

Abstract
A type of monophasic group B Salmonella enterica with the antigenic formula 4,12:a:- (“Fulica-like”) has been described as associated with harbour porpoises (Phocoena phocoena), most frequently recovered from lung samples. In the present study, lung tissue samples from 47 porpoises found along the Swedish coast or as bycatch in fishing nets were analysed, two of which were positive for S. enterica. Pneumonia due to the infection was considered the likely cause of death for one of the two animals. The recovered isolates were whole genome sequenced and found to belong to sequence type (ST) 416 and to be closely related to ST416/ST417 porpoise isolates from UK waters as determined by core-genome MLST. Serovars Bispebjerg, Fulica and Abortusequi were identified as distantly related to the porpoise isolates, but no close relatives from other host species were found. All ST416/417 isolates had extensive loss of function mutations in key Salmonella pathogenicity islands, but carried accessory genetic elements associated with extraintestinal infection such as iron uptake systems. Gene ontology and pathway analysis revealed reduced secondary metabolic capabilities and loss of function in terms of signalling and response to environmental cues, consistent with adaptation for the extraintestinal niche. A classification system based on machine learning identified ST416/417 as more invasive than classical gastrointestinal serovars. Genome analysis results are thus consistent with ST416/417 as a host-adapted and extraintestinal clonal population of S. enterica, which while found in porpoises without associated pathology can also cause severe opportunistic infections.
Supplementary Information
The online version contains supplementary material available at 10.1186/s13567-021-01001-0.
Keywords: Pangenome analysis, cgMLST, salmonellosis, marine mammals, cetaceans, host bias, molecular epidemiology, extraintestinal infection, porpoise, Phocoena
Introduction
The harbour porpoise (Phocoena phocoena) is a species of small cetaceans that inhabit coastal waters of the northern hemisphere. While it is abundant in the North Atlantic and the North Pacific, the Baltic Sea and Black Sea populations are critically endangered [1]. Because of their near-shore habitat, harbour porpoises are vulnerable to anthropogenic environmental disturbance including chemical and noise pollution and prey depletion as well as entanglement in fishing nets, especially in semi-enclosed seas such as the Baltic [1]. Such environmental stressors can weaken the immune system and leave animals more vulnerable to infection [2].
Harbour porpoises in Swedish waters can be classified into three populations: The North Sea, Belt Sea and Baltic Proper populations. Animals that are found dead are collected and examined to identify causes of death and other threats [3]. In a study of 98 stranded porpoises collected from 2008 to 2019, infectious disease was the second most common cause of death (17%) after bycatch and probable bycatch (25%). Infectious diseases included several cases of bacterial pneumonia in the presence of nematodes [3]. This is consistent with reports of a high frequency of pneumonia associated with parasites, bacterial infection, or both, among porpoises from the North Atlantic examined post-mortem [4–8].
A particular type of monophasic group B Salmonella enterica subsp. enterica, most often of sequence type (ST) 416 and with the antigenic formula 4,12:a:- (“Fulica-like”) has been described as exclusively associated with harbour porpoises in Scotland and England [9, 10]. This type of Salmonella occurs more frequently in lung samples than other tissues or faeces [11], and has also been recovered from porpoise lung worms [12] suggesting a possible mode of transmission. As low-grade Salmonella infections in the lungs appear to be prevalent among porpoises, it has been hypothesized that the infection becomes severe or life-threatening primarily in weakened individuals [11]. While most S. enterica are host generalists and cause self-limiting gastroenteritis in infected animals or humans, host adapted Salmonella serovars tend to infect one or a few host species, have a higher likelihood of causing extraintestinal infection, and more frequently establish a chronic carrier state in otherwise healthy infected animals, typical examples being S. Dublin in cattle, S. Choleraesuis in pigs and S. Gallinarum in poultry [13]. The evolution of a host adapted lineage of Salmonella from generalist ancestors appears to involve extensive degradation of genes not needed in the specialist niche with a corresponding loss of metabolic capability, as well as acquisition of mobile genetic elements with genes providing new traits [14–18]. Host and niche adaptation of the proposed porpoise-associated strains of Salmonella should therefore be evident in their genome sequences. In the present study we report the occurrence of S. enterica ST416 in lung samples from Swedish harbour porpoises and show extensive genomic evolution in these strains consistent with host adaptation and an increased capacity for causing extraintestinal infection.
Materials and methods
Animals, samples, and bacterial isolates
In Sweden, porpoises and several other mammalian and avian species of conservation interest are state property (“statens vilt” or “wildlife of the state”) and must be reported to the authorities if they are found dead. From 2008 to 2020, 142 porpoises found along the Swedish coast or incidentally killed as bycatch in fishing nets have been collected for necropsy examination within a collaborative program delivered by the Swedish National Veterinary Institute (SVA) and the Swedish Museum of Natural History. Almost all animals were stored frozen before investigation. Porpoises were examined following standardized protocols [19] to determine cause of death, investigate any abnormalities and collect tissues and data for other studies and archives [3]. Representative pieces of lung were fixed in 10% neutrally buffered formalin and processed and embedded in paraffin for microscopic examination. Sections (3–4 µm) were stained using Mayer’s hematoxylin and eosin [20]. Prior to 2018, any lesions consistent with possible bacterial infection were submitted for aerobic bacterial culture (n = 9 animals, of which lung was cultured in 5 animals) as described below. Since 2018, lung tissue from all porpoises that were not more than moderately decomposed were submitted for aerobic bacterial culture only (n = 21), for selective culture of Salmonella sp. (n = 15) or for both (n = 8). In total, lung tissue was cultured from 47 porpoises in this study. All bacteriology was performed by the Department of Microbiology, SVA. For aerobic bacterial culture samples were inoculated onto blood agar plates containing 5% horse blood and bromocresol purple lactose agar plates and held at 37 °C under aerobic conditions. In parallel, horse blood agar plates were grown at 37 °C in a 5% ± 1% CO2 incubator. Plates were inspected for growth at 24 and 48 h after inoculation. For selective culture, ISO 6579-1:2017 was followed with samples enriched in buffered peptone water and grown on modified semi-solid Rappaport–Vassiliadis (MSRV) agar, horse blood agar, brilliant green (BG) agar and xylose lysine deoxycholate (XLD) agar. Suspected Salmonella colonies were confirmed to species level by either real-time PCR [21] (in 2017) or by MALDI-TOF MS on a Bruker Biotyper instrument (in 2020) according to manufacturer instructions. Serotyping was performed with biochemical testing and slide agglutination, also according to ISO 6579-1:2017.
Sequencing and reference data
Libraries were generated from 17-VLT002652 with a Nextera XT kit and sequencing performed on an Illumina MiSeq instrument as 2 × 250 bp paired-end. Additional long read data was generated using an Oxford Nanopore MinION sequencing device run with a R9.4.1 flow cell and a library created using the Rapid PCR barcoding kit. Sequencing of the second isolate 20-VLT001389 was performed by Illumina technology in the same way but as 250 + 60 bp paired-end due to technical issues with the reverse read. All three sequence datasets were a minimum of 100×. Short read data from 59 close relatives of the Swedish porpoise strains were identified by core genome multi-locus sequence typing (cgMLST) via Enterobase [22], together forming group 4372 at the hierarchical clustering level HC2000 as defined by the HierCC algorithm. Short read data from these isolates were downloaded from the European Nucleotide Archive for inclusion in downstream analysis. Short read data was quality checked using FastQC v0.11.8 and trimmed using Trimmomatic v0.39, with a sliding window of 4 bp and quality threshold of 20. Long read data was quality checked using FastQC and NanoPlot v1.32.1 [23] and filtered using Filtlong v0.2.0 keeping the best 900 Mbp of reads. The S. enterica type strain LT2 (S. Typhimurium) genome, GenBank NC_003197, was used as reference for read mapping, annotation, and analysis of Salmonella pathogenicity islands.
Data analysis and visualization
Bowtie2 v2.3.5.1 was used to map short read data to the LT2 reference genome, and Samtools v1.9 was used for single nucleotide polymorphism (SNP) calling. The resulting VCF files were filtered using in-house R scripts (Additional file 4) and quality checked using the VCFR package v1.8.0. SNPEff v4.3 [24] was used to evaluate the impact of each SNP found. Gene Ontology (GO) categories impacted by the mutations were found using the Panther GO Enrichment tool [25] and the KEGG Mapper tool [26]. A machine learning algorithm developed by Wheeler et al. [27] was applied to read data from all 61 strains to assess the invasiveness of each and how it compared to known enteric and invasive Salmonella serovars. Unicycler v0.4.8 [28] with default settings was used to create a hybrid assembly for 17-VLT002652, and assemblies from short reads only for all other isolates. Read data were downsampled to 100× before assembly. Assemblies were inspected using Bandage [29] when necessary. Annotation of assemblies was performed using Prokka v1.14.5 [30] using the S. enterica type strain S. Typhimurium LT2 as a reference and default settings. Roary v3.13.0 [31] was run on the annotated assemblies, with a 90% identity cutoff and otherwise default settings, to identify the core and pan genomes. Scoary v1.6.16 [32] was run on the results to identify genes that were significantly more frequently observed in porpoise isolates compared to the reference isolate genomes. Structural information for the interpretation of results was accessed via the hybrid assembly of 17-VLT002652. 7-gene MLST and cgMLST were performed via Enterobase [22]. A minimum-spanning tree based on cgMLST data was generated with GrapeTree via Enterobase. Boxplots and gene maps were drawn in R 2.5.0 [33] with the gggenes package [34].
Results
Necropsy findings associated with Salmonella infection
The first Salmonella isolate in the present study (17-VLT002652) was recovered from a porpoise found stranded in Varberg municipality (N 57°7.1′, E 12°11.5′) on the Swedish west coast in July 2017. This mature, 18-year-old male was in very poor nutritional condition and suffered from severe bronchopneumonia in which 50–60% of the lung tissue was necrotic and inflamed (Figure 1). There was a concurrent, moderate to heavy nematode infection in the airways with few parasites in the heart and pulmonary vessels. Microscopically, large areas of acute pyogranulomatous inflammation and necrosis consistent with acute bacterial pneumonia were seen and within some areas, bacteria could also be identified (Figure 2). Parasites were not associated with these lesions. Given the pathological changes seen associated with bacteria and the abundant growth of Salmonella from lung tissue, pneumonia caused by Salmonella infection was determined to be the cause of death. The recovered Salmonella isolate could not be serotyped and was biochemically atypical (lysine decarboxylase negative). The second isolate (20-VLT001389) was cultured from an animal found dead in February 2020 in Helsingborg (N 56°2.7′, E 12°41.2′), further south on the west coast. This immature female porpoise was in good nutritional condition and deemed to be in good health. Linear marks on the head and extremities were consistent with net marks and death was attributed to incidental capture in fishing gear. This animal had a moderate burden of nematodes in the airways and scattered areas of caseous inflammation in the lung tissue. Microscopically, focal areas of chronic eosinophilic and granulomatous pneumonia typical of parasitic infection were seen associated with nematodes. No lesions consistent with bacterial pneumonia were observed and only weak growth of Salmonella was observed following culture of the lung. The presence of Salmonella bacteria in the lungs of this animal was not associated with any pathology. This second isolate was also lysine decarboxylase negative and was serotyped as 4:a:-.
Figure 1.
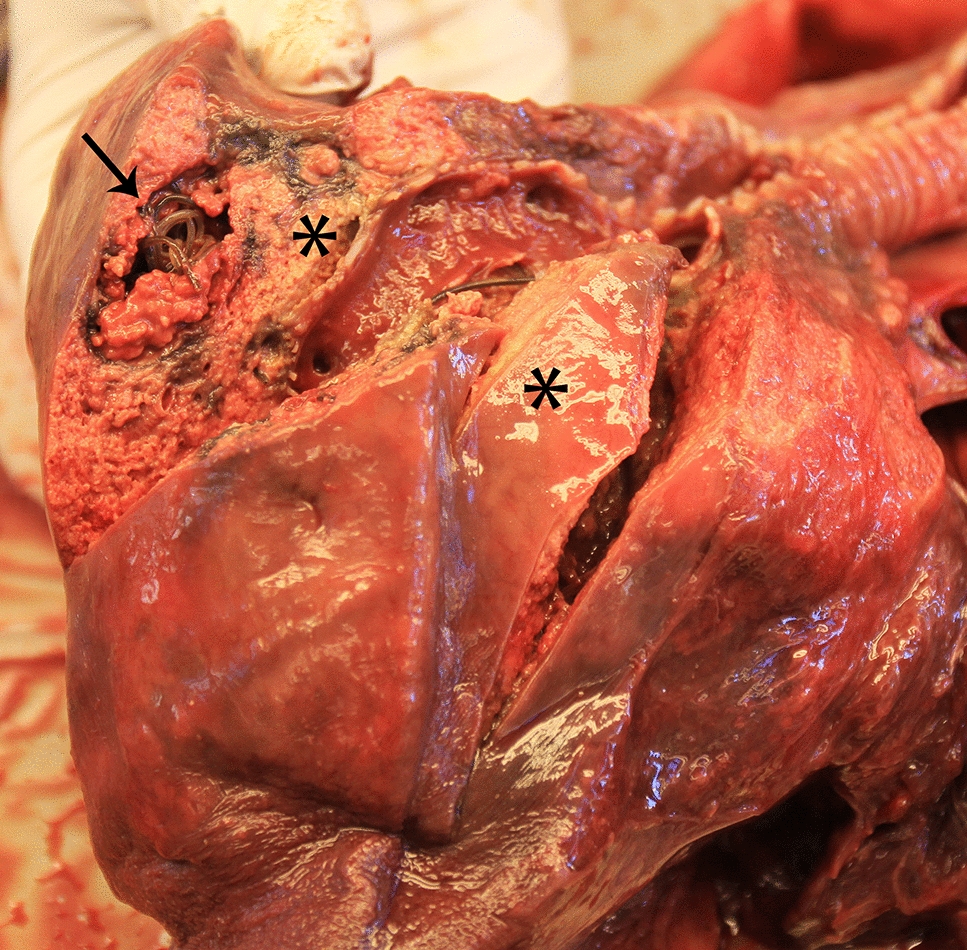
Lung from a stranded harbour porpoise (Phocoena phocoena) 17-VLT002652 from which abundant growth of Salmonella bacteria were cultured. Severe necrotizing pneumonia (asterisk) and moderate to heavy lungworm infestation (arrow) are evident.
Figure 2.
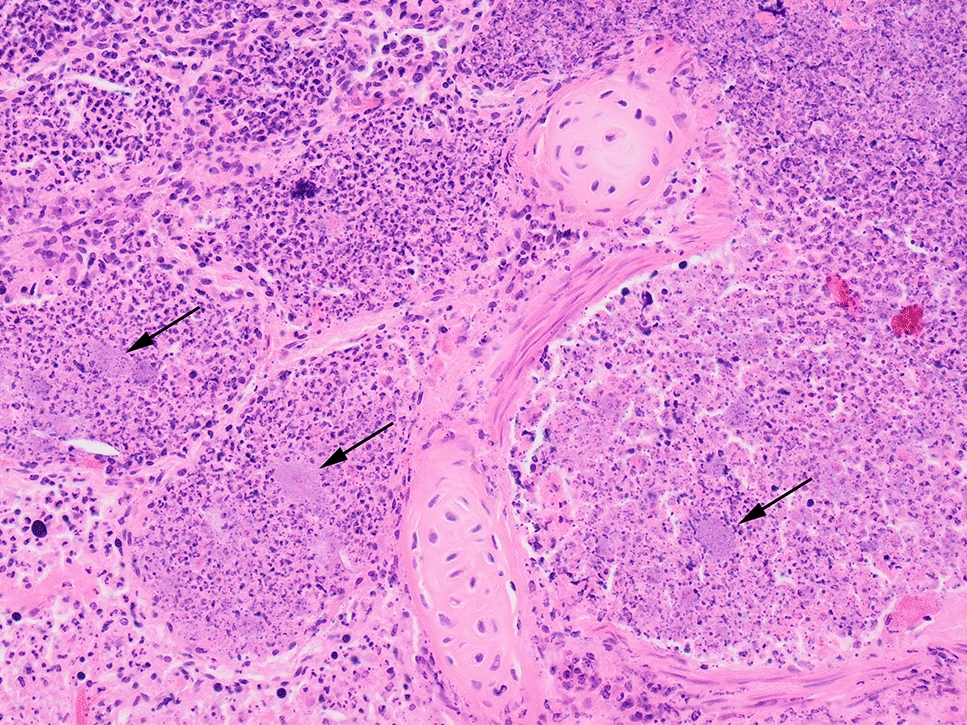
Photomicrograph of the lung from a stranded porpoise (Phocoena phocoena) 17-VLT002652 displaying severe, suppurative and necrotizing bronchopneumonia. Mats of bacteria (arrows) can be seen amongst abundant, degenerate neutrophils which fill alveoli and airways. Hematoxylin and eosin stain, ×200 magnification.
Core-genome MLST comparison of porpoise Salmonella and identification of close relatives
Both Swedish porpoise isolates were ST416. Phylogenetic analysis based on cgMLST indicated that the Swedish isolates most closely matched 17 “Fulica-like” or “Fulica-like rough” ST416 or ST417 isolates in Enterobase, of which 16 were from harbour porpoises in Scotland and 1 was from Scotland but lacked metadata. The Swedish isolates were most similar to each other but clustered closely with the main group of Scottish isolates. Isolates of serovars Bispebjerg (n = 23), Fulica (n = 2), Abortusequi (n = 10) and of unknown serovars (n = 7) were identified as the best matches from sources other than porpoises based on belonging to the cgMLST group 4372 at the hierarchical clustering level HC2000 but were far less closely related (Figure 3). Most included Bispebjerg isolates were from humans (n = 12), with additional isolates from turtles (n = 4), food (n = 2), a falcon (n = 1) or unspecified sources. The only two isolates of Fulica, both isolated from humans, clustered together but as part of the Bispebjerg cgMLST cluster. The Abortusequi isolates were from horses (n = 2) or unspecified sources. The isolates without specified serotypes were from humans (n = 2), palm nuts (n = 2), a donkey (n = 1) or unspecified sources.
Figure 3.
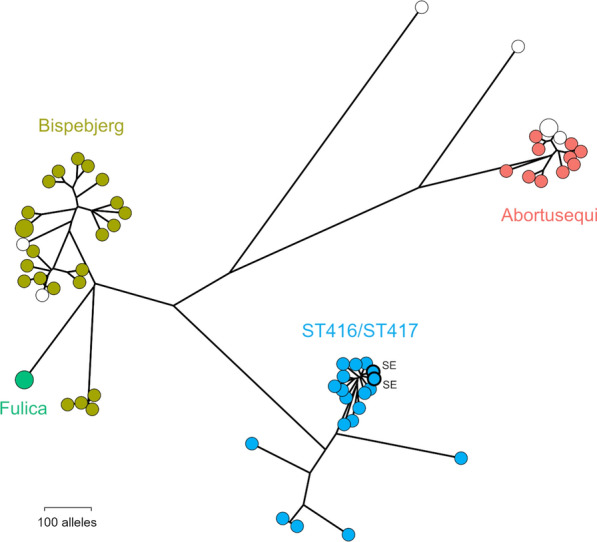
Phylogenetic relationship between harbour porpoise isolates of S. enterica ST416/ST417 and their nearest neighbours as determined by cgMLST. The closest relatives identified include isolates identified as serovars Bispebjerg, Fulica and Abortusequi. Swedish isolates sequenced in the present study are labelled SE. White circles indicate isolates for which no serotypes were reported.
Alterations in major virulence associated genomic regions
The spvR,A,B,C,D genes forming the signature locus of the Salmonella virulence plasmids [35] were absent in all ST416/417 isolates. The spv locus was present in most S. Abortusequi isolates, and a single isolate with no serovar assigned, while absent from the remaining genomes analysed. Major structural differences were observed between ST416/417 isolate pathogenicity islands SPI1-5 and those of the related isolates (Figure 4, Additional file 1). SPI-1 was found to be partially deleted with hilA which is a transcriptional regulator necessary for SPI-1 expression [36] missing together with iagB and the translocated effector proteins sptP, sipA and sipD. The majority of SPI-2 was also absent, leaving only ssrB and a segment containing ssaU,T,S,R. SPI-3 was completely absent together with a ~15kbp downstream genomic region present in LT2. SPI-4, which encodes a T1SS and a single large secreted adhesin, was present in the 17-VLT002652 genome, but both the adhesin gene (siiE, annotated as a hypothetical protein with locus tag STM4261 in the LT2 genome) and an essential gene for the T1SS (siiF, locus tag STM4262) were disrupted by IS-element insertions in coding regions. SPI-5 was also partially absent, with only sopB and pipA,B,C remaining. A short fragment consistent with a partial SPI-5 pipD gene was present elsewhere in the genome flanked by insertion sequence (IS)-element transposase genes. IS256 family ISEae1 or ISSod4 element transposase genes were present flanking the described deletions. The deletions observed in SPI-1, SPI-2, SPI-3 and SPI-5 were shared among ST416/417 isolates with variation in the extent of the deletions in a few isolates (Additional file 1). In contrast, the elements were mostly conserved in Bispebjerg, Fulica and Abortusequi isolates with only minor variation in SPI-3 and SPI-5 (Additional file 1). Consistent with the changes in SPIs described above, KEGG pathway analysis indicated overrepresentation of alterations in the categories “Salmonella infection”, “Bacterial secretion system” and “Bacterial invasion of epithelial cells” (Table 1) in the ST416 isolate 17-VLT002652 compared to the S. enterica type strain LT2.
Figure 4.

Comparison between Salmonella pathogenicity islands in S. Typhimurium and S. enterica ST416/ST417. SPI1–SPI5 (arrows) structure in the S. Typhimurium strain LT2 with highlighted regions present in the ST416 isolate 17-VLT2652 (blue) and IS-element insertions in 17-VLT2652 (red).
Table 1.
KEGG pathways altered in ST416/ST417
| Pathway | Pathway name | Pan-genome | SNP |
|---|---|---|---|
| stm01100 | Metabolic pathways | 46 | 30 |
| stm01120 | Microbial metabolism in diverse environments | 35 | 9 |
| stm02020 | Two-component system | 20 | 9 |
| stm05132 | Salmonella infection | 13 | 5 |
| stm02060 | Phosphotransferase system (PTS) | 12 | – |
| stm01110 | Biosynthesis of secondary metabolites | 12 | 7 |
| stm03070 | Bacterial secretion system | 12 | – |
| stm00350 | Tyrosine metabolism | 10 | 2 |
| stm00030 | Pentose phosphate pathway | 9 | – |
| stm01220 | Degradation of aromatic compounds | 7 | – |
| stm01200 | Carbon metabolism | 6 | 2 |
| stm02024 | Quorum sensing | 6 | – |
| stm02010 | ABC transporters | 5 | 5 |
| stm00051 | Fructose and mannose metabolism | 5 | 4 |
| stm00520 | Amino sugar and nucleotide sugar metabolism | 5 | 4 |
| stm00910 | Nitrogen metabolism | 5 | – |
| stm00010 | Glycolysis/gluconeogenesis | 5 | – |
| stm01230 | Biosynthesis of amino acids | 4 | – |
| stm00310 | Lysine degradation | 4 | 3 |
| stm05100 | Bacterial invasion of epithelial cells | 3 | – |
| stm00330 | Arginine and proline metabolism | 3 | – |
| stm00630 | Glyoxylate and dicarboxylate metabolism | 3 | 2 |
| stm00650 | Butanoate metabolism | 3 | 2 |
| stm00680 | Methane metabolism | 3 | 2 |
| stm00620 | Pyruvate metabolism | 3 | – |
| stm00260 | Glycine, serine and threonine metabolism | 3 | – |
| stm03430 | Mismatch repair | 3 | – |
| stm00760 | Nicotinate and nicotinamide metabolism | – | 3 |
| stm00250 | Alanine, aspartate and glutamate metabolism | – | 3 |
| stm01503 | Cationic antimicrobial peptide (CAMP) resistance | - | 3 |
| stm03010 | Ribosome | 2 | – |
| stm00071 | Fatty acid degradation | 2 | – |
| stm00400 | Phenylalanine, tyrosine and tryptophan biosynthesis | 2 | – |
| stm00633 | Nitrotoluene degradation | 2 | – |
| stm00640 | Propanoate metabolism | 2 | – |
| stm00500 | Starch and sucrose metabolism | 2 | 4 |
| stm02030 | Bacterial chemotaxis | 2 | - |
| stm00564 | Glycerophospholipid metabolism | 2 | – |
| stm00562 | Inositol phosphate metabolism | 2 | – |
| stm00541 | O-Antigen nucleotide sugar biosynthesis | – | 2 |
| stm00540 | Lipopolysaccharide biosynthesis | – | 2 |
| stm02060 | Phosphotransferase system (PTS) | – | 2 |
| stm00052 | Galactose metabolism | – | 2 |
| stm00230 | Purine metabolism | – | 2 |
KEGG pathways significantly enriched for presence/absence variation in isolates of ST416/ST417 compared to the S. enterica type strain LT2 as determined by pan-genome analysis and identification of high impact (i.e., presumed loss of function) SNPs.
Alterations in genes encoding metabolic pathways and other biological functions
ST416/417 isolates were shown by pangenome and SNP analysis to have enriched loss-of-function alterations in several metabolic pathways when compared to LT2 (Table 1). Genes associated with microbial metabolism in diverse environments were particularly strongly affected, as were pathways involved in sensing and responding to environmental cues such as two-component systems and quorum sensing. Contrasting ST416/417 with its close relatives by pangenome analysis revealed that several of the ST416/417 vs. LT2 differences were not shared with the rest of group 4372 (Additional file 2). For example, ST416/417 appear to consistently lack the gene encoding glucarate dehydratase (gudD) which is part of a locus essential for fermentation of galactarate and glucarate in S. Typhimurium. Loss of function in this locus is a trait known to be common only among serovars associated with extraintestinal infection [37]. Several genes of the 4-hydroxyphenylacetate catabolic pathway were missing in ST416/417, consistent with pseudogene formation in this locus in S. Typhi [17], as were the formate hydrogenlyase genes hycA,B,C,D which are active under anaerobic conditions [38]. Also missing was the transcription factor eutR, which is involved in niche recognition and utilization of ethanolamine in the inflamed host intestine by acting as a sensor for this substance [39]. Loss of eutR has previously been linked to host adapted extraintestinal pathovars of Salmonella [18]. Furthermore, multiple genes involved in adhesion, chemotaxis and motility (stdA,B,C; fljA; tsr, hin) which have been described as frequently disrupted in extraintestinal serovars [18] were absent or significantly altered in ST416/417, as was interestingly the aerotaxis receptor gene aer.
Acquired accessory genes
ST416/417 were found to be in possession of a number of genomic regions absent elsewhere in group 4372 (Additional file 2). Several plasmid and prophage-associated genes were included in this unique set, and PlasmidFinder-2 identified three contigs as putative plasmids of incompatibility types IncI1-I(Gamma) (contig 26), IncFIB (contig 3) and IncFII (contig 19). Bandage analysis confirmed that contigs 26 and 19 were indeed consistent with circular plasmids, whereas contig 3 appears to be chromosomal. Notably, the iucA,C,D/iutA aerobactin gene cluster encoding a siderophore iron uptake system associated with extraintestinal infections by both avian pathogenic E. coli (APEC) [40] and poultry-associated Salmonella [41] was uniquely present in all ST416/417. The same was true for the tsh gene encoding a temperature-sensitive hemagglutinin which is also linked to APEC virulence [42], and which was co-located with iucA,C,D/iutA in 17-VLT002652. ST416/417 also uniquely carried a version of the hemR hemin receptor gene, encoding another putative iron uptake system.
Model-based estimation of extraintestinal niche adaptation
A random forest classifier model previously trained to identify strains associated with extraintestinal disease based on well-characterised gastrointestinal and extraintestinal Salmonella serovars [27] was used to assess all ST416/417 isolates and their closest relatives. The results indicate that all group 4372 have genetic adaptations previously observed in extraintestinal serotypes, with ST416/417 and Abortusequi scoring higher than Bispebjerg and Fulica (Figure 5). However, all tested isolates scored lower than the reference set of extraintestinal serovars.
Figure 5.
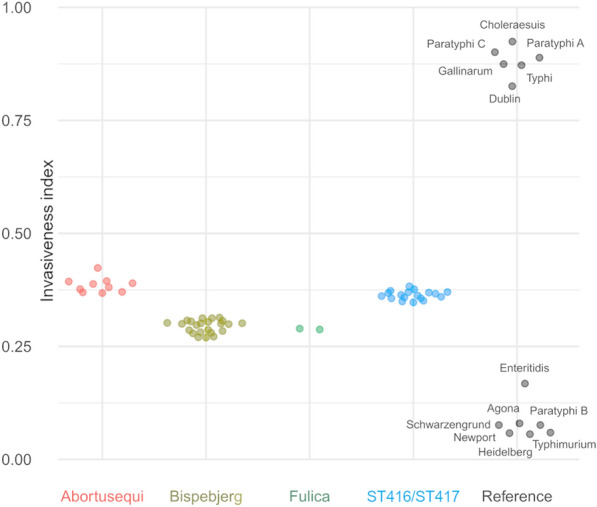
Invasiveness index for Salmonella isolates analysed in the present study compared to reference serotypes. Reference isolates considered to be associated with gastrointestinal (low score) or extraintestinal (high score) infection shown to the right.
Discussion
We here report the first observations of putatively host-adapted group B Salmonella enterica ST416 in harbour porpoises from Swedish waters. Both infected animals were found in a region occupied by porpoises of the Belt Sea population, but genetic analysis would be necessary to confirm their origin. Unlike studies on porpoises from waters around Great Britain where confirmed or presumed ST416/417 have been isolated from 20 to 35% of porpoises analysed [11, 12, 43], this bacterium was found in only 2 of the 47 lung samples (4%) examined in this study. However, relatively few porpoises were examined, and isolation rates may have been influenced by isolation protocols, freezing of carcasses prior to examination, and the effects of post-mortem decomposition. The occurrence of closely related strains in Swedish and British porpoises supports the theory of ST416/417 as specifically adapted for cetacean hosts. To our knowledge and with the exception of one possible case described from the Netherlands [6], host-adapted Salmonella has not been reported in harbour porpoises elsewhere. In a previous study, a few Salmonella sp. isolates were recovered from porpoises in the German Baltic Sea, but no infection was observed in animals from the German North Sea or in the waters around Norway and Greenland [44]. The recovered Salmonella isolates were not typed. Interestingly, Valderrama Vasquez et al. [43] found a significant increase in the frequency of Salmonella infection among porpoises over time from 6% in 1990–1994 to 27% in 2000–2002. The reason for this increase is not known but change in immune competence of the host, for example due to anthropogenic environmental disturbances, cannot be excluded. Further surveillance is needed to better estimate the prevalence of ST416/417 among Swedish porpoises and to monitor changes over time, as well as to investigate possible links between parasite load and risk of salmonellosis for these animals. Recovering more isolates for whole-genome analysis would also be beneficial to determine if ST416/417 has been introduced recently in Swedish waters, as we would expect a long-term established wildlife reservoir to host a higher degree of genetic variation among the bacteria.
Our results show that ST416/417 is not necessarily pathogenic for porpoises, as it was cultured from the lungs of an apparently healthy animal in the absence of associated pathology. However, it did cause serious disease in the other case. This is consistent with findings in other studies [10, 11, 43]. These researchers propose that this type of Salmonella is part of the commensal flora in the lung of porpoises but may become an opportunistic pathogen under the right conditions, including immunosuppression. The major genetic virulence factors of S. enterica are encoded on pathogenicity islands, SPIs, and we here show that the key SPIs 1–5 appear to be largely inactivated in ST416/417 in contrast to those of near relatives. These SPIs are believed to have been acquired early in the evolution of S. enterica and most of them are generally considered to be highly conserved among well studied serovars [15, 45]. It is possible that extensive loss of virulence factors is a common adaptation among commensal and opportunistically pathogenic strains of S. enterica, which are less likely to be sampled and sequenced. We note that the inactivation of these elements seems to have been the result of IS element activity, a mechanism of virulence modulation known to occur in a wide range of bacterial species [46].
Gastrointestinal serovars of S. enterica have extensive metabolic capabilities evolved e.g. to exploit nutrients available in the inflamed host gut. The complex genetic network needed for this tends to be degraded in previously studied extraintestinal serovars [17, 18]. Pathway analysis indicates reduced metabolic function in ST416/417, consistent with the fact that this type of Salmonella has in most cases been recovered from extraintestinal samples and in particular from the lungs of infected porpoises. ST416/417 also share other traits common among extraintestinal serovars such as loss of capabilities for sensing and responding to environmental cues. Directly linking alterations contributing to virulence or host adaptation on a gene-by-gene basis is complicated by our limited understanding of the complex networks of gene interaction that determine the fitness of a strain under a particular set of circumstances. Approaches based on machine learning show great promise in solving this type of problem and have recently been applied to Salmonella classification and source attribution [27, 47, 48]. The system implemented in the present study classified ST416/417 isolates as moderately invasive, but the small size and diversity of the training dataset is likely to be a limiting factor. Future improvement of predictions of host bias or disease phenotype based on genomics will strongly depend on the continued collection of high-quality epidemiological and clinical outcome data as well as sequences.
Unsuccessful attempts have been made in the past to find close relatives of the porpoise Salmonella strains using MLST [9], and other molecular methods such as ribotyping, insertion sequence element fingerprinting and PCR-based profiling [49]. Our results using cgMLST largely reflect the results of these previous studies in not being able to find any closely related isolates from other sources, despite the substantial number of genome sequences now available for cgMLST comparison in Enterobase. Again in agreement with results from the aforementioned studies, the closest relatives we do find include some but not all serotypes with similar antigenic profiles to 4,12:a:- (Figure 3). Bispebjerg (1,4,[5],12:a:e,n,x) and the rare Fulica (4,[5],12:a[1,5]:) are both poorly characterized and seem to occur in diverse sources including cases of illness in humans. The somewhat less closely related Abortusequi (4,12:-:e,n,x) is host restricted to horses [13] and known for causing extraintestinal infections in the form of equine paratyphoid characterized by abortions in mares and septicaemia in young foals [50]. Interestingly, Abortusequi has been recovered in high numbers from parasitic aneurysms in apparently healthy and otherwise Salmonella-negative horses [51], with the hypothesis raised that the lesions provided a niche for long-term carriage of the bacteria. An outlier without serotype assignment among the identified related strains was recovered by metagenomic sequencing from the tooth pulp of a Neolithic hunter-gatherer, presumably suffering from systemic infection [52]. In general, a parallel can perhaps be drawn between this lineage of S. enterica and that containing the generalist serovar Enteritidis, the host-adapted Dublin (cattle) and the host-restricted Gallinarum (poultry) [15] with a set of shared adaptations providing evolutionary opportunities for further specialization for different hosts. We intend to further investigate this by targeted sequencing of historical Salmonella isolates with antigen profiles similar to 4,12:a:- from other wildlife sources.
As previously mentioned, further studies will also be necessary to determine the relevance of extraintestinal salmonellosis as a health threat for porpoises and the possible emergent health effect of the combination of microbial infections, parasite load and environmental stressors. While the public health relevance of porpoise Salmonella strains is likely to be low due to limited exposure and likely reduced virulence, other host adapted serovars of Salmonella including Dublin [53], Choleraesuis [54], and certain Typhimurium [55] commonly cause illness in humans. Adaptation for causing extraintestinal infection in some of these serovars in fact increases the risk of severe and life-threatening systemic Salmonella infections, generally affecting the elderly or otherwise vulnerable [53, 54]. A better understanding of the genetic changes underlying the evolution of host adaptation and extraintestinal niche specialization therefore has the potential to be highly valuable both for improving animal and human health.
Supplementary Information
{kind=link}
Additional file 1. Deletions in Salmonella pathogenicity islands in S. enterica ST416/ST417. Presence (black) or absence (grey) of genes in Salmonella pathogenicity islands SPI-1,2,3,5 as determined by pangenome (Roary) analysis. The two Swedish isolates from the present study are labelled SE. Extensive deletions are evident in all ST416/ST417 isolates, while the islands are highly conserved among other related strains.
Additional file 2. Pan-genome analysis output, presence/absence and significance testing. Number of isolates of ST416/ST417 (referred to as “positive”) and other related Salmonella isolates (referred to as “negative”) that were found by pan-genome analysis to posess each identified sequence, i.e. gene or gene variant, and significance testing results for comparison of the frequence of occurrence in the two groups.
Additional file 3. Pan-genome analysis output, FASTA reference sequences. FASTA format sequences for the “groups” of sequence variants compared in Additional file 2.
Additional file 4. R script for processing SNP data. A short script for the R software environment used to process and filter SNP data.
Acknowledgements
The authors would like to thank Erik Ågren, Tobias Lilja and the SVA Salmonella laboratory for technical assistance.
Abbreviations
- bp
base pairs
- BG agar
brilliant green agar
- cgMLST
core genome multi-locus sequence typing
- IS
insertion sequence
- MALDI-TOFMS
matrix assisted laser desorption ionization-time of flight mass spectrometry
- MLST
multi-locus sequence typing
- MSRV agar
modified semi-solid Rappaport–Vassiliadis agar
- PCR
polymerase chain reaction
- SNP
single nucleotide polymorphism
- SPI
Salmonella Pathogenicity island
- ST
sequence type
- SVA
Swedish National Veterinary Institute
- T1SS/T3SS
type 1/type 3 secretion system
- XLD agar
xylose lysine deoxycholate agar
Authors’ contributions
AN and AR secured funding, organized the collection of animals and performed and interpreted post-mortem examinations. JE performed and interpreted microbiological analyses. AKSS and RS organized sequencing, performed the bioinformatic analyses and wrote the first manuscript draft. All authors read and approved the final manuscript.
Funding
Funding support for the collection and examination of harbour porpoises was provided by The Swedish Agency for Marine and Water Management and The Swedish Environmental Protection Agency. Funders had no influence over how the study was performed, the manuscript text or the decision to publish.
Availability of data and materials
Sequence data from long and short read sequencing with metadata have been deposited in the European Nucleotide Archive [56] under project accession number PRJEB40444. Typing data from cgMLST analysis has been deposited in Enterobase under isolate IDs (uberstrains) SAL_EB4912AA and SAL_EB4913AA. Pan-genome analysis output is presented in Additional files 2 and 3.
Declarations
Ethics approval and consent to participate
All animals were examined post-mortem as part of routine national wildlife surveillance. Therefore, this study required no ethics approval by Swedish law.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Braulik GM, Minton G, Amano M, Bjørge A (2020) Phocoena phocoena. The IUCN Red List of Threatened Species 2020. IUCN
- 2.Van Bressem MF, Raga JA, Di Guardo G, Jepson PD, Duignan PJ, Siebert U, Barrett T, Santos MC, Moreno IB, Siciliano S, Aguilar A, Van Waerebeek K. Emerging infectious diseases in cetaceans worldwide and the possible role of environmental stressors. Dis Aquat Organ. 2009;86:143–157. doi: 10.3354/dao02101. [DOI] [PubMed] [Google Scholar]
- 3.Neimane A, Stavenow J, Ågren E, Wikström E, Roos A (2020) Hälso-och sjukdomsövervakning av marina däggdjur Del 2. Hälsa, sjukdomar och dödsorsaker hos tumlare (Phocoena phocoena) i Sverige de senaste 10 åren. National Veterinary Institute (SVA), Sweden
- 4.Jauniaux T, Petitjean D, Brenez C, Borrens M, Brosens L, Haelters J, Tavernier T, Coignoul F. Post-mortem findings and causes of death of harbour porpoises (Phocoena phocoena) stranded from 1990 to 2000 along the coastlines of Belgium and Northern France. J Comp Pathol. 2002;126:243–253. doi: 10.1053/jcpa.2001.0547. [DOI] [PubMed] [Google Scholar]
- 5.Jepson PD, Baker JR, Kuiken T, Simpson VR, Kennedy S, Bennett PM. Pulmonary pathology of harbour porpoises (Phocoena phocoena) stranded in England and Wales between 1990 and 1996. Vet Rec. 2000;146:721–728. doi: 10.1136/vr.146.25.721. [DOI] [PubMed] [Google Scholar]
- 6.van Elk CE, van de Bildt MWG, van Run P, Bunskoek P, Meerbeek J, Foster G, Osterhaus A, Kuiken T. Clinical, pathological, and laboratory diagnoses of diseases of harbour porpoises (Phocoena phocoena), live stranded on the Dutch and adjacent coasts from 2003 to 2016. Vet Res. 2019;50:88. doi: 10.1186/s13567-019-0706-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fenton H, Daoust PY, Forzan MJ, Vanderstichel RV, Ford JK, Spaven L, Lair S, Raverty S. Causes of mortality of harbor porpoises Phocoena phocoena along the Atlantic and Pacific coasts of Canada. Dis Aquat Organ. 2017;122:171–183. doi: 10.3354/dao03080. [DOI] [PubMed] [Google Scholar]
- 8.Reckendorf A, Everaarts E, Bunskoek P, Haulena M, Springer A, Lehnert K, Lakemeyer J, Siebert U, Strube C. Lungworm infections in harbour porpoises (Phocoena phocoena) in the German Wadden Sea between 2006 and 2018, and serodiagnostic tests. Int J Parasitol Parasites Wildl. 2021;14:53–61. doi: 10.1016/j.ijppaw.2021.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haase JK, Brown DJ, Weill FX, Mather H, Foster G, Brisse S, Wain J, Achtman M. Population genetic structure of 4,12:a:- Salmonella enterica strains from harbor porpoises. Appl Environ Microbiol. 2012;78:8829–8833. doi: 10.1128/AEM.02310-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davison NJ, Simpson VR, Chappell S, Monies RJ, Stubberfield EJ, Koylass M, Quinney S, Deaville R, Whatmore AM, Jepson PD. Prevalence of a host-adapted group B Salmonella enterica in harbour porpoises (Phocoena phocoena) from the south-west coast of England. Vet Rec. 2010;167:173–176. doi: 10.1136/vr.c3760. [DOI] [PubMed] [Google Scholar]
- 11.Foster G, Patterson IA, Munro DS. Monophasic group B Salmonella species infecting harbour porpoises (Phocoena phocoena) inhabiting Scottish coastal waters. Vet Microbiol. 1999;65:227–231. doi: 10.1016/s0378-1135(98)00296-x. [DOI] [PubMed] [Google Scholar]
- 12.Davison N, Barnett J, Rule B, Chappell S, Wise G. Group B Salmonella in lungworms from a harbour porpoise (Phocoena phocoena) Vet Rec. 2010;167:351–352. doi: 10.1136/vr.c4495. [DOI] [PubMed] [Google Scholar]
- 13.Uzzau S, Brown DJ, Wallis T, Rubino S, Leori G, Bernard S, Casadesus J, Platt DJ, Olsen JE. Host adapted serotypes of Salmonella enterica. Epidemiol Infect. 2000;125:229–255. doi: 10.1017/s0950268899004379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Parkhill J, Dougan G, James KD, Thomson NR, Pickard D, Wain J, Churcher C, Mungall KL, Bentley SD, Holden MT, Sebaihia M, Baker S, Basham D, Brooks K, Chillingworth T, Connerton P, Cronin A, Davis P, Davies RM, Dowd L, White N, Farrar J, Feltwell T, Hamlin N, Haque A, Hien TT, Holroyd S, Jagels K, Krogh A, Larsen TS, Leather S, Moule S, O’Gaora P, Parry C, Quail M, Rutherford K, Simmonds M, Skelton J, Stevens K, Whitehead S, Barrell BG. Complete genome sequence of a multiple drug resistant Salmonella enterica serovar Typhi CT18. Nature. 2001;413:848–852. doi: 10.1038/35101607. [DOI] [PubMed] [Google Scholar]
- 15.Langridge GC, Fookes M, Connor TR, Feltwell T, Feasey N, Parsons BN, Seth-Smith HM, Barquist L, Stedman A, Humphrey T, Wigley P, Peters SE, Maskell DJ, Corander J, Chabalgoity JA, Barrow P, Parkhill J, Dougan G, Thomson NR. Patterns of genome evolution that have accompanied host adaptation in Salmonella. Proc Natl Acad Sci USA. 2015;112:863–868. doi: 10.1073/pnas.1416707112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chiu CH, Tang P, Chu C, Hu S, Bao Q, Yu J, Chou YY, Wang HS, Lee YS. The genome sequence of Salmonella enterica serovar Choleraesuis, a highly invasive and resistant zoonotic pathogen. Nucleic Acids Res. 2005;33:1690–1698. doi: 10.1093/nar/gki297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Seif Y, Kavvas E, Lachance JC, Yurkovich JT, Nuccio SP, Fang X, Catoiu E, Raffatellu M, Palsson BO, Monk JM. Genome-scale metabolic reconstructions of multiple Salmonella strains reveal serovar-specific metabolic traits. Nat Commun. 2018;9:3771. doi: 10.1038/s41467-018-06112-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nuccio SP, Baumler AJ. Comparative analysis of Salmonella genomes identifies a metabolic network for escalating growth in the inflamed gut. MBio. 2014;5:e00929-14. doi: 10.1128/mBio.00929-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.IJsseldijk LL, Brownlow AC, Mazzariol S (2019) Best practice on cetacean post mortem investigation and tissue sampling. Joint ACCOBAMS and ASCOBANS document
- 20.Bancroft JD, Cook HC. Manual of histological techniques. London: Churchill Livingstone; 1984. [Google Scholar]
- 21.Hoorfar J, Ahrens P, Radstrom P. Automated 5′ nuclease PCR assay for identification of Salmonella enterica. J Clin Microbiol. 2000;38:3429–3435. doi: 10.1128/JCM.38.9.3429-3435.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Enterobase. https://enterobase.warwick.ac.uk/
- 23.De Coster W, D’Hert S, Schultz DT, Cruts M, Van Broeckhoven C. NanoPack: visualizing and processing long-read sequencing data. Bioinformatics. 2018;34:2666–2669. doi: 10.1093/bioinformatics/bty149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cingolani P, Platts A, le Wang L, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly. 2012;6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mi H, Muruganujan A, Casagrande JT, Thomas PD. Large-scale gene function analysis with the PANTHER classification system. Nat Protoc. 2013;8:1551–1566. doi: 10.1038/nprot.2013.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kanehisa M, Sato Y. KEGG Mapper for inferring cellular functions from protein sequences. Protein Sci. 2020;29:28–35. doi: 10.1002/pro.3711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wheeler NE, Gardner PP, Barquist L. Machine learning identifies signatures of host adaptation in the bacterial pathogen Salmonella enterica. PLoS Genet. 2018;14:e1007333. doi: 10.1371/journal.pgen.1007333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wick RR, Judd LM, Gorrie CL, Holt KE. Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput Biol. 2017;13:e1005595. doi: 10.1371/journal.pcbi.1005595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wick RR, Schultz MB, Zobel J, Holt KE. Bandage: interactive visualization of de novo genome assemblies. Bioinformatics. 2015;31:3350–3352. doi: 10.1093/bioinformatics/btv383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30:2068–2069. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 31.Page AJ, Cummins CA, Hunt M, Wong VK, Reuter S, Holden MT, Fookes M, Falush D, Keane JA, Parkhill J. Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics. 2015;31:3691–3693. doi: 10.1093/bioinformatics/btv421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brynildsrud O, Bohlin J, Scheffer L, Eldholm V. Rapid scoring of genes in microbial pan-genome-wide association studies with Scoary. Genome Biol. 2016;17:238. doi: 10.1186/s13059-016-1108-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.The R project for statistical computing. https://www.r-project.org/
- 34.gggenes. https://wilkox.org/gggenes/
- 35.Silva C, Puente JL, Calva E. Salmonella virulence plasmid: pathogenesis and ecology. Pathog Dis. 2017;75:ftx070. doi: 10.1093/femspd/ftx070. [DOI] [PubMed] [Google Scholar]
- 36.Lou L, Zhang P, Piao R, Wang Y. Salmonella pathogenicity island 1 (SPI-1) and its complex regulatory network. Front Cell Infect Microbiol. 2019;9:270. doi: 10.3389/fcimb.2019.00270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Faber F, Tran L, Byndloss MX, Lopez CA, Velazquez EM, Kerrinnes T, Nuccio SP, Wangdi T, Fiehn O, Tsolis RM, Baumler AJ. Host-mediated sugar oxidation promotes post-antibiotic pathogen expansion. Nature. 2016;534:697–699. doi: 10.1038/nature18597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lamichhane-Khadka R, Benoit SL, Miller-Parks EF, Maier RJ. Host hydrogen rather than that produced by the pathogen is important for Salmonella enterica serovar Typhimurium virulence. Infect Immun. 2015;83:311–316. doi: 10.1128/IAI.02611-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Anderson CJ, Clark DE, Adli M, Kendall MM. Ethanolamine signaling promotes Salmonella niche recognition and adaptation during infection. PLoS Pathog. 2015;11:e1005278. doi: 10.1371/journal.ppat.1005278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gao Q, Wang X, Xu H, Xu Y, Ling J, Zhang D, Gao S, Liu X. Roles of iron acquisition systems in virulence of extraintestinal pathogenic Escherichia coli: salmochelin and aerobactin contribute more to virulence than heme in a chicken infection model. BMC Microbiol. 2012;12:143. doi: 10.1186/1471-2180-12-143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wellawa DH, Allan B, White AP, Koster W. Iron-uptake systems of chicken-associated Salmonella serovars and their role in colonizing the avian host. Microorganisms. 2020;8:1203. doi: 10.3390/microorganisms8081203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dozois CM, Dho-Moulin M, Bree A, Fairbrother JM, Desautels C, Curtiss R., 3rd Relationship between the Tsh autotransporter and pathogenicity of avian Escherichia coli and localization and analysis of the tsh genetic region. Infect Immun. 2000;68:4145–4154. doi: 10.1128/iai.68.7.4145-4154.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Valderrama Vasquez CA, Macgregor SK, Rowcliffe JM, Jepson PD. Occurrence of a monophasic strain of Salmonella group B isolated from cetaceans in England and Wales between 1990 and 2002. Environ Microbiol. 2008;10:2462–2468. doi: 10.1111/j.1462-2920.2008.01651.x. [DOI] [PubMed] [Google Scholar]
- 44.Siebert U, Prenger-Berninghoff E, Weiss R. Regional differences in bacterial flora in harbour porpoises from the North Atlantic: environmental effects? J Appl Microbiol. 2009;106:329–337. doi: 10.1111/j.1365-2672.2008.04006.x. [DOI] [PubMed] [Google Scholar]
- 45.Hensel M. Evolution of pathogenicity islands of Salmonella enterica. Int J Med Microbiol. 2004;294:95–102. doi: 10.1016/j.ijmm.2004.06.025. [DOI] [PubMed] [Google Scholar]
- 46.Vandecraen J, Chandler M, Aertsen A, Van Houdt R. The impact of insertion sequences on bacterial genome plasticity and adaptability. Crit Rev Microbiol. 2017;43:709–730. doi: 10.1080/1040841X.2017.1303661. [DOI] [PubMed] [Google Scholar]
- 47.Lupolova N, Lycett SJ, Gally DL. A guide to machine learning for bacterial host attribution using genome sequence data. Microb Genom. 2019;5:e000317. doi: 10.1099/mgen.0.000317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang S, Li S, Gu W, den Bakker H, Boxrud D, Taylor A, Roe C, Driebe E, Engelthaler DM, Allard M, Brown E, McDermott P, Zhao S, Bruce BB, Trees E, Fields PI, Deng X. Zoonotic source attribution of Salmonella enterica serotype Typhimurium using genomic surveillance data, United States. Emerg Infect Dis. 2019;25:82–91. doi: 10.3201/eid2501.180835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Old DC, Crichton PB, Taylor A, Mather H. An attempt to identify the evolutionary origin of a novel serotype of Salmonella enterica isolated from harbour porpoises. J Med Microbiol. 2001;50:415–420. doi: 10.1099/0022-1317-50-5-415. [DOI] [PubMed] [Google Scholar]
- 50.Grandolfo E, Parisi A, Ricci A, Lorusso E, de Siena R, Trotta A, Buonavoglia D, Martella V, Corrente M. High mortality in foals associated with Salmonella enterica subsp. enterica Abortusequi infection in Italy. J Vet Diagn Invest. 2018;30:483–485. doi: 10.1177/1040638717753965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Niwa H, Hobo S, Kinoshita Y, Muranaka M, Ochi A, Ueno T, Oku K, Hariu K, Katayama Y. Aneurysm of the cranial mesenteric artery as a site of carriage of Salmonella enterica subsp. enterica serovar Abortusequi in the horse. J Vet Diagn Invest. 2016;28:440–444. doi: 10.1177/1040638716649640. [DOI] [PubMed] [Google Scholar]
- 52.Key FM, Posth C, Esquivel-Gomez LR, Hubler R, Spyrou MA, Neumann GU, Furtwangler A, Sabin S, Burri M, Wissgott A, Lankapalli AK, Vagene AJ, Meyer M, Nagel S, Tukhbatova R, Khokhlov A, Chizhevsky A, Hansen S, Belinsky AB, Kalmykov A, Kantorovich AR, Maslov VE, Stockhammer PW, Vai S, Zavattaro M, Riga A, Caramelli D, Skeates R, Beckett J, Gradoli MG, Steuri N, Hafner A, Ramstein M, Siebke I, Losch S, Erdal YS, Alikhan NF, Zhou Z, Achtman M, Bos K, Reinhold S, Haak W, Kuhnert D, Herbig A, Krause J. Emergence of human-adapted Salmonella enterica is linked to the Neolithization process. Nat Ecol Evol. 2020;4:324–333. doi: 10.1038/s41559-020-1106-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Harvey RR, Friedman CR, Crim SM, Judd M, Barrett KA, Tolar B, Folster JP, Griffin PM, Brown AC. Epidemiology of Salmonella enterica serotype Dublin infections among humans, United States, 1968–2013. Emerg Infect Dis. 2017;23:1493–1501. doi: 10.3201/eid2309.170136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chiu CH, Su LH, Chu C. Salmonella enterica serotype Choleraesuis: epidemiology, pathogenesis, clinical disease, and treatment. Clin Microbiol Rev. 2004;17:311–322. doi: 10.1128/cmr.17.2.311-322.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Soderlund R, Jernberg C, Tronnberg L, Paajarvi A, Agren E, Lahti E. Linked seasonal outbreaks of Salmonella Typhimurium among passerine birds, domestic cats and humans, Sweden, 2009 to 2016. Euro Surveill. 2019;24:1900074. doi: 10.2807/1560-7917.ES.2019.24.34.1900074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.European nucleotide archive. https://www.ebi.ac.uk/ena
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1. Deletions in Salmonella pathogenicity islands in S. enterica ST416/ST417. Presence (black) or absence (grey) of genes in Salmonella pathogenicity islands SPI-1,2,3,5 as determined by pangenome (Roary) analysis. The two Swedish isolates from the present study are labelled SE. Extensive deletions are evident in all ST416/ST417 isolates, while the islands are highly conserved among other related strains.
Additional file 2. Pan-genome analysis output, presence/absence and significance testing. Number of isolates of ST416/ST417 (referred to as “positive”) and other related Salmonella isolates (referred to as “negative”) that were found by pan-genome analysis to posess each identified sequence, i.e. gene or gene variant, and significance testing results for comparison of the frequence of occurrence in the two groups.
Additional file 3. Pan-genome analysis output, FASTA reference sequences. FASTA format sequences for the “groups” of sequence variants compared in Additional file 2.
Additional file 4. R script for processing SNP data. A short script for the R software environment used to process and filter SNP data.
Data Availability Statement
Sequence data from long and short read sequencing with metadata have been deposited in the European Nucleotide Archive [56] under project accession number PRJEB40444. Typing data from cgMLST analysis has been deposited in Enterobase under isolate IDs (uberstrains) SAL_EB4912AA and SAL_EB4913AA. Pan-genome analysis output is presented in Additional files 2 and 3.