

SUMMARY
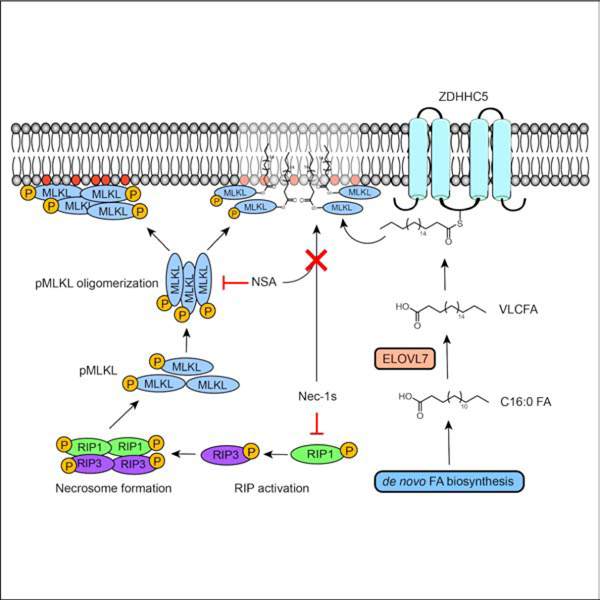
Necroptosis is a form of cell death characterized by receptor interacting protein kinase (RIPK) activity and plasma membrane permeabilization via mixed lineage kinase like protein (MLKL). This permeabilization is responsible for the inflammatory properties of necroptosis. We previously showed that very long chain fatty acids (VLCFAs) are functionally involved in necroptosis, potentially through protein fatty acylation. Here, we define the scope of protein acylation by saturated VLCFAs during necroptosis. We show that MLKL and phosphoMLKL, key for membrane permeabilization, are exclusively acylated during necroptosis. Reducing the levels of VLCFAs decreases their membrane recruitment, suggesting that acylation by VLCFAs contributes to their membrane localization. Acylation of phosphoMLKL occurs downstream of phosphorylation and oligomerization and appears to be, in part, mediated by ZDHHC5 (a palmitoyl transferase). We also show that disruption of endosomal trafficking increases cell viability during necroptosis, possibly by preventing recruitment, or removal, of phosphoMLKL from the plasma membrane.
Graphical Abstract
eTOC
Pradhan et al. show that MLKL and pMLKL are acylated by saturated very long chain fatty acids during necroptosis and that the disruption of endocytosis decreases the levels of membrane bound pMLKL and MLKL, resulting a rescue from cell death during this process.
INTRODUCTION
Programmed cell death is a critical component for maintaining cellular homeostasis. Imbalance between cell proliferation and cell death is linked to many phenotypes, including cancer formation, neurodegenerative disorders and inflammation (Thompson, 1995). Multiple forms of regulated cell death that are executed by distinct pathways have recently been characterized. A fundamental phenotypic difference between apoptosis and other regulated forms of cell death is the permeabilization of the plasma membrane, which leads to release of intracellular components and results in an inflammatory phenotype (Zhang et al., 2017).
Necroptosis is one of these inflammatory programmed cell death pathways. It is dependent on the activity of receptor interacting protein kinase 1, 3 (RIPK1, RIPK3) and mixed lineage kinase like protein (MLKL) phosphorylation when caspase activity is reduced (Holler et al., 2000; Degterev et al., 2005). Necroptosis exhibits phenotypic hallmarks of necrosis including cellular swelling, loss of plasma membrane integrity, and the release of intracellular content (Holler et al., 2000; Degterev et al., 2005). Closely linked to this membrane permeabilization and associated inflammatory response, necroptotic cell death has been observed in many diseases, including ischemia/reperfusion injuries (Lau et al., 2013) and myocardial infarction (Luedde et al., 2014).
MLKL oligomerization and translocation to the plasma membrane is required for necroptosis (Cai et al., 2014). MLKL consists of a C-terminal kinase-like domain that interacts with RIPK3, which results in the phosphorylation of MLKL (Sun et al., 2012). This phosphorylation triggers the oligomerization and translocation of pMLKL to the plasma membrane (Wang et al., 2014). The membrane recruitment of the oligomer is mediated by electrostatic interactions between positively charged regions of pMLKL and negatively charged phosphatidylinositol phosphate-rich domains of the plasma membrane (Quarato et al., 2016). While it seems clear from a number of studies that the membrane association of pMLKL oligomers are important for membrane permeabilization (Cai et al., 2014; Chen et al., 2014; Xia et al., 2016), the mechanism of membrane permeabilization induced by pMLKL oligomers is not completely understood. It is unclear how pMLKL oligomers impact membrane packing, whether other chemical transformations that pMLKL undergo impact membrane association, and if the presence of the oligomers induces further phosphatidylinositol recruitment to the protein binding site.
In parallel to its plasma membrane recruitment via canonical RIPK3/RIPK1 activity during necroptosis, vesicular trafficking of MLKL plays a critical role in its cellular localization. MLKL can modulate endosomal trafficking, including constitutive early and late endosomes that can fuse with lysosomes or the plasma membrane (Yoon et al., 2017). This trafficking modulation can be independent of the MLKL phosphorylation state, but is enhanced after phosphorylation and may result in the removal of pMLKL from the plasma membrane leading to delay in membrane permeabilization and cell death during necroptosis (Yoon et al., 2017).
Motivated by lipids’ role in numerous membrane-related transformations, our laboratory has investigated the role of lipids in necroptosis, specifically during membrane permeabilization. We found that saturated very long chain fatty acids (VLCFAs) accumulated during necroptosis via activation of fatty acid biosynthesis and elongation in two different necroptosis models (Parisi et al., 2017). We also showed that VLCFAs have a higher propensity to disturb membrane packing in liposomes and in cells most likely due to interleaflet interdigitation (Parisi et al., 2019). One intriguing observation was that in liposomes and during molecular dynamics simulations, high concentrations of VLCFAs were required to induce membrane permeabilizations whereas the intracellular concentration required for plasma membrane permeabilization was much lower. These observations suggest that VLCFAs can achieve high local membrane concentrations in cells during necroptosis. We envisioned that one way in which VLCFAs can reach high local membrane concentration was through their incorporation to proteins and the recruitment of the protein-lipid complex to distinct membrane locales. Since fatty acylation by saturated VLCFAs is understudied, we utilized ω-alkynyl VLCFAs to investigate the covalent modification of proteins with VLCFA in both control and necroptotic cells and found that certain proteins may exhibit increased labeling with VLCFAs during necroptosis (Parisi et al., 2019).
In this work, we identify the proteins that are acylated by VLCFAs through the use of a clickable fatty acid analog and investigated the involvement of acylation by VLCFAs in necroptosis. We first carried out quantitative proteomics with a unique IonStar approach (Shen et al., 2018b) to identify proteins that are differentially fatty acylated during this process. We observed that there was an overall decrease in proteins that are acylated by VLCFAs (referred to as VLCFAcylated here on) during necroptosis. However, importantly, MLKL and pMLKL were among the proteins that were VLCFAcylated in necroptotic cells, but not in control cells. Enrichment analysis highlighted the involvement of the endocytic/lysosomal pathway during necroptosis. Perturbing the endocytic trafficking using a small molecule inhibitor resulted in increased cell viability and decreased the levels of membrane localized pMLKL suggesting endosomal trafficking plays a role in localization of pMLKL. Our results show the differential protein fatty acylation by a VLCFA during necroptosis and suggests an acylation-dependent mechanism of membrane recruitment and maintenance of pMLKL in this process.
RESULTS
Differential incorporation of ω-alkynyl C20:0 fatty acid during necroptosis.
Our previous studies on establishing the changes in the lipid landscape during necroptosis showed that saturated VLCFAs are strongly upregulated during this process (Parisi et al., 2017) and are involved in membrane permeabilization (Parisi et al., 2019). We have also shown that these VLCFAs could be incorporated into proteins and this fatty acylation might play a role in necroptosis, for instance by targeting of VLCFAs to certain membrane regions resulting in high local concentrations, mediated by the protein-membrane interactions (Parisi et al., 2019). Here, in order to investigate the role of fatty acylation by VLCFAs and identify the proteins modified by VLCFAs during necroptosis, we analyzed the proteins that are modified by a representative VLCFA during this process using quantitative proteomics. We induced necroptosis in HT-29 human colorectal adenocarcinoma epithelial cells with BV6/zVAD-FMK/TNF-α treatment, confirmed by the decrease in cell viability (Figure 1A) and membrane localization of pMLKL (Figure 1B, Figure S1A). Following that, we used ω-alkynyl fatty acid probes to study fatty acylation during necroptosis. We previously reported that C20:0 FA is the shortest saturated VLCFA that accumulated during necroptosis (Parisi et al., 2017) and it is the shortest fatty acid that accumulated when we overexpressed ELOVL7, which induces membrane permeabilization (Parisi et al., 2019). We have also shown that qualitatively C20 and C22 alkFAs labeling are similar to each other (Parisi et al., 2019). Hence, in this study, we chose ω-alkynyl C20:0 fatty acid (C20 alkFA) as a representative VLCFA due to its improved solubility among VLCFAs to identify proteins that are modified with this probe using delivery and enrichment protocols we described previously (Parisi et al., 2019) (Figure 1C, n=3 for each condition, see STAR methods). The synthesis and characterization of C20 alkFA were described in our earlier work (Parisi et al., 2019). After collecting cell pellets, samples were normalized based on their protein content using Bradford assay and membrane fractions were isolated using ultracentrifugation. We installed a biotin reporter by copper catalyzed alkyne-azide cycloaddition (CuAAC) and captured proteins that are covalently modified by C20 alkFA using neutravidin resin (referred to as enriched portion from here on). We note a second normalization step prior to loading the neutravidin resin to ensure equal amounts of protein are used for each condition using BCA assay. We then carried out quantitative proteomics in order to identify proteins that are acylated by C20 alkFA and compared their levels in control and necroptotic cells. Briefly, once the proteins were enriched on the neutravidin beads, they were resuspended in detergent containing buffers and were then subjected to a surfactant-aided precipitation/on-bead digestion procedure modified from a recently published method (Shen et al., 2018a) (see Materials and Methods for details, Figure 1C). Derived tryptic peptides were analyzed by a well-optimized trapping nano LC-Orbitrap mass spectrometry system. Each sample was analyzed twice, once by Orbitrap (OT) and once by Ion Trap (IT) to allow accurate and sensitive peptide detection as well as cross validation of protein identification results. Proteomic quantification was accomplished by IonStar, an in-house developed MS1 ion current-based quantitative proteomics method (Figure 1C, see Materials and methods for details) (Shen et al., 2018b). A total of 1672 proteins were quantified with high precision and no missing data across samples in the same condition.
Figure 1. Induction of necroptosis and workflow of C20 fatty acid acylation and proteomics.
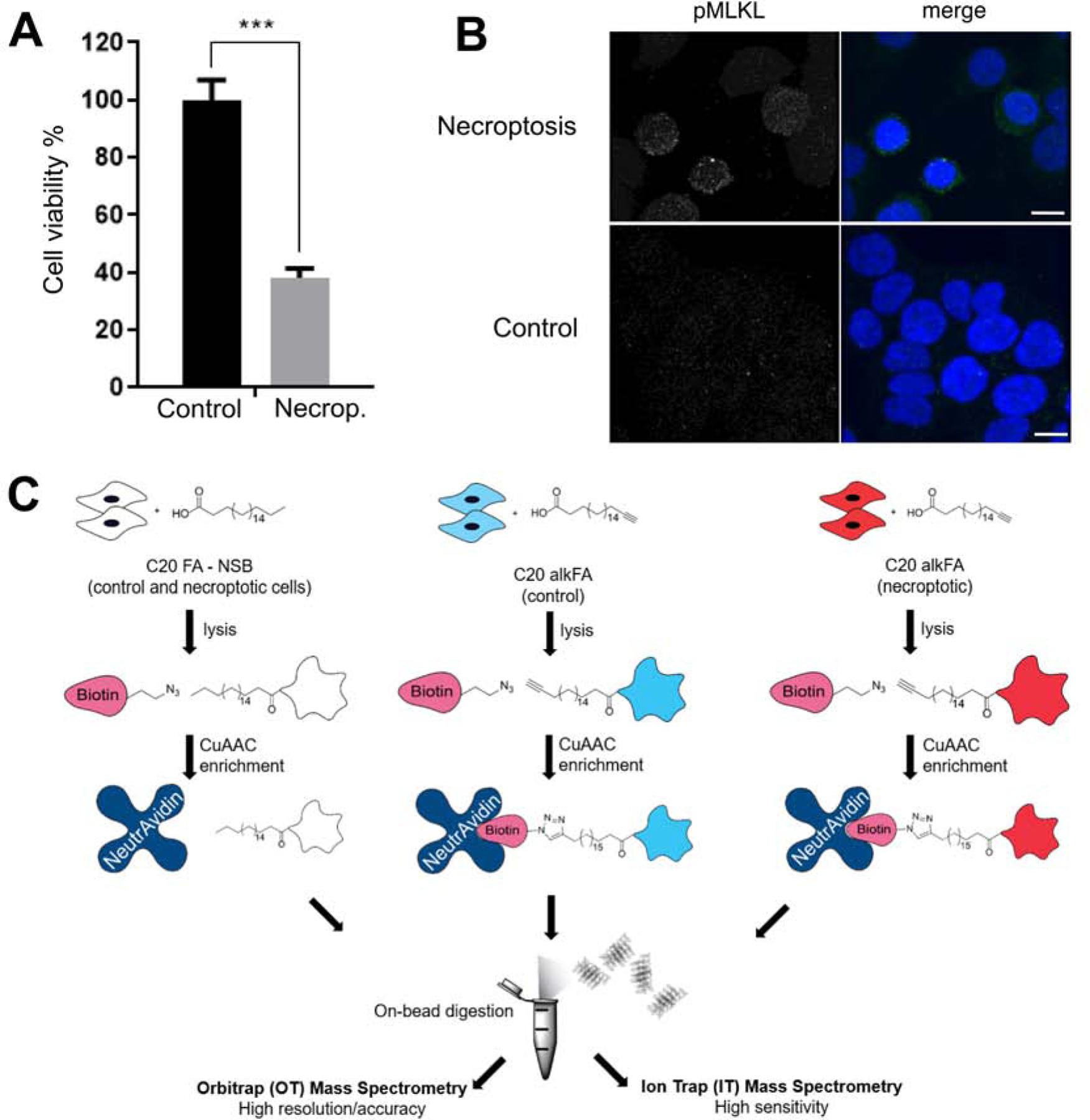
(A) Cell viability decreases as HT-29 cells undergo necroptosis. Necroptosis is induced with BV6/zVAD-FMK/TNF-α treatment. Data represent mean ± 1 SD; n=5. *** represents p < 0.001. (B) Cellular distribution of pMLKL during necroptosis. Sample 3D project images of pMLKL immunostaining in necroptotic (top) or control (bottom) cells show an increase in pMLKL staining, mainly concentrated at the plasma membrane. Merge images include DAPI nuclear staining (blue) and pMLKL (green). Scalebar represents 10 μm. (C) HT-29 cells were treated with endogenous C20:0 FA or C20:0 alkFA for 3h to account for non-specific and specific interactions respectively. Control and necroptotic cells were then membrane fractionated, subjected to CuAAC and installed with a biotin reporter. Biotinylated proteins were captured on neutravidin and subjected to on-bead trypsin digestion followed by quantitative proteomics. See also Figure S1.
The scatter plots in Figure S1 show the correlation between the mean intensities (calculated by summing up the area under the curve of all peptides inferred to a protein) of different biological replicates of each protein detected in control and necroptotic cells (Figure S1B). Based on the strong correlation between OT and IT dataset (R2>0.99 for control and necroptotic samples), we concluded that our measurements reflect biological rather than technical variation.
In order to eliminate proteins that are enriched due to non-specific interactions between the proteins and neutravidin resin, we treated cells with endogenous C20:0 FA and subjected them to the same enrichment and analysis protocols (Figure 1C, C20 FA-NSB). We excluded any proteins that we identified in these non-specific binding samples as they should not interact with neutravidin resin due to the lack of biotin modification. Out of the total 1672 proteins that were quantified in samples enriched from control and necroptotic cells, 1267 were detected with higher abundance in control cells and 405 were detected with higher abundance in necroptotic condition (see Table S1 for complete list of proteins identified and Table S2 for proteins that show differential acylation in necroptosis). Figure 2A shows the difference in mean intensities of proteins detected (total of 1672 proteins) in control and necroptotic cells in OT (x axis) and IT (y axis) platforms. Consistent with the high correlations we report in Figure S1 for the intensities detected in these platforms, we also observe a strong correlation between the differences in mean intensities for detected proteins in control and necroptotic conditions showing that these differences reflect biological variation rather than technical ones. Furthermore, an interesting observation was that a large number of proteins appeared to be only acylated in control (561, dark blue in Figure 2B) or in necroptotic (158, dark red in Figure 2B) cells (Table S1). Based on these results, it is clear that overall there are fewer proteins acylated by VLCFA during necroptosis (Figure 2B, Figure S2), however there is a distinct group of 158 proteins that are VLCFAcylated exclusively in necroptotic cells.
Figure 2. Quantitative analysis of acylated proteome in necroptosis.
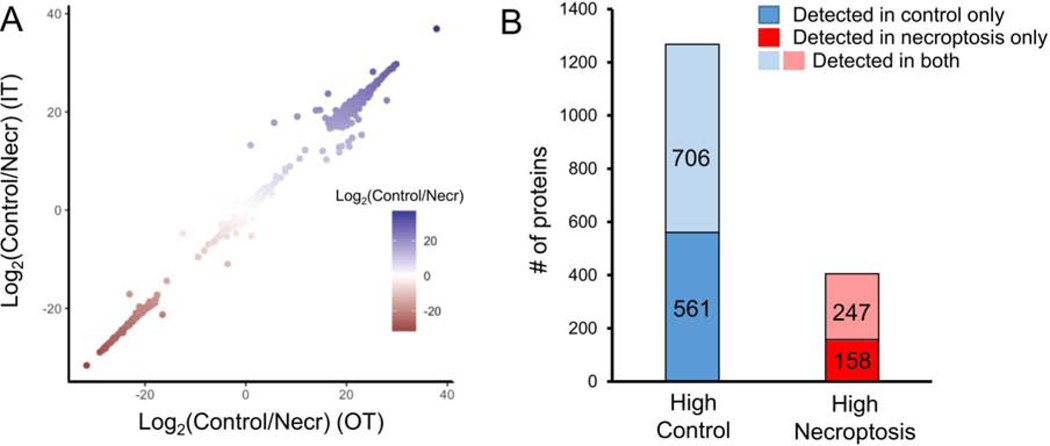
(A) High correlation between the mean intensities of acylated proteins detected in control and necroptotic cells in Orbitrap Lumos (x axis) and Ion trap (y axis) platforms. Strong correlation between difference in mean intensities indicates biological variation rather than technical variation among the proteins detected in control and necroptotic cells. (B) Bar plot shows the total number of proteins (1672) undergoing acylation in either control, necroptotic or both conditions. Blue and red bars represent the number of proteins with high abundance in control or necroptotic cells, respectively. A higher number of proteins appear acylated in only control (561) as compared to only necroptosis (158). Overall, fewer proteins undergo acylation in necroptosis (405) as compared to control (1267). See also Figure S2 and Table S1.
MLKL and pMLKL are acylated during necroptosis.
A total of 1332 proteins showed differential acylation by VLCFAs significantly (p < 0.01, Table S2) during necroptosis: the acylation of 285 proteins were upregulated during necroptosis whereas 1047 showed a reduction in VLCFAcylation in the process (Table S2). Among the 285 proteins with increased VLCFAcylation during necroptosis, 158 were only detected in necroptotic cells. Based on our previous work suggesting increased VLCFA levels and protein fatty acylation could be involved in necroptosis (Parisi et al., 2017; Parisi et al., 2019), we initially turned our attention to proteins with increased VLCFAcylation during necroptosis.
Intriguingly, MLKL was one of these proteins which appeared to be modified by C20:0 alkFA only during necroptosis. Our proteomics data set is limited in distinguishing phosphopeptides from unmodified ones, therefore little information was obtained whether MLKL and/or pMLKL was acylated. Hence, we investigated the fatty acylation of MLKL and pMLKL using western blotting. We enriched fatty acylated proteins from control and necroptotic cells as described above and analyzed protein content using western blotting. We detected both MLKL and pMLKL in necroptotic cells but not in control cells suggesting that both MLKL and pMLKL can be fatty acylated during necroptosis and that their acylation is not appreciable in the absence of necroptotic activity (Figure 3A). In order to understand the nature of acylation, we treated control and necroptotic cells with C20:0 alkFA, fractionated membrane proteins and installed Biotin-azide. We then treated the lysates with hydroxylamine (NH2OH) which cleaves thioester linkages (Thinon et al., 2018). We note that NH2OH treatment did not affect the overall levels of MLKL, pMLKL and calnexin (Figure S3A), showing that the treatment did not cause protein degradation. We then carried out protein enrichment through neutravidin beads. When we subjected the bead-bound proteins to western blotting, MLKL and pMLKL signal from necroptotic samples completely disappeared with NH2OH treatment, suggesting that MLKL and pMLKL may be acylated via S-linkage (Figure 3B).
Figure 3. MLKL and pMLKL are acylated downstream of RIPK1 activation.
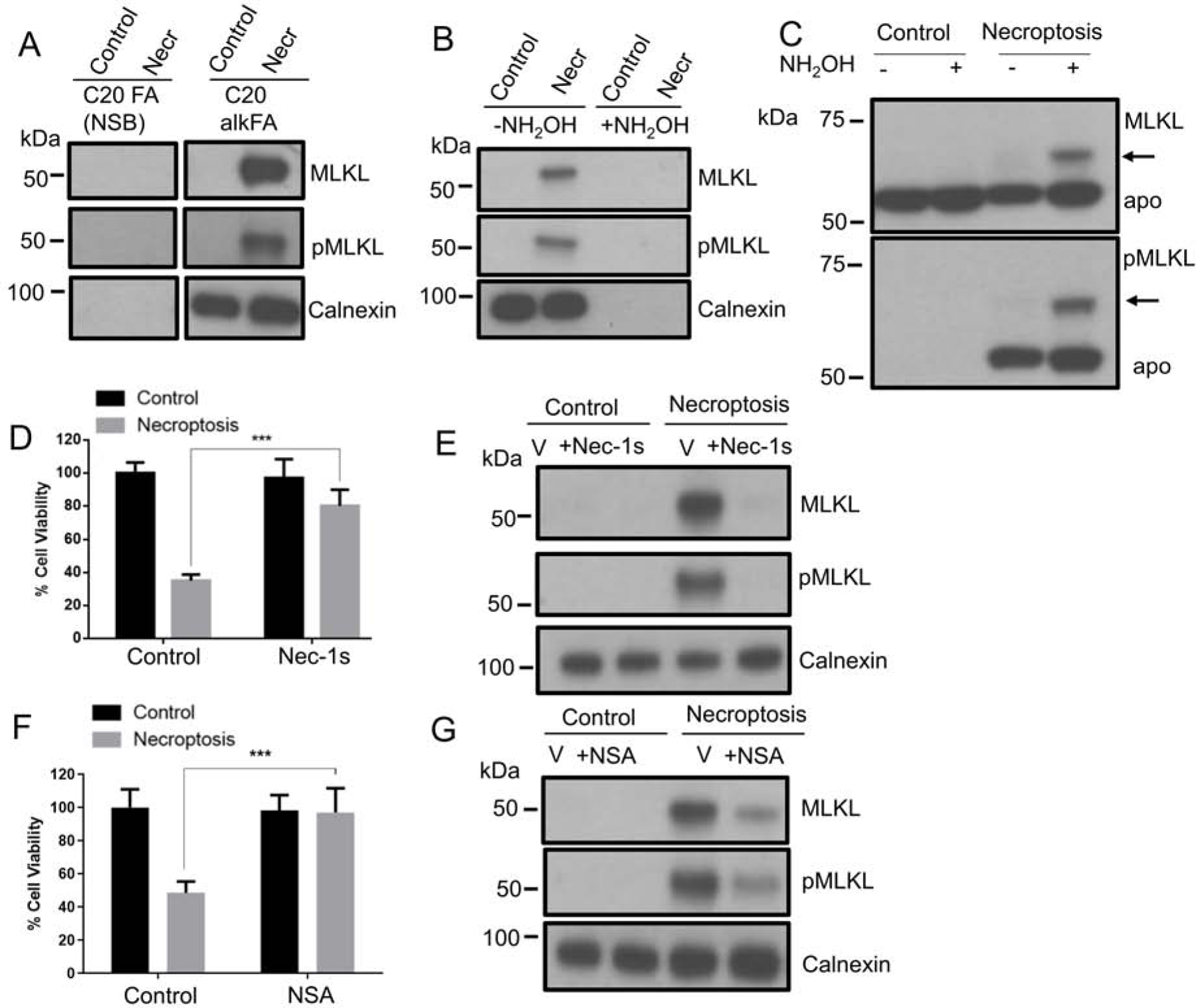
(A) Western blot analysis of proteins from C20 alkFA treated control and necroptosis cells. After cell lysis and Biotin-azide attachment via CuAAC, acylated proteins are enriched on neutravidin resin. The samples are blotted for MLKL and pMLKL and Calnexin. Calnexin is used as a loading control. Equal amounts of protein are used for enrichment of each condition (see Materials and Methods for details). We note that the treatments with C20 FA and C20 alkFA and the western blot analysis were carried out during the same experiment. Uncropped western blot image in shown in Figure S3F. (B) Acylation of MLKL and pMLKL with C20 alkFA show NH2OH sensitivity. Control and necroptotic cells treated C20 alkFA were membrane fractionated and subjected to CuAAC with Biotin-azide. The proteins were treated with 0.3 M NH2OH for 30 min, enriched using neutravidin beads, then blotted for MLKL and pMLKL and Calnexin. Calnexin is used as a positive control for NH2OH sensitivity. We note that NH2OH treatment does not cause protein degradation (Figure S3A). Loss of signal with NH2OH treatment indicates that MLKL and pMLKL acylation occurs primarily through S-linkages. (C) HT-29 cells were lysed and total cell lysates were subjected to APE with NEM, NH2OH, and mPEG-Mal, and compared with negative controls. The samples are blotted for MLKL and pMLKL. (D) 1 μM Nec-1s and (F) 1 μM NSA reduce necroptotic cell death. Percent cell viability of control, Nec-1s or NSA-treated and necroptotic cells are shown. Necroptosis is induced with BV6/zVAD-FMK/TNF-α treatment. Data represent mean ± 1 SD; n=5. *** represents p < 0.001. (E and G) Western blot analysis of C20 alkFA treated control and necroptotic cells in the presence of Nec-1s and NSA. Control and necroptotic cells treated with C20 alkFA and Nec-1s/NSA were membrane fractionated and subjected to CuAAC with Biotin-azide and enrichment using neutravidin beads. The proteins on the beads were blotted for MLKL and pMLKL. Equal amounts of protein are used for enrichment of each condition. Calnexin is used as loading control (see Materials and Methods for details). Nec-1s (E) and NSA (G) treatment significantly reduce VLCFAcylation of MLKL and pMLKL (see Figure S3B and D for quantification of band intensities). See also Figure S3.
To analyze the levels of acylated MLKL and pMLKL by endogenous fatty acids in cells we used acyl-PEG exchange (APE), a method in which NH2OH-sensitive thioesters are cleaved and exchanged with a mass tag (Percher et al., 2016; Percher et al., 2017). Using APE, we show that endogenous MLKL and pMLKL are S-fatty acylated (indicated by arrows on Figure 3C). Based on the mass shift we observe on SDS-PAGE (Percher et al., 2016; Percher et al., 2017), MLKL and pMLKL appear to be S-fatty acylated predominantly at a single cysteine residue. However, we cannot exclude the presence of multiple acylated forms that might be below our detection using western blotting. We note that the mass shift we observe in the absence of NH2OH (Figure 3C, lane 3) is likely due to the nonspecific alkylation as previously reported (Percher et al., 2017; Percher et al., 2016).
pMLKL acylation occurs downstream of RIPK1 activity and its membrane localization.
RIPK1 activity is critical for MLKL phosphorylation and its translocation to plasma membrane during necroptosis (Cai et al., 2014). In order to investigate whether the acylation of pMLKL occurs downstream of RIPK1 activation during necroptosis, we used a small molecule inhibitor of RIPK1, Necrostatin-1s (Nec-1s), which increased cell viability in the presence of necroptotic activity (Figure 3D). First, we demonstrate using APE that Nec-1s treatment completely blocks fatty acylation during necroptosis (Figure S3C). Second, to study VLCFAcylation we treated necroptotic cells and Nec-1s-protected cells with C20 alkFA, fractionated membrane proteins and installed Biotin-azide. We then enriched for proteins modified with C20 alkFA using neutravidin as described above. Figure 3E shows the levels of MLKL and pMLKL in the enriched portion in necroptotic and Nec-1s-protected cells compared to control cells. We detected very low MLKL and pMLKL in the protected group (~95% reduction in band intensity, p < 0.001, Figure S3B), suggesting that no appreciable fatty acylation of pMLKL takes place in protected cells. These results are important because it suggests that pMLKL is acylated downstream of RIPK1 activity (Figure 3E).
The next step in canonical necroptosis signaling after MLKL phosphorylation is the oligomerization of pMLKL, membrane recruitment, and binding. Using a similar approach as we describe above for inhibiting RIPK1 activity, we used a small molecule necrosulfonamide (NSA) (Sun et al., 2012) which prevents the oligomerization of pMLKL, membrane translocation and rescues cell death in necroptosis (Figure 3F). We treated necroptotic cells and NSA-protected cells with C20 alkFA and enriched for proteins that are acylated with C20 alkFA using neutravidin. Figure 3G shows the levels of acylated MLKL and pMLKL in necroptotic and NSA-protected cells compared to control cells. We detected a significant decrease in acylation of MLKL and pMLKL in the protected group (Figure 3G and Figure S3D, ~60% reduction in band intensity, p < 0.01) whereas their cellular levels did not change (as detected in whole lysates, Figure S3E), suggesting that the acylation of pMLKL primarily occurs downstream to its oligomerization.
ELOVL7 knockdown reduces the membrane recruitment of pMLKL during necroptosis.
Our results on the acylation of MLKL and pMLKL are based on exogenous delivery of fatty acid of clickable fatty acid analogs. We wanted to confirm that VLCFAcylation occurs in cells and during necroptosis without perturbing lipid levels by exogenous delivery. To achieve this, we tested whether the reduction of VLCFAs would impact the membrane recruitment of pMLKL during necroptosis. If our hypothesis that VLCFAcylation of pMLKL is functionally important for cell death during necroptosis is correct, we should see a decrease in pMLKL acylation (hence its membrane recruitment) and a rescue in cell death when the levels of VLCFAs are depleted. In our previous work (Parisi et al. 2017), we investigated the expression levels of different ELOVLs that might be responsible for the accumulation of saturated VLCFAs and found that only ELOVL1 and ELOVL7 were significantly upregulated. We focused here on ELOVL7 based on the magnitude of its upregulation increase (Parisi et al., 2017; Parisi et al., 2019) and its substrate specificity (Purdy et al., 2015; Tamura et al., 2009). We have shown a reduction of ELOVL7 (by shRNA) leads to reduced levels of saturated VLCFAs during necroptosis while its overexpression results in the accumulation of saturated VLCFAs (Parisi et al., 2019). As such, perturbing ELOVL7 levels is an ideal system to modulate the levels of saturated VLCFAs. We reduced the levels of VLCFAs by lentivirus-based ELOVL7 knockdown and tested the membrane recruitment of MLKL and pMLKL. The knockdown of ELOVL7 increased the viability of necroptotic cells as expected (Figure 4A) and significantly reduced the levels of membrane-associated MLKL and pMLKL during necroptosis (p < 0.05, Figure 4B, Figure S4A) supporting that VLCFAcylation of MLKL and pMLKL is be important for necroptotic cell death.
Figure 4. Membrane recruitment of MLKL and pMLKL in shELOVL7 and shZDHHC5 cells during necroptosis.
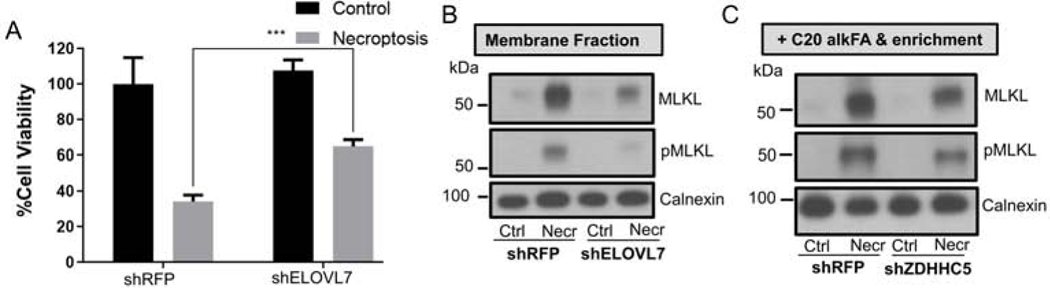
(A) ELOVL7 knockdown reduces necroptotic cell death. Percent cell viability of shRFP (used as control) and shELOVL7 transduced cells treated with BV6/zVAD-FMK/TNF-α to induce necroptosis compared to vehicle-treated control cells. Data represent mean ± 1 SD; n=5. *** represents p < 0.001 (B) Western blot analysis of membrane fraction from untreated and necroptotic shRFP (used as control) and shELOVL7 cells. Cell lysates were fractionated by ultracentrifugation and membrane fractions analyzed. Decrease in MLKL and pMLKL were observed in necroptotic shELOVL7 as compared to necroptotic control cells suggesting that membrane recruitment is reduced in shELOVL7 cells (see Figure S4A for quantification of band intensities). (D) Control and necroptotic shRFP and shZDHHC5 cells treated with C20 alkFA were membrane fractionated and subjected to CuAAC with Biotin-azide and enrichment using neutravidin beads. Equal amounts of protein were used for enrichment of each condition (see Materials and Methods for details). The proteins on the beads were blotted for MLKL, pMLKL and calnexin. An approximate 50% decrease in the enriched pMLKL was observed as compared to necroptotic control cells suggesting a role for ZDHHC5 in VLCFAcylation during necroptosis. (see Figure S4B for quantification of band intensities). See also Figure S4.
ZDHHC5 is in part responsible for MLKL acylation.
Our results suggest that the incorporation of VLCFAs to MLKL and pMLKL is S-linked (Figure 3B and 4C). S-linked acylation is a post-translational modification where fatty acids are covalently attached onto proteins via thioester bond (Jiang et al., 2018). These modifications are catalyzed by a family of 23 protein acyltransferases (ZDHHCs). All proteins of this family have a conserved catalytic zinc binding site, aspartic acid-histidine-histidine-cysteine (DHHC) cytosolic domain and several transmembrane domains (Jiang et al., 2018; Philippe and Jenkins, 2019). The family members share less homology within the N-and C-terminal cytosolic domains which are responsible for their recognition with specific substrates, although redundancy between different members have been reported (Jiang et al., 2018; Greaves et al., 2017). The substrate specificity of ZDHHCs are also greatly impacted by the cellular (co)localization with substrates at the plasmalemma, ER, Golgi or endosomal membranes. Based on the spatial organization of ZDHHCs, these enzymes acylate a variety of target proteins and regulate their targeting to distinct cellular membranes (Jiang et al., 2018).
Many members of the ZDHHC S-acyltransferases reside at the endoplasmic reticulum and Golgi apparatus; ZDHHC5 is one of the few localized to the plasma membrane and endosomal compartments (Ohno et al., 2006; He et al., 2014). A study using clickable fatty acid probes to determine the fatty acyl substrate selectivity of ZDHHCs showed that ZDHHC5 can accept C14, C16 and C18 FA but has preference for C16 FA (Greaves et al., 2017). Other studies have shown that the acylation of target proteins by ZDHHC5 takes place at the plasma membrane and retains them at this location, controlling their subcellular localization (Howie et al., 2014; Wang et al., 2019; He et al., 2014). ZDHHC5 is also involved in regulation of the localization of endocytosis-related proteins that have been linked to MLKL recycling including flotillin 2 (Li et al., 2012) (Kwiatkowska et al., 2020). Based on these observations and the fact that pMLKL acylation occurs downstream to its oligomerization, potentially at the plasma membrane, we focused on investigating ZDHHC5 as an acyltransferase that might be responsible for the VLCFA incorporation of MLKL/pMLKL. We knocked down ZDHHC5 using lentiviral shRNA (see Methods, Figure S4B) and tested whether ZDHHC5 could be responsible for the incorporation of VLCFAs to pMLKL during necroptosis by addition of C20 alkFA to knockdown cells and subsequent enrichment of proteins containing this probe via click chemistry and neutravidin beads. We observed a decrease in the levels of enriched pMLKL in shZDHHC5 cells (Figure 4C and S4B), suggesting that VLCFAcylation of pMLKL during necroptosis is, at least in part, mediated by ZDHHC5. The incomplete inhibition of MLKL and pMLKL acylation in shZDHHC5 cells suggests the involvement of additional ZDHHCs in processing MLKL and pMLKL. The knockdown of ZDHHC5 did not have a significant effect on the level of cell death in necroptotic cells (Figure S4C), further supporting the involvement of other ZDHHCs in this process. Overall, our results support that ZDHHC5 activity might play a role in acylation and membrane recruitment and retainment of pMLKL during necroptosis. However, the involvement of additional ZDHHCs in pMLKL acylation is likely.
Based on the preference of ZDHHC5 for C16 FA (Greaves et al., 2017), we next investigated palmitoylation of MLKL and pMLKL during necroptosis (Figure S3F). When delivered at the same concentration, both C16 alkFA and C20 alkFA are incorporated at similar levels based on the quantification of MLKL and pMLKL that are retained on neutravidin resin (p > 0.05, Figure S3G). However given the difference in intracellular concentration of endogenous C16 FA and C20 FA in necroptosis, it is possible that higher proportion of the proteins are palmitoylated.
SwissPalm (Blanc et al., 2019) predicts two palmitoylation site for MLKL and pMLKL (Cys18 and Cys24). Using high resolution LC-MS/MS, we investigated the site and type of fatty acylation of MLKL in necroptosis. We obtained enriched MLKL/pMLKL using immunoprecipitation, subjected the solution to SDS-PAGE and analyzed the band corresponding to the molecular weight of MLKL (~50 kDa) after in gel digestion (see Methods). LC-MS/MS data suggests that Cys24 could be modified by palmitoyl or C20 FA during necroptosis (Figure S3H-I), although the search engine score was low likely due to low abundance. While these results corroborate well with SwissPalm predictions and indicates the acylation of MLKL and pMLKL during necroptosis, further studies are necessary to unambiguously identify acylation sites and the corresponding type of acylation with high confidence.
Enrichment analysis suggests an involvement of endocytosis in necroptosis.
Next, in order to gain insights on whether proteins from a specific pathway are targets of VLCFAcylation during necroptosis, we carried out enrichment analysis using the Database for Annotation, Visualization and Integrated Discovery (DAVID) Bioinformatics Resource (Huang da et al., 2009). We input proteins that showed differential fatty acylation (p < 0.01) during necroptosis into DAVID which then mapped these genes against the KEGG database (Kanehisa and Goto, 2000). Eight pathways showed significant enrichments (p < 0.05, Benjamini Hochberg FDR, Figure 5A, Table S3) in this dataset. Three of the eight pathways (SNARE interactions, lysosome and endocytosis) were vesicular trafficking related (highlighted in gray in Table S3).
Figure 5. Inhibition of endocytic pathway specifically ameliorate necroptotic cell death.

(A) Pathway enrichment analysis of proteins that show differential fatty acylation during necroptosis. 8 pathways significantly enriched (corrected p value < 0.05). See also Table S3. (B) Small molecule inhibitor targeting endocytic pathway rescues necroptotic cell death, but not apoptotic cell death, in HT-29 cells. HT-29 cells were pretreated with 100 μM dynasore for 2 hours. Vehicle-treated control cells were treated with DMSO. For induction of necroptosis, cells were treated with BV6/zVAD-FMK/TNF-α. For induction of apoptosis, cells were treated with BV6/TNF-α. The percent cell viability in the presences of inhibitor during necroptosis was compared to the vehicle-treated control cells. Data represent mean ± 1 SD; n ≥ 3. ** represents p < 0.01, *** represents p < 0.001. (C-D) Western blot analysis of membrane fraction (C) and whole lysate (D) of vehicle-treated cells and dynasore treated cells with or without necroptosis. Cell lysates are fractionated by ultracentrifugation and membrane fractions are analyzed. The decrease in pMLKL observed in necroptosis when dynasore is present suggesting endocytosis maybe involved in necroptosis.
Endocytic and lysosomal activity affect MLKL and pMLKL trafficking and recycling from the plasma membrane. For example, the association of pMLKL and MLKL with flotillin 1 and 2 leads to their endocytosis, with pMLKL subsequently targeted for lysosomal degradation (Yoon et al., 2017; Fan et al., 2019). Flotillins were also found to associate with pMLKL in exosomes which then results in pMLKL extrusion from cells (Yoon et al., 2017; Meister and Tikkanen, 2014). Studies have also found that MLKL and pMLKL associate with proteins of the ESCRT (endosomal sorting complex required for transport) machinery, which causes a delay in membrane permeabilization during necroptosis through the scission and extracellular release of plasma membrane-derived vesicles (Gong et al., 2017).
S-acylation of proteins impacts endocytic activity (Linder and Deschenes, 2007; Fraser et al., 2020). Hence based on the differential acylation of endocytic proteins during necroptosis, we hypothesized that endocytic pathway may regulate necroptosis. As an initial test for this hypothesis, we took a pharmacological approach of targeting endocytic pathway using dynasore. Dynasore is a cell permeable and rapidly acting small molecule inhibitor for dynamin 1, dynamin 2 and dynamin-related protein 1, which promote fission and budding of the endocytic vesicles on the plasma membrane into the cytosol (Ferguson and De Camilli, 2012; Macia et al., 2006). We pre-treated HT-29 cells with dynasore, induced necroptosis via addition of BV6 and Z-VAD-FMK followed by TNF-α treatment for 3h, and assessed the changes in cell viability in the presence of the small molecule. Dynasore pretreatment prevented cell death significantly (p < 0.01) compared to non-treated necroptotic cells (Figure 5B). We note that the rescue is specific to necroptosis and is not observed for apoptosis suggesting that it is not the inhibition of canonical TNF-α signaling that mediated the rescue (Figure 5B). We then investigated the effect of dynasore treatment in membrane-localized and overall pMLKL levels in necroptotic cells and observed that dynasore treatment strongly reduced the levels of pMLKL in membranes (Figure 5C) and in whole lysate, without affecting MLKL levels (Figure 5D). These results suggest that the rescue we observe in cell death could be due to blocking the phosphorylation of MLKL, or prevention of pMLKL trafficking to the plasma membrane (Figure 5).
DISCUSSION
There are multiple signaling pathways that can be activated in response to TNF-α. Activation in the presence of low caspase-8 activity and low activity of the of the cellular inhibitor of apoptosis results in necroptotic signaling: activation of RIPK3 and phosphorylation of MLKL in necrosomes (Vandenabeele et al., 2010). A recent study illustrated that downstream to its phosphorylation, MLKL translocates to the plasma membrane via Golgi-, actin- and microtubule-dependent trafficking, a new rate-limiting factor in necroptosis (Samson et al., 2020). Upon binding to the plasma membrane, pMLKL oligomers induce membrane permeabilization (Vandenabeele et al., 2010) which does not appear to occur via the formation of regularly-structured membrane pores (Samson et al., 2020). Based on these observations and other studies (Fan et al., 2019), it is likely that pMLKL might undergo other transformations that contribute to membrane permeabilization or stabilization of pMLKL at the plasma membrane.
We have previously studied the involvement of lipids in necroptosis and showed that saturated VLCFAs were upregulated via activated biosynthesis and mediate membrane permeabilization (Parisi et al., 2019; Parisi et al., 2017). Our results suggested that these VLCFAs might be incorporated into proteins during necroptosis and targeted to certain membrane regions as a result of specific protein-bilayer interactions, allowing high local concentrations of these VLCFAs on the plasma membrane (Parisi et al., 2019). In this work, we investigate the role of VLCFAcylation during necroptosis and present two key findings.
First, we show that pMLKL and MLKL are acylated by a representative saturated VLCFA during necroptosis, and that the acylation of pMLKL occurs downstream to MLKL phosphorylation and plasma membrane localization, suggesting that the acylation takes place at the plasma membrane. MLKL consists of three domains: the 4-helix-bundle which mediates the interactions with the membrane; the brace domain, and the pseudokinase domain that is phosphorylated by RIPK3 (Murphy et al., 2013). This phosphorylation is critical for membrane recruitment during necroptosis (Johnston and Wang, 2018). Recently, it has been suggested that the 4-helix-bundle also contributes to the membrane recruitment and binding, in addition to the phosphorylation of the pseudokinase domain (Petrie et al., 2020).
Our results suggest that the acylation of pMLKL and MLKL is S-linked and, in part, mediated by ZDHHC5, a member palmitoyl transferase family, indicating the acylation of 4-helix-bundle domain during necroptosis. The integral ZDHHCs undergo an autoacylation step on the cytosolic DHHC motif. The acylated enzyme intermediate then transfers the acyl group to its substrate (Jiang et al., 2018). As one of the few plasma membrane-localized ZDHHC members, ZDHHC5 mediates the acylation of substrate at the plasma membrane and their plasma membrane recruitment (Philippe and Jenkins, 2019), supporting our findings that pMLKL and MLKL are acylated at the plasma membrane during necroptosis. Several studies have suggested increased binding of fatty acylated proteins to phosphoinositide lipids (Yang et al., 2020; Chopard et al., 2018). As such, it is possible that the membrane binding of pMLKL is enhanced after its fatty acylation.
Recruitment of certain proteins to the cellular membranes via S-acylation is involved in numerous vesicle and membrane fusion events, including endocytosis (Chamberlain and Shipston, 2015). For a variety of proteins, endocytosis can be triggered by partitioning of these proteins to cholesterol and sphingomyelin-rich membrane microdomains mediated by their S-acylation (Hilgemann et al., 2013).
ZDHHC5 activity is also involved in recruitment of its targets to certain membrane microdomains which impact processing of the substrates (Sergeeva and van der Goot, 2019), and has been shown to regulate endocytosis. Specifically, its presence at cell surface and endosome enriched lysates of various cell types have been shown (Zhang et al., 2011; Diaz-Vera et al., 2017). Screening based approached have also linked ZDHHC5 activity to endosome-to-Golgi trafficking (Breusegem and Seaman, 2014) and in massive endocytosis (Lin et al., 2013). ZDHHC5 also acts on substrates that are directly involved in endocytosis, including flotillins (Kwiatkowska et al., 2020; Li et al., 2012). Flotillins mediate endocytic uptake of pMLKL for targeted lysosomal degradation (Yoon et al., 2017; Fan et al., 2019). Flotillins were also found to associate with pMLKL in exosomes which then results in its extrusion from cells (Yoon et al., 2017; Meister and Tikkanen, 2014). In parallel, the association of pMLKL with the ESCRT machinery results in the extracellular release of pMLKL-containing plasma membrane-derived vesicles (Gong et al., 2017).
It is important to note that similar to what we have observed for modification with C20 alkFA, MLKL and pMLKL can also be acylated by C16 alkFA during necroptosis (Figure S3F). However, we believe that modifications by C16 FA and VLCFAs contribute to membrane permeabilization differently during necroptosis: We previously showed that VLCFAs interact with membranes and disrupt their integrity in model liposomes and molecular dynamics simulations (Parisi et al., 2019). Based on the modest increases of VLCFAs during necroptosis, we proposed that the incorporation of VLCFAs into proteins can facilitate their recruitment to defined membrane domains and allow high local concentrations of these lipids at the membrane and perturb membrane integrity (Parisi et al., 2019). Translocation of pMLKL oligomers to the plasma membrane is driven by electrostatic interactions between the oligomer interface and negatively charged phosphatidylinositol phosphate (PIP)-rich membrane domains (Dondelinger et al., 2014). Several PIP species are known to be actively involved in cell signaling cascades, both in endogenous processes as well as in mechanisms of action of viruses and other pathogens (Bunney and Katan, 2010; Hammond and Burke, 2020; Mucksch et al., 2019). Protein-lipid interactions can alter lipid patterns on the membrane via electrostatic interactions or amino acid insertion into the bilayer. In turn, lateral lipid sorting alters the local mechanical and structural properties of the membrane (Sengupta and Lippincott-Schwartz, 2020; Callan-Jones et al., 2011; Monje-Galvan and Klauda, 2015). Binding of pMLKL oligomers to defined regions of the plasma membrane and further rearrangement of the local lipid distribution at their binding site allows high concentration of fatty acids that covalently modify pMLKL. Acylation of pMLKL oligomers would allow the local recruitment of VLCFAs to membrane regions that already possess different mechanical and structural properties, which could facilitate their membrane disruption action. This perturbation may be important in allowing MLKL to form pores by assisting formation of non-lamellar lipid structures of a pore (Paz Ramos et al., 2016; Gilbert, 2016; Akimov et al., 2017; Flores-Romero et al., 2020), though this remains to be formally tested.
Second, we define the scope of protein VLCFAcylation during necroptosis using state-of-the-art quantitative proteomics methods. Overall, we observe a decreased number of proteins are modified by a representative VLCFA during necroptosis. Pathway analysis conducted using proteins that show different levels of VLCFAcylation during necroptosis highlighted the enrichment of proteins belonging to the endocytic and lysosomal pathways.
Previous studies have shown that MLKL translocates to the plasma membrane via Golgi-dependent trafficking downstream to its phosphorylation (Samson et al., 2020) and that pMLKL can associate with ESCRT proteins and flotillins, which is suggested to result in the removal of pMLKL from the plasma membrane, either through endocytosis, leading to degradation, or by direct excision into the extracellular milieux (Yoon et al., 2017; Meister and Tikkanen, 2014; Gong et al., 2017). Based on these observations, it is clear that vesicular trafficking is key for the maintenance of pMLKL and MLKL at the plasma membrane. We show that the use of dynasore resulted in a strong rescue from cell death during necroptosis and decreased the levels of membrane bound pMLKL, whereas the whole cellular MLKL levels remained unchanged. While dynasore inhibits dynamin-mediated budding of endocytic vesicles at the plasma membrane (Ferguson and De Camilli, 2012; Macia et al., 2006), it can also have other effects, including on Golgi vesiculation and subsequent cholesterol accumulation in the ER (Preta et al., 2015). As we show MLKL is modified by VLCFAcylation at the plasma membrane (Figure 4C), the dynasore results further support the idea that proper endocytic trafficking is required for MLKL function. As we have previously shown (Parisi et al., 2019), VLCFAs can disrupt lipid bilayers, and may be important in the pore formation function of MLKL. The results presented here suggest that targeting VLCFAcylation at the plasma membrane, or the endomembrane transport machinery required for proper MLKL localization, could be a way to delay membrane permeability during necroptosis. Overall, our results provide insights on the role of protein acylation by VLCFAs during necroptosis and endocytic trafficking in maintaining pMLKL at the plasma membrane during this process.
SIGNIFICANCE
Necroptosis is a well-characterized form of caspase-independent regulated cell death that is involved in numerous diseases. A hallmark of necroptosis is the membrane permeabilization and rupture that is mediated by mixed linage kinase like protein (MLKL). This membrane rupture is linked to the inflammatory properties of necroptosis and is critical for disease states involving this process. It is established that electrostatic interactions between phosphatidyl inositol phosphates and phosphorylated MLKL (pMLKL) oligomers facilitate the membrane recruitment of pMLKL. In efforts to understand how lipids might contribute to necroptosis, we previously showed that saturated very long chain fatty acids (VLCFAs) accumulate and that they are functionally involved in this process. Our results also indicated that protein fatty acylation by VLCFAs can be a mechanism by which VLCFAs contribute to this process. Here we define the scope of protein acylation by saturated VLCFAs during necroptosis. First, we show that, although there is an overall decrease in acylation, some proteins, including MLKL and pMLKL, are exclusively acylated during necroptosis. Reducing the levels of VLCFAs decreases the membrane recruitment of MLKL and pMLKL and rescues from cell death, suggesting that acylation by VLCFAs contributes to membrane localization of pMLKL and subsequent membrane permeabilization. Acylation of MLKL and pMLKL occur downstream of phosphorylation and oligomerization and appear to be S-linked. Second, we show that disruption of the endosomal trafficking pathway results in a strong rescue in cell viability during necroptosis, highlighting the importance of endosomal trafficking in the proper localization and maintenance of pMLKL at the plasma membrane. Altogether, we show that MLKL and pMLKL can be acylated by ZDHHC5 during necroptosis, and that this acylation occurs at the plasma membrane, which in turn depends on endosomal trafficking.
EXPERIMENTAL PROCEDURES
Detailed information on the materials used can be found in the Supplemental Experimental Procedures section, as well as detailed procedures for cell culture and other analyses.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to the Lead Contact, G. Ekin Atilla-Gokcumen (ekinatil@buffalo.edu)
Materials Availability
Plasmids psPAX2 (gift from Didier Trono) and pCMV-VSV-G (Stewart et al., 2003) used in this study were obtained from Addgene plasmid repository or Millipore Sigma as bacterial glycerol stocks and were used as received or used as described in the methods section. The Addgene accession and MilliporeSigma information is provided in the Key Resources Table. The bacterial glycerol stock for shRFP in pLKO.1 vector was obtained from the Genetic Perturbation Platform, Broad Institute with details in Key Resources table. Mammalian cell lines were obtained from ATCC and the information is provided in the Key Resources Table. See Key Resources Table for other sources and reagents.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-MLKL (phospho S358) | abcam | Cat# ab187091; RRID:AB_2619685 |
| Rabbit monoclonal anti-MLKL | Cell Signaling Technology | Cat# 14993S; RRID:AB_2721822 |
| Rabbit monoclonal anti-calnexin | Cell Signaling Technology | Cat# 2679S; RRID:AB_2228381 |
| Rabbit polyclonal anti-MLKL | Novus Biological | Cat# NBP1–56729; RRID:AB_11025736 |
| Rabbit polyclonal anti-ZDHHC5 | MilliporeSigma | Cat# SAB4301861; RRID AB_2889073 |
| Rabbit monoclonal anti-phospho S358-MLKL | Cell Signaling Technology | Cat# 91689S; RRID:AB_2732034 |
| Mouse monoclonal anti-α-tubulin | MilliporeSigma | Cat# T9026; RRID:AB_477593 |
| Goat anti-rabbit HRP conjugate | Promega | Cat# W4011; RRID: AB_430833 |
| Goat anti-mouse HRP conjugate | Jackson Immunoresearch Lab | Cat# 115–035-174; RRID: AB_2338512 |
| Alexa Fluor® 488 AffiniPure Goat Anti-Rabbit IgG | Jackson Immunoresearch Lab | Cat# 111–545-144; RRID:AB_2338052 |
| Bacterial and Virus Strains | ||
| ZDHHC5 MISSION shRNA Bacterial Glycerol Stock | MilliporeSigma | NM_015457; TRCN0000162357 |
| ELOVL7 MISSION shRNA Bacterial Glycerol Stock | MilliporeSigma | NM_024930; TRCN0000116231 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT reagent) | Alfa Aesar | Cat# L11939; CAS: 298–93-1 |
| Z-VAD-FMK | Enzo Life Sciences | Cat# ALX-260–020; CAS: 220644–02-0 |
| Necrostatin-1s (Nec-1s) | EMD Millipore | Cat# 504297; CAS: 852391–15-2 |
| BV6 | Selleck Chemicals | Cat# S7597; CAS: 1001600–56-1 |
| TNF-α | R&D Systems | Cat# 210-TA/CF |
| Pierce™ High Capacity NeutrAvidin™ Agarose | ThermoFisher Scientific | Cat# 29202 |
| Pierce™ Protease Inhibitor Mini Tablets, EDTA-free | ThermoFisher Scientific | Cat# A32955 |
| Puromycin | MilliporeSigma | Cat# P8833; CAS: 58–58-2 |
| Polybrene | MilliporeSigma | Cat# TR-1003 |
| X-tremeGENE 9 transfection reagent | MilliporeSigma | Cat# XTG9-RO |
| Mammalian Protein Extraction Reagent | ThermoFisher Scientific | Cat# 78501 |
| Dynasore | Cayman Chemical | Cat# 14062; CAS: 304448–55-3 |
| Necrosulfonamide | Cayman Chemical | Cat# 20844; CAS: 1360614–48-7 |
| Tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine (TBTA) | Cayman Chemical | Cat# 18816; CAS: 510758–28-8 |
| Tris(2-carboxyethyl)phosphine (TCEP) hydrochloride | VWR | Cat# K831; CAS: 51805–45-9 |
| Azide-PEG3-biotin conjugate | MilliporeSigma | Cat# 762024; CAS: 875770–34-6 |
| Fatty acid-free BSA | MilliporeSigma | Cat# A7030; CAS: 9048–46-8 |
| Hydroxylammonium chloride | Alfa Aesar | Cat# A15398; CAS: 5470–11-1 |
| Methoxypolyethylene glycol maleimide (mPEG) | MilliporeSigma | Cat# 63187; CAS: 99126–64-4 |
| N-ethylmaleimide (NEM) | MilliporeSigma | Cat# E3876; CAS: 128–53-0 |
| Dynabeads™ Proten A for Immunoprecipitation | ThermoFisher Scientific | Cat# 10001D |
| Triton™ X-100 | MilliporeSigma | Cat# T9284 |
| Triethanolamine | Acros Organics | Cat# 139560010; CAS: 102–71-6 |
| Sodium dodecyl sulfate (SDS) | MilliporeSigma | Cat# L3771; CAS: 151–21-3 |
| Tween 20 | VWR | Cat# VWRV0777–1L; CAS: 9005–64-5 |
| Dithiothreitol | MilliporeSigma | Cat# GE17–1318-02; CAS: 3483–12-3 |
| Iodoacetamide Bioultra | MilliporeSigma | Cat# I1149–100G; CAS: 144–48-9 |
| Tris, Hydrochloride, ULTROL Grade | MilliporeSigma | Cat# 648313–1KG; CAS: 1185–53-1 |
| Trypsin from bovine pancreas | MilliporeSigma | Cat# T6567–5X20UG; CAS: 9002–07-7 |
| Formic acid, for mass spectrometry | Fluka | Cat# 56302–50ML-F; CAS: 64–18-6 |
| Methanol, HPLC/ACS Grade | Fisher Chemical | Cat# A452–4; CAS: 67–56-1 |
| Acetonitrile, HPLC/ACS Grade | Fisher Chemical | Cat# A998–4; CAS: 75–05-8 |
| Acetone, HPLC/ACS Grade | Fisher Chemical | Cat# A949–4; CAS: 67–64-1 |
| Sodium lauryl sulfate | Fisher Scientific | Cat# S529–500; CAS: 151–21-3 |
| Critical Commercial Assays | ||
| Coomassie (Bradford) Protein Assay Kit | ThermoFisher Scientific | Cat# 23200 |
| Supersignal West Pico Chemiluminescence Substrate | ThermoFisher Scientific | Cat# 34080 |
| Pierce™ BCA Protein Assay Kit | ThermoFisher Scientific | Cat# 23225 |
| BCA Protein Assay | G-Biosciences | Cat# 786–572 |
| Deposited Data | ||
| Proteomics Data | ProteomeXchange | PXD022527 |
| Experimental Models: Cell Lines | ||
| HT-29 | ATCC | ATCC HTB38 |
| Recombinant DNA | ||
| psPAX2 | Gift from Didier Trono | Addgene plasmid catalog#: 12260 |
| pCMV-VSV-G | Gift from Bob Weinberg (Stewart et al., 2003) |
Addgene plasmid catalog#: 8854 |
| shRFP lentiviral construct | Genetic Perturbation Platform (GPP), Broad Institute | rfp_59s1c1; TRCN0000072205 https://portals.broadinstitute.org/gpp/public/clone/details?cloneId=TRCN0000072205 |
| Software and Algorithms | ||
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| GraphPad Prism 7 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Proteome Discoverer 1.4 | Thermo Fisher Scientific | https://planetorbitrap.com/relatedsoftware |
| Scaffold v4.8.4 | Proteome Software, Inc. | http://www.proteomesoftware.com/products/scaffold/ |
| SIEVE v2.2 | ThermoFisher Scientific | https://planetorbitrap.com/relatedsoftware |
| Other | ||
| TLS-55 Swinging-Bucket Rotor | Beckman Coulter Life Science | Cat# 346936 |
| Fused silica Column Waters xselect csh C18, 75um ID x 65 cm, 2.5um | Waters | Self pack |
| Agilent ZORBAX 300SB-C18, 0.3mm ID x 5mm, 3.5um, 300 A | Agilent | Cat# 5065–9913 |
| SUN-SRI Vial, Std. Screw Thread, 12×32, 250uL, PP w/Glass Insert, Standard Opening | SUN-Sri | Cat# 500 214 |
| SUN-SRI Cap Kit, Threaded, PP, 8–425, Black, PTFE/Silicone, 0.060” | SUN-Sri | Cat# 500 062 |
| SilicaTip™ Emitters | New Objective | FS360–20-8-N-20-C4 |
Data and Code Availability
The Proteomics experiment data are available via ProteomeXchange with identifier PXD022527. The project name is Quantitative proteomics characterization of proteins modified by very long chain fatty acids (VLCFAs) under necroptosis with username: reviewer_pxd022527@ebi.ac.uk and password: MisAktBm. Project DOI: Not applicable
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture and culture conditions.
Human colorectal adenocarcinoma HT-29 cell (ATCC® HTB-38™, adult Caucasian female origin) were cultured at 37 °C in 5% CO2 atmosphere in DMEM supplemented with 10% (v/v) fetal bovine serum and 1% (v/v) penicillin/streptomycin solution. Cells were cultured for approximately 2 months and were routinely checked for mycoplasma infection. Cells were plated according to the requirement of each experiment as described below.
Knockdown of ELOVL7 and ZDHHC5 in HT-29 cells were performed similarly as described previously (Parisi et al., 2017). Cells were transduced with lentiviral particles packaged with shELOVL7, shZDHHC5 or shRFP in the pLKO.1-Puro vector and stably transduced cells were selected with puromycin.
METHOD DETAILS
Necroptosis treatment.
To induce necroptosis cell death, HT-29 cells were initially sensitized to TNF-dependent cell death pathway by SMAC mimetic BV6 (1 μM), and were co-treated with pan-caspase inhibitor zVAD-FMK (25 μM) and incubated for 30 min at 37°C. Cells were then treated with TNF-α (10ng/mL) and was incubated for 3 h.
Apoptosis treatment.
Cells were sensitized to TNF-dependent apoptosis cell death by using SMAC mimetic BV6. HT-29 cells were treated with BV6 (1 μM) for 30 min. TNF-alpha (10 ng/mL) was added after the 30 min BV6 treatment to initiate apoptosis for 24h.
Cell Viability Assays.
HT-29 cells were seeded 96-well plates with 30,000 cells per well for 20 h before pretreatment with small molecule inhibitors. After the designated pretreatment time and/or induction of cell death, the 96-well plate was centrifuged for 2 min at 200 rcf at room temperature. The media was removed from each well and replaced with 200 μL of fresh media that contains 5 mg/mL of MTT reagent. The plate was incubated at 37°C for 2.5 h and then centrifuged for 2 min at room temperature. 155 μL of media was removed from each well and 90 μL of DMSO was added back. The plate was then incubated at 37°C for 10 min. Each well was then re-suspended to solubilize the formazan crystal and was centrifuged at 1000 rcf for 2 min at room temperature. Absorbance was measured at 550 nm using Biotek Synergy H1 plate reader. In order to calculate the percent viability of treated cells compared to control cells, the raw absorbance values of cells were subtracted with the average absorbance values of blanks (n ≥ 3). All corrected absorbance values were normalized to the average absorbance values of vehicle control cells and was expressed as percentage cell viability (n ≥ 3). Results are representative of at least two independent experiments.
C20 alkFA delivery and protein enrichment.
HT-29 cells (1×107) were plated in 10 cm dishes for ~16h. For proteomics, 30 dishes were used so that there were enough to combine 2 plates per sample for control, 4 plates per sample for necroptosis for 4 replicates per condition treated with alkFA, and one replicate per condition treated with FA to assess non-specific binding (NSB). For Western blotting experiments, 1 plate per sample for control/small molecule-treated/small molecule-treated necroptotic samples and 2 plates per sample for necroptosis were plated. Cells were starved for 2 h and treated to induce necroptosis (final concentrations of 1 μM BV6, 25 μM zVAD, 10 ng/mL TNF-α) then alkFA was added for 3 h before collection.
The complete media was removed and replaced with 7 mL DMEM with no additives. 20 mM arachidic acid (C20 FA) and ω-alkynyl C20 fatty acid (C20 alkFA) were prepared in ethanol. C20 alkFA was diluted to 0.4 mM by adding 100 μL of 20 mM solution in ethanol in pre-warmed sterile-filtered 10% fatty acid-free BSA in DMEM with no additives and sonicated at 50°C until use (~1–2 h). After 90 min starvation, DMSO was added to control flasks or BV6 and zVAD added to the necroptosis flasks. After 30 min, TNF-α was added to necroptosis flasks followed by 1 mL of alkFA solution to achieve a final concentration of 50 μM alkFA. Plates were gently rocked to further mix and incubated for 3 h before collection.
Cells were scraped from the plates on ice and transferred to 50 mL centrifuge tubes. Plates were rinsed with cold 1x PBS after scraping, which was also transferred to the centrifuge tube. Cells were centrifuged for 5 min at 500 rcf at 4°C. The supernatant was decanted and the cell pellets were washed two more times with cold 1xPBS. The supernatant was decanted and the cells were resuspended in 1050 μL 1xPBS, from which 50 μL was transferred to an Eppendorf tube for protein measurements using Bradford assay, and the remainder pellet was stored at −80°C.
Samples were thawed on ice, resuspended in 1xPBS with protease inhibitor with volumes normalized based on protein concentration, then sonicated on a cold block, mixed and transferred into 1 mL ultracentrifuge tube. Approximately 1.2 mg of protein was used for each condition. Lysates were centrifuged using TLS-55 swing bucket rotor (Beckman Coulter) at 35,000 rpm for 45 min at 4°C. The supernatant was decanted and 4% SDS in 1xPBS was added to the cell pellets, which were partially resuspended using a pipette and transferred to an Eppendorf tube, then sonicated. They were placed on ice and subjected to protein precipitation using cold 1xPBS, ice-cold methanol and ice-cold chloroform in 1:2:1.5 ratio. The tubes were vortexed, then centrifuged for 10 min at maximum speed (16,900 rcf) at 4°C. The liquid layers were carefully decanted and one volume of methanol was added to the pellet, which was then sonicated and centrifuged for 5 min at maximum speed at 4°C. The supernatant was decanted and the pellets were resuspended in 4% SDS in 1xPBS with sonication.
For the CuAAC reaction, CuSO4 (1 mM, stock 25 mM in water), TBTA (250 μM, stock 2.5 mM in DMSO), biotin-PEG3-azide (250 μM, stock 10 mM in DMSO) and TCEP (1 mM, stock 50 mM in 1xPBS) were used. A mastermix of all the regents except TCEP was prepared and added. TCEP was freshly prepared and added last. The samples were vortexed and incubated at 37 °C for 1 h.
After the CuAAC reaction, the samples were again subjected to protein precipitation as described before to remove excess CuAAC reagents. Samples were vortexed, then centrifuged for 10 min at maximum speed at 4°C. The liquid layers were carefully decanted and one volume methanol was added to the pellet, which was then sonicated and centrifuged for 5 min at maximum speed at 4°C. This step was repeated once. The pellets were then dissolved by sonication in 2% SDS in 1xPBS. Aliquots were taken for protein measurement using BCA assay.
The samples were normalized according to protein concentration. For proteomics, approximately 600 μg of protein were taken into a new Eppendorf tube for enrichment. For Western blot analysis, normalized samples containing approximately 200 μg of protein were used for enrichment. The samples within each experiment were normalized to have the same amount of protein and then subjected to enrichment. High capacity neutravidin-agarose resin was resuspended and for each sample, 20 μg slurry was transferred into a 15 mL centrifuge tube and subjected to three 0.2% SDS in 1xPBS washes. The tubes were capped and inverted several times to mix, then centrifuged again for 2 min at 2000 rcf at room temperature. After the third wash, the resin was resuspended in 150 μL 1% Brij-97 in 1xPBS then transferred to the sample. The samples were rotated end-over-end for 90 min at room temperature, then centrifuged for 2 min at 2000 rcf at room temperature. The enriched resin was subjected to three washes of 0.2% SDS in 1xPBS followed by three washes with regular 1xPBS. The samples were centrifuged for 2 min at 2000 rcf at room temperature, then the supernatant was carefully removed with suction and the resin was stored at −80°C for proteomics analysis. For Western blotting samples, resin after washing was boiled in 50 μL 2x loading dye containing 5mM of TCEP.
Quantitative proteomics.
Protein digestion.
A surfactant-aided precipitation/on-bead digestion method was modified from our previous sample preparation protocol(Shen et al., 2018a) and employed in the current study. In brief, beads were re-suspended with 60 uL 0.5% SDS, and were sequentially incubated with 10 mM dithiothreitol (DTT) at 56°C for 30 min and 25 mM iodoacetamide (IAM) at 37°C for 30 min to denature protein and dissociate protein disulfide bonds. Both steps were conducted with rigorous oscillation in darkness using a thermomixer (Eppendorf). A total of 7 volumes of chilled acetone was then added to the beads with constant vortexing, and the mixture was incubated at −20°C for 3 hr. After centrifugation at 20,000 g, 4°C for 30 min, liquid was removed and the beads was rinsed by 500 uL methanol and left to air-dry. A volume of 55 uL 50 mM pH 8.4 Tris-formic acid (FA) was then added to the beads, and a total volume of 5 uL trypsin (Sigma Aldrich) dissolved in 50 mM pH 8.4 Tris-FA was added for 6-hr digestion at 37°C with rigorous oscillation in a thermomixer. Digestion was terminated by addition of 0.6 uL FA, and beads were pelleted by centrifugation at 20,000 g, 4°C for 30 min. Supernatant was carefully transferred to LC vials for analysis.
LC-MS analysis.
The LC-MS system consists of a Dionex Ultimate 3000 nano LC system, a Dinex Ultimate 3000 micro LC system with a WPS-3000 autosampler, and an Orbitrap Fusion Lumos mass spectrometer. A large-inner diameter (i.d.) trapping column (300-um i.d. x 5 mm) was implemented before the nano LC column (75-um i.d. x 65 cm, packed with 2.5-um Xselect CSH C18 material) for high-capacity sample loading, cleanup and delivery. For each sample, 4 uL derived peptides was injected twice consecutively for LC-MS analysis. Mobile phase A and B were 0.1% FA in 2% acetonitrile (ACN) and 0.1% FA in 88% ACN. The 180-min LC gradient profile was: 4% for 3 min, 4–11 for 5 min, 11–32% B for 117 min, 32–50% B for 10 min, 50–97% B for 5 min, 97% B for 7 min, and then equilibrated to 4% for 27 min. The mass spectrometer was operated under data-dependent acquisition (DDA) mode with a maximal duty cycle of 3 s. MS1 spectra was acquired by Orbitrap (OT) under 120k resolution for ions within the m/z range of 400–1,500. Automatic Gain Control (AGC) and maximal injection time was set at 120% and 50 ms, and dynamic exclusion was set at 45 s, ± 10 ppm. Precursor ions were isolated by quadrupole using a m/z window of 1.2 Th, and were fragmented by high-energy collision dissociation (HCD). MS2 spectra was acquired differently in the two LC-MS injections. One was by OT under 15k resolution with a maximal injection time of 50 ms, and the other was by Ion trap (IT) under Rapid scan rate with a maximal injection time of 50 ms. Detailed LC-MS settings and relevant information are enclosed in a previous publication by Shen et al (Shen et al., 2017).
Data processing.
LC-MS files were searched against Homo Sapiens Swiss-Prot protein sequence database (20,350 entries) using Sequest HT embedded in Proteome Discoverer 1.4 (Thermo Fisher Scientific). Target-decoy searching approach using a concatenated forward and reverse protein sequence database was applied for FDR estimation purposes. Searching parameters include: 1) Precursor ion mass tolerance: 20 ppm; 2) Product ion mass tolerance: 0.02 Da for OT, 0.8 Da for IT; 3) Maximal missed cleavages per peptide: 2; 4) Fixed modifications: carbamidomethylation of cysteine; 5) Dynamic modifications: Oxidation of methionine, Acetylation of peptide N-terminals. Peptide filtering, protein inference and grouping, and FDR control were accomplished by Scaffold v4.8.4 (Proteome Software, Inc.) The filtered peptide-spectrum match (PSM) list was exported. Proteomic quantification was performed by an in-house developed MS1-based quantitative method, IonStar to achieve high-precision, low-missing data quantification (Shen et al., 2018b). IonStar involves SIEVE v2.2 (ThermoFisher Scientific) and an in-house developed R package IonStarStat. The major procedures include: i) Chromatographic alignment with ChromAlign for inter-run calibration of retention time (RT) shift. Quality control and selection of the optimal reference for alignment were accomplished by monitoring alignment scores (>0.8) and base-peak ion chromatogram intensity; ii) Data-independent MS1 quantitative feature generation using the DICE method, which extracts ion chromatograms for all precursor ions with corresponding MS2 scans in the aligned dataset with a defined m/z-RT window (10 ppm, 1 min). iii) Integration of the SQL database exported from SIEVE (containing corresponding intensities of all quantitative features in the sample set) and the filtered PSM list by MS2 scan number using a customized R script. Frames with valid PSMs were then subjected to dataset-wide normalization, multivariate outlier detection/removal, and aggregation to protein level using IonStarStat. Detailed steps of the IonStar method can be found in the user manual (available at https://github.com/shichens1989).
Acyl-Peg Exchange (APE).
APE procedure was modifed based on previous protocols (Percher et al., 2017). Control and necroptotic pellets were resuspended in 200 μL lysis buffer containing 5 mM EDTA. Protein content was measured using Bradford Assay and samples were normalized. Approximately 1000 μg of proteins were taken for further procedures. Each sample was treated with freshly prepared 200 mM neutralized TCEP to achieve a final concentration of 10 mM. The samples were rotated for 30 minutes at RT. The cysteines were treated to freshly prepared 1 M NEM in ethanol to achieve final concentration of 25 mM. The samples were rotated for 2 h at room temperature. The reaction was terminated by methanol-chloroform-water protein precipitations in the ratio of 2:1.5:1. Appropriate volumes of the prechilled solvents were added and the samples were gently vortexed. They were centrifuged for 10 minutes at 16900 rcf at 4°C. This process was repeated thrice. The solvents were gently decanted every time without disturbing the pellet.
After the last protein precipitation, the pellets were resuspended in 1xTEA buffer with 4% SDS and 5 mM EDTA. They were sonicated in a cool water bath and gently vortexed in between till the pellet was solubilized. The samples were then split into two equal fractions for treatment with and without NH2OH. The final concentration of NH2OH used was 0.75 M. A stock of 1 M in 1xTEA buffer with 0.2% Triton X-100 was freshly prepared and neutralized with NaOH. The portion of the samples undergoing NH2OH treatment an appropriate volume of 1 M stock of NH2OH to achieve a final concentration of 0.75 M and the samples without NH2OH treatment received an equal volume of 1xTEA buffer with 0.2% Triton X-100. The samples were rotated for 1 h at room temperature. After hydrolysis with NH2OH, the samples were subjected to a methanol-chloroform-water protein precipitation as described before. The samples were gently vortexed and again centrifuged for 10 minutes at 16900 rcf at 4°C. The solvents were decanted without disturbing the protein pellet. The pellets were resuspended in 1xTEA buffer with 4% SDS and 5 mM EDTA. They were sonicated in a cool water bath and gently vortexed till the pellet was solubilized. A 1.33 mM stock solution of mPEG-Mal (5 kDa) was freshly prepared in 1xTEA buffer with 0.2% Triton X-100 and an appropriate amount of volume of the stock was added to the sample to achieve a final concentration of 1 mM. The samples were rotated for 2 h at room temperature. After 2 h, the samples were subjected to a final methanol-chloroform-water protein precipitation. The protein disc obtained after final protein precipitation was resuspended in 50 μL of 2x loading dye containing 5 mM TCEP. 50 mM stock TCEP was prepared in 1x TEA buffer with 4% SDS. The samples were vortexed to disperse the protein disc and were boiled for 5 minutes at 100°C.
Western blotting.
Samples were loaded and separated with sodium dodecyl sulfate polyacrylamide gel (10%) electrophoresis at 120 V. Polyvinyl difluoride (PVDF) membranes were then activated in methanol for five minutes. Once the membrane was activated, the separated proteins were transferred onto the PVDF membranes at 50 V for 2 hours. After transfer, the membranes were blocked in 10% non-fat dry milk in tris-buffered saline (TBS)-Tween [10 mM Tris-base, 100 mM NaCl, 0.1% Tween 20 (pH 7.5)] at room temperature for 1 hour. Membranes were washed three times at 10 minute intervals in TBS-Tween. The corresponding membranes were incubated overnight at 4°C with primary antibodies (1:1000 dilution for MLKL, 1:500 for pMLKL, 1:10000 for calnexin, 1:5000 for α-tubulin). After incubation, the membranes were then washed three times with TBS-Tween for 10 minute each time. The secondary antibodies used were 1:2000 anti-rabbit and 1:1000 anti-mouse. Secondary antibodies were diluted with 5% non-fat dry milk in TBS-Tween and incubated for one hour at room temperature. The membranes were then washed again with TBS-Tween three times, 10 min each time, prior to developing with Super Signal West Pico kit (Thermo Scientific).
Quantification of western blots.
Quantitative analysis of Western Blot images was performed by using ImageJ software. A frame for measurement was developed by using the rectangle tool of ImageJ to cover the largest band of the protein of interest. The same frame size was then applied for measuring the intensity of the other protein bands. The background was also measured with the same frame to obtain background intensity measurements. For the background measurement, a region near the protein of interest was used. The measured intensities from the protein of interest and background were then inverted by deducting measurements from 255. 255 is the pixel value assigned to white background. The inverted intensities of the protein bands of interest were corrected with the inverted intensities of the background. The relative intensities were obtained by dividing the corrected intensity of MLKL or pMLKL to the corrected intensity of the loading control (calnexin, n=3).
Microscopy.
HT-29 cells were seeded onto glass coverslips in 24-well plates and treated as above to induce necroptosis. Following fixation with 4% paraformaldehyde, the cells were permeabilized and immunostained with rabbit monoclonal anti-phospho S358-MLKL, (Cell Signaling, Cat. #91689S) followed by Alexa-488 goat anti-rabbit secondary, (Jackson ImmunoResearch, Cat. #111–545-144) along with the nuclear DNA stain DAPI (Croft et al., 2018). After mounting on slides, microscopy was performed on a Dragonfly spinning disk confocal (Andor) and analyzed with FIJI (Schindelin et al., 2012) to determine the average pMLKL fluorescence intensity.
Immunoprecipitation (IP).
Necroptosis was induced in a plate of HT-29 cells (~1×107) and collected as described previously. The pellet was resuspended in 1 mL 1x PBS and protein content was measured using Bradford Assay. The cell suspension was centrifuged and resuspended in an appropriate volume of 1x PBS-0.02% Tween®−20 to get final concentration of 2 μg/μL protein. The cell suspension was sonicated and 100 μL aliquot (~200 μg of protein) was taken out and incubated with ~1.5 μg of anti-MLKL antibody solution suitable for IP overnight at 4 °C with rotation. The lysate was incubated with 50 uL of Dynabeads® for 1 h at room temperature with rotation. The enriched beads were separated from the lysate using a magnet and washed thrice with 1x PBS-0.02% Tween®−20. The beads were boiled with 40 uL of 2x loading dye containing 5 mM of TCEP. SDS-PAGE was used for separation and gel bands were excised at the approximate location of MLKL (~54 kDa) and subjected to mass spectrometry.
In-gel digestion and LC-MS on MLKL enriched lysates
Excised gel bands were first cut into smaller cubes (1–2 mm in each dimension) using a clean scalpel, and transferred to new LoBind tubes (Eppendorf, Hauppauge, NY). Gel cubes were dehydrated by incubating in 500 μL acetonitrile (ACN) for 5 min with constant vortexing and liquid was discarded (all dehydration steps below followed the same procedure if not specified). Gel de-staining was performed by incubating in 500 μL 50% ACN in 50 mM Tris-FA (pH 8.4) overnight. Gel cubes were then dehydrated three times and kept at 37°C in a Thermomixer for 5 min to completely evaporate remaining ACN. Protein reduction was performed by incubating dehydrated gel cubes in 100 μL 10 mM DTT (in Tris-FA) at 56°C for 30 min with constant shaking. After dehydration, protein alkylation was performed by incubating dehydrated gel cubes in 100 μL 25 mM IAM (in Tris-FA) at 37°C for 30 min with constant shaking (in darkness). Gel cubes were then dehydrated three times and incubated in 200 μL 0.0125 μg/μL trypsin (in Tris-FA) on ice for 30 min. Excess trypsin was then removed and replaced by 200 μL Tris-FA, and samples were incubated at 37°C overnight with constant shaking. Tryptic digestion was terminated by addition of 20 μL 5% FA and incubation for 15 min with constant vortexing, and liquid was transferred to new LoBind tubes. Gel bands were dehydrated by sequential incubation with 500 μL 50% ACN in 50 mM Tris-FA and 500 μL ACN, each for 15 min with constant vortexing, and liquid from all three steps was combined. Protein digest was dried in a Speed-Vac and reconstituted in 50 μL 1% ACN and 0.05% trifluoracetic acid (TFA) in ddH2O with 10-min gentle vortexing. Samples were centrifuged at 18,000 g, 4°C for 30 min, and transferred to LC vials for analysis.
LC-MS analysis and data processing.
LC-MS files were searched against Homo Sapiens MLKL protein sequence using Sequest HT embedded in Proteome Discoverer 1.4 (Thermo Fisher Scientific). Searching parameters include: 1) Precursor ion mass tolerance: 20 ppm; 2) Product ion mass tolerance: 0.8 Da; 3) Maximal missed cleavages per peptide: 2; 4) Fixed modifications: carbamidomethylation of cysteine; 5) Dynamic modifications: Oxidation of methionine, Phosphorylation of serine/threonine/methionine. C16:0/C20:0/C22:0/C24:0 modification of cysteine was included as dynamic modifications in separate searches. Search results and MS spectra were exported from Proteome Discoverer.
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical analysis for the western blot quantifications, cell viability experiments and proteomics were performed using unpaired Student’s t-test. p values and numbers of replicates in all figures are indicated in figure legends, where *** p < 0. 001, ** p < 0.01, * p < 0.05, and ns is p > 0.05.
Supplementary Material
This table is provided as a separate spread sheet.
Columns E-J show OT and IT abundances (area under the curve) in control samples (Control 1–3, n=3) in log2 scale.
Column K-F show OT and IT abundances (area under the curve) in necroptotic samples (Nec1–3, n=3) in log2 scale
Columns Q-V show OT and IT abundances (area under the curve) in control samples (Control 1–3, n=3) in non-transformed scale.
Columns W-AB show OT and IT abundances (area under the curve) in necroptotic samples (Nec1–3, n=3) in non-transformed scale
Columns AC and AD report that average abundances in control and necroptotic samples in non-transformed scale.
Column AE represents the ratio of protein abundance between necroptotic and control cells.
Column AF reports p values calculated by student’s t-test.
Entries with “0” abundance indicate no detection.
This table is provided as a separate spread sheet.
Columns E-J show OT and IT abundances (area under the curve) in control samples (Control 1–3, n=3) in log2 scale.
Columns K-F show OT and IT abundances (area under the curve) in necroptotic samples (Nec1–3, n=3) in log2 scale
Columns Q-V show OT and IT abundances (area under the curve) in control samples (Control 1–3, n=3) in non-transformed scale.
Columns W-AB show OT and IT abundances (area under the curve) in necroptotic samples (Nec1–3, n=3) in non-transformed scale
Columns AC and AD report average abundances in control and necroptotic samples in non-transformed scale.
Column AE represents the ratio of protein abundance between necroptotic and control cells.
Column AF reports p values calculated by student’s t-test.
Entries with “0” abundance indicate no detection.
HIGHLIGHTS.
Saturated VLCFAs are differentially incorporated to proteins during necroptosis.
pMLKL/MLKL exhibit acylation by VLCFAs in necroptosis.
pMLKL/MLKL acylation is, in part, mediated by ZDHHC5.
Perturbing trafficking to the plasma membrane rescues cell death during necroptosis.
ACKNOWLEDGMENTS
We acknowledge the support from the National Science Foundation grant (MCB1817468 to G.E.A.G.), the National Institutes of Health (GM078383 to S.R.C., DE028307 to J.G.K. and support from GM095459 for K.B.), and the Research Foundation for The State University of New York (J. Solo funds to G.E.A.G.). We thank P. Gollnick (UB Biological Sciences) for ultracentrifuge usage. Microscopy was performed in the Optical Imaging and Analysis Facility (UB Dental Sciences).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
SUPPLEMENTAL INFORMATION
Supplemental Information includes 4 figures, 3 tables, materials used, experimental procedures and can be found with this article online.
The authors declare no conflict of interest.
REFERENCES
- Akimov SA, Volynsky PE, Galimzyanov TR, Kuzmin PI, Pavlov KV & Batishchev OV (2017). Pore formation in lipid membrane I: Continuous reversible trajectory from intact bilayer through hydrophobic defect to transversal pore. Sci. Rep 7, 12152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc M, David FPA & Van Der Goot FG (2019). SwissPalm 2: Protein S-Palmitoylation Database. Methods Mol. Biol 2009, 203–214. [DOI] [PubMed] [Google Scholar]
- Breusegem SY & Seaman MNJ (2014). Genome-wide RNAi screen reveals a role for multipass membrane proteins in endosome-to-golgi retrieval. Cell Rep 9, 1931–1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunney TD & Katan M (2010). Phosphoinositide signalling in cancer: beyond PI3K and PTEN. Nat. Rev. Cancer 10, 342–52. [DOI] [PubMed] [Google Scholar]
- Cai Z, Jitkaew S, Zhao J, Chiang HC, Choksi S, Liu J, Ward Y, Wu LG & Liu ZG (2014). Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat. Cell Biol 16, 55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callan-Jones A, Sorre B & Bassereau P (2011). Curvature-driven lipid sorting in biomembranes. Cold Spring Harb. Perspect. Biol 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain LH & Shipston MJ (2015). The physiology of protein S-acylation. Physiol. Rev 95, 341–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Li W, Ren J, Huang D, He W-T, Song Y, Yang C, Li W, Zheng X, Chen P & Han J (2014). Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res 24, 105–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chopard C, Tong PBV, Toth P, Schatz M, Yezid H, Debaisieux S, Mettling C, Gross A, Pugniere M, Tu A, Strub JM, Mesnard JM, Vitale N & Beaumelle B (2018). Cyclophilin A enables specific HIV-1 Tat palmitoylation and accumulation in uninfected cells. Nat. Commun 9, 2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croft AJ, Metcalfe S, Honma K & Kay JG (2018). Macrophage Polarization Alters Postphagocytosis Survivability of the Commensal Streptococcus gordonii. Infect. Immun 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA & Yuan J (2005). Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol 1, 112–119. [DOI] [PubMed] [Google Scholar]
- Diaz-Vera J, Palmer S, Hernandez-Fernaud JR, Dornier E, Mitchell LE, Macpherson I, Edwards J, Zanivan S & Norman JC (2017). A proteomic approach to identify endosomal cargoes controlling cancer invasiveness. J. Cell Sci 130, 697–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dondelinger Y, Declercq W, Montessuit S, Roelandt R, Goncalves A, Bruggeman I, Hulpiau P, Weber K, Sehon CA, Marquis RW, Bertin J, Gough PJ, Savvides S, Martinou JC, Bertrand MJ & Vandenabeele P (2014). MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep 7, 971–81. [DOI] [PubMed] [Google Scholar]
- Fan W, Guo J, Gao B, Zhang W, Ling L, Xu T, Pan C, Li L, Chen S, Wang H, Zhang J & Wang X (2019). Flotillin-mediated endocytosis and ALIX–syntenin-1–mediated exocytosis protect the cell membrane from damage caused by necroptosis. Sci. Signal 12, eaaw3423. [DOI] [PubMed] [Google Scholar]
- Ferguson SM & De Camilli P (2012). Dynamin, a membrane-remodelling GTPase. Nat. Rev. Mol. Cell Biol 13, 75–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores-Romero H, Ros U & Garcia-Saez AJ (2020). Pore formation in regulated cell death. EMBO J 39, e105753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser NJ, Howie J, Wypijewski KJ & Fuller W (2020). Therapeutic targeting of protein S-acylation for the treatment of disease. Biochem. Soc. Trans 48, 281–290. [DOI] [PubMed] [Google Scholar]
- Gilbert RJC (2016). Protein–lipid interactions and non-lamellar lipidic structures in membrane pore formation and membrane fusion. Biochim. Biophys. Acta, Biomembr 1858, 487–499. [DOI] [PubMed] [Google Scholar]
- Gong Y-N, Guy C, Olauson H, Becker JU, Yang M, Fitzgerald P, Linkermann A & Green DR (2017). ESCRT-III Acts Downstream of MLKL to Regulate Necroptotic Cell Death and Its Consequences. Cell 169, 286–300.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greaves J, Munro KR, Davidson SC, Riviere M, Wojno J, Smith TK, Tomkinson NC & Chamberlain LH (2017). Molecular basis of fatty acid selectivity in the zDHHC family of S-acyltransferases revealed by click chemistry. Proc. Natl. Acad. Sci. U.S.A 114, E1365–e1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond GRV & Burke JE (2020). Novel roles of phosphoinositides in signaling, lipid transport, and disease. Curr. Opin. Cell Biol 63, 57–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He M, Abdi KM & Bennett V (2014). Ankyrin-G palmitoylation and betaII-spectrin binding to phosphoinositide lipids drive lateral membrane assembly. J. Cell Biol 206, 273–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilgemann DW, Fine M, Linder ME, Jennings BC & Lin MJ (2013). Massive endocytosis triggered by surface membrane palmitoylation under mitochondrial control in BHK fibroblasts. Elife 2, e01293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S, Bodmer JL, Schneider P, Seed B & Tschopp J (2000). Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat. Immunol 1, 489–95. [DOI] [PubMed] [Google Scholar]
- Howie J, Reilly L, Fraser NJ, Vlachaki Walker JM, Wypijewski KJ, Ashford ML, Calaghan SC, Mcclafferty H, Tian L, Shipston MJ, Boguslavskyi A, Shattock MJ & Fuller W (2014). Substrate recognition by the cell surface palmitoyl transferase DHHC5. Proc. Natl. Acad. Sci. U.S.A 111, 17534–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Da W, Sherman BT & Lempicki RA (2009). Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Zhang X, Chen X, Aramsangtienchai P, Tong Z & Lin H (2018). Protein Lipidation: Occurrence, Mechanisms, Biological Functions, and Enabling Technologies. Chem. Rev 118, 919–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston A & Wang Z (2018). Necroptosis: MLKL Polymerization. J. Nat. Sci 4, e513. [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M & Goto S (2000). KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28, 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowska K, Matveichuk OV, Fronk J & Ciesielska A (2020). Flotillins: At the Intersection of Protein S-Palmitoylation and Lipid-Mediated Signaling. Int. J. Mol. Sci 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau A, Wang S, Jiang J, Haig A, Pavlosky A, Linkermann A, Zhang ZX & Jevnikar AM (2013). RIPK3-mediated necroptosis promotes donor kidney inflammatory injury and reduces allograft survival. Am. J. Transplant 13, 2805–18. [DOI] [PubMed] [Google Scholar]
- Li Y, Martin BR, Cravatt BF & Hofmann SL (2012). DHHC5 Protein Palmitoylates Flotillin-2 and Is Rapidly Degraded on Induction of Neuronal Differentiation in Cultured Cells. J. Bio. Chem 287, 523–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MJ, Fine M, Lu JY, Hofmann SL, Frazier G & Hilgemann DW (2013). Massive palmitoylation-dependent endocytosis during reoxygenation of anoxic cardiac muscle. Elife 2, e01295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linder ME & Deschenes RJ (2007). Palmitoylation: policing protein stability and traffic. Nat. Rev. Mol. Cell Biol 8, 74–84. [DOI] [PubMed] [Google Scholar]
- Luedde M, Lutz M, Carter N, Sosna J, Jacoby C, Vucur M, Gautheron J, Roderburg C, Borg N, Reisinger F, Hippe HJ, Linkermann A, Wolf MJ, Rose-John S, Lullmann-Rauch R, Adam D, Flogel U, Heikenwalder M, Luedde T & Frey N (2014). RIP3, a kinase promoting necroptotic cell death, mediates adverse remodelling after myocardial infarction. Cardiovasc. Res 103, 206–16. [DOI] [PubMed] [Google Scholar]
- Macia E, Ehrlich M, Massol R, Boucrot E, Brunner C & Kirchhausen T (2006). Dynasore, a Cell-Permeable Inhibitor of Dynamin. Dev. Cell 10, 839–850. [DOI] [PubMed] [Google Scholar]
- Meister M & Tikkanen R (2014). Endocytic Trafficking of Membrane-Bound Cargo: A Flotillin Point of View. Membranes 4, 356–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monje-Galvan V & Klauda JB (2015). Modeling Yeast Organelle Membranes and How Lipid Diversity Influences Bilayer Properties. Biochemistry 54, 6852–6861. [DOI] [PubMed] [Google Scholar]
- Mucksch F, Citir M, Luchtenborg C, Glass B, Traynor-Kaplan A, Schultz C, Brugger B & Krausslich HG (2019). Quantification of phosphoinositides reveals strong enrichment of PIP2 in HIV-1 compared to producer cell membranes. Sci. Rep 9, 17661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy JM, Czabotar PE, Hildebrand JM, Lucet IS, Zhang JG, Alvarez-Diaz S, Lewis R, Lalaoui N, Metcalf D, Webb AI, Young SN, Varghese LN, Tannahill GM, Hatchell EC, Majewski IJ, Okamoto T, Dobson RC, Hilton DJ, Babon JJ, Nicola NA, Strasser A, Silke J & Alexander WS (2013). The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity 39, 443–53. [DOI] [PubMed] [Google Scholar]
- Ohno Y, Kihara A, Sano T & Igarashi Y (2006). Intracellular localization and tissue-specific distribution of human and yeast DHHC cysteine-rich domain-containing proteins. Biochim. Biophys. Acta 1761, 474–83. [DOI] [PubMed] [Google Scholar]
- Parisi LR, Li N & Atilla-Gokcumen GE (2017). Very Long Chain Fatty Acids Are Functionally Involved in Necroptosis. Cell Chem. Biol 24, 1445–1454.e8. [DOI] [PubMed] [Google Scholar]
- Parisi LR, Sowlati-Hashjin S, Berhane IA, Galster SL, Carter KA, Lovell JF, Chemler SR, Karttunen M & Atilla-Gokcumen GE (2019). Membrane Disruption by Very Long Chain Fatty Acids during Necroptosis. ACS Chem. Biol 14, 2286–2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paz Ramos A, Lagüe P, Lamoureux G & Lafleur M (2016). Effect of Saturated Very Long-Chain Fatty Acids on the Organization of Lipid Membranes: A Study Combining 2H NMR Spectroscopy and Molecular Dynamics Simulations. J. Phys. Chem. B 120, 6951–6960. [DOI] [PubMed] [Google Scholar]
- Percher A, Ramakrishnan S, Thinon E, Yuan X, Yount JS & Hang HC (2016). Mass-tag labeling reveals site-specific and endogenous levels of protein S-fatty acylation. Proc. Natl. Acad. Sci. U.S.A 113, 4302–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Percher A, Thinon E & Hang H (2017). Mass-Tag Labeling Using Acyl-PEG Exchange for the Determination of Endogenous Protein S-Fatty Acylation. Curr Protoc Protein Sci 89, 14.17.1–14. 17.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrie EJ, Birkinshaw RW, Koide A, Denbaum E, Hildebrand JM, Garnish SE, Davies KA, Sandow JJ, Samson AL, Gavin X, Fitzgibbon C, Young SN, Hennessy PJ, Smith PPC, Webb AI, Czabotar PE, Koide S & Murphy JM (2020). Identification of MLKL membrane translocation as a checkpoint in necroptotic cell death using Monobodies. Proc. Natl. Acad. Sci. U.S.A 117, 8468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippe JM & Jenkins PM (2019). Spatial organization of palmitoyl acyl transferases governs substrate localization and function. Mol. Membr. Biol 35, 60–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preta G, Cronin JG & Sheldon IM (2015). Dynasore - not just a dynamin inhibitor. Cell Commun. Signal 13, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purdy JG, Shenk T & Rabinowitz JD (2015). Fatty acid elongase 7 catalyzes lipidome remodeling essential for human cytomegalovirus replication. Cell Rep 10, 1375–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quarato G, Guy CS, Grace CR, Llambi F, Nourse A, Rodriguez DA, Wakefield R, Frase S, Moldoveanu T & Green DR (2016). Sequential Engagement of Distinct MLKL Phosphatidylinositol–Binding Sites Executes Necroptosis. Mol. Cell 61, 589–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samson AL, Zhang Y, Geoghegan ND, Gavin XJ, Davies KA, Mlodzianoski MJ, Whitehead LW, Frank D, Garnish SE, Fitzgibbon C, Hempel A, Young SN, Jacobsen AV, Cawthorne W, Petrie EJ, Faux MC, Shield-Artin K, Lalaoui N, Hildebrand JM, Silke J, Rogers KL, Lessene G, Hawkins ED & Murphy JM (2020). MLKL trafficking and accumulation at the plasma membrane control the kinetics and threshold for necroptosis. Nat. Commun 11, 3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P & Cardona A (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta P & Lippincott-Schwartz J (2020). Revisiting Membrane Microdomains and Phase Separation: A Viral Perspective. Viruses 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sergeeva OA & Van Der Goot FG (2019). Anthrax toxin requires ZDHHC5-mediated palmitoylation of its surface-processing host enzymes. Proc. Natl. Acad. Sci. U.S.A 116, 1279–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen S, An B, Wang X, Hilchey SP, Li J, Cao J, Tian Y, Hu C, Jin L, Ng A, Tu C, Qu M, Zand MS & Qu J (2018a). Surfactant Cocktail-Aided Extraction/Precipitation/On-Pellet Digestion Strategy Enables Efficient and Reproducible Sample Preparation for Large-Scale Quantitative Proteomics. Anal.Chem 90, 10350–10359. [DOI] [PubMed] [Google Scholar]
- Shen X, Shen S, Li J, Hu Q, Nie L, Tu C, Wang X, Orsburn B, Wang J & Qu J (2017). An IonStar Experimental Strategy for MS1 Ion Current-Based Quantification Using Ultrahigh-Field Orbitrap: Reproducible, In-Depth, and Accurate Protein Measurement in Large Cohorts. J. Proteome Res 16, 2445–2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X, Shen S, Li J, Hu Q, Nie L, Tu C, Wang X, Poulsen DJ, Orsburn BC, Wang J & Qu J (2018b). IonStar enables high-precision, low-missing-data proteomics quantification in large biological cohorts. Proc. Natl. Acad. Sci. U.S.A 115, E4767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart SA, Dykxhoorn DM, Palliser D, Mizuno H, Yu EY, An DS, Sabatini DM, Chen IS, Hahn WC, Sharp PA, Weinberg RA & Novina CD (2003). Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA 9, 493–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Wang H, Wang Z, He S, Chen S, Liao D, Wang L, Yan J, Liu W, Lei X & Wang X (2012). Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148, 213–227. [DOI] [PubMed] [Google Scholar]
- Tamura K, Makino A, Hullin-Matsuda F, Kobayashi T, Furihata M, Chung S, Ashida S, Miki T, Fujioka T, Shuin T, Nakamura Y & Nakagawa H (2009). Novel Lipogenic Enzyme ELOVL7 Is Involved in Prostate Cancer Growth through Saturated Long-Chain Fatty Acid Metabolism. Cancer Res 69, 8133. [DOI] [PubMed] [Google Scholar]
- Thinon E, Fernandez JP, Molina H & Hang HC (2018). Selective Enrichment and Direct Analysis of Protein S-Palmitoylation Sites. J. Proteome Res 17, 1907–1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson CB 1995. Apoptosis in the pathogenesis and treatment of disease. Science 267, 1456–62. [DOI] [PubMed] [Google Scholar]
- Vandenabeele P, Galluzzi L, Vanden Berghe T & Kroemer G 2010. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat. Rev. Mol. Cell Biol 11, 700–14. [DOI] [PubMed] [Google Scholar]
- Wang H, Sun L, Su L, Rizo J, Liu L, Wang L-F, Wang F-S & Wang X (2014). Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol. Cell 54, 133–46. [DOI] [PubMed] [Google Scholar]
- Wang J, Hao JW, Wang X, Guo H, Sun HH, Lai XY, Liu LY, Zhu M, Wang HY, Li YF, Yu LY, Xie C, Wang HR, Mo W, Zhou HM, Chen S, Liang G & Zhao TJ (2019). DHHC4 and DHHC5 Facilitate Fatty Acid Uptake by Palmitoylating and Targeting CD36 to the Plasma Membrane. Cell Rep 26, 209–221 e5. [DOI] [PubMed] [Google Scholar]
- Xia B, Fang S, Chen X, Hu H, Chen P, Wang H & Gao Z (2016). MLKL forms cation channels. Cell Res 26, 517–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang HQ, Martinez-Ortiz W, Hwang J, Fan X, Cardozo TJ & Coetzee WA (2020). Palmitoylation of the KATP channel Kir6.2 subunit promotes channel opening by regulating PIP2 sensitivity. Proc. Natl. Acad. Sci. U.S.A 117, 10593–10602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon S, Kovalenko A, Bogdanov K & Wallach D (2017). MLKL, the Protein that Mediates Necroptosis, Also Regulates Endosomal Trafficking and Extracellular Vesicle Generation. Immunity 47, 51–65.e7. [DOI] [PubMed] [Google Scholar]
- Zhang L, Katselis GS, Moore RE, Lekpor K, Goto RM, Lee TD & Miller MM (2011). Proteomic analysis of surface and endosomal membrane proteins from the avian LMH epithelial cell line. J. Proteome Res 10, 3973–82. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Chen X, Gueydan C & Han J (2017). Plasma membrane changes during programmed cell deaths. Cell Res 28, 9–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This table is provided as a separate spread sheet.
Columns E-J show OT and IT abundances (area under the curve) in control samples (Control 1–3, n=3) in log2 scale.
Column K-F show OT and IT abundances (area under the curve) in necroptotic samples (Nec1–3, n=3) in log2 scale
Columns Q-V show OT and IT abundances (area under the curve) in control samples (Control 1–3, n=3) in non-transformed scale.
Columns W-AB show OT and IT abundances (area under the curve) in necroptotic samples (Nec1–3, n=3) in non-transformed scale
Columns AC and AD report that average abundances in control and necroptotic samples in non-transformed scale.
Column AE represents the ratio of protein abundance between necroptotic and control cells.
Column AF reports p values calculated by student’s t-test.
Entries with “0” abundance indicate no detection.
This table is provided as a separate spread sheet.
Columns E-J show OT and IT abundances (area under the curve) in control samples (Control 1–3, n=3) in log2 scale.
Columns K-F show OT and IT abundances (area under the curve) in necroptotic samples (Nec1–3, n=3) in log2 scale
Columns Q-V show OT and IT abundances (area under the curve) in control samples (Control 1–3, n=3) in non-transformed scale.
Columns W-AB show OT and IT abundances (area under the curve) in necroptotic samples (Nec1–3, n=3) in non-transformed scale
Columns AC and AD report average abundances in control and necroptotic samples in non-transformed scale.
Column AE represents the ratio of protein abundance between necroptotic and control cells.
Column AF reports p values calculated by student’s t-test.
Entries with “0” abundance indicate no detection.
Data Availability Statement
The Proteomics experiment data are available via ProteomeXchange with identifier PXD022527. The project name is Quantitative proteomics characterization of proteins modified by very long chain fatty acids (VLCFAs) under necroptosis with username: reviewer_pxd022527@ebi.ac.uk and password: MisAktBm. Project DOI: Not applicable