

Abstract
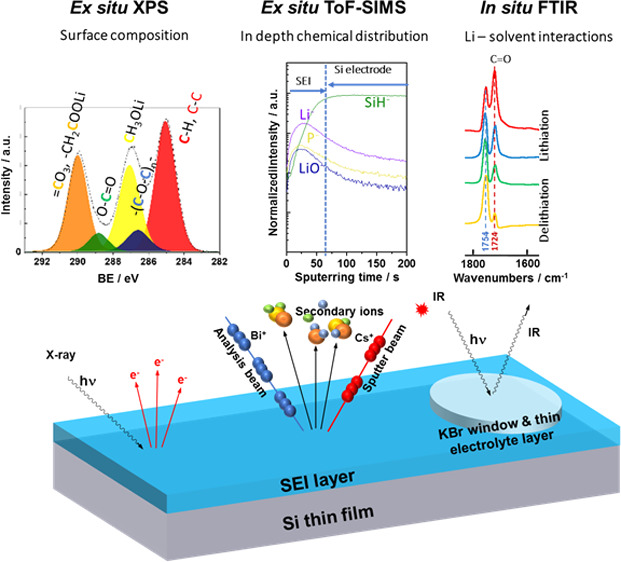
A solid electrolyte interphase (SEI) layer on Si-based anodes should have high mechanical properties to adapt the volume changes of Si with low thickness and good ionic conductivity. To better understand the influence of carbonate solvents on the SEI composition and mechanism of formation, systematic studies were performed using dimethyl carbonate (DMC) or propylene carbonate (PC) solvent and LiPF6 as a salt. A 1 M LiPF6/EC-DMC was used for comparison. The surface chemical composition of the Si electrode was analyzed at different potentials of lithiation/delithiation and after a few cycles. Ex situ X-ray photoelectron spectroscopy and time-of-flight secondary ion mass spectrometry results demonstrate that a thinner and more stable SEI layer is formed in LiPF6/DMC. The in situ Fourier transform infrared spectroscopy proves that the coordination between Li+ and DMC is weaker, and fewer DMC molecules take part in the formation of the SEI layer. The higher capacity retention during 60 cycles and less significant morphological modifications of the Si electrode in 1 M LiPF6/DMC compared to other electrolytes were demonstrated, confirming a good and stable interfacial layer. The possible surface reactions are discussed, and the difference in the mechanisms of formation of SEI in these three various electrolytes is proposed.
1. Introduction
Silicon is one of the most promising candidates for the next generation of negative electrodes for Li-ion batteries due to its remarkably high specific capacity (reaching 3579 mAh g–1 at room temperature, which corresponds to the formation of Li3.75Si alloy), high safety, and low cost.1−5 However, the cycle stability of the Si anode is still limited by the huge volume expansion of Si (around 280% upon lithiation) resulting from the high ratio of Li in a Li–Si alloy.6−9 The volume expansion causes material pulverization,10 electrical contact loss, and poor cycle performance.11−13 Moreover, the volume expansion/contraction during each cycle of lithiation/delithiation process also damages the solid electrolyte interphase (SEI) layer formed on the surface of the Si electrode.14 Thus, the SEI layer has to reconstruct on an uneven electrode surface and consumes an extra amount of lithium and electrolyte components during every cycle, which leads to irreversible capacity.3,15 The irreversible capacity observed during the first lithiation can be as high as 30%.12 The composition of the SEI depends on the electrode material,16 the nature of the electrolyte (salt, solvent, additive),17,18 and the conditions of lithiation/delithiation.11 The SEI is formed by the electrochemical reduction of the electrolyte at low potentials (close to 0.5 V).11 The properties of the SEI can significantly influence the reversible capacity (columbic efficiency and energy efficiency) and battery cycle life.16,19 Therefore, understanding the SEI formation mechanism and interfacial properties are considered as one of the key parameters for Li-ion battery design. The formation and composition of SEI have been widely studied on graphite electrode materials but much less on Si-based electrode materials. For commercial graphite anode, ethylene carbonate (EC) is an indispensable electrolyte co-solvent because it plays a key role in the formation of passive layer on the graphite surface to protect the graphite from Li-solvent co-intercalation, graphite exfoliation, and continuous decomposition of the electrolyte.20 Thus, the other carbonate solvents such as DMC, PC, and diethyl carbonate (DEC) cannot be applied without EC for graphite anode.20,21 However, they could be used as the electrolyte solvents alone for the Si anode because Si is not a layered structure. The electrolyte for the Si anode is required to form the SEI layer that has better mechanical properties to adapt the volume changes and has lower thickness allowing better electrode conductivity.19 Therefore, different kinds of electrolytes were employed on the Si-based anodes. Ohara et al.(22) applied two different electrolytes 1 M LiClO4/PC and 1 M LiClO4/EC-DMC (1:1) on Si film electrodes. The electrodes in both electrolytes exhibit long stable cycle life.22 Schroder et al.(23) applied the 1 M LiPF6/EC DEC (1:1) electrolyte on Si wafer electrodes and tested the cells by linear sweep voltammetry (LSV), cyclic voltammetry (CV), and chronoamperometry (CA) methods. X-ray photoelectron spectroscopy (XPS) data showed that the ratios of different species in the SEI layers became quite different after these electrochemical treatments.23 Our research group previously employed 1 M LiClO4/PC and 1 M LiPF6/EC-DMC electrolytes on a-Si:H and Si nanowire electrodes and analyzed the SEI layers by XPS and time-of-flight secondary ion mass spectrometry (ToF-SIMS).24 These studies allowed to conclude that the SEI layer formed in the PC-based electrolytes was thicker and richer in alkyl carbonate species than that formed in EC:DMC-based electrolytes, which was mainly composed of salt decomposition products (LiCl or LiF).
In order to improve the Si-based electrode performances and the SEI properties on these electrodes, some electrolyte additives were also widely investigated for Si-based electrode materials such as vinylene carbonate (VC)25−31 and fluoroethylene carbonate (FEC).32−42 These additives have some advantages of decomposition potentials higher than the main electrolyte solvents. However, the addition of the VC leads to higher cell impedance due to the formation of the thicker SEI layer.43 The FEC in the electrolyte results in gas evolution at high temperatures, fast FEC consumption, and finally a cell failure.34 Therefore, the understanding of the reaction mechanisms of each single electrolyte component, the formulation of new electrolytes, and a good approach in terms of analytical methods and fundamental studies of surface chemistry are necessary to optimize the performance of the Si electrode.
The efficient approach in the chemical and structural characterization of the SEI layer is very important but not straightforward. For example, a lot of information about the organic species on the SEI layer could be obtained by the Fourier transform infrared spectroscopy (FTIRS) method.44,45In situ FTIRS was already applied for SEI studies on the Si electrode.41,46−49 Another powerful technique in characterization of the surface chemistry of electrode materials (organic and inorganic species) is XPS.15,24,45,50−54 However, XPS has also different limitations: it is an ultrahigh vacuum ex situ technique, i.e., the sample needs to be rinsed and dried before analysis, which can lead to some surface modifications. Moreover, some of the peaks corresponding to different species can overlap (e.g., the Si–O or Si–F bonds in the region of the Si 2p core level like demonstrated hereafter). ToF-SIMS, an extremely surface-sensitive analytical method, can be used to analyze both surface and bulk electrode modifications (by means of ion in-depth profiling).24,33,55,56 However, ToF-SIMS as a technique under vacuum has similar limitations as XPS.
The ex situ techniques such as scanning electron microscopy (SEM) and atomic force microscopy (AFM) are common techniques to observe the surface morphology of the electrode and the SEI layer.16,57,58 As the surface topography might be changed after solvent washing and the sample exposition to the ambient air, several in situ techniques such as in situ electrochemical-AFM (EC-AFM) and in situ scanning transmission electron microscopy (STEM) can be applied to follow the surface modifications induced by lithiation/delithiation processes.59−62 It should be noticed that to have a good insight into the formation of the SEI layer, the combination of different ex situ and in situ methods is necessary. Moreover, a good choice of model electrode materials (e.g., thin films without binders) is necessary to avoid the ambiguities in the surface chemical and morphological characterization and to better understand the influence of the intrinsic properties of electrode materials on the SEI layer formation.
In this work, the systematic studies of the surface characterization of the Si thin film electrode in three electrolytes (1 M LiPF6/EC-DMC, 1 M LiPF6/DMC, and 1 M LiPF6/PC) were performed in order to better understand the influence of solvent on the SEI layer composition. The application of a model thin film electrode allowed to avoid interference from other electrode components and additives such as a binder and to determine more easily the different surface reaction mechanisms. It should be noticed that the surface modifications of commercial composite Si-based electrodes can vary slightly. The analyses of the Si thin film were performed at different lithiation and delithiation potentials and after several cycles (5 cycles) by XPS, in situ microscope FTIRS, and ToF-SIMS. The morphology of the Si thin film electrode was also examined by SEM. The capacity and Coulombic efficiency induced by cycling of the Si thin film electrode were also tested as a function of electrolyte composition.
2. Results and Discussion
2.1. Influence of Lithiation/Delithiation on the Surface Modifications of Si by XPS
2.1.1. Cyclic Voltammetry of the Si Thin Film Electrode
The first cycle (CV curves) performed on Si thin film electrodes in different electrolytes (1 M LiPF6/EC-DMC, 1 M LiPF6/DMC, and 1 M LiPF6/PC) is shown in Figure 1. The two broad cathodic peaks particularly visible for the EC-DMC electrolyte between 0.3 and 2.0 V (also shown in the inset of Figure 1) correspond principally to the decomposition of the electrolyte and the formation of the SEI layer.63 The peak corresponding to the electrolyte decomposition on the Si-based electrodes is the peak observed frequently at around 0.75 V.11,24,41,49,64 Here, for the EC-DMC electrolyte, this peak is observed between the C1 and C2 points. A reduction of a small quantity of surface contaminations and/or traces of water occurring at higher potentials of around 1.5–1.0 V cannot be completely ruled out.24,41 The most intense, narrow cathodic peaks between 0.01 and 0.4 V correspond to the formation of Li–Si amorphous phases (LixSi),65−67 whereas the high intensity two anodic peaks between 0.1 and 0.6 V corresponds to the phase transfer from the Li–Si alloy to amorphous Si.10,68−71 It should be noted that CV curves do not show a significant influence of the electrolyte on their shapes. To demonstrate the differences in the surface composition of the Si electrode cycled in these three different electrolytes, six points of lithiation (cathodic, C1–C4) and delithiation (anodic, A1 to A2) were chosen for XPS analysis:
-
(i)
point C1 (∼0.9 V) corresponding to the state after electrolyte decomposition and SEI formation and point C2 (∼0.25 V) to the state before lithiation,
-
(ii)
points C3 (∼0.08 V) and C4 (∼0.01 V) corresponding to the state after the first and the second lithation peak, and
-
(iii)
point A1 (at ∼0.015 V) corresponding to the beginning of the delithiation process and point A2 (∼1.0 V) to the end of the delithiation process.
Figure 1.

First CV cycle for Si thin film electrodes performed in different electrolytes: 1 M LiPF6/EC-DMC, 1 M LiPF6/DMC, or 1 M LiPF6/PC (scan rate v = 0.02 mV s–1).
.
2.1.2. SEI Layer Formation in DMC-Based Electrolyte
The high-resolution XPS spectra of Si 2p, C 1s, F 1s, and O 1s for pristine Si electrodes and for the Si electrode after the formation of the SEI layer (point C2) and before delithiation (point A1) and after delithiation (point A2) in a 1 M LiPF6/DMC electrolyte are shown in Figure 2. For clarity of presentation, the detailed decomposition was only presented here for selected stages of lithiation/delithiation of Si in 1 M LiPF6/DMC. The XPS decompositions (C 1s, F 1s, and O 1s peaks) for all electrolytes and at all stages of lithiation/delithiation (points C1 to C4 and A1, A2) are presented in the Supporting Information (Figures S2–S4). All carbon, oxygen, fluorine, and silicon characteristic species, their binding energies (BE), and full width at half-maximum (FWHM) used in the peak decomposition are presented in Table 1.
Figure 2.

High resolution Si 2p, C 1s, F 1s, and O 1s core level spectra obtained on the pristine surface of the Si thin film electrode and on Si thin film after different stages of lithiation/delithiation (points C2, A1, and A2) in 1 M LiPF6/DMC.
Table 1. Binding Energies (BE) and Full Width at Half-Maximum (FWHM) of Different Species in C 1s, F 1s, O 1s, and Si 2p XPS Spectra.
| C species C 1s | C–H, C–C | C–O ether linkage | –OCH3 | O–C=O | –CH2OCO2–, Li2CO3 |
|---|---|---|---|---|---|
| BE (±0.1 eV) | 285.0 | 286.5 | 287.1 | 288.8 | 290.0 |
| FWHM = 1.5 ± 0.2 eV | |||||
| O species O 1s | Li–O | ROLi, LixSiOy | C=O, P=O | Si–O, C–O | RCH2OCO2Li |
|---|---|---|---|---|---|
| BE (±0.1 eV) | 528.1 | 530.5 | 531.8 | 532.9 | 533.6 |
| FWHM = 1.6 ± 0.2 eV | |||||
| F species F 1s | Li–F | SiOF | P–F |
|---|---|---|---|
| BE (±0.1 eV) | 685.0 | 686.8 | 688.1 |
| FWHM = 1.75 ± 0.25 eV | |||
| S species Si 2p3/2 | Si0 | Si+ | Si2+ | Si4+ | SiOxFy |
|---|---|---|---|---|---|
| BE (±0.1 eV) pristine sample | 99.1 | 100.2 ΔBE = BESi+ – BESi0 = 1.1 | 101.8 ΔBE = BESi2+ – BESi0 = 2.7 | 103.5 ΔBE = BESi4+ – BESi0 = 4.4 | |
| BE (± 0.1 eV) cycled samples | 97.8 | 99.3 ΔBE = BESi+ – BESi0 = 1.5 | 100.8 ΔBE = BESi2+ – BESi0 = 3.0 | 102.9 ΔBE = BESi4+ – BESi0 = 5.1 | 104.7 ΔBE = BESiOxFy – BESi0 = 6.9 |
| FWHM (eV) | 1.0 ± 0.2 | 1.6 ± 0.2 | |||
In Figure 2, the Si 2p spectrum displays Si 2p3/2 and Si 2p1/2 spin–orbit doublet. A theoretical value (1:2) of the area ratio of the Si 2p1/2:Si 2p3/2 peak was fixed for decomposition. The decomposition of the Si 2p core level peak shows that the surface of the pristine Si thin film electrode prepared by magnetron sputtering is covered by a thin native Si oxide layer. The lowest binding energy peak at ∼99.1 eV (Si 2p3/2) can be attributed to Si064,72 and three higher binding energy peaks (Si 2p3/2) to different Si oxides (Si+ at ∼100.2 eV, Si2+ at ∼101.8 eV, and Si4+ at ∼103.5 eV, Table 1).69,72−75 After the lithiation (C2) and delithiation (A1 and A2), the SEI layer was formed on the surface of the Si thin film. The intensity of the Si signal decrease significantly, but it could still be detected from the electrode bulk, probably due to the fractures, pores in the SEI layer, or its not homogeneous formation. After correction of the binding energy versus carbon peak (C–C at 285.0 eV), the Si 2p peaks shift to lower binding energies because of the differential charging effects on different compounds formed on the surface of the Si electrode.24,69 Therefore, after electrochemical processes, these Si 2p peaks were assigned to different Si compounds by calculating the difference in the binding energies corresponding to Si0 and other Si peaks (ΔBE = BESi0 – BESi2+ or 4+ as shown in Table 1). Then, the Si 2p3/2 peak at ∼97.8 eV was assigned to Si0, a peak at ∼100.8 eV to Si2+ (Si–O, Si–F, or LiSixOy).69,73,74,76 The Si 2p3/2 peak at ∼102.9 eV could be assigned to Si4+ oxide and fluoride.73,76 The peak at ∼104.7 eV corresponds to SiOxFy.72,73,77
The C 1s profile (Figure 2) shows large C–H, C–C peak (285.0 eV) and small peaks corresponding to the −OCH3 (∼287.1 eV) and O–C=O (∼288.9 eV) contaminations on the pristine Si sample.3,4,24,78,79 After the electrolyte decomposition and SEI formation processes (point C2), the −OCH3 peak becomes very intense principally due to the electrochemical reduction of DMC solvent according to reaction (3) shown in Scheme 1.20,41,80 A small amount of −CH2OCO2–, Li2CO3 (∼290.0 eV) can also be observed.3,4,11,24,79,81,82 The most probable principal component here is Li2CO3 due to the decomposition of DMC according to reaction (5). The relative intensities of O–C=O and −CH2OCO2–, Li2CO3 peaks becomes much higher after lithiation (at point A1). This phenomenon could be related to the changes of SEI layer composition during the lithiation process. After delithiation (at point A2), the −CH2OCO2–, Li2CO3 peak disappears and the intensity of O–C=O peak decreases, while the intensity of the −OCH3 peak increases. It probably indicates that the −CH2OCO2– and Li2CO3 species decomposed or some new C–H, C–C, and −OCH3 species formed and covered the surface during the delithiation process on the Si electrode in the 1 M LiPF6/DMC electrolyte.
Scheme 1. Reduction Reactions on the Si Thin Film Electrode with Different Electrolytes (1 M LiPF6/EC-DMC, 1 M LiPF6/DMC, and 1 M LiPF6/PC).

There is no F on the pristine Si sample. However, the high intensity of the F 1s peak appears already after the first stage of lithiation (point C2). This peak can be decomposed in three components: Li–F (at ∼685.1 eV),3,4,11,24,79,82 SiOF (at ∼686.8 eV),73,74,76,83 and P–F (at ∼688.1 eV)3,4,24,79,82 as shown in Table 1. A very high intensity of the SiOF component (e.g., SiOxFy) at different stages of lithiation/delithiation illustrates that the SEI layer formed on the Si electrode in 1 M LiPF6/DMC samples is not very thick. The P–F signal (at ∼688.1 eV) keeps at a relatively low intensity during the whole cycle, and the Li–F signal (∼685.1 eV) increases during the lithiation process and decreases during the delithiation process, demonstrating a formation and decomposition of LiF salt according to reactions (11)–(15).31,41,84,85
The O 1s profile for the pristine Si sample shows as a major component the peak at ∼532.9 eV corresponding to the Si–O bonding, which is in agreement of the Si 2p core level peak where the presence of SiOx is observed.3,4,24,81,82 The other two peaks at around 533.6 and 531.8 eV correspond to −CH2OCO2– and C=O surface organic compounds and/or organic surface contaminations, respectively (Table 1). After electrolyte decomposition and SEI formation processes (point C2), RCH2OCO2Li, P=O species, and Li2CO3 were formed. Thus, the RCH2OCO2Li (∼533.5 eV) and C=O, P=O peaks show the higher intensity. After the lithiation process, a new peak corresponding to ROLi, Li4SiO4 (∼530.5 eV) appears.4,20,73,82,86,87 Then, after the delithiation process, this peak still exists but shows a slightly lower intensity. The formation of silicates (Li4SiO4) according to reaction (18) cannot be ruled out. The RCH2OCO2Li (∼533.6 eV) peak decreases, which is in agreement with the C 1s core level peak where the −CH2OCO2– peak is observed.
2.1.3. SEI Layer Evolution in EC-DMC, DMC and PC-Based Electrolytes
To discuss the surface modifications of the Si electrode, the evolution of the amount of Si was measured by XPS on the Si thin film pristine electrode surface and at different stages of lithiation and delithiation of Si in three electrolytes. To do so, the atomic percentage of Si (a total Si 2p core level signal) was calculated. From the Si 2p intensity variation (as observed in Figure 2 and Figure S5), the SEI thickness modifications can be deduced. As discussed above after the formation of the SEI layer (C2) or at a lithiated state (A1), the principal Si 2p components are the Si oxide and Si fluoride species, which are difficult to be distinguished due to their close binding energies and problems of overlapping.73,76 Thus, the changes of Si-related species on the Si electrode as a function of different electrolytes are not straightforward. However, it can be concluded that the SEI layer is not stable during the first cycle of lithiation and delithiation in these three electrolytes and undergoes thickening and thinning, respectively (as shown in Figure S5). At the end of the delithiation process, the Si 2p signal is considerably attenuated compared with the C1 point confirming the irreversible surface modifications after the first cycle. A similar behavior was observed on the nanosilicon composite electrodes cycled in LiPF6/EC-DEC.3,11,64,88 This dynamic behavior of the SEI layer, showing its thickening and thinning during lithiation and delithiation, respectively, was also observed on the other types of negative electrode materials.52
To compare other surface components with the Si-related components formed on Si electrodes cycled in three different electrolytes (1 M LiPF6/EC-DMC, 1 M LiPF6/DMC, and 1 M LiPF6/PC), the relative intensity ratios for C 1s, F 1s, and O 1s peaks were calculated for different lithiation/delithiation potentials (C1–C4, A1, A2) (Figure 3). The details of peak decompositions are shown in the Supporting Information (Figures S2–S4). From Figure 3 for the C 1s peak, it can be observed that the major species formed on the Si electrode cycled in 1 M LiPF6/EC-DMC are RCH2OCO2Li, Li2CO3, −OCH3, and a C–O ether link, which are typical species originated from an electrochemical decomposition of EC and DMC solvents according to reactions (1)–(4), (9), (10), (19), and (20).20,41,80,84,85,87 The main species on the Si electrode cycled in 1 M LiPF6/DMC is −OCH3 due to the electrochemical decomposition of DMC according to reaction (3).20,41 The quantity of RCH2OCO2Li species is lower and no signal of the C–O ether link (∼286.5 eV) is observed on the Si sample cycled in 1 M LiPF6/DMC in comparison to the other two electrolytes. The ether species are clearly evidenced in PC- and EC-DMC-based electrolytes in agreement with previous works.82,89 It is reported that LiPF6 could react with solvents EC and DMC differently according to reactions (8), (11), (12), (19), and (20).84,85 A component with a C–O ether link could form in the electrolyte with a ring EC structure but could not form with only linear DMC molecules, which supports the XPS results. The electrode in 1 M LiPF6/PC also shows the C–O ether link during the cycling. Thus, reaction (21) between LiPF6 and ring structure PC molecules could be inferred. On the surface of the Si electrode cycled in 1 M LiPF6/PC, the peaks corresponding to RCH2OCO2Li, Li2CO3, according to electrochemical reactions (6) and (7), increase during the lithiation and then decrease during the delithiation, indicating that the thickness of the SEI layer changes.
Figure 3.

A comparative analysis of the relative intensity ratios of different species for C 1s, F 1s, and O 1s core levels peaks obtained for Si electrodes lithiated and delithiated at different potentials (C1–C4, A1, A2) in three different electrolytes (1 M LiPF6/EC-DMC, 1 M LiPF6/DMC, and 1 M LiPF6/PC).
Looking at the presence and relative intensity ratios of F-like components (F 1s profiles in Figure 3), the SiOF signal is the principle peak on the electrode cycled in 1 M LiPF6/DMC, while the Li–F is the principle peak on the electrode cycled in 1 M LiPF6/EC-DMC and 1 M LiPF6/PC. The SiOF signal can be attributed to the formation of SiOxFy on the bulk electrode material.90,91 Therefore, the SiOF signal decrease can be attributed to the increase of the SEI layer thickness. On the other hand, the volume variations of Si during the lithiation and delithiation processes could lead to the fracture of the SEI layer and the exposition of the Si electrode not covered by the SEI layer. The SiOF signal can be a fingerprint of these new revealed parts of the Si electrode. Therefore, it is difficult to firmly conclude about the changes of the SiOF signal as a function of lithiation/delithiation as the signal intensity can be influenced by a modified morphology of the Si electrode (e.g., the increased roughness). However, a clear tendency for PC and EC-DMC electrolytes can be observed, showing a decrease of the SiOF signal intensity with lithiation and an increase with delithiation, confirming a thickening and thinning of the SEI layer, respectively, as discussed above. The highest relative intensity of the SiOF signal observed for the DMC electrolyte can confirm the formation of the thinner SEI layer compared to the other two electrolytes.
In the O 1s profiles (Figure 3), the Si–O signals could be only observed at the beginning of lithiation corresponding to decomposition of the electrolyte (point C1) on the electrodes cycled in 1 M LiPF6/EC-DMC or 1 M LiPF6/PC. Then, the Si–O signal disappears, suggesting that the surface of the Si electrode is covered by a thick SEI layer, while the Si–O signal is well observed during the whole cycle on the electrode cycled in 1 M LiPF6/DMC, demonstrating the formation of a thinner SEI layer. After the full lithiation (C4), the Si electrode can undergo a huge volume change, which results in some cracks on the SEI layer. Thus, a small Li2O signal near the Si surface is detected and then disappears when the volume change decreased after delithiation (A2) in these three different electrolytes. The formation of Li2O can be according to reaction (16).88 On the electrode with 1 M LiPF6/DMC, −OCH3 becomes the main species at the lithiated state (point C4) as already confirmed from the decomposition of the C 1s peak, while the Si–O signal becomes lower, indicating the thicker SEI layer at this point.
2.1.4. SEI Layer Evolution with Cycling
Figure 4 shows the C 1s, F 1s, and O 1s core level spectra for Si electrodes obtained after 5 cycles (CV) at a delithated state in three different electrolytes (1 M LiPF6/EC-DMC, 1 M LiPF6/DMC, and 1 M LiPF6/PC). Comparing with the XPS spectra after the delithation stage of the first cycle (point A2, shown in Figures S2–S4), the shapes of the spectra on the electrode with 1 M LiPF6/PC do not show significant changes, but the intensities of the F 1s peak decrease significantly. It demonstrates that the ratios of fluorine-related components decrease in the SEI layer after 5 cycles. On the electrode with 1 M LiPF6/DMC, the shape of the F 1s and O 1s spectra after 5 cycles are similar to those after the first cycle, illustrating that the thickness of the SEI layer on the electrode does not change significantly. The C 1s peak shows a significant difference with a −OCH3 relative intensity decrease, while a C–O ether linkage peak (∼286.5 eV) and a small −CH2OCO2–, Li2CO3 peak (∼290.0 eV) appear, which can be attributed to reactions (5) and (8).41,80,87 On the electrode with 1 M LiPF6/EC-DMC, the shape of O 1s after 5 cycles is nearly the same as that after the first cycle, while the SiOF and P–F relative intensities decrease in the F 1s region, indicating that the SEI layer becomes thicker and more LiF is generated instead of P–F. The C 1s peak shows that the C–O ether linkage ratio increases, while the −OCH3 and −CH2OCO2–, Li2CO3 relative intensities decrease. It demonstrates that more polymers with C–O ether linkage are formed instead of CH3OLi, RCH2OCO2Li, and Li2CO3 on the surface.84,85
Figure 4.

High-resolution C 1s, F 1s, and O 1s core level spectra obtained for Si electrodes after 5 CV cycles in three different electrolytes (1 M LiPF6/EC-DMC, 1 M LiPF6/DMC, and 1 M LiPF6/PC).
2.2. Surface and Bulk Thin Film Si Electrode Modifications by ToF-SIMS Ion Depth Profiles
To have a better insight into the surface and bulk modifications of the Si thin film electrode material induced by lithiation and delithiation processes in the three different electrolytes, a ToF-SIMS ion depth profile analysis was performed at fully lithiated (0.01 V) and delithiated (3.0 V) states. First, the ToF-SIMS ion depth profiles (expressed as a function of sputtering time) are presented for the pristine Si thin film (Figure 5a) and then compared with fully lithiated (Figure 5b) and delithiated (Figure 5c) samples in the LiPF6/DMC electrolyte. The used sputtering conditions and specifically the sputtered area (300 μm*300 μm) are optimized to get a sufficiently high sputtering rate to be able to reach the Cu substrate, while maintaining a sufficiently high depth resolution to have a good overview of surface as well as bulk modifications of the Si electrode material. The intensities are shown with a logarithmic scale in order to magnify the low intensity signals.
Figure 5.

ToF-SIMS negative ion depth profiles for the (a) pristine Si thin film; (b) Si electrode after half CV cycle in LiPF6/DMC; and (c) Si electrode after a full CV cycle in LiPF6/DMC (sputtering area 300 μm × 300 μm, CV scan rate 0.2 mV s–1).
For the pristine Si thin film electrode (Figure 5a), the stable intensities of SiH– and SiO– in the beginning 255 s of sputtering can be attributed to a homogenous Si thin film distribution over the sputtered depth. Then, between 255 and 400 s, one enters the interfacial region between the Si thin film and the Cu substrate, where an obvious increase of the Cu– signal and decrease of SiH– and SiO– signals are observed. In the interfacial region, the maximum intensity for SiC–, P–, and CH2– signals (attributed to Si electrode material and some electrode and/or Cu substrate contaminations) can be observed. These intensities that increase in the interfacial region can be explained by enrichment in carbon, hydrocarbon, and phosphorus-like contaminations on the surface of Cu substrate (current collector). For longer sputtering times (over 400 s), the Cu– intensity becomes high and stable while all other signals are decreasing, indicating that the Cu substrate is reached.
Figure 5b shows the ion depth profiles of the Si thin film electrode after a half CV cycle performed in LiPF6/DMC. Comparing with the pristine Si, the significant increased intensities of organic (CH3O– and CH2–), lithium (Li–, LiF–, LiO–, and LiSi–), and P–-related species, illustrate the formation of SEI and the diffusion of electrolyte decomposition products into the Si film. The presence of organic (C–H–-like components) and inorganic (Li-containing components) components is in agreement with XPS results discussed above. The high intensity of organic and inorganic components is particularly observed for around the first 200 s of sputtering time. The appearance of a new LiSi– signal showing a high intensity over the long sputtering time (up to ∼1500 s) confirms the formation of a LixSi alloy. The decrease in the SiO– and SiC– intensity can be also attributed to the formation of the LixSi alloy.
The sputtering time to reach the interfacial region increases from about 255 s (for pristine Si electrode, Figure 5a) to about 1400 s (for lithiated Si electrode, Figure 5b), demonstrating the increase of thickness of the Si thin film electrode material related to volume change and the pulverization of the Si electrode after lithiation. This strong increase of sputtering time can be also related to changes in the electrode composition (formation of the LixSi alloy), i.e., a slower sputtering yield of the lithiated sample than that of the pristine sample. The significant increase in the sputtering time is also observed for the interfacial region (from around 150 s for the pristine sample to over 1000 s for the lithiated sample), which can be related to the roughness increase induced by the formation of the LixSi alloy. A similar huge increase of the interfacial region of the Si thin film electrode after electrochemical treatment has also been observed by energy-dispersive X-ray (EDX) analysis according to a previously published paper.92
Figure 5c shows the Si electrode after a full CV cycle performed in LiPF6/DMC. The sputtering time to reach the interface region decreases to around 400 s, indicating the Si electrode contraction. However, the electrode contraction is not completely reversible as it can be deduced from the lower sputtering time of the Si pristine electrode (around 255 s). The irreversible process of delithiation can be explained by some Li trapping in the Si thin film electrode material,33,56 which can also be confirmed from the relatively high intensity of LiSi– signal in the delithiated sample. The intensities of CH3O– and CH2– decrease in the bulk Si in comparison to the lithiated Si electrode (Figure 5b) but not on the surface, confirming the presence of the SEI layer on the electrode surface after delithiation. A significant decrease of SEI sputtering time for the delithiated sample (around 30 s) in comparison to the lithiated sample (200 s) is observed, indicating the thinning of the SEI layer. This dynamic behavior of the SEI layer with the increase and successive decrease of the SEI layer was previously reported for Si-based64,93 and conversion-type negative electrodes.94
In order to better observe and understand the surface modifications and the composition of SEI layer on the Si electrodes cycled in different electrolytes after half and full CV cycles, ToF-SIMS depth profiling with a lower sputtering rate (by enlarging the sputtering area from 300 μm × 300 μm to 500 μm × 500 μm) are obtained, as shown in Figure 6. The ion depth-profile intensities obtained after the half and full CV cycles were normalized by the Cu– signal, which becomes stable in the Si electrode region, assuming that the Cu signal is not affected by the lithiation and delithiation processes. The variations of SiC–, SiO–, Cu–, and P– ion depth profiles are clearly visible for faster sputtering rates (Figure 5) than for slower sputtering rates (Figure 6), and thus these profiles are not shown here in Figure 6. The results presented in Figure 6 show significant differences in the ion profile shapes and their intensities after half and full cycle.
Figure 6.

ToF-SIMS negative ion depth profiles of the Si thin film electrode after half and full CV cycles (scan rate 0.2 mV s–1) in LiPF6/EC-DMC, LiPF6/DMC, and LiPF6/PC electrolytes. Sputtering area 500 μm × 500 μm.
For the lithiated (after half cycle, Figure 6) Si electrodes, a high intensity of Li– signals can be observed, indicating the formation of the Li–Si phase. The high Li– signal intensity over 300 s indicates that the Cu substrate was not reached, contrary to the Li– ion profiles showed in Figure 5b where a decrease of this ion signal can be observed at around 2300 s. In the first 20–25 s of sputtering of lithiated samples (Figure 6), the peaks and then the abrupt decreases of lithiated species (LiO–, LiSi–, and LiF2–) are observed. However, the Li– signal has a different shape compared to LiO–, LiSi–, and LiF2– signals at the beginning of sputtering. Thus, these LiO–, LiSi–, and LiF2– peaks can be ascribed to the uneven morphology of the Si electrode induced by lithiation and huge volume changes of Si, resulting in a patchy surface layer. The LiO– profile shows a small peak in the first 25 s, indicating the presence of an oxide layer on the extreme surface. After 20–25 s, the Li-related (Li–, LiO–, LiSi–, and LiF2–) and organic-related (CH2– and CH3O–) signals exhibit different shapes as a function of sputtering time, indicating different species distributions in the SEI layer. In the inner part of the SEI layer, a slow intensity decrease of the LiO– signal (more or less important depending on the electrolyte) indicates a relatively homogenous distribution of LiO-related species over the SEI. At the minimum intensity of the LiSi– signal (at around 25 s of sputtering), the maximum intensity of CH2– and CH3O– signals corresponding to organic species can be observed, indicating that the concentration of organic species decreases and is smaller in the inner part of the SEI layer close to the lithiated Si electrode. The LiF2– profiles corresponding to LiF salt show the maximum intensity peak at the same sputtering time as the maximum intensity of LiO– and LiSi– signals, which indicates the uneven morphology caused by the huge volume changes leading to the revelation of some species attributed to the inner layer (LiO– and LiSi–). After reaching the maximum intensity, a significant slope in the LiF– profile can be observed with a higher decreasing rate than in the LiO– profile, indicating that these species are present principally in the outer part rather than in the inner part of SEI layer. After 200–250 s, SiH– and LiSi– profiles become relatively stable, indicating that the sputtering reaches the Si-lithiated electrode.
For the lithiated samples, it is difficult to discuss the surface and bulk differences in chemical composition induced by different electrolytes. However, some differences in the signal ratios can be observed. The CH2– signal is higher than the LiO– signal up to 200 s on the electrode with LiPF6/EC-DMC and LiPF6/PC, while the CH2– signal becomes lower than the LiO– signal at 55 s on the electrode cycled in LiPF6/DMC, illustrating the differences of the SEI layer composition. More important differences can be observed as function of the sputtering time in the different electrodes. In the case of the Si electrode cycled in LiPF6/DMC, a more rapid increase of the SiH– signal to a high intensity than in the other two electrolytes, leading to intersection with the Li– ion profile at around 200 s, is observed. This intersection point can be considered as the limit of the SEI layer, and the beginning of the lithiated Si electrode where a more stable LiSi– signal intensity is observed. The intersection of SiH– and Li– ion profiles is shifted to higher sputtering time (∼250 s) for the Si electrode cycled in LiPF6/PC, and no intersection can be observed for Si cycled LiPF6/EC-DMC, indicating the much thicker SEI layer in these two electrolytes. However, it should be noted that the sputtering time increase of the interface region can be also affected by more significant morphological modifications induced by lithiation and material pulverization as already demonstrated before.24,95
It can be concluded from these results that the SEI layer has a LiF-like and organic-rich thin outer layer. The inorganic LiO-species can be found in SEI layers as well as in the bulk of the lithiated LixSi phase. The thinnest SEI layer was observed on the surface of the Si electrode cycled in the LiPF6/DMC electrolyte.
After the full cycle of lithiation and delithiation (Figure 6), the principal high intensity signal observed in the bulk delithiated Si electrode is SiH–. In the case of all three electrolytes, similar shapes and relative intensity signals can be observed in the second region corresponding to the area of Si delithiated electrode and they are similar to the pristine electrode (Figure 5a). In comparison to the pristine electrode, in the bulk of delithiated samples, the higher intensities of Li–, LiO–, LiF2–, and most particularly LiSi– are observed. However, delithiation leads to lowering the LiSi– signal intensity when comparing to the lithiated samples due to the removal of most of the lithium. The other compounds related to products of electrolyte decomposition (CH2– and CH3O–) diffuse into the bulk of the electrode and are trapped in the electrode’s structure.
More significant modifications of the delithiated samples comparing with the lithiated samples can be observed in the first region related to the SEI layer. The LiO– signal shows the same shape as the Li– signal, confirming the presence of Li2O in the SEI layer. The higher quantity of Li2O can be observed on the surface of the Si electrode cycled in LiPF6/EC-DMC and LiPF6/PC than on the Si electrode cycled in LiPF6/DMC. The presence of Li2O is a result of reaction (16), and it was already confirmed by XPS on the lithiated samples.
Together with a high intensity of Li–, LiF2–, and LiO– signals, a decrease of organic species such as CH3O- and CH2-like species can be observed after delithiation with more homogenous in-depth distribution (in the SEI layer and in the delithiated Si electrode region), indicating the presence of some organic species in the SEI layer after delithiation.
Here, from the intensity changes of signals as a function of sputtering time, the apparent differences in the SEI layer thickness can be also evidenced. As aforementioned, the influence of the increased roughness induced by the morphological changes (cracks, pulverization) on the shape of ion profiles and then related to its increased sputtering time cannot be ruled out. Considering that the delithiated Si electrode was reached after reaching the stable intensity of the SiH– signal, the region before the plateau of SiH– can be attributed to the SEI layer. This region is characterized by the high intensity of Li–, LiF2–, and LiO– signals and also slightly higher signal of CH3O– and CH2–. As illustrated on the lithiated samples, the lowest sputtering time for the SEI layer present on the delithiated sample is observed for the Si electrode cycled in the LiPF6/DMC electrolyte (∼45 s) in comparison to ∼60–70 s for the other two electrolytes. The SEI layer on all delithiated samples is considerably thinner than that on the lithiated samples. These results are in agreement with previously published research.24,95 The SEI double-layered structure (with the outer and inner layer) is less evident on the surface of delithiated than lithiated samples.
2.3. Influence of Lithiation/Delithiation to the Electrolyte Components by In Situ MFTIRS
2.3.1. Cyclic Voltammetry of Si Thin Film Electrode during In Situ MFTIRS
The in situ MFTIRS was adopted to illustrate the molecular level of the interface chemistry. The shapes of the CV performed in three different electrolytes (1 M LiPF6/EC-DMC, 1 M LiPF6/DMC, and 1 M LiPF6/PC) are similar, and thus only the result for the Si electrode cycled in 1 M LiPF6/EC-DMC is shown in Figure 7a as an example. This CV curve exhibits more peaks during the cathodic process than those curves shown in Figure 1 because of its lower scan rate and different cell geometry. Before lithiation takes place, a single beam spectrum was recorded at the reference potential ER and then the other single beam spectra were collected at different cathodic (C1–C3) and anodic (A1–A4) potentials during lithiation and delithiation processes, respectively. The selected in situ MFTIR spectra (at C1, C3, A1, and A3 points) focusing on the changes of C=O and C–H bonds are shown in Figure 7b–d. The detail in situ MFTIR spectra from 800–2000 cm–1 at all cathodic and anodic potentials are shown in Figures S7–S9, respectively.
Figure 7.

(a) First cycle CV of the Si electrode tested in the three-electrode in situ MFTIRS cell in 1 M LiPF6/EC-DMC (scan rate v = 0.05 mV s–1). The in situ MFTIR spectra of the Si electrode in (b) 1 M LiPF6/PC (ER = 0.611 V), (c) 1 M LiPF6/DMC (ER = 0.590 V), and (d) 1 M LiPF6/EC-DMC (ER = 0.671 V) at C1, C3, A1, and A3 points during the first CV cycle.
2.3.2. In Situ MFTIRS
Previous research shows that Li ions could solvate with carbonate solvent molecules, resulting in the formation of Li(sol)n+ species (sol: EC, DMC and PC).21,96,97 The schematic molecular structure of Li+ solvated carbonates are shown in Figure S6. During this process, the coordination of Li+...O=C may lead to the weakening of the C=O bond in the Li(sol)n+ species, resulting in the red shift of the νC=O of Li(sol)n+ related to the free solvent molecules.21,98,99 During the lithiation process, the desolvated Li+ ions from the Li(sol)n+ complex could alloy with Si, and as a result, the free solvent molecules will increase and the amount of Li(sol)n+ species decrease.
Figure 7b shows the spectra of the Si electrode with cyclic carbonate solvent PC during the first CV cycle. The spectra are calculated according to the definition of ΔR/R recorded at different ES values during the first lithiation and subsequent delithiation processes. Here, ER was chosen at 0.611 V. The lithiation and delithiation was accompanied with the reduction of the thin-layer electrolyte between the IR window and the Si electrode, indicated by the presence of obvious downward/upward IR bands in the spectra. The band at 1780 cm–1 is assigned to the stretching vibration of C=O. Due to the solvation of PC with Li ions, this band is shifted to 1760 cm–1 toward the low frequency end.48,100 The intensity of the upward band at 1780 cm–1 correlating to free PC decreases during the lithiation process and then increases during the delithiation process. The intensity of the upward band at 1760 cm–1 correlating to Li(PC)n+ shows an opposite trend. It increases during the lithiation process, decreases during the delithiation process, and becomes downward at 0.660 V of delithiation. The peak at 1386 cm–1 is attributed to the scissoring vibration of CH3 and the wagging vibration of O–CH2 of PC. The peak at 1354 cm–1 is assigned to the bending vibration of the PC ring and the symmetric bending vibration of CH3 of PC. Due to the solvation of PC with Li ions, the original bands at 1386 and 1354 cm–1 are shifted to 1408 and 1362 cm–1 toward the high-frequency end.100 The upward peaks during the lithiation process at 1408 and 1362 cm–1 correlating to Li(PC)n+ change to downward peaks during the delithiation process. The downward peaks during the lithiation process at 1386 and 1354 cm–1 corresponding to free PC change to upward peaks during the delithiation process. It demonstrates that during the lithiation, Li(PC)n+ decreases with a decrease in potential, while the free PC increases. During the delithiation process, free PC decreases with an increase in potential, while Li(PC)n+ increases. In a diffusion double layer, it has been assumed that a decrease in the potential causes an increase in the cation concentration, as well as a decrease in the anion concentration, and the ion concentrations change with an opposite trend when the potential increases. Therefore, an intensity growth at 844 cm–1 (PF6–) is observed in Figure S7 during the lithiation process. It decreases during the delithiation.
Similar experiment has been performed on the Si electrode with linear carbonate solvent DMC, as shown in Figure 7c. The peaks at 1754 cm–1 and 1724 cm–1 can be correlated with the νC=O of unsolvated DMC and Li(DMC)n+, respectively.46,101 The upward band of 1724 cm–1 correlating with the νC=O of Li(DMC)n+ decrease obviously during the delithiation process, indicating the increase of Li(DMC)n+ concentration. When the Li ions extract from the Li–Si alloy, the Li ions will solvate with free solvent molecules. In the same case, the peak corresponding to C–H of Li(DMC)n+ at 1468 cm–1 increases during the lithiation process and decrease during the subsequent delithiation, since Li(DMC)n+ concentration decreases when Li+ alloys with Si and then increase when Li ions extract from the Li–Si alloy. The peak at 1453 cm–1 corresponding to C–H of free DMC shows an opposite trend of intensity variety compared with the peak at 1468 cm–1. In Figure S8, the peaks correlating toνC–O of free DMC and the peak of Li(DMC)n+ species can be found at 1274 and 1319 cm–1, respectively.101 The upward peak of νC–O for Li(DMC)n+ at 1319 cm–1 is increasing and decreasing during the lithiation and delithiation process, respectively. The peak correlating to PF6– can be observed at 844 cm–1.
For the experiment performed on the Si electrode with mixed solvent EC-DMC, ER was chosen as 0.617 V. The results are shown in Figure 7d. The upward peak at 1775 cm–1 corresponding to νC=O of free EC and the downward peak at 1764 cm–1 corresponding to νC=O of Li(EC)n+ decrease during the lithiation process102 and then increase during the delithiation process. It demonstrates that the concentration of free EC increases during the lithiation and decrease during the delithiation, while the concentration of Li(EC)n+ exhibits the opposite changing trend during the first CV cycle. However, the upward peak corresponding to Li(DMC)n+ at 1724 cm–1 decreases during the lithiation and changes to be a downward peak during the delithiation.102 Thus, EC plays the main role to coordinate with Li ions in the electrolyte. The upward peaks at 1409 and 1468 cm–1 correspond to C–H of Li(sol)n+.46,101 The downward peaks at 1390 and 1453 cm–1 correspond to C–H of solvent molecules. To ensure the balance of electric during the Li+ alloying process with the Si electrode, an obvious 842 cm–1 upward peak was found in Figure S9, correlating with PF6– anions.29
Comparing the intensities of in situ MFIRS (ΔR/R) on the Si electrode with these three electrolytes, the Si electrode with LiPF6/DMC shows less significant change during the lithiation and delithiation processes, illustrating that fewer DMC molecules take part in the interfacial reactions, which probably results in a thinner SEI layer on the electrode.
2.4. Morphological Modifications of Si Thin Electrodes as a Function of Electrolyte
The morphology and composition of the SEI layer depend strongly on the electrolyte components. Figure 8 shows SEM micrographs of the Si thin film electrode after 1 CV cycle and 5 CV cycles in different electrolytes and for comparison the pristine sample is shown in Figure S10a. After one complete cycle in LiPF6/EC-DMC, the Si electrode (Figure 8a) exhibits several slight cracks. A homogeneous surface is shown on the Si electrode cycled in LiPF6/DMC (Figure 8c), while the surface of the Si electrode cycled in LiPF6/PC (Figure 8e) is more uneven. After 5 CV cycles, the Si electrode cycled in LiPF6/DMC still exhibits a homogeneous surface (Figure 8d), while the Si electrodes cycled in LiPF6/EC-DMC (Figure 8b) and LiPF6/PC (Figure 8f) show many deep cracks.
Figure 8.

Morphology of Si electrodes with (a, b) LiPF6/EC-DMC, (c, d) LiPF6/DMC, and (e, f) LiPF6/PC after the 1 CV cycle (left) and 5 CV cycles (right) in the Swagelok cells (scan rate 0.2 mV s–1).
2.5. Galvanostatic Charge–Discharge Test of Si Thin Film Electrode in Different Electrolytes
Galvanostatic charge–discharge tests of the Si electrode with different electrolytes were performed to investigate the influence of the electrode solvents on capacity retention (Figure 9). The Si electrode cycled in the LiPF6/DMC electrolyte exhibits an initial charge capacity of 2704.5 mAh g–1 and a capacity retention of 77.2% after 60 cycles. However, the employment of the LiPF6/EC-DMC electrolyte resulted in lower initial charge capacity (2532.3 mAh g–1) and capacity retention (47.7%). A higher initial capacity of 3074.1 mAh g–1 was observed in LiPF6/PC however with a low capacity retention of 40.4% after 60 cycles. The electrode cycled in LiPF6/EC-DMC shows an unstable Coulombic efficiency, while the electrodes cycled in other two electrolytes show relatively stable and similar Coulombic efficiencies. The better cycle performance in LiPF6/DMC also demonstrates that this electrolyte is beneficial to form a good interface layer on the Si electrode.
Figure 9.

Cycle performance of the Si electrode in different electrolytes at a current density of 0.1 C.
2.6. Summarizing Discussion
To summarize, the ToF-SIMS data converge well with the XPS data showing the existence and content changes of −OCH3, −SiO, and −LiF species on the surface of the Si thin film electrode. At the same time in the electrolyte, the in situ MFTIRs also illustrate the species changes. The combination of these ex situ and in situ methods enables us to demonstrate different aspects of the Si electrode surface chemistry, which are schematically presented in Figure 10. Figure 10 shows the structure, composition, and morphological modifications of the SEI thin film on the Si thin film electrode after the complete cycle of lithiation and delithiation as a function of electrolyte composition. It can be summarized that Li2CO3, RCH2OCO2Li, −OCH3, Li2O, and LiF species are commonly observed on the Si surface but the quantity can vary depending on type of electrolyte. It should be noted that −OCH3 is one of the main species formed in LiPF6/DMC due to presence of two −OCH3 functional groups in DMC. The species with C–O ether linkage are only found on the electrodes with LiPF6/EC-DMC and LiPF6/PC due to the ring structures of solvent molecules. Moreover, the ToF-SIMS ion depth profiles demonstrated that components are not homogenously distributed in depth. The inhomogeneous, double-layered structure of the SEI was easily observed after the half cycle (at the lithiation state) by ToF-SIMS profiles (Figure 6), but after the full-cycle (at the delithiation state), this double-layered structure is less evident. It indicates that the SEI layer is not stable and strongly evolves during lithiation/delithiation process. Moreover, the composition is also changing as a function of lithiation/delithiation and types of electrolyte as discussed above. The SEI layer schema (Figure 10) demonstrates the principle findings, and thus a formation of double-layered structure is highlighted with the outer part enriched in LiF for all electrolytes and inner part enriched in other components. The presence of these components varies as a function of solvent. The SEI layer is also less enriched in Li2O after cycling in the DMC-based electrolyte.
Figure 10.
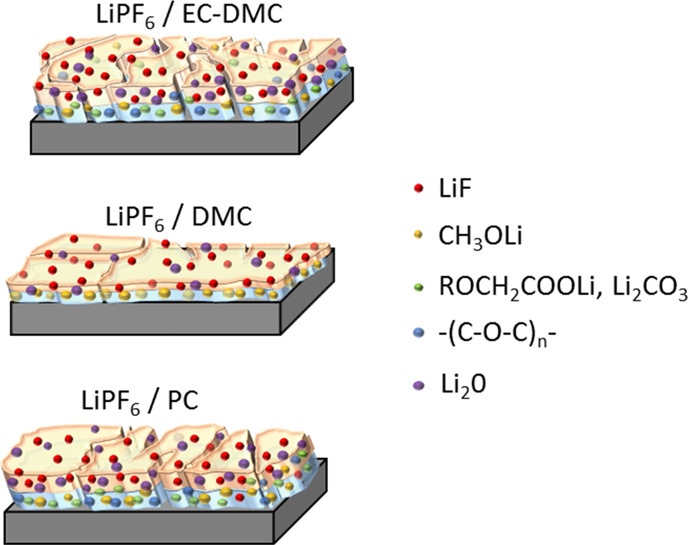
Schematic representation of the SEI structure and composition on Si thin film electrodes after a full CV cycle in different electrolytes (1 M LiPF6/EC-DMC, 1 M LiPF6/DMC, and 1 M LiPF6/PC).
The higher intensities of SiOF and Si–O signals in F 1 s, O 1s, and Si 2p XPS spectra on the electrode cycled in LiPF6/DMC indicate the formation of a thinner SEI layer. These results were well confirmed by differences in the sputtering time by ToF-SIMS. The dynamic behavior of the SEI layer is not shown here in a schema (Figure 10) as only the lithiated state is demonstrated. More significant morphological modifications (cracks formation) were also observed on the electrodes cycled in LiPF6/EC-DMC and LiPF6/PC in Figure 10.
The electrode surface state is not stable after the first cycle, and the next cycles lead to further modifications, which can vary as a function of electrolyte. After 5 CV cycles, more LiF species are generated in LiPF6/EC-DMC and LiPF6/DMC. The ratio of the species with C–O ether linkage increases significantly in LiPF6/EC-DMC. The peaks corresponding to Li2CO3, RCH2OCO2Li, and C–O ether linkage appear on the electrode cycled in LiPF6/DMC, while the ratio of −OCH3 species decreases. The intensities of the peaks related to LiF and P–F species decrease in LiPF6/PC. In LiPF6/DMC, the obvious SiOF and Si–O signals can be observed, indicating that the SEI layer does not increase after 5 cycles. In this work, a significant influence of single DMC solvent on the formation of the SEI layer in comparison with EC-DMC and PC-based electrolytes is demonstrated and this shows the importance of electrolyte optimization for Si-negative electrode materials.
3. Conclusions
By means of ex situ XPS, ToF-SIMS, and in situ FTIRS characterization of the Si thin film electrode/electrolyte interface together with SEM and galvanostatic tests, the influence of different LiPF6-based carbonate solvents on the surface chemistry of the Si electrode was revealed. The ex situ XPS and ToF-SIMS data proved a dynamic behavior of the SEI layer, showing increased and decreased thicknesses, induced by lithiation and delithiation, respectively, in these three different electrolytes. The stronger Si-related signals such as SiOF and Si–O in XPS profiles (Si 2p, F 1s, and O 1s) showed that a thinner and more stable SEI layer is formed when the Si thin film electrode is cycled in LiPF6/DMC. The in situ FTIRS, focused on the changes in the electrolyte near the electrode surface, demonstrated that the coordination between Li+ and DMC was weaker and fewer DMC molecules took part in the formation of the SEI layer. The morphological characterization by SEM showed also much less cracks and lower electrode degradation when cycled in the DMC-based electrolyte. Finally, the Si electrode with LiPF6/DMC exhibited better cycle performance than the electrodes with other two electrolytes, in agreement with the surface characterization results.
4. Experimental Part
4.1. Sample Preparation
To prepare the Si electrode, a JS3X-100B magnetron sputtering system was adopted to deposit a 500 nm-thick Si layer on the Cu foil (20 μm thick). Before deposition, the Cu substrate was washed with deionized water and acetone (Sinopharm, Chemical Reagent Co., Ltd., China). Then, the morphological characterizations of the Si anode were performed by SEM (Zeiss Ultra55 microscope with high-resolution field emission gun, Schottky SEM-FEG). The crystal structure of the Si layer was checked by means of X-ray diffraction (XRD, PANalytical X’pert PRO, Philips, Cu Kα radiation).
4.2. Electrochemical Tests
Three types of electrolytes were used in this study to illustrate the influence of different organic electrolytes on the formation and stability of the SEI layer: 1 M LiPF6 in EC- DMC (1:1), 1 M LiPF6 in DMC, and 1 M LiPF6 in PC. The electrolytes were purchased from Sigma Aldrich (battery grade) without further treatment.
The electrochemical measurements were performed in the Swagelok cell with the Si thin film electrode as the working electrode and lithium foil (99.9% purity, Alfa Aesar) as the counter electrode, which were used for the sample preparation. The CV measurements were performed at scan rate of 0.2 mV s–1 using a VMP3 Biologic multi-channel potentiostat/galvanostat. The lithiation or delithiation was stopped at different potentials during the CV tests (as detailed in results and discussion), and then these potentials were fixed for more than 10 h in order to form a stable surface layer suitable for XPS and ToF-SIMS analyses. After the cell disassembling in the Ar-filled glove box, the cycled Si electrodes were rinsed by DMC, dried with Ar-flow, and transferred directly to the XPS or ToF-SIMS analysis chamber under anaerobic and anhydrous conditions.
The in situ microscope FTIRS (in situ MFTIRS) cell (schematically illustrated in Figure S1) consisting of a KBr disk as the IR window, Si electrode as the working electrode, Li metal foil (China Energy Lithium Co., Ltd. China) as the reference and counter electrodes, and electrolyte was assembled in the Ar-filled glove box. The battery-grade electrolytes were purchased from Dodochem, Suzhou, China. CV measurements were performed using a CHI660E electrochemical working station (Chenhua, China) simultaneously with the measurements of reflectance MFTIRS at different voltages of the first cycle being recorded.
The long-term galvanostatic cycle performance was carried out between 0.01 and 1.2 V with a current density of 0.1 C (1 C = 3580 mAh g–1) at 30 °C in 2025 type coin cells using a LAND battery testing instrument. The cells were assembled with the Si thin film electrode, a Celgard 2400 membrane as the separator, a metallic lithium wafer as the counter electrode, and different electrolytes.
4.3. X-ray Photoelectron Spectroscopy
XPS analysis was conducted on a ThermoElectron ESCALAB 250 spectrometer under a ultrahigh-vacuum (UHV) condition (under ∼10–9 mbar) equipped with an Al Kα X-ray radiation source (hν = 1486.6 eV). Spectra were taken at 90° photoelectron take-off angle. Pass energies of 100 and 20 eV were used to record the survey spectrum and high-resolution spectra (Si 2p, C 1s, O 1s, F 1s), respectively. Charge effects were corrected by setting the lower energy component of the C 1s peak at 285.0 eV (corresponding to C–C bonding). Peak fitting and decomposition were performed with CASA software, using a Shirley-type background and Gaussian/Lorentzian peak shapes with a fixed ratio of 70/30.
4.4. In Situ Microscope Fourier Transform Infrared Spectroscopy
In situ MFTIRS was carried out on a Thermo Nicolet Nexus iN10 FTIR spectrometer equipped with a microscope and a liquid-N2-cooled HgCdTe detector. A total of 400 interferograms were collected and coadded at a spectral resolution of 4 cm–1 to obtain every single beam spectrum. The resulting spectrum was defined according to ΔR/R = (R(ES) – R(ER))/ER, in which R(ES) represents the single beam spectrum collected at potential ES and R(ER) corresponds to the single beam spectrum recorded at reference potential ER.44 The three-electrode cell for in situ MFTIRS is shown in Figure S1. The CV was performed starting with a negative scan at the scan rate of 0.05 mV s–1.
4.5. Time-of-Flight Secondary Ion Mass Spectrometry
ToF-SIMS ion depth profiles were carried out at an operating pressure of 10–9 mbar using the ToF-SIMS 5 spectrometer (IONTOF GmbH, Munster, Germany). A 25 keV Bi+ beam (100 × 100 μm2) was used for analysis, delivering a target current of 1.2 pA. The 2 keV Cs+ beam (300 × 300 μm2 or 500 × 500 μm2) was employed for sputtering, delivering a target current of 90 nA. Data acquisition and post-processing analysis were performed with Ion-Spec software (version 6.8).
Acknowledgments
Région Ile de-France is acknowledged for partial support for the XPS and ToF-SIMS equipment. This work is supported in part by the scholarship from China Scholarship Council (CSC) under the grant CSC N°201706310139.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.1c04226.
Figures such as schematic diagram of in situ FTIR cell, high-resolution XPS spectra (C 1s, F1s, and O1s), and in situ FTIR spectra obtained in three different electrolytes, molecular structure of three different solvents, and SEM, EDS, and XRD of the thin film electrode (PDF.)
The authors declare no competing financial interest.
Supplementary Material
References
- Meister P.; Jia H.; Li J.; Kloepsch R.; Winter M.; Placke T. Best Practice: Performance and Cost Evaluation of Lithium Ion Battery Active Materials with Special Emphasis on Energy Efficiency. Chem. Mater. 2016, 28, 7203–7217. 10.1021/acs.chemmater.6b02895. [DOI] [Google Scholar]
- Obrovac M. N.; Christensen L. Structural Changes in Silicon Anodes during Lithium Insertion/Extraction. Electrochem. Solid-State Lett. 2004, 7, A93. 10.1149/1.1652421. [DOI] [Google Scholar]
- Philippe B.; Dedryvère R.; Gorgoi M.; Rensmo H.; Gonbeau D.; Edström K. Role of the LiPF 6 Salt for the Long-Term Stability of Silicon Electrodes in Li-Ion Batteries – A Photoelectron Spectroscopy Study. Chem. Mater. 2013, 25, 394–404. 10.1021/cm303399v. [DOI] [Google Scholar]
- Verma P.; Maire P.; Novák P. A Review of the Features and Analyses of the Solid Electrolyte Interphase in Li-Ion Batteries. Electrochim. Acta 2010, 55, 6332–6341. 10.1016/j.electacta.2010.05.072. [DOI] [Google Scholar]
- Zhang W.-J. A Review of the Electrochemical Performance of Alloy Anodes for Lithium-Ion Batteries. J. Power Sources 2011, 196, 13–24. 10.1016/j.jpowsour.2010.07.020. [DOI] [Google Scholar]
- Beattie S. D.; Larcher D.; Morcrette M.; Simon B.; Tarascon J.-M. Si Electrodes for Li-Ion Batteries—A New Way to Look at an Old Problem. J. Electrochem. Soc. 2008, 155, A158. 10.1149/1.2817828. [DOI] [Google Scholar]
- McDowell M. T.; Lee S. W.; Nix W. D.; Cui Y. 25th Anniversary Article: Understanding the Lithiation of Silicon and Other Alloying Anodes for Lithium-Ion Batteries. Adv. Mater. 2013, 25, 4966–4985. 10.1002/adma.201301795. [DOI] [PubMed] [Google Scholar]
- Wang K.-X.; Li X.-H.; Chen J.-S. Surface and Interface Engineering of Electrode Materials for Lithium-Ion Batteries. Adv. Mater. 2015, 27, 527–545. 10.1002/adma.201402962. [DOI] [PubMed] [Google Scholar]
- Eshetu G. G.; Figgemeier E. Confronting the Challenges of Next-Generation Silicon Anode-Based Lithium-Ion Batteries: Role of Designer Electrolyte Additives and Polymeric Binders. ChemSusChem 2019, 12, 2515–2539. 10.1002/cssc.201900209. [DOI] [PubMed] [Google Scholar]
- Green M.; Fielder E.; Scrosati B.; Wachtler M.; Moreno J. S. Structured Silicon Anodes for Lithium Battery Applications. Electrochem. Solid-State Lett. 2003, 6, A75. 10.1149/1.1563094. [DOI] [Google Scholar]
- Chan C. K.; Ruffo R.; Hong S. S.; Cui Y. Surface Chemistry and Morphology of the Solid Electrolyte Interphase on Silicon Nanowire Lithium-Ion Battery Anodes. J. Power Sources 2009, 189, 1132–1140. 10.1016/j.jpowsour.2009.01.007. [DOI] [Google Scholar]
- Ryu J. H.; Kim J. W.; Sung Y.-E.; Oh S. M. Failure Modes of Silicon Powder Negative Electrode in Lithium Secondary Batteries. Electrochem. Solid-State Lett. 2004, 7, A306. 10.1149/1.1792242. [DOI] [Google Scholar]
- Obrovac M. N.; Krause L. J. Reversible Cycling of Crystalline Silicon Powder. J. Electrochem. Soc. 2007, 154, A103. 10.1149/1.2402112. [DOI] [Google Scholar]
- Jin Y.; Zhu B.; Lu Z.; Liu N.; Zhu J. Challenges and Recent Progress in the Development of Si Anodes for Lithium-Ion Battery. Adv. Energy Mater. 2017, 7, 1700715. 10.1002/aenm.201700715. [DOI] [Google Scholar]
- Peled E.; Menkin S. Review—SEI: Past, Present and Future. J. Electrochem. Soc. 2017, 164, A1703–A1719. 10.1149/2.1441707jes. [DOI] [Google Scholar]
- Chu Y.; Shen Y.; Guo F.; Zhao X.; Dong Q.; Zhang Q.; Li W.; Chen H.; Luo Z.; Chen L. Advanced Characterizations of Solid Electrolyte Interphases in Lithium-Ion Batteries. Electrochem. Energy Rev. 2020, 187. 10.1007/s41918-019-00058-y. [DOI] [Google Scholar]
- Chagnes A.; Swiatowska J.. Electrolyte and Solid-Electrolyte Interphase Layer in Lithium-Ion Batteries. In Lithium Ion Batteries - New Developments; Belharouak I., Ed.; InTech, 2012; pp. 145–172. 10.5772/31112. [DOI] [Google Scholar]
- Chagnes A.; Carré B.; Willmann P.; Dedryvère R.; Gonbeau D.; Lemordant D. Cycling Ability of γ-Butyrolactone-Ethylene Carbonate Based Electrolytes. J. Electrochem. Soc. 2003, 150, A1255. 10.1149/1.1597882. [DOI] [Google Scholar]
- Zhang S.; He M.; Su C.-C.; Zhang Z. Advanced Electrolyte/Additive for Lithium-Ion Batteries with Silicon Anode. Curr. Opin. Chem. Eng. 2016, 13, 24–35. 10.1016/j.coche.2016.08.003. [DOI] [Google Scholar]
- Xu K.; von Cresce A. Interfacing Electrolytes with Electrodes in Li Ion Batteries. J. Mater. Chem. 2011, 21, 9849. 10.1039/c0jm04309e. [DOI] [Google Scholar]
- Nie M.; Abraham D. P.; Seo D. M.; Chen Y.; Bose A.; Lucht B. L. Role of Solution Structure in Solid Electrolyte Interphase Formation on Graphite with LiPF6 in Propylene Carbonate. J. Phys. Chem. C 2013, 117, 25381–25389. 10.1021/jp409765w. [DOI] [Google Scholar]
- Ohara S.; Suzuki J.; Sekine K.; Takamura T. Li Insertion/Extraction Reaction at a Si Film Evaporated on a Ni Foil. J. Power Sources 2003, 119-121, 591–596. 10.1016/S0378-7753(03)00301-X. [DOI] [Google Scholar]
- Schroder K. W.; Celio H.; Webb L. J.; Stevenson K. J. Examining Solid Electrolyte Interphase Formation on Crystalline Silicon Electrodes: Influence of Electrochemical Preparation and Ambient Exposure Conditions. J. Phys. Chem. C 2012, 116, 19737–19747. 10.1021/jp307372m. [DOI] [Google Scholar]
- Pereira-Nabais C.; Światowska J.; Chagnes A.; Ozanam F.; Gohier A.; Tran-Van P.; Cojocaru C.-S.; Cassir M.; Marcus P. Interphase Chemistry of Si Electrodes Used as Anodes in Li-Ion Batteries. Appl. Surf. Sci. 2013, 266, 5–16. 10.1016/j.apsusc.2012.10.165. [DOI] [Google Scholar]
- Chen L.; Wang K.; Xie X.; Xie J. Effect of Vinylene Carbonate (VC) as Electrolyte Additive on Electrochemical Performance of Si Film Anode for Lithium Ion Batteries. J. Power Sources 2007, 174, 538–543. 10.1016/j.jpowsour.2007.06.149. [DOI] [Google Scholar]
- Dalavi S.; Guduru P.; Lucht B. L. Performance Enhancing Electrolyte Additives for Lithium Ion Batteries with Silicon Anodes. J. Electrochem. Soc. 2012, 159, A642. 10.1149/2.076205jes. [DOI] [Google Scholar]
- Ulldemolins M.; Le Cras F.; Pecquenard B.; Phan V. P.; Martin L.; Martinez H. Investigation on the Part Played by the Solid Electrolyte Interphase on the Electrochemical Performances of the Silicon Electrode for Lithium-Ion Batteries. J. Power Sources 2012, 206, 245–252. 10.1016/j.jpowsour.2012.01.095. [DOI] [Google Scholar]
- Jaumann T.; Balach J.; Langklotz U.; Sauchuk V.; Fritsch M.; Michaelis A.; Teltevskij V.; Mikhailova D.; Oswald S.; Klose M.; Stephani G.; Hauser R.; Eckert J.; Giebeler L. Lifetime vs. Rate Capability: Understanding the Role of FEC and VC in High-Energy Li-Ion Batteries with Nano-Silicon Anodes. Energy Storage Mater. 2017, 6, 26–35. 10.1016/j.ensm.2016.08.002. [DOI] [Google Scholar]
- Yohannes Y. B.; Lin S. D.; Wu N.-L. In Situ DRIFTS Analysis of Solid Electrolyte Interphase of Si-Based Anode with and without Fluoroethylene Carbonate Additive. J. Electrochem. Soc. 2017, 164, A3641–A3648. 10.1149/2.0681714jes. [DOI] [Google Scholar]
- Rezqita A.; Sauer M.; Foelske A.; Kronberger H.; Trifonova A. The Effect of Electrolyte Additives on Electrochemical Performance of Silicon/Mesoporous Carbon (Si/MC) for Anode Materials for Lithium-Ion Batteries. Electrochim. Acta 2017, 247, 600–609. 10.1016/j.electacta.2017.06.128. [DOI] [Google Scholar]
- Profatilova I. A.; Stock C.; Schmitz A.; Passerini S.; Winter M. Enhanced Thermal Stability of a Lithiated Nano-Silicon Electrode by Fluoroethylene Carbonate and Vinylene Carbonate. J. Power Sources 2013, 222, 140–149. 10.1016/j.jpowsour.2012.08.066. [DOI] [Google Scholar]
- Gao H.; Xiao L.; Plümel I.; Xu G.-L.; Ren Y.; Zuo X.; Liu Y.; Schulz C.; Wiggers H.; Amine K.; Chen Z. Parasitic Reactions in Nanosized Silicon Anodes for Lithium-Ion Batteries. Nano Lett. 2017, 17, 1512–1519. 10.1021/acs.nanolett.6b04551. [DOI] [PubMed] [Google Scholar]
- Nakai H.; Kubota T.; Kita A.; Kawashima A. Investigation of the Solid Electrolyte Interphase Formed by Fluoroethylene Carbonate on Si Electrodes. J. Electrochem. Soc. 2011, 158, A798. 10.1149/1.3589300. [DOI] [Google Scholar]
- Schiele A.; Breitung B.; Hatsukade T.; Berkes B. B.; Hartmann P.; Janek J.; Brezesinski T. The Critical Role of Fluoroethylene Carbonate in the Gassing of Silicon Anodes for Lithium-Ion Batteries. ACS Energy Lett. 2017, 2, 2228–2233. 10.1021/acsenergylett.7b00619. [DOI] [Google Scholar]
- Jung R.; Metzger M.; Haering D.; Solchenbach S.; Marino C.; Tsiouvaras N.; Stinner C.; Gasteiger H. A. Consumption of Fluoroethylene Carbonate (FEC) on Si-C Composite Electrodes for Li-Ion Batteries. J. Electrochem. Soc. 2016, 163, A1705–A1716. 10.1149/2.0951608jes. [DOI] [Google Scholar]
- Kim K.; Park I.; Ha S.-Y.; Kim Y.; Woo M.-H.; Jeong M.-H.; Shin W. C.; Ue M.; Hong S. Y.; Choi N.-S. Understanding the Thermal Instability of Fluoroethylene Carbonate in LiPF6 -Based Electrolytes for Lithium Ion Batteries. Electrochim. Acta 2017, 225, 358–368. 10.1016/j.electacta.2016.12.126. [DOI] [Google Scholar]
- Park J. M.; Kim S.; Ha J. H.; Kim S. W.; Lee J.; Park S.; Cho B.-W.; Choi H.-J. Enhancing the Stability of Silicon Nanosheets Electrodes by Fluoroethylene Carbonate. Chem. Phys. Lett. 2017, 684, 383–389. 10.1016/j.cplett.2017.07.009. [DOI] [Google Scholar]
- Jin Y.; Kneusels N.-J. H.; Magusin P. C. M. M.; Kim G.; Castillo-Martínez E.; Marbella L. E.; Kerber R. N.; Howe D. J.; Paul S.; Liu T.; Grey C. P. Identifying the Structural Basis for the Increased Stability of the Solid Electrolyte Interphase Formed on Silicon with the Additive Fluoroethylene Carbonate. J. Am. Chem. Soc. 2017, 139, 14992–15004. 10.1021/jacs.7b06834. [DOI] [PubMed] [Google Scholar]
- Horowitz Y.; Han H.-L.; Soto F. A.; Ralston W. T.; Balbuena P. B.; Somorjai G. A. Fluoroethylene Carbonate as a Directing Agent in Amorphous Silicon Anodes: Electrolyte Interface Structure Probed by Sum Frequency Vibrational Spectroscopy and Ab Initio Molecular Dynamics. Nano Lett. 2018, 18, 1145–1151. 10.1021/acs.nanolett.7b04688. [DOI] [PubMed] [Google Scholar]
- Choi N.-S.; Yew K. H.; Lee K. Y.; Sung M.; Kim H.; Kim S.-S. Effect of Fluoroethylene Carbonate Additive on Interfacial Properties of Silicon Thin-Film Electrode. J. Power Sources 2006, 161, 1254–1259. 10.1016/j.jpowsour.2006.05.049. [DOI] [Google Scholar]
- Etacheri V.; Haik O.; Goffer Y.; Roberts G. A.; Stefan I. C.; Fasching R.; Aurbach D. Effect of Fluoroethylene Carbonate (FEC) on the Performance and Surface Chemistry of Si-Nanowire Li-Ion Battery Anodes. Langmuir 2012, 28, 965–976. 10.1021/la203712s. [DOI] [PubMed] [Google Scholar]
- Bordes A.; Eom K.; Fuller T. F. The Effect of Fluoroethylene Carbonate Additive Content on the Formation of the Solid-Electrolyte Interphase and Capacity Fade of Li-Ion Full-Cell Employing Nano Si–Graphene Composite Anodes. J. Power Sources 2014, 257, 163–169. 10.1016/j.jpowsour.2013.12.144. [DOI] [Google Scholar]
- Hu Y.-S.; Demir-Cakan R.; Titirici M.-M.; Müller J.-O.; Schlögl R.; Antonietti M.; Maier J. Superior Storage Performance of a Si@SiOx/C Nanocomposite as Anode Material for Lithium-Ion Batteries. Angew. Chem., Int. Ed. 2008, 47, 1645–1649. 10.1002/anie.200704287. [DOI] [PubMed] [Google Scholar]
- Gong H.; Sun S.-G.; Li J.-T.; Chen Y.-J.; Chen S.-P. Surface Combinatorial Studies of IR Properties of Nanostructured Ru Film Electrodes Using CO as Probe Molecule. Electrochim. Acta 2003, 48, 2933–2942. 10.1016/S0013-4686(03)00358-X. [DOI] [Google Scholar]
- Delpuech N.; Mazouzi D.; Dupré N.; Moreau P.; Cerbelaud M.; Bridel J. S.; Badot J.-C.; De Vito E.; Guyomard D.; Lestriez B.; Humbert B. Critical Role of Silicon Nanoparticles Surface on Lithium Cell Electrochemical Performance Analyzed by FTIR, Raman, EELS, XPS, NMR, and BDS Spectroscopies. J. Phys. Chem. C 2014, 118, 17318–17331. 10.1021/jp503949y. [DOI] [Google Scholar]
- Yang J.; Solomatin N.; Kraytsberg A.; Ein-Eli Y. In-Situ Spectro-Electrochemical Insight Revealing Distinctive Silicon Anode Solid Electrolyte Interphase Formation in a Lithium-Ion Battery. ChemistrySelect 2016, 1, 572–576. 10.1002/slct.201600119. [DOI] [Google Scholar]
- Dalla Corte D. A.; Gouget-Laemmel A. C.; Lahlil K.; Caillon G.; Jordy C.; Chazalviel J.-N.; Gacoin T.; Rosso M.; Ozanam F. Molecular Grafting on Silicon Anodes: Artificial Solid-Electrolyte Interphase and Surface Stabilization. Electrochim. Acta 2016, 201, 70–77. 10.1016/j.electacta.2016.03.105. [DOI] [Google Scholar]
- Koo B. M.; Corte D. A. D.; Chazalviel J.-N.; Maroun F.; Rosso M.; Ozanam F. Lithiation Mechanism of Methylated Amorphous Silicon Unveiled by Operando ATR-FTIR Spectroscopy. Adv. Energy Mater. 2018, 8, 1702568. 10.1002/aenm.201702568. [DOI] [Google Scholar]
- Nguyen C. C.; Song S.-W. Characterization of SEI Layer Formed on High Performance Si–Cu Anode in Ionic Liquid Battery Electrolyte. Electrochem. Commun. 2010, 12, 1593–1595. 10.1016/j.elecom.2010.09.003. [DOI] [Google Scholar]
- Li J.-T.; Maurice V.; Swiatowska-Mrowiecka J.; Seyeux A.; Zanna S.; Klein L.; Sun S.-G.; Marcus P. XPS, Time-of-Flight-SIMS and Polarization Modulation IRRAS Study of Cr2O3 Thin Film Materials as Anode for Lithium Ion Battery. Electrochim. Acta 2009, 54, 3700–3707. 10.1016/j.electacta.2009.01.052. [DOI] [Google Scholar]
- Li J.-T.; Swiatowska J.; Seyeux A.; Huang L.; Maurice V.; Sun S.-G.; Marcus P. XPS and ToF-SIMS Study of Sn–Co Alloy Thin Films as Anode for Lithium Ion Battery. J. Power Sources 2010, 195, 8251–8257. 10.1016/j.jpowsour.2010.07.043. [DOI] [Google Scholar]
- Liao F.; Światowska J.; Maurice V.; Seyeux A.; Klein L. H.; Zanna S.; Marcus P. Electrochemical Lithiation and Passivation Mechanisms of Iron Monosulfide Thin Film as Negative Electrode Material for Lithium-Ion Batteries Studied by Surface Analytical Techniques. Appl. Surf. Sci. 2013, 283, 888–899. 10.1016/j.apsusc.2013.07.039. [DOI] [Google Scholar]
- Ota H.; Sakata Y.; Inoue A.; Yamaguchi S. Analysis of Vinylene Carbonate Derived SEI Layers on Graphite Anode. J. Electrochem. Soc. 2004, 151, A1659. 10.1149/1.1785795. [DOI] [Google Scholar]
- Tian B.; Światowska J.; Maurice V.; Zanna S.; Seyeux A.; Marcus P. Binary Iron-Chromium Oxide as Negative Electrode for Lithium-Ion Micro-Batteries – Spectroscopic and Microscopic Characterization. Appl. Surf. Sci. 2015, 353, 1170–1178. 10.1016/j.apsusc.2015.07.041. [DOI] [Google Scholar]
- Ota H.; Akai T.; Namita H.; Yamaguchi S.; Nomura M. XAFS and TOF–SIMS Analysis of SEI Layers on Electrodes. J. Power Sources 2003, 119-121, 567–571. 10.1016/S0378-7753(03)00291-X. [DOI] [Google Scholar]
- Bordes A.; De Vito E.; Haon C.; Boulineau A.; Montani A.; Marcus P. Multiscale Investigation of Silicon Anode Li Insertion Mechanisms by Time-of-Flight Secondary Ion Mass Spectrometer Imaging Performed on an In Situ Focused Ion Beam Cross Section. Chem. Mater. 2016, 28, 1566–1573. 10.1021/acs.chemmater.6b00155. [DOI] [Google Scholar]
- Ling M.; Xu Y.; Zhao H.; Gu X.; Qiu J.; Li S.; Wu M.; Song X.; Yan C.; Liu G.; Zhang S. Dual-Functional Gum Arabic Binder for Silicon Anodes in Lithium Ion Batteries. Nano Energy 2015, 12, 178–185. 10.1016/j.nanoen.2014.12.011. [DOI] [Google Scholar]
- Liu J.; Galpaya D. G. D.; Yan L.; Sun M.; Lin Z.; Yan C.; Liang C.; Zhang S. Exploiting a Robust Biopolymer Network Binder for an Ultrahigh-Areal-Capacity Li–S Battery. Energy Environ. Sci. 2017, 10, 750–755. 10.1039/C6EE03033E. [DOI] [Google Scholar]
- Huang S.; Cheong L.-Z.; Wang S.; Wang D.; Shen C. In-Situ Study of Surface Structure Evolution of Silicon Anodes by Electrochemical Atomic Force Microscopy. Appl. Surf. Sci. 2018, 452, 67–74. 10.1016/j.apsusc.2018.05.020. [DOI] [Google Scholar]
- Huang S.; Cheong L.-Z.; Wang D.; Shen C. Nanostructured Phosphorus Doped Silicon/Graphite Composite as Anode for High-Performance Lithium-Ion Batteries. ACS Appl. Mater. Interfaces 2017, 9, 23672–23678. 10.1021/acsami.7b04361. [DOI] [PubMed] [Google Scholar]
- Harks P. P. R. M. L.; Mulder F. M.; Notten P. H. L. In Situ Methods for Li-Ion Battery Research: A Review of Recent Developments. J. Power Sources 2015, 288, 92–105. 10.1016/j.jpowsour.2015.04.084. [DOI] [Google Scholar]
- Hou C.; Han J.; Liu P.; Yang C.; Huang G.; Fujita T.; Hirata A.; Chen M. Operando Observations of SEI Film Evolution by Mass-Sensitive Scanning Transmission Electron Microscopy. Adv. Energy Mater. 2019, 9, 1902675. 10.1002/aenm.201902675. [DOI] [Google Scholar]
- Tang H.; Zhang J.; Zhang Y. J.; Xiong Q. Q.; Tong Y. Y.; Li Y.; Wang X. L.; Gu C. D.; Tu J. P. Porous Reduced Graphene Oxide Sheet Wrapped Silicon Composite Fabricated by Steam Etching for Lithium-Ion Battery Application. J. Power Sources 2015, 286, 431–437. 10.1016/j.jpowsour.2015.03.185. [DOI] [Google Scholar]
- Philippe B.; Dedryvère R.; Allouche J.; Lindgren F.; Gorgoi M.; Rensmo H.; Gonbeau D.; Edström K. Nanosilicon Electrodes for Lithium-Ion Batteries: Interfacial Mechanisms Studied by Hard and Soft X-Ray Photoelectron Spectroscopy. Chem. Mater. 2012, 24, 1107–1115. 10.1021/cm2034195. [DOI] [Google Scholar]
- Cui L.-F.; Ruffo R.; Chan C. K.; Peng H.; Cui Y. Crystalline-Amorphous Core–Shell Silicon Nanowires for High Capacity and High Current Battery Electrodes. Nano Lett. 2009, 9, 491–495. 10.1021/nl8036323. [DOI] [PubMed] [Google Scholar]
- Laïk B.; Ung D.; Caillard A.; Sorin Cojocaru C.; Pribat D.; Pereira-Ramos J.-P. An Electrochemical and Structural Investigation of Silicon Nanowires as Negative Electrode for Li-Ion Batteries. J. Solid State Electrochem. 2010, 14, 1835–1839. 10.1007/s10008-010-1045-5. [DOI] [Google Scholar]
- Hou X.; Zhang M.; Wang J.; Hu S.; Liu X.; Shao Z. High Yield and Low-Cost Ball Milling Synthesis of Nano-Flake Si@SiO2 with Small Crystalline Grains and Abundant Grain Boundaries as a Superior Anode for Li-Ion Batteries. J. Alloys Compd. 2015, 639, 27–35. 10.1016/j.jallcom.2015.03.127. [DOI] [Google Scholar]
- Pollak E.; Salitra G.; Baranchugov V.; Aurbach D. In Situ Conductivity, Impedance Spectroscopy, and Ex Situ Raman Spectra of Amorphous Silicon during the Insertion/Extraction of Lithium. J. Phys. Chem. C 2007, 111, 11437–11444. 10.1021/jp0729563. [DOI] [Google Scholar]
- Gauthier M.; Danet J.; Lestriez B.; Roué L.; Guyomard D.; Moreau P. Nanoscale Compositional Changes during First Delithiation of Si Negative Electrodes. J. Power Sources 2013, 227, 237–242. 10.1016/j.jpowsour.2012.11.047. [DOI] [Google Scholar]
- Chen L. B.; Xie J. Y.; Yu H. C.; Wang T. H. An Amorphous Si Thin Film Anode with High Capacity and Long Cycling Life for Lithium Ion Batteries. J. Appl. Electrochem. 2009, 39, 1157–1162. 10.1007/s10800-008-9774-1. [DOI] [Google Scholar]
- Baranchugov V.; Markevich E.; Pollak E.; Salitra G.; Aurbach D. Amorphous Silicon Thin Films as a High Capacity Anodes for Li-Ion Batteries in Ionic Liquid Electrolytes. Electrochem. Commun. 2007, 9, 796–800. 10.1016/j.elecom.2006.11.014. [DOI] [Google Scholar]
- Jeschull F.; Lindgren F.; Lacey M. J.; Björefors F.; Edström K.; Brandell D. Influence of Inactive Electrode Components on Degradation Phenomena in Nano-Si Electrodes for Li-Ion Batteries. J. Power Sources 2016, 325, 513–524. 10.1016/j.jpowsour.2016.06.059. [DOI] [Google Scholar]
- Ermolieff A.; Martin F.; Amouroux A.; Marthon S.; Westendorp J. F. M. Surface Composition Analysis of HF Vapour Cleaned Silicon by X-Ray Photoelectron Spectroscopy. Appl. Surf. Sci. 1991, 48-49, 178–184. 10.1016/0169-4332(91)90327-G. [DOI] [Google Scholar]
- Cardinaud C.; Rhounna A.; Turban G.; Grolleau B. Analyse XPS Des Surfaces de Si et SiO2 Exposées Aux Plasmas de CHF3 et CHF3—C2F6. Polymérisation et Gravure. Rev. Phys. Appl. 1989, 24, 309–321. 10.1051/rphysap:01989002403030900. [DOI] [Google Scholar]
- Lehner A.; Steinhoff G.; Brandt M. S.; Eickhoff M.; Stutzmann M. Hydrosilylation of Crystalline Silicon (111) and Hydrogenated Amorphous Silicon Surfaces: A Comparative x-Ray Photoelectron Spectroscopy Study. J. Appl. Phys. 2003, 94, 2289–2294. 10.1063/1.1593223. [DOI] [Google Scholar]
- Gruntz K. J.; Ley L.; Johnson R. L. Photoelectron Spectra of Fluorinated Amorphous Silicon ( a -Si: F). Phys. Rev. B 1981, 24, 2069–2080. 10.1103/PhysRevB.24.2069. [DOI] [Google Scholar]
- Philippe B.; Dedryvère R.; Gorgoi M.; Rensmo H.; Gonbeau D.; Edström K. Improved Performances of Nanosilicon Electrodes Using the Salt LiFSI: A Photoelectron Spectroscopy Study. J. Am. Chem. Soc. 2013, 135, 9829–9842. 10.1021/ja403082s. [DOI] [PubMed] [Google Scholar]
- Konstadinidis K.; Zhang P.; Opila R. L.; Allara D. L. An In-Situ X-Ray Photoelectron Study of the Interaction between Vapor-Deposited Ti Atoms and Functional Groups at the Surfaces of Self-Assembled Monolayers. Surf. Sci. 1995, 338, 300–312. 10.1016/0039-6028(95)80048-4. [DOI] [Google Scholar]
- Philippe B.; Hahlin M.; Edström K.; Gustafsson T.; Siegbahn H.; Rensmo H. Photoelectron Spectroscopy for Lithium Battery Interface Studies. J. Electrochem. Soc. 2016, 163, A178–A191. 10.1149/2.0051602jes. [DOI] [Google Scholar]
- Etacheri V.; Geiger U.; Gofer Y.; Roberts G. A.; Stefan I. C.; Fasching R.; Aurbach D. Exceptional Electrochemical Performance of Si-Nanowires in 1,3-Dioxolane Solutions: A Surface Chemical Investigation. Langmuir 2012, 28, 6175–6184. 10.1021/la300306v. [DOI] [PubMed] [Google Scholar]
- Andersson A. M.; Edström K. Chemical Composition and Morphology of the Elevated Temperature SEI on Graphite. J. Electrochem. Soc. 2001, 148, A1100. 10.1149/1.1397771. [DOI] [Google Scholar]
- Malmgren S.; Ciosek K.; Hahlin M.; Gustafsson T.; Gorgoi M.; Rensmo H.; Edström K. Comparing Anode and Cathode Electrode/Electrolyte Interface Composition and Morphology Using Soft and Hard X-Ray Photoelectron Spectroscopy. Electrochim. Acta 2013, 97, 23–32. 10.1016/j.electacta.2013.03.010. [DOI] [Google Scholar]
- Zazzera L. A.; Moulder J. F. XPS and SIMS Study of Anhydrous HF and UV/Ozone-Modified Silicon (100) Surfaces. J. Electrochem. Soc. 1989, 136, 484. 10.1149/1.2096659. [DOI] [Google Scholar]
- Campion C. L.; Li W.; Lucht B. L. Thermal Decomposition of LiPF6-Based Electrolytes for Lithium-Ion Batteries. J. Electrochem. Soc. 2005, 152, A2327. 10.1149/1.2083267. [DOI] [Google Scholar]
- Wilken S.; Treskow M.; Scheers J.; Johansson P.; Jacobsson P. Initial Stages of Thermal Decomposition of LiPF6-Based Lithium Ion Battery Electrolytes by Detailed Raman and NMR Spectroscopy. RSC Adv. 2013, 3, 16359. 10.1039/c3ra42611d. [DOI] [Google Scholar]
- Dupré N.; Moreau P.; De Vito E.; Quazuguel L.; Boniface M.; Bordes A.; Rudisch C.; Bayle-Guillemaud P.; Guyomard D. Multiprobe Study of the Solid Electrolyte Interphase on Silicon-Based Electrodes in Full-Cell Configuration. Chem. Mater. 2016, 28, 2557–2572. 10.1021/acs.chemmater.5b04461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dedryvère R.; Gireaud L.; Grugeon S.; Laruelle S.; Tarascon J.-M.; Gonbeau D. Characterization of Lithium Alkyl Carbonates by X-Ray Photoelectron Spectroscopy: Experimental and Theoretical Study. J. Phys. Chem. B 2005, 109, 15868–15875. 10.1021/jp051626k. [DOI] [PubMed] [Google Scholar]
- Radvanyi E.; De Vito E.; Porcher W.; Jouanneau Si Larbi S. An XPS/AES Comparative Study of the Surface Behaviour of Nano-Silicon Anodes for Li-Ion Batteries. J. Anal. At. Spectrom. 2014, 29, 1120–1131. 10.1039/C3JA50362C. [DOI] [Google Scholar]
- Kang S.-H.; Abraham D. P.; Xiao A.; Lucht B. L. Investigating the Solid Electrolyte Interphase Using Binder-Free Graphite Electrodes. J. Power Sources 2008, 175, 526–532. 10.1016/j.jpowsour.2007.08.112. [DOI] [Google Scholar]
- Zhuk S. I.; Isaev V. A.; Grishenkova O. V.; Isakov A. V.; Apisarov A. P.; Zaykov Y. P. Silicon Electrodeposition from Chloride-Fluoride Melts Containing K2SiF6 and SiO2. J. Serb. Chem. Soc. 2017, 82, 51–62. 10.2298/JSC160712109Z. [DOI] [Google Scholar]
- Gaudig M.; Naumann V.; Hirsch J.; Werner M.; Großer S.; Hagendorf C.; Bernhard N.; Lausch D.. Formation and Evolution of the SiOxFy Masking Layer Caused by Plasma Texturing; IEEE, 2018, pp. 0036–0040. [Google Scholar]
- Maranchi J. P.; Hepp A. F.; Evans A. G.; Nuhfer N. T.; Kumta P. N. Interfacial Properties of the A-Si/Cu:Active–Inactive Thin-Film Anode System for Lithium-Ion Batteries. J. Electrochem. Soc. 2006, 153, A1246. 10.1149/1.2184753. [DOI] [Google Scholar]
- Cao C.; Abate I. I.; Sivonxay E.; Shyam B.; Jia C.; Moritz B.; Devereaux T. P.; Persson K. A.; Steinrück H.-G.; Toney M. F. Solid Electrolyte Interphase on Native Oxide-Terminated Silicon Anodes for Li-Ion Batteries. Joule 2019, 3, 762–781. 10.1016/j.joule.2018.12.013. [DOI] [Google Scholar]
- Rezvani S. J.; Nobili F.; Gunnella R.; Ali M.; Tossici R.; Passerini S.; Di Cicco A. SEI Dynamics in Metal Oxide Conversion Electrodes of Li-Ion Batteries. J. Phys. Chem. C 2017, 121, 26379–26388. 10.1021/acs.jpcc.7b08259. [DOI] [Google Scholar]
- Li J.-T.; Światowska J.; Maurice V.; Seyeux A.; Huang L.; Sun S.-G.; Marcus P. XPS and ToF-SIMS Study of Electrode Processes on Sn–Ni Alloy Anodes for Li-Ion Batteries. J. Phys. Chem. C 2011, 115, 7012–7018. 10.1021/jp201232n. [DOI] [Google Scholar]
- Morita M.; Asai Y.; Yoshimoto N.; Ishikawa M. A Raman Spectroscopic Study of Organic Electrolyte Solutions Based on Binary Solvent Systems of Ethylene Carbonate with Low Viscosity Solvents Which Dissolve Different Lithium Salts. J. Chem. Soc., Faraday Trans. 1998, 94, 3451–3456. 10.1039/a806278a. [DOI] [Google Scholar]
- Fukushima T.; Matsuda Y.; Hashimoto H.; Arakawa R. Studies on Solvation of Lithium Ions in Organic Electrolyte Solutions by Electrospray Ionization-Mass Spectroscopy. Electrochem. Solid-State Lett. 2001, 4, A127. 10.1149/1.1383428. [DOI] [Google Scholar]
- Masia M.; Probst M.; Rey R. Ethylene Carbonate–Li + : A Theoretical Study of Structural and Vibrational Properties in Gas and Liquid Phases. J. Phys. Chem. B 2004, 108, 2016–2027. 10.1021/jp036673w. [DOI] [Google Scholar]
- Wang Y.; Balbuena P. B. Theoretical Studies on Cosolvation of Li Ion and Solvent Reductive Decomposition in Binary Mixtures of Aliphatic Carbonates. Int. J. Quantum Chem. 2005, 102, 724–733. 10.1002/qua.20466. [DOI] [Google Scholar]
- Ikezawa Y.; Ariga T. In Situ FTIR Spectra at the Cu Electrode/Propylene Carbonate Solution Interface. Electrochim. Acta 2007, 52, 2710–2715. 10.1016/j.electacta.2006.09.050. [DOI] [Google Scholar]
- Joho F.; Novák P. SNIFTIRS Investigation of the Oxidative Decomposition of Organic-Carbonate-Based Electrolytes for Lithium-Ion Cells. Electrochim. Acta 2000, 45, 3589–3599. 10.1016/S0013-4686(00)00480-1. [DOI] [Google Scholar]
- Aurbach D.; Markovsky B.; Shechter A.; Ein-Eli Y.; Cohen H. A Comparative Study of Synthetic Graphite and Li Electrodes in Electrolyte Solutions Based on Ethylene Carbonate-Dimethyl Carbonate Mixtures. J. Electrochem. Soc. 1996, 143, 3809. 10.1149/1.1837300. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.