

Abstract
Exertional heat stroke (EHS), the most severe manifestation of heat illness, is accompanied by a marked inflammatory response. The release of acute phase proteins (APPs) is an important component of inflammation, which can assist in tissue survival/repair. The time course of APPs in recovery from EHS is unknown. Furthermore, skeletal muscles produce APPs during infection, but it is unknown whether they can produce APPs after EHS. Our objective was to determine the time course of representative APPs in liver, plasma and skeletal muscle during recovery from EHS. Male C57BL6/J mice ran in a forced running wheel at 37.5°C, 40% relative humidity until symptom limitation. Exercise control (EXC) mice ran for the same duration and intensity at 22.5°. Samples were collected (n = 6–12 per group) over 14 days of recovery. Protein abundance was quantified using immunoblots. Total and phosphorylated STAT3 (pSTAT3) at Tyr705, responsible for APP activation, increased in liver at 0.5 h after EHS compared with EXC, (P < 0.05 and P < 0.001, respectively). In contrast, in tibialis anterior (TA) muscle, total STAT3 increased at 3 h (P < 0.05) but pSTAT3 (Tyr705) did not. Liver serum amyloid A1 (SAA1) increased at 3 and 24 h after EHS (P < 0.05), whereas plasma SAA1 increased only at 3 h (P < 0.05). SAA1 was not detected in TA muscle. In liver and plasma, fibrinogen decreased at 3 h (P < 0.01) and increased in TA muscle (P < 0.05). Lipocalin-2 was undetectable in liver or TA muscle. Recovery from EHS is characterized by a transient acute phase response in both liver and skeletal muscle. However, APP expression profiles and subtypes differ between skeletal muscle and liver.
Keywords: exercise, fibrinogen, hyperthermia, lipocalin, SAA1
1 |. INTRODUCTION
Exertional heat stroke (EHS) is the most severe manifestation of heat-related illness, which is characterized by CNS dysfunction (e.g. delirium and seizures) in hyperthermic subjects (Laitano, Leon, Roberts, & Sawka, 2019; Leon & Bouchama, 2015). Exertional heat stroke is the third leading cause of death among athletes during physical activity (Howe & Boden, 2007), but those surviving the episode undergo an array of inflammatory responses and tissue abnormalities during recovery (Bouchama & Knochel, 2002; Casa et al., 2015) resulting in long-term health consequences (Laitano et al., 2020; Wallace, Kriebel, Punnett, Wegman, & Amoroso, 2007; Wang et al., 2019). Although the inflammatory response during recovery from EHS has been well characterized (Garcia et al., 2018; King, Leon, Morse, & Clanton, 2017), the cellular and systemic events leading to the acute phase response (APR) are unknown. This is important because the APR constitutes one of the main mechanisms by which the body re-establishes homeostasis after major inflammatory insults. During recovery from EHS, the time course of major proteins that participate in the APR remains unexplored.
A hallmark of recovery from EHS in mice involves elevations in circulatory interleukin-6 (IL-6), which peaks very rapidly, at ~0.5 h after collapse (Garcia et al., 2018; King et al., 2017). Sustained high levels of circulating IL-6 are associated with increased risk of mortality and morbidity in models of classic heat stroke (Bouchama, al-Sedairy, Siddiqui, Shail, & Rezeig, 1993), whereas transient elevations in circulating IL-6 have been shown to be protective in a model of heat injury (Phillips, Welc, Wallet, King, & Clanton, 2015). Moreover, IL-6 has been shown to regulate the hepatic APR response after severe infection via phosphorylation (at Tyr705) of signal transducer and activator of transcription-3 (STAT3) on the IL-6–GP130 receptor. This is a key pathway in cytokine signalling that results in the gene expression of key acute phase proteins (APPs) in the liver and perhaps other organ systems (Castell et al., 1988; Hagihara et al., 2004). Therefore, it is reasonable to expect that phosphorylation of STAT3 (Tyr705) would occur during recovery from EHS, as has been shown in the liver in passive models of heat stroke (Leon, Dineen, Blaha, Rodriguez-Fernandez, & Clarke, 2013).
Skeletal muscles participate in immune regulation and function (Pedersen & Febbraio, 2008; Steensberg et al., 2001). They synthesize APP mRNA in murine models of cancer cachexia (Bonetto et al., 2011, 2012) and septic shock (Langhans et al., 2014). Nevertheless, APP responsiveness has never been demonstrated in skeletal muscle after other acute illnesses, such as heat stroke. Given that skeletal muscles constitute ~40% of the total body mass in humans and rodents, it is logical to hypothesize that if they produce APPs, they could contribute substantially to recovery from severe heat illness.
In this study, we targeted three representative APPs that are particularly relevant to outcomes in EHS. For instance, fibrinogen participates in coagulation events (Hayakawa, 2017), and coagulation is a well-known problem in severe human heat stroke (Bouchama et al., 1996). Serum amyloid A1 (SAA1) opsonizes bacteria and initiates M1 to M2 macrophage shifts (Linke, Meinel, Chalcroft, & Urieli-Shoval, 2017), which could help to prevent or help in recovery from infection arising from a leaky gastrointestinal tract. Lipocalin-2 responds to renal injury and inflammation (Sultan et al., 2012) and is known to be elevated during exercise in the heat (Junglee et al., 2013). Therefore, our main goal was to determine the abundance of these APPs during EHS recovery in liver, plasma and skeletal muscle. We hypothesized that the APR after EHS would follow the IL-6 response previously characterized in our model (King et al., 2017) and that skeletal muscle would contribute to the APP response to EHS.
2 |. METHODS
2.1 |. Ethical approval
All animal protocols were approved by the University of Florida Institutional Animal Care and Use Committee (#201807422 and #201808999). We adhered to the Guide for the Care and Use of Laboratory Animals as prepared by the Committee for the Update of the Guide for the Care and Use of Laboratory Animals of the Institute for Laboratory Animal Research, National Research Council. Reported information about the procedures conformed to the Animal Research Reporting of In Vivo Experiments (ARRIVE) guidelines (Kilkenny, Browne, Cuthill, Emerson, & Altman, 2010). The investigators understand the ethical principles under which Experimental Physiology operates, and the work complies with the animal ethics checklist used by the journal (Grundy, 2015).
2.2 |. Animal care
Sixty-two C57BL/6J male mice (Jackson Laboratories, Bar Harbor, ME, USA) were studied. All mice were 4–6 months of age. A standard chow diet and water were provided ad libitum until experimentation. Mice were housed in groups until they were implanted with telemetry devices, after which they were housed individually, and maintained on a 12 h–12 h light–dark cycle at 19–22°C and 30–60% relative humidity (RH).
2.3 |. Animal surgery and exercise training
A laparotomy was conducted under isoflurane (USP – Patterson Veterinary, St. Greeley, CO, USA) /oxygen anaesthesia (5% for induction and 1.5% for maintenance, Eagle Eye Anesthesia, Jacksonville, FL, USA) and in sterile conditions to allow the placement of a temperature telemetry emitter into the abdominal cavity (G2 E-Mitter; Starr Life Sciences, Oakmont, PA, USA) for measurements of core temperature (Tc) during the experimental protocol. After surgery, mice were given buprenorphine (Patterson Veterinary, St. Greeley, CO, USA) injections S.C. (0.1 mg/kg) every 12 h for 48 h and then monitored for a minimum of 2 weeks until fully healed. After this recovery period, mice were given in-cage running wheels (model 0297-0521; Columbus Instruments, Columbus, OH, USA) to allow for voluntary exercise training for 3 weeks. Animals were brought to the laboratory in the third week to begin familiarization with the set-up (e.g. chamber and forced wheel system) and training period. A forced running wheel (model 80840; Lafayette, Lafayette, IN, USA) powered by a DC power supply was used to train the animals. Four training sessions were performed, as described previously (Garcia et al., 2018; King, Leon, Mustico, Haines, & Clanton, 2015). After the training sessions, mice were given 2 days of recovery, before initiation of the EHS protocol.
2.4 |. Exertional heat stroke protocol
Animals were brought from the vivarium to the laboratory 1 day before the EHS procedure in their own cages, remaining on the same light cycle. This was done to adapt the animal to the laboratory and allow for recovery from the stress of transport. The Tc was monitored over time in 30 s intervals to ensure that temperature profiles were normal before EHS. The EHS protocol began in the early- to mid-morning. Mice remained in their cages and were placed in environmental chambers (Thermo Forma, 3940; Thermo-Fisher, Waltham, MA, USA) set to 37.5°C and 30–40% RH. Chamber temperature and humidity were measured, recorded and controlled at the location of the running wheel. After the temperature of the environmental chamber equilibrated (30–45 min), mice were quickly placed in the enclosed running wheel. Once the mouse Tc stabilized at 36–37.5°C (~5 min), the forced running wheel protocol was initiated. The preprogrammed protocol began at 2.5 m min−1 and increased by 0.3 m min−1 every 10 min until the mouse reached a Tc of 41°C. The speed was then maintained constant until the mouse reached the end-point criteria. The end-point, i.e. EHS, was defined by loss of consciousness. A loss of consciousness was determined when the mouse underwent three consecutive revolutions with no physical response. Mice were returned to their original cage for recovery at room temperature.
Mice were allowed to recover for varying time periods. Recovery time points were 0.5 h, 3 h, 24 h, 4 days, 9 days and 14 days, with six males per group. A separate set of mice was euthanized at 4 days as exercise controls (EXC). These mice were treated identically to the EHS mice except that there was no elevation of temperature within the chamber. The exercise time and speed of the forced running protocol for the EXC mice was matched to the mean value of the EHS mice previously described (King et al., 2015).
For collection of samples, mice were anaesthetized with isoflurane at their respective recovery time points. Immediately after induction and attainment of steady-state anaesthesia, heparinized blood was collected by transcardiac puncture, using a caudal approach, immediately left of the xiphoid process. After tissue collection, the mice were euthanized by removal of the heart. Whole blood was centrifuged for 10 min at 3000 g at 4°C. The supernatant (plasma) was removed, aliquoted and stored at −80°C until further analyses. The liver and tibialis anterior (TA) muscle were harvested, snap frozen, and stored at −80°C until further analyses.
2.5 |. Caecal slurry injection
In order to test whether skeletal muscles produce acute phase proteins (positive control), we induced sepsis in a subset of mice by i.p. injection of caecal slurry (CS) collected previously from donor mice. The batch of CS used in the present study was prepared and stored according to the method described by Starr et al. (2014). In brief, caecal contents were filtered and mixed with 15% glycerol solution in PBS. The CS was then aliquoted in 2 ml cryovials and kept in freezing containers (Nalgene Mr. Frosty; Thermo Scientific, Waltham, MA, USA) at −80°C. Immediately before injection, cryovials were thawed to room temperature, mixed well, and then injected at a dose that would induce a 40–60% survival rate over the course of 5 days. Animals were restrained briefly by tail and scruff handling, and CS was injected i.p. into the lower left quadrant of the peritoneal cavity while the animal was held in the supine position with the head below the body as previously demonstrated (Laitano et al., 2018). Under deep isoflurane anaesthesia, skeletal muscles were harvested 24 h after the caecal slurry injections and snap frozen in liquid nitrogen for later Western blot analyses.
2.6 |. SDS-PAGE and Western blot analyses
Skeletal muscle and liver samples were homogenized using a Kontess Duall Homogenizer in 1× cell lysis buffer (no. 9803; Cell Signaling Technology) containing the following: 20 mM Tris–HCl (pH 7.5), 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% Triton, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, 1 μg ml−1 leupeptin, and 1× protease and phosphatase inhibitor cocktail (no. 5872; Cell Signaling Technology). We performed a detergent-compatible assay (DC Assay, Bio-Rad Laboratories) to determine the total protein content of the lysates and plasma. Lysates and plasma were mixed 1:1 with 2× Laemmli sample buffer (Bio-Rad Laboratories). We loaded approximately equal amounts of protein (15 μg per lane) into 4–20% stain-free TGX gels (Bio-Rad Laboratories). The desired concentration loaded was determined beforehand using a linearity Western blot. After loading the protein, we performed electrophoresis at 200 V for 50 min with the chamber surrounded by ice. We scanned the gel to quantify the total protein (Gel Doc EZ Imager; Bio-Rad Laboratories) and transferred the proteins to a nitrocellulose membrane (GE Healthcare Bio-Sciences) at 100 mA overnight at 4°C. We scanned the membrane to determine protein transfer and proceeded by blocking the membrane using Li-COR Blocking Buffer (Li-COR, Lincoln, NE, USA) for 1 h at room temperature. Subsequently, the membrane was probed with primary antibodies for total STAT3, pSTAT3 (Tyr705), fibrinogen, lipocalin-2 and SAA1 (Table 1). After primary antibody incubation, we washed the membranes in TBS-T (4 × 5 min), incubated in secondary antibody (IR Dye; Li-COR) for 45 min at room temperature, washed in TBS-T (4 × 5 min), rinsed in 1× TBS, and scanned the membrane using an Odyssey Infrared Imaging system (Li-COR). The signal intensity was quantified and normalized to the total protein signal measured in the corresponding membrane lanes. Homogenate samples were aliquoted and added to the same test tube. This was referred to as the pooled sample and was used as an internal control to make comparisons amongst immunoblots.
TABLE 1.
Antibodies used for detection of protein abundance
| Antibody | Type | Molecularweight (kDa) | Host species | Company (catalogue no.) |
|---|---|---|---|---|
| Fibrinogen | Polyclonal | 340 | Rabbit | Dako (A0080) |
| Lipocalin | Polyclonal | 24 | Goat | R&D (AF1857) |
| STAT3/pSTAT3 (Tyr705) | Polyclonal | 79/86 | Rabbit | Cell Signaling (9131S) |
| Serum amyloid A1 (SAA1) | Polyclonal | 12 | Rabbit | Abcam (Ab171030) |
2.7 |. Statistical analysis
The data are presented as box plots, because our study design consisted of independent groups and also based on the fact that the time points studied (reported on the x-axis) were not continuous. Data distributions were tested for normality using the Shapiro–Wilk test. The majority of the data displayed non-parametric distributions and were reported as the median ± interquartile range, and parametric data were reported as means ± SD. We used the Kruskal–Wallis test to compare populations among treatments of multiple groups. Steel–Dwass post hoc analysis was used to confirm where the differences occurred between groups. We used a value of α< 0.05 for statistical difference. All analyses were made using SAS JMP Pro v.12.0 (SAS, Cary, NC, USA) and GraphPad Prism v.8.0 software (GraphPad Software, San Diego, CA, USA).
3 |. RESULTS
3.1 |. Thermoregulatory responses to EHS
The results for the thermoregulatory profile and descriptive information of this cohort of animals during EHS have been published before and can be verified (Garcia et al., 2018). In this cohort, mice ran 557 ± 204.6 m and took 124.1 ± 22.6 min to collapse (i.e. reach heat stroke). The maximal core temperature recorded at the time of collapse was 42.2 ± 0.2°C. A typical and representative core temperature profile throughout the EHS protocol is shown in Figure 1.
FIGURE 1.

Core temperature profile of a representative male mouse during the exertional heat stroke (EHS) protocol (shaded red area) and during immediate recovery from EHS (shaded blue area)
3.2 |. Acute phase response in liver and plasma
To assess whether IL-6 signalling was involved in the APR in liver, we analysed the abundance of total and phosphorylated STAT3 (pSTAT3), at Tyr705, at 0.5, 3 and 24 h after EHS. Protein abundance of both total STAT3 and pSTAT3 (Tyr705) in the liver increased at 0.5 h after EHS (P < 0.05 and P < 0.001, respectively; Figure 2).
FIGURE 2.

(a,b) Abundance of total STAT3 (a) and pSTAT3 (Tyr705; b) in liver samples from exertional heat stroke (n = 6 per time point) and exercise control (EXC) mice (n = 6). (c) Total protein content used for normalization. *P < 0.05. ***P < 0.001
Of the acute phase proteins studied, SAA1 had the most noteworthy and unique pattern of expression in liver after EHS. At 3 and 24 h of recovery, we observed an increase in SAA1 protein abundance compared with EXC (P < 0.05; Figure 3). To determine whether SAA1 secretion resulted in elevated plasma levels, we measured the abundance in plasma at 3 h after EHS. SAA1 protein was significantly elevated (P < 0.01; Figure 4). The timing of the sample collection is consistent with the expression of SAA1 in the liver contributing to the plasma SAA1 at 3 h after EHS.
FIGURE 3.
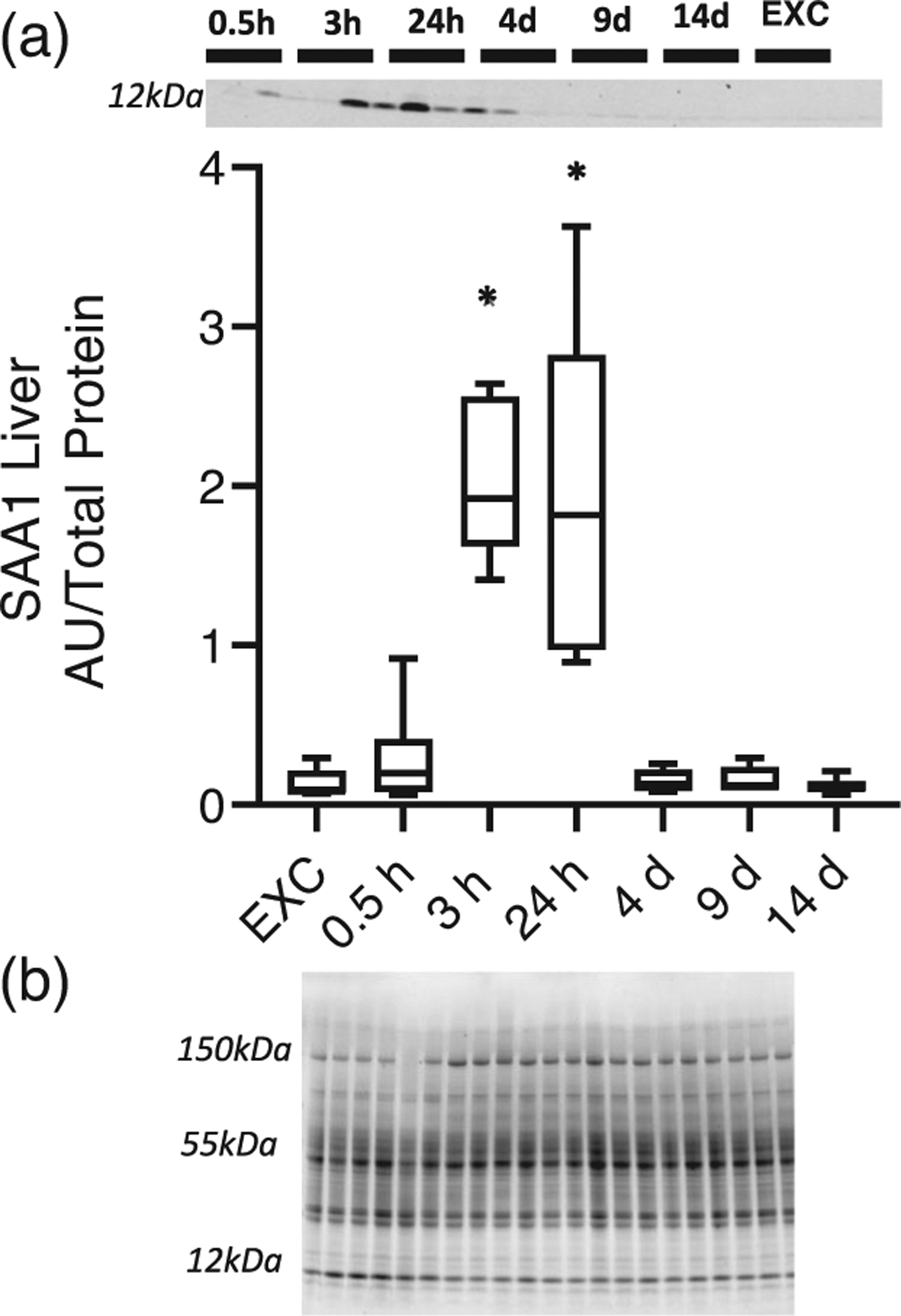
(a) Abundance of serum amyloid A1 (SAA1) in liver after exertional heat stroke compared with exercise control (EXC) mice (n = 6 per group). (b) total protein used for normalization. *P < 0.05
FIGURE 4.
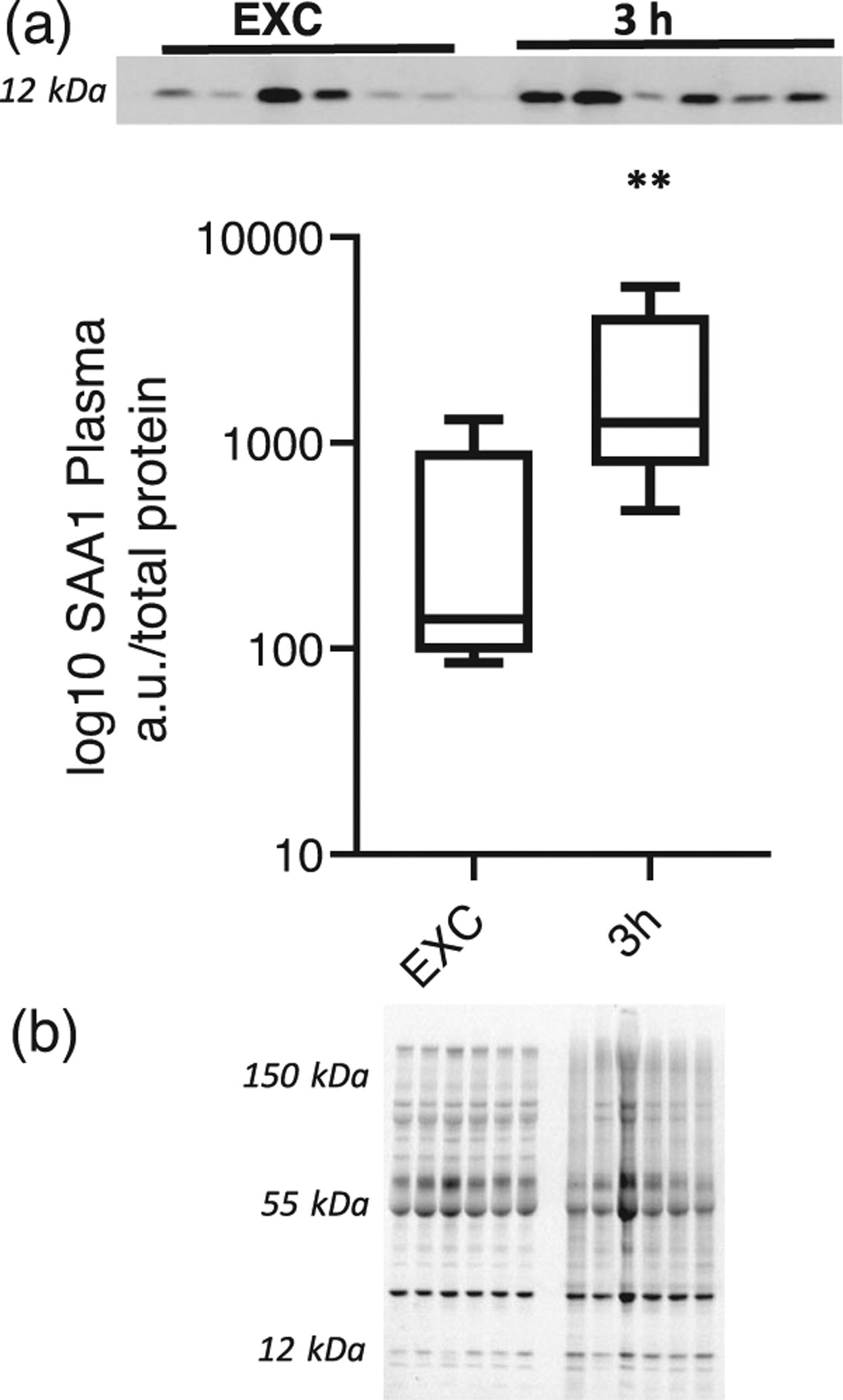
(a) Abundance of serum amyloid A1 (SAA1) in plasma in exercise control (EXC) mice (n = 6) and 3 h after exertional heat stroke (n = 6). (b) Total protein used for normalization. **P < 0.01
We also determined the time course of fibrinogen and lipocalin-2 production in the liver and plasma. Fibrinogen protein abundance in liver significantly decreased at 3 h after EHS when compared with EXC (P < 0.01; Figure 5). Similar findings were seen in plasma (Figure 6). Lipocalin-2 was not detected in the liver at any of the time points measured (Table 2).
FIGURE 5.

(a) Abundance of fibrinogen in liver after exertional heat stroke compared with exercise control (EXC). (b) Total protein used for normalization. *P < 0.05
FIGURE 6.

(a) Abundance of fibrinogen in plasma in exercise control (EXC) mice (n = 10) and 3 h after exertional heat stroke (n = 6). (b) Total protein used for normalization. ***P < 0.001
TABLE 2.
Arbitrary units/total protein of the 24 kDa band for lipocalin-1 abundance in the liver
| Protein | Control | 0.5 h | 3 h | 24 h | 4 days | 9 days | 14 days |
|---|---|---|---|---|---|---|---|
| Lipocalin-1 | 0.03 ± 0.02 | 0.06 ± 0.02 | 0.06 ± 0.04 | 0.02 ± 0.02 | 0.04 ± 0.02 | 0.03 ± 0.03 | 0.04 ± 0.04 |
No differences were observed in lipocalin-1 abundance over the course of the recovery. Data are means ± SD.
3.3 |. Acute phase response in skeletal muscle
To assess whether the skeletal muscle APR was stimulated by EHS exposure, we analysed the abundance of total and pSTAT3 (Tyr705), at 0.5, 3 and 24 h after EHS. In the TA, total STAT3 expression was elevated at 3 h after EHS (P < 0.05; Figure 7). However, expression of pSTAT3 was unchanged at these time points (Table 3).
FIGURE 7.

(a) STAT3 abundance in the tibialis anterior muscle in exertional heat stroke (n = 6) and exercise control (EXC) mice (n = 6). (b) Total protein used for normalization. *P < 0.05
TABLE 3.
Arbitrary units/total protein of the 85 kDa band for pSTAT3 (Tyr705) abundance in the tibialis anterior muscle
| Protein | Control | 0.5 h | 3 h |
|---|---|---|---|
| pSTAT3 (Tyr705) | 0.04 ± 0.03 | 0.09 ± 0.04 | 0.09 ± 0.04 |
pSTAT3 (Tyr705) was not different amongst groups studied. Data are means ± SD.
Expression of SAA1 was not detected in the TA at any of the recovery time points after EHS (Table 4). However, the TA retrieved from septic animals 24 h after CS injection revealed that SAA1 expression was elevated, serving as a positive control for this measurement (Figure 8). In contrast, fibrinogen expression in the TA was significantly elevated by 3 h after EHS, returning to control values by 24 h (Figure 9), a response that was the reverse of what was observed in the liver (Figure 5).
TABLE 4.
Arbitrary units/total protein of the 12 kDa band for serum amyloid A1 (SAA1) abundance in the tibialis anterior muscle
| Protein | Control | 0.5 h | 3 h | 24 h | 4 days | 9 days | 14 days |
|---|---|---|---|---|---|---|---|
| SAA1 | 0.14 ± 0.01 | 0.06 ± 0.02 | 0.04 ± 0.03 | 0.02 ± 0.03 | 0.11 ± 0.04 | 0.07 ± 0.01 | 0.11 ± 0.01 |
SAA1 was not different amongst groups studied. Data are means ± SD.
FIGURE 8.

(a) Serum amyloid A1 (SAA1) abundance in tibialis anterior muscle from sham-treated mice injected with vehicle (n = 2) and septic mice (n = 6) injected with caecal slurry. (b) Total protein used for normalization
FIGURE 9.

(a) Fibrinogen abundance in tibialis anterior muscle after exertional heat stroke compared with exercise control (EXC) mice (n = 6 per group). (b) Total protein used for normalization. *P < 0.05
4 |. DISCUSSION
Our main objective in this study was to delineate the time course of major acute phase proteins in liver, plasma and skeletal muscle after exposure to EHS. Our results demonstrate that SAA1, which is one of the main proteins involved in the APR in rodents, is upregulated in the liver and plasma 3–24 h after EHS. The observed peak of SAA1 in liver at 3–24 h after EHS is consistent with the timing of IL-6 elevation in the plasma, previously observed in this model (King et al., 2017), and with the timing of pSTAT3 (Tyr705) abundance in the liver (Figure 2). We also observed a depletion of fibrinogen in liver and plasma, which contrasted with acute elevations in fibrinogen in skeletal muscle during this same interval. Comparison of the different outcomes in liver and skeletal muscle suggests that different regulatory mechanisms are activated during heat stress in these two secretory tissues. A schematic summary of the findings is reported in Figure 10.
FIGURE 10.
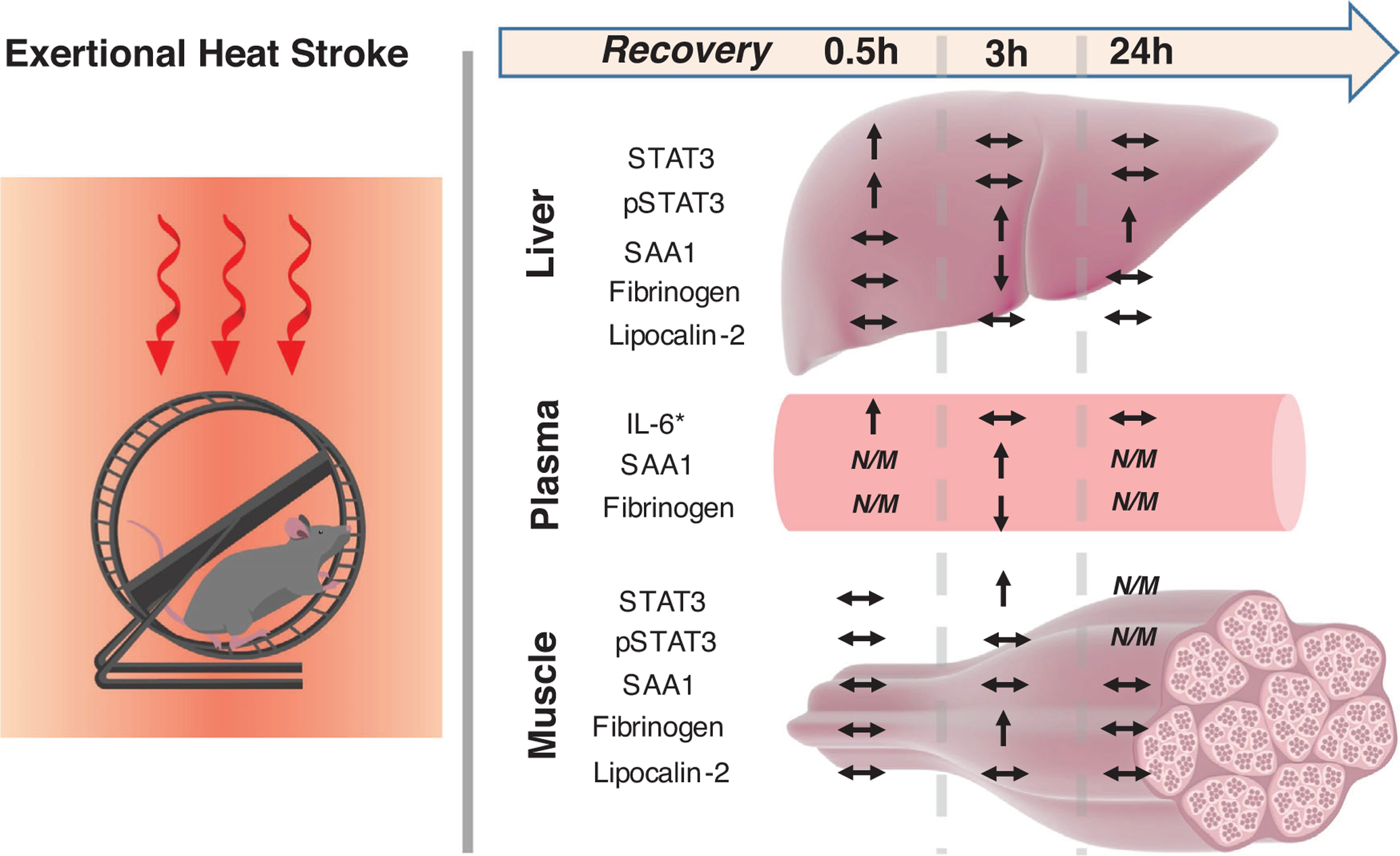
Schematic summary of the findings in liver, plasma and skeletal muscle. Abbreviations and symbols: ↑, increase; ↓, decrease; ↔, unchanged in relationship to control; N/M, not measured at that time point. IL-6* data are as presented by Garcia et al. (2018)
The APR to heat stroke in either humans or animal models has received little attention in the heat illness literature, although it is likely to play an important role in pathophysiology. Previously, Bouchama et al. (1993) reported that humans exposed to heat stroke exhibited a ~10-fold elevation in plasma C-reactive protein. Other evidence in animal models includes a marked increase in mRNA for liver SAA3 gene in response to passive heat stroke in mice (Leon et al., 2013). Dogs exposed to naturally occurring heat stroke (that do not survive) exhibit progressive elevations in plasma fibrinogen within 24 h, but dogs that survive do not show this response (Bruchim, Horowitz, & Aroch, 2017). One new contribution of the present study is that the time course and tissue distribution of APP production differs in liver versus skeletal muscle and must follow a different set of regulatory signals compared with conditions of septic shock, where the APR is the most commonly studied.
4.1 |. Interleukin-6 signalling and APP synthesis
Previous studies from our group have demonstrated the time course of circulating IL-6 and other cytokines during recovery from EHS. King et al. (2017) observed that plasma IL-6 peaked at 0.5 h after loss of consciousness in male mice undergoing EHS. More recently, Garcia et al. (2018) confirmed these findings in males and showed a similar response in females. Induction of IL-6 in severe hyperthermia is thought to come from two sources. Some evidence from models of classic (i.e. passive) heat stroke suggests that during severe hyperthermia there is endotoxin leakage from the gut, which could trigger a cytokine response (Leon & Bouchama, 2015). However, we do not have evidence for major endotoxaemia in our model of EHS. For instance, a recent EHS study performed by the US Army, using the same preclinical model described herein, demonstrated an increase in 4 kDa fluorescein isothiocyanate–dextran permeability and increased transfer across gut epithelial membranes. However, circulating 16S RNA, which is a sensitive measure of bacterial release, was undetectable (M. A. King, S. Dineen, J. A. Ward, M. L. Plamper, S. Vidyasagar, and L. R. Leon, unpublished observations).
Endotoxaemia, however, is not the only stimulus for IL-6. Hyperthermia per se has been shown to induce IL-6 mRNA in isolated skeletal muscle (Welc et al., 2012, 2016). This stress-induced IL-6 response is mediated, in part, by heat shock factors associated with the heat shock response (Welc, Clanton, Dineen, & Leon, 2013). This is relevant, because IL-6 regulates the hepatic APR during severe infection (Castell et al., 1988; Jain, Gautam, & Naseem, 2011).
A key downstream component of the IL-6 signalling pathway is the phosphorylation of STAT3 at Tyr705, which results in the gene transcription for acute phase proteins. Our data for increased pSTAT3 (Tyr705) (Figure 2) in the liver are consistent with the possibility of IL-6 being the major driver of the observed SAA1 response described herein.
4.2 |. Significance of SAA1 abundance
Serum amyloid A proteins are a highly conserved family of apolipoproteins (Benditt & Eriksen, 1977). Different isoforms of SAA are expressed constitutively in response to inflammatory stimuli (De Buck et al., 2016). Concentrations of SAA1 are upregulated three-to fivefold in patients with inflammatory and infectious diseases, locally and systemically (De Buck et al., 2016; Sack, 2018). It is accepted that the transient APR, which includes SAA1, contributes to innate immunity to defend the host against pathological damage and restoration of homeostasis (De Buck et al., 2016; Sack, 2018). Although controversial, many studies have demonstrated that SAA1 has functions that are either pro-inflammatory (Hatanaka, Furlaneto, Ribeiro, Souza, & Campa, 2004; He, Shepard, Chen, Pan, & Ye, 2006; Sun et al., 2015) when chronically elevated or anti-inflammatory (Lavie et al., 2006; Linke, Bock, Valet, & Rothe, 1991; Shah, Hari-Dass, & Raynes, 2006) when transiently elevated. This response is similar to IL-6, at least in heat injury settings, where it is considered pro-inflammatory when chronically elevated (Bouchama et al., 1993) or anti-inflammatory when it is transiently elevated (Phillips et al., 2015). On contrast, adverse properties of SAA1 include, but are not limited to, the sustained induction of cytokines (Furlaneto & Campa, 2000; Patel, Fellowes, Coade, & Woo, 1998), chemokines (Gouwy et al., 2015; Yang et al., 2006) and matrix-degrading enzymes (Lee et al., 2006; Migita et al., 1998), because SAA1 can function as a damage-associated molecular pattern (DAMP) (Sack, 2018).
Given the observed transient elevation in both liver and plasma during recovery from EHS, it is reasonable to conjecture that SAA1 is acting as an anti-inflammatory APP in EHS settings. The anti-inflammatory or regenerative effects of SAA1 include M2 macrophage polarization (Sun et al., 2015; Tidball & Wehling-Henricks, 2007), opsonization of bacteria (Shah et al., 2006) and angiogenesis (Lee et al., 2006). M2 macrophages are considered to be pro-resolving immune cells, which play a role in satellite cell activation and tissue repair after injury (Merly, Lescaudron, Rouaud, Crossin, & Gardahaut, 1999; Sun et al., 2015; Tidball & Wehling-Henricks, 2007). Whether SAA1 is indeed anti-inflammatory in EHS settings, however, warrants further investigation.
4.3 |. Unique fibrinogen expression
Fibrinogen is a soluble APP that is essential for haemostasis and wound healing via coagulation (Weisel & Litvinov, 2017). Fibrinogen is converted into insoluble fibrin monomers that aggregate to form a blood clot. Coagulopathy is a well-known response to EHS (Bouchama et al., 1996; Proctor et al., 2019). Furthermore, in cases of severe trauma, systemic levels of fibrinogen are depleted upon arrival at the emergency room (Hayakawa, 2017), and after severe tissue injury the expression of thrombin increases, which acts to cleave fibrin peptides from fibrinogen glycoproteins, forming an excess of fibrin for clotting (Hayakawa, 2017; Weisel & Litvinov, 2017). Interestingly, we observed an acute depletion of fibrinogen in liver after an episode of EHS (Figure 4), which could be indicative of fibrinogen being released by the liver and used to promote clotting through fibrin formation in response to the tissue injury from EHS. This response might contribute to disseminated intravascular coagulation, which is a condition commonly observed in heat stroke and used recently to predict the severity of organ damage in classic human heat stroke victims (Jilma & Derhaschnig, 2012).
The decrease in liver fibrinogen observed during recovery from EHS seems to be independent of STAT3. For instance, IL-6 has been shown to regulate fibrinogen gene expression in the liver. However, STAT3 has no direct role in regulating IL-6-mediated inducible expression of the fibrinogen gene (Ray, 2000). The marked reduction in plasma fibrinogen at 3 h (hyperfibrinogenaemia) is likely to be a reflection of consumption attributable to clot formation and is a common response to trauma (Hagemo et al., 2014)
To our knowledge, this is the first study to demonstrate fibrinogen protein production in skeletal muscle. The significance of this finding is that a pro-haemostatic influence of damaged muscle has been expressed in the surgical literature for decades (Monroe, 2016), but has been poorly understood. Although myosin release has been identified as one muscle-derived mediator of clotting (Deguchi et al., 2016), the demonstration that skeletal muscle is a potential source of fibrinogen could provide an additional mechanism for this phenomenon. Muscle fibrinogen might play important roles in blocking local muscle haematomas, because muscle haematomas are one of the most common symptoms of congenital afibrinogenaemia (Peyvandi, Haertel, Knaub, & Mannucci, 2006).
4.4 |. Acute phase protein and STAT3 abundance in skeletal muscle
Contrary to our original hypothesis, we did not detect SAA1 or lipocalin-2 in skeletal muscle after the EHS episode. We have previously demonstrated that skeletal muscle contributes to the immune response via secretion of IL-6 in response to EHS (Welc et al., 2013). Others have observed an upregulation of IL-6 and SAA1 mRNA in skeletal muscle after systemic infection and inflammation (Langhans et al., 2014). Indeed, immunohistochemical analysis demonstrates that SAA1 accumulates on the outer membrane of muscle in patients with critical-illness myopathy (Langhans et al., 2014). This evidence led us to the hypothesis that a similar response would be observed in skeletal muscle in response to EHS exposure. However, we did not see it. To be certain of the validity of our assay, we used skeletal muscles harvested from a more severe inflammatory model as a positive control, and there we observed a clear elevation in expression of SAA1 (Figure 7). This might indicate that the inflammatory response observed after EHS is either not severe enough to elicit SAA1 expression in skeletal muscle or that IL-6 signalling is not adequate for activation.
In support of the latter possibility, we observed no expression of pSTAT3 in muscle during EHS, which is essential for IL-6-mediated SAA1 transcription. However, we did observe elevated expression of total STAT3 protein 3 h after EHS. This is indicative of a secondary signalling pathway for STAT3 transcriptional control, which might involve other transcriptional modifiers, such as the cyclic-AMP response element (Qi & Yang, 2014). The fact that a fibrinogen signal was evident in muscle after EHS despite there being no apparent SAA1 signal might be attributable to the distinctly different signalling pathways, e.g. fibrinogen can be regulated transcriptionally by glucocorticoids (Grieninger, Hertzberg, & Pindyck, 1978), which are drastically increased in this model of EHS (Garcia et al., 2018).
In conclusion, our results demonstrate that a single episode of EHS transiently increases the expression of SAA1 in mouse liver and plasma, at least in this preclinical model. Furthermore, EHS results in a depletion of liver and plasma fibrinogen, a typical response to severe trauma. In contrast, skeletal muscle fibrinogen production is stimulated in EHS, which might provide an additional reservoir for fibrinogen supply in a crisis, thus maintaining the clotting potential throughout the body and locally within the muscle. This study has established that a multi-organ APR must be included as an important aspect of the stress-induced inflammatory response accompanying EHS exposure.
New Findings.
• What is the central question of this study?
Exertional heat stroke is accompanied by a marked inflammatory response. In this study, we explored the time course of acute phase proteins during recovery from severe heat stress in mice and the potential role of skeletal muscles as their source.
• What is the main finding and its importance?
Exertional heat stroke transiently increased expression of acute phase proteins in mouse liver and plasma and depleted liver and plasma fibrinogen, a typical response to severe trauma. In contrast, skeletal muscle fibrinogen production was stimulated by heat stroke, which can provide an additional reservoir for fibrinogen supply to maintain the clotting potential throughout the body and locally within the muscle.
FUNDING INFORMATION
This study was funded by contracts from the Department of Defense, W81XWH-15-2-0038 (T.L.C.) and BA180078 (T.L.C.), and the National Institutes of Health GMS RO1GM118895 (T.L.C.), with supplemental support from the B. K. and Betty Stevens Endowment at the University of Florida (T.L.C.).
Funding information
HHS; NIH; National Institute of General Medical Sciences, Grant/Award Number: RO1GM118895; U.S. Department of Defense, Grant/Award Number: W81XWH-15-2-0038
AUTHOR BIOGRAPHY
John Iwaniec completed his Master’s of Science degree at the University of Florida under the guidance of Professors Thomas Clanton and Orlando Laitano, where he investigated the impact of exertional heat stroke and sepsis on the acute phase response and epigenome. He is currently a Senior Research Support Specialist at SUNY Upstate Medical University, working with Professor Joel Wilmore on the pathophysiology of induced microbiome immunity and immunoglobulin-mediated protection in models of septicaemia.
Footnotes
Publisher's Disclaimer: DISCLAIMER
Publisher's Disclaimer: The opinions or assertions contained herein are the private views of the author(s) and are not to be construed as official or as reflecting the views of the Army or the Department of Defense. Citations of commercial organizations and trade names in this report do not constitute an official Department of the Army endorsement or approval of the products or services of these organizations.
COMPETING INTERESTS
None declared.
REFERENCES
- Benditt EP, & Eriksen N (1977). Amyloid protein SAA is associated with high density lipoprotein from human serum. Proceedings of the National Academy of Sciences of the United States of America, 74, 4025–4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonetto A, Aydogdu T, Jin X, Zhang Z, Zhan R, Puzis L, … Zimmers TA (2012). JAK/STAT3 pathway inhibition blocks skeletal muscle wasting downstream of IL-6 and in experimental cancer cachexia. American Journal of Physiology-Endocrinology and Metabolism, 303, E410–E421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonetto A, Aydogdu T, Kunzevitzky N, Guttridge DC, Khuri S, Koniaris LG, & Zimmers TA (2011). STAT3 activation in skeletal muscle links muscle wasting and the acute phase response in cancer cachexia. PLoS ONE, 6, e22538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchama A, al-Sedairy S, Siddiqui S, Shail E, & Rezeig M (1993). Elevated pyrogenic cytokines in heatstroke. Chest, 104, 1498–1502. [DOI] [PubMed] [Google Scholar]
- Bouchama A, Bridey F, Hammami MM, Lacombe C, Al-Shail E, Al-Ohali Y, … deProst D (1996). Activation of coagulation and fibrinolysis in heatstroke. Thrombosis and Haemostasis, 76, 909–915. [PubMed] [Google Scholar]
- Bouchama A, & Knochel JP (2002). Heat stroke. New England Journal of Medicine, 346, 1978–1988. [DOI] [PubMed] [Google Scholar]
- Bruchim Y, Horowitz M, & Aroch I (2017). Pathophysiology of heatstroke in dogs – revisited. Temperature, 4, 356–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casa DJ, DeMartini JK, Bergeron MF, Csillan D, Eichner ER, Lopez RM, … Yeargin SW (2015). National Athletic Trainers’ Association position statement: Exertional heat illnesses. Journal of Athletic Training, 50, 986–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castell JV, Gómez-Lechón MJ, David M, Hirano T, Kishimoto T, & Heinrich PC (1988). Recombinant human interleukin-6 (IL-6/BSF-2/HSF) regulates the synthesis of acute phase proteins in human hepatocytes. FEBS Letters, 232, 347–350. [DOI] [PubMed] [Google Scholar]
- De Buck M, Gouwy M, Wang JM, Van Snick J, Opdenakker G, Struyf S, & Van Damme J (2016). Structure and expression of different serum amyloid A (SAA) variants and their concentration-dependent functions during host insults. Current Medicinal Chemistry, 23, 1725–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deguchi H, Sinha RK, Marchese P, Ruggeri ZM, Zilberman-Rudenko J, McCarty OJT, … Griffin JH (2016). Prothrombotic skeletal muscle myosin directly enhances prothrombin activation by binding factors Xa and Va. Blood, 128, 1870–1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furlaneto CJ, & Campa A (2000). A novel function of serum amyloid A: A potent stimulus for the release of tumor necrosis factor-α, interleukin-1β, and interleukin-8 by human blood neutrophil. Biochemical and Biophysical Research Communications, 268, 405–408. [DOI] [PubMed] [Google Scholar]
- Garcia CK, Mattingly AJ, Robinson GP, Laitano O, King MA, Dineen SM, … Clanton TL (2018). Sex-dependent responses to exertional heat stroke in mice. Journal of Applied Physiology, 125, 841–849. [DOI] [PubMed] [Google Scholar]
- Gouwy M, De Buck M, Pörtner N, Opdenakker G, Proost P, Struyf S, & Van Damme J (2015). Serum amyloid A chemoattracts immature dendritic cells and indirectly provokes monocyte chemotaxis by induction of cooperating CC and CXC chemokines. European Journal of Immunology, 45, 101–112. [DOI] [PubMed] [Google Scholar]
- Grieninger G, Hertzberg KM, & Pindyck J (1978). Fibrinogen synthesis in serum-free hepatocyte cultures: Stimulation by glucocorticoids. Proceedings of the National Academy of Sciences of the United States of America, 75, 5506–5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundy D (2015). Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology. The Journal of Physiology, 593, 2547–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagemo JS, Stanworth S, Juffermans NP, Brohi K, Cohen M, Johansson PI, … Gaarder C (2014). Prevalence, predictors and outcome of hypofibrinogenaemia in trauma: A multicentre observational study. Critical Care, 18, R52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagihara K, Nishikawa T, Isobe T, Song J, Sugamata Y, & Yoshizaki K (2004). IL-6 plays a critical role in the synergistic induction of human serum amyloid A (SAA) gene when stimulated with proinflammatory cytokines as analyzed with an SAA isoform real-time quantitative RTPCR assay system. Biochemical and Biophysical Research Communications, 314, 363–369. [DOI] [PubMed] [Google Scholar]
- Hatanaka E, Furlaneto CJ, Ribeiro FP, Souza GM, & Campa A (2004). Serum amyloid A-induced mRNA expression and release of tumor necrosis factor-alpha (TNF-α) in human neutrophils. Immunology Letters, 91, 33–37. [DOI] [PubMed] [Google Scholar]
- Hayakawa M (2017). Dynamics of fibrinogen in acute phases of trauma. Journal of Intensive Care, 5, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He R, Shepard LW, Chen J, Pan ZK, & Ye RD (2006). Serum amyloid A is an endogenous ligand that differentially induces IL-12 and IL-23. Journal of Immunology, 177, 4072–4079. [DOI] [PubMed] [Google Scholar]
- Howe AS, & Boden BP (2007). Heat-related illness in athletes. American Journal of Sports Medicine, 35, 1384–1395. [DOI] [PubMed] [Google Scholar]
- Jain S, Gautam V, & Naseem S (2011). Acute-phase proteins: As diagnostic tool. Journal of Pharmacy & Bioallied Sciences, 3, 118–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jilma B, & Derhaschnig U (2012). Disseminated intravascular coagulation in heat stroke: A hot topic. Critical Care Medicine, 40, 1370–1372. [DOI] [PubMed] [Google Scholar]
- Junglee NA, Di Felice U, Dolci A, Fortes MB, Jibani MM, Lemmey AB, … Macdonald JH (2013). Exercising in a hot environment with muscle damage: Effects on acute kidney injury biomarkers and kidney function. American Journal of Physiology-Renal Physiology, 305, F813–F820. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne WJ, Cuthill IC, Emerson M, & Altman DG (2010). Improving bioscience research reporting: The ARRIVE guidelines for reporting animal research. PLoS Biology, 8, e1000412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King MA, Leon LR, Morse DA, & Clanton TL (2017). Unique cytokine and chemokine responses to exertional heat stroke in mice. Journal of Applied Physiology, 122, 296–306. [DOI] [PubMed] [Google Scholar]
- King MA, Leon LR, Mustico DL, Haines JM, & Clanton TL (2015). Biomarkers of multiorgan injury in a preclinical model of exertional heat stroke. Journal of Applied Physiology, 118, 1207–1220. [DOI] [PubMed] [Google Scholar]
- Laitano O, Garcia CK, Mattingly AJ, Robinson GP, Murray KO, King MA, … Clanton TL (2020). Delayed metabolic dysfunction in myocardium following exertional heat stroke in mice. The Journal of Physiology, 598, 967–985. [DOI] [PubMed] [Google Scholar]
- Laitano O, Leon LR, Roberts WO, & Sawka MN (2019). Controversies in exertional heat stroke diagnosis, prevention, and treatment. Journal of Applied Physiology, 127, 1338–1348. [DOI] [PubMed] [Google Scholar]
- Laitano O, Van Steenbergen D, Mattingly AJ, Garcia CK, Robinson GP, Murray KO, … Nunamaker EA (2018). Xiphoid surface temperature predicts mortality in a murine model of septic shock. Shock, 50, 226–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langhans C, Weber-Cartens S, Schmidt S, Hamati J, Kny M, Zhu X, & Fielitz J (2014). Inflammation-induced acute phase response in skeletal muscle and critical illness myopathy. PLoS ONE, e92048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavie M, Voisset C, Vu-Dac N, Zurawski V, Duverlie G, Wychowski C, & Dubuisson J (2006). Serum amyloid A has antiviral activity against hepatitis C virus by inhibiting virus entry in a cell culture system. Hepatology, 44, 1626–1634. [DOI] [PubMed] [Google Scholar]
- Lee M-S, Yoo S-A, Cho C-S, Suh P-G, Kim W-U, & Ryu SH (2006). Serum amyloid A binding to formyl peptide receptor-like1 induces synovial hyperplasia and angiogenesis. Journal of Immunology, 177, 5585–5594. [DOI] [PubMed] [Google Scholar]
- Leon LR, & Bouchama A (2015). Heat stroke. Comprehensive Physiology, 5, 611–647. [DOI] [PubMed] [Google Scholar]
- Leon LR, Dineen S, Blaha MD, Rodriguez-Fernandez M, & Clarke DC (2013). Attenuated thermoregulatory, metabolic, and liver acute phase protein response to heat stroke in TNF receptor knockout mice. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology, 305, R1421–R1432. [DOI] [PubMed] [Google Scholar]
- Linke RP, Bock V, Valet G, & Rothe G (1991). Inhibition of the oxidative burst response of N-formyl peptide-stimulated neutrophils by serum amyloid-A protein. Biochemical and Biophysical Research Communications, 176, 1100–1105. [DOI] [PubMed] [Google Scholar]
- Linke RP, Meinel A, Chalcroft JP, & Urieli-Shoval S (2017). Serum amyloid A (SAA) treatment enhances the recovery of aggravated poly-microbial sepsis in mice, whereas blocking SAA’s invariant peptide results in early death. Amyloid, 24, 149–150. [DOI] [PubMed] [Google Scholar]
- Merly F, Lescaudron L, Rouaud T, Crossin F, & Gardahaut MF (1999). Macrophages enhance muscle satellite cell proliferation and delay their differentiation. Muscle & Nerve, 22, 724–732. [DOI] [PubMed] [Google Scholar]
- Migita K, Kawabe Y, Tominaga M, Origuchi T, Aoyagi T, & Eguchi K (1998). Serum amyloid A protein induces production of matrix metalloproteinases by human synovial fibroblasts. Laboratory Investigation, 78, 535–539. [PubMed] [Google Scholar]
- Monroe DM (2016). Adding some muscle to blood coagulation. Blood, 128, 1786–1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel H, Fellowes R, Coade S, & Woo P (1998). Human serum amyloid A has cytokine-like properties. Scandinavian Journal of Immunology, 48, 410–418. [DOI] [PubMed] [Google Scholar]
- Pedersen BK, & Febbraio MA (2008). Muscle as an endocrine organ: Focus on muscle-derived interleukin-6. Physiological Reviews, 88, 1379–1406. [DOI] [PubMed] [Google Scholar]
- Peyvandi F, Haertel S, Knaub S, & Mannucci PM (2006). Incidence of bleeding symptoms in 100 patients with inherited afibrinogenemia or hypofibrinogenemia. Journal of Thrombosis and Haemostasis, 4, 1634–1637. [DOI] [PubMed] [Google Scholar]
- Phillips NA, Welc SS, Wallet SM, King MA, & Clanton TL (2015). Protection of intestinal injury during heat stroke in mice by interleukin-6 pretreatment. The Journal of Physiology, 593, 739–752; discussion 753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proctor EA, Dineen SM, Nostrand SCV, Kuhn MK, Barrett CD, Brubaker DK, … Leon LR (2019). Coagulopathy signature precedes and predicts severity of end-organ heat stroke pathology in a mouse model. bioRxiv, 771410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Q-R, & Yang Z-M (2014). Regulation and function of signal transducer and activator of transcription3. World Journal of Biological Chemistry, 5, 231–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray A (2000). A SAF binding site in the promoter region of human γ-fibrinogen gene functions as an IL-6 response element. Journal of Immunology, 165, 3411–3417. [DOI] [PubMed] [Google Scholar]
- Sack GH (2018). Serum amyloid A – a review. Molecular Medicine, 24, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah C, Hari-Dass R, & Raynes JG (2006). Serum amyloid A is an innate immune opsonin for Gram-negative bacteria. Blood, 108, 1751–1757. [DOI] [PubMed] [Google Scholar]
- Starr ME, Steele AM, Saito M, Hacker BJ, Evers BM, & Saito H (2014). A new cecal slurry preparation protocol with improved long-term reproducibility for animal models of sepsis. PLoS ONE, 9, e115705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steensberg A, Febbraio MA, Osada T, Schjerling P, vanHall G, Saltin B, & Pedersen BK (2001). Interleukin-6 production in contracting human skeletal muscle is influenced by pre-exercise muscle glycogen content. The Journal of Physiology, 537, 633–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sultan S, Pascucci M, Ahmad S, Malik IA, Bianchi A, Ramadori P, … Ramadori G (2012). LIPOCALIN-2 is a major acute-phase protein in a rat and mouse model of sterile abscess. Shock, 37, 191–196. [DOI] [PubMed] [Google Scholar]
- Sun L, Zhou H, Zhu Z, Yan Q, Wang L, Liang Q, & Ye RD (2015). Ex vivo and in vitro effect of serum amyloid a in the induction of macrophage M2 markers and efferocytosis of apoptotic neutrophils. Journal of Immunology, 194, 4891–4900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tidball JG, & Wehling-Henricks M (2007). Macrophages promote muscle membrane repair and muscle fibre growth and regeneration during modified muscle loading in mice in vivo. The Journal of Physiology, 578, 327–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace RF, Kriebel D, Punnett L, Wegman DH, & Amoroso PJ (2007). Prior heat illness hospitalization and risk of early death. Environmental Research, 104, 290–295. [DOI] [PubMed] [Google Scholar]
- Wang J-C, Chien W-C, Chu P, Chung C-H, Lin C-Y, & Tsai S-H (2019). The association between heat stroke and subsequent cardiovascular diseases. PLoS ONE, 14, e0211386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisel JW, & Litvinov RI (2017). Fibrin formation, structure and properties. Sub-Cellular Biochemistry, 82, 405–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welc SS, Clanton TL, Dineen SM, & Leon LR (2013). Heat stroke activates a stress-induced cytokine response in skeletal muscle. Journal of Applied Physiology, 115, 1126–1137. [DOI] [PubMed] [Google Scholar]
- Welc SS, Morse DA, Mattingly AJ, Laitano O, King MA, & Clanton TL (2016). The impact of hyperthermia on receptor-mediated interleukin-6 regulation in mouse skeletal muscle. PLoS ONE, 11, e0148927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welc SS, Phillips NA, Oca-Cossio J, Wallet SM, Chen DL, & Clanton TL (2012). Hyperthermia increases interleukin-6 in mouse skeletal muscle. American Journal of Physiology-Cell Physiology, 303, C466–C466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang R-Z, Lee M-J, Hu H, Pollin TI, Ryan AS, Nicklas BJ, … Gong D-W (2006). Acute-phase serum amyloid A: An inflammatory adipokine and potential link between obesity and its metabolic complications. PLoS Medicine, 3, e287. [DOI] [PMC free article] [PubMed] [Google Scholar]