

Abstract
The synucleinopathies Parkinson disease (PD), multiple system atrophy (MSA), and pure autonomic failure (PAF) are characterized by intra-cytoplasmic deposition of the protein alpha-synuclein and by catecholamine depletion. PAF, which manifests with neurogenic orthostatic hypotension (nOH) and no motor signs of central neurodegeneration, can evolve into PD+nOH. Cerebrospinal fluid (CSF) levels of catecholamine metabolites may indicate central catecholamine deficiency in these synucleinopathies, but the literature is inconsistent and incomplete. In this retrospective cohort study we reviewed data about CSF catecholamines, the dopamine metabolites 3,4-dihydroxyphenylacetic acid (DOPAC) and homovanillic acid (HVA), and the norepinephrine metabolites 3,4-dihydroxyphenylglycol (DHPG) and 3-methoxy-4-hydroxyphenylglycol (MHPG). The compounds were measured in 36 PD, 37 MSA, and 19 PAF patients and in 38 controls. Compared to the control group, the PD, MSA, and PAF groups had decreased CSF MHPG (p<0.0001 each by Dunnett’s post-hoc test), DHPG (p=0.004; p<0.0001; p<0.0001) and norepinephrine (p=0.017; p=0.0003; p=0.044). CSF HVA and DOPAC were decreased in PD (p<0.0001 each) and MSA (p<0.0001 each) but not in PAF. The three synucleinopathies therefore have in common in vivo evidence of central noradrenergic deficiency but differ in extents of central dopaminergic deficiency—prominent in PD and MSA, less apparent in PAF. Data from putamen 18F-DOPA and cardiac 18F-dopamine neuroimaging in the same patients, post-mortem tissue catecholamines in largely separate cohorts, and review of the neuropathology literature fit with these distinctions. The results suggest a “norepinephrine first” ascending pathogenetic sequence in synucleinopathies, with degeneration of pontine locus ceruleus noradrenergic neurons preceding loss of midbrain substantia nigra dopaminergic neurons.
Keywords: norepinephrine, Parkinson, multiple system atrophy, pure autonomic failure, MHPG, HVA
INTRODUCTION
The neurochemical hallmark of Parkinson’s disease (PD) is depletion of the catecholamine dopamine in the nigrostriatal system—especially in the putamen (Kish et al. 1988). By the time the characteristic motor symptoms manifest clinically it is likely that a substantial proportion of striatal dopaminergic terminals have already been lost (DelleDonne et al. 2008). Comparable if not greater (Zarow et al. 2003) loss of neurons occurs in the pontine locus ceruleus (LC), possibly before the loss of substantia nigra (SN) neurons (Del Tredici & Braak 2013).
Multiple system atrophy (MSA) can be difficult to distinguish clinically from PD (Rajput et al. 1991). Both conditions are characterized by cytoplasmic deposition of the protein alpha-synuclein (α-syn)—in Lewy bodies in PD (Spillantini et al. 1997) and in glial cytoplasmic inclusions in MSA (Wakabayashi et al. 1998). PD and MSA are now considered to be forms of synucleinopathy (Bras et al. 2020; Yamasaki et al. 2019).
Pure autonomic failure (PAF) is a rare but scientifically important disease that manifests with neurogenic orthostatic hypotension (nOH) related to sympathetic noradrenergic deficiency (Ziegler et al. 1977), without motor signs of central neurodegeneration (Kaufmann 1996). Post-mortem studies of PAF patients have consistently reported Lewy body pathology (van Ingelghem et al. 1994; Bannister et al. 1967) or intra-neuronal α-syn deposition (Hague et al. 1997; Arai et al. 2000; Kaufmann et al. 2001) in the sympathetic nervous system or brainstem, justifying including PAF in the synucleinopathy family.
PAF can evolve into PD+nOH (Kaufmann et al. 2017), in line with the notion of a “body-first” pathogenetic process (Horsager et al. 2020) beginning in the autonomic nervous system and followed by ascending central catecholaminergic neurodegeneration in the brainstem (Del Tredici & Braak 2012; Van Den Berge et al. 2019).
There has long been interest in cerebrospinal fluid (CSF) levels of catecholamine metabolites as biomarkers of central catecholamine deficiency, and many studies over the past half century have compared PD, MSA, or PAF groups with controls (Table 1). Several publications have described reduced CSF levels of 3,4-dihydroxyphenylacetic acid (DOPAC), the main intra-neuronal metabolite of dopamine, in PD (Eldrup et al. 1995; Andersen et al. 2017; Engelborghs et al. 2003). We reported that PD, MSA, and PAF entail decreased CSF DOPAC (Goldstein et al. 2012b), with a lower DOPAC in PAF than in PD. More recently we found that in individuals with multiple PD risk factors, low CSF levels of DOPAC and 3,4-dihydroxyphenylalanine (DOPA), the precursor of the catecholamines, predict later development of the disease (Goldstein et al. 2018); however, DOPAC undergoes extensive extra-neuronal metabolism by catechol-O-methyltransferase (COMT) to form homovanillic acid (HVA), the main end-product of dopamine metabolism (see concept diagram in Figure 1), and individual differences in COMT activity could affect the utility of CSF DOPAC as a biomarker.
Table 1:
Published reports about cerebrospinal fluid (CSF) levels of catecholamines and catecholamine metabolites in Parkinson disease (PD), multiple system atrophy (MSA), or pure autonomic failure (PAF) and control groups.
| Disease | N | CSF DOPA | CSF DA | CSF DOPAC | CSF HVA | CSF NE | CSF DHPG | CSF MHPG | First Author | Year | PMID |
|---|---|---|---|---|---|---|---|---|---|---|---|
| PD | 1 | Decreased | Bernheimer | 1966 | 4229554 | ||||||
| 2 | Decreased | Guldberg | 1967 | 5588903 | |||||||
| 3 | Decreased | Johansson | 1967 | 6035772 | |||||||
| 4 | Normal | Olsson | 1968 | 5668438 | |||||||
| 5 | Decreased | Gottfries | 1969 | 5808100 | |||||||
| 6 | Normal | Wilk | 1971 | 5571114 | |||||||
| 7 | Decreased | Granerus | 1974 | 4854088 | |||||||
| 8 | Normal | Davidson | 1977 | 591981 | |||||||
| 9 | Decreased | Cunha | 1983 | 6839227 | |||||||
| 10 | Normal | Mann | 1983 | 6644314 | |||||||
| 11 | Normal | Mena | 1984 | 6204498 | |||||||
| 12 | Normal | Normal | Turkka | 1987 | 3816883 | ||||||
| 13 | Decreased | Normal | Hartikainen | 1992 | 1347220 | ||||||
| 14 | Decreased | Normal | Martignoni | 1992 | 1320891 | ||||||
| 15 | Decreased | Strittmatter | 1992 | 1378766 | |||||||
| 16 | Lewitt | 1993 | 8420188 | ||||||||
| 17 | Decreased | Normal | Normal | Chia | 1993 | 8336158 | |||||
| 18 | Normal | Normal | Normal | González-Quevedo | 1993 | 7521168 | |||||
| 19 | Decreased | Decreased | Normal | Normal | Mashige | 1994 | 7914240 | ||||
| 20 | Normal | Normal | Decreased | Decreased | Eldrup | 1995 | 7484057 | ||||
| 21 | Decreased | Cheng | 1996 | 9617787 | |||||||
| 22 | Decreased | Kanemaru | 1998 | 9605500 | |||||||
| 23 | Normal | Decreased | Normal | Engelborghs | 2003 | 12834252 | |||||
| 24 | Normal | Decreased | Goldstein | 2008 | 18325818 | ||||||
| 25 | Decreased | Decreased | Ishibashi | 2010 | 20002007 | ||||||
| 26 | Normal | Lewitt | 2011 | 21784416 | |||||||
| 27 | Decreased | Normal | Decreased | Normal | Decreased | Goldstein | 2012 | 22451506 | |||
| 28 | Decreased | Normal | Decreased | Normal | Decreased | Engelborghs | 2012 | 22451506 | |||
| 29 | Decreased | Normal | Herbert | 2013 | 24122060 | ||||||
| 30 | Decreased | Decreased | Decreased | Decreased | Normal | Andersen | 2017 | 28244186 | |||
| 31 | Decreased | Normal | Decreased | Decreased | Decreased | Decreased | Decreased | Goldstein | This study | ||
| MSA | 1 | Decreased | Decreased | Polinsky | 1984 | 6539879 | |||||
| 2 | Decreased | Decreased | Martignoni | 1992 | 1320891 | ||||||
| 3 | Goldstein | 2003 | 12540289 | ||||||||
| 4 | Decreased | Decreased | Abdo | 2007 | 17448720 | ||||||
| 5 | Normal | Decreased | Goldstein | 2008 | 18325818 | ||||||
| 6 | Decreased | Decreased | Decreased | Normal | Decreased | Goldstein | 2012 | 22451506 | |||
| 7 | Decreased | Normal | Decreased | Decreased | Decreased | Decreased | Decreased | Goldstein | This study | ||
| PAF | 1 | Decreased | Polinsky | 1984 | 6539879 | ||||||
| 2 | Decreased | Goldstein | 2003 | 12540289 | |||||||
| 3 | Decreased | Decreased | Decreased | Decreased | Decreased | Goldstein | 2012 | 22451506 | |||
| 4 | Normal | Normal | Normal | Normal | Decreased | Decreased | Decreased | Goldstein | This study |
Abbreviations: DA=dopamine; DHPG=3,4-dihydroxyphenylglycol; DOPA=3,4-dihydroxyphenylalanine; DOPAC=3,4-dihydroxyphenylacetic acid; HVA=homovanillic acid; MHPG=3-methoxy-4-hydroxyphenylglycol; NE=norepinephrine; PMID=PubMed ID number.
Figure 1: Concept diagram depicting sources of levels of catecholamines and their metabolites in cerebrospinal fluid.
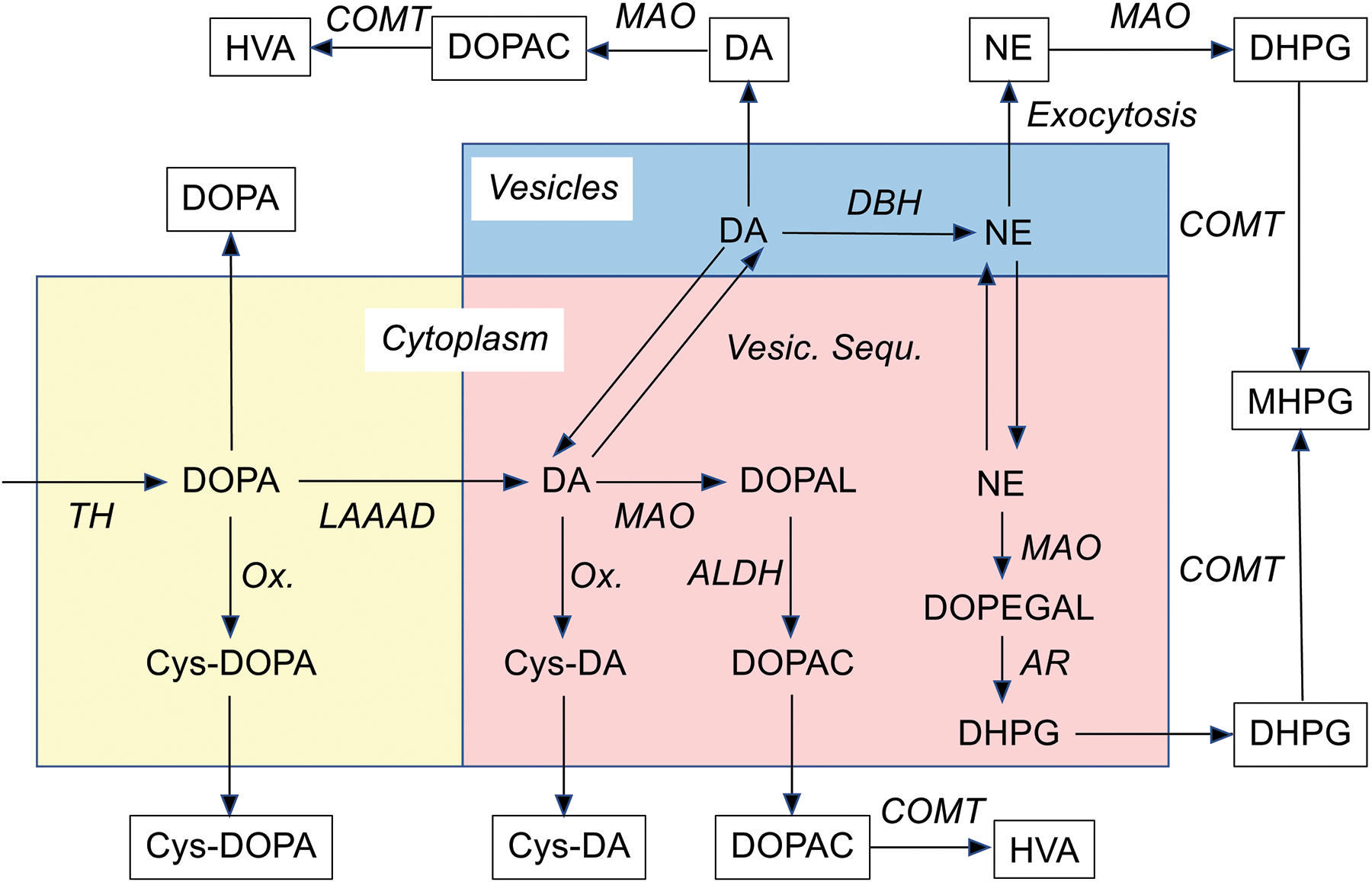
Biochemicals are in plain text and processes in italics. The colored rectangles represent (blue) vesicles and two cytoplasmic compartments, one involving processes proximal to cytoplasmic dopamine (banana color) and the other distal to cytoplasmic dopamine production (pink). Abbreviations: ALDH=aldehyde dehydrogenase; AR=aldehyde/aldose reductase; COMT=catechol-O-methyltransferase; Cys-DOPA=5-S-cysteinyldopa; Cys-DA=5-S-cysteinyldopamine; DA=dopamine; DHPG=3,4-dihydroxyphenylglycol; DOPA=3,4-dihydroxyphenylalanine; DOPAL=3,4-dihydroxyphenylacetaldehyde; DOPEGAL=3,4-dihydroxyphenyglycolaldehyde; HVA=homovanillic acid; LAAAD=L-aromatic-amino-acid decarboxylase; MAO=monoamine oxidase; MHPG=3-methoxy-4-hydroxyphenylglycol; NE=norepinephrine; Ox.=spontaneous oxidation; TH=tyrosine hydroxylase; VMAT=vesicular monoamine transporter.
The literature about whether synucleinopathies entail CSF neurochemical evidence of central norepinephrine deficiency is inconsistent and incomplete. Eldrup et al. noted decreased CSF norepinephrine in PD (Eldrup et al. 1995), but Turkka et al. and Chia et al. (Turkka et al. 1987; Chia et al. 1993) did not observe decreases in either norepinephrine or 3-hydroxy-4-hydroxyphenylglycol (MHPG), the main end-product of central norepinephrine metabolism. Martignoni et al. reported low CSF norepinephrine in PD and MSA and reduced MHPG in MSA but not in PD (Martignoni et al. 1992). Herbert et al. also did not find decreased MHPG in PD (Herbert et al. 2013). Polinsky et al. found decreased CSF MHPG in MSA and in PAF (then called idiopathic orthostatic hypotension) (Polinsky et al. 1984). Our group reported decreased CSF levels of norepinephrine and 3,4-dihydroxyphenylglycol (DHPG), the main intra-neuronal metabolite of norepinephrine, in PAF and in a subgroup of MSA patients with orthostatic hypotension but not in MSA without orthostatic hypotension (Goldstein et al. 2003a). In our 2012 study we reported decreased CSF DHPG in PD, MSA, and PAF (Goldstein et al. 2012b); however, just as DOPAC is metabolized to HVA by COMT, DHPG is metabolized to MHPG, and our study was limited in that it did not include data about MHPG.
These considerations led us to revisit the issue of CSF biomarkers of central catecholamine deficiency in synucleinopathies, by comprehensively assaying CSF DOPA, DHPG, MHPG, DOPAC, HVA, and the catecholamines themselves in relatively large cohorts of PD, MSA, and PAF patients and controls. We assessed the efficacy of CSF levels of each of these neurochemicals for distinguishing the synucleinopathy group from the control group.
We were especially interested in measuring indices of central dopamine deficiency (CSF HVA and DOPAC) and norepinephrine deficiency (CSF MHPG, DHPG, and norepinephrine) in PAF. Since PAF does not entail clinical evidence of parkinsonism, we predicted that PAF patients would have normal CSF HVA and DOPAC levels. Meanwhile, noradrenergic deficiency, which in the periphery is a well established feature of PAF (Goldstein et al. 2003b; Ziegler et al. 1977), might extend to the brain, in which case CSF MHPG, DHPG, and norepinephrine would all be low.
To examine directly whether central noradrenergic deficiency is a common theme in synucleinopathies, in a mainly different cohort we assayed tissue catecholamine contents post-mortem in frontal cortex and putamen. Frontal cortex receives noradrenergic innervation exclusively from the pontine LC (Itoi et al. 2011), while putamen receives dopaminergic innervation from the mesencephalic SN. In PD and MSA we predicted decreases in both cortical norepinephrine and putamen dopamine. In PAF we predicted decreases in cortical norepinephrine and, based on post-mortem literature about SN neuronal loss (Table 2), variably decreased putamen dopamine.
Table 2:
Autopsy findings in idiopathic orthostatic hypotension or pure autonomic failure.
| Reference | No. | SNS LBs | LC Neuronal Loss | SN Neuronal Loss |
|---|---|---|---|---|
| (Johnson et al. 1966) | Case 1 | Eosinophilic bodies | 0 | 0 |
| Case 2 | 1 | 0 | 0 | |
| (Roessmann et al. 1971) | Case 1 | 1 | 1 | 1 |
| (Schober et al. 1975) | Case 2 | 1 | 1 | 1 |
| (Black & Petito 1976) | Case C | 1 | Blank TH | Normal TH |
| (Petito & Black 1978) | Patient 3 | Hyaline bodies | 1 | 1 |
| (van Ingelghem et al. 1994) | 1 | 1 | 0 | 0 |
| (Hague et al. 1997) | 1 | 1 | Mild, WNL | Mild, WNL |
| (Arai et al. 2000) | 1 | 1 | No comment | 0 |
| (Miura et al. 2001) | 1 | No ganglia found | 1 | 1 |
| (Terao et al. 1993) | 1 | 1 | 1 | 1 |
| Case 2 | 1 | 1 (Pallor) | 1 (Pallor) |
Abbreviations: Brstem=brainstem; LB=Lewy body; TH=tyrosine hydroxylase; WNL=within normal limits.
METHODS
Subjects
The Intramural Research Board of the National Institute of Neurological Disorders and Stroke (NINDS) at the National Institutes of Health approved the protocols for this study (NIH Clinical Protocols 94N0186, 03N0004, and 18N0140). The participants gave informed written consent before any research procedures.
Data were reviewed for all subjects who had undergone lumbar puncture (LP) from 2004 to 2020 at the NIH Clinical Center and had CSF assayed for levels of catechols, MHPG, and HVA in the laboratory of the Autonomic Medicine Section (formerly Clinical Neurocardiology Section) in intramural NINDS.
The study was not pre-registered. No randomization was performed to allocate subjects in the study. No sample calculation was performed. The study was exploratory. The retrospective study included data for all subjects studied under the relevant protocols during the specified time period. There was no prospective estimation of the number of subjects that would be required. For all subjects the following comorbid conditions were exclusionary for the present analysis: symptomatic coronary artery disease, diabetes mellitus requiring drug treatment, history of stroke with residual symptoms, symptomatic cerebrovascular disease, and renal or hepatic parenchymal failure. Subject data were excluded in the retrospective analyses, rather than subjects being excluded according to predetermined criteria.
The patients in the synucleinopathy group had been referred based on signs of central neurodegeneration (parkinsonism or cerebellar ataxia) or OH. PD was diagnosed according to UK Parkinson’s Disease Society Brain Bank Diagnostic Criteria (https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/GetPdf.cgi?id=phd000042), with the exception that early severe autonomic involvement was not considered to be exclusionary (Kaufmann & Goldstein 2013; Goldstein 2006). Diagnostic categorization in terms of MSA and PAF was based on previously published consensus criteria (Kaufmann 1996). Data from MSA patients were included regardless of classification as parkinsonian or cerebellar types.
Control subjects were studied over about the same time period (2006–2019). The control subjects were either healthy volunteers or were patients who had been referred for autonomic testing, did not have clinical evidence of neurodegenerative parkinsonism or cerebellar ataxia, and were not diagnosed with a form of chronic autonomic failure. Patients were included in the control group based on retrospective analyses of their clinical and laboratory findings. The healthy volunteers were not recruited by advertisement but were self-referred by word of mouth or through the NIH Normal Volunteer Program.
CSF Neurochemicals
To obtain CSF for neurochemical assays, subjects at the NIH Clinical Center underwent LP under fluoroscopic guidance. In patients on levodopa, the drug was withheld for at least 72 hours before the LP while the patients were inpatients at the NIH Clinical Center. A total of 12 1-mL aliquots of fluid were collected into chilled 1.5 mL plastic sample tubes, which were frozen immediately in dry ice and then stored at or below −70°C until the samples were assayed. The sixth aliquot was assayed for catechols by personnel (C.H.) who were blinded as to clinical diagnosis, using batch alumina extraction followed by liquid chromatography with electrochemical detection (LCED) (Holmes et al. 1994; Goldstein et al. 2003a; Goldstein et al. 2008). The closest available stored aliquot was thawed and assayed for MHPG and HVA. The person carrying out the assay for MHPG and HVA (P.S.) was also blinded as to clinical diagnosis.
For assaying MHPG and HVA, 150 μL of thawed, centrifuged CSF was injected directly into the LCED system. For HVA, better reproducibility was obtained by quantifying peak areas than peak heights, and so for both metabolites the former method was used.
Putamen dopaminergic neuroimaging
The putamen/occipital ratio (PUT/OCC) of 18F-DOPA-derived radioactivity was used as a model-independent measure of striatal dopaminergic innervation (Goldstein et al. 2008; Hoshi et al. 1993; Jokinen et al. 2009). The proportionate loss of putamen radioactivity between the peak value (at about 30 minutes after tracer injection) and 2 hours after tracer injection (“washout”) provided an inverse index of vesicular retention of 18F-dopamine derived from 18F-DOPA. The percent washout was calculated for the period between the peak value (at about 30 minutes after tracer injection) and the value at about 2 hours after tracer injection (Goldstein et al. 2008). 18F-DOPA scanning was done without carbidopa pre-treatment. 18F-DOPA was synthesized by the NIH PET Department and administered under Investigational New Drug (IND) #35,513. There is no Research Resource Identifier for this drug.
Cardiac noradrenergic neuroimaging
To evaluate peripheral noradrenergic innervation, cardiac 18F-dopamine PET scanning was done as described previously (Lamotte et al. 2019). PET scans were acquired on a GE Advance Tomograph (GE Healthcare) prior to January 2016 and on a Siemens PET/CT scanner after the GE Advance scanner was retired in January 2016. 18F-Dopamine was synthesized by the NIH PET Department and administered under IND #33,866. There is no Research Resource Identifier for this drug.
Post-mortem tissue catecholamines
Putamen and frontal cortex samples were from the University of Miami Brain Endowment Bank or the Pathology Department of the NIH Clinical Center. Brain tissue samples were obtained with a post-mortem interval less than 24 hours. Because of the direct injection used for measuring HVA and MHPG in CSF, these compounds were not measured in tissue samples.
Data Reduction, Analysis, and Statistics
Levodopa treatment would be expected to increase CSF HVA and DOPAC levels. To eliminate influences of outlying data from artifactual effects of treatment, we included data only from levodopa-treated patients who had CSF DOPA less than 6.79 pmol/mL (the upper limit of normal).
The graphics and statistical package was GraphPad Prism 9.0.0 (GraphPad Software LLC, San Diego, CA). Mean values for CSF levels of DOPA, dopamine, norepinephrine, DOPAC, DHPG, MHPG, and HVA in the synucleinopathy group were compared to those in the control group by independent-means t-tests. Mean values for across PD, MSA, PAF, and control groups were compared by analyses of variance with post-hoc group comparisons by Dunnett’s test. For scatter plots of individual data, Pearson correlation coefficients were calculated. Receiver operating characteristic (ROC) curves were generated for distinguishing the synucleinopathy from control groups were generated using GraphPad Prism for each neurochemical measure. An assessment of the normality of data was carried out as part of the testing using GraphPad Prism. Outlying data points were not excluded, but a few were not displayed in Figures. All clinical, neuroimaging, and neurochemical mean values were expressed ± 1 SEM. A p value less than 0.05 defined statistical significance.
RESULTS
Subject groups
CSF catechols, MHPG, and HVA data were analyzed from 130 subjects, including 92 synucleinopathy patients (36 PD, 37 MSA, 19 PAF). Mean (±SEM) ages and sex makeups were: PD 65±2 years, 22 men, 14 women; MSA 57±1 years, 25 men, 12 women; and PAF 65±2 years, 14 men, 5 women. Detailed clinical information about the 3 synucleinopathy groups, including treatment status for a variety of potentially interfering medications (e.g., a monoamine oxidase inhibitors (MAOI), catechol-O-methyltransferase inhibitor (COMTI), or selective serotonin reuptake inhibitor (SSRI)) are in a Supplementary Table.
There were 38 control subjects (mean age 52±2 years, 20 men, 18 women). Among the controls, 24 were healthy volunteers (46 ± 3 years old, 14 men, 10 women), and 14 were referred for autonomic testing and did not have parkinsonism or cerebellar ataxia or evidence of chronic autonomic failure (62 ± 3 years old, 6 men, 8 women).
CSF catecholamines and metabolites
Compared to the control group, the synucleinopathy group had lower mean CSF levels of DOPA (by 15%, t=4.04, p<0.0001), norepinephrine (by 35%, t=3.99, p<0.0001), DHPG (by 25%, t=4.41, p<0.0001), MHPG (by 21%, 4.39, p<0.0001), DOPAC (by 36%, t=5.30, p<0.0001), and HVA (by 36%, t=4.62, p<0.0001) (Figure 2). The groups did not differ in mean dopamine levels (t=0.22, p=0.83).
Figure 2: Individual values for cerebrospinal fluid (CSF) concentrations of DOPA, DA, DOPAC, HVA, NE, DHPG, and MHPG in synucleinopathy patients (N=98, red) and controls (N=32, gray).
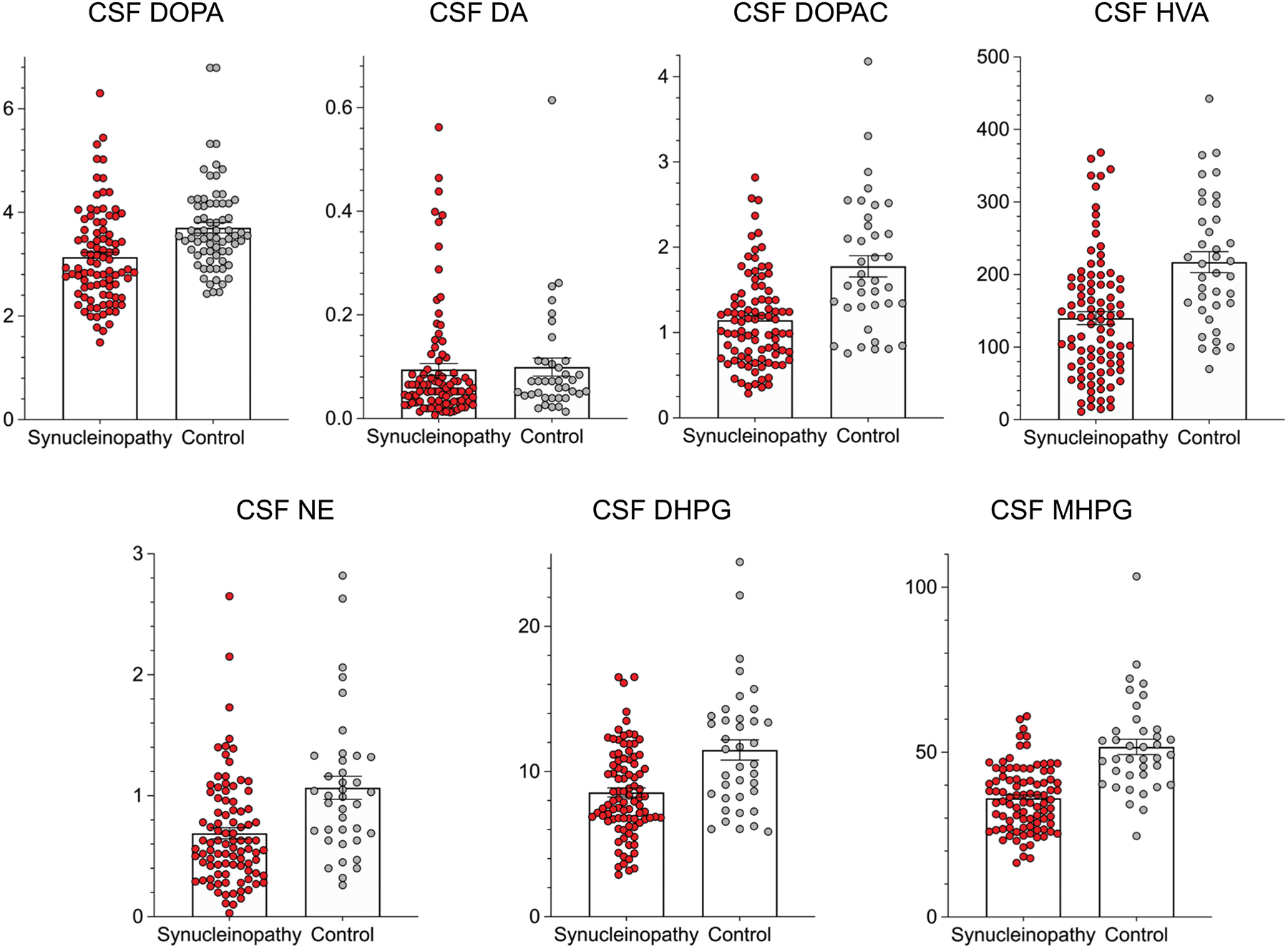
Rectangles show group mean values. Error bars are SEMs. Abbreviations: DA=dopamine; DHPG=3,4-dihydroxyphenylglycol; DOPA=3,4-dihydroxyphenylalanine; DOPAC=3,4-dihydroxyphenylacetic acid; HVA=homovanillic acid; MHPG=3-methoxy-4-hydroxyphenylglycol; NE=norepinephrine. The population distributions in the synucleinopathy group (with the except of DA) are shifted downward with respect to those in the controls.
CSF concentrations of norepinephrine, DHPG, and MHPG were positively inter-correlated (r=0.57 for DHPG vs. NE, r=0.59 for MHPG vs. NE, r=0.79 for MHPG vs. DHPG, p<0.0001 each). In the scatter plots relating levels of MHPG or DHPG vs. norepinephrine, the y-intercept values for the lines of best fit were above the origin (Figure 3). The y-intercept value for MHPG, 28.4 pmol/mL, had a 95% confidence interval of 24.9–31.9 pmol/mL, and the y-intercept value for DHPG, 6.17 pmol/mL, had a 95% confidence interval of 5.20–7.15 pmol/mL. For the relationship between MHPG and DHPG, the y-intercept value for MHPG was 13.6 pmol/mL, with a 95% confidence interval of 9.66–17.6 pmol/mL.
Figure 3: Scatter plots comparing individual values for (A) CSF DHPG vs. NE, (B) CSF MHPG vs. NE and (C) CSF MHPG vs. DHPG across all subjects (N=130).
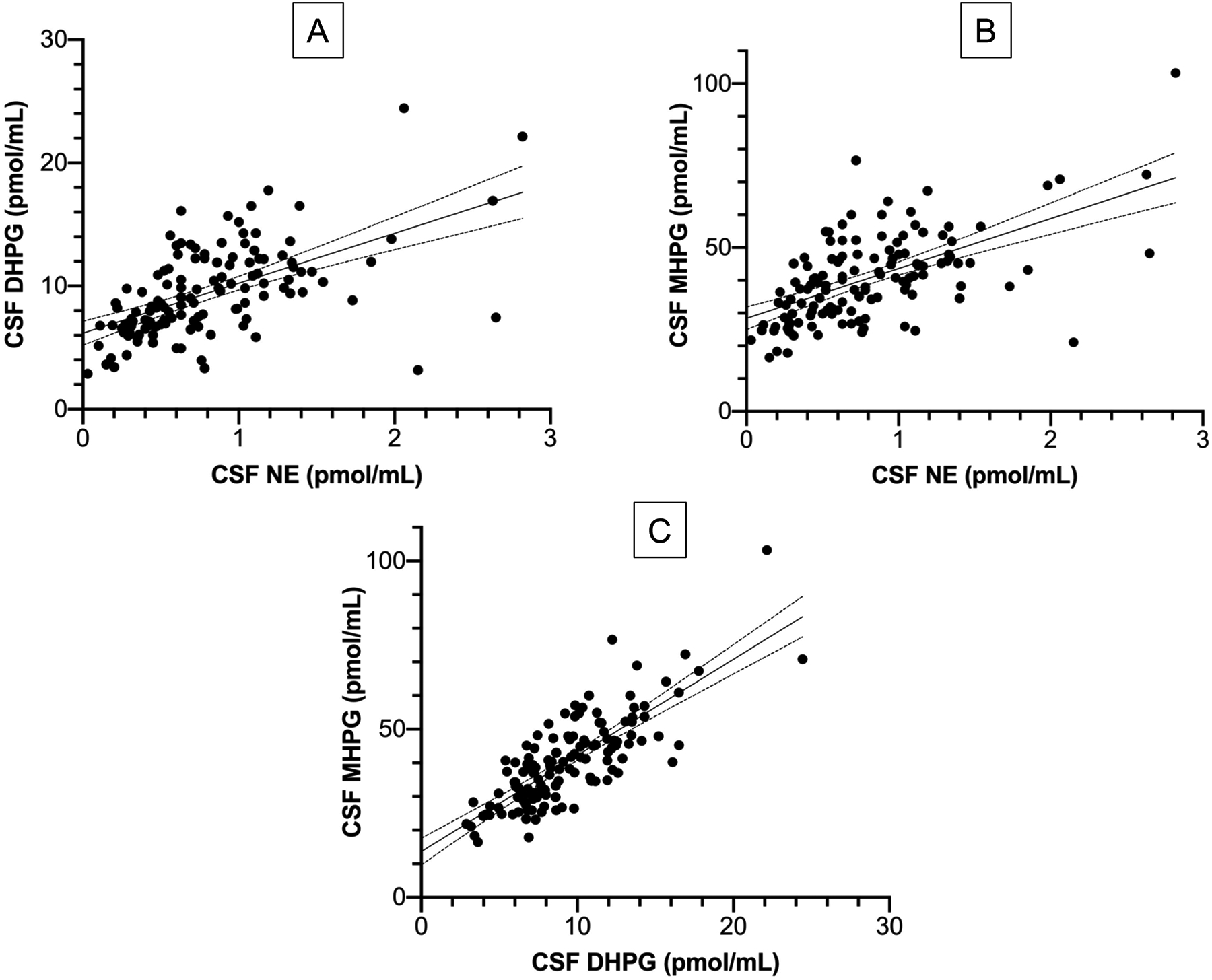
Lines of best fit with 95% confidence intervals are shown. Abbreviations: DHPG=3,4-dihydroxyphenylglycol; MHPG=3-methoxy-4-hydroxyphenylglycol; NE=norepinephrine. There are strong positive inter-correlations among the 3 central noradrenergic indices. The lines of best fit have their y-intercept values above the origin.
Compared to the control group, the PD, MSA, and PAF groups had decreased mean CSF levels of MHPG (p<0.0001 each; Figure 4A), DHPG (p=0.004; p<0.0001; p<0.0001; Figure 4B), and norepinephrine (p=0.017; p=0.0003; p=0.044). With regard to central dopaminergic indices CSF HVA and DOPAC levels were decreased in the PD and MSA groups but not in the PAF group (Figure 4C, 4D). The PD, MSA, and PAF groups did not differ from the controls in CSF dopamine levels.
Figure 4: Individual values for cerebrospinal fluid (CSF) levels of (A) MHPG, (B) DHPG, (C) HVA, and (D) DOPAC in synucleinopathies and control subjects.
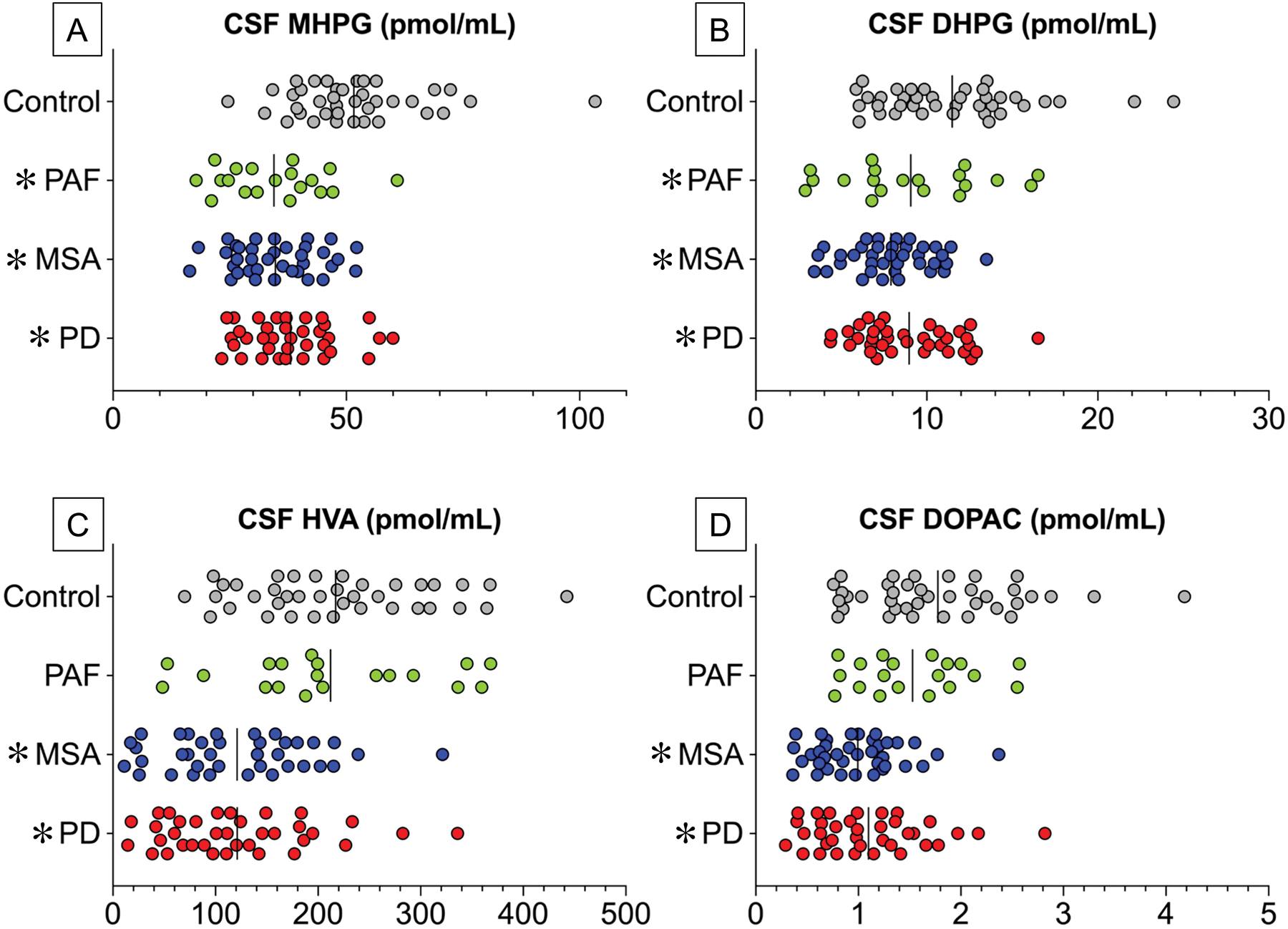
Abbreviations: DHPG=3,4-dihydroxyphenylglycol; DOPAC=3,4-dihydroxyphenylacetic acid; HVA=homovanillic acid; MHPG=3-methoxy-4-hydroxyphenylglycol; MSA=multiple system atrophy (N=32, no CSF MHPG in 1 MSA patient) ; PAF=pure autonomic failure (N=19). PD=Parkinson disease (N=36). PD data are in red, MSA in blue, PAF in green, and control subjects in gray (N=32). Vertical lines indicate group mean values. CSF MHPG and DHPG are decreased in the 3 synucleinopathy groups, whereas CSF HVA and DOPAC are decreased in PD and MSA but not PAF.
ROC curves were constructed to examine the efficiency of CSF DOPA, catecholamines, MHPG, and HVA for separating the synucleinopathy and control groups (Figure 5). The ROC area for DOPA was 0.70 (p<0.0001), dopamine 0.57 (not significant), DOPAC 0.76 (p<0.0001), HVA 0.74 (p<0.0001), norepinephrine 0.71 (p<0.0001), DHPG 0.70 (p<0.0001), and MHPG 0.83 (p<0.0001). Thus, the largest ROC area was for CSF MHPG. Based on the ROC curve for MHPG, at a specificity of 80% the sensitivity was 65% (blue dashed lines in Figure 5G).
Figure 5: Receiver operating characteristic (ROC) curves for distinguishing synucleinopathy patients (N=92) from controls (N=24) by CSF levels of (A) DOPA, (B) DA, (C) DOPAC, (D) HVA, (E) NE, (F) DHPG, and (G) MHPG.
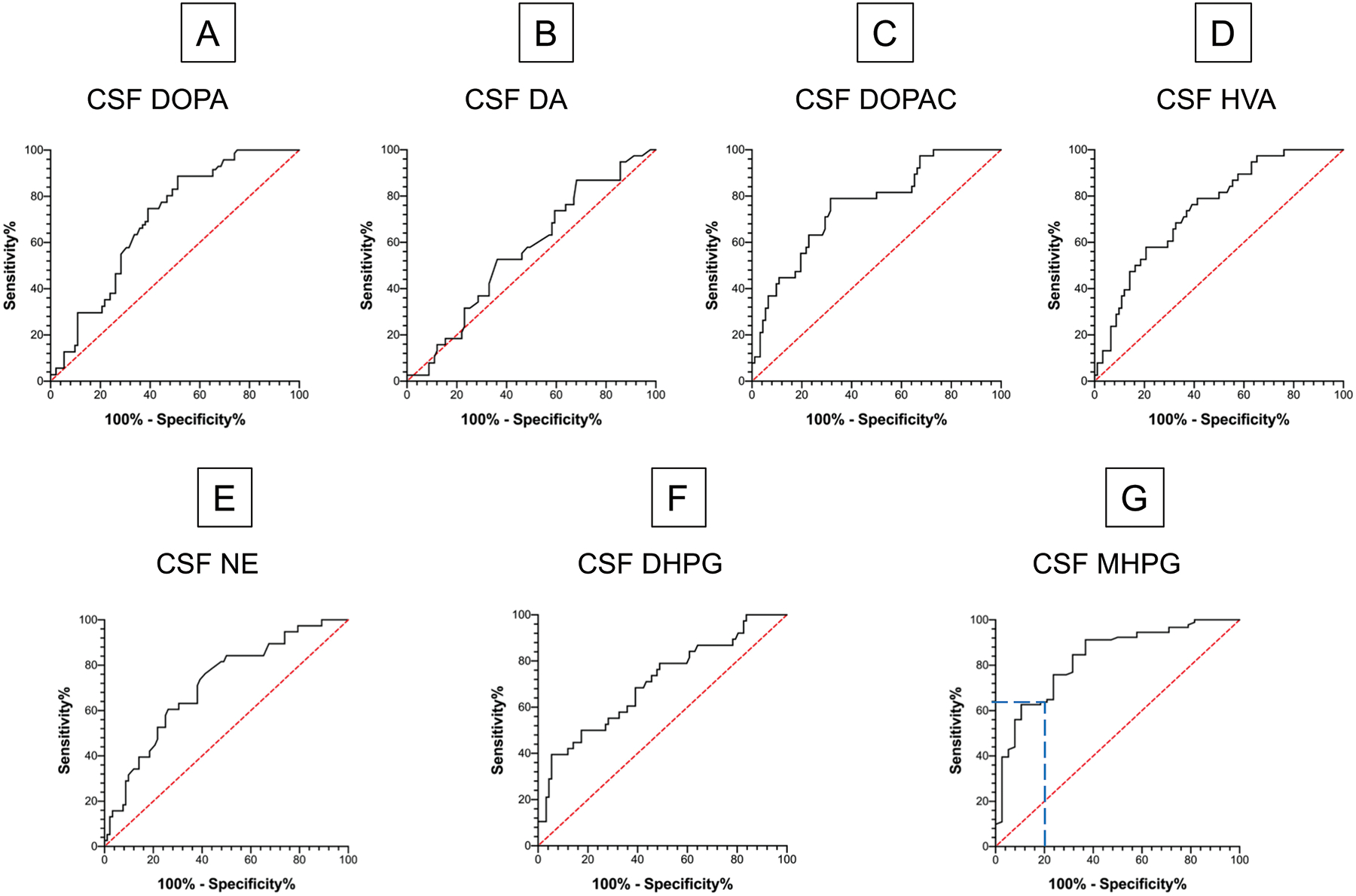
Abbreviations: DA=dopamine; DHPG=3,4-dihydroxyphenylglycol; DOPA=3,4-dihydroxyphenylalanine; DOPAC=3,4-dihydroxyphenylacetic acid; HVA=homovanillic acid; MHPG=3-methoxy-4-hydroxyphenylglycol; NE=norepinephrine. Red line indicates predictions from the null hypothesis of no distinction between the groups. Blue dashed line shows that for CSF MHPG sensitivity is 65% and specificity 80%.
CSF levels of the norepinephrine metabolite MHPG were positively correlated with those of the dopamine metabolite HVA (r=0.45, p<0.0001; Figure 6A). Similarly, CSF levels of deaminated norepinephrine metabolite DHPG were positively correlated with those of the deaminated dopamine metabolite DOPAC (r=0.45, p<0.0001). Across all subjects, CSF HVA was weakly positively correlated with CSF dopamine (r=0.21, p=0.045); however, within the PD, MSA, PAF, and control groups CSF HVA was unrelated to CSF dopamine.
Figure 6: Individual values for cerebrospinal fluid (CSF) levels of (A) MHPG as a function of HVA, (B) HVA as a function of the putamen/occipital (PUT/OCC) ratio of 18F-DOPA-derived radioactivity, (C) HVA as a function of the washout percent of 18F-DOPA-derived radioactivity, and (D) MHPG as a function of septal myocardial 18F-dopamine- (18F-DA-) derived radioactivity in synucleinopathies and control subjects.
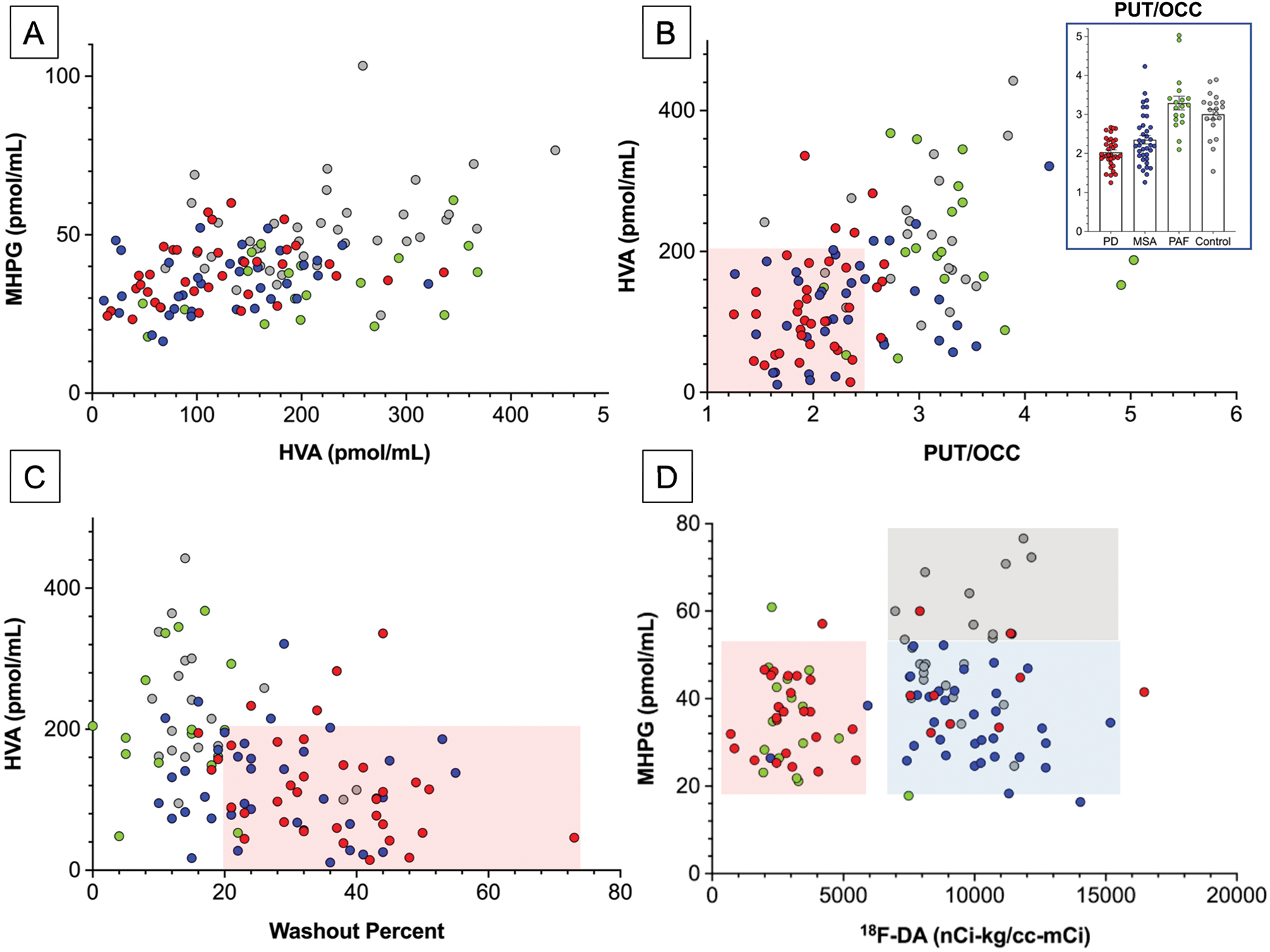
Abbreviations: HVA=homovanillic acid; MSA=multiple system atrophy (total N=37, blue); PAF=pure autonomic failure (total N=19). PD=Parkinson disease (Total N=36). PD data are in red, multiple system atrophy, MSA blue, PAF green, and control subjects gray (Total N=24). Inset in (B) shows individual PUT/OCC ratios, with means ± SEM. Pink rectangles in (B) and (C) placed visually to emphasize low HVA, PUT/OCC ratios, and increased 18F-DOPA washout precents in PD and MSA. In (D), gray rectangle placed visually to depict normal values. The pink and blue rectangles in (D) are placed visually to indicate low 18F-DA-derived radioactivity in PD and PAF and normal radioactivity in MSA.
Several synucleinopathy patients were on potentially interfering drugs. Among 6 patients on a MAO-B inhibitor, CSF MHPG and HVA did not differ from those in the remaining patients (40.1 ± 4.9 vs. 35.7 ± 1.0 pmol/mL, 95.3 ± 18.9 vs. 143.0 ± 9.4 pmol/mL); however, CSF DOPAC was lower in the subgroup on a MAOI (0.61 ± 0.14 vs. 1.18 ± 0.06 pmol/mL, p=0.012). Among 6 patients on a COMTI, none of the analyte levels were different from those in the remaining patients (data not shown). Among 22 patients on a SSRI, CSF MHPG and HVA did not differ from those in the remaining patients (33.3 ± 2.2 vs. 36.6 ± 1.2 pmol/mL, 116.5 ± 15.8 vs. 145.4 ± 10.7 pmol/mL); however, CSF DHPG was lower in the subgroup on a SSRI (7.2 ± 0.7 vs. 9.0 ± 0.4 pmol/mL, p=0.017).
Associations with other clinical laboratory data
A total of 110 subjects underwent CSF sampling and 18F-DOPA positron emission tomographic scanning. CSF HVA levels were positively correlated with PUT/OCC ratios of 18F-DOPA-derived radioactivity (r=0.43, p<0.0001; Figure 6B), as were CSF DOPAC levels (r=0.32, p=0.0005). Across 109 subjects CSF HVA levels were negatively correlated with washout percents of putamen 18F-DOPA-derived radioactivity (r=−0.43, p<0.0001; Figure 6C), as were DOPAC levels (r=−0.33, p=0.0005). Among subjects who had the combination of low CSF HVA levels, low PUT/OCC ratios, and increased washout of putamen 18F-DOPA-derived radioactivity, virtually all had PD or MSA, as indicated by the pink rectangles in Figures 6B and 6C.
PD and MSA patients had lower PUT/OCC ratios of 18F-DOPA-derived radioactivity (N=35, mean 2.03±0.06; N=37, mean 2.35±0.11) than did the control subjects (N=20, mean 3.01±0.13, p<0.0001 each). In contrast, PUT/OCC ratios were normal in the PAF group (N=18, mean 3.29±0.18). The mean washout percent of 18F-DOPA-derived radioactivity was higher in the PD (38±3%) and MSA (28±2%) groups than in the control group (18±2%; p<0.0001, p=0.0002) and was normal in the PAF group. Across the PD and MSA patients, CSF HVA levels were weakly positively correlated with PUT/OCC ratios (N=72, r=0.27, p=0.025) and tended to be negatively correlated with washout percents (N=71, r=−0.22, p=0.068). The PD and MSA groups did not differ in terms of either PUT/OCC ratios or washout percents (data not shown).
To address whether central noradrenergic deficiency is related to peripheral noradrenergic deficiency in synucleinopathies, we examined correlations of CSF MHPG with results of cardiac sympathetic neuroimaging in the same subjects. A total of 112 subjects had data for 18F-dopamine-derived radioactivity in the interventricular septal myocardium. 18F-Dopamine-derived radioactivity varied as a function of the type of synucleinopathy (F=35.6, p<0.0001), with radioactivity in the PD and PAF groups being lower (p<0.0001 each) than in the control group, while radioactivity in the MSA group did not differ from that in the control group.
CSF MHPG was also unrelated to cardiac 18F-dopamine-derived radioactivity (Figure 6D). Synucleinopathy patients with decreased CSF MHPG levels could be divided into two groups, those with PD or PAF (pink rectangle in Figure 6D) and those with MSA (blue rectangle).
CSF MHPG and HVA levels were unrelated to scores on the University of Pennsylvania Smell Identification Test (UPSIT, N=93, r=0.04; N=94, r=−0.06). CSF MHPG and HVA levels were also unrelated to Montreal Cognitive Assessment scores (N=32, r=0.23, p=0.21; N=33, r=0.00).
On the other hand, CSF MHPG was related to the magnitude of OH. CSF MHPG was lower in 64 subjects with an orthostatic fall in systolic blood pressure ≥ 20 mmHg than in 39 subjects with a fall in blood pressure <20 mm Hg (t=4.08, p<0.0001). Across all subjects, the magnitude of orthostatic decrease in blood pressure was correlated with CSF MHPG (r=0.37, p<0.0001). Across 36 PD patients, those with OH (N=17) had lower CSF MHPG than did those without OH (N=19, t=2.25, p=0.031). Too few MSA and PAF patients had no OH (N=6, N=3) to carry out meaningful statistics.
Post-mortem tissue catecholamines and their metabolites
Autopsy samples of putamen were assayed from 73 subjects (24 PD, 22 MSA, 4 PAF, 23 controls). The synucleinopathy group had decreased putamen norepinephrine concentrations compared to the control group (by 78%, t=6.33, p<0.0001). The PD and MSA groups had lower mean putamen norepinephrine concentrations than did the controls (p<0.0001; Figure 7C), whereas among the 4 PAF patients putamen norepinephrine was variable, with 2 having low levels. Putamen dopamine was lower in the synucleinopathy than control group (by 89%, p<0.0001). Putamen dopamine levels in the PD and MSA groups were lower than in the controls (p<0.0001 each, Figure 7D). As for putamen norepinephrine, putamen dopamine was variable among the 4 PAF patients, with 2 having low levels.
Figure 7: Individual data for frontal cortex (CTX) and putamen (PUT) concentrations of dopamine (DA) and norepinephrine (NE) in synucleinopathy patients and control subjects.
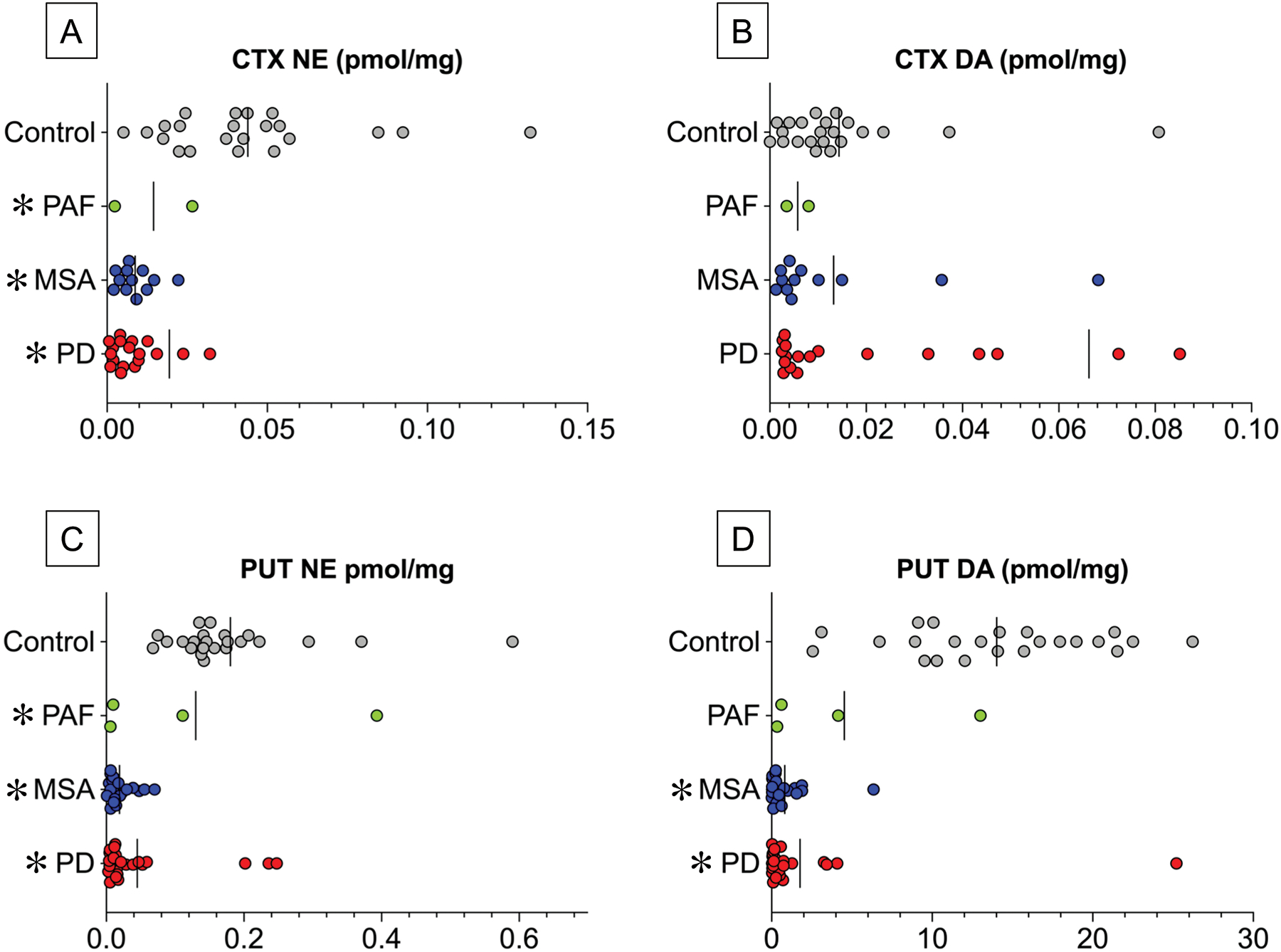
Patients with Parkinson disease (PD) are in red (Total N=19), multiple system atrophy (MSA) blue (Total N=12), pure autonomic failure (PAF) green (Total N=2 for CTX, 4 for PUT), and controls gray (Total N=22). Vertical lines indicate group mean values. Abbreviations: DA=dopamine; NE=norepinephrine. In (A) 1 data point for CTX NE in a control subject is outside the axis limit, and in (B) 1 data point for CTX DA in a PD patient is outside the axis limit. Note decreased CTX NE and normal CTX DA in the 3 synucleinopathy groups, whereas PUT DA is decreased in the PD and MSA groups and variable in the PAF group.
Frontal cortical samples were assayed from 55 subjects (19 PD, 12 MSA, 2 PAF, 22 controls). The synucleinopathy group had decreased cortical norepinephrine concentrations compared to the control group (by 65%, t=3.05, p=0.0036; Figure 7). The PD and MSA groups had lower mean norepinephrine concentrations than did the controls (by 79% in PD, p<0.0001 and 80% in MSA, p<0.0001; Figure 7A). There were only 2 PAF patients with cortex norepinephrine data. Cortical dopamine levels in the 3 synucleinopathy groups did not differ from those in the controls (Figure 7B).
DISCUSSION
The concept that CSF levels of catecholamines or their metabolites provide biomarkers of central catecholamine deficiency is important but not novel. On the contrary, there is abundant relevant literature over more than a half century, as documented by the contents of Table 1; however, the literature has been inconsistent, incomplete, and unpersuasive. We believe the present results are consistent and tell a convincing story in terms of differential abnormalities of CSF dopaminergic vs. noradrenergic indices across synucleinopathies.
Novel aspects of this study include the comparisons of 3 synucleinopathy groups in terms of CSF levels of the deaminated and O-methylated deaminated metabolites of dopamine and norepinephrine and levels of the parent catecholamines in the same participants; relationships between levels of the deaminated and O-methylated deaminated metabolites; cross-correlations of CSF HVA with central 18F-DOPA- and of CSF MHPG with cardiac 18F-dopamine-derived radioactivity; post-mortem tissue concentrations of dopamine and norepinephrine in cortex and putamen in synucleinopathy and control groups; and, most importantly, in vivo and post-mortem neurochemical evidence for central noradrenergic deficiency across synucleinopathy groups, in contrast with evidence for central dopaminergic deficiency in PD and MSA but not in PAF.
Central noradrenergic deficiency: A common theme in synucleinopathies
Two general types of findings from the present study support the view that PD, MSA, and PAF have in common central noradrenergic deficiency. First, in all three synucleinopathy groups CSF levels of MHPG (the main end-product of norepinephrine metabolism in the brain), DHPG (the main intra-neuronal metabolite of norepinephrine), and norepinephrine itself were decreased compared to controls. Second, in a largely different cohort, post-mortem assays of tissue catecholamines revealed decreased norepinephrine contents in the frontal cortex and putamen in the synucleinopathy group.
Validation of CSF indices
Strongly positive inter-correlations among norepinephrine, DHPG, and MHPG levels and between DOPAC and HVA levels cross-validated these neurochemical indices. Moreover, virtually all the PD and MSA patients also underwent brain 18F-DOPA positron emission tomographic scanning and had decreased PUT/OCC ratios of 18F-DOPA-derived radioactivity, accelerated loss of putamen radioactivity, and low CSF HVA (Figure 6B, 6C), confirming these neuroimaging and neurochemical modalities as in vivo biomarkers of central dopamine deficiency.
Differential abnormalities of CSF dopaminergic vs. noradrenergic indices
In the PAF group, mean CSF DOPAC and HVA levels, PUT/OCC ratios of 18F-DOPA-derived radioactivity, and washout percents of putamen 18F-DOPA-derived radioactivity all were not decreased from those in controls. These results contrasted strikingly with those in the PD and MSA groups, in which values for these dopaminergic indices clearly were decreased. The results therefore indicate differential abnormalities of central dopaminergic vs. noradrenergic indices across these synucleinopathies.
Axons emanating from the LC arborize widely and are the main source of norepinephrine in the brain—in particular, they are the sole source of norepinephrine in the cerebral cortex (Itoi et al. 2011). A LC lesion might contribute to early non-motor manifestations such as cognitive or olfactory dysfunction (Zweig et al. 1993; Ross et al. 2006; Cash et al. 1987; Del Tredici & Braak 2013) even before locomotor abnormalities become evident. Considering that PAF can evolve into PD+nOH (Kaufmann et al. 2017), PAF might represent not only a “body-first” (Goldstein et al. 2012a; Van Den Berge et al. 2019; Horsager et al. 2020) but also a “norepinephrine-first” process. Thus, in this study all but one of the PAF patients had neuroimaging evidence of cardiac noradrenergic deficiency, and the sole exception developed low 18F-dopamine-derived radioactivity during follow-up testing. As of this writing he has not developed motor signs of central neurodegeneration.
Results of post-mortem assessments of PAF cases also fit with the norepinephrine-first interpretation. All of 14 PAF patients in whom data have been reported for sympathetic ganglion tissue have had Lewy bodies (or eosinophilic or hyaline bodies) in this tissue (Table 2). Of the 14, 8 had SN neuronal loss, and of these all 8 also had LC neuronal loss. One patient had no detectable TH activity in LC and normal TH activity in SN (Black & Petito 1976). Of 5 PAF patients with normal SN neurons, 4 also had normal LC neurons, and in 1 there was no comment about the LC neuron number. In the present study, although the number of data points for PAF patients was small, frontal cortical norepinephrine content was significantly decreased from control, while putamen dopamine content was variable. Our findings in PAF seem to fit with Braak’s stage 2 (pontine LC lesion) and in PD with stage 3 (mesencephalic SN lesion) in the spatiotemporal progression of Lewy body forms of synucleinopathy (Braak et al. 2004). A prospective study of LC and SN melanin neuroimaging in PAF patients could test the norepinephrine-first idea (Knudsen et al. 2018b; Sommerauer et al. 2018).
Clinical significance of central noradrenergic deficiency
The clinical significance of decreased central noradrenergic innervation in synucleinopathies is unknown. Extensive animal research has indicated a variety of roles of norepinephrine derived from the LC in neurobehavioral phenomena such as vigilance, sleep, olfaction, memory of distressing events, emotional eating, nociception, mood, and social appropriateness; and it is suspected that loss of LC neurons may be causally related to non-motor manifestations such as cognitive dysfunction, anxiety/depression, inattention, and pain (Del Tredici & Braak 2013). The advent of central noradrenergic neuroimaging (Belfort-DeAguiar et al. 2018; Pietrzak et al. 2013; Knudsen et al. 2018a) offers an opportunity to determine whether abnormalities in these neuropsychological realms are related to loss of central noradrenergic innervation and if so in which brain regions.
Across all subjects, CSF levels of MHPG were associated with the magnitude of OH. In particular, within the PD group the subgroup with nOH had lower CSF levels than did the subgroup without nOH. There were too few MSA patients without nOH in the present study to conduct meaningful statistics; however, it has been reported that LC 18F-DOPA-derived radioactivity is lower in MSA with than without OH (Lewis et al. 2012), and we reported previously that patients with MSA+OH have decreased CSF DHPG (Goldstein et al. 2003a).
It is possible that central noradrenergic deficiency, reflecting a lesion of the LC, contributes to baroreflex-sympathoneural failure that in turn is a determinant of nOH. The LC is known to project to the nucleus of the solitary tract, where all baroreceptor afferents initially synapse in the brain, and to the rostral ventrolateral medulla (RVLM), a major source of descending projections to sympathetic pre-ganglionic neurons. In PD there are decreases in LC neurons (Zarow et al. 2003) and variable decreases in RVLM (Halliday et al. 1990; Benarroch et al. 2000) neurons. MSA is associated with decreases in LC neurons (Benarroch 2003; Benarroch et al. 2002) as well as in RVLM and A5 noradrenergic neurons (Benarroch et al. 2008; Benarroch et al. 2000). Detailed brainstem immunohistochemistry in these regions has not been described in PAF.
Determinants of CSF MHPG
Lines of best fit for scatter plots relating MHPG to norepinephrine and to DHPG were above the origin. These findings suggest that even if there were no norepinephrine release there would still be intra-neuronal metabolism to DHPG, due to passive leakage of norepinephrine from vesicular stores, followed by extra-neuronal metabolism of DHPG to MHPG (see concept diagram in Figure 1). In the periphery, ongoing norepinephrine metabolism reflects net leakage from vesicular stores, a process that occurs independently of pathway traffic-induced exocytosis (Eisenhofer et al. 2004). Based on the present results the same may hold true for norepinephrine metabolism in the brain. We propose that CSF MHPG has two main sources—norepinephrine that is released by exocytosis in response to nerve pathway traffic and norepinephrine that leaks passively into the cytoplasm from vesicular stores independently of pathway traffic. Other likely determinants of CSF MHPG levels are circulating MHPG (Kopin et al. 1983) and sulfoconjugation (Tyce et al. 1989).
Utility as diagnostic biomarkers
Areas under ROC curves were significantly above 0.5 for most of the CSF neurochemical measures in this study. The largest area was for CSF MHPG (area=0.83), with sensitivity 65% at a specificity of 80%. These results indicate moderate efficiency in separating synucleinopathy from control groups but do not support the application of these measures in individual diagnosis.
STUDY LIMITATIONS
Steps in intra-neuronal catecholamine synthesis, storage, release, reuptake, and metabolism are complex (Figure 1), and the catecholamine metabolites that were assayed in CSF provide only indirect indices of central dopaminergic and noradrenergic innervation and function. The combined measurements of norepinephrine, DHPG, and MHPG, with strongly positive inter-correlations and highly significant differences between the synucleinopathy and control groups for all three analytes, help buttress the inferences drawn; however, even with careful attention to the conditions at the time of sampling there was substantial inter-individual variability in the data in all the subject groups. Possibly this variability reflects genetic differences for several enzymes and transporters.
CSF HVA and DOPAC were only weakly correlated with neuroimaging indices of putamen dopamine deficiency. This can be explained by the indirectness of both types of measures in evaluating loss and dysfunctions of catecholaminergic neurons.
Since the CSF and post-mortem neurochemical data were obtained in largely separate cohorts, the CSF neurochemical indices could not be validated by comparison with tissue catecholamine contents.
We did not assay HVA or MHPG in brain tissue. Our assay method involved direct injection of CSF into the LCED system. Liquid chromatography with tandem mass spectroscopy offers an opportunity in the future to obtain data about catecholamines, HVA, and MHPG in the same brain tissue samples, but so far there are no published methods demonstrating adequate validity and reliability for assaying levels of HVA, and MHPG in human brain tissue.
There were relatively few data points about brain tissue catecholamines in PAF patients. PAF is a rare disease, and to our knowledge there is no availability of banked tissue from PAF patients.
In LCED chromatographs, dopamine has a relatively long retention time and therefore appears as a short, wide peak. In CSF samples there often are contaminating small peaks that can make it difficult to quantify dopamine. These factors, combined with low dopamine concentrations in CSF, question the validity and reliability of CSF dopamine in our study. In contrast, norepinephrine has a much shorter chromatographic retention time, the peak is far more spikey, and CSF norepinephrine levels are normally higher than dopamine levels.
Because of the unusual referral pattern of patients to the NIH, our research focus on autonomic disorders, and the comprehensive inpatient testing that was done, the synucleinopathy groups in this study may not reflect those in the general population. In particular, PD+nOH probably was over-represented.
CONCLUSIONS AND PERSPECTIVE
The results of this study indicate that three synucleinopathies—PD, MSA, and PAF—entail central norepinephrine deficiency. We report clear distinctions among these diseases, in that by both CSF neurochemical and PET neuroimaging approaches there is more extensive central dopamine deficiency in PD and MSA than in PAF. The retrospective results of this study should be confirmed in a more formally designed cohort study, especially to assess clinical correlates of the CSF neurochemical abnormalities reported here. Prospective studies of brainstem neuroimaging by PET or MRI in PAF patients could test the “norepinephrine-first” concept.
Supplementary Material
ACKNOWLEDGEMENTS
The research reported here was supported (in part) by the research Division of Intramural Research, National Institutes of Health (NINDS).
Tandees Najimi assisted with culling relevant literature.
FINANCIAL SUPPORT:
Division of Intramural Research, NINDS, NIH.
ABBREVIATIONS
- α-syn
alpha-synuclein
- ALDH
aldehyde dehydrogenase
- AR
aldehyde/aldose reductase
- COMT
catechol-O-methyltransferase
- COMTI
catechol-O-methyltransferase inhibitor
- CSF
cerebrospinal fluid
- CTX
cerebral cortex
- Cys-DOPA
5-S-cysteinyldopa
- Cys-DA
5-S-cysteinyldopamine
- DA
dopamine
- DHPG
3,4-dihydroxyphenylglycol
- DOPA
3,4-dihydroxyphenylalanine
- DOPAL
3,4-dihydroxyphenylacetaldehyde
- DOPEGAL
3,4-dihydroxyphenyglycolaldehyde
- HVA
homovanillic acid
- LAAAD
L-aromatic-amino-acid decarboxylase
- IND
Investigational New Drug
- LCED
liquid chromatography with electrochemical detection
- LP
lumbar puncture
- MAO
monoamine oxidase
- MAOI
monoamine oxidase inhibitor
- MHPG
3-methoxy-4-hydroxyphenylglycol
- MSA
multiple system atrophy
- NE
norepinephrine
- nOH
neurogenic orthostatic hypotension
- OCC
occipital cortex
- OH
orthostatic hypotension
- Ox.
spontaneous oxidation
- PAF
pure autonomic failure
- PD
Parkinson disease
- PMID
PubMed ID number
- PUT
putamen
- PUT/OCC
putamen/occipital cortex ratio
- ROC
receiver operating characteristic
- RVLM
rostral ventrolateral medulla
- SN
substantia nigra
- SSRI
selective serotonin reuptake inhibitor
- TH
tyrosine hydroxylase
- VMAT
vesicular monoamine transporter
Footnotes
CONFLICTS OF INTEREST: The authors have no conflicts of interest to disclose.
REFERENCES
- Andersen AD, Blaabjerg M, Binzer M, Kamal A, Thagesen H, Kjaer TW, Stenager E and Gramsbergen JBP (2017) Cerebrospinal fluid levels of catecholamines and its metabolites in Parkinson’s disease: effect of l-DOPA treatment and changes in levodopa-induced dyskinesia. J. Neurochem 141, 614–625. [DOI] [PubMed] [Google Scholar]
- Arai K, Kato N, Kashiwado K and Hattori T (2000) Pure autonomic failure in association with human alpha-synucleinopathy. Neurosci. Lett 296, 171–173. [DOI] [PubMed] [Google Scholar]
- Bannister R, Ardill L and Fentem P (1967) Defective autonomic control of blood vessels in idiopathic orthostatic hypotension. Brain 90, 725–746. [DOI] [PubMed] [Google Scholar]
- Belfort-DeAguiar R, Gallezot JD, Hwang JJ, Elshafie A, Yeckel CW, Chan O, Carson RE, Ding YS and Sherwin RS (2018) Noradrenergic Activity in the Human Brain: A Mechanism Supporting the Defense Against Hypoglycemia. J Clin Endocrinol Metab 103, 2244–2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benarroch EE (2003) Brainstem in multiple system atrophy: clinicopathological correlations. Cell. Mol. Neurobiol 23, 519–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benarroch EE, Schmeichel AM, Low PA, Sandroni P and Parisi JE (2008) Loss of A5 noradrenergic neurons in multiple system atrophy. Acta Neuropathol 115, 629–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benarroch EE, Schmeichel AM and Parisi JE (2000) Involvement of the ventrolateral medulla in parkinsonism with autonomic failure. Neurology 54, 963–968. [DOI] [PubMed] [Google Scholar]
- Benarroch EE, Schmeichel AM and Parisi JE (2002) Depletion of mesopontine cholinergic and sparing of raphe neurons in multiple system atrophy. Neurology 59, 944–946. [DOI] [PubMed] [Google Scholar]
- Black IB and Petito CK (1976) Catecholamine enzymes in the degenerative neurological disease idiopathic orthostatic hypotension. Science 192, 910–912. [DOI] [PubMed] [Google Scholar]
- Braak H, Ghebremedhin E, Rub U, Bratzke H and Del Tredici K (2004) Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res 318, 121–134. [DOI] [PubMed] [Google Scholar]
- Bras IC, Dominguez-Meijide A, Gerhardt E, Koss D, Lazaro DF, Santos PI, Vasili E, Xylaki M and Outeiro TF (2020) Synucleinopathies: Where we are and where we need to go. J Neurochem 153, 433–454. [DOI] [PubMed] [Google Scholar]
- Cash R, Dennis T, L’Heureux R, Raisman R, Javoy-Agid F and Scatton B (1987) Parkinson’s disease and dementia: norepinephrine and dopamine in locus ceruleus. Neurology 37, 42–46. [DOI] [PubMed] [Google Scholar]
- Del Tredici K and Braak H (2012) Lewy pathology and neurodegeneration in premotor Parkinson’s disease. Mov. Disord 27, 597–607. [DOI] [PubMed] [Google Scholar]
- Del Tredici K and Braak H (2013) Dysfunction of the locus coeruleus-norepinephrine system and related circuitry in Parkinson’s disease-related dementia. J. Neurol. Neurosurg. Psychiatry 84, 774–783. [DOI] [PubMed] [Google Scholar]
- DelleDonne A, Klos KJ, Fujishiro H et al. (2008) Incidental Lewy body disease and preclinical Parkinson disease. Arch. Neurol 65, 1074–1080. [DOI] [PubMed] [Google Scholar]
- Eisenhofer G, Kopin IJ and Goldstein DS (2004) Catecholamine metabolism: a contemporary view with implications for physiology and medicine. Pharmacol. Rev 56, 331–349. [DOI] [PubMed] [Google Scholar]
- Eldrup E, Mogensen P, Jacobsen J, Pakkenberg H and Christensen NJ (1995) CSF and plasma concentrations of free norepinephrine, dopamine, 3,4-dihydroxyphenylacetic acid (DOPAC), 3,4-dihydroxyphenylalanine (DOPA), and epinephrine in Parkinson’s disease. Acta Neurol. Scand 92, 116–121. [DOI] [PubMed] [Google Scholar]
- Engelborghs S, Marescau B and De Deyn PP (2003) Amino acids and biogenic amines in cerebrospinal fluid of patients with Parkinson’s disease. Neurochem. Res 28, 1145–1150. [DOI] [PubMed] [Google Scholar]
- Goldstein DS (2006) Orthostatic hypotension as an early finding in Parkinson disease. Clin. Auton. Res 16, 46–64. [DOI] [PubMed] [Google Scholar]
- Goldstein DS, Holmes C, Bentho O, Sato T, Moak J, Sharabi Y, Imrich R, Conant S and Eldadah BA (2008) Biomarkers to detect central dopamine deficiency and distinguish Parkinson disease from multiple system atrophy. Parkinsonism Relat. Disord 14, 600–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein DS, Holmes C, Lopez GJ, Wu T and Sharabi Y (2018) Cerebrospinal fluid biomarkers of central dopamine deficiency predict Parkinson’s disease. Parkinsonism Relat. Disord 50, 108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein DS, Holmes C, Patronas N and Kopin IJ (2003a) Cerebrospinal fluid levels of catechols in patients with neurogenic orthostatic hypotension. Clin. Sci 104, 649–654. [DOI] [PubMed] [Google Scholar]
- Goldstein DS, Holmes C, Sewell L, Park MY and Sharabi Y (2012a) Sympathetic noradrenergic before striatal dopaminergic denervation: relevance to Braak staging of synucleinopathy. Clin. Auton. Res 22, 57–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein DS, Holmes C and Sharabi Y (2012b) Cerebrospinal fluid biomarkers of central catecholamine deficiency in Parkinson’s disease and other synucleinopathies. Brain 135, 1900–1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein DS, Holmes C, Sharabi Y, Brentzel S and Eisenhofer G (2003b) Plasma levels of catechols and metanephrines in neurogenic orthostatic hypotension. Neurology 60, 1327–1332. [DOI] [PubMed] [Google Scholar]
- Hague K, Lento P, Morgello S, Caro S and Kaufmann H (1997) The distribution of Lewy bodies in pure autonomic failure: autopsy findings and review of the literature. Acta Neuropathol 94, 192–196. [DOI] [PubMed] [Google Scholar]
- Halliday GM, Li YW, Blumbergs PC, Joh TH, Cotton RG, Howe PR, Blessing WW and Geffen LB (1990) Neuropathology of immunohistochemically identified brainstem neurons in Parkinson’s disease. Ann. Neurol 27, 373–385. [DOI] [PubMed] [Google Scholar]
- Herbert MK, Kuiperij H, Bloem BR and Verbeek MM (2013) Levels of HVA, 5-HIAA, and MHPG in the CSF of vascular parkinsonism compared to Parkinson’s disease and controls. J Neurol 260, 3129–3133. [DOI] [PubMed] [Google Scholar]
- Holmes C, Eisenhofer G and Goldstein DS (1994) Improved assay for plasma dihydroxyphenylacetic acid and other catechols using high-performance liquid chromatography with electrochemical detection. J. Chromatogr. B Biomed. Appl 653, 131–138. [DOI] [PubMed] [Google Scholar]
- Horsager J, Andersen KB, Knudsen K et al. (2020) Brain-first versus body-first Parkinson’s disease: a multimodal imaging case-control study. Brain 143, 3077–3088. [DOI] [PubMed] [Google Scholar]
- Hoshi H, Kuwabara H, Leger G, Cumming P, Guttman M and Gjedde A (1993) 6-[18F]fluoro-L-dopa metabolism in living human brain: a comparison of six analytical methods. J. Cereb. Blood Flow Metab 13, 57–69. [DOI] [PubMed] [Google Scholar]
- Itoi K, Sugimoto N, Suzuki S, Sawada K, Das G, Uchida K, Fuse T, Ohara S and Kobayashi K (2011) Targeting of locus ceruleus noradrenergic neurons expressing human interleukin-2 receptor alpha-subunit in transgenic mice by a recombinant immunotoxin anti-Tac(Fv)-PE38: a study for exploring noradrenergic influence upon anxiety-like and depression-like behaviors. J Neurosci 31, 6132–6139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RH, Lee Gde J, Oppenheimer DR and Spalding JM (1966) Autonomic failure with orthostatic hypotension due to intermediolateral column degeneration. A report of two cases with autopsies. Q J Med 35, 276–292. [PubMed] [Google Scholar]
- Jokinen P, Helenius H, Rauhala E, Bruck A, Eskola O and Rinne JO (2009) Simple ratio analysis of 18F-fluorodopa uptake in striatal subregions separates patients with early Parkinson disease from healthy controls. J. Nucl. Med 50, 893–899. [DOI] [PubMed] [Google Scholar]
- Kaufmann H (1996) Consensus statement on the definition of orthostatic hypotension, pure autonomic failure and multiple system atrophy. Clin. Auton. Res 6, 125–126. [DOI] [PubMed] [Google Scholar]
- Kaufmann H and Goldstein DS (2013) Autonomic dysfunction in Parkinson disease. Handb. Clin. Neurol 117, 259–278. [DOI] [PubMed] [Google Scholar]
- Kaufmann H, Hague K and Perl D (2001) Accumulation of alpha-synuclein in autonomic nerves in pure autonomic failure. Neurology 56, 980–981. [DOI] [PubMed] [Google Scholar]
- Kaufmann H, Nahm K, Purohit D and Wolfe D (2004) Autonomic failure as the initial presentation of Parkinson disease and dementia with Lewy bodies. Neurology 63, 1093–1095. [DOI] [PubMed] [Google Scholar]
- Kaufmann H, Norcliffe-Kaufmann L, Palma JA et al. (2017) Natural history of pure autonomic failure: A United States prospective cohort. Ann. Neurol 81, 287–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kish SJ, Shannak K and Hornykiewicz O (1988) Uneven pattern of dopamine loss in the striatum of patients with idiopathic Parkinson’s disease. Pathophysiologic and clinical implications. The New England Journal of Medicine 318, 876–880. [DOI] [PubMed] [Google Scholar]
- Knudsen K, Fedorova TD, Hansen AK et al. (2018a) In-vivo staging of pathology in REM sleep behaviour disorder: a multimodality imaging case-control study. Lancet Neurol 17, 618–628. [DOI] [PubMed] [Google Scholar]
- Knudsen K, Fedorova TD, Hansen AK et al. (2018b) In-vivo staging of pathology in REM sleep behaviour disorder: a multimodality imaging case-control study. Lancet Neurol 17, 618–628. [DOI] [PubMed] [Google Scholar]
- Kopin IJ, Gordon EK, Jimerson DC and Polinsky RJ (1983) Relation between plasma and cerebrospinal fluid levels of 3-methoxy-4- hydroxyphenylglycol. Science 219, 73–75. [DOI] [PubMed] [Google Scholar]
- Lamotte G, Holmes C, Wu T and Goldstein DS (2019) Long-term trends in myocardial sympathetic innervation and function in synucleinopathies. Parkinsonism Relat. Disord 67, 27–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis SJ, Pavese N, Rivero-Bosch M, Eggert K, Oertel W, Mathias CJ, Brooks DJ and Gerhard A (2012) Brain monoamine systems in multiple system atrophy: a positron emission tomography study. Neurobiol. Dis 46, 130–136. [DOI] [PubMed] [Google Scholar]
- Martignoni E, Blandini F, Petraglia F, Pacchetti C, Bono G and Nappi G (1992) Cerebrospinal fluid norepinephrine, 3-methoxy-4-hydroxyphenylglycol and neuropeptide Y levels in Parkinson’s disease, multiple system atrophy and dementia of the Alzheimer type. J Neural Transm Park Dis Dement Sect 4, 191–205. [DOI] [PubMed] [Google Scholar]
- Miura H, Tsuchiya K, Kubodera T, Shimamura H and Matsuoka T (2001) An autopsy case of pure autonomic failure with pathological features of Parkinson’s disease. Rinsho Shinkeigaku 41, 40–44. [PubMed] [Google Scholar]
- Petito CK and Black IB (1978) Ultrastructure and biochemistry of sympathetic ganglia in idiopathic orthostatic hypotension. Ann. Neurol 4, 6–17. [DOI] [PubMed] [Google Scholar]
- Pietrzak RH, Gallezot JD, Ding YS, Henry S, Potenza MN, Southwick SM, Krystal JH, Carson RE and Neumeister A (2013) Association of posttraumatic stress disorder with reduced in vivo norepinephrine transporter availability in the locus coeruleus. JAMA Psychiatry 70, 1199–1205. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Polinsky RJ, Jimerson DC and Kopin IJ (1984) Chronic autonomic failure: CSF and plasma 3-methoxy-4- hydroxyphenylglycol. Neurology 34, 979–983. [DOI] [PubMed] [Google Scholar]
- Rajput AH, Rozdilsky B and Rajput A (1991) Accuracy of clinical diagnosis in parkinsonism--a prospective study. Can. J. Neurol. Sci 18, 275–278. [DOI] [PubMed] [Google Scholar]
- Roessmann U, Van den Noort S and McFarland DE (1971) Idiopathic orthostatic hypotension. Arch Neurol 24, 503–510. [DOI] [PubMed] [Google Scholar]
- Ross GW, Abbott RD, Petrovitch H et al. (2006) Association of olfactory dysfunction with incidental Lewy bodies. Mov. Disord 21, 2062–2067. [DOI] [PubMed] [Google Scholar]
- Schober R, Langston JW and Forno LS (1975) Idiopathic orthostatic hypotension. Biochemical and pathologic observations in 2 cases. Eur. Neurol 13, 177–188. [DOI] [PubMed] [Google Scholar]
- Sommerauer M, Fedorova TD, Hansen AK et al. (2018) Evaluation of the noradrenergic system in Parkinson’s disease: an 11C-MeNER PET and neuromelanin MRI study. Brain 141, 496–504. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R and Goedert M (1997) Alpha-synuclein in Lewy bodies. Nature 388, 839–840. [DOI] [PubMed] [Google Scholar]
- Terao Y, Takeda K, Sakuta M, Nemoto T, Takemura T and Kawai M (1993) Pure progressive autonomic failure: a clinicopathological study. Eur. Neurol 33, 409–415. [DOI] [PubMed] [Google Scholar]
- Tyce GM, Ahlskog JE, Carmichael SW, Chritton SL, Stoddard SL, van Heerden JA, Yaksh TL and Kelly PJ (1989) Catecholamines in CSF, plasma, and tissue after autologous transplantation of adrenal medulla to the brain in patients with Parkinson’s disease. J Lab Clin Med 114, 185–192. [PubMed] [Google Scholar]
- Van Den Berge N, Ferreira N, Gram H et al. (2019) Evidence for bidirectional and trans-synaptic parasympathetic and sympathetic propagation of alpha-synuclein in rats. Acta Neuropathol 138, 535–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Ingelghem E, van Zandijcke M and Lammens M (1994) Pure autonomic failure: a new case with clinical, biochemical, and necropsy data. J. Neurol. Neurosurg. Psychiatry 57, 745–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderhaeghen JJ, Perier O and Sternon JE (1970) Pathological findings in idiopathic orthostatic hypotension. Its relationship with Parkinson’s disease. Arch. Neurol 22, 207–214. [DOI] [PubMed] [Google Scholar]
- Wakabayashi K, Yoshimoto M, Tsuji S and Takahashi H (1998) Alpha-synuclein immunoreactivity in glial cytoplasmic inclusions in multiple system atrophy. Neurosci. Lett 249, 180–182. [DOI] [PubMed] [Google Scholar]
- Yamasaki TR, Holmes BB, Furman JL, Dhavale DD, Su BW, Song ES, Cairns NJ, Kotzbauer PT and Diamond MI (2019) Parkinson’s disease and multiple system atrophy have distinct alpha-synuclein seed characteristics. J Biol Chem 294, 1045–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarow C, Lyness SA, Mortimer JA and Chui HC (2003) Neuronal loss is greater in the locus coeruleus than nucleus basalis and substantia nigra in Alzheimer and Parkinson diseases. Arch. Neurol 60, 337–341. [DOI] [PubMed] [Google Scholar]
- Ziegler MG, Lake CR and Kopin IJ (1977) The sympathetic-nervous-system defect in primary orthostatic hypotension. N. Engl. J. Med 296, 293–297. [DOI] [PubMed] [Google Scholar]
- Zweig RM, Cardillo JE, Cohen M, Giere S and Hedreen JC (1993) The locus ceruleus and dementia in Parkinson’s disease. Neurology 43, 986–991. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.