

Abstract
Mitochondrial malate dehydrogenase (MDH2) deficiency (MDH2D) is an ultra-rare disease with only three patients described in literature to date. MDH2D leads to an interruption of the tricarboxylic acid (TCA) cycle and malate-aspartate shuttle (MAS) and results in severe early onset encephalopathy. Affected infants suffer from psychomotor delay, muscular hypotonia and frequent seizures. Laboratory findings are unspecific, including elevated lactate in blood and cerebrospinal fluid. Brain magnetic resonance imaging reveals delayed myelination and brain atrophy. Currently there is no curative therapy to treat this devastating disease.
Here, we present a female patient diagnosed with MDH2D after a stroke-like episode at 18 months. Trio-whole exome sequencing revealed compound heterozygous missense variants in the MDH2 gene: c.398C>T, p.(Pro133Leu) and c.445delinsACA, p.(Pro149Hisfs*22). MDH2 activity assay and oxygraphic analysis in patient's fibroblasts confirmed the variants were pathogenic. At the age of 36 months, a drug trial with triheptanoin was initiated and well tolerated. The patient's neurologic and biochemical phenotype improved and she had no further metabolic decompensations during the treatment period suggesting a beneficial effect of triheptanoin on MDH2D.
Further preclinical and clinical studies are required to evaluate triheptanoin treatment for MDH2D and other TCA cycle and MAS defects.
Keywords: Mitochondrial malate dehydrogenase deficiency, Encephalopathy, Inborn errors of the tricarboxylic acid cycle, Malate-aspartate shuttle, Triheptanoin
List of abbreviations
| CGI | clinical global impression |
| CSF | cerebrospinal fluid |
| GMFCS | gross motor functions classification system |
| LC-FAOD | long-chain fatty acid oxidation disorders |
| LLN | lower limit of normal |
| MACS | manual ability classification system |
| MAS | malate-aspartate shuttle |
| MCTs | medium-chain triglycerides |
| MDH2 | mitochondrial malate dehydrogenase |
| MDH2D | MDH2 deficiency |
| MRI | magnetic resonance imaging |
| OXPHOS | oxidative phosphorylation |
| PEG tube | percutaneous endoscopic gastrostomy tube |
| RC | respiratory chain |
| TCA cycle | tricarboxylic acid cycle |
| trio-WES | trio-whole-exome sequencing |
| ULN | upper limit of normal |
1. Introduction
Ultra-rare genetic defects causing enzyme and transporter deficiencies of the tricarboxylic acid (TCA) cycle and the malate-aspartate shuttle (MAS) are devastating diseases associated with an energy undersupply of organs with a high-energy demand such as brain and muscle [1], [2]. Mitochondrial malate dehydrogenase (MDH2; EC 1.1.1.37; OMIM *154100) deficiency (MDH2D; OMIM # 617339) is caused by mutations in the MDH2 gene and has been described in only three patients thus far (1). The enzyme is part of the TCA cycle and the MAS [3], [4], [5] and it catalyses the reversible oxidation of malate to oxaloacetate using the NAD+/NADH cofactor system (6).
Here we describe a four year-old girl suffering from MDH2D. To date there is no treatment for MDH2D. Patients suffered from severe early-onset epileptic encephalopathy and were treated with anticonvulsant drugs and ketogenic diet to reduce seizure frequency (1).
Our main aim is to bring forward a potential new treatment strategy for MDH2D by using triheptanoin in this young patient. Triheptanoin is a medium-chain triglyceride (MCT) – the product of esterification of glycerol with three molecules of heptanoate (C7). Glycerol is either metabolized via glycolysis or it is shuttled into gluconeogenesis. C7 is degraded in the β-oxidation giving rise to acetyl-CoA and/or propionyl-CoA which replenish the TCA cycle. Alternatively, C7 is utilized for the production of ketone bodies [7], [8], [9]. Triheptanoin has been used in various metabolic disorders and refractory epilepsy [9], [10], [11] and has recently been approved in the USA for the treatment of patients with long-chain fatty acid oxidation disorders LC-FAOD (12). In a patient cohort with LC-FAOD, triheptanoin significantly reduced hospitalization days/year and the hospitalization rate (13).
The mode of action of triheptanoin is not yet fully understood. Acetyl-CoA and propionyl-CoA generated by triheptanoin help maintain an appropriate substrate balance for the TCA cycle, supplying peripheral tissues with an alternative fuel and improving energy generation for gluconeogenesis (9).
Here, triheptanoin treatment was given as an individual clinical trial for a young girl suffering from MDH2D. It was well tolerated during the 12 months observation period so that the therapy was continued beyond the clinical trial for more than two years now. Under triheptanoin therapy the patient's overall health, neurologic development and several biochemical parameters improved. Triheptanoin prevented metabolic decompensations. Further studies including more MDH2D patients are required to evaluate efficiency of this promising treatment approach.
2. Material and methods
2.1. Patient work-up
2.1.1. Brain magnetic resonance imaging
Our patient underwent three brain magnetic resonance (MR) imaging (MRI) examinations at the age of 19, 20 and 24 months, respectively, performed in a 1.5-Tesla magnetic resonance unit (Siemens Magnetom Aera or Siemens Magnetom Avanto, Siemens, Erlangen, Germany). Sequence acquisitions included diffusion-weighted imaging; time-of-flight MR angiography; contrast-enhanced MR-angiography; 3D sagittal T1-weighted images; axial T2-weighted images; susceptibility-weighted imaging; coronal FLAIR sequences and proton MR spectroscopy.
2.1.2. Molecular genetic analysis
Blood samples from parents and index patient were obtained for DNA extraction using chemagic Prepito (PerkinElmer, Waltham, MA, USA). Exome sequencing using Agilent SureSelect Human All Exon v6 was performed on an Illumina Hiseq 4000 instrument. Reads were mapped to human reference genome (hg19) and variants were identified using CLC Biomedical Genomics Workbench v5 (Qiagen, Hildesheim, Germany). MDH2-variants were confirmed using bi-directional Sanger sequencing.
2.2. Mitochondrial malate dehydrogenase activity assay
To determine MDH2 activity mitochondria were isolated from fibroblasts of four controls and the patient and lysed by sonification as described previously (14). Isolation of mitochondrial fraction was validated in our accredited routine laboratory. Contamination of the mitochondrial fraction with cytosolic and peroxisomal proteins was excluded by Western Blots revealing absence of a cytosolic p70 S6 Kinase and peroxisomal PEX19. MDH2 activity was determined by measuring the linear decrease in absorbance at 340 nm at 30 °C resulting from the oxidation of NADH. One unit oxidizes one μmol of NADH per minute at pH 7.4 under the specified conditions (8 mM K2PO4, 0.2 mM NADH, 2 mM oxaloacetate and 10 μg mitochondrial protein in a total volume of 1 mL in cuvettes).
2.3. Galactose stress test and respiratory chain enzyme analysis
Patient's dermal fibroblasts were grown from a skin punch biopsy and cultured in either standard glucose or galactose containing medium (“oxidative” stress test) as described previously (15). Oxidative phosphorylation (OXPHOS) was assessed by analyses of the respiratory chain (RC) complexes I (NADH-coenzyme Q oxidoreductase), II (succinate-Q oxidoreductase), III (Q-cytochromec oxidoreductase), IV (cytochrome c oxidase) and V (ATP synthase) in patient's fibroblast homogenates as described previously (15). The activities of complexes were related to citrate synthase (CS). Oxygraphic analysis of patient fibroblasts were assessed by OROBOROS as described previously (15).
2.4. Triheptanoin trial
Triheptanoin oil (Ultragenyx pharmaceutical Inc., CA, USA) was administered orally by nasogastric and/or percutaneous endoscopic gastrostomy (PEG) tube. The treatment scheme was based on previous studies with triheptanoin (13). The initial therapy was designated for 12 months after which its effects were assessed. Triheptanoin was continued thereafter and has now been administered for more than 24 months.
Daily caloric requirements are approximately 90 cal per kilogram (kg) bodyweight (bw). Recommended target dose of triheptanoin equates 30–35% of total daily caloric intake divided into three to five portions resulting in a final dose of 55 mL. Triheptanoin was gradually introduced over a period of one month. During the 12 months treatment period regular and frequent clinical and laboratory parameters were assessed. Small amounts of regular child food under strict avoidance of rapidly absorbable carbohydrates were taken up by the patient. The rest of the required daily calories was administered over the tube in form of Nutrini Energy Multi Fiber which is a nutritionally complete tube feed.
3. Results
3.1. Case report
Our patient is the second, female child of healthy, non-consanguineous Caucasian parents. Pregnancy, birth and the neonatal period were unremarkable. In the first months of life, she suffered from several febrile seizures in the context of common viral infections. During the first year of life she failed to thrive (Supplementary Fig. 1) and her head circumference dramatically dropped from the 97th percentile at 4 months to the 10th-25th percentile at 19 months of age (Supplementary Fig. 1). She developed psychomotor delay and muscular hypotonia.
At the age of 18 months, she was hospitalized and presented with acute signs of a stroke including a dyskinetic and choreatic movement disorder predominantly affecting her right extremities. Her speech was slurred and she fell to the right side while sitting and crawling. Plasma amino acid and acylcarnitine profiles were normal. Urinary organic acids were unremarkable. In contrast, blood and cerebrospinal fluid (CSF) lactate levels were elevated to 3.0 mmol/L and 2.25 mmol/L (reference <1.7), respectively. The brain MRI scan revealed a subacute stroke in the left nucleus lentiformis highly suspicious of a metabolic origin (Fig. 1A). On follow-up, there were neither signs of ischemia, vasculitis nor haemorrhage (Fig. 1B and C). Strikingly, myelination was delayed and there was a symmetric atrophy of the frontal lobes (Fig. 1D). Atrophy was of subcortical nature with a slight enlargement of the frontal horns of the lateral ventricles with adjacent germinolytic cysts (pseudocysts) (Fig. 1E). In addition, there was atrophy of the culmen of the cerebellum (Fig. 1F and G). Proton MR spectroscopy revealed accumulation of lactate in CSF and the brain parenchyma (Fig. 1H) – overall findings highly suspicious of being of metabolic origin.
Fig. 1.

Magnetic resonance imaging (MRI) features. (A) Axial diffusion-weighted imaging (DWI) shows restricted diffusion within the left globus pallidus. (B) Follow-up imaging 3 months post stroke shows normalization of DWI and complete resolution with no residual changes on T2-weighed imaging (T2WI, (C)). (D) Axial T2WI at the age of 2 years shows slightly delayed myelination in the centrum semiovale, not only posteriorly and superiorly to the occipital horns but also extending anteriorly around the bodies of the lateral ventricles. Axial T1WI (E) shows enlargement of the frontal horns and adjacent germinolytic cysts. Coronal FLAIR (F) and sagittal T1W images (G) show selective atrophy of the superior vermis (culmen). (H) Long-TE MR spectroscopy chemical shift imaging demonstrates a lactate peak at 1.33 ppm within the cerebrospinal fluid.
Taken together, despite the unspecific nature of our clinical, laboratory and radiographic findings, they were evocative of a metabolic disorder. A genetic analysis with trio-whole exome sequencing (trio-WES) revealed compound heterozygous missense variants in the MDH2 gene: c.398C>T, p.(Pro133Leu) inherited from her mother and c.445delinsACA, p.(Pro149Hisfs*22) inherited from her father. According to the ACMG guidelines (16) both variants were classified pathogenic. While the maternally inherited variant has been described previously in all three published cases (1), the c.445delinsACA variant has not been described before (Supplementary Table 1).
The first months following the stroke she developed epileptic seizures (both generalized and absence type). She suffered from several episodes of metabolic decompensations in the context of febrile infections, which responded well to anti-catabolic therapy. Prior to initiating the clinical trial with triheptanoin she received antiepileptic treatment with lamotrigine and levetiracetam.
3.2. Mitochondrial malate dehydrogenase activity assay
Enzymatic MDH2 activity in patient fibroblasts was 37 ± 9.7 mU/mg protein (range: 18–56 mU/mg protein) and thus significantly decreased compared to control fibroblasts showing an activity of 160 ± 43 mU/mg protein (range: 107–211 mU/mg protein, n = 4) confirming MDH2D.
3.3. Galactose stress test and respiratory chain enzyme analysis
MDH2D interrupts the TCA cycle and leads to a decreased production of reducing equivalents for oxidative ATP production. However, patient fibroblasts survived cell culture in a galactose-containing medium indicating a normal function of the RC [15], [17]. In line with this, all OXPHOS activities were normal (Table 1). All oxygraphic analysis were normal except for a decreased ratio of pyruvate/succinate respiration (Table 2).
Table 1.
OXPHOS activities in the patient's fibroblasts. Normal findings.
| Oxidoreductases | Patient(mU/mU CS) | Reference values (mU/mU CS) |
|---|---|---|
| Complex I | 0.55 | 0.27–0.66 |
| Complex II | 0.39 | 0.34–0.69 |
| Complex III | 0.75 | 0.68–1.49 |
| Complex IV | 0.89 | 0.52–1.32 |
| Complex V | 0.22 | 0.21–0.62 |
| Citrate synthase (CS) (mU/mg protein) | 150.00 | 92–247 |
Table 2.
Oxygraphic analysis of patient's fibroblasts. Results of oxygraphic analysis were normal except for a decreased pyruvate/succinate respiration rate (bold).
| Oxygraphic analysis | Patient (pmol O2/s) | Reference values (pmol O2/s) |
|---|---|---|
| Pyruvate respiration (complex I dependent respiration) | 44.2 | 40–86 |
| Succinate respiration (complex II dependent respiration) | 62.9 | 35–70 |
| Max. respiration | 85.9 | 62–110 |
| Max. uncoupled respiration | 120.1 | 75–143 |
| Complex IV dependent respiration | 129.5 | 67–162 |
| Pyruvate/succinate respiration rate | 0.7 | 0.86–1.42 |
3.4. Triheptanoin trial
3.4.1. Safety of triheptanoin
Triheptanoin was well tolerated by our patient. Except for frequent hiccups no adverse events were reported. During the entire treatment period she did not suffer from any metabolic decompensation. Prior to receiving triheptanoin she suffered from several metabolic decompensations leading to six emergency consultations and/or hospitalisations. During the fifth month of the trial, she was hospitalized only once to install a PEG tube.
3.4.2. Clinical efficacy of triheptanoin
Under treatment with triheptanoin, the patient's weight normalized, while microcephaly persisted (Supplementary Fig. 1). According to the “Manual Ability Classification System (MACS)” and the „Gross Motor Functions Classification System (GMFCS)“ her manual skills and gross motor functions remained constant over time and slightly improved, respectively (data not shown) (18). To receive a more detailed view on changes of her choreatic and hyperkinetic movement disorders during the treatment period with triheptanoin, video ratings were performed and analysed (Fig. 2 and Supplementary Table 2). Her movements (e.g., reaching, grasping, use of both arms) got more purposeful over time. During treatment, trunk control as well as her ability to sit and stand improved. Furthermore, progress in verbal skills and functional play was observed. “Clinical Global Impression (CGI)” assessments revealed an improvement over time. Taken together several objective criteria revealed a beneficial effect of triheptanoin on the patient's clinical, i.e. neurologic condition. These findings were further substantiated by the parents' observations. The girl's progress could be supported with the aid of lower leg orthosis, Norsk Funktion-walker orthosis and a special wheel chair. According to the parents, the patient steadily improved her behaviour during the treatment period. They reported an increased attention span and more interactions with her environment.
Fig. 2.

Video ratings during the first twelve months of trial. Her overall motor skills got better over time. For details, including improved items, see supplementary table 2.
3.4.3. Biochemical effects of triheptanoin
Before and upon triheptanoin treatment, repeated laboratory investigations revealed several normal liver and renal function tests. Creatinine kinase was occasionally elevated irrespective of treatment (n = 23; range 79–304 U/L; reference <149). Plasma ammonia levels remained (close to) normal throughout the observation period (n = 14; range 31–62 μmol/L; reference range 11–51 μmol/L).
Plasma acylcarnitine profiles are reflective of the intracellular metabolization of triheptanoin (19). Analysis of plasma acylcarnitine profiles (n = 15; 3 measurements before and 12 under triheptanoin treatment) revealed slightly increased C3 acylcarnitine levels under triheptanoin treatment indicating cellular uptake and metabolism (Fig. 3). Free carnitine (C0) levels as well as the most abundant short chain acylcarnitines C2, C4 and C5 did not change significantly after introduction of triheptanoin, whereas C18 modestly decreased (Fig. 3). Total carnitine levels remained stable throughout the triheptanoin treatment period (data not shown).
Fig. 3.

Analysis of plasma acylcarnitine profiles (n = 15; 3 measurements before and 12 under triheptanoin treatment). Shown is a scatter dot plot and the median concentration of free carnitine (C0) and some of the most abundant acylcarnitines C2, C3, C4, C5 and C18. Plasma C3 acylcarnitine levels increased in response to triheptanoin treatment indicating cellular uptake and metabolism (p = 0.0566; Student's t-test) and C18 decreased significantly (p = 0.0297). In contrast, none of the other acylcarnitines changed significantly. ULN, upper limit of normal; LLN, lower limit of normal.
Analysis of urinary organic acid profiles (n = 28; 12 measurements before and 16 under triheptanoin treatment) revealed normal concentrations of malate, fumarate and succinate despite several metabolic decompensations before triheptanoin treatment (Fig. 4). Malate excretion marginally increased upon triheptanoin treatment but remained in the normal range (Fig. 4). Except for pimelate, which slightly increased upon triheptanoin treatment as expected, the vast majority of the other 63 analysed organic acids showed no (relevant) increase or change (data not shown).
Fig. 4.

Analysis of urinary organic acid profiles (n = 28; 12 measurements before and 16 under triheptanoin treatment). Shown is a scatter dot plot and the median excretion of malate, fumarate and succinate. While malate excretion marginally increased upon triheptanoin treatment (p = 0.0116; Student's t-test), fumarate and succinate remained unchanged. ULN, upper limit of normal.
Analysis of plasma amino acid profiles (n = 14; 3 measurements before and 11 under triheptanoin treatment) revealed normal levels of serine and glycine throughout the observation period whereas glutamine and threonine levels slightly increased upon triheptanoin treatment (Fig. 5). None of the other 38 analysed amino acids showed any significant abnormalities or response to triheptanoin (data not shown).
Fig. 5.

Analysis of plasma amino acid profiles (n = 14; 3 measurements before and 11 under triheptanoin treatment). Shown is a scatter dot plot and the median concentration of serine, glycine, glutamine and threonine. While all serine and glycine values were in the reference range and no change was observed in response to triheptanoin treatment, glutamine and threonine levels increased slightly above the reference range. ULN, upper limit of normal; LLN, lower limit of normal.
Strikingly, plasma lactate levels decreased significantly under triheptanoin treatment while the ratio of lactate/pyruvate did not alter significantly (Fig. 6).
Fig. 6.

Plasma lactate concentrations before and under treatment with triheptanoin. Shown is a scatter dot plot and the median lactate levels which significantly decreased under treatment with triheptanoin (p = 0.001; Student's t-test) while the ratio lactate/pyruvate did not change significantly. ULN, upper limit of normal; LLN, lower limit of normal.
4. Discussion
MDH2D is an ultra-rare disease which has been described in only three patients to date (1). Metabolic decompensation occurred in context of infections and other catabolic situations. Our patient's phenotype is in accordance with the above-mentioned patients (1). Clinical presentation of MDH2D is rather unspecific and resembles mitochondrial defects. Brain MRI revealed nonspecific findings including atrophy of the brain and delayed myelination (Fig. 1). A sensitive but unspecific biochemical marker for MDH2D is an elevated lactate in plasma and CSF. Interestingly all four patients described thus far (including our patient) share a common mutation on one allele of the MDH2 gene (c.398C>T, p.(Pro133Leu); see Supplementary Table 1) (1).
Defects in the TCA cycle such as MDH2D limits the flux of pyruvate to complex I and consequently reduces complex I dependent respiration (20). In contrast succinate as a complex II substrate can be directly fed into complex II avoiding the TCA cycle (21). This is in line with the decreased pyruvate-/succinate-respiration-ratio detected in oxygraphic analysis in our patient's cultured fibroblasts (Table 2).
MDH2D causes an energy undersupply due to the different role of the enzyme. On one hand it leads to an interruption of the TCA cycle and mitochondrial NADH production. As part of the MAS, MDH2 is furthermore essential for the intracellular NADH/NAD+ homeostasis. The net effect of the MAS is the transfer of NADH generated in the cytosol into the mitochondria. By doing so the MAS restores the cytosolic NAD+ pool and simultaneously enables the intramitochondrial re-oxidation of NADH to NAD+. Mutations in MDH2 therefore also impair the transfer of cytosolic NADH into the mitochondria and they impair the cytosolic regeneration of NAD+ from NADH [1], [2]. One of the main biochemical consequences of MDH2D is an increased conversion of pyruvate into lactate by the lactate dehydrogenase which not only enables (compensatory) re-oxidation of cytosolic NADH into NAD+ but also increases lactate levels (as observed in our and the three previously reported patients, Fig. 6) [1], [2].
A therapeutic strategy to attenuate the effects of MDH2D is to increase the amount of available substrates in the mitochondria to ensure a proper flux of energy through the TCA cycle and to improve the intracellular NADH/NAD+ homeostasis by bypassing the MAS (Fig. 7). Using MCTs as a treatment for MAS defects has been suggested (2).
Fig. 7.
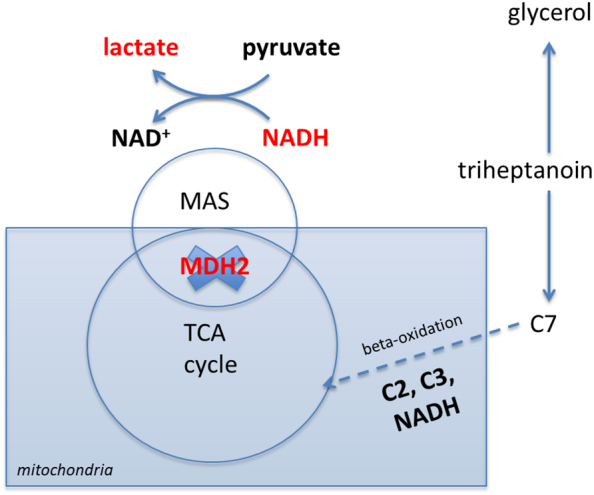
Scheme illustrating MDH2 deficiency (MDH2D) and selected effects of triheptanoin. MDH2 is located in the mitochondria and is part of the TCA cycle and the malate-aspartate shuttle (MAS). MDH2 as part of the MAS is required for the transfer of NADH generated in the cytosol into the mitochondria. MDH2 as part of the TCA cycle converts malate into oxaloacetate thereby generating (intramitochondrial) NADH (not shown). In MDH2D (illustrated as a blue cross through the MDH2 enzyme) cytosolic NADH cannot be transferred into the mitochondria and less NADH is generated intramitochondrially. As a consequence, NADH is accumulating in the cytosol. One cellular adaptation in MDH2D is a compensatory and increased conversion of pyruvate into lactate in order to re-oxidize cytosolic NADH into NAD+. Triheptanoin has several potential modes of action (for details see discussion). Here, we schematically illustrate the breakdown of triheptanoin into glycerol and C7 fatty acid. While glycerol has several mechanisms of action (not illustrated here), C7 enters the mitochondria directly and is degraded in the beta-oxidation to C2 and C3 thereby enhancing the intramitochondrial NADH generation. C2 and C3 are anaplerotic by entering the TCA cycle.
Therefore, a clinical trial with the odd-chain triglyceride triheptanoin known for its anaplerotic effect was introduced and well tolerated by our patient. The results of triheptanoin treatment were considered satisfactory. Video ratings showed an overall improvement in motor skills during the treatment period (Fig. 2 and Supplementary Table 2). After 12 months of therapy, parents reported an increased attention span, more constructive interactions with her environment as well as an improvement in her playing habits. On the other hand, clinical assessment applying MACS and GMFCS revealed only modest improvements. These classification systems are not sensitive enough to capture subtle skill improvements (18). In addition, the hyperkinetic movement disorder resulting from a metabolic stroke, is probably irreversible irrespective of the applied treatment.
While it is difficult to differentiate between the effects of triheptanoin treatment and the spontaneous remission the patient might have undergone during the observation period, it is impressive how her medical condition improved. Most strikingly during the entire treatment period she did not suffer from any metabolic decompensation unlike the 6 metabolic decompensations she experienced prior to treatment with triheptanoin. From a biochemical point of view the most striking result was a significant decrease in plasma lactate levels in response to triheptanoin (Fig. 6). A decrease in plasma lactate is most likely reflective of an improved cytoplasmic NADH/NAD+ ratio related to the therapy. Further studies are needed to evaluate the effect of triheptanoin in MDH2D.
An additional and potentially beneficial effect of triheptanoin treatment is the utilization of heptanoate for gluconeogenesis (22). Further, glycerol as part of triheptanoin can also be used for pyruvate production (9). In the mitochondria pyruvate may be used for anaplerosis via the pyruvate carboxylase to yield oxaloacetate [20], [23] entering the TCA cycle. Another anaplerotic effect of triheptanoin could be the increased amount of acetyl-CoA entering the TCA cycle thereby generating more intramitochondrial NADH. This explanation has been previously suggested by others and can be best illustrated when splitting the TCA cycle into two functionally complementary mini-cycles [1], [17], [24]. Last but not least, the glycerol could be entering the glycerol-3-phosphate shuttle, another mechanism allowing re-oxidation of NAD+ from NADH in the cytosol (2).
Taken together, triheptanoin has a therapeutic benefit on brain energy deficit caused by impaired TCA cycle and/or glucose metabolism [8], [9]. In MDH2D it directly supplies the TCA cycle with intermediates (acetyl-CoA, propionyl-CoA and glycerol). Further, triheptanoin partially bypasses the MAS defect by beta-oxidation of C7 and consecutive intramitochondrial generation of NADH (Fig. 5). Another mechanism of action related to triheptanoin is the provision of an alternative source of energy in terms of ketone bodies. These help stabilizing the membrane potential in epileptic brains and modulate the balance between catabolic and anabolic pathways through anaplerosis of the TCA cycle in different diseases [7], [8], [9], [11], [19], [25].
Other MDH deficient patients have been treated with a ketogenic diet, which reduced the frequency of epileptic seizures. Two of the patients described in literature are still alive. One died due to a metabolic decompensation (2). Compared to ketogenic diet, triheptanoin is far less restrictive and very simple in its administration leading to a better compliance [7], [8], [9], [10], [11], [25].
5. Conclusion
The MDH2 deficient patient described in this case study benefited from a drug trial with triheptanoin. A target dose of 55 mL was well tolerated by the patient. Weight and height development normalized under treatment, her social abilities and medical condition improved. Triheptanoin might be a therapeutic approach for MDH2D. More research is needed to clarify how triheptanoin interferes with the disease, to consolidate the present findings and to further support its therapeutic effectiveness.
Declaration of Competing Interest
None.
Acknowledgments
Our thank goes to the patient and her parents for their trust in us and their participation in this drug trial. We thank Ultragenyx Pharmaceutical Inc. for providing us with triheptanoin (Compassionate Use Program number UX007-CU302).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ymgmr.2021.100814.
Appendix A. Supplementary data
Supplementary material
References
- 1.Ait-El-Mkadem S., Dayem-Quere M., Gusic M., Chaussenot A., Bannwarth S., Francois B. Mutations in MDH2, encoding a Krebs cycle enzyme, cause early-onset severe encephalopathy. Am. J. Hum. Genet. 2017;100(1):151–159. doi: 10.1016/j.ajhg.2016.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van Karnebeek C.D.M., Ramos R.J., Wen X.Y., Tarailo-Graovac M., Gleeson J.G., Skrypnyk C. Bi-allelic GOT2 mutations cause a treatable malate-aspartate shuttle-related encephalopathy. Am. J. Hum. Genet. 2019;105(3):534–548. doi: 10.1016/j.ajhg.2019.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lu M., Zhou L., Stanley W.C., Cabrera M.E., Saidel G.M., Yu X. Role of the malate-aspartate shuttle on the metabolic response to myocardial ischemia. J. Theor. Biol. 2008;254(2):466–475. doi: 10.1016/j.jtbi.2008.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Borst P. The malate-aspartate shuttle (Borst cycle): how it started and developed into a major metabolic pathway. IUBMB Life. 2020;72(11):2241–2259. doi: 10.1002/iub.2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Broeks M.H., van Karnebeek C.D.M., Wanders R.J.A., Jans J.J.M., Verhoeven-Duif N.M. Inborn disorders of the malate aspartate shuttle. J. Inherit. Metab. Dis. 2021;44(4):792–808. doi: 10.1002/jimd.12402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Minarik P., Tomaskova N., Kollarova M., Antalik M. Malate dehydrogenases–structure and function. Gen. Physiol. Biophys. 2002;21(3):257–265. [PubMed] [Google Scholar]
- 7.Roe C.R., Mochel F. Anaplerotic diet therapy in inherited metabolic disease: therapeutic potential. J. Inherit. Metab. Dis. 2006;29(2–3):332–340. doi: 10.1007/s10545-006-0290-3. [DOI] [PubMed] [Google Scholar]
- 8.Wehbe Z., Tucci S. Therapeutic potential of triheptanoin in metabolic and neurodegenerative diseases. J. Inherit. Metab. Dis. 2020;43(3):385–391. doi: 10.1002/jimd.12199. [DOI] [PubMed] [Google Scholar]
- 9.Mochel F. Triheptanoin for the treatment of brain energy deficit: a 14-year experience. J. Neurosci. Res. 2017;95(11):2236–2243. doi: 10.1002/jnr.24111. [DOI] [PubMed] [Google Scholar]
- 10.Calvert S., Barwick K., Par M., Ni Tan K., Borges K. A pilot study of add-on oral triheptanoin treatment for children with medically refractory epilepsy. Eur. J.Paediatr. Neurol. 2018;22(6):1074–1080. doi: 10.1016/j.ejpn.2018.07.014. [DOI] [PubMed] [Google Scholar]
- 11.Tan K.N., Simmons D., Carrasco-Pozo C., Borges K. Triheptanoin protects against status epilepticus-induced hippocampal mitochondrial dysfunctions, oxidative stress and neuronal degeneration. J. Neurochem. 2018;144(4):431–442. doi: 10.1111/jnc.14275. [DOI] [PubMed] [Google Scholar]
- 12.Shirley M. Triheptanoin: first approval. Drugs. 2020;80(15):1595–1600. doi: 10.1007/s40265-020-01399-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vockley J., Marsden D., McCracken E., DeWard S., Barone A., Hsu K. Long-term major clinical outcomes in patients with long chain fatty acid oxidation disorders before and after transition to triheptanoin treatment–A retrospective chart review. Mol. Genet. Metab. 2015;116(1–2):53–60. doi: 10.1016/j.ymgme.2015.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jackson C.B., Nuoffer J.M., Hahn D., Prokisch H., Haberberger B., Gautschi M. Mutations in SDHD lead to autosomal recessive encephalomyopathy and isolated mitochondrial complex II deficiency. J. Med. Genet. 2014;51(3):170–175. doi: 10.1136/jmedgenet-2013-101932. [DOI] [PubMed] [Google Scholar]
- 15.Hertig D., Felser A., Diserens G., Kurth S., Vermathen P., Nuoffer J.M. Selective galactose culture condition reveals distinct metabolic signatures in pyruvate dehydrogenase and complex I deficient human skin fibroblasts. Metabolomics. 2019;15(3):32. doi: 10.1007/s11306-019-1497-2. [DOI] [PubMed] [Google Scholar]
- 16.Richards S., Aziz N., Bale S., Bick D., Das S., Gastier-Foster J. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet.Med. 2015;17(5):405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rustin P., Bourgeron T., Parfait B., Chretien D., Munnich A., Rotig A. Inborn errors of the Krebs cycle: a group of unusual mitochondrial diseases in human. Biochim. Biophys. Acta. 1997;1361(2):185–197. doi: 10.1016/s0925-4439(97)00035-5. [DOI] [PubMed] [Google Scholar]
- 18.Paulson A., Vargus-Adams J. Overview of four functional classification systems commonly used in cerebral palsy. Children. 2017;4(4) doi: 10.3390/children4040030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Willis S., Stoll J., Sweetman L., Borges K. Anticonvulsant effects of a triheptanoin diet in two mouse chronic seizure models. Neurobiol. Dis. 2010;40(3):565–572. doi: 10.1016/j.nbd.2010.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gray L.R., Tompkins S.C., Taylor E.B. Regulation of pyruvate metabolism and human disease. Cell.Mol. Life Sci. 2014;71(14):2577–2604. doi: 10.1007/s00018-013-1539-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jodeiri Farshbaf M., Kiani-Esfahani A. Succinate dehydrogenase: prospect for neurodegenerative diseases. Mitochondrion. 2018;42:77–83. doi: 10.1016/j.mito.2017.12.002. [DOI] [PubMed] [Google Scholar]
- 22.Marin-Valencia I., Good L.B., Ma Q., Malloy C.R., Pascual J.M. Heptanoate as a neural fuel: energetic and neurotransmitter precursors in normal and glucose transporter I-deficient (G1D) brain. J. Cerebral Blood Flow Metabol. 2013;33(2):175–182. doi: 10.1038/jcbfm.2012.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li M., Zhou S., Chen C., Ma L., Luo D., Tian X. Therapeutic potential of pyruvate therapy for patients with mitochondrial diseases: a systematic review. Therap.Adv.Endocrinol.Metabol. 2020;11 doi: 10.1177/2042018820938240. 2042018820938240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yudkoff M., Nelson D., Daikhin Y., Erecinska M. Tricarboxylic acid cycle in rat brain synaptosomes. Fluxes and interactions with aspartate aminotransferase and malate/aspartate shuttle. J. Biol. Chem. 1994;269(44):27414–27420. [PubMed] [Google Scholar]
- 25.Reid C.A., Mullen S., Kim T.H., Petrou S. Epilepsy, energy deficiency and new therapeutic approaches including diet. Pharmacol. Ther. 2014;144(2):192–201. doi: 10.1016/j.pharmthera.2014.06.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material