked by two cysteine residues.
(B) Chemical structures of designed fatty acylated EETI-II peptides.
(C) Strategy for generation of Alexa 488-labeled lipid EETI-II molecules.
(D) Fatty acylated EETI-II peptides show the same trypsin inhibition activity as wild-type (WT) EETI-II. A reduced linear version of EETI-II (lin-EETI) was used as negative control. V/V0 is normalized proteolytic activity.
(E and F) Concentration- and time-dependent uptake of Alexa 488 labeled WT and fatty acylated EETI-II assayed by fluorescence microscopy. NIH3T3 cells were treated with E) increasing concentrations (1, 5, 10, 20, 30 μM) of Alexa 488-labeled WT (EETI-II) or fatty acylated EETI-II for 2 h, or f) 5 μM of Alexa 488-labeled WT or fatty acylated EETI-II for 1, 2, 3, 6 h.
(G) Representative images of cells treated with 20 μM Alexa 488 labeled WT and fatty acylated EETI-II for 2 h. Green: WT or fatty acylated EETI-II; blue: nuclei. Scale bar, 20 μm. Cells were processed as described in methods. Cells were washed with PBS and fixed with 4% PFA. All samples (E–G) were imaged on a high throughput ImageXpress Micro XL imaging system (Molecular Devices) with a 40× objective and images were analyzed by MetaXpress 4.0. Integrated fluorescence intensity values above a threshold defined using the DMSO-treated samples were measured and normalized to samples with the highest signal. Values represent mean ± SD. n = 1,000 cells. Representative images from at least three independent experiments are shown.
We then examined whether fatty acyl modifications impacted the enzymatic inhibitory activity of EETI-II. In a trypsin activity assay, acylated EETI-II peptides exhibited the same inhibition as wild-type EETI-II (Kiapp, 0.106 ± 0.01, 0.102 ± 0.004, 0.1 ± 0.01, 0.132 ± 0.047, or 0.125 ± 0.011 nM for WT-, C12-, C14-, C16-, or C18-EETI-II, respectively) (Figure 1D), indicating that incorporation of fatty acyl moieties did not alter the native trypsin inhibitory activity of EETI-II. As expected, a control linear EETI-II peptide did not show trypsin inhibitor activity (Figure 1D), in agreement with the tertiary folded structure being required for trypsin binding. Altogether, the synthetic strategy described here enables efficient and site-specific conjugation of fatty acids to peptides without impairing CKP function.
Fatty acylation enhances cellular uptake of peptides
To investigate whether fatty acylation influences the cellular uptake of EETI-II, we treated mouse NIH3T3 cells with varying concentrations of fluorescently-labeled EETI-II conjugates for 2 h, or at a fixed concentration (5 μM) for varying periods of time, and then monitored their cellular uptake by fluorescence microscopy. All the fatty acyl-modified EETI-II peptides were internalized into cells in a concentration- and time-dependent manner and they demonstrated a strong increase in cellular uptake compared to the parent unconjugated EETI-II peptide (Figures 1E–1G and S3). At 10 μM, we observed 78-, 97-, 8-, or 2-fold improvement in cellular uptake of C12-, C14-, C16-, or C18-tagged EETI-II, respectively, as measured by total cellular fluorescence intensity after 2 h of treatment (Figure 1E). Although the majority of fatty acylated EETI-II peptides exhibited a punctate pattern similar to EETI-II, there was a pool of internalized fatty acylated peptides, in particular C12-, C14-, and C16-EETI-II, that exhibited cytosolic and plasma membrane staining, in sharp contrast to the phenotype observed for internalized EETI-II (Figures 2 and S4–S7). At 4°C, a condition in which energy-dependent cellular uptake is suppressed, the fatty acylated peptides C12-, C14-, and C16-EETT-II still demonstrated cellular uptake, albeit reduced, with a strong plasma membrane and cytosolic staining pattern observed after 1 h (Figures 2 and S4–S7). In sharp contrast, the uptake of wild-type EETI-II was mostly abolished, as observed by the complete loss in cellular fluorescence (Figure 2, lower panel and S4–S7) (see sections below for more detailed studies). C18-EETI-II also exhibited a strong reduction in uptake at 4°C.
Figure 2.

Fatty acylated peptides localize in cellular vesicles, plasma membrane and cytosol
NIH3T3 cells were treated with 5 μM peptides for 2 h at 37°C or 4°C. Cells were washed with PBS at the end of the study and fixed with 4% PFA (20 min at RT for 37°C treated cells, and 20 min at 4°C then 20 min at RT for 4°C treated cells to avoid internalization of membrane bound peptides). Cells were then incubated with tetramethylrhodamine conjugated Concanavalin A (10 μg/mL, shown in red) for 10 min at RT to label the plasma membrane. Images of samples on coverslips were captured on an upright AX10 LEICA SPE laser scanning confocal microscope and images were analyzed with the LAS AF image processing software (Leica Microsystems). Representative images from two independent experiments are shown. Scale bar, 20 μm.
Altogether, these results demonstrate that fatty acyl modification with C12, C14 or C16 enhances the uptake of EETI-II into the cytosol as evidenced by the fluorescence signal observed at 4°C, potentially enabling peptide entry through the plasma membrane.
Fatty acylation increases cytosolic accumulation of peptides and their membrane association
To corroborate the above findings, we used quantitative immunoEM to investigate how the different fatty acyl-modified EETI-II conjugates enter mammalian cells and distribute into different membrane-lined compartments as well as the cytosol. To optimize the signal for immunoEM studies, the concentrations of the individual molecules tested were adjusted to yield comparable signal intensities as observed by immunofluorescence microscopy (Figure S8A).
All peptides including wild-type EETI-II could be detected in the lumen of endosomes and lysosomes. In addition, they were seen in small vesicles and tubules that were likely of endosomal origin (Figures 3A and S8B). Both endosomal and lysosomal vacuoles (marked by yellow false color, Figure 3A) as well as the vesiculo-tubular structures contained Alexa 488 gold particles in their lumen or on their limiting membrane. Gold particles at the cytosolic face of these membranes but within a distance of 20 nm (the size of an antibody-Protein A-gold particle complex) were considered as membrane-associated. However, C12-EETI-II, C14-EETI-II, and C16-EETI-II were also associated with ER, Golgi mitochondria, and with the plasma membrane (Figure S8C). More importantly, they were detected in the cytosol, well outside the endocytic membranes (Figures 3A and S8D), suggesting that they could permeate into the cytosol. Such cytosolic distribution was virtually absent in cells incubated with EETI-II or C18-EETI-II (Figure 3A), even though these molecules were tested at 2.5 – 5-fold higher concentration than C12, C14, and C16-EETI-II and they showed comparable signal by fluorescence microscopy (Figure S8A). It is noteworthy that the background (anti-Alexa488 antibody) is little to none in the control sections. We then compared the levels of cytosolic distribution by quantifying the numbers of Alexa 488 gold particles per cytosol area, which we defined as the cellular region that is at least 20 nm away from any cellular membranes including the plasma membrane, endosomes/lysosomes, ER, nuclear envelope, Golgi apparatus, or mitochondria (see quantification criteria in STAR Methods section). The data demonstrate that C14-EETI-II showed the highest level of cytosolic distribution, followed by C12- and C16-EETI-II. Minimal cytosolic peptide signal was observed in cells treated with C18-EETI-II or EETI-II (Figure 3B). Moreover, C14- and C16-EETI-II displayed higher levels of plasma membrane signal compared to the rest of the peptides tested (Figure 3C). Taken together, the data indicate that although the majority of internalized acylated peptides localize in endosomes/lysosomes, an increased fraction was found on the plasma membrane and in the cytosol compared to the un-acylated peptide.
Figure 3.

Cellular distribution of fatty acylated EETI-II across the cytosol, plasma membrane and intracellular membranes of the ER, Golgi and mitochondria
(A) EM images of NIH3T3 cells treated with Alexa 488-conjugated peptides (50 μM for WT EETI-II; 10 μM for C12- and C14-EETI-II; 20 μM for C16-EETI-II; 40 μM for C18-EETI-II; 6 h incubation at 37°C). Cells were washed with PBS and fixed with 4% PFA in 0.1 M Sorensen phosphate buffer, pH 7.4 for 3 h at RT. Ultrathin cryosections were immunolabelled with anti-Alexa488 antibodies, detected with Protein-A-gold particles (10 nm). Samples were subjected to EM analysis as described in the STAR Methods section. Yellow shade (false colored): endosomal and lysosomal vacuoles. Circles: Alexa 488 gold particles in the cytosol. Black arrowheads: Alexa 488 gold particles associated with small membrane-lined structures, i.e. vesicles and tubules (probably endosomal vesiculo-tubular structures), or on the plasma membrane. Green arrowheads: Alexa 488 gold particles associated with mitochondria. Purple arrowheads: Alexa 488 gold particles associated with ER or Golgi. See Figure S8D for original pictures without annotations. PM: plasma membrane; L: lysosomes; E: early/late endosomes; M: mitochondrion; G: Golgi; ER: ER N: nuclei. Scale bar, 200 nm.
(B) Statistics of cytosolic distribution of the peptides. The numbers of gold particles in the “true cytosol” (cellular regions at least 20 nm away from any cellular membranes including plasma membrane, endosomes/lysosomes, small vesicles and tubules, ER, Golgi apparatus or mitochondria) were counted and divided by the “true cytosol” area.
(C) Statistics of plasma membrane (P.M.) bound peptides. The numbers of gold particles on the plasma membrane were counted and divided by P.M. area (area extending over plasma membrane and neighboring 20 nm zones on both extracellular and intracellular side, in arbitrary units). Values represent mean ± S.E.M. n > 10 images per treatment.
Fatty acylated peptides show improved membrane association and resistance to detergent extraction
ImmunoEM studies established that C14- and C16-EETI-II demonstrated a high level of intracellular localization with ER, Golgi and mitochondrial membranes (Figures 3A, S8, and S9), in contrast to EETI-II which was predominantly in the lumen of endosomes and lysosomes (Figures S8B and S8C). These observations suggest that internalized C14- and C16-EETI-II could be associated with endomembranes. To validate this hypothesis, we assessed the effect of various detergents such as Triton X-100, saponin and digitonin on peptide intracellular localization. Triton X-100 is a non-selective membrane permeabilization reagent which permeabilizes all cellular membranes. On the other hand, saponin and digitonin are mild non-ionic detergents that form holes in the membrane by extracting the membrane cholesterol without damaging intracellular membrane compartments such as endosomes (Diaz et al., 1989; Scheffler et al., 2014). Permeabilization of cells with Triton X-100 led to a substantial loss in cellular fluorescence signal, with the fatty acylated peptides showing more residual signal compared to EETI-II (Figure S10), consistent with the notion that the majority of internalized EETI-II localized in the lumen of intracellular vesicles. Under milder permeabilization conditions in the presence of digitonin, the fluorescence signal of internalized fatty acylated peptides was not affected (Figure S10). Permeabilization of cells with saponin (0.04%) led to a reduction in intracellular fluorescence signal, with C16-EETI-II retaining the most cellular staining (∼50% cells, Figure S9), followed by C14-EETI-II and C12-EETI-II. The observed difference in peptide retention signal indicates that there is more loss of cytosolic constituents upon treatment with saponin compared to digitonin. In sum, the findings demonstrate that fatty acylation of peptide leads to higher retention compared to the non-acylated parent in cells under detergent permeabilization conditions, consistent with the immunoEM observations above and indicating tight membrane association. It is conceivable that fatty acid binding proteins could facilitate the exchange of lipidated CKPs between membranes and cytosol.
Uptake of fatty acylated CKPs is influenced by the presence of serum in the media
Since fatty acids bind to serum albumin, we sought to assess whether serum levels could affect the cellular uptake of fatty acylated peptides. NIH3T3 cells were treated with various peptides in growth medium containing either 10% or 0.2% fetal bovine serum (FBS) and they were then imaged by fluorescence microscopy. Whereas reducing the serum level did not alter the cellular uptake of EETI-II or C12-EETI-II, a marked increase in the cellular uptake of C14-, C-16, and C18-EETI-II peptides was observed in the presence of 0.2% FBS compared to 10% FBS, as demonstrated by the 5- to 20-fold increase in cellular fluorescence intensity (Figures 4A–4C). No apparent changes in the intracellular distribution profiles of fatty acylated peptides were observed upon changes in serum levels (Figure 4A). We hypothesize that under reduced serum levels, the concentration of BSA is not sufficient to fully bind and capture the majority of the fatty acylated peptide population in the medium; therefore, a higher proportion of free peptide is readily available to enter cells. Biophysical measurements established that fatty acylated peptides bound to human serum albumin (HSA) immobilized on a chip, with C16- and C-18-EETI-II demonstrating the highest binding capacity followed by C14-EETI-II and C12-EETI-II (Figure 4D). Under the same conditions, EETI-II did not show any binding to HSA even at high concentrations (30 μM) (Figure 4D). Altogether, the biophysical and cellular data establish that cellular uptake of C14-EETI-II, C16-EETI-II, and C18-EETI-II is influenced by the presence of serum albumin in the growth media, with C16- and C18-conjugated peptides demonstrating the biggest improvement in cellular uptake under reduced serum levels (Figures 4A–4C), likely because of their strong association to serum albumin.
Figure 4.

Fatty acylated EETI-II peptides bind to serum albumin
(A–C) Cellular uptake of C14, C16, and C18-EETI-II is enhanced with reduced serum. NIH3T3 cells were preincubated with 10% or 0.2% FBS supplemented medium then incubated with WT or fatty acylated EETI-II-A488 (5 μM) for 2 h at 37°C in 10% or 0.2% FBS supplemented medium. Cells were washed, fixed with 4% PFA and processed as described in the STAR Methods section. Fluorescence images were captured on a high throughput ImageXpress Micro XL imaging system (Molecular Devices). Representative images from four independent experiments are shown. Scale bar, 20 μm.
(D) Surface Plasmon Resonance (SPR) shows fatty acylated EETI-II peptides bind to human serum albumin. SPR sensor chip SA was coated with biotinylated human serum albumin and subjected to the peptides at varying concentrations (30, 10, 3.33, 1.11, 0.37, 0.123 μM) in PBST (0.05% Tween 20). Shown are measured binding responses (black) and curve fits (red) to a two-state model, assuming two preferred fatty acid binding sites on albumin. Representative data from three independent experiments are shown. Binding constant values represent mean ± S.E.M.
Fatty acylated CKPs undergo clathrin-mediated endocytosis and macropinocytosis
To further understand the cellular uptake mechanism of fatty acylated peptides, we analyzed their intracellular distribution in NIH3T3 cells with various endocytic markers including Texas red-conjugated 3 kDa dextran (a marker of fluid phase uptake and macropinocytosis), Alexa 555-conjugated transferrin (Tf-A555; a marker of early endosomes), Alexa 647-conjugated cholera toxin subunit B (CTxB-A647; a marker for dynamin-dependent caveolae-mediated endocytosis) and LysoTracker red DND-99, a marker for lysosomes. Fluorescence microscopy established that fatty acylated EETI-II peptides co-localized with 3 kDa dextran, similar to wild-type EETI-II (Gao et al., 2016), suggesting that their cellular uptake involves macropinocytosis (Figures S11A and S11C). Moreover, all the peptides were found to be at least partially co-localized with transferrin, indicating that they are targeted to the early/recycling endosome via a clathrin-mediated pathway (Figures S11B and S11D). However, there was little or no co-localization observed with internalized cholera toxin (Figure S13), suggesting that the uptake of the fatty acylated peptides is not mediated by caveolae. After 5 h, the fatty acylated EETI-II peptides were found partially co-localized with LysoTracker-positive lysosomes, similar to internalized EETI-II (Figure S14). Altogether, our findings indicate that conjugation of fatty acids to the N terminus of EETI-II does not appear to substantially perturb its internalization mechanism utilizing clathrin-mediated and macropinocytosis pathways, even though there are marked differences observed in their intracellular distributions.
Endocytosis inhibitors disrupt uptake of fatty acylated peptides
To corroborate the above findings, we assessed the effect of endocytosis inhibitors on cellular uptake of fatty acylated peptides. Treatment of cells with nocodazole, an inhibitor of microtubule polymerization which disrupts transport from early endosome to late endosome but not from the plasma membrane to early endosomes (Gruenberg and Howell, 1989; Mukhopadhyay et al., 1997), reduced the internalization of all peptides by 40–60% (Figure S15), suggesting that microtubules are critical for the endocytic mechanism of fatty acylated peptides from the plasma membrane to intracellular vesicles. Furthermore, the internalized peptides were largely found proximal to the plasma membrane (Figure S15B), supporting the notion that their transport to late endosomes is perturbed upon treatment with nocodazole. Treatment of cells with ikarugamycin, an inhibitor of clathrin-mediated endocytosis (Elkin et al., 2016), or EIPA, an inhibitor of macropinocytosis (Koivusalo et al., 2010), also perturbed the cellular internalization of all peptides (Figure S15). These results confirm that the cellular trafficking route of fatty acylated EETI-II peptides is at least partially dependent on clathrin-mediated endocytosis, macropinocytosis and intact microtubules. There could be other yet unidentified mechanisms that mediate uptake and subcellular distribution of the fatty acylated peptides including fatty acid binding proteins that could facilitate transition to the cytosol.
Discussion and conclusion
In this study, we systematically investigated the effect of fatty acylation on the cellular uptake and distribution of peptides. By conjugating fatty acids of different hydrocarbon chain lengths to the N terminus of a model cystine-knot peptide, we generated a panel of fatty acylated peptides without compromising their trypsin inhibition activity, and explored their mechanisms of cellular uptake and distribution. Fluorescence microscopy experiments established that fatty acyl modification drastically improved the cellular permeability of EETI-II. Cellular uptake was time- and concentration-dependent and occurred via clathrin-mediated and macropinocytosis pathways, with fatty acylated peptides eventually accumulating in endosomes and lysosomes. Importantly, a fraction of fatty acylated EETI-II peptides exhibited plasma membrane and cytosolic distribution, a phenotype that was not observed for wild-type EETI-II. ImmunoEM studies further confirmed the presence of fatty acylated EETI-II in both the cytosol and on the plasma membrane, as well on the ER, Golgi and mitochondrial membranes (Figure 5).
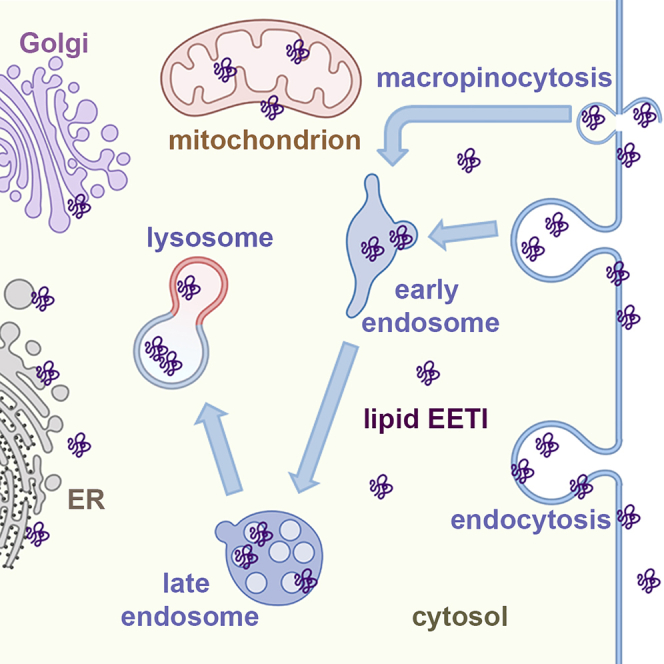
Figure 5.

Model of fatty acylated EETI-II cellular uptake
Fatty acylated EETI-II can enter cells through endocytosis and macropinocytosis. Importantly, a pool of fatty acylated peptides, primarily with C14 and C16 modifications, is found on the plasma membrane and in the cytosol. Although the majority of fatty acylated EETI-II localizes in the lumen of endosomes/lysosomes, a fraction can be detected on the ER, Golgi and mitochondrial membranes. The cartoon was created with BioRender.com.
The mechanism by which fatty-acylated peptides translocate to the cytosol remains unclear. From our studies, there is no evidence that the fatty acylated peptides induce endosomal leakage as all the intracellular organelles and markers investigated appear to be intact. Fluorescence and electron microscopy data indicate that enhanced association with the cell membrane seems to be the first step during cellular uptake of the fatty acylated peptides. This is further corroborated by earlier findings demonstrating that the myristoyl group inserts into the lipid bilayer via hydrophobic interactions (McLaughlin and Aderem, 1995), explaining the increased cell surface signal observed for the fatty acylated peptides. We hypothesize that the enhanced localization of peptides on the membranes leads to a higher overall cellular uptake via active endocytosis, resulting in increased cytosolic signals. We cannot rule out at this point whether endosomal escape efficiency could be enhanced by fatty acylation. However, the rapid increase in peptide distribution throughout the cell at early time points (within 1 h) would be inconsistent with this notion. Furthermore, the difference in cellular uptake observed for the various hydrocarbon chain lengths indicates that the nature of the fatty acid impacts membrane association and its translocation to the cytosol.
Another potential mechanism that has been proposed to explain translocation into the cell is flip-flop diffusion of myristoylated peptides (Eisele et al., 2002). However, this process would require significant energy expenditure and could also be influenced by many factors including temperature (Sankaram, 1994). Future studies to clarify the molecular mechanism of peptide translocation across the plasma membrane will help to elucidate this issue. In addition, the solubility, length and amino acid composition of the peptide cargo in physiologic buffers is a key factor in mediating cellular uptake, with less soluble peptides likely resulting in reduced cellular uptake in general. Studies aimed at exploring the broad application of fatty acylation in cellular delivery, in particular elucidating how fatty acylation mediates peptide delivery in different cell types and how it influences the uptake of different types of cargo, are underway in our laboratories.
Our studies support the potential of fatty acyl modification as a method to improve the cellular uptake of peptides, as exemplified by EETI-II, and enhance their subcellular distribution pattern. Our data also demonstrate that it is feasible to introduce albumin binding properties into cystine-knot peptides without altering their nascent trypsin inhibition activity, thereby generating bifunctional CKPs. The studies described in this work provide the first proof-of-concept of partial cytosolic delivery of EETI-II through fatty acylation and could have future applications in intracellular targeting of proteins. Fatty acylation could also have a potential utility in enhancing the delivery of other drug modalities such as oligonucleotides and macrocycles.
Synthetic chemical modification of cystine-knot peptides with fatty acids has not been demonstrated previously, nor their binding to human serum albumin. The set of fatty acylated CKPs generated in this study demonstrated effective binding to human serum albumin as measured by SPR, with C18 and C16 fatty acyl-modified peptides being the most preferred binders. Consistent with this notion, these peptides demonstrated the largest improvement in cellular uptake under reduced serum levels (Figures 4A–4C). As such, this trend seems to be consistent with the fatty acid binding preferences of human serum albumin, in which palmitic and stearic acids are preferred over lauric (C12:0) and myristic (C14:0) acid (Spector, 1975). The panel of albumin binding CKPs generated in this study serve as useful tools to characterize how fatty acylation could improve the in vivo half-life of CKPs and fine-tune their pharmacokinetics properties through binding to serum albumin. Investigating the in vivo half-lives of fatty-acylated cystine-knot peptides, and further evaluation of the relationship between their albumin binding capacities and systemic half-life is a subject of ongoing studies in our laboratories. Moreover, the bifunctional CKPs generated in this study demonstrate that new biological functions could be introduced into the CKP framework without compromising its nascent bioactivity. Finally, the simplicity and robustness of our synthetic strategy, which does not involve chemical conjugation steps post-folding, makes it suitable for engineering bioactive cystine-knot peptides with improved cellular uptake efficiency and distribution.
Limitation of the study
One limitation of the current study is the resolution of the fluorescence microscopy technique. To validate our conclusions from these studies, we used electron microscopy and additional detergent permeabilization methods to demonstrate cytosolic distribution of the lipidated peptides. Future exploration of additional types of fatty acids, conjugated to bioactive peptides that bind to cytosolic protein targets, is warranted.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Alexa Fluor 488 Polyclonal Antibody | ThermoFisher Scientific | Cat#: A11094; RRID: AB_221544 |
| Chemicals, peptides, and recombinant proteins | ||
| Trypsin | Promega | Cat#V5280 |
| Concanavalin A, TMA Conjugate | ThermoFisher Scientific | Cat#C860 |
| Alexa Fluor 488 NHS Ester | ThermoFisher Scientific | Cat#A20000 |
| EZ-Link NHS-PEG4-Biotin | ThermoFisher Scientific | Cat#A39259 |
| Human Albumin Biotin Conjugated | Rockland Immunochemicals | Cat#009-0633 |
| Fetal Bovine Serum | VWR | Cat#TXL97068077GEN |
| Gibco GlutaMAX Supplement | ThermoFisher Scientific | Cat#Gibco 35050061 |
| Dulbecco’s Modified Eagle’s Medium | ThermoFisher Scientific | Cat#11965084 |
| DMSO | Sigma | Cat#D2650, CAS: 67-68-5 |
| Hoechst 333421 | ThermoFisher Scientific | Cat#62249, CAS: 875756-97-1 |
| 16% formaldehyde | ThermoFisher Scientific | Cat#28906, CAS: 50-00-0 |
| nocodazole | Sigma | Cat#M1404, CAS: 31430-18-9 |
| ikarugamycin | Sigma | Cat#SML0188, CAS: 36531-78-9 |
| EIPA | Sigma | Cat#A3085, CAS: 1154-25-2 |
| Experimental models: Cell lines | ||
| Mouse: NIH-3T3 cells | ATCC | Cat#CRL-1658 |
| Software and algorithms | ||
| MetaXpress 4.0 | Molecular Devices | https://www.moleculardevices.com/products/cellular-imaging-systems/acquisition-and-analysis-software/metaxpress#gref |
| MassHunter Qualitative Analysis B.06.00 | Agilent | https://www.agilent.com/en/product/software-informatics/mass-spectrometry-software/data-analysis/qualitative-analysis |
| LAS AF image processing software | Leica Microsystems | https://www.leica-microsystems.com/products/microscope-software/p/leica-las-x-ls/ |
| ChemDraw | PerkinElmer Informatics | https://perkinelmerinformatics.com/products/research/chemdraw/ |
| Kaleidagraph | Synergy Software | https://www.synergy.com/plots/?gclid=CjwKCAjwhOyJBhA4EiwAEcJdcZOcr5fVjNmOXV65fB9tVPY2piiLif06U-oP9aI2BINMd3UdeIZCyxoCk6wQAvD_BwE |
| ImageJ | Schindelin et al., 2012 | https://imagej.nih.gov/ij/ |
| Other | ||
| Corning CellBIND 96-well Microplates | Corning | Cat#3340 |
| Nunc Lab-Tek II 8-chamber slides | ThermoFisher Scientific | Cat#154534 |
| Series S SA Sensor chip | GE Healthcare | Cat#BR100531 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Rami N. Hannoush (hannoush.rami@gene.com).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
NIH 3T3 cells were grown in high glucose Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10 % FBS and 2 mM Glutamax™. All cells were incubated in a 5 % CO2 humidified incubator at 37°C for 24 h before experiments. Cells were seeded onto Corning CellBIND 96 well plates (10,000 cells per well) or Nunc Lab-Tek II 8-chamber slides (20,000 cells per well) and were grown for 24 h at 37°C / 5 % CO2 before experiments.
Method details
Generation of fluorescently labeled fatty acylated EETI-II
Linear fatty acylated EETI-II peptides were synthesized and folded in the optimal folding buffer (0.1 M ammonium bicarbonate, pH 9.0, 2 mM reduced glutathione, 0.5 mM oxidized glutathione, 4 % DMSO for EETI-II, 0.1 M ammonium bicarbonate, pH 9.0, 1 mM reduced glutathione, 50 % DMSO for lipid EETI-II) at 0.5 mg/ml for 24 h at RT with shaking. Excess salt was removed by C18 Sep-Pak (Waters, cat # WAT043345). Folded peptides were lyophilized, reconstituted in 50 % DMSO (in H2O) and purified with a C18 (for EETI-II) or C4 (for fatty acylated EETI-II) reversed phase HPLC column. The final products were confirmed by mass spectrometry using a LC-MS system (Agilent Technologies). Purified folded EETI-II was labeled with NHS-AlexaFluor488 (Thermo Fisher Scientific). All labeling reactions were done with a molar ratio of 1:3 (CKP: dye) in 0.1 M sodium bicarbonate (pH 7.5) and incubated with gentle rocking at room temp for 18 hours in the dark. Labeled CKP was lyophilized and purified with a C18 reversed phase HPLC column (Gao et al., 2016). The final product (Alexa488 labeled EETI-II) was confirmed by mass spectrometry using a LC-MS system with a fluorescence detector (Agilent Technologies) and comprises either single-labeled EETI-II (at the lysine residue), or a mixture of single- and dual-labeled EETI-II (at the lysine residue and the N-terminus).
Fluorescence imaging
For cell permeabilization experiments, NIH 3T3 cells were incubated with 5 μM EETI-II-A488 for 60 min at 37°C / 5 % CO2, washed three times with cold PBS. Cells were then fixed with 4 % PFA for 20 min at room temperature, washed three times with PBS, and incubated with either PBS, or detergent containing PBS for 5 min at room temperature. Cells were washed again. To stain nuclei, cells were incubated with Hoechst 333421 (5 μg/ml in PBS) for 10 min and washed three times with PBS then stored in 100 μl PBS in the dark until image acquisition. Fluorescence images were captured on a high throughput ImageXpress Micro XL imaging system (Molecular Devices) and images were analyzed by MetaXpress 4.0. Fluorescence intensity values above a threshold defined using the DMSO-treated samples were measured and used to quantify integrated fluorescence intensity per cell or percentage of dye-positive cells. For endocytic inhibitor treatment, NIH 3T3 cells were treated with DMSO, nocodazole (10 μM; Sigma, cat # M1404), ikarugamycin (4 μM; Sigma, cat # SML0188), or EIPA (50 μM; Sigma, cat # A3085) for 30 min then with 5 μM WT or lipid EETI-II-A488, in the presence of DMSO or inhibitors for 60 min at 37°C. Cells were washed three times with cold PBS, fixed with 4 % PFA for 20 min at room temperature, and washed three times with PBS. Cells were incubated with Hoechst 333421 (5 μg/ml in PBS) for 10 min and washed three times with PBS then imaged and analyzed by MetaXpress 4.0. For confocal imaging, images of samples on coverslips were captured on an upright AX10 LEICA SPE laser scanning confocal microscope and images were analyzed with the LAS AF image processing software (Leica Microsystems).
Electron microscopy
NIH 3T3 cells were incubated with the Alexa488-conjugated peptides (50 μM for WT EETI-II; 10 μM for C12- and C14-EETI-II; 20 μM for C16-EETI-II; 40 μM for C18-EETI-II) for 6 h at 37 ºC. Cells were then washed with PBS and fixed with 4 % PFA in 0.1 M Sorensen phosphate buffer, pH 7.4 for 3 h at RT. Samples were washed with PBS for 5 min once and stored in 1 % PFA in 0.1 M Sorensen phosphate buffer, pH 7.4 until EM analysis. For EM analysis, the cells were rinsed in PBS, blocked in 0.15 % glycine in PBS, scraped in 1% gelatin in PBS, pelleted at 800 RCF, and embedded in 12 % gelatin. Small blocks of cell pellet were cryoprotected with 2.3 M sucrose, mounted on aluminum pins and frozen in liquid nitrogen. Ultrathin cryosections were cut at -120 ºC, incubated with PBS at 37 ºC to dissolve gelatin, then at room temperature with rabbit anti-Alexa488 antibody (Molecular Probes, A-11094, 1:50) followed by Protein A-conjugated 10 nm gold particles (CMC Utrecht). Blocking of background labeling was done with a mixture of fish skin gelatin (Sigma) and acetylated BSA (Aurion). Final staining of the sections was performed with uranyl acetate followed by a uranyl-acetate methylcellulose mixture. Sections were examined in a JEOL JEM-1011 or a FEI T12 electron microscope. For the quantitative EM analysis, random section areas were recorded at 50000x for analysis by a Grids plugin in Fiji or ImageJ software (Schindelin et al., 2012). An orthogonal line grid with square mesh areas of 0.25 μm2 as arbitrary area unit was placed over the recorded images. Areas of the different types of subcellular compartments were measured by counting the number of intersections of the grid overlapping with the compartments inclusive a 20 nm wide border of cytosol around their limiting membrane. For the assignment of Alexa488 immunogold particles to either membrane-bounded compartments or cytosol, we took into account that the anti-Alexa488 antibody-Protein A-10-nm gold particle complex is approximately 25 nm long. Hence, 10 nm gold particles located with their centers at a maximum distance of 20 nm from a membrane could still label a membrane-bound epitope. Gold particles were therefore scored as associated with membrane-bounded organelles (endosomes, lysosomes, small vesicles-tubules, ER, nuclear envelope, Golgi, mitochondria) if they were located with their center inside the organelle, on its limiting membrane, or within a range of 20 nm outside the limiting membrane. Gold particles were assigned to the plasma membrane if their centers were located on the membrane or within a distance of 20 nm from either surface of the plasma membrane. All gold particles located with their center at a distance larger than 20 nm from a membrane were assigned as “true cytosol”. The Alexa488 gold particle density on subcellular compartments was calculated as the number of gold particles per arbitrary unit of compartment area.
Surface Plasmon Resonance (SPR)
All measurements were done on a Biacore S200 instrument (GE Healthcare) with PBS-T (PBS with 0.05 % Tween 20, pH 7.4) as the running buffer. For kinetic measurements, biotinylated human serum albumin (2 μg/ml, Rockland Immunochemicals Inc.) was captured on three separate sensor channels of a Series S SA Sensor chip (GE Healthcare). The chip (all four channels) was then blocked with 0.1 mg/ml free biotin. A concentration series of EETI-II peptides (0.123 – 30 μM) were injected to the chip. All data were double-referenced by subtracting the signal from a control sensor channel (coated with biotin only) and the signal from a buffer injection. Referenced datasets that showed binding were fitted using the manufacturer's software to a two-state kinetic binding model to account for the two fatty acid preferred binding sites on albumin.
Trypsin enzymatic assay
Normalized proteolytic activity (vi/vo) of trypsin (2 nM) toward L-Arg-AMC substrate (75 μ M) was measured for 60 min after pre-incubation of the peptides at different concentrations (Io) with trypsin (Eo) for 30 min at room temperature. Data were plotted and fitted using GraphPad Prism 9 software. Apparent inhibition constant (Kiapp) was determined from at least three independent experiments. Apparent inhibition constants (Kiapp) were calculated by fitting the Morrison Equation 1 for tight-binding inhibitors using non-linear regression. Substrate-independent inhibition constants Ki were calculated from Kiapp and the measured KM values according to Equation 2 (Gao et al., 2016; Stanger et al., 2014).
| (Equation 1) |
| (Equation 2) |
Quantification and statistical analysis
Fluorescent images were analyzed by MetaXpress 4.0. For each sample, integrated fluorescence intensity values above a threshold defined using the DMSO-treated samples were measured and averaged from 9 different sites. For EM analysis, random section areas were recorded at 50000x for analysis by a Grids plugin in Fiji or ImageJ software. An orthogonal line grid with square mesh areas of 0.25 μm2 as arbitrary area unit was placed over the recorded images. Areas of the different types of subcellular compartments were measured by counting the number of intersections of the grid overlapping with the compartments inclusive a 20 nm wide border of cytosol around their limiting membrane.
Acknowledgments
We thank Kristin Wucherer for purification of Alexa647-conjugated EETI-II and C14-EETI-II, Suzanne van Dijk and Cecilia de Heus for EM preparations, René Scriwanek for help with the EM figures and Suzie Scales for discussions. The EM infrastructure of UMC Utrecht is partially funded by the Netherlands Electron Microscopy Infrastructure (NEMI), project number 184.034.014 of the National Roadmap for Large-Scale Research Infrastructure of the Dutch Research Council (NWO).
Author contributions
R.N.H, X.G. and J.K. designed the experiments. X.G., A.D.M. and R.B. performed the described experiments. X.G. and A.D.M. analyzed the data. R.N.H and X.G. drafted the manuscript, with input from A.D.M. and J.K.. All authors approved the final version of the manuscript. R.N.H. conceived and supervised the research strategy.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
One or more of the authors of this paper self-identifies as a member of the LGBTQ + community.
Published: November 19, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.103220.
Supplemental information
Data and code availability
No new datasets were generated in this study. This paper does not report original code. Any additional information reported in this paper is available from the lead contact upon request.
References
- Blaskovic S., Blanc M., van der Goot F.G. What does S-palmitoylation do to membrane proteins? FEBS J. 2013;280:2766–2774. doi: 10.1111/febs.12263. [DOI] [PubMed] [Google Scholar]
- Čemažar M., Daly N.L., Häggblad S., Lo K.P., Yulyaningsih E., Craik D.J. Knots in Rings. J. Biol. Chem. 2006;281:8224–8232. doi: 10.1074/jbc.M513399200. [DOI] [PubMed] [Google Scholar]
- Chamberlain L.H., Shipston M.J. The physiology of protein S-acylation. Physiol. Rev. 2015;95:341–376. doi: 10.1152/physrev.00032.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M.H., Li Y.J., Kawakami T., Xu S.M., Chuang P.T. Palmitoylation is required for the production of a soluble multimeric Hedgehog protein complex and long-range signaling in vertebrates. Genes Development. 2004;18:641–659. doi: 10.1101/gad.1185804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craik D.J., Du J. Cyclotides as drug design scaffolds. Curr. Opin. Chem. Biol. 2017;38:8–16. doi: 10.1016/j.cbpa.2017.01.018. [DOI] [PubMed] [Google Scholar]
- Darling J.E., Zhao F., Loftus R.J., Patton L.M., Gibbs R.A., Hougland J.L. Structure-activity analysis of human ghrelin O-acyltransferase reveals chemical determinants of ghrelin selectivity and acyl group recognition. Biochemistry. 2015;54:1100–1110. doi: 10.1021/bi5010359. [DOI] [PubMed] [Google Scholar]
- Diaz R., Wileman T.E., Anderson S.J., Stahl P. The use of permeabilized cells to study the ion requirements of receptor-ligand dissociation in endosomes. Biochem. J. 1989;260:127–134. doi: 10.1042/bj2600127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisele F., Kuhlmann J., Waldmann H. Synthesis and membrane binding properties of a lipopeptide fragment from influenza virus a hemagglutinin. Chemistry (Weinheim an der Bergstrasse, Germany) 2002;8:3362–3376. doi: 10.1002/1521-3765(20020802)8:15<3362::AID-CHEM3362>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Elkin S.R., Oswald N.W., Reed D.K., Mettlen M., MacMillan J.B., Schmid S.L. Ikarugamycin: a natural product inhibitor of clathrin-mediated endocytosis. Traffic. 2016;17:1139–1149. doi: 10.1111/tra.12425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enseñat-Waser R., Martin F., Barahona F., Vazquez J., Soria B., Reig J.A. Direct visualization by confocal fluorescent microscopy of the permeation of myristoylated peptides through the cell membrane. IUBMB life. 2002;54:33–36. doi: 10.1080/15216540213823. [DOI] [PubMed] [Google Scholar]
- Gao X., De Maziere A., Iaea D.B., Arthur C.P., Klumperman J., Ciferri C., Hannoush R.N. Visualizing the cellular route of entry of a cystine-knot peptide with Xfect transfection reagent by electron microscopy. Scientific Rep. 2019;9:6907. doi: 10.1038/s41598-019-43285-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X., Hannoush R.N. Single-cell imaging of Wnt palmitoylation by the acyltransferase porcupine. Nat. Chem. Biol. 2014;10:61–68. doi: 10.1038/nchembio.1392. [DOI] [PubMed] [Google Scholar]
- Gao X., Hannoush R.N. Single-cell in situ imaging of palmitoylation in fatty-acylated proteins. Nat. Protoc. 2014;9:2607–2623. doi: 10.1038/nprot.2014.179. [DOI] [PubMed] [Google Scholar]
- Gao X., Hannoush R.N. A decade of Click chemistry in protein palmitoylation: impact on Discovery and new Biology. Cell Chem. Biol. 2018;25:236–246. doi: 10.1016/j.chembiol.2017.12.002. [DOI] [PubMed] [Google Scholar]
- Gao X., Stanger K., Kaluarachchi H., Maurer T., Ciepla P., Chalouni C., Franke Y., Hannoush R.N. Cellular uptake of a cystine-knot peptide and modulation of its intracellular trafficking. Scientific Rep. 2016;6:35179. doi: 10.1038/srep35179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetz J.A., Singh S., Suber L.M., Kull F.J., Robbins D.J. A highly conserved amino-terminal region of sonic hedgehog is required for the formation of its freely diffusible multimeric form. J. Biol. Chem. 2006;281:4087–4093. doi: 10.1074/jbc.M511427200. [DOI] [PubMed] [Google Scholar]
- Gruenberg J., Howell K.E. Membrane traffic in endocytosis: insights from cell-free assays. Annu. Rev. Cell Biol. 1989;5:453–481. doi: 10.1146/annurev.cb.05.110189.002321. [DOI] [PubMed] [Google Scholar]
- Hannoush R.N., Sun J. The chemical toolbox for monitoring protein fatty acylation and prenylation. Nat. Chem. Biol. 2010;6:498–506. doi: 10.1038/nchembio.388. [DOI] [PubMed] [Google Scholar]
- Hantschel O., Nagar B., Guettler S., Kretzschmar J., Dorey K., Kuriyan J., Superti-Furga G. A myristoyl/phosphotyrosine switch regulates c-Abl. Cell. 2003;112:845–857. doi: 10.1016/s0092-8674(03)00191-0. [DOI] [PubMed] [Google Scholar]
- Home P., Kurtzhals P. Insulin detemir: from concept to clinical experience. Expert Opin. Pharmacother. 2006;7:325–343. doi: 10.1517/14656566.7.3.325. [DOI] [PubMed] [Google Scholar]
- Jackson S.H., Martin T.S., Jones J.D., Seal D., Emanuel F. Liraglutide (victoza): the first once-daily incretin mimetic injection for type-2 diabetes. P T : a Peer-Reviewed J. Formulary Management. 2010;35:498–529. [PMC free article] [PubMed] [Google Scholar]
- Janda C.Y., Waghray D., Levin A.M., Thomas C., Garcia K.C. Structural basis of Wnt recognition by frizzled. Science. 2012;337:59–64. doi: 10.1126/science.1222879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudsen L.B., Nielsen P.F., Huusfeldt P.O., Johansen N.L., Madsen K., Pedersen F.Z., Thogersen H., Wilken M., Agerso H. Potent derivatives of glucagon-like peptide-1 with pharmacokinetic properties suitable for once daily administration. J. Med. Chem. 2000;43:1664–1669. doi: 10.1021/jm9909645. [DOI] [PubMed] [Google Scholar]
- Koivusalo M., Welch C., Hayashi H., Scott C.C., Kim M., Alexander T., Touret N., Hahn K.M., Grinstein S. Amiloride inhibits macropinocytosis by lowering submembranous pH and preventing Rac1 and Cdc42 signaling. J. Cell Biol. 2010;188:547–563. doi: 10.1083/jcb.200908086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima M., Kangawa K. Ghrelin: structure and function. Physiol. Rev. 2005;85:495–522. doi: 10.1152/physrev.00012.2004. [DOI] [PubMed] [Google Scholar]
- Kowalczyk R., Harris P.W.R., Williams G.M., Yang S.H., Brimble M.A. Peptide lipidation - a synthetic strategy to Afford peptide based therapeutics. Adv. Exp. Med. Biol. 2017;1030:185–227. doi: 10.1007/978-3-319-66095-0_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Floch J.P. Critical appraisal of the safety and efficacy of insulin detemir in glycemic control and cardiovascular risk management in diabetics. Diabetes Metab. Syndr. Obes. : Targets Ther. 2010;3:197–213. doi: 10.2147/dmsott.s7315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin S., Aderem A. The myristoyl-electrostatic switch: a modulator of reversible protein-membrane interactions. Trends Biochem. Sci. 1995;20:272–276. doi: 10.1016/s0968-0004(00)89042-8. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay A., Funato K., Stahl P.D. Rab7 regulates transport from early to late endocytic compartments in Xenopus oocytes. J. Biol. Chem. 1997;272:13055–13059. doi: 10.1074/jbc.272.20.13055. [DOI] [PubMed] [Google Scholar]
- Myers S.R., Yakubu-Madus F.E., Johnson W.T., Baker J.E., Cusick T.S., Williams V.K., Tinsley F.C., Kriauciunas A., Manetta J., Chen V.J. Acylation of human insulin with palmitic acid extends the time action of human insulin in diabetic dogs. Diabetes. 1997;46:637–642. doi: 10.2337/diab.46.4.637. [DOI] [PubMed] [Google Scholar]
- Nelson A.R., Borland L., Allbritton N.L., Sims C.E. Myristoyl-based transport of peptides into living cells. Biochemistry. 2007;46:14771–14781. doi: 10.1021/bi701295k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nile A.H., Hannoush R.N. Fatty acylation of Wnt proteins. Nat. Chem. Biol. 2016;12:60–69. doi: 10.1038/nchembio.2005. [DOI] [PubMed] [Google Scholar]
- Pepinsky R.B., Zeng C., Wen D., Rayhorn P., Baker D.P., Williams K.P., Bixler S.A., Ambrose C.M., Garber E.A., Miatkowski K. Identification of a palmitic acid-modified form of human Sonic hedgehog. J. Biol. Chem. 1998;273:14037–14045. doi: 10.1074/jbc.273.22.14037. [DOI] [PubMed] [Google Scholar]
- Resh M.D. Myristylation and palmitylation of Src family members: the fats of the matter. Cell. 1994;76:411–413. doi: 10.1016/0092-8674(94)90104-x. [DOI] [PubMed] [Google Scholar]
- Resh M.D. Membrane targeting of lipid modified signal transduction proteins. Sub-Cellular Biochem. 2004;37:217–232. doi: 10.1007/978-1-4757-5806-1_6. [DOI] [PubMed] [Google Scholar]
- Resh M.D. Fatty acylation of proteins: the long and the short of it. Prog. Lipid Res. 2016;63:120–131. doi: 10.1016/j.plipres.2016.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocks O., Peyker A., Kahms M., Verveer P.J., Koerner C., Lumbierres M., Kuhlmann J., Waldmann H., Wittinghofer A., Bastiaens P.I. An acylation cycle regulates localization and activity of palmitoylated Ras isoforms. Science. 2005;307:1746–1752. doi: 10.1126/science.1105654. [DOI] [PubMed] [Google Scholar]
- Sankaram M.B. Membrane interaction of small N-myristoylated peptides: implications for membrane anchoring and protein-protein association. Biophysical J. 1994;67:105–112. doi: 10.1016/S0006-3495(94)80459-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheffler J.M., Schiefermeier N., Huber L.A. Mild fixation and permeabilization protocol for preserving structures of endosomes, focal adhesions, and actin filaments during immunofluorescence analysis. Methods Enzymol. 2014;535:93–102. doi: 10.1016/B978-0-12-397925-4.00006-7. [DOI] [PubMed] [Google Scholar]
- Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shipston M.J. Ion channel regulation by protein palmitoylation. J. Biol. Chem. 2011;286:8709–8716. doi: 10.1074/jbc.R110.210005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smotrys J.E., Linder M.E. Palmitoylation of intracellular signaling proteins: regulation and function. Annu. Rev. Biochem. 2004;73:559–587. doi: 10.1146/annurev.biochem.73.011303.073954. [DOI] [PubMed] [Google Scholar]
- Spector A.A. Fatty acid binding to plasma albumin. J. Lipid Res. 1975;16:165–179. [PubMed] [Google Scholar]
- Stanger K., Maurer T., Kaluarachchi H., Coons M., Franke Y., Hannoush R.N. Backbone cyclization of a recombinant cystine-knot peptide by engineered Sortase A. FEBS Lett. 2014;588:4487–4496. doi: 10.1016/j.febslet.2014.10.020. [DOI] [PubMed] [Google Scholar]
- Willert K., Brown J.D., Danenberg E., Duncan A.W., Weissman I.L., Reya T., Yates J.R., 3rd, Nusse R. Wnt proteins are lipid-modified and can act as stem cell growth factors. Nature. 2003;423:448–452. doi: 10.1038/nature01611. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
No new datasets were generated in this study. This paper does not report original code. Any additional information reported in this paper is available from the lead contact upon request.