

Abstract
Fundamental organelles that occur in every cell type with the exception of mammal erythrocytes, the mitochondria are required for multiple pivotal processes that include the production of biological energy, the biosynthesis of reactive oxygen species, the control of calcium homeostasis, and the triggering of cell death. The disruption of anyone of these processes has been shown to impact strongly the function of all cells, but especially of neurons. In this review, we discuss the role of the mitochondria impairment in the development of the neurodegenerative diseases Amyotrophic Lateral Sclerosis, Parkinson’s disease and Alzheimer’s disease. We highlight how mitochondria disruption revolves around the processes that underlie the mitochondria’s life cycle: fusion, fission, production of reactive oxygen species and energy failure. Both genetic and sporadic forms of neurodegenerative diseases are unavoidably accompanied with and often caused by the dysfunction in one or more of the key mitochondrial processes. Therefore, in order to get in depth insights into their health status in neurodegenerative diseases, we need to focus into innovative strategies aimed at characterizing the various mitochondrial processes. Current techniques include Mitostress, Mitotracker, transmission electron microscopy, oxidative stress assays along with expression measurement of the proteins that maintain the mitochondrial health. We will also discuss a panel of approaches aimed at mitigating the mitochondrial dysfunction. These include canonical drugs, natural compounds, supplements, lifestyle interventions and innovative approaches as mitochondria transplantation and gene therapy. In conclusion, because mitochondria are fundamental organelles necessary for virtually all the cell functions and are severely impaired in neurodegenerative diseases, it is critical to develop novel methods to measure the mitochondrial state, and novel therapeutic strategies aimed at improving their health.
Key Words: Alzheimer's disease, amyotrophic lateral sclerosis, mitochondria, mitochondria modulation, mitochondrial dysfunction, mitochondrial health, Mitostress, Mitotracker, neurodegenerative disease, Parkinson's disease
Introduction
Neurodegenerative diseases are an heterogeneous class of incurable and debilitating disorders characterized by the progressive degeneration and death of central nervous system cells (Gao et al., 2017). Such diseases occur with multiple underlying mechanisms, and although some of them are specific for each disease, ultimately leading to the degeneration of a specific subclass of neurons, they share common degenerative features as mitochondrial dysfunction (Gao et al., 2017). Mitochondria are cellular organelles required for multiple processes such as production of biological energy and of reactive oxygen species, control of calcium homeostasis, and cell death. It is easy to imagine how disrupting any of these processes strongly impacts neuronal functions, as neurons are polarized cells that require an incredibly high amount of energy, and must therefore rely on calcium availability for synaptic transmission and multiple other functions. This makes it clear that any alteration in the above processes leads to numerous abnormalities common to a wide range of neurodegenerative diseases.
Search Strategy and Selection Criteria
This narrative review work reports recent advances in the field of mitochondrial degeneration in neurodegenerative diseases. We performed a bibliographic reserch in PubMed (1990–2020) focusing on the most relevant review articles in the field, along with pre-clinical and clinical experimental evidence, in order to provide the reader with a clear and comprehensive analysis of the topic.
Mitochondrial Dysfunctions in Neurodegenerative Diseases: Current Understanding of Common and Divergent Mechanisms
The mitochondria life cycle encompasses four stages: biogenesis, fusion, fission and degradation (Gao et al., 2017) (Figure 1). Biogenesis, which is triggered in response to increased cell energy needs, requires the co-expression of both nuclear and mitochondrial DNA, along with the action of transcription factor A (TFAM), nuclear respiratory factors 1 and 2 (NRF1 and NRF2), the PPARγ coactivator-1 family of transcription coactivators, sirtuins and AMPK (Wang et al., 2019). After synthesis, mitochondria may undergo fusion and/or fission in a delicate balance that responds to cell energy need fluctuations (Burté et al., 2015). During the fusion process, which takes place under stressing conditions, two or more mitochondria mix their contents thereby preventing permanent loss of essential components. Specifically, Optic atrophy 1 (OPA1), Mitofusin 1 and 2 (MFN1 and MFN2) coordinate this process through modulation of outer and inner membrane fusion of adjacent mitochondria (Burté et al., 2015). By contrast, fission takes place when mitochondria divide through a process that needs dynamin-related protein 1 (DRP1), whose recruitment from the cytosol enables constriction of mitochondrial tubules (Burté et al., 2015). Dysfunctional mitochondria are finally removed through an autophagic process called mitophagy, in which impaired mitochondria are selected for fragmentation followed by degradation in lysosomes (Wang et al., 2019). A summary of the mechanisms altered in neurodegenerative diseases is depicted in Figure 1.
Figure 1.
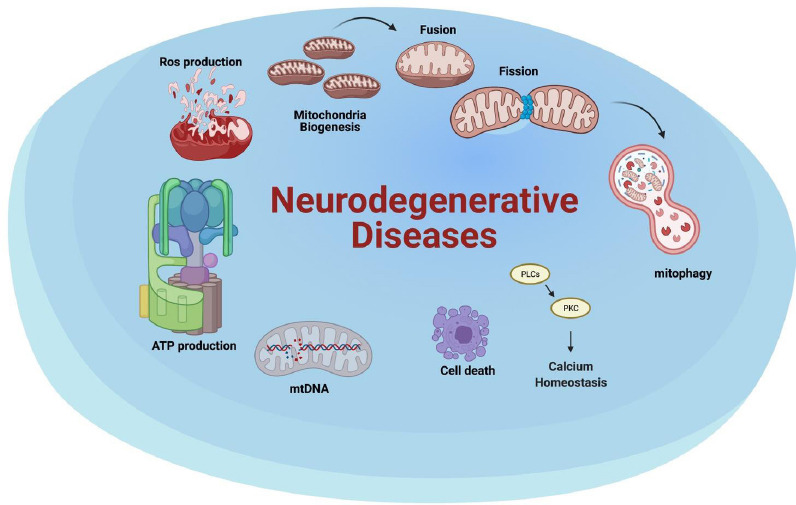
Neurodegenerative diseases impact mitochondrial health at different stages.
The altered processes may affect the mitochondria life cycle (biogenesis, fusion, fission and mitophagy), calcium homeostasis, cell death, mtDNA, ATP production and ROS production. ATP: Adenosine triphosphate; mtDNA: mitochondrial DNA; ROS: reactive oxygen species. Created with BioRender.com.
The most common neurodegenerative diseases include Parkinson’s disease (PD), amyotrophic lateral sclerosis (ALS), and Alzheimer’s disease (AD), all of which are strongly influenced by alterations in mitochondria function. Here, we will provide a brief overview of recent advances in these dysfunctions, which have been extensively addressed in multiple studies (Burté et al., 2015; Gao et al., 2017; Wang et al., 2019; Kodavati et al., 2020; Monzio Compagnoni et al., 2020). In PD, the mitochondria dysfunction, undisputed responsible for the disease insurgence, can be mediated by either environmental factors or gene mutations (Monzio Compagnoni et al., 2020). One of the most relevant proof of the involvement of mitochondria in PD, self-intravenous injection by drug users of 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine, an inhibitor of mitochondrial chain complex I, leads to the development of parkinsonism (Langston et al., 1983). Numerous studies revealed impaired function of mitochondria complex I in PD patients Substantia Nigra, skeletal muscles, platelets and leukocytes (Monzio Compagnoni et al., 2020). The implication of mitochondria in PD development has been further reinforced by the discovery that mutations in PINK1 and Parkin, two proteins involved in mitochondria dynamics, lead to familial forms of PD (Hattori et al., 1998; Valente et al., 2004; Monzio Compagnoni et al., 2020). Specifically, the activation of PINK1, responsible for mitochondria health monitoring and damage detection, enables the recruitment of Parkin, which conjugates ubiquitin onto outer mitochondrial membrane proteins, thereby leading to mitophagy (Valente et al., 2004; Monzio Compagnoni et al., 2020). Other PD distinctive factors involved in mitochondria processes as transport, fusion-fission, and clearance are α-synuclein, which leads to complex I deficiency and interacts with the mitochondrial protein import machinery, DJ-1, which prevents neuronal death induced by oxidative stress, and leucine-rich repeat kinase 2 (LRRK2), which potentiates the pro-fission activity of DRP1 (Burté et al., 2015; Wu et al., 2019; Kodavati et al., 2020; Monzio Compagnoni et al., 2020). Controversial findings on PD and mitochondria concern the alterations in mitochondrial DNA (mtDNA): while some studies indicate increased susceptibility due to more frequent mtDNA mutations, others do not show such correlation (Lin et al., 2012; Coxhead et al., 2016; Wei et al., 2017; Monzio Compagnoni et al., 2020).
The oxidative stress is one of the driving causes of motor neuron degeneration underlying ALS. The mitophagy process is markedly involved in ALS pathogenesis, with reduced number of phagosomes at the neuromuscular junction, increased activity of adenosine triphosphate (ATP) synthase, and augmented number of mitochondria (Wang et al., 2019). Numerous mitochondria-associated proteins have been linked to both familial and sporadic ALS, including FUS, TDP-43, SOD1, and C9ORF72 (Kim et al., 2020). The first to be discovered, the SOD1 gene, currently enlists more than 170 gene mutations responsible for ALS (Deng et al., 1993; Gao et al., 2017; Kodavati et al., 2020). SOD1 encodes for Cu-Zn superoxide dismutase, which catalyses the conversion of free superoxide radicals to molecular oxygen and hydrogen peroxide (Deng et al., 1993; Kodavati et al., 2020). In ALS patients, this protein aggregates in both the cytosolic and nuclear compartments (Bordoni et al., 2019), losing the capacity of protecting DNA from damage (Bordoni et al., 2019) and influencing mitochondrial fusion and fission, thereby modulating fragmentation of the mitochondrial network (Kodavati et al., 2020). Besides negatively affecting mitochondria transport and calcium homeostasis, SOD1 mutants reduce the activity of respiratory chain complexes II and IV (Kodavati et al., 2020). Together with TDP-43 and FUS, SOD1 contributes to mitochondria morphology fragmentation, impaired endoplasmic reticulum-mitochondria contacts, defective mitochondria transport and alterations in calcium exchange (Kodavati et al., 2020). TDP-43, that localizes and is overexpressed in the mitochondria in response to stress stimuli, has the double function activating the unfolded protein response, thereby leading to mitochondria degradation, and influencing calcium homeostasis, thereby impairing endoplasmic reticulum-mitochondria association (Kodavati et al., 2020). Mutations in FUS augment both mitochondrial fragmentation with loss of membrane potential, and reactive oxygen species (ROS) production with defective mitochondrial axonal transport (Kodavati et al., 2020). Lastly, the repeat expansion in C9ORF72, the major genetic cause of ALS worldwide, results in poly-GR dipeptides that bind mitochondria ribosomal proteins compromising its function and increasing the oxidative stress (Kodavati et al., 2020). Moreover, this mutation increases mtDNA and mitochondrial mass, with structural abnormalities such as swollen mitochondria (Kodavati et al., 2020).
Several mechanisms related to mitochondrial dysfunction are known to underlie the development of neurodegeneration in AD, including impaired glucose and oxygen metabolism in the brain, mtDNA alterations, Ab and tau accumulation, altered mitochondria morphology, and respiratory chain dysfunction. One of the current hypotheses, based on the observations that AD brains display reduced mitochondria metabolism, impaired activity of pyruvate dehydrogenase, ketoglutarate dehydrogenase and cytochrome oxidase, impaired oxygen consumption and altered morphology, implies that mitochondrial dysfunction is the initial trigger for AD (Swerdlow et al., 2010; Swerdlow, 2018; Monzio Compagnoni et al., 2020). Nevertheless, it is still not yet clear whether mitochondrial dysfunction represents a primary cause for AD or if it is rather a secondary consequence of amyloidogenic pathology originated from tau accumulation (Hardy and Selkoe, 2002; Monzio Compagnoni et al., 2020).
In the neurodegenerative diseases so far identified, mitochondria dysfunctions, although primarily involved, are part of a complex network of degenerating processes that ultimately lead to neuronal death. It is worth mentioning that a panel of human disorders is linked to mitochondrial dynamics alterations that are mainly caused by autosomal mitochondrial genes mutations (Burté et al., 2015). The main implied genes include MFN2, involved in Charcot-Marie-Tooth disease and sensory neuropathy, OPA1 and Optic atrophy 3 (OPA3), implicated in optic atrophy, DRP1, implicated in severe infantile neurodegenerative diseases, and ganglioside-induced differentiation associated protein 1 (GDAP1), also implicated in Charcot-Marie-Tooth disease (Burté et al., 2015). Among the mechanisms that lead to mitochondria dysfunction, mtDNA point mutations have a prominent role because they target proteins related to mitochondrial tRNAs, rRNAs, and perhaps mtDNA (Szczepanowska et al., 2012). Such mutations are involved in the development of MELAS (Mitochondrial Encephalopathy, Lactic Acidosis, and Stroke-like episodes), MERRF (Myoclonic Epilepsy and Ragged Red Fibres), LHON (Leber’s Hereditary Optic Neuropathy) and NARP (Neuropathy, Ataxia, and Retinitis Pigmentosa), extensively reviewed by Szczepanowska and colleagues (Szczepanowska et al., 2012). The most common mtDNA mutation in these diseases is the A3243G point mutation in the tRNALeu gene, which accounts for 80–90% of patients with MELAS. In general, mtDNA point mutations affect mitochondrial physiology by altering the mitochondrial membrane potential, ATP level, calcium homeostasis, ROS production, mitochondria morphology and dynamics (Szczepanowska et al., 2012).
Approaches to Investigate Mitochondrial Health and Metabolism
Given that mitochondria dysfunctions are widely implicated in several neurodegenerative diseases, there is a growing need to investigate mitochondria functions through development of innovative strategies as known techniques often imply several disadvantages along with important plus points (Figure 2). At present, mitochondrial activity can be successfully evaluated in real time using Seahorse XF analyzer and Mitotracker assays.
Figure 2.
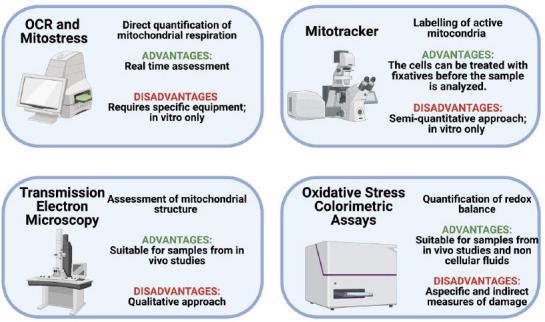
Techniques for mitochondria analyses.
Innovative techniques are needed in order to assess degeneration status and potential repair strategies. The figure reports the main techniques used with the respective advantages and disadvantages. OCR: Oxygen consumption rate. Created with BioRender.com.
With a functionality focused approach, the Seahorse XF analyzer can be used for the real time estimation of O2 consumption rate in cell models in vitro. As the mitochondrion represents the main consumer of O2 in the cell, O2 consumption rate is one of the main available measures to monitor mitochondrial activity in real time. With the same analyzer, a Mitostress assay can give still more detailed information on the metabolic state of the cells. It consists of the sequential injection of the following modulators of the mitochondrial respiratory chain: oligomycin, an inhibitor of ATP synthase and ATP linked mitochondrial respiration, carbonyl cyanide-4-(trifluoromethoxy) phenylhydrazone, an uncoupler of mitochondrial respiration, and rotenone/antimycin A, inhibitors of complex I and complex III, respectively, that cause a complete block of mitochondrial function. This approach allows the quantification of basal respiration, ATP-linked respiration, proton leak, maximal respiratory capacity and non-mitochondrial respiration (Chacko et al., 2014). With this method, 1-methyl-4-phenylpyridinium iodide (MPP+), the active form of 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine which is usually used to reproduce in vitro the oxidative damage in models of PD, was observed to exert mitochondrial toxicity in SH-SY5Y cells, by targeting complex I (Giordano et al., 2012). The main limitation of this technique is that it is confined for in vitro analysis and requires specific equipment (Giordano et al., 2012; Chacko et al., 2014).
The Mitotracker staining is one of the most useful and practical methods to assess mitochondrial damage in vitro with a colorimetric approach (Cottet-Rousselle et al., 2011; Rey et al., 2021). This technique is based upon the binding of a fluorescent dye to living cells mitochondria and measurement by confocal microscopy of the degree of staining which is thus proportional to the mitochondrial membrane potential. In this case, cells are treated with fixatives before analysis. As for Seahorse XF analyzer, Mitotracker staining analysis can be performed semi-quantitatively only in vitro (Rey et al., 2021). Transmission electronic microscopy enables qualitative mitochondria morphology analysis, but with the ability to monitor the deformation in mitochondrial crista and membranes, for example when mitochondria are exposed to MPP+ (Rey et al., 2021).
The here mentioned direct mitochondrial assays can be integrated with the assessment of the redox balance, which can be performed in both cell models and blood samples. The oxidative stress can be assessed by several methods including the thiobarbituric acid reactive substances and the ferric reducing ability of plasma to demonstrate lipid peroxidation and consumption of antioxidant barrier (Medeiros et al., 2016). The main limitation of these techniques is the non-specificity of the measures that therefore reflect an indirect evaluation (Medeiros et al., 2016). In addition, canonical approaches such as real-time polymerase chain reaction and western blot analysis enable assessing the expression of genes and proteins that control mitochondria health (Giordano et al., 2012; Rey et al., 2021).
Therapeutic Advances in Modulating Mitochondria
Therapeutic interventions aimed at modulating mitochondria dynamics are heterogeneous. They include canonical drugs as well as novel gene therapies, which have been revised in numerous studies that are summarized here. Up to 171 studies that include biomarker studies, genetic analyses and therapeutic interventions, appear in the NIH Clinical Trials Database when searching for “mitochondria” and “neurodegenerative diseases” (Medicine USNLo, 2020). The number of canonical drugs that target mitochondria in neurodegenerative diseases is remarkably high: Olanzapine, Mitoquinone, Melatonin, Riluzole, Dichloroacetate, Z-LEHD-FMK, N-acetylcysteine, Clozapine, Z-FA-MK, Olesoxime, Cholest-4-en-3-one and erythropoietin (Wu et al., 2019; Rey et al., 2021). By reducing the oxidative stress and improving the overall mitochondria health, such drugs have a general neuroprotective effect. Specifically, they exert anti-oxidant protection in AD, ALS, PD, and neuropathies, as well as neuroprotective activity in Huntington disease, schizophrenia, multiple sclerosis with improved motor symptoms in Huntington Disease (Wu et al., 2019). Among the drugs cited above, erythropoietin has been widely investigated for its protective effects in neurodegenerative diseases through the regulation of oxygen balance, mitochondrial dynamics, and glycolysis (Rey et al., 2019, 2021).
A crucial deregulation to which mitochondrial dysfunction strongly contributes in neurodegenerative diseases is represented by altered brain energetics, which is the target of pharmacological strategies aimed at modulating this process (Cunnane et al., 2020). Examples are CP2, a proprietary tricyclic pyrone that improves mitochondrial bioenergetics, the antioxidant MitoQ that reduces oxidative stress in mitochondria, and resveratrol that stimulates mitochondrial biogenesis (Cunnane et al., 2020). Noticeably, some of these studies are moving to the clinic, as that involving a selective oestrogen receptor-β agonist that improves mitochondrial function by selective partial inhibition of complex I (Cunnane et al., 2020). The redox state modulation may represent an alternative strategy for therapy, as for example the use of the NAD+ precursor nicotinamide riboside that mitigates cognitive impairment, synaptic degeneration and neuronal death, as well as that of pyruvate that could improve brain energetics by stimulating pyruvate dehydrogenase (Cunnane et al., 2020). Other molecules have been developed to counteract the dysfunctions found in neurodegenerative diseases targeting glycolysis and the TCA cycle (Cunnane et al., 2020; Rey et al., 2021). The nutrients vitamins E and C, α-Lipoic acid, resveratrol and other flavonoids exert protective effect on mitochondria wellbeing (Wu et al., 2019; Cunnane et al., 2020). Mitochondria biogenesis can also be enhanced in order to treat neurodegenerative diseases (Wang et al., 2019). Examples include rosiglitazone and bezafibrate that activate the PPAR-PGC-1α axis. Specifically, rosiglitazone attenuates mitochondrial dysfunction by increasing the mitochondrial mass, whereas bezafibrate increases the mitochondrial ATP generating capacity by augmenting mitochondrial proteins (Wang et al., 2019). Moreover, the SIRT1 agonists quercetin and resveratrol activate sirtuins and AMPK that stimulate mitochondria biogenesis (Wang et al., 2019).
By acting through similar mechanisms, numerous natural drugs have a beneficial effects on mitochondrial modulation, including the plants curcuma longa, Smallanthus sonchifolius, Ginkgo biloba, Camellia sinensis, Ginseng, Glycyrrhiza, Herpestis monniera, Withania somnifera, Centella asiatica, Celastrus regelii, Trehalose, Lycopene, Sesamum indicum (Wu et al., 2019). These natural extracts typically possess strong antioxidant and anti-inflammatory properties that counteract key several neurodegenerative diseases (Cunnane et al., 2020). Among the plant extracts, the Ginkgo biloba is one of the most studied natural remedy for AD and PD, while curcuma longa has amyloid disaggregating properties in AD, modulating the proteotoxic formation of aggregates with are primary cause of neurodegeneration (Wu et al., 2019).
Today, novel genome editing technologies are paving the road for gene therapy in numerous diseases. Hopefully, such technologies will be applied to genes involved in mitochondrial dynamics. Advances are currently being made in neurodegeneration associated to visual failure, with intravitreal injection of adeno-associated virus vector that harbors the correct form of the OPA1 gene (Burté et al., 2015). A further innovative approach to treat mitochondrial dysfunctions concerns mitochondria transplantation, a strategy successfully used for cardiac ischemia that is proving to be effective in neurodegenerative diseases also (Chang et al., 2019). Mitochondrial delivery has been tested in multiple PD models, resulting in rescued mitochondrial respiratory function, improved cell viability and relieve of mitochondrial oxidative stress (Chang et al., 2019). Moreover, this strategy has the potentiality to modulate the proteins involved in mitochondrial fusion and fission, such as Mfn2, OPA1 and Drp1 (Chang et al., 2019). Mitochondrial fission may be modulated through gene therapy and canonical drugs. Indeed, recombinant adeno-associated virus expressing the dominant negative Drp1 mutant or Mdivi-1 and a small molecular inhibitor of Drp1, can modulate mitochondrial fragmentation and improve overall health in PD animal models (Gao et al., 2017).
Other approaches addressing the correction of mitochondrial dysfunction concern exercise and caloric restriction (Wang et al., 2019). Interestingly, exercise is beneficial to delay neurodegenerative diseases progression because it increases the mRNA expression of TFAM and Ndufa6, subunits of mitochondrial complex I (Wang et al., 2019). Moreover, exercise improves mtDNA repair capacity, activates autophagy and modulates mitochondrial proliferation and ROS production, ultimately reducing the cell oxidative stress (Wang et al., 2019). Another alternative approach concerns caloric restriction, which decreases ROS production and oxidative DNA damage, probably through activation of sirtuins. By reducing excitotoxicity, this approach increases the number of mitochondria cristae and of mitochondria themselves (Wang et al., 2019). A summary of all recent therapeutic advances is reported in Figure 3.
Figure 3.
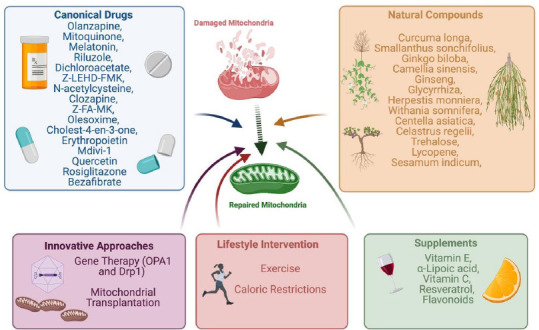
Summary of therapeutic strategies to protect mitochondria health.
These include canonical drugs, nutrients, herbal extracts and alternative approaches as gene therapy, exercise, caloric restrictions and mitochondria transplantation. Drp-1: Dynamin related protein-1; OPA1: optic athropy 1 gene. Created with BioRender.com.
Conclusions
Mitochondrial degeneration is a crucial process strongly impaired in neurodegenerative diseases. Indeed, this organelle is involved in multiple processes fundamental for homeostasis maintenance, such as energy production, ROS production, apoptosis, calcium signalling, and mitochondrial dynamics. There is thus the need to develop innovative strategies for the study of this organelle, which to this day include both morphological studies and functional assessments of mitochondria functionality. Moreover, there is an incredibly high number of drugs aimed at targeting this organelle, both conventional and natural extracts. Nevertheless, further advances are needed in order to develop innovative therapeutic strategies as for example gene therapy approaches.
Additional file:
Additional file: Open peer review reports 1 (84.8KB, pdf) and 2 (85.1KB, pdf) .
Footnotes
P-Reviewers: Correia SC, Kumar KR; C-Editors: Zhao M, Liu WJ, Qiu Y; T-Editor: Jia Y
Conflicts of interest: The authors declare no conflicts of interest.
Financial support: None.
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open peer reviewers: Sónia C. Correia, Center for Neuroscience and Cell Biology, Portugal; Kishore Raj Kumar, Garvan Institute of Medical Research, Australia.
References
- 1.Bordoni M, Pansarasa O, Dell’Orco M, Crippa V, Gagliardi S, Sproviero D, Bernuzzi S, Diamanti L, Ceroni M, Tedeschi G, Poletti A, Cereda C. Nuclear phospho-SOD1 protects DNA from oxidative stress damage in amyotrophic lateral sclerosis. J Clin Med. 2019;8:729. doi: 10.3390/jcm8050729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Burté F, Carelli V, Chinnery PF, Yu-Wai-Man P. Disturbed mitochondrial dynamics and neurodegenerative disorders. Nat Rev Neurol. 2015;11:11–24. doi: 10.1038/nrneurol.2014.228. [DOI] [PubMed] [Google Scholar]
- 3.Chacko BK, Kramer PA, Ravi S, Benavides GA, Mitchell T, Dranka BP, Ferrick D, Singal AK, Ballinger SW, Bailey SM, Hardy RW, Zhang J, Zhi D, Darley-Usmar VM. The Bioenergetic Health Index: a new concept in mitochondrial translational research. Clin Sci (Lond) 2014;127:367–373. doi: 10.1042/CS20140101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chang CY, Liang MZ, Chen L. Current progress of mitochondrial transplantation that promotes neuronal regeneration. Transl Neurodegener. 2019;8:17. doi: 10.1186/s40035-019-0158-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cottet-Rousselle C, Ronot X, Leverve X, Mayol JF. Cytometric assessment of mitochondria using fluorescent probes. Cytometry A. 2011;79:405–425. doi: 10.1002/cyto.a.21061. [DOI] [PubMed] [Google Scholar]
- 6.Coxhead J, Kurzawa-Akanbi M, Hussain R, Pyle A, Chinnery P, Hudson G. Somatic mtDNA variation is an important component of Parkinson’s disease. Neurobiol Aging. 2016;38:217.e211–217. doi: 10.1016/j.neurobiolaging.2015.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cunnane SC, Trushina E, Morland C, Prigione A, Casadesus G, Andrews ZB, Beal MF, Bergersen LH, Brinton RD, de la Monte S, Eckert A, Harvey J, Jeggo R, Jhamandas JH, Kann O, la Cour CM, Martin WF, Mithieux G, Moreira PI, Murphy MP, et al. Brain energy rescue: an emerging therapeutic concept for neurodegenerative disorders of ageing. Nat Rev Drug Discov. 2020;19:609–633. doi: 10.1038/s41573-020-0072-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deng HX, Hentati A, Tainer JA, Iqbal Z, Cayabyab A, Hung WY, Getzoff ED, Hu P, Herzfeldt B, Roos RP. Amyotrophic lateral sclerosis and structural defects in Cu, Zn superoxide dismutase. Science. 1993;261:1047–1051. doi: 10.1126/science.8351519. [DOI] [PubMed] [Google Scholar]
- 9.Gao J, Wang L, Liu J, Xie F, Su B, Wang X. Abnormalities of mitochondrial dynamics in neurodegenerative diseases. Antioxidants (Basel) 2017;6:25. doi: 10.3390/antiox6020025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Giordano S, Lee J, Darley-Usmar VM, Zhang J. Distinct effects of rotenone, 1-methyl-4-phenylpyridinium and 6-hydroxydopamine on cellular bioenergetics and cell death. PLoS One. 2012;7:e44610. doi: 10.1371/journal.pone.0044610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 12.Hattori N, Kitada T, Matsumine H, Asakawa S, Yamamura Y, Yoshino H, Kobayashi T, Yokochi M, Wang M, Yoritaka A, Kondo T, Kuzuhara S, Nakamura S, Shimizu N, Mizuno Y. Molecular genetic analysis of a novel Parkin gene in Japanese families with autosomal recessive juvenile parkinsonism: evidence for variable homozygous deletions in the Parkin gene in affected individuals. Ann Neurol. 1998;44:935–941. doi: 10.1002/ana.410440612. [DOI] [PubMed] [Google Scholar]
- 13.Kim G, Gautier O, Tassoni-Tsuchida E, Ma XR, Gitler AD. ALS genetics: gains, losses, and implications for future therapies. Neuron. 2020;108:822–842. doi: 10.1016/j.neuron.2020.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kodavati M, Wang H, Hegde ML. Altered mitochondrial dynamics in motor neuron disease: an emerging perspective. Cells. 2020;9:1065. doi: 10.3390/cells9041065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Langston JW, Ballard P, Tetrud JW, Irwin I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science. 1983;219:979–980. doi: 10.1126/science.6823561. [DOI] [PubMed] [Google Scholar]
- 16.Lin MT, Cantuti-Castelvetri I, Zheng K, Jackson KE, Tan YB, Arzberger T, Lees AJ, Betensky RA, Beal MF, Simon DK. Somatic mitochondrial DNA mutations in early Parkinson and incidental Lewy body disease. Ann Neurol. 2012;71:850–854. doi: 10.1002/ana.23568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Medeiros MS, Schumacher-Schuh A, Cardoso AM, Bochi GV, Baldissarelli J, Kegler A, Santana D, Chaves CM, Schetinger MR, Moresco RN, Rieder CR, Fighera MR. Iron and oxidative stress in Parkinson’s disease: an observational study of injury biomarkers. PLoS One. 2016;11:e0146129. doi: 10.1371/journal.pone.0146129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Medicine USNLo. ClinicalTrials.gov. 2020. [Accessed March 31, 2021]. https://clinicaltrials.gov/
- 19.Monzio Compagnoni G, Di Fonzo A, Corti S, Comi GP, Bresolin N, Masliah E. The role of mitochondria in neurodegenerative diseases: the lesson from Alzheimer’s disease and Parkinson’s disease. Mol Neurobiol. 2020;57:2959–2980. doi: 10.1007/s12035-020-01926-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rey F, Balsari A, Giallongo T, Ottolenghi S, Di Giulio AM, Samaja M, Carelli S. Erythropoietin as a neuroprotective molecule: an overview of its therapeutic potential in neurodegenerative diseases. ASN Neuro. 2019;11:1759091419871420. doi: 10.1177/1759091419871420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rey F, Ottolenghi S, Giallongo T, Balsari A, Martinelli C, Rey R, Allevi R, Giulio AMD, Zuccotti GV, Mazzucchelli S, Foresti R, Samaja M, Carelli S. Mitochondrial metabolism as target of the neuroprotective role of erythropoietin in Parkinson’s disease. Antioxidants (Basel) 2021;10:121. doi: 10.3390/antiox10010121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Swerdlow RH. Mitochondria and mitochondrial cascades in Alzheimer’s disease. J Alzheimers Dis. 2018;62:1403–1416. doi: 10.3233/JAD-170585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Swerdlow RH, Burns JM, Khan SM. The Alzheimer’s disease mitochondrial cascade hypothesis. J Alzheimers Dis 20 Suppl. 2010;2:S265–279. doi: 10.3233/JAD-2010-100339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Szczepanowska J, Malinska D, Wieckowski MR, Duszynski J. Effect of mtDNA point mutations on cellular bioenergetics. Biochim Biophys Acta. 2012;1817:1740–1746. doi: 10.1016/j.bbabio.2012.02.028. [DOI] [PubMed] [Google Scholar]
- 25.Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG, Albanese A, Nussbaum R, González-Maldonado R, Deller T, Salvi S, Cortelli P, Gilks WP, Latchman DS, Harvey RJ, Dallapiccola B, et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science. 2004;304:1158–1160. doi: 10.1126/science.1096284. [DOI] [PubMed] [Google Scholar]
- 26.Wang Y, Xu E, Musich PR, Lin F. Mitochondrial dysfunction in neurodegenerative diseases and the potential countermeasure. CNS Neurosci Ther. 2019;25:816–824. doi: 10.1111/cns.13116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wei W, Keogh MJ, Wilson I, Coxhead J, Ryan S, Rollinson S, Griffin H, Kurzawa-Akanbi M, Santibanez-Koref M, Talbot K, Turner MR, McKenzie CA, Troakes C, Attems J, Smith C, Al Sarraj S, Morris CM, Ansorge O, Pickering-Brown S, Ironside JW, et al. Mitochondrial DNA point mutations and relative copy number in 1363 disease and control human brains. Acta Neuropathol Commun. 2017;5:13. doi: 10.1186/s40478-016-0404-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu Y, Chen M, Jiang J. Mitochondrial dysfunction in neurodegenerative diseases and drug targets via apoptotic signaling. Mitochondrion. 2019;49:35–45. doi: 10.1016/j.mito.2019.07.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.