

Abstract
Community-acquired Staphylococcus aureus infections often present as serious skin infections in otherwise healthy individuals, and have become a world-wide epidemic problem fueled by the emergence of strains with antibiotic resistance, such as methicillin-resistant S. aureus (MRSA). The cytokine, thymic stromal lymphopoeitin (TSLP), is highly expressed in the skin and other barrier surfaces and plays a deleterious role by promoting T helper type 2 (TH2) responses during allergic diseases; however, its role in host-defense against bacterial infections has not been well-elucidated. Here, we describe a previously unrecognized non-TH2 role for TSLP in enhancing neutrophil killing of MRSA during an in vivo skin infection. Specifically, we demonstrate that TSLP acts directly on both mouse and human neutrophils to augment control of MRSA. Additionally, we show that TSLP also enhances killing of Streptococcus pyogenes, another clinically important cause of human skin infections. Unexpectedly, mechanistically TSLP mediates its anti-bacterial effect by directly engaging the complement C5 system to modulate production of reactive oxygen species by neutrophils. Thus, TSLP increases MRSA killing in a neutrophil- and complement-dependent manner, revealing a key connection between TSLP and the innate complement system, with potentially important therapeutic implications for control of MRSA infection.
Introduction
The gram positive bacterium, Staphylococus aureus, is the most common cause of bacterial skin infections, causing millions of outpatient and emergency room visits per year (1). Whereas S. aureus infections are endemic in hospitals world-wide and were once mainly considered to be hospital-acquired, the emergence of new and more virulent antibiotic-resistant strains, such as methicillin-resistant S. aureus (MRSA), has resulted in an increase in community-acquired infections in otherwise healthy people, which are not limited to the hospital setting. USA300 is the most prevalent community-acquired MRSA strain in the United States and presents as a skin infection in approximately 90% of cases (2). Despite typically beginning as skin infections, MRSA infections can lead to life threatening and invasive infections, including pneumonia, sepsis, and meningitis. Overall, MRSA infections cause more deaths in the United States than HIV, viral hepatitis, and tuberculosis combined, with 94,360 severe invasive infections and 18,650 deaths in 2005 (2–4). To combat this increasing epidemic, particularly in an era of decreased antibiotic efficacy, an understanding of the factors governing the protective immune response is required to help develop immunotherapies to combat these infections.
Neutrophils are the first line of defense against bacterial infections and play a vital role in host-defense against S. aureus by employing multiple mechanisms to kill bacteria, including phagocytosis, production of reactive oxygen species (ROS), anti-microbial peptides, and proteinases and acid hydrolases that degrade bacterial components (4). The importance of neutrophils in combating S. aureus infection is underscored by the recurrent S. aureus infections in patients with chronic granulomatous disease, which is characterized by defects in neutrophil respiratory burst and NADPH oxidase (5). Additionally, neutropenic cancer patients have an increased incidence of S. aureus infections, resulting in increased mortality and morbidity (6). In the mouse, neutrophil abscess formation is required for bacterial clearance in the skin, and mice depleted of neutrophils have a non-healing skin infection with increased bacteremia (7). Thus, neutrophils play a critical role in host-defense against S. aureus infections.
TSLP was originally described as a stromal factor with actions on B and T cells (8) but subsequently was shown to be produced by a broad range of non-hematopoietic cells, with additional actions on dendritic cells, basophils, eosinophils, macrophages, smooth muscle cells, macrophages, and mast cells (9, 10). TSLP has been most extensively studied in the context of allergic diseases, including asthma and atopic dermatitis, where it promotes disease in atopic individuals in a TH2 dependent manner(10–14). A recent study reported that TSLP inhibits production of IL-22 by type 3 innate lymphoid cells (ILC3 cells) in the gut during Citrobacter rodentium infection, thereby reducing the host’s ability to control this bacterial infection (15); however, little is known of the role of TSLP in host defense to other bacterial infections, including those in the skin. Better understanding the role of TSLP in host defense is crucial, as responses and the role(s) of specific cytokines can differ greatly based on the site of disease/infection, with for example, contrasting clinical outcomes to IL-17 blockade in patients with psoriasis versus Crohn’s disease (16, 17). Since TSLP is highly expressed in the skin (18) and there is an increasing prevalence of MRSA skin infections (19), we investigated whether TSLP can contribute to host-defense against skin MRSA infection and now demonstrate that TSLP acts directly on both human and mouse neutrophils to enhance MRSA clearance in a complement- and ROS-dependent manner.
Results
TSLP enhances MRSA killing in a whole blood assay
We initially assessed whether TSLP promotes MRSA killing in an in vitro whole blood assay. Incubating TSLP together with MRSA in mouse blood significantly increased bacterial killing at both 2 and 3 hours, as compared to that observed with the addition of PBS and MRSA (assayed by colony forming units, CFU) (Fig. S1, A and B, and Fig. 1, A). We excluded the possibility that the increased killing of MRSA by TSLP resulted from a direct action of TSLP on the bacteria (Fig. S1C), and we thus sought to define the cell type that mediated TSLP-induced killing of the bacteria. Neutrophils are critical for host-defense against S. aureus (20), and we found that mouse bone marrow neutrophils not only express the TSLP binding protein (receptor), TSLPR, but that TSLPR expression was further increased upon in vitro stimulation with heat-killed S. aureus (HKSA) in these cells (Fig. 1B). These data suggested that mouse neutrophils might exhibit enhanced responsiveness to TSLP during MRSA infection. To determine if neutrophils were required for the action of TSLP, we depleted mice of neutrophils by using anti-Ly6G (Fig. 1C). When neutrophil-depleted blood was used in the whole blood killing assay, TSLP no longer augmented MRSA killing (Fig. 1D), demonstrating that the increased killing of MRSA induced by TSLP was neutrophil-dependent. Importantly, neutrophils are potent killers of bacteria and while depletion of neutrophils in the blood resulted in reduced control of bacteria in general, in line with the important role neutrophils play in bacterial clearance, it did not result in complete loss of bacterial control (Fig. S1D), consistent with the contributions of other cell types, such as macrophages, to MRSA clearance. Taken together, these data demonstrate that TSLP-enhanced killing of MRSA is neutrophil-dependent.
Fig. 1. TSLP increases MRSA killing in mouse blood in a neutrophil-dependent manner.
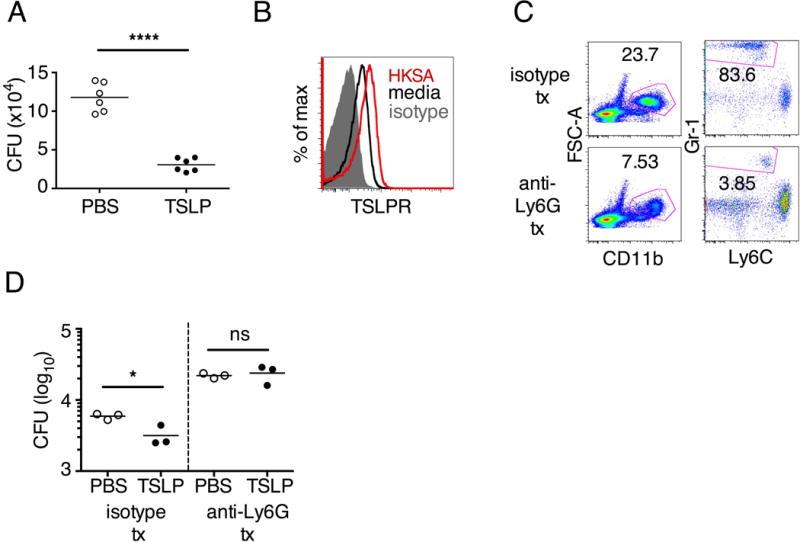
(A, C, D) Mouse blood was incubated with PBS or TSLP and MRSA for 3 h. (A) CFU analysis (shown is a representative experiment from blood from 2 mice performed in triplicate). (B) TSLPR expression on mouse neutrophils incubated with medium or HKSA. (C) Flow-cytometric staining of blood neutrophils in mice treated with a control antibody or depleted of neutrophils using anti-Ly6G antibody. (D) A representative experiment showing CFU of MRSA after an in vitro whole blood killing assay performed with blood from mice treated with an isotype control or anti-Ly6G antibodies (blood from 2–3 mice were combined for each treatment condition and assayed in triplicate). *, p < 0.05; ***, p < 0.001; ns = not significant, two-tailed Student’s t-test. Data are representative of 6 independent experiments for (A), 2 experiments for (B) and 3 experiments for (C) and (D).
TSLP acts directly on both mouse and human neutrophils to increase killing of MRSA
To determine whether TSLP could act directly on neutrophils, we next purified thioglycollate-elicited mouse peritoneal neutrophils, as less mature bone marrow neutrophils are incapable of killing MRSA in vitro (Fig. S2A), and first demonstrated that they expressed TSLPR (Fig. 2A). Moreover, when these neutrophils were incubated with MRSA and TSLP for 2 h, they exhibited increased killing as compared to cells incubated with MRSA and PBS (Fig. 2B), demonstrating that TSLP can act directly on mouse neutrophils in vitro to enhance MRSA killing. This direct effect of TSLP on neutrophils was TSLPR-dependent, as TSLP did not increase the killing of MRSA by Tslpr−/− neutrophils (Fig. S2B).
Fig. 2. TSLP acts directly on both mouse and human neutrophils to increase MRSA killing in vitro.
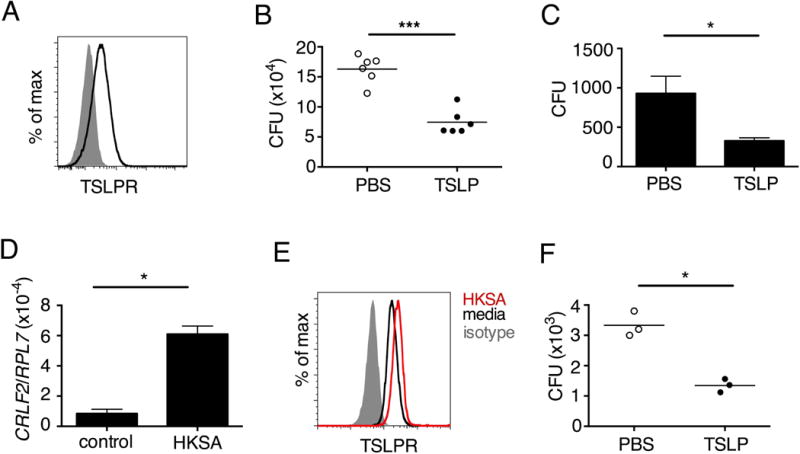
(A,B) Thioglycollate-elicited mouse neutrophils were used. (A) Representative flow-cytometric staining of TSLPR expression on neutrophils. (B) CFU after purified neutrophils were incubated with PBS or TSLP and MRSA for 2 h. (C) Whole human blood was incubated with MRSA and PBS or TSLP for 3 h and CFU determined (representative graph of 1 donor shown in triplicate, statistics shown are of two-tailed paired t-test from 6 donors). (D–F) Purified human blood neutrophils were used. (D) CRLF2 expression by human blood neutrophils determined by RT-PCR after 4 h treatment with medium (control) or HKSA (representative donor shown) and normalized to expression of RPL7. (E) Representative flow-cytometric staining of TSLPR on human blood neutrophils. (F) CFU after neutrophils were incubated with MRSA and PBS or TSLP for 3 h (shown is a representative graph of 1 donor done in triplicate; statistics shown are of two-tailed paired t-test from 7 donors). *, p < 0.05; ***, p < 0.001; ns = not significant, using two-tailed Student’s t-test for unless indicated. Data are representative of at least 3 independent experiments.
We next investigated whether TSLP exerts similar effects on human neutrophils. Indeed, TSLP treatment resulted in increased killing of MRSA in a whole blood killing assay (a representative donor is shown in Fig. 2C, with all donors shown in Fig. S2C). Although two previous studies reported that a synthetic short form of human TSLP could have direct antimicrobial activities on some pathogens, they observed little or no killing with S. aureus (21, 22). Consistent with this, we found that the increased killing of MRSA by TSLP did not result from a direct action of TSLP on the bacteria, as MRSA and TSLP incubated together with serum alone (i.e., in the absence of cells) resulted in a similar bacterial titer to that observed when control PBS was used in place of TSLP (Fig. S2D). To determine whether TSLP-induced killing of MRSA in human whole blood was mediated by neutrophils, analogous to what we found for the mouse, we purified neutrophils from whole blood from healthy donors. These human neutrophils expressed mRNA for CRLF2 (encoding TSLPR), and its expression was significantly enhanced by stimulation with heat killed S. aureus (HKSA), ranging from 5–76 fold enhancement depending on the donor (one donor shown in Fig. 2D). This increase in CRLF2 expression by HKSA was likely due to TLR2 activation, as we found that stimulation of neutrophils with peptidoglycan, a TLR2 agonist present on gram positive bacteria including S. aureus, also increased CRLF2 expression (Fig. S2E). Consistent with these mRNA expression data, the purified human neutrophils also expressed TSLPR protein, with higher expression upon HKSA stimulation (Fig. 2E), indicating that human neutrophils might also be able to respond to TSLP. Indeed, when freshly isolated human neutrophils were incubated with PBS or TSLP and MRSA for 3 h, TSLP markedly lowered the CFU (a representative donor is shown in Fig. 2F and all donors tested are depicted in Fig. S2F). Since priming of neutrophils can enhance their function (23, 24), we primed freshly isolated human neutrophils with HKSA and either PBS or TSLP and found that TSLP increased the ability of primed neutrophils to kill MRSA in vitro (Fig. S2G), analogous to unprimed neutrophils. Consistent with our experiments in mice, these data demonstrate that TSLP acts directly on both unprimed and primed human neutrophils to increase their killing of MRSA.
Tslpr-deficient mice have increased MRSA titers during an in vivo skin infection
We next investigated whether the TSLP-neutrophil axis also enhanced MRSA killing in vivo by using a skin infection model in which MRSA was injected intradermally (i.d.) into the mouse ear. Interestingly, TSLP protein was potently increased in the ears at days 1 and 2 post-infection (p.i.) with MRSA, as compared to naïve PBS-injected controls (Fig. 3A). Additionally, TSLPR was expressed by ear neutrophils (Fig. 3B). To elucidate the role of TSLP in skin MRSA infection, we infected mice with MRSA i.d. and found that Tslpr-deficient (Tslpr−/−) mice had a significantly higher bacterial burden than did wild-type (WT) mice (Fig. 3C), indicating that TSLP helps to control MRSA in vivo. The increased bacterial burden in Tslpr−/− mice was not due to reduced recruitment of neutrophils to the ear, as Tslpr−/− and WT mice had similar percentages (Fig. 3, D and E) and numbers (Fig. 3F) of neutrophils in their infected ears. To eliminate the possibility that the in vivo results we had observed resulted from compensatory mechanisms in Tslpr−/− mice, we treated WT mice with either a human IgG1 Fc isotype control or TSLPR-Fc fusion protein i.d. at the time of MRSA infection and found that the mice with in vivo TSLP blockade (TSLPR-Fc treated) had significantly increased MRSA titers in the ear compared to isotype control treated mice, confirming that TSLP enhances bacterial control during in vivo MRSA skin infection (Fig 3G).
Fig. 3. Tslpr-deficient mice have increased MRSA burden during in vivo skin infection.
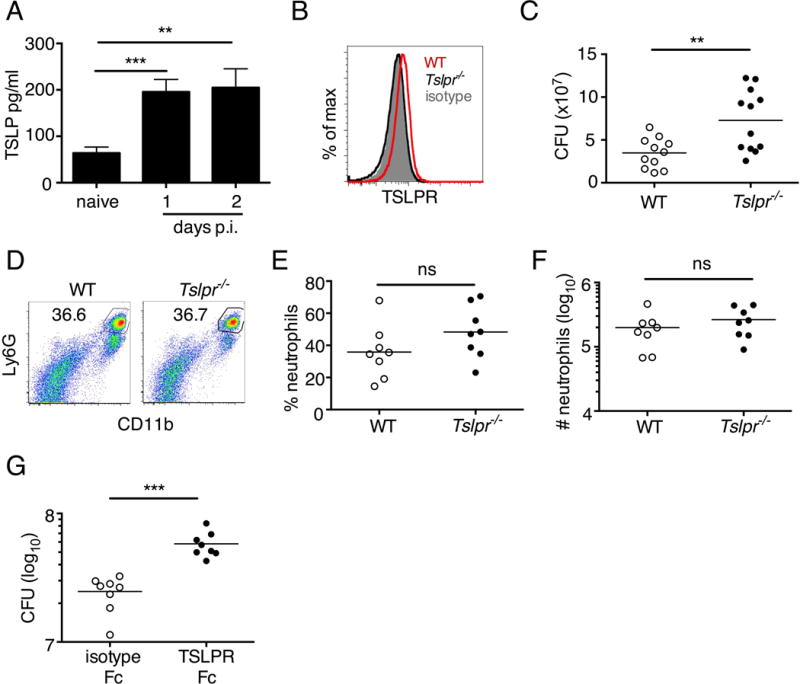
(A–G) Mice were infected with MRSA i.d. in the ear. (A) TSLP protein expression in the ear after i.d. MRSA infection (n=4), naïve controls were mock-infected with PBS only. (B–G) Ears were analyzed on day 1 p.i. (B) Representative TSLPR expression on neutrophils. (C) Analysis of CFU in the ear of WT (n=11) and Tslpr−/− mice (n=12 ears). (D–F) Analysis of neutrophils in the ear. Shown are representative FACS plots (D) and percent (E) and total number (F) of neutrophils from WT and Tslpr−/− mice (n=8 ears). (G) CFU of MRSA in the ear on day 2 post-i.d. ear infection of WT mice treated with human IgG1 Fc isotype control or TSLPR Fc (two-tailed Mann-Whitney test) (n=8 ears). *, p < .05; **, p < .01; ***, p < .001; ns = not significant. a–f, two-tailed Student’s t-test. Data are representative of 3 (or 2 for (G)) independent experiments.
TSLP treatment enhances MRSA killing in vivo in normal WT hosts
We next sought to determine whether increased TSLP signaling could augment MRSA killing in the skin of normal hosts and therefore injected PBS or TSLP plus MRSA i.d. into the ears of WT mice. TSLP treatment significantly reduced the bacterial burden in the ears at day 2 p.i. (Fig. 4A), and this effect was sustained as one injection of TSLP at the time of infection resulted in significantly reduced titers even at days 3 and 6 p.i.(Fig. S3A). Because bacterial titers can only be assessed at one time-point per mouse we also evaluated if TSLP has a more cumulative effect by assessing pathology in these mice and found that TSLP also decreased pathological changes, with significantly decreased inflammation in the skin after MRSA infection compared to that observed in PBS-treated animals (Fig. 4, B and C). Moreover, the effect of TSLP was mediated by its functional receptor rather than an off-target effect, as Tslpr−/− mice treated with TSLP had similar MRSA titers to those treated with PBS (Fig. S3B). To determine whether TSLP’s ability to increase in vivo killing of bacteria was limited to MRSA, we tested whether TSLP could also enhance the killing of both a non-MRSA strain of S. aureus (MW2) and Streptococcus pyogenes, another bacterial strain that causes clinically significant human skin infections (25). Indeed, WT mice treated with TSLP had significantly lower S. aureus MW2 and S. pyogenes titers compared to PBS-treated control mice (Figs. S3C and 4D). Thus, treatment with TSLP not only can decrease MRSA burden in vivo but also can kill a non-MRSA strain of S. aureus and another pathogenic bacterial strain (S. pyogenes) in the skin as well.
Fig. 4. TSLP treatment enhances in vivo MRSA and S. pyogenes killing during a skin infection.

Mice were infected with MRSA i.d. in the ear. (A) CFU of MRSA at day 2 p.i. in the ears of WT mice treated with PBS or TSLP (two-tailed Mann-Whitney test) (n=10 ears). (B) Representative images of hematoxylin and eosin (H&E)-stained ear sections on day 2 p.i. (5X magnification: bar indicates 200 microns). (C) Inflammation score according to blinded histological analysis (n=9 ears). (D) CFU of S. pyogenes in the ears of WT mice treated with PBS or TSLP on day 1 post-i.d infection (two-tailed Mann-Whitney test) (n=17 ears, shown are results of two combined independent experiments). (E) CFU in the ear of WT, neutrophil-depleted WT, and Tslpr−/− mice on day 1 p.i. (two-tailed Mann-Whitney test) (n=8 ears for WT and Tslpr−/−, n=6 for neutrophil depleted WT) (F) CFU of MRSA in the ears of WT mice or neutrophil-depleted WT mice treated with PBS or TSLP on day 2 p.i. (two-tailed Mann-Whitney test) (n=10 ears). *, p < .05; **, p < .01; ***, p < .001; ns = not significant. Data are representative of 2 (B, C, E, F) or 4 (A) independent experiments.
Additionally, Tslpr−/− infected mice had a similar bacterial burden to that observed in neutrophil-depleted WT mice (Fig. 4E), suggesting that TSLP-enhanced MRSA killing in vivo might be dependent upon neutrophils. Importantly, in contrast to its ability to enhance MRSA control in mice treated with an isotype control antibody, TSLP treatment did not increase MRSA control in neutrophil-depleted (anti-Ly6G treated) WT mice, thus demonstrating that TSLP-enhanced MRSA killing in vivo was dependent on neutrophils (Fig. 4F).
TSLP acts directly on neutrophils in vivo to decrease MRSA burden
Having shown above that TSLP acts directly on both mouse and human neutrophils to enhance MRSA killing in vitro and that TSLP effects in vivo were neutrophil-dependent, we next investigated whether TSLP acts directly on neutrophils in vivo. Unfortunately, a neutrophil-specific Cre is not available, and LysM-Cre affects monocytes/macrophages as well as neutrophils (26). We thus used a cell transfer approach in which we co-transferred equal numbers of purified WT and Tslpr−/− bone marrow neutrophils into naïve mice, which could be distinguished by their expression of different isoforms of the congenic marker CD45. After infection i.d. with MRSA in the ear, transferred Tslpr−/− neutrophils were recruited to the infection site and accumulated there equally well as WT neutrophils (Fig. 5A). We next adoptively transferred an equal number of CMDFA-labeled WT or Tslpr−/−purified bone marrow neutrophils into Tslpr−/− mice and then injected these mice with MRSA and TSLP i.d. in the ear, as outlined in Fig. 5B. In these experiments, only the transferred WT neutrophils can respond to TSLP. On day 1 p.i., the Tslpr−/− mice that received WT neutrophils exhibited significantly greater MRSA killing (i.e., lower CFU) than mice receiving Tslpr−/− neutrophils (Fig. 5C). Importantly, this difference in MRSA titer was not due to less efficient recruitment of Tslpr−/− neutrophils than of WT neutrophils, as the percent of transferred Tslpr−/− neutrophils was even slightly higher than for WT neutrophils (Fig. 5, D and E), and the overall numbers of Tslpr−/− and WT transferred neutrophils in the ear were similar (Fig. 5F). Given that TSLP does not directly act on MRSA (Figs. S1C and S2C) and requires TSLPR signals to act both in vitro (Fig. S2B) and in vivo (Fig. S3B), these data together demonstrate that TLSP acts directly on neutrophils in vivo to enhance MRSA clearance.
Fig. 5. TSLP acts directly on neutrophils in vivo to enhance killing of MRSA during a skin infection.
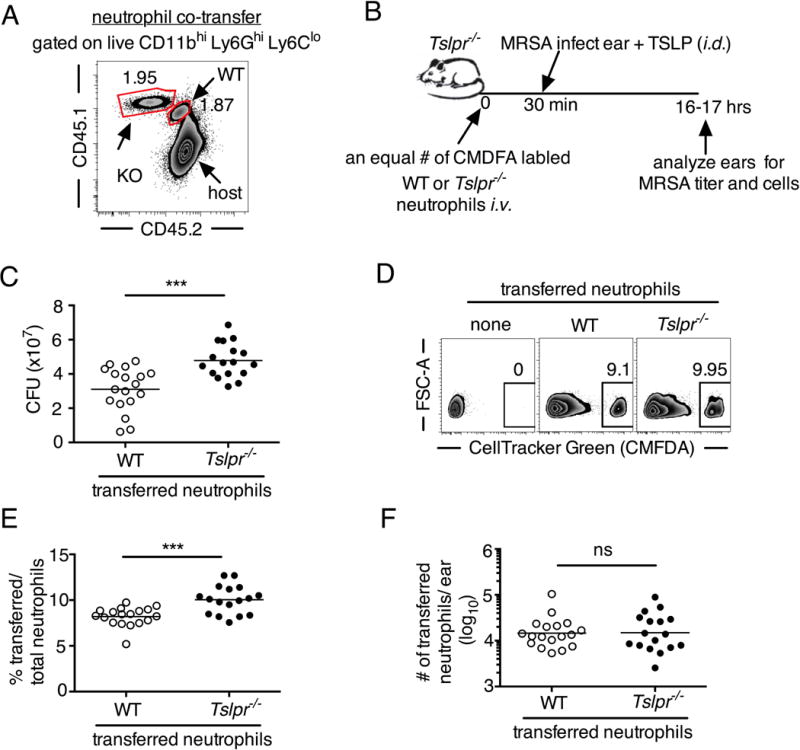
(A) Equal numbers of purified bone marrow neutrophils from WT (CD45.1+/2+) and Tslpr−/− (KO; CD45.1+/1+) mice were co-transferred i.v. into WT C57BL/6 host mice (CD45.2+/2+) and were then infected with MRSA i.d. in the ear. Shown is a representative flow cytometric plot of the neutrophil populations in the ear on day 1 p.i. (n=10, gated on neutrophils = live CD11bhiLy6GhiLy6Clo cells). (B) Experimental design for (C–F), where an equal number of purified CellTracker Green (CMDFA) labeled WT or Tslpr−/− bone marrow neutrophils were transferred i.v. into Tslpr−/− host mice, which were subsequently injected with MRSA + TSLP i.d. in the ear. (C) CFU of MRSA in the ears 16–18 h p.i.(n=17–18). (D) Representative flow-cytometric plot showing the percent of transferred neutrophils (CMDFA+) out of total neutrophils in the ears of Tslpr−/− mice receiving no cells, WT neutrophils, or Tslpr−/− neutrophils (n=17–18 from two combined individual experiments, gated on total neutrophils). (E) Percent and (F) number of transferred neutrophils per ear (n=17–18). *, p < .05; **, p < .01; ***, p < .001; ns = not significant, using a two-tailed Mann-Whitney test.
A non-transcriptional mechanism for TSLP-mediated MRSA killing by neutrophils
We next sought to elucidate the mechanism underlying TSLP-mediated killing of MRSA. We initially performed RNA sequencing (RNA-Seq) on purified human neutrophils treated with PBS or TSLP with or without HKSA for 4 and 24 h. We found that TSLP did not significantly alter the transcriptional profile of human neutrophils at either 4 or 24 h, whereas HKSA greatly increased the number of differential expressed genes (1394 genes common to both donors at 4 h and 1252 at 24 h). As compared to HKSA alone, the addition of TSLP plus HKSA resulted in the common induction in both donors of only a single gene (CCL22) at 24 h (Table S1). These data suggest that TSLP-mediated neutrophil killing of MRSA is not due to transcriptional activation of new gene expression during the time of the killing assays and that proximal signaling events instead might be involved. Indeed, studies using inhibitors of mitogen-activated protein kinase/extracellular signal regulated kinase (MAPK/ERK) kinase or phosphatidyl inositol 3-kinase (PI3K) showed that both of these pathways are necessary for TSLP-mediated killing of MRSA by human neutrophils, as pre-treatment with these inhibitors blocked TSLP-increased MRSA killing (Fig. S4, A and B for MAP/ERK inhibition and Fig. S4, A and C for PI3K inhibition) but did not eliminate the basal ability of human neutrophils to kill MRSA (Fig. S4D). Given the rapid TSLP-induced neutrophil-mediated killing of MRSA (2–3 h for the in vitro assay) and the fact that the MAPK/ERK and PI3K pathways can mediate non-transcriptional effects in neutrophils (27–29), our results indicate that TSLP-mediated MRSA killing by neutrophils is a rapid response that does not require de novo gene induction.
TSLP-enhanced killing of MRSA in both mouse and human is ROS-dependent
As phagocytosis of microbes is an important rapid response of neutrophils, we hypothesized that TSLP might increase neutrophil phagocytosis. Pathogen uptake is likely necessary for TSLP-enhanced killing of MRSA as treatment of neutrophils with cytochalasin D, an inhibitor of phagocytosis, eliminated TSLP-enhanced killing of MRSA in vitro (Fig S5A). To our surprise, however, TSLP treatment did not affect expression of CD11b (a component of the phagocytic CR3 receptor) on human neutrophils in vitro (Fig. S5B) or on mouse neutrophils in vivo (Fig. S5C). Moreover, TSLP did not augment the phagocytic uptake of S. aureus by either human (Fig. S5D) or mouse (Fig. S5, E and F) neutrophils.
A major mechanism used by human and mouse neutrophils to eliminate bacteria is the production of reactive oxygen species (ROS) (20), and we therefore evaluated the role of ROS in TSLP-driven MRSA killing in vivo utilizing the mouse skin infection model. Strikingly, neutrophils from infected Tslpr−/− mice had lower ROS levels (Fig. 6, A and B) compared to neutrophils from infected WT mice, indicating that ROS might contribute to TSLP-enhanced neutrophil killing of MRSA. Consistent with this notion, TSLP treatment did not enhance MRSA killing when a ROS scavenger, N-acetyl-L-cysteine, (NAC) (30) was administered i.d. (Fig. 6C), demonstrating that ROS is essential for TSLP-induced neutrophil-mediated killing of MRSA in vivo. To eliminate the possibility that these data resulted from non-specific actions of NAC, we used Gp91phox−/− (Nos2−/−) mice, which are deficient in an integral component of the NADPH oxidase complex that generates ROS. TSLP treatment did not increase the killing of MRSA in Gp91phox−/− mice infected i.d. in vivo with MRSA, unlike its effect in WT controls (Fig. 6D), demonstrating that ROS is essential for TSLP-induced neutrophil-mediated killing of MRSA in vivo. Consistent with an essential role for ROS in TSLP-enhanced MRSA killing in the mouse skin infection model, pre-treatment of purified human neutrophils with Diphenyliodonium (DPI), an NADPH-oxidase inhibitor (36), eliminated the ability of TSLP to enhance their killing of MRSA (Fig. 6E), demonstrating that ROS is also essential for TSLP-augmented control of MRSA by human neutrophils.
Fig. 6. TSLP induced killing of MRSA is mediated by reactive oxygen species.
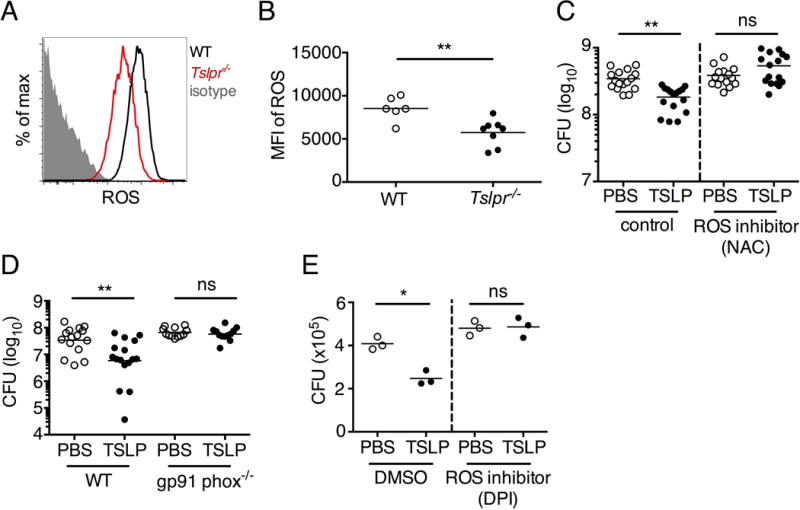
(A–C) Day 1 p.i. of mice infected with MRSA i.d. in the ear. (A–B) ROS production of mouse neutrophils after staining with CellROX deep red. Shown are a representative FACS plot (A) and mean fluorescence intensity (MFI) (B) of WT (n=6) and Tslpr−/− mice (n=8 ears). (C) Mice were injected i.d. in the ear with MRSA and either PBS or TSLP along with either control (PBS) or a ROS inhibitor (NAC). CFU on day 1 p.i. (two-tailed Mann-Whitney test, n=16 ears). (D) CFU in the ear on day 2 p.i. of WT and Gp91 phox−/− mice infected with MRSA and PBS or TSLP (two-tailed Mann-Whitney test, n=12–16 ears). (E) Purified human neutrophils were pretreated with DMSO or DPI, treated with PBS or TSLP, and incubated for 2 h with MRSA. CFU was then determined (representative donor shown in triplicate, statistics shown are using a two-tailed paired-t test of 3 donors, 3 independent experiments). *, p < .05; **, p < .01; ***, p < .001; ns = not significant. (b) Two-tailed Student’s t-test. Data are representative of 3 independent experiments (A, B) or are combined data from two independent experiments (C&D).
TSLP-enhanced killing of MRSA is complement-dependent
The complement system is a highly conserved innate defense system poised to rapidly respond to invading pathogens (31, 32), and binding of the complement activation fragment C5a to the C5a receptor 1 (CD88, C5aR1) expressed on neutrophils drives ROS production in these cells (33, 34). In our whole blood assays above where TSLP promotes the killing of MRSA, we had collected blood with sodium citrate; however, we observed that treatment of mouse blood with EDTA, which prevents complement activation and C5a generation (35), eliminated TSLP-mediated MRSA killing in neutrophils (Fig. S6A). We therefore investigated whether a complement-dependent mechanism was involved in this process. Importantly, local injection of WT mice with a C5-blocking antibody (anti-C5) during i.d. MRSA ear infection decreased ROS production by neutrophils as compared to ROS production by neutrophils from isotype control treated animals (Fig. 7A), showing that C5 can drive neutrophil ROS production in this model. To elucidate the potential role of C5 in TSLP-mediated MRSA killing in vivo, we treated WT mice with an isotype control or anti-C5 antibody along with TSLP or PBS during i.d. MRSA infection. Strikingly, whereas TSLP enhanced MRSA killing in isotype control antibody-treated animals, it had no effect in animals treated with anti-C5 (Fig. 7B), demonstrating that C5 is necessary for TSLP-induced neutrophil killing of MRSA in vivo. Although the C5a fragment of C5 is an anaphylotoxin that can act as a chemotactic factor for neutrophils (23, 33, 34), local blockade of C5 did not affect neutrophil recruitment to the site of infection, as animals treated i.d. with either anti-C5 or control antibodies had similar numbers of neutrophils in the ear after MRSA infection (Fig. S6, B and C). Additionally, TSLP treatment of WT mice increased C5aR1 expression on neutrophils during MRSA skin infection (Fig. 7, C and D). Expression of C5aR1 indeed appeared critical, as TSLP treatment did not increase MRSA killing in C5ar1−/− mice (Fig. 7E). Thus, TSLP induces in vivo killing of MRSA by neutrophils in a C5- and C5aR1-dependent fashion, via induction of anti-bacterial ROS generation in neutrophils.
Fig. 7. TSLP induced killing of MRSA is mediated by reactive oxygen species and complement.
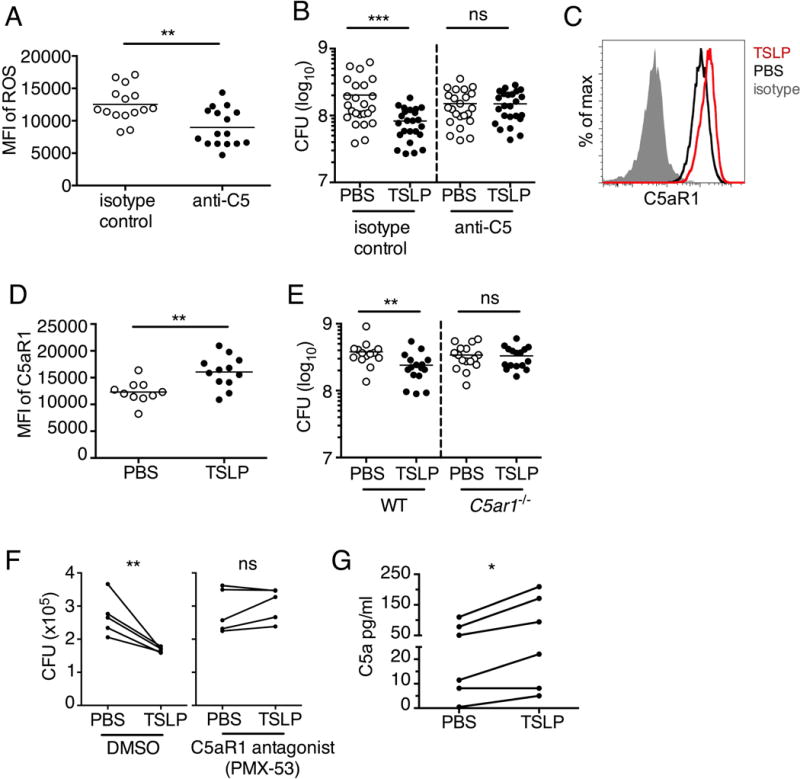
(A–G) Day 1 p.i. of mice infected with MRSA i.d. in the ear. (A) MFI of ROS production of mouse neutrophils after day 1 p.i. with MRSA plus isotype control (n=15) or anti-C5 antibodies injected i.d. (n=16 ears). (B) Mice were infected with MRSA and PBS or TSLP with isotype control or anti-C5 antibodies given i.d. in the ear. CFU on day 1 p.i. (two-tailed Mann-Whitney test, n=15 (PBS isotype) or n=16 ears). (C–D) C5aR1 expression on mouse neutrophils as assessed by flow cytometry. Shown are a representative FACS plot (C) and MFI for multiple animals (D) (n=10 (WT) or 12 (TSLP) ears. (E) CFU at day 1 p.i. of WT or C5ar1−/− mice infected with MRSA and PBS or TSLP (n=10 (PBS WT) or 16 (all other groups) ears. (F) Purified human neutrophils were treated with DMSO or PMX-53 and PBS or TSLP and incubated for 2 h with MRSA. CFU was then determined (n=5 donors, two-tailed paired-t test of 4 independent experiments). (G) Purified human neutrophils were incubated with PBS or TSLP for 30 min and supernatants were assessed for C5a protein (n=6 donors, ratio-paired two-tailed student’s t-test, 3 independent experiments). *, p < .05; **, p < .01; ***, p < .001; ns = not significant. (A) Two-tailed Student’s t-test. Data are representative of 3 independent experiments (A, C–D) or 3 combined independent experiments shown in (B) and 2 in (E).
We next investigated whether complement C5 was also necessary for the TSLP-enhanced neutrophil killing of MRSA by human neutrophils. Importantly, the C5-axis was also required for TSLP-enhanced killing by human neutrophils, as incubating purified human neutrophils with PMX-53, a peptide antagonist of C5aR1 that has been used in clinical trials (37, 38), prevented the TSLP-induced MRSA killing (Fig. 7F). Thus, TSLP enhanced MRSA killing by human neutrophils is both ROS- and complement C5-dependent, in agreement with our in vivo mouse data. Of note, neither the ROS inhibitor (DPI) nor C5aR1 antagonist (PMX-53) abolished the overall killing of MRSA by human neutrophils (Fig. S6, D and E) but only eliminated the ability of TSLP to enhance neutrophil-killing of MRSA (Figs. 6E and 7F), indicating the critical engagement of this “C5-ROS axis” by TSLP. Moreover, incubation of purified human neutrophils with TSLP increased their secretion of C5a (Fig. 7G), indicating that TSLP may increase conversion of C5 to C5a or cycling of C5a, thereby creating more ligand for C5aR1. These data demonstrate that TSLP engages the C5 system for MRSA killing in both mouse neutrophils in an in vivo skin infection and in human neutrophils in vitro.
Discussion
TSLP has been well-characterized as a cytokine that promotes TH2-type responses, with detrimental effects in asthma, atopic dermatitis, and other allergic diseases (10–14). Analogously, TSLP modulates intestinal inflammation by enhancing TH2-type responses and limiting proinflammatory TH1-type responses (39). In the context of infection, however, the role of TSLP is less well known. Respiratory syncytial virus can stimulate TSLP production by lung epithelial cells, promoting an immunopathogenic TH2-type response that may be further enhanced in asthmatics, leading to disease exacerbation (40). Consistent with this, TSLP is important for immunity to the nematode Trichuris muris due to its promoting a TH2-type response and worm expulsion (39), and a recent study demonstrated that TSLP lowers the ability to control Citrobacter rodentium infection in the gut (15). Thus, previous studies have generally focused on TSLP’s enhancement of TH2-type responses. In contrast, our experiments reveal a key unexpected non-TH2 function for TSLP that can be protective in vivo, with TSLP acting directly on neutrophils to enhance control of both methicillin resistant and sensitive strains of S. aureus and S. pyogenes in the skin. Conceivably, these results may extend to the control of other bacterial species in the skin, an area for future investigation.
We hypothesize that multiple factors together likely determine whether TSLP plays a beneficial or detrimental role in infection, potentially including the specific infectious agent, the context (e.g., if there is a chronic ongoing TH2 response, such as in allergy/asthma), and the site of infection. This notion aligns with the growing understanding that immune responses and their regulation can be drastically distinct in different organs. For example, IL-17 inhibitors, anti-IL-17 and anti-IL17R, are therapeutic for psoriatic skin lesions (16), whereas a neutralizing antibody to IL-17 did not have therapeutic benefit for Crohn’s disease and even exacerbated disease in some patients(17). Thus, our identification of a host protective role for TSLP by its enhancing killing of MRSA, S. aureus MW2 and S. pyogenes in the skin, in contrast to the detrimental effect of TSLP observed in the gut observed with Citrobacter rodentium (15), underscores the importance of carefully assessing the impact of TSLP in different organs and in the context of different infections. This idea is further supported by two recent studies that highlight the controversial role of TSLP in sepsis (41, 42). While one study demonstrated that TSLP may play a detrimental role by increasing morbidity and mortality during cecal ligation and puncture (CLP)-induced sepsis in mice (42), a recent study showed the opposite, indicating that TSLP reduces mortality and morbidity by decreasing inflammation (41). However, both studies show that blockade of TSLP (or use of Tslpr−/− mice) decreases bacterial titers during CLP-sepsis, implying that TSLP is detrimental for septic bacterial clearance and again demonstrating that the role of TSLP in bacterial infections may depend greatly on either, or both, the location of infection and the infectious agent.
Finally, we identify an unanticipated critical crosstalk between TSLP and the complement system, with therapeutic implications for MRSA skin infections. Given that anti-TSLP (43) and anti-C5 (eculizumab or Soliris) (44) are currently being used therapeutically, our study indicates that one should be cognizant of possible diminished host defense in these settings.
Material and Methods
Study Design
The aim of this study was to elucidate and characterize the role of TSLP in neutrophil function, including for MRSA skin infections. The experimental design involved in vivo (mouse) and in vitro (mouse and human) experiments, including flow cytometric, histological, RNA-seq, and RT-PCR analysis along with bacterial colony enumeration. The animal experiments were not randomized. The investigators were not blinded to allocation during experiments and analyses unless otherwise indicated. Experimental replication is indicated in the figure legends.
Mice
In experiments where only WT mice were used, 6–9 week old WT BALB/c mice or C57/BL6 mice were obtained from The Jackson Laboratory. Tslpr−/− (14) and C5aR1−/− (Jackson Laboratory) mice were bred in our facility. Gp91phox−/− mice were purchased from the Jackson Laboratory. For experiments, using both knockout mice and WT mice, littermate control WT mice were used. 6–9 week old strain-, age- and sex- matched mice were used for experiments. All experiments were performed under protocols approved by the National Heart, Lung, and Blood Institute Animal Care and Use Committee and followed National Institutes of Health guidelines for the use of animals in intramural research.
Bacteria
The USA 300 clinical isolate (FPR3757) of MRSA was used in these studies, except where indicated. For whole blood killing assays, MRSA was plated overnight on a blood agar plate, 1 colony was picked and grown overnight at 37° C shaking in 2 ml of Tryptic Soy Broth (TSB) (Fisher Scientific) and then washed 2 times with PBS. The non-MRSA S. aureus strain, MW2, and S. pyogenes strain, NZ131, (both from ATCC) were used in the same way as MRSA, except NZ131 was grown in Todd Hewitt Broth under static culture conditions. For intradermal (i.d.) ear infections, bacteria in logarithmic growth were used.
Whole blood killing assays
Whole blood killing assays were adapted from Kaplan et al. (45) In brief, whole mouse blood was collected into 4% sodium citrate, and whole human blood from healthy donors was collected into sodium citrate tubes. 75 μl of whole blood, 5 μl of 4% sodium citrate, 10 μl of PBS or mouse or human TSLP (100 ng/ml final concentration; both from R&D Systems) and 25 μl of MRSA (at a 1:1800 dilution of OD600= 0.25) were sequentially added to capped 2 ml skirted tubes and the tubes were slowly rotated in a 37° C incubator for 3 h. Serial 10-fold dilutions were then made, and the blood was spread on blood agar plates and incubated overnight. Colonies were counted the following day to determine the colony forming units (CFU)/tube. Experiments were performed with triplicate samples.
Mouse neutrophil isolation
For elicitating peritoneal neutrophils, mice were injected i.p. with 1 ml of 3% thioglycollate and 4 h later their peritoneums were lavaged with 10 ml cold PBS and cells were collected. For bone marrow neutrophils, femurs from mice were excised under sterile conditions and the cells were flushed out using 2% FBS in PBS + 1 mM EDTA. Both peritoneal and bone marrow neutrophils were purified using either a Miltenyi Biotech negative selection Neutrophil Isolation Kit, or by a 55%/65%/ 75% percoll gradient.
Human neutrophil isolation and in vitro MRSA killing assays
Whole blood from healthy donors was collected in EDTA tubes, and neutrophils were isolated directly from the blood by negative selection using a kit (Stem Cell). For neutrophil killing assays, 3–4 × 105 neutrophils (either purified human blood neutrophils or thioglycollate-elicited mouse peritoneal neutrophils) were added to a capped 2 ml skirted tube in RPMI medium. PBS or TSLP (100 ng/ml final), and/or PMX-53 (5 pM; Tocris Bioscience) were added and incubated for 5 min. 50 μl of coated MRSA or S. pyogenes (bacteria at a 1:50 dilution of OD600= 0.25) pre-incubated in 10% autologous human or mouse serum) was added/tube, for a final total volume of 200 μl. In some experiments, neutrophils were primed with HKSA (Invivogen) plus either PBS or TSLP for 2 h before addition of MRSA. The tubes were slowly rotated in a 37° C incubator for 2–3 h as indicated in the figure legends. For DPI treatment, neutrophils were incubated with 2 μM DPI for 30 min, washed, counted, and then used in the killing assay as described above. Each treatment was done in triplicate. Whole blood from healthy human NIH blood bank volunteer donors was obtained without donor identification and met the criteria for exemption from informed consent and institutional review board review as defined in The Code of Federal Regulations Title 45 (Public Welfare), Department of Health and Human Services, Part 46 (Protection of Human Subjects), and their distribution was in accord with National Institutes of Health guidelines for the research of human subjects.
Neutrophil depletion
Neutrophil-depleted blood was obtained by injecting mice i.p. with 0.5 mg of anti-Ly6G antibody (1A8, Bioxcel) two days before blood was collected. For infection studies of neutrophil-depleted mice, mice were injected i.p. with 0.5 mg of anti-Ly6G antibody two days before and again on the day of infection. Neutrophil depletion was ~93–98% efficient as assessed by flow cytometric staining with Gr-1 and Ly6C antibodies (Biolegend).
Intradermal ear infection
6–9 week old WT, Tslpr−/−, or C5ar1−/− BALB/c mice or neutrophil-depleted WT BALB/c mice were injected intradermally (i.d.) using a 29 ½ -gauge 3/10 ml insulin syringe (BD Biosciences) with MRSA or S. pyogenes mixed with either TSLP (2 μg) or PBS (final OD600=0.125 in a total volume of 10 μl). In some experiments, 10 μg of anti-mouse C5 blocking antibody (BB5.1, Hycult Biotech) or mouse IgG1 isotype control (MOPC-21, Bioxcel) was additionally added, but the total volume injected was still 10 μl. For in vivo ROS inhibition, 1.3 μg of NAC (N-acetyl-L-cysteine, Sigma-Aldrich) was co-injected with PBS or TSLP and MRSA i.d. into the ears. Each experiment included 6–12 ears per group. Some samples were excluded at the time of infection due to a poor injection.
Neutrophil in vivo transfer experiments
Equal numbers of purified WT and Tslpr−/− bone marrow neutrophils were either co-transferred (~3 ×106 of each) into WT mice or labeled with 5 μM CMDFA, as previously described (46), and transferred separately (~15 ×106) into Tslpr−/− mice i.v. 30 min prior to infection with MRSA i.d. in the ear.
Ex-vivo detection of ROS
Mouse ear samples were processed as described above, and cells were incubated in medium with 5 μM of Cell Rox® Deep Red reagent (Life Technologies) for 30 min at 37° C, washed 3 times with PBS, and fixed with 4% paraformaldehyde before staining for CD11b+ Ly6G+ (Ly6Clow).
CRLF2 RT-PCR
Human neutrophils were isolated and stimulated with medium or 109 HKSA/ml (Heat killed S. aureus or 10 μg/ml peptidoglycan (InvivoGen) for 4 h. Probes for CRLF2 (Hs00845692_m1) and RPL7 (Hs02596927_g1) were from Life technologies.
Statistics
Statistical significance was calculated as indicated in the figure legends, using GraphPad Prism 6 software. For all statistical analyses, data were considered significant when P ≤ 0.05 (*), P ≤ 0.01 (**), P ≤ 0.001 (***) or P ≤ 0.0001 (****). Variances were similar between groups in all experiments, as determined by the F test using GraphPad Prism 6 software.
Supplementary Material
Fig. S1. TSLP does not directly kill MRSA, and normal neutrophil-depleted blood can still reduce MRSA burden.
Fig. S2. TSLP requires TSLPR and acts on human neutrophils to increase control of MRSA.
Fig. S3. TSLP is TSLPR-dependent and enhances the killing of both MRSA and S. aureus in vivo.
Fig. S4. TSLP treatment increases killing of MRSA by human neutrophils in a PI3K- and MAPK/ERK-dependent manner.
Fig. S5. TSLP treatment of mouse or human neutrophils does not affect phagocytosis.
Fig. S6. ROS and complement-dependent TSLP-enhanced neutrophil killing.
Table S1. TSLP does not alter the transcriptional profile of human neutrophils.
Editor’s summary.
‘Complement’ary MRSA Fight
Thymic stromal lymphopoeitin (TSLP) is a cytokine thought to promote allergic responses; however, its role in fighting infectious diseases is less clear. Now West et al. report that TSLP in the skin can enhance killing of methicillin-resistant S. aureus (MRSA). TSLP enhances killing in both mouse and human neutrophils, in part through interactions with the complement system that induce reactive oxygen species in neutrophils. This enhanced killing is not limited to MRSA, as TSLP also boosts killing of Streptococcus pyogenes. These data suggest that TSLP may augment innate immune cells and complement to fight bacterial infection.
Acknowledgments
We thank Melanie Faivre-Charmoy for instruction on how to perform intradermal ear injections and ear cell processing and Phillip A. Swanson II for critical comments.
Funding: This work was supported by the Division of Intramural Research, National Heart, Lung, and Blood Institute, NIH, the Wellcome Trust (CK), an MRC Centre grant (MR/J006742/1), and the King’s Bioscience Institute at King’s College London, the National Institute for Health Research (NIHR) Biomedical Research Centre based at Guy’s and St Thomas’ NHS Foundation Trust and King’s College London.
Footnotes
Author contributions: E.E.W. designed and performed experiments, analysed and interpreted data, and wrote the paper; R.S. designed and performed experiments, interpreted data, and wrote the paper; M.K. analysed and interpreted data; Z.Y. prepared and analyzed histology and wrote the paper; C.K. designed experiments, analysed and interpreted data, and wrote the paper; W.J.L. designed, interpreted data, and wrote the paper.
Competing interests: E.E.W., R.S., and W.J.L. are inventors on an NIH patent application related to this study. The other authors declare no competing interests.
Data and materials availability: primary accessions deposited in Gene Expression Omnibus: GSE73313.
References and Notes
- 1.McCaig LF, McDonald LC, Mandai S, Jernigan DB. Staphylococcus aureus-associated skin and soft tissue infections in ambulatory care. Emerging infectious diseases. 2006 Nov;12:1715. doi: 10.3201/eid1211.060190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.DeLeo FR, Chambers HF. Reemergence of antibiotic-resistant Staphylococcus aureus in the genomics era. The Journal of clinical investigation. 2009 Sep;119:2464. doi: 10.1172/JCI38226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Klevens RM, et al. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. Jama. 2007 Oct 17;298:1763. doi: 10.1001/jama.298.15.1763. [DOI] [PubMed] [Google Scholar]
- 4.Miller LS, Cho JS. Immunity against Staphylococcus aureus cutaneous infections. Nature reviews Immunology. 2011 Aug;11:505. doi: 10.1038/nri3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Heyworth PG, Cross AR, Curnutte JT. Chronic granulomatous disease. Current opinion in immunology. 2003 Oct;15:578. doi: 10.1016/s0952-7915(03)00109-2. [DOI] [PubMed] [Google Scholar]
- 6.Gonzalez-Barca E, Carratala J, Mykietiuk A, Fernandez-Sevilla A, Gudiol F. Predisposing factors and outcome of Staphylococcus aureus bacteremia in neutropenic patients with cancer. European journal of clinical microbiology & infectious diseases: official publication of the European Society of Clinical Microbiology. 2001 Feb;20:117. doi: 10.1007/pl00011241. [DOI] [PubMed] [Google Scholar]
- 7.Moine L, Verdrengh M, Tarkowski A. Role of neutrophil leukocytes in cutaneous infection caused by Staphylococcus aureus. Infection and immunity. 2000 Nov;68:6162. doi: 10.1128/iai.68.11.6162-6167.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Friend SL, et al. A thymic stromal cell line supports in vitro development of surface IgM+ B cells and produces a novel growth factor affecting B and T lineage cells. Experimental hematology. 1994 Mar;22:321. [PubMed] [Google Scholar]
- 9.Ziegler SF, Artis D. Sensing the outside world: TSLP regulates barrier immunity. Nature immunology. 2010 Apr;11:289. doi: 10.1038/ni.1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.West EE, Kashyap M, Leonard WJ. TSLP: A Key Regulator of Asthma Pathogenesis. Drug discovery today Disease mechanisms. 2012 Dec 1;9 doi: 10.1016/j.ddmec.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Divekar R, Kita H. Recent advances in epithelium-derived cytokines (IL-33, IL-25, and thymic stromal lymphopoietin) and allergic inflammation. Current opinion in allergy and clinical immunology. 2015 Feb;15:98. doi: 10.1097/ACI.0000000000000133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yoo J, et al. Spontaneous atopic dermatitis in mice expressing an inducible thymic stromal lymphopoietin transgene specifically in the skin. The Journal of experimental medicine. 2005 Aug 15;202:541. doi: 10.1084/jem.20041503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou B, et al. Thymic stromal lymphopoietin as a key initiator of allergic airway inflammation in mice. Nature immunology. 2005 Oct;6:1047. doi: 10.1038/ni1247. [DOI] [PubMed] [Google Scholar]
- 14.Al-Shami A, Spolski R, Kelly J, Keane-Myers A, Leonard WJ. A role for TSLP in the development of inflammation in an asthma model. The Journal of experimental medicine. 2005 Sep 19;202:829. doi: 10.1084/jem.20050199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Giacomin PR, et al. Epithelial-intrinsic IKKalpha expression regulates group 3 innate lymphoid cell responses and antibacterial immunity. The Journal of experimental medicine. 2015 Sep 21;212:1513. doi: 10.1084/jem.20141831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Garber K. Anti-IL-17 mAbs herald new options in psoriasis. Nature biotechnology. 2012 Jun;30:475. doi: 10.1038/nbt0612-475. [DOI] [PubMed] [Google Scholar]
- 17.Hueber W, et al. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn’s disease: unexpected results of a randomised, double-blind placebo-controlled trial. Gut. 2012 Dec;61:1693. doi: 10.1136/gutjnl-2011-301668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Soumelis V, et al. Human epithelial cells trigger dendritic cell mediated allergic inflammation by producing TSLP. Nature immunology. 2002 Jul;3:673. doi: 10.1038/ni805. [DOI] [PubMed] [Google Scholar]
- 19.David MZ, Daum RS. Community-associated methicillin-resistant Staphylococcus aureus: epidemiology and clinical consequences of an emerging epidemic. Clinical microbiology reviews. 2010 Jul;23:616. doi: 10.1128/CMR.00081-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rigby KM, DeLeo FR. Neutrophils in innate host defense against Staphylococcus aureus infections. Seminars in immunopathology. 2012 Mar;34:237. doi: 10.1007/s00281-011-0295-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bjerkan L, et al. The short form of TSLP is constitutively translated in human keratinocytes and has characteristics of an antimicrobial peptide. Mucosal immunology. 2015 Jan;8:49. doi: 10.1038/mi.2014.41. [DOI] [PubMed] [Google Scholar]
- 22.Sonesson A, et al. Thymic stromal lymphopoietin exerts antimicrobial activities. Experimental dermatology. 2011 Dec;20:1004. doi: 10.1111/j.1600-0625.2011.01391.x. [DOI] [PubMed] [Google Scholar]
- 23.Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nature reviews Immunology. 2013 Mar;13:159. doi: 10.1038/nri3399. [DOI] [PubMed] [Google Scholar]
- 24.McPhail LC, Clayton CC, Snyderman R. The NADPH oxidase of human polymorphonuclear leukocytes. Evidence for regulation by multiple signals. The Journal of biological chemistry. 1984 May 10;259:5768. [PubMed] [Google Scholar]
- 25.Cunningham MW. Pathogenesis of group A streptococcal infections. Clinical microbiology reviews. 2000 Jul;13:470. doi: 10.1128/cmr.13.3.470-511.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Clausen BE, Burkhardt C, Reith W, Renkawitz R, Forster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic research. 1999 Aug;8:265. doi: 10.1023/a:1008942828960. [DOI] [PubMed] [Google Scholar]
- 27.Hawkins PT, Stephens LR, Suire S, Wilson M. PI3K signaling in neutrophils. Current topics in microbiology and immunology. 2010;346:183. doi: 10.1007/82_2010_40. [DOI] [PubMed] [Google Scholar]
- 28.Zhong B, et al. Human neutrophils utilize a Rac/Cdc42-dependent MAPK pathway to direct intracellular granule mobilization toward ingested microbial pathogens. Blood. 2003 Apr 15;101:3240. doi: 10.1182/blood-2001-12-0180. [DOI] [PubMed] [Google Scholar]
- 29.Hommes DW, Peppelenbosch MP, van Deventer SJ. Mitogen activated protein (MAP) kinase signal transduction pathways and novel anti-inflammatory targets. Gut. 2003 Jan;52:144. doi: 10.1136/gut.52.1.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Allen IC, et al. The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity. 2009 Apr 17;30:556. doi: 10.1016/j.immuni.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nature immunology. 2010 Sep;11:785. doi: 10.1038/ni.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kolev M, Le Friec G, Kemper C. Complement-tapping into new sites and effector systems. Nature reviews Immunology. 2014 Dec;14:811. doi: 10.1038/nri3761. [DOI] [PubMed] [Google Scholar]
- 33.Sarma JV, Ward PA. New developments in C5a receptor signaling. Cell health and cytoskeleton. 2012 Jul 1;4:73. doi: 10.2147/CHC.S27233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Klos A, et al. The role of the anaphylatoxins in health and disease. Molecular immunology. 2009 Sep;46:2753. doi: 10.1016/j.molimm.2009.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Muller-Eberhard HJ. Complement. Annual review of biochemistry. 1969;38:389. doi: 10.1146/annurev.bi.38.070169.002133. [DOI] [PubMed] [Google Scholar]
- 36.Bauernfeind F, et al. Cutting edge: reactive oxygen species inhibitors block priming, but not activation, of the NLRP3 inflammasome. Journal of immunology. 2011 Jul 15;187:613. doi: 10.4049/jimmunol.1100613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Woodruff TM, Nandakumar KS, Tedesco F. Inhibiting the C5-C5a receptor axis. Molecular immunology. 2011 Aug;48:1631. doi: 10.1016/j.molimm.2011.04.014. [DOI] [PubMed] [Google Scholar]
- 38.Tamamis P, et al. Insights into the mechanism of C5aR inhibition by PMX53 via implicit solvent molecular dynamics simulations and docking. BMC biophysics. 2014;7:5. doi: 10.1186/2046-1682-7-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Taylor BC, et al. TSLP regulates intestinal immunity and inflammation in mouse models of helminth infection and colitis. The Journal of experimental medicine. 2009 Mar 16;206:655. doi: 10.1084/jem.20081499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee HC, et al. Thymic stromal lymphopoietin is induced by respiratory syncytial virus-infected airway epithelial cells and promotes a type 2 response to infection. The Journal of allergy and clinical immunology. 2012 Nov;130:1187. doi: 10.1016/j.jaci.2012.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Piliponsky AM, et al. Thymic Stromal Lymphopoietin Improves Survival and Reduces Inflammation in Sepsis. American journal of respiratory cell and molecular biology. 2016 Mar 2; doi: 10.1165/rcmb.2015-0380OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kuethe JW, et al. Thymic stromal lymphopoietin mediates the host response and increases mortality during sepsis. The Journal of surgical research. 2014 Sep;191:19. doi: 10.1016/j.jss.2014.05.024. [DOI] [PubMed] [Google Scholar]
- 43.Gauvreau GM, et al. Effects of an anti-TSLP antibody on allergen-induced asthmatic responses. The New England journal of medicine. 2014 May 29;370:2102. doi: 10.1056/NEJMoa1402895. [DOI] [PubMed] [Google Scholar]
- 44.Morgan BP, Harris CL. Complement, a target for therapy in inflammatory and degenerative diseases. Nature reviews Drug discovery. 2015 Oct 23; doi: 10.1038/nrd4657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kaplan A, et al. Failure to induce IFN-beta production during Staphylococcus aureus infection contributes to pathogenicity. Journal of immunology. 2012 Nov 1;189:4537. doi: 10.4049/jimmunol.1201111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Swamydas M, Lionakis MS. Isolation, purification and labeling of mouse bone marrow neutrophils for functional studies and adoptive transfer experiments. Journal of visualized experiments: JoVE. 2013:e50586. doi: 10.3791/50586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ribeiro-Gomes FL, et al. Site-dependent recruitment of inflammatory cells determines the effective dose of Leishmania major. Infection and immunity. 2014 Jul;82:2713. doi: 10.1128/IAI.01600-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. TSLP does not directly kill MRSA, and normal neutrophil-depleted blood can still reduce MRSA burden.
Fig. S2. TSLP requires TSLPR and acts on human neutrophils to increase control of MRSA.
Fig. S3. TSLP is TSLPR-dependent and enhances the killing of both MRSA and S. aureus in vivo.
Fig. S4. TSLP treatment increases killing of MRSA by human neutrophils in a PI3K- and MAPK/ERK-dependent manner.
Fig. S5. TSLP treatment of mouse or human neutrophils does not affect phagocytosis.
Fig. S6. ROS and complement-dependent TSLP-enhanced neutrophil killing.
Table S1. TSLP does not alter the transcriptional profile of human neutrophils.