

Abstract
Tragically, the death toll from the COVID-19 pandemic continues to rise, and with variants being observed around the globe new therapeutics, particularly direct-acting antivirals that are easily administered, are desperately needed. Studies targeting the SARS-CoV-2 3CL protease, which is critical for viral replication, with different peptidomimetics and warheads is an active area of research for development of potential drugs. To date, however, only a few publications have evaluated the nitrile warhead as a viral 3CL protease inhibitor, with only modest activity reported. This article describes our investigation of P3 4-methoxyindole peptidomimetic analogs with select P1 and P2 groups with a nitrile warhead that are potent inhibitors of SARS-CoV-2 3CL protease and demonstrate in vitro SARS-CoV-2 antiviral activity. A selectivity for SARS-CoV-2 3CL protease over human cathepsins B, S and L was also observed with the nitrile warhead, which was superior to that with the aldehyde warhead. A co-crystal structure with SARS-CoV-2 3CL protease and a reversibility study indicate that a reversible, thioimidate adduct is formed when the catalytic sulfur forms a covalent bond with the carbon of the nitrile. This effort also identified efflux as a property limiting antiviral activity of these compounds, and together with the positive attributes described these results provide insight for further drug development of novel nitrile peptidomimetics targeting SARS-CoV-2 3CL protease.
This article describes peptidomimetic SARS-CoV-2 3CLpro inhibitors with a nitrile warhead with in vitro antiviral inhibition. Superior selectivity was observed for the nitrile warhead compared to the aldehyde against 3 human cathepsins (B, S and L).
Introduction
The outbreak of coronavirus disease 2019 (COVID-19) was first reported in December 2019 in Wuhan, China. To date, over 3.8 million people have died from this ongoing pandemic.1 Although vaccines have been developed and are being administered as rapidly as possible, the emergence of variants has underscored the need for additional modalities of treatment. Remdesivir, which was originally in clinical trials as an Ebola treatment, is the only direct-acting antiviral that has been approved.2 It is administered intravenously and has demonstrated only modest efficacy for moderate cases of COVID-19.3 Novel therapeutics to treat coronavirus infections, especially ones with a different mechanism of action that are orally administered, are desperately needed.
COVID-19 is caused by a severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2).1 Other coronaviruses are responsible for a portion of common seasonal colds as well as the 2003 severe acute respiratory syndrome (SARS, caused by the SARS-CoV-1), and 2012 Middle East respiratory syndrome (MERS, caused by the MERS-CoV). Coronaviruses have four genera with two, the alpha- and beta-coronaviruses, descending from the bat viral gene pool with SARS-CoV-1, MERS-CoV and SARS-CoV-2 being beta-coronaviruses.4
During the replication cycle of the coronavirus, after endocytosis into the cell, cellular machinery is utilized to express two overlapping polyproteins, pp1a and pp1b, from the viral RNA.5 These polyproteins must be processed by two proteases known as 3-chymotrypsin-like protease (3CLpro, Mpro or 3CLP) and papain-like protease (PLPro) to liberate the viral proteins required for replication. The majority of the cleavages are performed by 3CLpro, however, the exact number cleaved by each protease is still being debated.6 Both 3CLpro and PLPro are cysteine proteases with different site specificities. The peptide sequence cleaved by a protease is labeled as P3P2P1↓P1′P2′P3′ where the amide bond cleavage occurs between P1 and P1′ amino acid as indicated by the arrow, and the corresponding sites on the protease protein are referred to as S3S2S1S1′S2′S3′. Coronavirus 3CLpro hydrolyses proteins predominantly between a P1 glutamine and a small P1′ amino acid, such as alanine, serine or glycine. For the P2 position, leucine is the most common in the sequence specificity for coronaviruses.7 Given the importance of 3CLpro to viral replication, it is a promising drug target.
Peptidomimetic inhibitors of proteases mimic the substrate with similar labels (P3, P2, P1, etc.) and typically include a moiety that interacts with the catalytic site. That group is known as the warhead, and for cysteine proteases it is typically an electrophile that forms a covalent bond with the catalytic sulfur.5 The exploration of peptidomimetic inhibitors against SARS-CoV-2 3CLpro has unsurprisingly exploded since the COVID-19 pandemic began. Repurposing peptidomimetic drugs, such as boceprevir, telaprevir, and ritonavir, that were developed against other viruses is one approach that has been utilized.8–10 Peptidomimetics that are tailored for SARS-CoV-2 3CLpro are increasingly being reported, and many build upon SARS-CoV-1 findings or those of viruses that have a similar protease to the coronavirus 3CLpro, such as enterovirus 71 and rhinovirus.11–14 For these SARS-CoV-2 targeted peptidomimetics, most utilize a P1 glutamine mimic with a gamma-lactam (5-membered) being the most common, with N,N-dimethylglutamine, delta-lactam (6-membered) and non-glutamine mimics being less frequently reported. For P2, consistent with the consensus sequence, the most common utilized group is leucine, although related ones, such as cyclopropylalanine, tert-butylalanine and cyclohexylalanine, are also reported.11,14–22 The main variations being explored by different researchers are at either P3 or the warhead. A wide range of P3 capping groups, such as benzyl carbamate and indole, in addition to tetra- (P4) and pentapeptides (P4 and P5) with capping groups are utilized. The warhead is critical as it makes a covalent bond with the catalytic sulfur and must be reactive enough in the protease active site, but not too reactive to be sequestered by glutathione or indiscriminately react with other thiols. The warhead selection tends to differentiate the focus of investigators as different groups study distinct warheads and variations thereof. Recent SARS-CoV-2 targeted warheads that have been examined are: aldehydes and their prodrugs,14–16 ketoamides,11,17 halomethylketones,18 enoates,19 α-hydroxymethylketones20 (HMK) and α-acyloxymethylketones (AMK).20–22 As the research into SARS-CoV-2 peptidomimetics matures, like prior HCV and HIV protease efforts, the structural elements will be adjusted to allow good pharmacokinetics and the ability to demonstrate in vivo efficacy. A noteworthy recent example was reported by Qiao et al. that found an aldehyde warhead with the P2 leucine being replaced by a bicyclic proline (from HCV protease drug development) was able to demonstrate efficacy in a murine model of COVID-19 by intraperitoneal and oral administration.23 Further research into understanding the roles the different moieties play in not only protease inhibition and anti-viral activity, but also other attributes, such as permeability, efflux, and metabolism, will allow inhibitors to be refined to discover drugs for the treatment of coronavirus infections. In addition, assessing selectivity for these peptidomimetics and their warheads for SARS-CoV-2 3CLpro over human cysteine proteases with a similar sequence specificity will be important to avoid potential toxicity; however, only a few articles include selectivity considerations.20,22,24
To date no cysteine protease inhibitors for any indication have made it to regulatory approval.25 Inspired by the clinical success of odanacatib (Fig. 1), a human cathepsin K inhibitor with a nitrile warhead, that progressed to phase III clinical trials, we decided to explore the nitrile group as a warhead on peptidomimetics to inhibit SARS-CoV-2 3CLpro.26 A search of the literature in March 2020 for nitrile warhead utilization for 3CLpro inhibition produced only a couple of examples. These were against related enterovirus 71 and SARS-CoV-1, with all the compounds having at best either single or double digit micromolar IC50 values.27,28 Representative compounds 1 and 2 from those publications and their reported IC50 values are shown in Fig. 1. Undeterred by the high IC50 values we decided to examine nitrile analogs of an active compound 3 that was discovered upon screening compounds from our legacy norovirus protease program.
Fig. 1. Structures of odanacatib and compounds 1 to 8.

After our effort commenced, GC-376 (4) and PF-07304814 (5) were disclosed as SARS-CoV-2 3CLpro inhibitors that were under development to enter clinical trials to treat COVID-19.29,30,31
Prodrug 5, a more water soluble form of active compound 6, is undergoing clinical trials as an intravenously administered drug.20 Compound 6 and related compounds, such as aldehyde 7, were initially disclosed in 2005 and 2006 and were some of the published sources of inspiration for our prior norovirus protease exploratory effort.32,33 In April 2021, a new potential COVID-19 treatment, PF-07321332 (8), targeting 3CLpro was disclosed by Pfizer and is reported to be dosed orally and has initiated clinical trials.34 Compound 8 contains a nitrile warhead and to date we have observed no publication with its biological results. A recent literature search uncovered an investigation by Breidenbach et al. of a few tetra peptides with azaglutamine P1, nitrile warhead and benzylcarbamate capping group as a SARS-CoV-2 3CLpro inhibitors.35 Other than the nitrile, these were structurally very different from our compounds and Breidenbach et al. only reported a modest kobs/I and no antiviral data. Herein, we report our results for nitrile warhead analogs of compound 3, 6 and 7.
Results
Chemistry
Compound 9 and 10 were prepared as described by Shang and coworkers.27 Conversion of 9 to 11 and 10 to 13 (Scheme 1) followed literature procedures and 10 was converted to 13 using those sample protocols, but substituting N-(tert-butoxycarbonyl)-4-methyl-l-leucine for N-(tert-butoxycarbonyl)-l-leucine.36,37 Compound 11–13 were converted to the ester version of the target by removal of the t-butyl carbamate with HCl in a mixture of dioxane and dichloromethane followed by HATU coupling of the corresponding indol-2-ylcarboxylic acid (a to e). Conversion of the methyl ester to the nitrile target was achieved by a saponification with LiOH, then carbonyldiimidazole mediated coupling of ammonia and dehydration of the primary amide with trifluoroacetic acid anhydride.
Scheme 1. Synthesis of compounds. i) TFA in DCM or HCl in dioxane, DCM ii) N-(tert-butoxycarbonyl)-l-leucine (R = H) or N-(tert-butoxycarbonyl)-4-methyl-l-leucine (R = CH3), HATU, NMM or Et3N, DMF, 94% yield (2 steps, R = H), 91% yield (2 steps, R = CH3); iii) corresponding (a–e) indolecarboxylic acid, HATU, NMM or Et3N, DMF, 30–98% yield (2 steps); iv) LiOH, THF, v) CDI, THF, NH3·H2O; vi) trifluoroacetic acid anhydride, Et3N, THF, 0 °C, 12–63% yield (3 steps).

Late stage diversification is always preferred to generate the same number of targets in fewer overall steps. The nitrile version of 10 proved challenging to efficiently generate, deprotect and couple to N-t-Boc-l-leucine producing low yields of impure material. The route in Scheme 1 was thus employed for compound production, despite the added number of steps required.
Biology
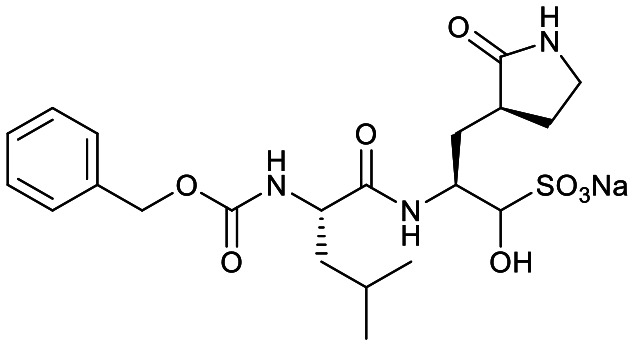
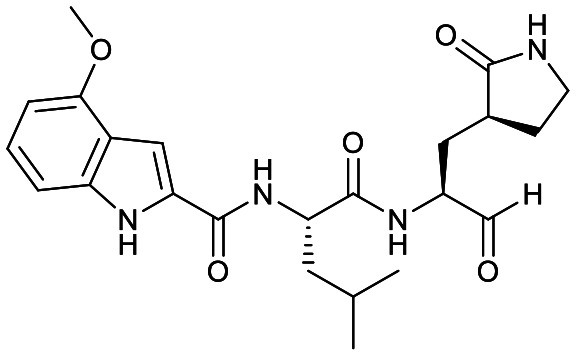
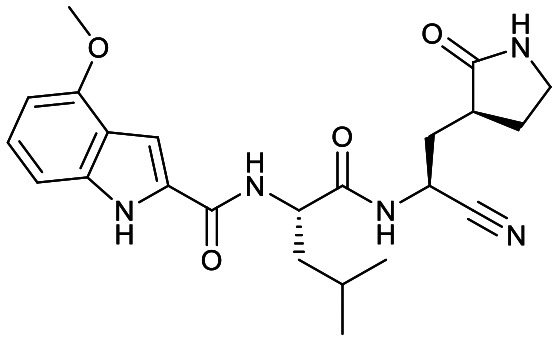
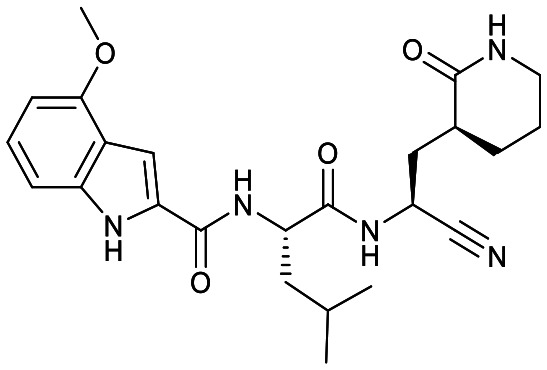
Studies by a subset of authors of this manuscript reported last year the discovery that GC-376 (4 in Fig. 1), which is an aldehyde prodrug, displayed good SARS-CoV-2 3CLpro and antiviral activity.29 Exploring the structure–activity relationship of the aldehyde and bisulfate derivatives generated a series of compounds, including 7 (Fig. 1), that showed better inhibition of SARS-CoV-2 3CLpro, although similar antiviral activity was obtained.15,38 For this study we wanted to explore the utility of the nitrile warhead with the gamma- versus delta-lactam in P1, and examine different indole substituents with the more active P1. The same protocols were used as detailed previously, see also ESI.†29 In Table 1, the SARS-CoV-2 3CLpro FRET inhibition results are shown for our peptidomimetics with a nitrile warhead in comparison to 4 and 7. Compound 17a, which is the nitrile analog of 7, showed a comparable IC50 value to aldehyde 7 against SARS-CoV-2 3CLpro, but was considerably more active than 4. The α-hydroxymethylketone (HMK) 6 (Fig. 1) had an IC50 value of 18.9 nM in this FRET assay. The IC50 values for SARS-CoV-2 3CLpro of 6, 7 and 17a were only a few-fold of each other with HMK appearing slightly more active. Replacing the gamma-lactam in 17a with its 6-membered homologue (compound 18a) provided a slight increase in inhibition reducing the IC50 value to 13 nM for SARS-CoV-2 3CLpro. An IC50 value of 19.5 nM was obtained for the HMK analogue 3 (Fig. 1), which is similar to 18a reinforcing the observation that the peptidomimetics with the nitrile and HMK warheads show comparable inhibition towards SARS-CoV-2 3CLpro.
Results for protease inhibition and SARS-CoV-2 plaque reduction for compounds.
| Cmpd | Structure | SARS-CoV-2 3CLpro IC50a (nM) | EC50 PRAb (μM) | EC50 PRA + CPc (μM) | Inhibition at 1 μM and/ord [IC50 in nM] | ||
|---|---|---|---|---|---|---|---|
| Human CatB | Human CatS | Human CatL | |||||
| 4 |
|
190 ± 40 | 0.9 ± 0.2 | 0.25 ± 0.02 | 53% [1400 ± 10] | 99% [23 ± 3] | 100% [0.25 ± 0.08] |
| 7 |
|
60 ± 10 | 0.9 ± 0.1 | 0.25 ± 0.01 | NR [2000 ± 700] | NR [40 ± 10] | 53% [280 ± 60] |
| 17a |
|
40 ± 8 | >5 | NR | <1% [28 000 ± 8000] | 78% [700 ± 100] | 15% [1450 ± 300] |
| 18a |
|
13 ± 3 | 2.7 ± 0.7 | 0.23 ± 0.04 | <1% [32 000 ± 7000] | 73% [2500 ± 500] | 22% [640 ± 050] |
| 18b |
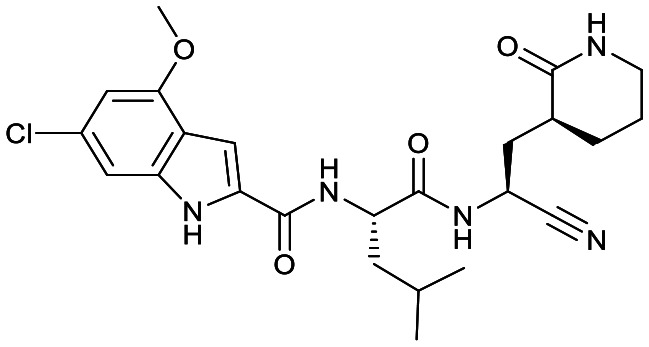
|
9.1 ± 2.5 | 2.2 ± 0.5 | 0.233 ± 0.007 | <1% [18 000 ± 3000] | 72% [400 ± 80] | 37% [470 ± 80] |
| 19b |
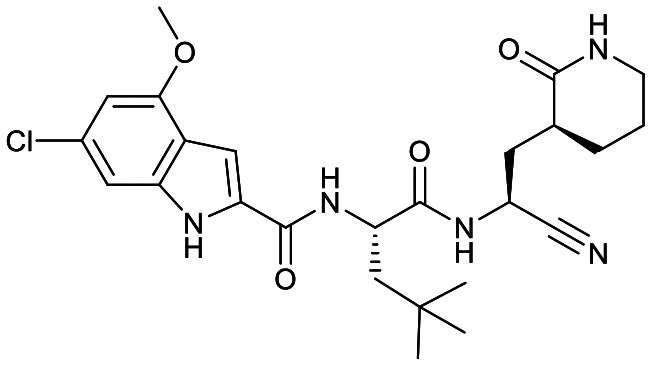
|
14 ± 2 | 1.5 ± 0.4 | 0.21 ± 0.05 | <1% | 64% | 18% |
| 18c |
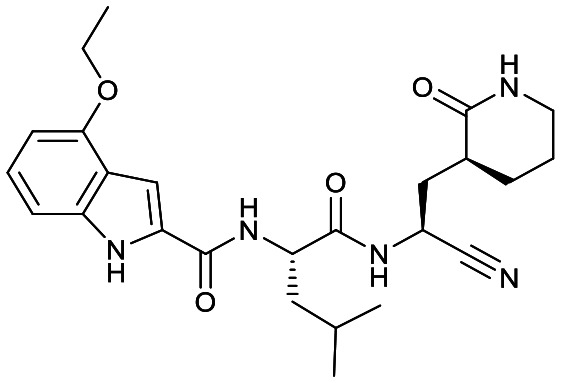
|
24 ± 4 | 4.8 ± 0.5 | 0.12 ± 0.04 | <1% | 74% | <1% |
| 18d |
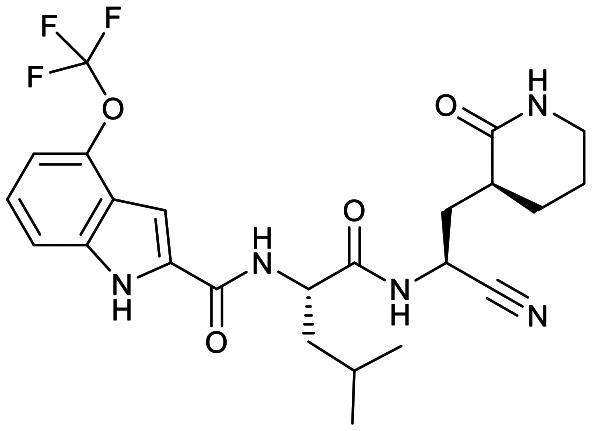
|
13.7 ± 1.5 | 2.6 ± 0.3 | 0.35 ± 0.09 | <1% | 67% | 8% |
| 18e |
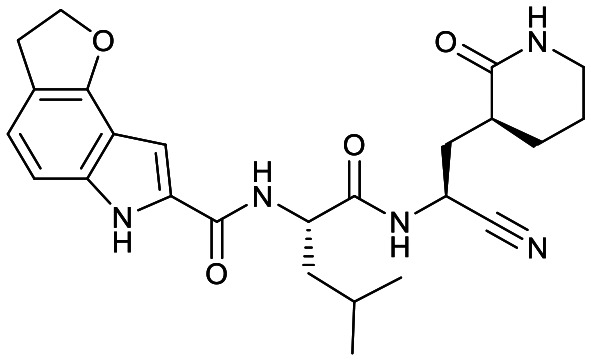
|
49 ± 10 | >10 | NR | <1% | 67% | 10% |
– See ESI,† determined from 8 concentrations performed in triplicate.
– Determined from 6 concentrations performed in at least triplicate (Vero E6 host cells); CC50 of all compounds (except 18d) was >200 μM (Vero E6 and A549).
– Same as b, but all wells contain 0.5 μM CP-100356.
– Run in duplicate or triplicate and at least 6 concentrations were utilized to determine IC50 values. NR – not run.
Based on a crystal structure obtained (vide infra) there appeared to be room available to further substitute the indole, and with 18a showing slightly better activity we decided to maintain the delta-lactam in P1 while exploring additional variations. The 6-chlorosubstitution, 18b, produced a similar, although possibly slightly improved inhibition of SARS-CoV-2 3CLpro compared to 18a. The t-butylalanine P2 version, 19b, produced inhibition in the same range as 18a and 18b, suggesting no activity benefit compared to P2 leucine. Utilizing a 4-ethoxy (18c) instead of 4-methoxy (18a) appears to result in a slightly reduced inhibition. The IC50 values for the trifluoromethyl (18d) and methyl (18a) ethers were virtually identical. The 4,5-dihydrofurano-fused indole 18e lost activity compared to both the methoxy and ethoxy versions, 18a and 18c.
To confirm the compounds were not cytotoxic, viability determinations were carried out on all 17–19 compounds using both CellTiter Glo and CCK8 readouts with both Vero E6 and A549 cells and all the compounds displayed a CC50 value greater than 200 μM in all 4 assays, except 18d. The outlier, 18d, had a CC50 of >100 μM against Vero E6 and against the more sensitive A549 cells had a CC50 value of approximately 90 μM. Compound 4 and 7 were reported previously and had CC50 values of greater than 200 μM.15
Compounds 17–19 were examined in a SARS-CoV-2 plaque reduction assay (PRA) using Vero E6 host cells (Table 1). They were initially screened for their antiviral activity at 10 μM and if 3-fold or greater reduction in plaques was observed for a compound it was tested in a concentration response curve to determine the EC50 value. If less than 2-fold reduction was observed the compound was assigned >10 μM, and if less than a 3-fold reduction was observed at 10 μM then >5 μM was assigned. Compounds 18a, 18b, 18c, 19b and 18d had low single digit micromolar EC50 values in the PRA. Compared to compound 4 and 7, these 5 compounds had lower IC50 values against SARS-CoV-2 3CLpro, but higher EC50 values in the PRA. One possible rationale is that these nitriles are more susceptible to efflux than structurally related 7, which is a recognised consideration for Vero cells.39 To determine if these compounds were undergoing efflux we repeated the PRA assay in the presence of a known efflux inhibitor CP-100356 (CP) at 0.5 μM to determine the extent of efflux (Table 1).20,40 As reported previously, the EC50 values for 4 and 7 decreased about 3-fold indicating some active transport out of the cell.15 For compounds 18a, 18b, 19b, 18c, and 18d, inhibiting efflux with CP in the PRA resulted in approximately a 10-fold improvement in their anti-viral activity resulting in EC50 values similar to 4 and 7 in the presence of CP. This strongly suggests that efflux plays a role in the higher EC50 values for these nitrile compounds in the absence of CP.
As these nitrile inhibitors demonstrated SARS-CoV-2 3CLpro inhibition and in vitro SARS-CoV-2 antiviral activity, we decided to examine their reactivity and selectivity. Some reactive warheads are susceptible to reaction with glutathione (GSH) and so we decided to test if GSH sequesters these nitrile inhibitors. We incubated 18a with GSH (10 mM) at pH 7.4 at 37 °C for 24 h and observed equally high recovery (≥90%) of 18a to one incubated in the phosphate buffer without GSH. The high recovery of 18a and lack of observed glutathione adduct (by LC/MS) demonstrate these nitrile warheads are stable in the presence of high levels of GSH, or that any reaction is readily reversible. We then turned our attention to selectivity over human cysteine proteases that have a similar sequence specificity for P2 and P1, leucine and glutamine, respectively. These are more likely to prove challenging to obtain selectivity over than a protease with a different catalytic mechanism and substrate specificity. The human cysteine proteases cathepsin B (CatB), cathepsin S (CatS) and cathepsin L (CatL) have similar specificities in P2 and P1 as SARS-CoV-2 3CLpro.41 Cathepsins are involved in numerous physiological and pathological processes and are themselves potential drug targets as they have suspected roles in numerous human diseases.41
To compare selectivity of another warhead we decided to also examine 4 and 7 for their ability to inhibit CatB, S and L. As the bisulfite adduct formed in 4 is reversible, both these compounds utilize aldehyde warheads.15 Both 4 and 7 were less potent inhibitors of CatB than CatS. The CatS IC50 values were 23 nM and 40 nM, respectively, which indicate that these two aldehyde warhead compounds show no selectivity for SARS-CoV-2 3CLpro. Compound 4 was 8-fold more active against CatS than SARS-CoV-2 3CLpro. The extremely potent inhibition of CatL by 4, with an IC50 value of 0.25 nM, was consistent with the recently reported value of 0.33 nM.24 Compound 7 was 1120-fold less active against CatL than 4, indicating a significant influence of the 4-methoxyindol-2-yl versus benzyloxy P3 groups. Compound 7 was, however, still less than 5-fold more selective for SARS-CoV-2 3CLpro than CatL.
All the nitriles 17–19 displayed low inhibition of CatB when screened at 1 μM, while CatS and CatL showed inhibition at that concentration (Table 1). Concentration response curves were obtained for select compounds to compare with the IC50 values obtained for 4 and 7. Comparing 7 to 17a, where the only change is the warhead, indicates that the nitrile warhead is over 10 times less active against CatB and CatS than the aldehyde warhead. Although for CatL the difference is reduced, the nitrile analog is still 5-fold less active compared to the aldehyde. This resulted in nitrile 17a displaying a significantly better selectivity for SARS-CoV-2 3CLpro over the human cathepsins B, S and L than aldehyde 7.
Comparison of the gamma- and delta-lactam, 17a and 18a, showed the IC50 value against CatB was within error for both, while for CatS 18a was a few-fold less active than 17a and for CatL the reverse was the case. With 18a having a low IC50 value for SARS-CoV-2 3CLpro, this compound obtained excellent selectivity over CatB and CatS of 2424-fold and 189-fold, respectively, and reasonable selectivity over CatL of 48-fold. The compound with the lowest IC50 value against SARS-CoV-2 3CLpro, 18b, also had increased inhibition of CatB, CatS and CatL in relation to 17a and 18a. Regardless, compared to 4 and 7, 18b still showed better selectivity for SARS-CoV-2 3CLpro over CatB, CatS and CatL than the aldehyde warhead containing peptidomimetics.
To determine the nature of the inhibition a reversibility study was done with 18a (Fig. 2). The dialysis of inhibited 3CLpro resulted in an increase of enzymatic activity over time, which is indicative of reversible binding of the inhibitor. We observed a recovery of 10% of activity after only 4 hours, and a 45% recovery of initial activity after 3 days. The enzyme 3CLpro loses activity overtime and the intersection of recovery of activity and loss of enzymatic activity is approximately 65 h for 18a. This recovery of activity as 18a is dialyzed away demonstrates the reversible nature of the inhibition of this nitrile warhead.
Fig. 2. Activity of SARS-CoV-2 3CLpro over time in media or after exposure to 18a (reversibility experiment).

An initial structure of SARS-CoV-2 3CLpro was solved by Zhang et al.11 The structure of 17a with SARS-CoV2 3CLpro was determined by molecular replacement, using the crystal structure of the free enzyme of the SARS-CoV-2 3CLpro (PDB entry 6WTM)29 as a search model. The structure obtained had a resolution of 2.15 Å and is shown in Fig. 3 (PDB: 7R7H). It crystalized as a dimer, and the active site of both protomers contained 17a bound in a very similar orientation. The nitrile reacted with the catalytic sulfur of Cys145 to generate a thioimidate adduct. This is a reversible process as seen in Fig. 2. This thioimidate has similar bond lengths, 1.8 Å C–S bond length, and geometries, sp2 carbon, to that observed with other cysteine proteases with nitrile warheads, such as odanacatib – cathepsin K complex (PDB: 5TDI). For both SARS-CoV-2 3CLpro-17a protomers the imine nitrogen of the thioimidate moiety is within hydrogen bonding distance of the NH for both Cys145 and Gly143, 3.3 Å and 3.4 Å, respectively for chain A, with 3.2 Å and 3.7 Å for chain B. The rest of the hydrogen bonding network is practically identical to that seen previously for aldehyde 7 and HMK 6 (PDB: 7LDL and 6XHM, respectively).15,20
Fig. 3. Structure of 17a bound to SARS-COV-2 3CLpro (PDB entry 7R7H) with A showing the hydrogen bonding network, S2 pocket and thioimidate (chain A) and B showing the electron density of the ligand (chain B).

Discussion
Despite the previous modest results against viral cysteine proteases in the literature, the nitrile warhead with improved peptidomimetic groups demonstrated potent SARS-CoV-2 3CLpro inhibition. The inhibition of SARS-CoV-2 3CLpro for the nitrile warhead containing peptidomimetic was similar to the corresponding aldehyde analog, comparing IC50 values of 17a to 7 (Table 1).
The delta-lactam had a lower IC50 than the gamma-lactam, 17a to 18a, which is consistent to Shang, Yin and coworkers observation for enterovirus-71 3C protease with an aldehyde warhead.27 It is worth noting that no difference in IC50 values for the gamma- and delta-lactams in P1 against SARS-CoV-2 3CLpro was observed in our previous study with an AMK warhead.22 Further studies with different warheads and proteases will be required to understand the factors influencing the activity of the delta-lactam compared to the gamma-lactam. In addition, the selectivity differences, such as seen in Table 1, and other attributes, such as pharmacokinetic properties, will also need to be evaluated to select the optimal lactam group.
Fig. 3 shows the co-crystal structure for SARS-CoV-2 3CLpro-17a that we obtained indicating that a covalent bond is formed between the sulfur of Cys145 and the nitrile generating a thioimidate adduct. Based on reversibility studies this is a reversible, covalent adduct. That structure did not reveal any close contacts around the end of the capping group (Fig. 3) suggesting the opportunity to add substitutents on the phenyl or methoxy of the 4-methyloxyindole. However, only the 6-chloro derivative 18b displayed a possibly modest increase in protease inhibition compared to 18a, but resulted in slightly lower selectivity attributes over CatB and CatS compared to the same compound. Other substitutions tested did not prove advantageous as the trifluoromethoxy group (18d) displayed toxicity (CC50 < 200 μM) and the ethoxy (18c) and dihydrofurano (18e) derivatives lost activity against SARS-CoV-2 3CLpro as well as in the antiviral assay.
All the nitrile analogs (17–19) have IC50 values less than 100 nM for SARS-CoV-2 3CLpro inhibition, but only those with values less than 25 nM showed an EC50 value of less than 5 μM in the PRA. Aldehydes 4 and 7 were both more active in the PRA. The nitrile analogs all had an approximately 10-fold reduction of EC50 value when the efflux inhibitor CP was added, suggesting that these peptidomimetic compounds were undergoing significant efflux from Vero E6 cells. Structural alterations to improve properties, such as H-bond donor removal and other depeptidization strategies could improve properties and overcome the efflux issue.42
Compound 4 demonstrated extremely potent inhibition of CatL, which was consistent with that recently reported in the literature.24 Compound 4 also had almost 10-fold higher inhibition of CatS than SARS-CoV-2 3CLpro. For CatL, the P3 capping group made a substantial difference when comparing 7 to 4, but only a modest difference was observed for CatS (Table 1).
All compounds in Table 1 were more selective for SARS-CoV-2 3CLpro than for CatB, with the nitriles 17a, 18a and 18b having >1000-fold selectivity. These compounds, 17a, 18a and 18b, also showed lower inhibition of CatS and CatL than aldehydes 4 and 7 and superior selectivity for SARS-CoV-2 3CLpro. Literature IC50 values for HMK 6 for CatB and CatL inhibition are 1.3 μM and 146 nM, respectively.20,24 Comparison of these values to the nitrile analog 17a (Table 1) for CatB and CatL suggests that the nitrile warhead is significantly less active at inhibiting these cathepsins than the HMK warhead (ca. 21- and 10-fold less), although a head-to-head comparison is required to confirm this higher selectivity. The selectivity criteria required for a drug still needs to be established. For example, CatS drugs in clinical trials were well-tolerated in healthy volunteers, but did show an increased respiratory tract infection rate in a small percentage of patients making potent CatS inhibition a concern for COVID-19 therapy.43 CatL activity is implicated in being important to the SARS-CoV-2 replication cycle, such as spike protein processing and possibly exit from cells, and its inhibition could also be a mechanism to inhibit viral replication.44,45,46 CatS and CatL are important lysosomal endopeptidase enzymes and further research is needed to understand the level of selectivity that is required to avoid side-effects in vivo.
The results herein demonstrate that these nitrile warhead containing peptidomimetics have good potency against SARS-CoV-2 3CLpro and display selectivity over human cysteine proteases (CatB, CatS and CatL) that have overlapping sequence specificity for P2 and P1. Further structural alterations to reduce efflux, for example by removing peptide character, is currently being investigated. The structure of Pfizer's recently revealed oral clinical candidate with a nitrile warhead (8 in Fig. 1) demonstrates one method that this can be successfully achieved, and alternate approaches could yield diverse oral SARS-CoV-2 3CLpro therapies with different favourable attributes, such as maintaining activity against clinically relevant SARS-CoV-2 3CLpro mutants.47 Continued research on this promising target and these peptidomimetics has the potential to add to the arsenal of direct-acting antivirals needed to combat the current COVID-19 pandemic, emerging variants and future coronavirus outbreaks.
Conclusions
Direct-acting antivirals to treat COVID-19 are desperately needed and the viral encoded protein 3CLpro, which is a cysteine protease required for viral replication, is a promising drug target. Treatment of proteases typically utilize peptidomimetics that mimic the substrate and this article demonstrated that a nitrile warhead provides compounds with good SARS-CoV-2 3CLpro inhibition and selectivity over human cysteine proteases (CatB, CatS and CatL) with overlapping sequence specificity in P1 and P2. For the same peptidomimetic, the nitrile warhead demonstrated superior activity (lower IC50) against SARS-CoV-2 3CLpro and greater selectivity over human CatB, CatS and CatL than the aldehyde warhead analog. Some nitrile warhead analogs with a P1 delta-lactam also displayed EC50 values of 1–3 micromolar in a SARS-CoV-2 plaque reduction assay, with CC50 values of greater than 200 μM. Efflux appears be an issue for these compounds and reducing the susceptibility of active transport of future nitrile warhead containing analogs, such as by removing peptide character, has the potential to improve both antiviral activity and generate favourable properties to enable development of novel oral therapeutics to treat COVID-19.
Author contributions
Authors that contributed to investigation and methodology are BB, EA, MJL, MBK, MAJ, HAS, JAS, ASK, AB, MH, and WV. BB, JL, ASK, WV and TL contributed to resources. Visualization was performed by EA, MBK, MAJ, MJL and JAN. The authors that supervised were HSY, JCV, DLT, MAJ, MJL and JAN. Funding acquisition and project administration were by MJL, DLT and JAN. JAN wrote the original draft and all authors reviewed the manuscript prior to publication.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
The authors would like to thank numerous colleagues at the University of Alberta including: Vishwa Somayaji (Faculty of Pharmacy and Pharmaceutical Sciences) for running 1H, 13C and 19FNMRs; Jack Moore (Alberta Proteomics and Mass Spectrometry Facility) for HRMS; Wayne Moffat and Madison Gauthier (Department of Chemistry) for IR. We thank the staff at SSRL beamline 12-1, in particular Dr. Silvia Russi and Lisa Dunn. Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (P30GM133894). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS or NIH. Funding for the project were provided by Alberta Innovates (RES27408) and Canadian Institutes of Health Research Rapid Research (VR3-172655).
Electronic supplementary information (ESI) available. See DOI: 10.1039/d1md00247c
Notes and references
- World Health Organization, http://www.who.int/emergencies/diseases/novel-coronavirus-2019, (accessed June 14, 2021)
- Pardo J. Shukla A. M. Chamarthi G. Gupte A. Drugs Context. 2020;9:1–9. doi: 10.7573/dic.2020-4-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- US Food and Drug Administration, https://www.fda.gov/news-events/press-announcements/fda-approves-first-treatment-covid-19, (accessed February 6, 2021)
- Lau S. K. P. Lee P. Tsang A. K. L. Yip C. C. Y. Tse H. Lee R. A. So L.-Y. Lau Y.-L. Chan K.-H. Woo P. C. Y. Yuen K.-Y. J. Virol. 2011;85(21):11325–11337. doi: 10.1128/JVI.05512-11. [DOI] [PMC free article] [PubMed] [Google Scholar]; Centers for Disease Control and Prevention, https://www.cdc.gov/coronavirus/types.html, (accessed February 6, 2021); Anirudhan V. Lee H. Cheng H. Cooper L. Rong L. J. Med. Virol. 2021;93(5):1–13. doi: 10.1002/jmv.26814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H. Yang J. RSC Med. Chem. 2021;12:1026–1036. doi: 10.1039/d1md00066g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan S. Wu G. FASEB J. 2021;35:e21573. doi: 10.1096/fj.202100280RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong M. Su H. Zhao W. Xie H. Shao Q. Xu Y. Med. Res. Rev. 2020;41(4):1–34. doi: 10.1002/med.21783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oerlemans R. Ruiz-Moreno A. J. Cong Y. Kumar N. D. Velasco-Velazquez D. A. Neochoritis C. G. Smith J. Reggiori F. Grovesa M. R. Dömling A. RSC Med. Chem. 2021;12:370–379. doi: 10.1039/d0md00367k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahdi M. Mótyán J. A. Szojka Z. I. Golda M. Miczi M. Tőzsér J. Virol. J. 2020;17:190. doi: 10.1186/s12985-020-01457-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citarella A. Scala A. Piperno A. Micale N. Biomolecules. 2021;11:607. doi: 10.3390/biom11040607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L. Lin D. Sun X. Curth U. Drosten C. Sauerhering L. Becker S. Rox K. Hilgenfeld R. Science. 2020;368:409–412. doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillaiyar T. Manickam M. Namasivayam V. Hayashi Y. Jung S.-H. J. Med. Chem. 2016;59:6595–6628. doi: 10.1021/acs.jmedchem.5b01461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui W. Yang K. Yang H. Front. Mol. Biosci. 2020;7:616341. doi: 10.3389/fmolb.2020.616341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai W. Zhang B. Jiang X.-M. Su H. Li J. Zhao Y. Xie X. Jin Z. Peng J. Liu F. Li C. Li Y. Bai F. Wang H. Cheng X. Cen X. Hu S. Yang X. Wang J. Liu X. Xiao G. Jiang H. Rao Z. Zhang L.-K. Xu Y. Yang H. Liu H. Science. 2020;368:1331–1335. doi: 10.1126/science.abb4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuong W. Fischer C. van Belkum M. J. Lamer T. Willoughby K. D. Khan M. B. Arutyunova E. Joyce M. A. Saffran H. A. Shields J. A. Young H. S. Nieman J. A. Tyrrell D. L. Lemieux M. J. Vederas J. C. Eur. J. Med. Chem. 2021;222:113584. doi: 10.1016/j.ejmech.2021.113584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dampalla C. S. Kim Y. Bickmeier N. Rathnayake A. D. Nguyen H. N. Zheng J. Kashipathy M. M. Baird M. A. Battaile K. P. Lovell S. Perlman S. Chang K.-O. Groutas W. C. J. Med. Chem. 2021;64(14):10047–10058. doi: 10.1021/acs.jmedchem.1c00319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L. Lin D. Kusov Y. Nian Y. Ma Q. Wang J. von Brunn A. Leyssen P. Lanko K. Neyts J. de Wilde A. Snijder E. J. Liu H. Hilgenfeld R. J. Med. Chem. 2020;63(9):4562–4578. doi: 10.1021/acs.jmedchem.9b01828. [DOI] [PubMed] [Google Scholar]
- Zhu W. Xu M. Chen C. Z. Guo H. Shen M. Hu X. Shinn P. Klumpp-Thomas C. Michael S. G. Zheng W. ACS Pharmacol. Transl. Sci. 2020;3:1008–1016. doi: 10.1021/acsptsci.0c00108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Z. Du X. Xu Y. Nature. 2020;582(7811):289–293. doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- Hoffman R. L. Kania R. S. Brothers M. A. Davies J. F. Ferre R. A. Gajiwala K. S. He M. Hogan R. J. Kozminski K. Li L. Y. Lockner J. W. Lou J. Marra M. T. Mitchell Jr. L. J. Murray B. W. Nieman J. A. Noell S. Planken S. P. Rowe T. Ryan K. Smith III G. J. Solowiej J. E. Steppan C. M. Taggart B. J. Med. Chem. 2020;63:12725–12747. doi: 10.1021/acs.jmedchem.0c01063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Plassche M. A. T. Barniol-Xicota M. Verhelst S. H. L. ChemBioChem. 2020;21:3383–3388. doi: 10.1002/cbic.202000371. [DOI] [PubMed] [Google Scholar]
- Bai B. Belovodskiy A. Hena M. Kandadai A. S. Joyce M. A. Saffran H. A. Shields J. A. Khan M. B. Arutyunova E. Lu J. Bajwa S. K. Hockman D. Fischer C. Lamer T. Vuong W. van Belkum M. J. Gu Z. Lin F. Du Y. Xu J. Rahim M. Young H. S. Vederas J. C. Tyrrell D. L. Lemieux M. J. Nieman J. A. J. Med. Chem. 2021 doi: 10.1021/acs.jmedchem.1c00616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao J. Li Y.-S. Zeng R. Liu F.-L. Luo R.-H. Huang C. Wang Y.-F. Zhang J. Quan B. Shen C. Mao X. Liu X. Sun W. Yang W. Ni X. Wang K. Xu L. Duan Z.-L. Zou Q.-C. Zhang H.-L. Qu W. Long Y.-H.-P. Li M.-H. Yang R.-C. Liu X. You J. Zhou Y. Yao R. Li W.-P. Liu J.-M. Chen P. Liu Y. Lin G.-F. Yang X. Zou J. Li L. Hu Y. Lu G.-W. Li W.-M. Wei Y.-Q. Zheng Y.-T. Lei J. Yang S. Science. 2021;371:1374–1378. doi: 10.1126/science.abf1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandyck K. Abdelnabi R. Gupta K. Jochmans D. Jekle A. Deval J. Misner D. Bardiot D. Foo C. S. Liu C. Ren S. Beigelman L. Blatt L. M. Boland S. Vangeel L. Dejonghe S. Chaltin P. Marchand A. Serebryany A. Stoycheva A. Chanda S. Symons J. A. Raboisson P. Neyts J. Biochem. Biophys. Res. Comm. 2021;555:134–139. doi: 10.1016/j.bbrc.2021.03.096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Turk B. Nat. Rev. Drug Discovery. 2006;5:785–799. doi: 10.1038/nrd2092. [DOI] [PubMed] [Google Scholar]; (b) Hamada Y. Kiso Y. Biopolymers. 2016;106:563–579. doi: 10.1002/bip.22780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClung M. R. O'Donoghue M. L. Papapoulos S. E. Bone H. Langdahl B. Saag K. G. Reid I. R. Kiel D. P. Cavallari I. Bonaca M. P. Wiviott S. D. de Villiers T. Ling X. Lippuner K. Nakamura T. Reginster J.-Y. Rodriguez-Portales J. A. Roux C. Zanchetta J. Zerbini C. A. F. Park J.-G. Im K. Cange A. Grip L. T. Heyden N. DaSilva C. Cohn D. Massaad R. Scott B. B. Verbruggen N. Gurner D. Miller D. L. Blair M. L. Polis A. B. Stoch S. A. Santora A. Lombardi A. Leung A. T. Kaufman K. D. Sabatine M. S. Lancet. 2019;7(12):899–911. doi: 10.1016/S2213-8587(19)30346-8. [DOI] [PubMed] [Google Scholar]
- Zhai Y. Zhao X. Cui Z. Wang M. Wang Y. Li L. Sun Q. Yang X. Zeng D. Liu Y. Sun Y. Lou Z. Shang L. Yin Z. J. Med. Chem. 2015;58:9414–9420. doi: 10.1021/acs.jmedchem.5b01013. [DOI] [PubMed] [Google Scholar]; Wang Y. Cao L. Zhai Y. Ma J. Nie Q. Li T. Yin A. Sun Y. Shang L. Antiviral Res. 2017;121:91–100. doi: 10.1016/j.antiviral.2017.01.002. [DOI] [PubMed] [Google Scholar]
- Chuck C.-P. Chen C. Ke Z. Wan D. C.-C. Chow H.-K. Wong K.-B. Eur. J. Med. Chem. 2013;59:1–6. doi: 10.1016/j.ejmech.2012.10.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuong W. Khan M. B. Fischer C. Arutyunova E. Lamer T. Shields J. Saffran H. A. McKay R. T. van Belkum M. J. Joyce M. A. Young H. S. Tyrrell D. L. Vederas J. C. Lemieux M. J. Nat. Commun. 2020;11:4282. doi: 10.1038/s41467-020-18096-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anivive Repurposes Veterinary Drug GC376 for COVID-19 And Submits Pre-IND to FDA, PRNewswire Press release May 26, 2020, Anivive Lifesciences Inc. [Google Scholar]; Vandyck K. Deval J. Curr. Opin. Virol. 2021;49:36–40. doi: 10.1016/j.coviro.2021.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halford B., Pfizer's novel COVID-19 antiviral heads to clinical trials, C&EN, 2020, vol. 98(37), p. 9 [Google Scholar]
- Hoffman R. L., Kania R. S., Nieman J. A., Planken S. P. and Smith G. J., WO2005/113580, 2005
- Kania R. S., Mitchell L. J. and Nieman J. A., WO2006/061714, 2006
- Halford B., Pfizer unveils its oral SARS-CoV-2 inhibitor, C&EN, 2021, vol. 99(13), p. 7 [Google Scholar]; Dolgin E. Nature. 2021;592:340–343. doi: 10.1038/d41586-021-00958-4. [DOI] [PubMed] [Google Scholar]
- Breidenbach J. Lemke C. Pillaiyar T. Schäkel L. Al Hamwi G. Diett M. Gedschold R. Geiger N. Lopez V. Mirza S. Namasivayam V. Schiedel A. C. Sylvester K. Thimm D. Vielmuth C. Vu L. P. Zyulina M. Bodem J. Gütschow M. Müller C. E. Angew. Chem., Int. Ed. 2021;60:10423–10429. doi: 10.1002/anie.202016961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priora A. M. Kimb Y. Weerasekaraa S. Morozea M. Alliston K. R. Uy R. A. Z. Groutas W. Chang K.-O. Hua D. H. Bioorg. Med. Chem. Lett. 2013;23:6317–6320. doi: 10.1016/j.bmcl.2013.09.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai Y. Ma Y. Ma F. Nie Q. Ren X. Wang Y. Shang L. Yin Z. Eur. J. Med. Chem. 2016;124:559–573. doi: 10.1016/j.ejmech.2016.08.064. [DOI] [PubMed] [Google Scholar]
- Arutyunova E. Khan M. B. Fischer C. Lu J. Lamer T. Vuong W. van Belkum M. J. McKay R. T. Tyrrell D. L. Vederas J. C. Young H. S. Lemieux M. J. J. Mol. Biol. 2021;433(13):167003. doi: 10.1016/j.jmb.2021.167003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalgutkar A. S. Frederick K. S. Chupka J. Feng B. Kempshall S. Mireles R. J. Fenner K. S. Troutman M. D. J. Pharm. Sci. 2009;98(12):4914–4927. doi: 10.1002/jps.21756. [DOI] [PubMed] [Google Scholar]
- Vero E6 cells treated with 0.5 μM CP in the absence of compound showed high viability (>80%) after 24 h using both CCK8 and CellTitreGlo determinations. The viability of Vero E6 cells in the presence 0.5 μM CP and 18a–18c and 19b at 50 μM and 200 μM was also determined. The viability of the Vero E6 cells by CCK8 and CellTitreGlo for these compounds was lower than without CP, but was still above 50% at 200 μM (CC50 > 200 μM).
- Biniossek M. L. Nägler D. K. Becker-Pauly C. Schilling O. J. Proteome Res. 2011;10:5363–5373. doi: 10.1021/pr200621z. [DOI] [PubMed] [Google Scholar]
- Tinworth C. P. Young R. J. J. Med. Chem. 2020;63:10091–10108. doi: 10.1021/acs.jmedchem.9b01596. [DOI] [PubMed] [Google Scholar]
- Brown R. Nath S. Lora A. Samaha G. Elgamal Z. Kaiser R. Taggart C. Weldon S. Geraghty R. Respir. Res. 2020;21:111. doi: 10.1186/s12931-020-01381-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T. Luo S. Libby P. Shi G.-P. Pharmacol. Ther. 2020;213:107587. doi: 10.1016/j.pharmthera.2020.107587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pišlar A. Mitrović A. Sabotič J. Fonović U. P. Nanut M. P. Jakoš T. Senjor E. Kos J. PLoS Pathog. 2020;16(11):e1009013. doi: 10.1371/journal.ppat.1009013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes C. P. Gernandes D. E. Casimiro F. da Mata G. F. Passos M. T. Varela P. Matroianni-Kirsztajn G. Pesquero J. B. Front. Cell. Infect. Microbiol. 2020;10:589505. doi: 10.3389/fcimb.2020.589505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross T. J. Takahashi G. R. Diessner E. M. Crosby M. G. Farahmand V. Zhuang S. Butts C. T. Martin R. W. Biochemistry. 2020;59(39):3741–3756. doi: 10.1021/acs.biochem.0c00462. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.