

Abstract
Graph neural networks (GNNs) constitute a class of deep learning methods for graph data. They have wide applications in chemistry and biology, such as molecular property prediction, reaction prediction, and drug–target interaction prediction. Despite the interest, GNN-based modeling is challenging as it requires graph data preprocessing and modeling in addition to programming and deep learning. Here, we present Deep Graph Library (DGL)-LifeSci, an open-source package for deep learning on graphs in life science. Deep Graph Library (DGL)-LifeSci is a python toolkit based on RDKit, PyTorch, and Deep Graph Library (DGL). DGL-LifeSci allows GNN-based modeling on custom datasets for molecular property prediction, reaction prediction, and molecule generation. With its command-line interfaces, users can perform modeling without any background in programming and deep learning. We test the command-line interfaces using standard benchmarks MoleculeNet, USPTO, and ZINC. Compared with previous implementations, DGL-LifeSci achieves a speed up by up to 6×. For modeling flexibility, DGL-LifeSci provides well-optimized modules for various stages of the modeling pipeline. In addition, DGL-LifeSci provides pretrained models for reproducing the test experiment results and applying models without training. The code is distributed under an Apache-2.0 License and is freely accessible at https://github.com/awslabs/dgl-lifesci.
1. Introduction
A large amount of the chemical and biological data corresponds to attributed graphs, e.g., molecular graphs, interaction networks, and biological pathways. Many of the machine learning (ML) tasks that arise in this domain can be formulated as learning tasks on graphs. For example, molecular property prediction can be formulated as learning a mapping from molecular graphs to real numbers (regression) or discrete values (classification);1 molecule generation can be formulated as learning a distribution over molecular graphs;2 reaction prediction can be formulated as learning a mapping from one set of graphs (reactants) to another set of graphs (products).3 A representation is a vector of a user-defined dimensionality. Graph neural networks (GNNs) combine graph structures and features in representation learning and they have been one of the most popular approaches for learning on graphs.4,5 GNNs have also attracted considerable attention in life science and researchers have applied them to many different tasks.1−3,6−12
Despite significant research, it is often challenging for experts in life science to use GNN-based approaches. To unlock the power of GNNs requires clean interfaces for custom datasets and robust and efficient pipelines. This is because developing GNN pipelines by oneself requires a combined skill set of programming, machine learning, and GNN modeling, which is time-consuming to obtain. This calls for a set of ready-to-run programs, which should make little assumption about users’ background.
Prior efforts have greatly lowered the bar for GNN-based modeling in life science, but none of them fully addresses the problem. DeepChem13 is a package for deep learning in drug discovery, materials science, quantum chemistry, and biology. While it implements several GNN models, it still requires users to program. Chainer Chemistry14 is a package for deep learning in biology and chemistry, based on Chainer.15 It only provides a command-line interface for GNN-based regression on molecules and requires users to write code for other tasks. PiNN16 implements a GNN variant for predicting potential energy surfaces and physicochemical properties of molecules and materials. It also requires users to program themselves.
Here, we present a python toolkit named Deep Graph Library (DGL)-LifeSci. It provides high-quality and robust implementations of seven models for molecular property prediction, one model for molecule generation, and one model for chemical reaction prediction. For all of these models and tasks, there is an associated command-line script for predictions on custom datasets without writing a single line of code. DGL-LifeSci also provides pretrained models for all experiments. Compared with previous implementations, it achieves a speed up by up to 6×.
In the following, we first provide a high-level overview of how graph neural networks work over molecules. Then, we discuss the implementation and package features of DGL-LifeSci. After that, we present the results of evaluating DGL-LifeSci in terms of robustness and efficiency. Finally, we conclude with a discussion on future work.
2. Graph Neural Networks over Molecules
GNNs perform graph-based representation learning by combining information from the topology of a graph and the features associated with its nodes and edges. They iteratively update the representation of a node by aggregating representations of its neighbors. As the number of iterations increases, the nodes gain information from an increasingly larger local subgraph.
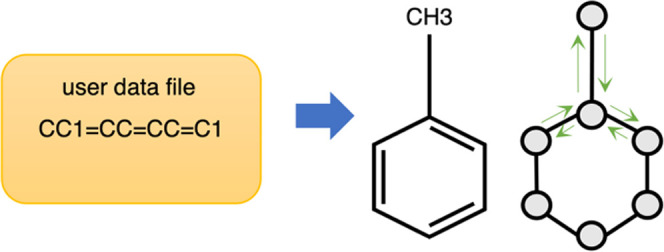
When applying GNNs to molecules as in molecular property prediction, there are two phases—a message passing phase and a readout phase. Figure 1 is an illustration of them.
Figure 1.

Illustration of the message passing phase (left) and readout phase (right).
2.1. Message Passing Phase
The message passing phase updates node representations simultaneously across the entire graph and consists of multiple rounds of message passing. In a round of message passing, the representation of a node is updated by applying learnable functions to its original representation, the representations of its adjacent nodes, and the representations of its incident edges. The operation is similar to gathering messages from adjacent nodes. By performing k rounds of message passing, we can aggregate information from all of the nodes/edges that are within k hops from each node.
2.2. Readout Phase
The readout phase computes a representation for the entire graph. This representation is computed by applying a potentially learnable function to the representations of all of the nodes in the graph, e.g., summation over them. Once we obtain graph representations, we can pass them to a multilayer perceptron (MLP) for final prediction.
3. Package Features
DGL-LifeSci contains four components: (i) a set of ready-to-run scripts for training and inference; (ii) programming APIs for allowing researchers to develop their own custom pipelines and models; (iii) a set of pretrained models that can either be fine-tuned or directly used to perform inference; and (iv) a set of built-in datasets for quick experimentation.
It provides models that can be used to solve three tasks. The first task is molecular property prediction or quantitative structure–activity relationship (QSAR) prediction. This can be formulated as a regression or classification task for single molecules. The second task is molecule generation. The third task is chemical reaction prediction.
3.1. Usage
DGL-LifeSci provides command-line
scripts for each task. They are responsible for invoking the modeling
pipeline, which handles model training and model evaluation. Users
need to prepare their data in a standard format. They can then use
the command-line interface by specifying the path to the data file
along with some additional arguments. For example, below is the command-line
interface for regression and classification problems in molecular
property prediction. Users need to prepare molecules in the form of
SMILES strings with their properties to predict in a CSV file. DGL-LifeSci
also provides an optional support for a hyperparameter
search other than using the default ones. It uses Bayesian optimization
based on hyperopt17 for a hyperparameter
search.
DGL-LifeSci
also provides an optional support for a hyperparameter
search other than using the default ones. It uses Bayesian optimization
based on hyperopt17 for a hyperparameter
search.
4. Implementation
4.1. Dependencies
DGL-LifeSci is developed using PyTorch18 and Deep Graph Library (DGL).19 PyTorch is a general-purpose deep learning framework and DGL is a high-performant GNN library. In addition, it uses RDKit20 for utilities related to cheminformatics.
4.2. Modeling Pipeline and Modules
A general GNN-based modeling pipeline consists of three stages: dataset preparation, model initialization, and model training. The dataset preparation stage involves data loading, graph construction, representation initialization for nodes and edges (graph featurization), and dataset interface construction. The model training stage involves model update, metric computation, and early stopping. As presented in Figure 2, DGL-LifeSci is modularized for these various stages and stage components so as to cater to the need of different uses. While DGL-LifeSci allows users to perform GNN-based modeling without programming, advanced users can also adapt these modules for their own development.
Figure 2.

Overview of modules in DGL-LifeSci and their usage.
4.3. Dataset Preparation
DGL-LifeSci provides dataset interfaces for supporting both built-in datasets and custom datasets. The interfaces are responsible for loading raw data files and invoking graph construction and featurization.
Graph construction and featurization are two important steps for GNN-specific data preparation. DGL-LifeSci provides built-in support for constructing three kinds of graphs for molecules—molecular graphs, distance-based graphs, and complete graphs. In all of these graphs, each node corresponds to an atom in a molecule. In a molecular graph, the edges correspond to chemical bonds in the molecule. The construction of a distance-based graph requires a molecule conformation and there is an edge between a pair of atoms if the distance between them is within a cutoff distance. In a complete graph, every pair of atoms is connected. For graph featurization, DGL-LifeSci allows initializing various node and edge features from atom and bond descriptors. Table 1 gives an overview of them.
Table 1. Descriptors for Feature Initializationa.
| descriptors | possible values |
|---|---|
| atom type | C, N, O, etc. |
| atom degree excluding hydrogen atoms | non-negative integers |
| atom degree including hydrogen atoms | non-negative integers |
| atom explicit valence | non-negative integers |
| atom implicit valence | non-negative integers |
| atom hybridization | S, SP, SP2, SP3, SP3D, SP3D2 |
| total number of hydrogen atoms attached | non-negative integers |
| atom formal charge | integers |
| number of radical electrons of an atom | non-negative integers |
| whether an atom is aromatic | 1 (true), 0 (false) |
| whether an atom is in a ring | 1 (true), 0 (false) |
| atom chiral tag | CW, CCW, unspecified, other |
| atom chirality type | R, S |
| atom mass | non-negative real numbers |
| whether an atom is chiral center | 1 (true), 0 (false) |
| bond type | single, double, triple, aromatic |
| whether a bond is conjugated | 1 (true), 0 (false) |
| whether a bond is in a ring | 1 (true), 0 (false) |
| stereo configuration of a bond | none, any, OZ, OE, CIS, TRANS |
| direction of a bond | none, end-up-right, end-down-right |
For non-numeric discrete-valued descriptors, one-hot encoding is used in featurization. For numeric discrete-valued descriptors, either raw number or one-hot encoding can be used in featurization.
Users can split the dataset into training/validation/test subsets or do so for k-fold cross-validation. DGL-LifeSci provides built-in support for random split, scaffold split, weight split, and stratified split.21 The random split performs a pure random split of a dataset. The scaffold split separates structurally different molecules into different subsets based on their Bemis–Murcko scaffolds.22 The weight split sorts molecules based on their weight and then splits them in order. The stratified split sorts molecules based on their label and ensures that each subset contains nearly the full range of provided labels.
4.4. Models Included
Table 2 lists the models implemented. Graph Convolutional Network (GCN)23 and Graph Attention Network (GAT)24 are two popular GNNs initially developed for node classification and we extend them for graph regression/classification. Originally, GCN and GAT output node representations. We compute graph-level representations from node-level representations by two operations. The first operation performs a weighted sum of the representations of nodes in a graph, where the weights are determined by passing the node representations to a linear layer followed by a sigmoid function. The second operation takes the element-wise maximum of the representations of nodes in a graph. We then concatenate the results of the two operations and pass them to an MLP for final prediction. Neural Fingerprint (NF)1 and Weave25 are among the earliest models that extend rule-based molecular fingerprints with graph neural networks. Message Passing Neural Network (MPNN)9 unifies multiple GNNs for quantum chemistry. AttentiveFP26 extends GAT with gated recurrent units.27
Table 2. Models Implemented.
One difficulty in developing learning-based approaches for molecular property prediction is the gap between an extremely large chemical space and extremely limited labels for molecular properties. It is estimated that the number of drug-like molecules is between 1023 and 1060 while most datasets have less than tens of thousands of molecules in MoleculeNet.21,28−31 Hu et al.32 propose to approach this problem by utilizing millions of unlabeled molecules in pretraining the weights of a GIN model for general molecule representations. One can then fine-tune the model weights for predicting particular properties. We directly include four models pretrained from their work in DGL-LifeSci. The details of the datasets used for pretraining can be found in Section 5.1 of their paper. The models were pretrained with a same strategy for supervised graph-level property prediction as described in Section 3.2.1 of their paper. Each model was pretrained with a different strategy for self-supervised learning as described in Sections 3.1 and 5.2 of their paper. We distinguish the models by the associated strategies for self-supervised learning, which are context prediction, deep graph infomax, edge prediction, and attribute masking.
Junction Tree Variational Autoencoder (JTVAE)2 is an autoencoder that utilizes both a junction tree and a molecular graph for the intermediate representation of a molecule. Weisfeiler–Lehman Network (WLN)3 is a two-stage model for chemical reaction prediction. It first identifies potential bond changes and then enumerates and ranks candidate products.
5. Results and Discussion
5.1. Molecular Property Prediction
We test against six binary classification datasets in MoleculeNet and evaluate the model performance by ROC-AUC averaged over all tasks.21 To evaluate the model performance on unseen structures, we employ the scaffold split and use 80, 10, and 10% of the dataset for training, validation, and test, respectively. We train six models (NF, GCN, GAT, Weave, MPNN, AttentiveFP) from scratch using the featurization proposed in DeepChem, which is described in Table 3. GCN and GAT take initial node features only and they do not take initial edge features. We also fine-tuned the four pretrained GIN models. For non-GNN baseline models, we train an MLP and a k nearest-neighbor classifier (KNN, k = 1) taking Extended-Connectivity Fingerprints (ECFPs).
Table 3. Descriptors Considered in DeepChem Featurization.
| descriptors | possible values |
|---|---|
| atom type (one-hot encoding) | C, N, O, S, F, Si, P, Cl, Br, Mg, Na, Ca, |
| Fe, As, Al, I, B, V K, Tl, Yb, Sb, Sn, | |
| Ag, Pd, Co, Se, Ti, Zn, H, Li, Ge, Cu, | |
| Au, Ni, Cd, In, Mn, Zr, Cr, Pt, Hg, Pb | |
| atom degree excluding hydrogen atoms (one-hot encoding) | 0–10 |
| atom implicit valence (one-hot encoding) | 0–6 |
| atom formal charge | integers |
| number of radical electrons of an atom | non-negative integers |
| whether an atom is aromatic | 1 (true), 0 (false) |
| atom hybridization (one-hot encoding) | SP, SP2, SP3, SP3D, SP3D2 |
| total number of hydrogen atoms attached (one-hot encoding) | 0 - 4 |
| bond type (one-hot encoding) | single, double, triple, aromatic |
| whether a bond is conjugated | 1 (true), 0 (false) |
| whether a bond is in a ring | 1 (true), 0 (false) |
| stereo configuration of a bond (one-hot encoding) | none, any, OZ, OE, CIS, TRANS |
For all of the settings, we perform a hyperparameter search for 32 trials and choose the best hyperparameters based on the performance on the validation set. Within each trial, we train a randomly initialized model and perform an early stopping if the performance on the validation set no longer improves for 30 epochs. Finally, we evaluate the model achieving the best validation performance on the test set. Table 4 presents the summary of the test performance. For reference, we also include the fine-tuning performance reported previously32 and the best performance achieved by GNNs reported in the MoleculeNet paper.21 For Tox21, ToxCast, and SIDER, the MoleculeNet paper reported performance numbers for a random dataset split and the dataset split cannot be reproduced.
Table 4. Test ROC-AUC on Six Datasets from MoleculeNeta.
| model | BBBP | Tox21 | ToxCast | SIDER | HIV | BACE |
|---|---|---|---|---|---|---|
| Models trained from scratch | ||||||
| GCN | 0.63 | 0.77 | 0.62 | 0.58 | 0.76 | 0.84 |
| GAT | 0.68 | 0.71 | 0.64 | 0.52 | 0.76 | 0.84 |
| NF | 0.66 | 0.75 | 0.60 | 0.53 | 0.74 | 0.80 |
| Weave | 0.67 | 0.56 | 0.62 | 0.58 | 0.73 | 0.79 |
| MPNN | 0.65 | 0.70 | 0.59 | 0.54 | 0.74 | 0.85 |
| AttentiveFP | 0.71 | 0.70 | 0.57 | 0.53 | 0.75 | 0.73 |
| Non-GNN baseline | ||||||
|---|---|---|---|---|---|---|
| MLP + ECFP | 0.67 | 0.70 | 0.58 | 0.63 | 0.76 | 0.80 |
| KNN + ECFP | 0.57 | 0.58 | 0.52 | 0.56 | 0.57 | 0.66 |
| Pretrained models fine-tuned | ||||||
|---|---|---|---|---|---|---|
| GIN + context prediction | 0.63 | 0.75 | 0.64 | 0.61 | 0.77 | 0.86 |
| GIN + deep graph infomax | 0.72 | 0.78 | 0.59 | 0.63 | 0.76 | 0.71 |
| GIN + edge prediction | 0.70 | 0.80 | 0.59 | 0.66 | 0.72 | 0.86 |
| GIN + attribute masking | 0.72 | 0.75 | 0.58 | 0.58 | 0.75 | 0.74 |
| Previously reported results | ||||||
|---|---|---|---|---|---|---|
| GIN + context prediction32 | 0.69 | 0.78 | 0.66 | 0.63 | 0.80 | 0.85 |
| GIN + deep graph infomax32 | 0.68 | 0.78 | 0.65 | 0.61 | 0.78 | 0.80 |
| GIN + edge prediction32 | 0.67 | 0.78 | 0.67 | 0.63 | 0.78 | 0.79 |
| GIN + attribute masking32 | 0.67 | 0.78 | 0.65 | 0.64 | 0.77 | 0.80 |
| Best GNN in MoleculeNet paper21 | 0.69 | 0.76 | 0.81 | |||
The best numbers are in bold.
For all datasets, the best ROC-AUC achieved by DGL-LifeSci models is higher than the ROC-AUC numbers achieved by the two baseline models. The best fine-tuned pretrained model either outperforms the best model trained from scratch or achieves a comparable performance, which suggests the effectiveness of pretraining on a large amount of unlabeled molecules. The performance of the fine-tuned models is comparable with the previously reported results. On BBBP, HIV, and BACE, the best ROC-AUC achieved by DGL-LifeSci models is consistently higher than the best ROC-AUC achieved by GNNs in the MoleculeNet paper.
5.2. Reaction Prediction
We test WLN against USPTO33 dataset following the setting in the original work.3 WLN is a two-stage model for reaction prediction. The first stage identifies candidate reaction centers, i.e., pairs of atoms that lose or form a bond in the reaction. The second stage enumerates candidate products from the candidate reaction centers and ranks them. We achieve comparable performance for both stages as in Table 5.
Table 5. Test Top-k Accuracy (%) of WLN on USPTO.
| reaction
center prediction |
candidate
ranking |
||||||
|---|---|---|---|---|---|---|---|
| implementations | top 6 | top 8 | top 10 | top 1 | top 2 | top 3 | top 5 |
| original | 89.8 | 92.0 | 93.3 | 85.6 | 90.5 | 92.8 | 93.4 |
| DGL-LifeSci | 91.2 | 93.8 | 95.0 | 85.6 | 90.0 | 91.7 | 92.9 |
5.3. Molecule Generation
We test JTVAE against a ZINC34 subset for reconstructing input molecules.2 We achieve an accuracy of 76.4% while the authors’ released code achieved an accuracy of 74.4%.
5.4. Training Speed
We compare the modeling efficiency of DGL-LifeSci against previous implementations, including original implementations and DeepChem. All experiments record the averaged training time of one epoch. The testbed is one AWS EC2 p3.2 × large instance (one NVidia V100 GPU with 16GB GPU RAM and 8 VCPUs). Due to the differences in the combinations of models and datasets across implementations, we only evaluate on a subset of the experiments. The preliminary results in Table 6 show that DGL-LifeSci achieves a comparable or superior training speed, up to 6×.
Table 6. Epoch Training Time in Seconds.
| experiment | dataset | previous implementation | DGL-LifeSci | speed up |
|---|---|---|---|---|
| Molecular property prediction | ||||
| NF | HIV | 5.8 (DeepChem 2.3.0) | 2.5 | 2.3× |
| attentiveFP | aromaticity26 | 6.0 | 1.0 | 6.0× |
| Reaction prediction | ||||
|---|---|---|---|---|
| WLN for reaction center prediction | USPTO | 11 657 | 2315 | 5.0× |
| Molecule generation | ||||
|---|---|---|---|---|
| JTVAE | ZINC subset | 44666 | 44 843 | 1.0× |
6. Conclusions
Here, we present DGL-LifeSci, an open-source Python toolkit for deep learning on graphs in life science. In the current version of DGL-LifeSci, we support GNN-based modeling for molecular property prediction, reaction prediction, and molecule generation.
With command-line interfaces, users can perform efficient modeling on custom datasets without programming a single line of code. Advanced users can also adapt highly modularized building blocks for their own development.
In the current implementations of DGL-LifeSci, the primary focus is on small molecules. In the future, we aim to extend the support to other graphs in life science like proteins and biological networks. This will open up a much richer set of tasks in life science.
7. Data and Software Availability
The datasets and models are publicly available at https://github.com/awslabs/dgl-lifesci. The scripts for reproducing the experiments are available in the following examples.
Molecular property prediction: examples/property_prediction/moleculenet.
Reaction prediction: examples/reaction_prediction/rexgen_direct.
Molecule generation: examples/generative_models/jtvae.
RDKit, PyTorch, and DGL are all open-source software.
Acknowledgments
Since its release, DGL-LifeSci has received many valuable suggestions and contributions from the community. The authors are grateful to the support of our users and contributors. For details on the community contributions not covered in this manuscript and in the future, one may refer to https://github.com/awslabs/dgl-lifesci/blob/master/CONTRIBUTORS.md.
The authors declare no competing financial interest.
References
- Duvenaud D. K.; Maclaurin D.; Iparraguirre J.; Bombarell R.; Hirzel T.; Aspuru-Guzik A.; Adams R. P.. Convolutional Networks on Graphs for Learning Molecular Fingerprints, Advances in Neural Information Processing Systems: Proceedings of the 28th International Conference on Neural Information Processing Systems, ACM, 2015; pp 2224–2232.
- Jin W.; Barzilay R.; Jaakkola T.. Junction Tree Variational Autoencoder for Molecular Graph Generation, Proceedings of the 35th International Conference on Machine Learning, PMLR, Stockholmsmässan, Stockholm, 2018; pp 2323–2332.
- Coley C.; Jin W.; Rogers L.; Jamison T. F.; Jaakkola T. S.; Green W. H.; Barzilay R.; Jensen K. F. A graph-convolutional neural network model for the prediction of chemical reactivity. Chem. Sci. 2019, 10, 370–377. 10.1039/C8SC04228D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z.; Pan S.; Chen F.; Long G.; Zhang C.; Yu P. S. A comprehensive survey on graph neural networks. IEEE Trans. Neural Networks Learn. Syst. 2021, 32, 4–24. 10.1109/TNNLS.2020.2978386. [DOI] [PubMed] [Google Scholar]
- Zhou J.; Cui G.; Hu S.; Zhang Z.; Yang C.; Liu Z.; Wang L.; Li C.; Sun M. Graph neural networks: A review of methods and applications. AI Open 2020, 1, 57–81. 10.1016/j.aiopen.2021.01.001. [DOI] [Google Scholar]
- Sun M.; Zhao S.; Gilvary C.; Elemento O.; Zhou J.; Wang F. Graph convolutional networks for computational drug development and discovery. Briefings Bioinf. 2019, 21, 919–935. 10.1093/bib/bbz042. [DOI] [PubMed] [Google Scholar]
- Zitnik M.; Agrawal M.; Leskovec J. Modeling polypharmacy side effects with graph convolutional networks. Bioinformatics 2018, 34, i457–i466. 10.1093/bioinformatics/bty294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokes J. M.; et al. A Deep Learning Approach to Antibiotic Discovery. Cell 2020, 180, 688–702. 10.1016/j.cell.2020.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmer J.; Schoenholz S. S.; Riley P. F.; Vinyals O.; Dahl G. E.. Neural Message Passing for Quantum Chemistry, Proceedings of the 34th International Conference on Machine Learning, PMLR, 2017; pp 1263–1272.
- Shui Z.; Karypis G.. Heterogeneous Molecular Graph Neural Networks for Predicting Molecule Properties, 2020 IEEE International Conference on Data Mining (ICDM), IEEE, Los Alamitos, CA, 2020; pp 492–500.
- Feinberg E. N.; Sur D.; Wu Z.; Husic B. E.; Mai H.; Li Y.; Sun S.; Yang J.; Ramsundar B.; Pande V. S. PotentialNet for Molecular Property Prediction. ACS Cent. Sci. 2018, 4, 1520–1530. 10.1021/acscentsci.8b00507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingraham J.; Garg V.; Barzilay R.; Jaakkola T.. Generative Models for Graph-Based Protein Design, Advances in Neural Information Processing Systems 32, PMLR, 2019; pp15820–15831.
- Ramsundar B.; Eastman P.; Walters P.; Pande V.. Deep Learning for the Life Sciences, O’Reilly Media, Inc., 2019. [Google Scholar]
- Chainer Chemistry: A Library for Deep Learning in Biology and Chemistry. https://github.com/chainer/chainer-chemistry, [Online; accessed 29-October-2020].
- Tokui S.; Oono K.; Hido S.; Clayton J. Chainer: a Next-Generation Open Source Framework for Deep Learning. Workshop on Systems for ML at NeurIPS. 2015.
- Shao Y.; Hellström M.; Mitev P. D.; Knijff L.; Zhang C. PiNN: A Python Library for Building Atomic Neural Networks of Molecules and Materials. J. Chem. Inf. Model. 2020, 60, 1184–1193. 10.1021/acs.jcim.9b00994. [DOI] [PubMed] [Google Scholar]
- Bergstra J.; Yamins D.; Cox D. D.. Making a Science of Model Search: Hyperparameter Optimization in Hundreds of Dimensions for Vision Architectures, Proceedings of the 30th International Conference on Machine Learning, PMLR, 2012; pp 115–123.
- Paszke A.et al. PyTorch: An Imperative Style, High-Performance Deep Learning Library, Advances in Neural Information Processing Systems 32, 2019; pp 8026–8037.
- Wang M.; Zheng D.; Ye Z.; Gan Q.; Li M.; Song X.; Zhou J.; Ma C.; Yu L.; Gai Y.; Xiao T.; He T.; Karypis G.; Li J.; Zhang Z.. Deep Graph Library: A Graph-Centric, Highly-Performant Package for Graph Neural Networks, arXiv.1909.01315, arXiv.org e-Print archive, https://arxiv.org/abs/1909.01315.2020.
- RDKit: Open-source cheminformatics. http://www.rdkit.org, [Online; accessed 30-October-2020].
- Wu Z.; Ramsundar B.; Feinberg E. N.; Gomes J.; Geniesse C.; Pappu A. S.; Leswing K.; Pande V. MoleculeNet: a benchmark for molecular machine learning. Chem. Sci. 2018, 9, 513–530. 10.1039/C7SC02664A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bemis G. W.; Murcko M. A. The Properties of Known Drugs. 1. Molecular Frameworks. J. Med. Chem. 1996, 39, 2887–2893. 10.1021/jm9602928. [DOI] [PubMed] [Google Scholar]
- Kipf T. N.; Welling M.. Semi-Supervised Classification with Graph Convolutional Networks, International Conference on Learning Representations, OpenReview.net, 2017.
- Veličković P.; Cucurull G.; Casanova A.; Romero A.; Lió P.; Bengio Y.. Graph Attention Networks, International Conference on Learning Representations, Apollo, 2018.
- Kearnes S.; McCloskey K.; Berndl M.; Pande V.; Riley P. Molecular graph convolutions: moving beyond fingerprints. J. Comput.-Aided Mol. Des. 2016, 30, 595–608. 10.1007/s10822-016-9938-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Z.; Wang D.; Liu X.; Zhong F.; Wan X.; Li X.; Li Z.; Luo X.; Chen K.; Jiang H.; Zheng M. J. Med. Chem. 2020, 63, 8749–8760. 10.1021/acs.jmedchem.9b00959. [DOI] [PubMed] [Google Scholar]
- Cho K.; van Merriënboer B.; Gulcehre C.; Bahdanau D.; Bougares F.; Schwenk H.; Bengio Y.. Learning Phrase Representations using RNN Encoder–Decoder for Statistical Machine Translation, Proceedings of the 2014 Conference on Empirical Methods in Natural Language Processing (EMNLP), Association for Computational Linguistics, 2014; pp 1724–1734.
- Polishchuk P. G.; Madzhidov T. I.; Varnek A. Estimation of the size of drug-like chemical space based on GDB-17 data. J. Comput.-Aided Mol. Des. 2013, 27, 675–679. 10.1007/s10822-013-9672-4. [DOI] [PubMed] [Google Scholar]
- Steve O’Hagan D. B. K. Analysing and Navigating Natural Products Space for Generating Small, Diverse, But Representative Chemical Libraries. Biotechnol. J. 2018, 13, 1700503 10.1002/biot.201700503. [DOI] [PubMed] [Google Scholar]
- Reymond J.-L. The Chemical Space Project. Acc. Chem. Res. 2015, 48, 722–730. 10.1021/ar500432k. [DOI] [PubMed] [Google Scholar]
- Dobson C. M. Chemical space and biology. Nature 2004, 432, 824–828. 10.1038/nature03192. [DOI] [PubMed] [Google Scholar]
- Hu W.; Liu B.; Gomes J.; Zitnik M.; Liang P.; Pande V.; Leskovec J.. Strategies for Pre-training Graph Neural Networks, International Conference on Learning Representations, OpenReview.net, 2020.
- Patent reaction extraction: downloads. https://bitbucket.org/dan2097/patent-reaction-extraction/downloads, 2014.
- Sterling T.; Irwin J. J. ZINC 15 – Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. 10.1021/acs.jcim.5b00559. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets and models are publicly available at https://github.com/awslabs/dgl-lifesci. The scripts for reproducing the experiments are available in the following examples.
Molecular property prediction: examples/property_prediction/moleculenet.
Reaction prediction: examples/reaction_prediction/rexgen_direct.
Molecule generation: examples/generative_models/jtvae.
RDKit, PyTorch, and DGL are all open-source software.