

Abstract
Mitochondria play a multidimensional role in the function and the vitality of the neurological system. From the generation of neural stem cells to the maintenance of neurons and their ultimate demise, mitochondria play a critical role in regulating our neural pathways’ homeostasis, a task that is critical to our cognitive health and neurological well-being. Mitochondria provide energy via oxidative phosphorylation for the neurotransmission and generation of an action potential along the neuron’s axon. This paper will first review and examine the molecular subtleties of the mitochondria’s role in neurogenesis and neuron vitality, as well as outlining the impact of defective mitochondria in neural aging. The authors will then summarize neurodegenerative diseases related to either neurogenesis or homeostatic dysfunction. Because of the significant detriment neurodegenerative diseases have on the quality of life, it is essential to understand their etiology and ongoing molecular mechanics. The mitochondrial role in neurogenesis and neuron vitality is essential. Dissecting and understanding this organelle’s role in the genesis and homeostasis of neurons should assist in finding pharmaceutical targets for neurodegenerative diseases.
Key Words: Alzheimer's disease, autophagy, mitochondria, mitophagy, neural stem cells, neurodegenerative diseases, neurogenesis, Parkin, Parkinson's disease, PINK1
Introduction
The study of neurogenesis in mature mammalian brains is a novel and innovative research area that may provide insight into the pathogenesis of neurodegenerative diseases. Studying neurogenesis in mature mammalian brains is considered complicated and controversial (Aimone et al., 2014; Katsimpardi and Lledo, 2018). While the recognition of neurogenesis within the adult brains of fish, birds and reptiles has been accepted, neurogenesis in adult human brains is still being disputed amongst some (Aimone et al., 2014; Katsimpardi and Lledo, 2018). Contrary to intuition, the adult brain is not static and is susceptible to change from external stimuli (Fares et al., 2019). One contributor to the brain’s plasticity is neurogenesis, which is the production of new neurons from neural stem cells (NSCs). In adults, neural stem cells are located in the subventricular zone of the olfactory bulb and dentate gyrus of the hippocampus, derived from neural stem/progenitor cells (NSPC). These stem cells provide a pool of self-renewing cells able to differentiate into neurons. In addition, the dentate gyrus of the hippocampus is essential for retaining memories and other cognitive functions (Horgusluoglu et al., 2017). Current research suggests that neurodegenerative diseases such as Parkinson’s and Alzheimer’s, arise from complications associated with neurogenesis in adults, making the study of neurogenesis imperative (Winner and Winkler, 2015).
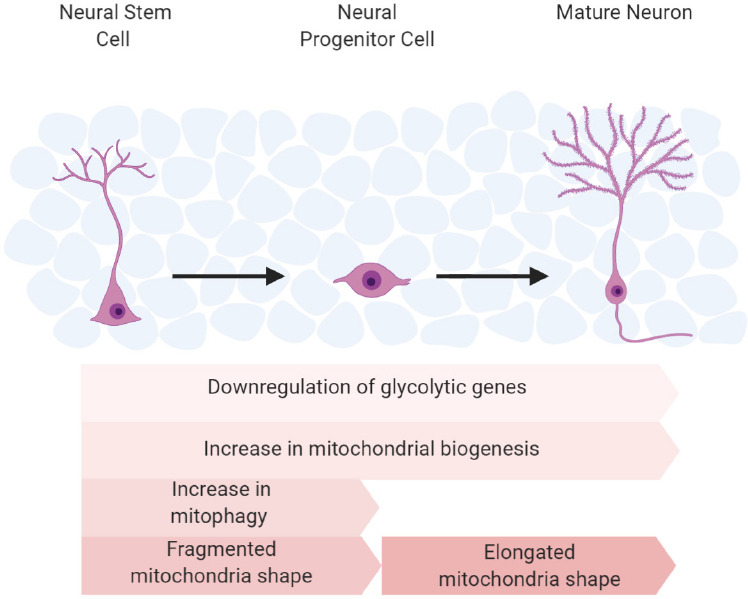
Though various factors contribute to neural stem cell differentiation, this paper will focus on the mitochondria’s role in neurogenesis, including the physical aspects of mitochondria such as shape and mass of the organelle, as well as the mitochondrial quality control mechanism, mitophagy (Figure 1). Additionally, stem cell differentiation requires substantial energy. Mitochondria play an essential role in neurogenesis, and a significant aspect of neural regulation and homeostasis hinges on mitophagy (Chen et al., 2012; Xavier et al., 2014; Beckervordersandforth, 2017; Bahat and Gross, 2019). Mitophagy is the physiological action that removes defective mitochondria from cells, providing the cell a method of self-regulation. In summation, there is a strong relationship between neurogenesis and mitophagy, as mitophagy ensures an abundance of operative mitochondria to supply energy for neurogenesis. Recent research suggests that aging and related pathologies such as Alzheimer’s, Parkinson’s, and Glaucoma are linked to an accumulation of dysfunctional mitochondria (Navarro and Boveris, 2007; Chen and Chan, 2009; Knobloch and Jessberger, 2017).
Figure 1.
A diagram that organizes the many factors that contribute to OXPHOS activity in neurogenesis.
These different factors take place at different stages of neurogenesis and are essential for the switch and establishment of OXPHOS, which contributes to neural stem cell commitment to a mature neuron. OXPHOS: Oxidative phosphorylation.
Defective mitophagy has been implicated in several neurodegenerative diseases (Navarro and Boveris, 2007; Chen and Chan, 2009). Functionally, impaired mitochondria contribute to reactive oxygen species (ROS) production and interruptions in cellular signaling pathways (Cai and Jeong, 2020). As a result, neurotransmission may be impaired, and cognitive dysfunction may occur (Khacho et al., 2017). The removal of defective mitochondria, is vital not just to neurogenesis but to neuron homeostasis and the brain’s cognitive function as a whole (Knobloch and Jessberger, 2017).
Search Strategy and Selection Criteria
Studies cited in this review published from 1985 to 2020 were searched on the National Library of Medicine database using the following keywords: Alzheimer’s disease, glaucoma, metabolic switch, mitochondrial regulation, mitophagy, neural stem cells, neurodegenerative diseases, neurogenesis, and Parkinson’s disease.
Mitochondrial Regulation in Neurogenesis
Genetic regulation of metabolic switch
Neurogenesis is a relatively new concept in neuroscience. The prevailing thought, based on canine and rodent adult models, is neurogenesis occurs in the dentate gyrus of the human adult hippocampus, and the subventricular (Spalding et al., 2013; Toda and Gage, 2018; Bekiari et al., 2020). NSC in these zones, can transition between quiescent and active states (Mohammad et al., 2019). When NSC is active, neurogenesis can be asymmetric or symmetric. Asymmetric division is self-renewing, and yields an NSC and a progenitor. Symmetric division yields two NSC or two progenitor cells (Morrison and Kimble, 2006; Kim and Hirth, 2009).
The process of neuronal differentiation/proliferation and neuron activity requires a substantial amount of energy. NSPCs rely on glycolysis for energy because they are in a hypoxic state (Ito and Di Polo, 2017). As NSPC differentiate to intermediate progenitor cells (IPCs) rather than remaining self-renewing NSPCs, their metabolism switches to oxidative phosphorylation (OXPHOS), reducing glycolytic machinery and lactate products (Beckervordersandforth, 2015; Wanet et al., 2015; Ottoboni et al., 2017; Colangelo et al., 2019). Once mature neurons form, they fully rely on an OXPHOS metabolic state for neurotransmission (Beckervordersandforth, 2015).
Zheng et al. (2016) documented the transition of metabolism from anaerobic glycolysis in neural progenitor cells to oxidative phosphorylation in mature neurons. They found that the shutdown of glycolysis was essential for neuronal differentiation and matured neuron survival (Zheng et al., 2016). Pyruvate kinase mRNA is altered via mRNA splicing, causing PKM2 to change to PKM1 (Zheng et al., 2016). The enzyme, PKM1 favors oxidative phosphorylation, and it is normally expressed in tissues with high aerobic activity (Zheng et al., 2016). Additionally, hexokinase (HK2) and lactate dehydrogenase A (LDHA), two key enzymes in anaerobic glycolysis, disappear upon differentiation. Importantly, the upregulation of HK2 and LDHA in neurons compromises the differentiation and causes cell death. Turning on HK2 and LDHA decreases the amount of pyruvate available for mitochondrial oxidative phosphorylation, as LDHA converts pyruvate to lactate in the cell (Zheng et al., 2016). Therefore, the metabolic switch from anaerobic glycolysis to oxidative phosphorylation is the critical process in providing substantial energy for neurogenesis.
Genetic regulation of mitochondrial biogenesis
Mitochondrial mass increases to assist the ETC machinery, in conjunction with OXPHOS metabolism required for neuronal differentiation (Agostini et al., 2016; Almeida and Vieira, 2017). The proteins Myc, E2F, and PGC1-α are regulatory components essential to mitochondrial biogenesis during neurogenesis. Myc and E2F are cell cycle transcription factors that also promote mitochondrial biogenesis during NPCs proliferation (Zheng et al., 2016). A study by Zheng et al. (2016) suggests that PGC1-α, the master regulator of mitochondria biogenesis, plays a larger role in differentiated cells than in NPCs. In addition, other studies have proven the increase of PGC1-α during neuronal differentiation (O’Brien et al., 2015; Khacho et al., 2016).
Additionally, the integrity of the mitochondrial electron transport chain and oxidative phosphorylation machinery is essential for the proliferation and survival of IPCs (Beckervordersandforth et al., 2017). The mitochondrial transcriptional factor A, downstream of PGC1-α, directly transcribes nuclear-encoded mitochondrial proteins that are involved in the translation and repair of mt-DNA (Kang et al., 2018). Deletion of mitochondrial transcriptional factor A disrupts the mitochondrial electron transport chain and provokes a depletion of IPCs (Hood, 2001; Beckervordersandforth et al., 2017). This data substantiates the significance of a stable supply of energy in neuronal differentiation. To summarize, mitochondrial biogenesis increases OXPHOS machinery, integral in the NSPC commitment to differentiation.
Genetic regulation of reactive oxygen species
The aforementioned genetic findings represent how metabolic strategy and mitochondrial mass contribute to the energy supply for neurogenesis. Mitochondrial morphology also regulates neurogenesis by regulating ROS. ROS, by-products of oxidative phosphorylation, can mediate signals like Batch and NRF2 to inhibit proliferation and promote differentiation during neurogenesis. Mitochondria can change shape via fusion and fission, resulting in altered function and ROS levels (Michelakis et al., 2010; Liesa and Shirihai, 2013; Shen et al., 2013; Jezek et al., 2018). When NSC commits to an NPC, the mitochondria transform from an elongated shape to a fragmented form; when converting to a mature neuron, the mitochondria shifts back to an elongated shape (Seo et al., 2018; Bahat and Gross, 2019). The fragmented shape contains reduced levels of complex I resulting in increased ROS production that is utilized as a signaling complex for NSC commitment (Bigarella et al., 2014; Khacho et al., 2016). Mitofusion 1 and 2 (MFN1/2) are fusion proteins that promote mitochondrial elongation (Hoppins et al., 2011). Through manipulation of Dynamin-related protein 1 (DRP1) and MFN1/2, changes in mitochondrial shape and ROS signaling can regulate NSC self-renewal. MFN1/2 double knock-out hinders the self-renewal of NSCs but enhances the commitment of differentiation. DRP1, a fission protein, induces mitochondrial fragmentation (Khacho et al., 2016). The loss of DRP1 promotes the self-renewal of NSCs. Furthermore, these changes are ROS dependent, indicating that mitochondrial morphology regulates NSC differentiation through mt-ROS signaling.
Mitophagy and neurogenesis
Metabolic switch, mitochondrial biogenesis, and mitochondrial fragmentation with increased ROS will result in the removal of “old” or defective mitochondria. Therefore, mitophagy, a mechanism that clears targeted mitochondria using autophagy machinery, plays a critical role in ensuring mitochondrial health and thus, neurogenesis (Priault et al., 2005; Khaminets et al., 2016). Once a damaged mitochondrion is tagged, it is engulfed by a double-membraned autophagosome and degraded by the lysosome (Kraft et al., 2009). Active mitophagy has been observed in NSC before differentiation, aiding in the clearance of faulty mitochondria generated by mitochondrial remodeling (Beckervordersandforth, 2017). Loss of PINK1, a mitophagy regulator, leads to metabolic deficits in NSCs and impedes differentiation (Agnihotri et al., 2017).
Furthermore, mitophagy is crucial in retinal ganglion cell differentiation. During development, mitophagy in retinal ganglion cells (RGCs) regulates metabolic reprogramming which in turn regulates RGC proliferation. This is seen through the BNIP3L/NIX mitophagy receptor which results in a glycolytic shift crucial for RGC differentiation (Esteben-Martínez et al., 2017). Tissue hypoxia can be observed in mouse embryonic retinal development resulting in increased expression of BIP3L/NIX mRNA. Deficient levels of BIP3L/NIX in retinas have been observed to contain reduced mRNA expression of glycolytic genes Pfkfb3, Hk2, Gapdh, and Pkm2 (Esteben-Martínez et al., 2017). Therefore, one can conclude NIX-dependent mitophagy is essential for RGC metabolic reprogramming and differentiation.
In conclusion, mitochondrial remodeling and regulation are essential for neurogenesis in both neuronal and RGC proliferation and differentiation. Elucidating these mechanisms will deepen our understanding of brain development and contribute to finding new pharmaceutical targets for neurodegenerative diseases.
Neurodegenerative Diseases
The following section is devoted to reviewing pathologies associated with mitochondrial dysfunction. Mitochondria are essential organelles for maintaining and supplying the energy needed for neuronal function and neurogenesis (Mandal and Drerup, 2019; Rangaraju et al., 2019). As earlier reviewed, errors in mitochondrial function due to the organelle’s age or biochemical disorder are tackled by mitophagy, thus eliminating the problem organelle. However, over time, often associated with the normal process of cellular aging, but occasionally due to genetic miscoding, the mitophagy process is impeded, resulting in a variety of neurodegenerative diseases such as Alzheimer’s, Parkinson’s, glaucoma, Huntington’s chorea, and amyotrophic lateral sclerosis (Hiona and Leeuwenburgh, 2008; Osborne, 2008; Frank et al., 2012; Ryan et al., 2015; Evans and Holzbaur, 2020). Understanding the mitochondria’s mechanics, pathways, and processes, as well as the organelle’s impact on neurogenesis and homeostasis, will continue to provide researchers with prospective drug targets and therapies to alleviate aging and neurodegenerative diseases (Khacho et al., 2019; Nicaise et al., 2020). This review will examine three diseases prevalent in aging populations, Alzheimer’s, Parkinson’s, and Glaucoma (Hiona and Leeuwenburgh, 2008; Osborne, 2008; Frank et al., 2012; Ryan et al., 2015; Evans and Holzbaur, 2020). These three neurological diseases are well known and affect a statistically significant proportion of the aging population (Moon et al., 2018; Licher et al., 2019).
Parkinson’s disease
Parkinson’s disease (PD) characterized by resting tremors, bradykinesia, postural instability, amnesia, and loss of motor skills over time results from a progressive loss of dopaminergic neurons in the substantia nigra (Mhyre et al., 2012; DeMaagd and Philip, 2015). PD was first described by James Parkinson in 1817 and was later clinically identified as intracellular accumulation of α-synuclein in aggregates forming Lewy bodies via histochemistry (Spillantini et al., 1998; Luk et al., 2009; Stefanis, 2012). However, researchers have had difficulties in targeting α-synuclein pharmaceutically (Stefanis, 2012). Several studies have examined the genetic aspects of the disease. Researchers have identified several genes, such as PINK1 and Parkin, in the regulation of mitophagy, making it a disease of mitochondria and aging and the numerous genes that regulate mitophagy in neurons, which may serve as potential pharmaceutical targets (Dernie, 2020).
Researchers have pinpointed dysfunction in the PINK1/Parkin pathway as a significant factor in autosomal recessive early-onset PD (Kitada et al., 1998; Valente et al., 2004). In the normally functioning pathway, PINK1 resides in the cytosol where it is chaperoned to the mitochondria by Hsp90 and Cdc37. PINK1 first enters the outer mitochondrial membrane (OMM) via translocase of the outer membrane (TOM) complex (Figure 2). The protein, PINK1 extends through the inner mitochondrial membrane (IMM) via Tim23 (translocase of the inner membrane) into the mitochondrial matrix (Jin et al., 2010; Sekine and Youle, 2018). Once the N-terminus of PINK1 reaches the matrix, it is cleaved off by the mitochondrial processing peptidase (MPP α/β) (Figure 2). The process of PINK1 import requires an electrochemical gradient along the mitochondrial membrane. Any compromise of the electrochemical gradient will impede the protein, Tim23’s ability to facilitate PINK1’s translocation into the mitochondrial matrix. A damaged electrochemical gradient will result in the full length of PINK1, to reside in the mitochondria’s outer membrane (OMM) (Barazzuol et al., 2020). Lack of electrical membrane potential is a symptom of a poorly functioning mitochondrion. In the normally functioning system, dysfunctional mitochondria are identified tagged and disposed of in a process known as ubiquitination (Jin et al., 2010). The protein PINK1 accumulates on the dysfunctional mitochondria, acting as a signal to the protein Parkin. Once PINK1 sufficiently accumulates on the OMM, Parkin is recruited to the OMM and ubiquitinates, or identifies, the damaged mitochondrion. This signaling device is referred to as Parkin dependent mitophagy (Ordureau et al., 2014). After the defective mitochondrion is ubiquitinated by PINK1/Parkin, an autophagosome is recruited by the nuclear domain10 protein52 and optineurin (Kim et al., 2007). The autophagosome attaches and engulfs the mitochondria, where it eventually binds to a lysosome forming an auto-phagolysosome. Once in the auto-phagolysosome, the damaged mitochondrion is degraded (He and Klionsky, 2009; Heo et al., 2015).
Figure 2.

A simplified schematic of the PINK1/Parkin pathway.
The PINK1/Parkin pathway is activated upon loss of mitochondrial membrane potential (Δψ). PINK1 attaches to TOM, and no membrane depolarization causes PINK1 to not be cleaved and extend into the matrix via TIM. This results in full length PINK1 to remain in the inner membrane. Parkin is then phosphorylated by PINK1 and recruited to damaged mitochondria, where it ubiquinates and targets the damaged mitochondria for autophagosome recruitment. The auto-phagolysosome then degrades the damaged mitochondria. IMM: Inner mitochondrial membrane; OMM: outer mitochondrial membrane; TIM: translocase of inner membrane; TOM: translocase of outer membrane; Ub: ubiquitin.
The PINK1/Parkin pathway may provide therapeutic answers to PD, as it is intricate with many possible pharmaceutical targets (Liu et al., 2020). One thought would be to increase mitophagy as a mechanism to control and decrease PD pathogenesis (Truban et al., 2017). A molecule of interest is celastrol, derived from the Chinese herb Tripterygium wilfordii, also known as the (Thunder God Vine) was discovered to activate mitophagy and inhibit dopaminergic apoptosis (Cascao et al., 2017; Lin et al., 2019). After treating PD mouse models with celastrol, the group found an increase in gene transcription of PINK1 and other genes related to the PINK1/Parkin pathway such as ATG7, ATG12, and LC3. Celastrol suppressed motor symptoms in the PD mouse models; additionally, these animals’ necropsies revealed diminished neurodegeneration in the substantial nigra and striatum (Lin et al., 2019). Another molecule, BC1464, increases PINK1 intracellular concentrations, resulting in an increase of mitophagy. It is hypothesized that the protein the BC1464 inhibits FBXO7 from degrading PINK1 contributing to the efficacy of ubiquitination so critical to the process of mitophagy (Liu et al., 2019). These studies show promising concepts for PD drugs and treatments targeting mitophagy.
In addition to rescuing PINK1/Parkin mitophagy mechanisms via drug treatment, utilizing other mitophagy pathways to increase mitophagy in PD patients may be an option as well. The BNIP3L/NIX mitophagy receptor has been found to restore PINK1/Parkin related mitophagy in some PD patients (Koentjoro et al., 2017). How BNIP3L/NIX can restore this function in PD is still unclear, but BNIP3L/NIX activity with PARK2 has been observed. The BNIP3L/NIX protein is a mitophagy receptor that is observed in reticulocyte maturation. BNIP3L/NIX can be found located on the outer-membrane of the mitochondria along with PINK1. PARK2 can ubiqutinate BNIP3L/NIX in response to deficient complex I activity. This then leads to the recruitment of autophagosome machinery such as NBR1 and LC3, and degradation of the targeted mitochondria (Gao et al., 2015). Therefore, BNIP3L/NIX shows capabilities of rescuing PINK1 function and a potential target for PD treatment.
Glaucoma
Glaucoma is the leading cause of blindness in older populations. The disease is insidious, frequently without pain or obvious symptom until irreparable damage to the optical nerve has occurred. The optic nerve also referred to as the second cranial nerve, sits in the posterior portion of the eye which is composed of several axons that come together to form a network of RGCs. There are a large number of mitochondria located in the axon (Lascaratos et al., 2012; Van Bergen et al., 2015). Retinal ganglion cells require a tremendous amount of energy; and RGCs depend on the mitochondria for synaptic energy as well as the generation of ROS as part of their cellular regulation (Lascaratos et al., 2012). Currant research suggests mitophagy, plays an important role in ocular health (Gupta and Yucel, 2007; Weinreb et al., 2014; Calkins et al., 2017).
The leading mechanical cause of glaucoma is either acute or chronic increase of intraocular pressure, but once started, it snowballs and leads to the destruction of RGC, damaged optic nerves, impaired vision, and subsequent blindness (Gupta and Yucel, 2007; Weinreb et al., 2014; Calkins et al., 2017). Although mitochondrial dysfunction has not been linked to causing glaucoma, mitochondrial dysfunction plays a role in the originating cellular damage associated with glaucoma’s pathogenesis (Ito and Di Polo, 2017). The premise is mtDNA mutations create a snowballing effect of mitochondrial dysfunction, which in turn, generates increases in ROS. The increase in ROS causes cell death and contributes in turn to further cellular and mitochondrial dysfunction, leading to RBG damage and rising intraocular pressure (Wu et al., 2015). To better understand the relationship between intraocular pressure and mitochondria, studies have been performed in glaucomatous mice predisposed to high intraocular pressure (Ju et al., 2008). Van Bergen and Ju’s electron microscopy indicated that the mitochondria in glaucoma mice exhibited structural changes, such as mitochondrial fission, smaller cristae, and expanded matrixes (Ju et al., 2008). Additionally, mitochondria damaged due to intraocular pressure exhibited lower rates of ATP synthesis from complex I despite higher mitochondrial numbers (Ju et al., 2008; Lee et al., 2012; Van Bergen et al., 2015). Low PGC1-α levels confirmed that the increased number of mitochondria was not a result of mitochondrial biogenesis (Coughlin et al., 2015). Other studies have correlated patient age and mitochondrial efficacy with glaucoma’s progression (Lascaratos et al., 2012).
In primary open angle glaucoma, the most prevalent form of glaucoma, the increase in ocular pressure is caused by an obstruction in trabecular meshwork that prevents the outflow of the aqueous humor, which results in an increase of ocular pressure damaging the RBGs. These axon heads are heavily populated by mitochondria which are responsible for the energy supplying nerve impulses. Defects in the PINK1/Parkin pathway have been implicated in glaucoma progression. Parkin overexpression has been found to protect RGCs in studies utilizing rats, receiving translimbal laser photocoagulation to the trabecular mesh to induce glaucoma (Dai et al., 2018). Two proteins, optineurin (OPTN), a multifunction protein that works in addition to other tasks as an autophagy receptor, and TANK binding kinase 1 are associated with the Parkin pathway and are intrinsic to the maintaining of homeostasis by assisting in the coordinating vesicle trafficking and removal of debris (Fingert et al., 2016; Minegishi et al., 2016; Weil et al., 2018). OPTN is involved in multiple autophagy pathways such as aggrephagy, mitophagy, and xenophagy. As part of mitophagy, after recruitment by Parkin, OPTN facilitates the final fusion of the autophagosome with the lysosome. Researchers have identified OPTN missense mutations in primary open angle glaucoma which is the prevalent form of the disease (Ying and Yue, 2012). Duplications of TANK binding kinase 1, a modifier of OPTN, are present in roughly 1% of normal tension glaucoma (Ritch et al., 2014). Together these two mutations account for 2–3% normal tension glaucoma (Wiggs and Pasquale, 2017).
Dysfunctional mitophagy contributes to the cycle of glaucoma as poorly functioning mitochondria can lead to an increased production of ROS. An increase in ROS levels causes oxidation and induces cell death by apoptosis (Jiang and Wang, 2004; McMonnies, 2018). Therefore, even though glaucoma is not initiated by the mitochondria, dysfunction can escalate the cycle of RGC destruction. Consequently, halting or controlling excess ROS production is critical to prevent irreversible damage to the RGC (Frost et al., 2014). PINK1/Parkin mitophagy can be induced by a loss of iron, so one possible candidate, is the iron chelator deferiprone which has been shown to induce autophagy (Rosignol et al., 2020). Rosignol et al. (2020) found that incubating primary RGC cells with the iron chelator deferiprone increased mitophagy, which increased the removal of damaged mitochondria. This in turn, resulted in an elimination of ROS, lessening the oxidative stress and appears to rescue the RGC with regards to some glaucoma-related stress factors.
Alzheimer’s disease
Alzheimer’s disease (AD) is a progressive neurodegenerative disease associated with aging populations. The disease is recognized as the leading cause of dementia in the elderly population, with a small subset afflicted with the autosomal familiar Alzheimer disease (FAD) that appears within families prior to the age of 65 (Reddy and Beal, 2008; Alzheimer’s, 2016). The aging process is typified by changes in cellular function and oxidative stress both associated with changes in mitochondrial function. Recent studies have indicated that targeting mitochondrial dysfunction in the initial phase of the disease could prove beneficial (Reddy and Beal, 2008; Hauptmann et al., 2009). AD is caused by accumulation of insoluble amyloid β (Aβ) peptide deposits and tau neurofibrillary tangles at the synaptic junction, which disrupts transmission between the neural synapses (Wong et al., 1985; Grundke-Iqbal et al., 1986; Erkkinen et al., 2018). In AD, the build-up is located primarily in the hippocampus whereas in the unafflicted but older individual (Aβ) peptide deposits and tau neurofibrillary tangles appear in the cerebellum (Wong et al., 1985; Grundke-Iqbal et al., 1986; Erkkinen et al., 2018). There are multiple hypotheses proposed regarding, how these insoluble proteins affect the development and progression of AD. Some researchers hypothesize the Aβ aggregates initiate the AD process while the malformed Tau proteins within the neurons further the disease (Selkoe and Hardy, 2016). Other researchers argue that the Tau proteins actually play a larger role in the development and progression of Alzheimer’s. A hypothesis stemming from data received regarding the drugs targeting Aβ aggregates have proved ineffective (Kametani and Hasegawa, 2018).
Tau proteins appear to play a significant role in the progression of AD. Tau acts as a structural stabilizer within the neuron. In AD, the protein becomes malformed, creating tangles that are no longer structurally relevant, which affect signaling. Additionally, the protein aggregates appear to impact the PINK1/Parkin mitophagy pathway, confirmed in Cummins et al.’s study (2019) involving Caenorhabditis elegans. In this study, the central nervous system of the Caenorhabditis elegans carried pathogenic Tau mutations, which were found to inhibit the proper translocation of Parkin to the mitochondria completely (Cummins et al., 2019). Mitophagy will not occur without the translocation of Parkin to the mitochondria, disrupting homeostasis and consequently shortening the cell’s life. Another contributor to mitochondrial dysfunction within the central nervous system is Aβ plaques. Aβ peptides are generated by the amyloid precursor protein (APP) and presenilins 1 or 2 (PS1/2). In FAD, an autosomal dominant form of Alzheimer’s that affects individuals 65 years of age or less, missense mutations in APP and PS1/2 contribute to the development of plaques that impede synaptic and mitochondrial function (Rovelet-Lecrux et al., 2006; Lanoiselee et al., 2017; Reddy et al., 2018; Arboleda-Velasquez et al., 2019). PS1/2, is a transmembrane protein complex that cleaves APP releasing Aβ peptides. In AD, the PS1/2 substrate structure changes and no longer effectively cleaves the APP, resulting in longer Aβ peptides (Szaruga et al., 2017). Over time, the longer Aβ peptides bind together to create plaques. Aβ plaques and mutant APP impact the neurons’ function and durability. The plaques inhibit mitochondrial biogenesis, synaptic protein production, dendritic spines, and mitophagy (Rovelet-Lecrux et al., 2006; Lanoiselee et al., 2017; Reddy et al., 2018; Arboleda-Velasquez et al., 2019).
Another significant contributor to the development of Aβ plaques is mutations in the PSEN1 gene. Mutations in the PSEN1 gene may kick-start plaque development by increasing Aβ42 (Kelleher and Shen, 2017). This protein is also responsible for encoding the protein Presenilin-1 (PS-1). PS-1 mutations are associated with early-onset Alzheimer’s (FAD) (Kelleher and Shen, 2017). An aspect that needs to be examined is the impact PS-1 has on Ca2+ levels as disruptions in signaling have been noted in FAD patients (Tu et al., 2006). Mechanistically, PS-1 regulates Ca2+ levels in mitochondria, which downstream affects ATP production and the ion level within the cytosol (Malli et al., 2003; Fairless et al., 2014). Ca2+ concentration in the cytosol contributes to neurotransmission capability and if the neuron is unable to properly regulate Ca2+ levels, transmission falters (Lee et al., 2018). The PS-1 mutation impacts other organelles within the cell. By opening 1,4,5-triophophate channels, Ca2+ is released from the endoplasmic reticulum. In response to the accumulation of Ca2+ within the cell, the mitochondria increase their Ca2+ uptake to the neuron’s detriment. This uptake leads to increased ROS levels, decreased ATP production, and subsequent death of the neuron (Toglia et al., 2016; Alzheimer’s Association Calcium Hypothesis, 2017).
Locating effective AD therapies has proven difficult as the mechanisms triggering plaque development, mitochondrial dysfunction, and general neural death are involved. There are two thoughts or paths to arresting the progression of AD: the first, targeting the proteins responsible for the network of tangles, and the second, unraveling the mitochondrial mechanisms and correcting pathway dysfunction. First, though Aβ and tau aggregates have been identified as contributing significantly to signal impairment, combating these specific proteins with targeted drug therapy has not been effective. The second implicates defective mitochondria and related products that contribute to the pathogenesis of AD. Currently, two treatments are being investigated that target the AD related mitochondrial dysfunction and excess ROS production (Oliver and Reddy, 2019). The first, methylene blue already has FDA approval for the treatment of malaria, cyanide poisoning, and ischemic brain injuries (Yang et al., 2020). Methylene blue can cross the blood-brain barrier to donate electrons to the ETC resulting in increased ATP production. In randomized, double-blind studies, both AD patients and AD animal models showed cognitive improvement and vascular blood flow within the brain following methylene blue treatment. The second, photobiomodulation, also known as low-level laser therapy, has been utilized to stimulate vascularization in damaged muscle tissue with minimal side effects (Yang et al., 2020). Photobiomodulation can target mitochondria because complex IV absorbs light between 665 nm and 810 nm. In AD, the premise is the low-level laser appears to directly target the mitochondria by donating photons to the Complex IV, which stimulates the consumption of excess oxygen, alleviating the excess ROS production generated by damaged mitochondria AD (Yang et al., 2020).
Summary
Within the field of neuroscience, there is growing interest in the mitochondria and its related pathways, as it becomes increasingly evident that mitochondrial function impacts the body’s neurons from development to destruction. As mentioned before, there are no cures at this time for Alzheimer’s, Parkinson’s, or Glaucoma. However, there is evidence that unraveling and identifying the mitochondria’s role in these pathologies may well serve as a source of information that will allow researchers to locate therapeutic targets to slow the progression of these diseases (Chaturvedi and Beal, 2013; Kamat et al., 2014).
Acknowledgments:
We apologize to the authors of several high-quality articles that we were not able to discuss and cite owing to space limitations.
Footnotes
C-Editors: Zhao M, Liu WJ, Qiu Y; T-Editor: Jia Y
Conflicts of interest: None declared.
Financial support: This work was supported by a grant from the National Institutes for Health (K22-HL135051, to NS).
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Funding: This work was supported by a grant from the National Institutes for Health (K22-HL135051, to NS).
References
- 1.Agnihotri SK, Shen R, Li J, Gao X, Bueler H. Loss of PINK1 leads to metabolic deficits in adult neural stem cells and impedes differentiation of newborn neurons in the mouse hippocampus. FASEB J. 2017;31:2839–2853. doi: 10.1096/fj.201600960RR. [DOI] [PubMed] [Google Scholar]
- 2.Agostini M, Romeo F, Inoue S, Niklison-Chirou MV, Elia AJ, Dinsdale D, Morone N, Knight RA, Mak TW, Melino G. Metabolic reprogramming during neuronal differentiation. Cell Death Differ. 2016;23:1502–1514. doi: 10.1038/cdd.2016.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aimone JB, Li Y, Lee SW, Clemenson GD, Deng W, Gage FH. Regulation and function of adult neurogenesis: from genes to cognition. Physiol Rev. 2014;94:991–1026. doi: 10.1152/physrev.00004.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Almeida AS, Vieira HLA. Role of cell metabolism and mitochondrial function during adult neurogenesis. Neurochem Res. 2017;42:1787–1794. doi: 10.1007/s11064-016-2150-3. [DOI] [PubMed] [Google Scholar]
- 5.Alzheimer’s A. 2016 Alzheimer’s disease facts and figures. Alzheimers Dement. 2016;12:459–509. doi: 10.1016/j.jalz.2016.03.001. [DOI] [PubMed] [Google Scholar]
- 6.Alzheimer’s Association Calcium Hypothesis W (2017). Calcium hypothesis of Alzheimer’s disease and brain aging: a framework for integrating new evidence into a comprehensive theory of pathogenesis. Alzheimers Dement. 13:178–182 e117. doi: 10.1016/j.jalz.2016.12.006. [DOI] [PubMed] [Google Scholar]
- 7.Arboleda-Velasquez JF, Lopera F, O’Hare M, Delgado-Tirado S, Marino C, Chmielewska N, Saez-Torres KL, Amarnani D, Schultz AP, Sperling RA, Leyton-Cifuentes D, Chen K, Baena A, Aguillon D, Rios-Romenets S, Giraldo M, Guzmán-Vélez E, Norton DJ, Pardilla-Delgado E, Artola A, et al. Resistance to autosomal dominant Alzheimer’s disease in an APOE3 Christchurch homozygote: a case report. Nat Med. 2019;25:1680–1683. doi: 10.1038/s41591-019-0611-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bahat A, Gross A. Mitochondrial plasticity in cell fate regulation. J Biol Chem. 2019;294:13852–13863. doi: 10.1074/jbc.REV118.000828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barazzuol L, Giamogante F, Brini M, Cali T. PINK1/Parkin mediated mitophagy, Ca(2+) signalling, and ER-mitochondria contacts in Parkinson’s disease. Int J Mol Sci. 2020;21:1772. doi: 10.3390/ijms21051772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beckervordersandforth R. Mitochondrial metabolism-mediated regulation of adult neurogenesis. Brain Plast. 2017;3:73–87. doi: 10.3233/BPL-170044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beckervordersandforth R, Ebert B, Schäffner I, Moss J, Fiebig C, Shin J, Moore DL, Ghosh L, Trinchero MF, Stockburger C, Friedland K, Steib K, von Wittgenstein J, Keiner S, Redecker C, Hölter SM, Xiang W, Wurst W, Jagasia R, Schinder AF, et al. Role of mitochondrial metabolism in the control of early lineage progression and aging phenotypes in adult hippocampal neurogenesis. Neuron. 2017;93:1518. doi: 10.1016/j.neuron.2017.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beckervordersandforth R, Häberle BM, Lie DC. Metabolic regulation of adult stem cell-derived neurons. Front Biol. 2015;10:107–116. [Google Scholar]
- 13.Bekiari C, Grivas I, Tsingotjidou A, Papadopoulos GC. Adult neurogenesis and gliogenesis in the dorsal and ventral canine hippocampus. J Comp Neurol. 2020;528:1216–1230. doi: 10.1002/cne.24818. [DOI] [PubMed] [Google Scholar]
- 14.Bigarella CL, Liang R, Ghaffari S. Stem cells and the impact of ROS signaling. Development. 2014;141:4206–4218. doi: 10.1242/dev.107086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cai Q, Jeong YY. Mitophagy in Alzheimer’s disease and other age-related neurodegenerative diseases. Cells. 2020;9:150. doi: 10.3390/cells9010150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Calkins DJ, Pekny M, Cooper ML, Benowitz L, Lasker IIoA, Glaucomatous Neurodegeneration P. The challenge of regenerative therapies for the optic nerve in glaucoma. Exp Eye Res. 2017;157:28–33. doi: 10.1016/j.exer.2017.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cascao R, Fonseca JE, Moita LF. Celastrol: a spectrum of treatment opportunities in chronic diseases. Front Med (Lausanne) 2017;4:69. doi: 10.3389/fmed.2017.00069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chaturvedi RK, Beal MF. Mitochondria targeted therapeutic approaches in Parkinson’s and Huntington’s diseases. Mol Cell Neurosci. 2013;55:101–114. doi: 10.1016/j.mcn.2012.11.011. [DOI] [PubMed] [Google Scholar]
- 19.Chen CT, Hsu SH, Wei YH. Mitochondrial bioenergetic function and metabolic plasticity in stem cell differentiation and cellular reprogramming. Biochim Biophys Acta. 2012;1820:571–576. doi: 10.1016/j.bbagen.2011.09.013. [DOI] [PubMed] [Google Scholar]
- 20.Chen H, Chan DC. Mitochondrial dynamics--fusion, fission, movement, and mitophagy--in neurodegenerative diseases. Hum Mol Genet. 2009;18:R169–176. doi: 10.1093/hmg/ddp326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Colangelo AM, Cirillo G, Alberghina L, Papa M, Westerhoff HV. Neural plasticity and adult neurogenesis: the deep biology perspective. Neural Regen Res. 2019;14:201–205. doi: 10.4103/1673-5374.244775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Coughlin L, Morrison RS, Horner PJ, Inman DM. Mitochondrial morphology differences and mitophagy deficit in murine glaucomatous optic nerve. Invest Ophthalmol Vis Sci. 2015;56:1437–1446. doi: 10.1167/iovs.14-16126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cummins N, Tweedie A, Zuryn S, Bertran-Gonzalez J, Gotz J. Disease-associated tau impairs mitophagy by inhibiting Parkin translocation to mitochondria. EMBO J. 2019;38:e99360. doi: 10.15252/embj.201899360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dai Y, Hu X, Sun X. Overexpression of parkin protects retinal ganglion cells in experimental glaucoma. Cell Death Dis. 2018;9:88. doi: 10.1038/s41419-017-0146-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.DeMaagd G, Philip A. Parkinson’s disease and its management: part 1: disease entity, risk factors, pathophysiology, clinical presentation, and diagnosis. P T. 2015;40:504–532. [PMC free article] [PubMed] [Google Scholar]
- 26.Dernie F. Mitophagy in Parkinson’s disease: from pathogenesis to treatment target. Neurochem Int. 2020;138:104756. doi: 10.1016/j.neuint.2020.104756. [DOI] [PubMed] [Google Scholar]
- 27.Erkkinen MG, Kim MO, Geschwind MD. Clinical neurology and epidemiology of the major neurodegenerative diseases. Cold Spring Harb Perspect Biol. 2018;10:a033118. doi: 10.1101/cshperspect.a033118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Evans CS, Holzbaur ELF. Quality control in neurons: mitophagy and other selective autophagy mechanisms. J Mol Biol. 2020;432:240–260. doi: 10.1016/j.jmb.2019.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fairless R, Williams SK, Diem R. Dysfunction of neuronal calcium signalling in neuroinflammation and neurodegeneration. Cell Tissue Res. 2014;357:455–462. doi: 10.1007/s00441-013-1758-8. [DOI] [PubMed] [Google Scholar]
- 30.Fares J, Bou Diab Z, Nabha S, Fares Y. Neurogenesis in the adult hippocampus: history, regulation, and prospective roles. Int J Neurosci. 2019;129:598–611. doi: 10.1080/00207454.2018.1545771. [DOI] [PubMed] [Google Scholar]
- 31.Fingert JH, Robin AL, Scheetz TE, Kwon YH, Liebmann JM, Ritch R, Alward WL. Tank-Binding Kinase 1 (TBK1) gene and open-angle glaucomas (An American Ophthalmological Society Thesis) Trans Am Ophthalmol Soc. 2016;114:T6. [PMC free article] [PubMed] [Google Scholar]
- 32.Frank M, Duvezin-Caubet S, Koob S, Occhipinti A, Jagasia R, Petcherski A, Ruonala MO, Priault M, Salin B, Reichert AS. Mitophagy is triggered by mild oxidative stress in a mitochondrial fission dependent manner. Biochim Biophys Acta. 2012;1823:2297–2310. doi: 10.1016/j.bbamcr.2012.08.007. [DOI] [PubMed] [Google Scholar]
- 33.Frost LS, Mitchell CH, Boesze-Battaglia K. Autophagy in the eye: implications for ocular cell health. Exp Eye Res. 2014;124:56–66. doi: 10.1016/j.exer.2014.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci U S A. 1986;83:4913–4917. doi: 10.1073/pnas.83.13.4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gupta N, Yucel YH. Glaucoma as a neurodegenerative disease. Curr Opin Ophthalmol. 2007;18:110–114. doi: 10.1097/ICU.0b013e3280895aea. [DOI] [PubMed] [Google Scholar]
- 36.Hauptmann S, Scherping I, Drose S, Brandt U, Schulz KL, Jendrach M, Leuner K, Eckert A, Muller WE. Mitochondrial dysfunction: an early event in Alzheimer pathology accumulates with age in AD transgenic mice. Neurobiol Aging. 2009;30:1574–1586. doi: 10.1016/j.neurobiolaging.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 37.He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. 2009;43:67–93. doi: 10.1146/annurev-genet-102808-114910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Heo JM, Ordureau A, Paulo JA, Rinehart J, Harper JW. The PINK1-PARKIN mitochondrial ubiquitylation pathway drives a program of OPTN/NDP52 recruitment and TBK1 activation to promote mitophagy. Mol Cell. 2015;60:7–20. doi: 10.1016/j.molcel.2015.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hiona A, Leeuwenburgh C. The role of mitochondrial DNA mutations in aging and sarcopenia: implications for the mitochondrial vicious cycle theory of aging. Exp Gerontol. 2008;43:24–33. doi: 10.1016/j.exger.2007.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hood DA. Invited review: contractile activity-induced mitochondrial biogenesis in skeletal muscle. J Appl Physiol (1985) 2001;90:1137–1157. doi: 10.1152/jappl.2001.90.3.1137. [DOI] [PubMed] [Google Scholar]
- 41.Hoppins S, Edlich F, Cleland MM, Banerjee S, McCaffery JM, Youle RJ, Nunnari J. The soluble form of Bax regulates mitochondrial fusion via MFN2 homotypic complexes. Mol Cell. 2011;41:150–160. doi: 10.1016/j.molcel.2010.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Horgusluoglu E, Nudelman K, Nho K, Saykin AJ. Adult neurogenesis and neurodegenerative diseases: a systems biology perspective. Am J Med Genet B Neuropsychiatr Genet. 2017;174:93–112. doi: 10.1002/ajmg.b.32429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ito YA, Di Polo A. Mitochondrial dynamics, transport, and quality control: A bottleneck for retinal ganglion cell viability in optic neuropathies. Mitochondrion. 2017;36:186–192. doi: 10.1016/j.mito.2017.08.014. [DOI] [PubMed] [Google Scholar]
- 44.Jezek J, Cooper KF, Strich R. Reactive oxygen species and mitochondrial dynamics: The Yin and Yang of mitochondrial dysfunction and cancer progression. Antioxidants (Basel) 2018;7:13. doi: 10.3390/antiox7010013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jiang X, Wang X. Cytochrome C-mediated apoptosis. Annu Rev Biochem. 2004;73:87–106. doi: 10.1146/annurev.biochem.73.011303.073706. [DOI] [PubMed] [Google Scholar]
- 46.Jin SM, Lazarou M, Wang C, Kane LA, Narendra DP, Youle RJ. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J Cell Biol. 2010;191:933–942. doi: 10.1083/jcb.201008084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ju WK, Kim KY, Lindsey JD, Angert M, Duong-Polk KX, Scott RT, Kim JJ, Kukhmazov I, Ellisman MH, Perkins GA, Weinreb RN. Intraocular pressure elevation induces mitochondrial fission and triggers OPA1 release in glaucomatous optic nerve. Invest Ophthalmol Vis Sci. 2008;49:4903–4911. doi: 10.1167/iovs.07-1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kamat PK, Kalani A, Kyles P, Tyagi SC, Tyagi N. Autophagy of mitochondria: a promising therapeutic target for neurodegenerative disease. Cell Biochem Biophys. 2014;70:707–719. doi: 10.1007/s12013-014-0006-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kametani F, Hasegawa M. Reconsideration of amyloid hypothesis and Tau hypothesis in Alzheimer’s disease. Front Neurosci. 2018;12:25. doi: 10.3389/fnins.2018.00025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kang I, Chu CT, Kaufman BA. The mitochondrial transcription factor TFAM in neurodegeneration: emerging evidence and mechanisms. FEBS Lett. 2018;592:793–811. doi: 10.1002/1873-3468.12989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Katsimpardi L, Lledo PM. Regulation of neurogenesis in the adult and aging brain. Curr Opin Neurobiol. 2018;53:131–138. doi: 10.1016/j.conb.2018.07.006. [DOI] [PubMed] [Google Scholar]
- 52.Kelleher RJ, 3rd, Shen J. Presenilin-1 mutations and Alzheimer’s disease. Proc Natl Acad Sci U S A. 2017;114:629–631. doi: 10.1073/pnas.1619574114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Khacho M, Harris R, Slack RS. Mitochondria as central regulators of neural stem cell fate and cognitive function. Nat Rev Neurosci. 2019;20:34–48. doi: 10.1038/s41583-018-0091-3. [DOI] [PubMed] [Google Scholar]
- 54.Khacho M, Clark A, Svoboda DS, MacLaurin JG, Lagace DC, Park DS, Slack RS. Mitochondrial dysfunction underlies cognitive defects as a result of neural stem cell depletion and impaired neurogenesis. Hum Mol Genet. 2017;26:3327–3341. doi: 10.1093/hmg/ddx217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Khacho M, Clark A, Svoboda DS, Azzi J, MacLaurin JG, Meghaizel C, Sesaki H, Lagace DC, Germain M, Harper ME, Park DS, Slack RS. Mitochondrial dynamics impacts stem cell identity and fate decisions by regulating a nuclear transcriptional program. Cell Stem Cell. 2016;19:232–247. doi: 10.1016/j.stem.2016.04.015. [DOI] [PubMed] [Google Scholar]
- 56.Khaminets A, Behl C, Dikic I. Ubiquitin-dependent and independent signals in selective autophagy. Trends Cell Biol. 2016;26:6–16. doi: 10.1016/j.tcb.2015.08.010. [DOI] [PubMed] [Google Scholar]
- 57.Kim DW, Hirth F. Genetic mechanisms regulating stem cell self-renewal and differentiation in the central nervous system of Drosophila. Cell Adh Migr. 2009;3:402–411. doi: 10.4161/cam.3.4.8690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kim I, Rodriguez-Enriquez S, Lemasters JJ. Selective degradation of mitochondria by mitophagy. Arch Biochem Biophys. 2007;462:245–253. doi: 10.1016/j.abb.2007.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605–608. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- 60.Knobloch M, Jessberger S. Metabolism and neurogenesis. Curr Opin Neurobiol. 2017;42:45–52. doi: 10.1016/j.conb.2016.11.006. [DOI] [PubMed] [Google Scholar]
- 61.Kraft C, Reggiori F, Peter M. Selective types of autophagy in yeast. Biochim Biophys Acta. 2009;1793:1404–1412. doi: 10.1016/j.bbamcr.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 62.Lanoiselée HM, Nicolas G, Wallon D, Rovelet-Lecrux A, Lacour M, Rousseau S, Richard AC, Pasquier F, Rollin-Sillaire A, Martinaud O, Quillard-Muraine M, de la Sayette V, Boutoleau-Bretonniere C, Etcharry-Bouyx F, Chauviré V, Sarazin M, le Ber I, Epelbaum S, Jonveaux T, Rouaud O, et al. APP, PSEN1, and PSEN2 mutations in early-onset Alzheimer disease: a genetic screening study of familial and sporadic cases. PLoS Med. 2017;14:e1002270. doi: 10.1371/journal.pmed.1002270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lascaratos G, Garway-Heath DF, Willoughby CE, Chau KY, Schapira AH. Mitochondrial dysfunction in glaucoma: understanding genetic influences. Mitochondrion. 2012;12:202–212. doi: 10.1016/j.mito.2011.11.004. [DOI] [PubMed] [Google Scholar]
- 64.Lee KS, Huh S, Lee S, Wu Z, Kim AK, Kang HY, Lu B. Altered ER-mitochondria contact impacts mitochondria calcium homeostasis and contributes to neurodegeneration in vivo in disease models. Proc Natl Acad Sci U S A. 2018;115:E8844–8853. doi: 10.1073/pnas.1721136115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lee S, Sheck L, Crowston JG, Van Bergen NJ, O’Neill EC, O’Hare F, Kong YX, Chrysostomou V, Vincent AL, Trounce IA. Impaired complex-I-linked respiration and ATP synthesis in primary open-angle glaucoma patient lymphoblasts. Invest Ophthalmol Vis Sci. 2012;53:2431–2437. doi: 10.1167/iovs.12-9596. [DOI] [PubMed] [Google Scholar]
- 66.Licher S, Darweesh SKL, Wolters FJ, Fani L, Heshmatollah A, Mutlu U, Koudstaal PJ, Heeringa J, Leening MJG, Ikram MK, Ikram MA. Lifetime risk of common neurological diseases in the elderly population. J Neurol Neurosurg Psychiatry. 2019;90:148–156. doi: 10.1136/jnnp-2018-318650. [DOI] [PubMed] [Google Scholar]
- 67.Liesa M, Shirihai OS. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab. 2013;17:491–506. doi: 10.1016/j.cmet.2013.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lin MW, Lin CC, Chen YH, Yang HB, Hung SY. Celastrol inhibits dopaminergic neuronal death of Parkinson’s disease through activating mitophagy. Antioxidants (Basel) 2019;9:37. doi: 10.3390/antiox9010037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Liu J, Liu W, Li R, Yang H. Mitophagy in Parkinson’s disease: from pathogenesis to treatment. Cells. 2019;8:712. doi: 10.3390/cells8070712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Liu Y, Lear TB, Verma M, Wang KZ, Otero PA, McKelvey AC, Dunn SR, Steer E, Bateman NW, Wu C, Jiang Y, Weathington NM, Rojas M, Chu CT, Chen BB, Mallampalli RK. Chemical inhibition of FBXO7 reduces inflammation and confers neuroprotection by stabilizing the mitochondrial kinase PINK1. JCI Insight. 2020;5:e131834. doi: 10.1172/jci.insight.131834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Luk KC, Song C, O’Brien P, Stieber A, Branch JR, Brunden KR, Trojanowski JQ, Lee VM. Exogenous alpha-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc Natl Acad Sci U S A. 2009;106:20051–20056. doi: 10.1073/pnas.0908005106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Malli R, Frieden M, Osibow K, Zoratti C, Mayer M, Demaurex N, Graier WF. Sustained Ca2+ transfer across mitochondria is Essential for mitochondrial Ca2+ buffering, sore-operated Ca2+ entry, and Ca2+ store refilling. J Biol Chem. 2003;278:44769–44779. doi: 10.1074/jbc.M302511200. [DOI] [PubMed] [Google Scholar]
- 73.Mandal A, Drerup CM. Axonal transport and mitochondrial function in neurons. Front Cell Neurosci. 2019;13:373. doi: 10.3389/fncel.2019.00373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.McMonnies C. Reactive oxygen species, oxidative stress, glaucoma and hyperbaric oxygen therapy. J Optom. 2018;11:3–9. doi: 10.1016/j.optom.2017.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mhyre TR, Boyd JT, Hamill RW, Maguire-Zeiss KA. Parkinson’s disease. Subcell Biochem. 2012;65:389–455. doi: 10.1007/978-94-007-5416-4_16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, Maguire C, Gammer TL, Mackey JR, Fulton D, Abdulkarim B, McMurtry MS, Petruk KC. Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med. 2010;2:31ra34. doi: 10.1126/scitranslmed.3000677. [DOI] [PubMed] [Google Scholar]
- 77.Minegishi Y, Nakayama M, Iejima D, Kawase K, Iwata T. Significance of optineurin mutations in glaucoma and other diseases. Prog Retin Eye Res. 2016;55:149–181. doi: 10.1016/j.preteyeres.2016.08.002. [DOI] [PubMed] [Google Scholar]
- 78.Mohammad K, Dakik P, Medkour Y, Mitrofanova D, Titorenko VI. Quiescence entry, maintenance, and exit in adult stem cells. Int J Mol Sci. 2019;20:2158. doi: 10.3390/ijms20092158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Moon JY, Kim HJ, Park YH, Park TK, Park EC, Kim CY, Lee SH. Association between open-angle glaucoma and the risks of Alzheimer’s and Parkinson’s diseases in South Korea: a 10-year nationwide cohort study. Sci Rep. 2018;8:11161. doi: 10.1038/s41598-018-29557-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Morrison SJ, Kimble J. Asymmetric and symmetric stem-cell divisions in development and cancer. Nature. 2006;441:1068–1074. doi: 10.1038/nature04956. [DOI] [PubMed] [Google Scholar]
- 81.Navarro A, Boveris A. The mitochondrial energy transduction system and the aging process. Am J Physiol Cell Physiol. 2007;292:C670–686. doi: 10.1152/ajpcell.00213.2006. [DOI] [PubMed] [Google Scholar]
- 82.Nicaise AM, Willis CM, Crocker SJ, Pluchino S. Stem cells of the aging brain. Front Aging Neurosci. 2020;12:247. doi: 10.3389/fnagi.2020.00247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.O’Brien LC, Keeney PM, Bennett JP., Jr Differentiation of human neural stem cells into motor neurons stimulates mitochondrial biogenesis and decreases glycolytic flux. Stem Cells Dev. 2015;24:1984–1994. doi: 10.1089/scd.2015.0076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Oliver DMA, Reddy PH. Small molecules as therapeutic drugs for Alzheimer’s disease. Mol Cell Neurosci. 2019;96:47–62. doi: 10.1016/j.mcn.2019.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ordureau A, Sarraf SA, Duda DM, Heo JM, Jedrychowski MP, Sviderskiy VO, Olszewski JL, Koerber JT, Xie T, Beausoleil SA, Wells JA, Gygi SP, Schulman BA, Harper JW. Quantitative proteomics reveal a feedforward mechanism for mitochondrial PARKIN translocation and ubiquitin chain synthesis. Mol Cell. 2014;56:360–375. doi: 10.1016/j.molcel.2014.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Osborne NN. Pathogenesis of ganglion “cell death” in glaucoma and neuroprotection: focus on ganglion cell axonal mitochondria. Prog Brain Res. 2008;173:339–352. doi: 10.1016/S0079-6123(08)01124-2. [DOI] [PubMed] [Google Scholar]
- 87.Ottoboni L, Merlini A, Martino G. Neural stem cell plasticity: advantages in therapy for the injured central nervous system. Front Cell Dev Biol. 2017;5:52. doi: 10.3389/fcell.2017.00052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Priault M, Salin B, Schaeffer J, Vallette FM, di Rago JP, Martinou JC. Impairing the bioenergetic status and the biogenesis of mitochondria triggers mitophagy in yeast. Cell Death Differ. 2005;12:1613–1621. doi: 10.1038/sj.cdd.4401697. [DOI] [PubMed] [Google Scholar]
- 89.Rangaraju V, Lewis TL, Jr, Hirabayashi Y, Bergami M, Motori E, Cartoni R, Kwon SK, Courchet J. Pleiotropic mitochondria: the influence of mitochondria on neuronal development and disease. J Neurosci. 2019;39:8200–8208. doi: 10.1523/JNEUROSCI.1157-19.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Reddy PH, Beal MF. Amyloid beta, mitochondrial dysfunction and synaptic damage: implications for cognitive decline in aging and Alzheimer’s disease. Trends Mol Med. 2008;14:45–53. doi: 10.1016/j.molmed.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Reddy PH, Yin X, Manczak M, Kumar S, Pradeepkiran JA, Vijayan M, Reddy AP. Mutant APP and amyloid beta-induced defective autophagy, mitophagy, mitochondrial structural and functional changes and synaptic damage in hippocampal neurons from Alzheimer’s disease. Hum Mol Genet. 2018;27:2502–2516. doi: 10.1093/hmg/ddy154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ritch R, Darbro B, Menon G, Khanna CL, Solivan-Timpe F, Roos BR, Sarfarzi M, Kawase K, Yamamoto T, Robin AL, Lotery AJ, Fingert JH. TBK1 gene duplication and normal-tension glaucoma. JAMA Ophthalmol. 2014;132:544–548. doi: 10.1001/jamaophthalmol.2014.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rosignol I, Villarejo-Zori B, Teresak P, Sierra-Filardi E, Pereiro X, Rodriguez-Muela N, Vecino E, Vieira HLA, Bell K, Boya P. The mito-QC reporter for quantitative mitophagy assessment in primary retinal ganglion cells and experimental glaucoma models. Int J Mol Sci. 2020;21:1882. doi: 10.3390/ijms21051882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rovelet-Lecrux A, Hannequin D, Raux G, Le Meur N, Laquerriere A, Vital A, Dumanchin C, Feuillette S, Brice A, Vercelletto M, Dubas F, Frebourg T, Campion D. APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat Genet. 2006;38:24–26. doi: 10.1038/ng1718. [DOI] [PubMed] [Google Scholar]
- 95.Ryan BJ, Hoek S, Fon EA, Wade-Martins R. Mitochondrial dysfunction and mitophagy in Parkinson’s: from familial to sporadic disease. Trends Biochem Sci. 2015;40:200–210. doi: 10.1016/j.tibs.2015.02.003. [DOI] [PubMed] [Google Scholar]
- 96.Sekine S, Youle RJ. PINK1 import regulation; a fine system to convey mitochondrial stress to the cytosol. BMC Biol. 2018;16:2. doi: 10.1186/s12915-017-0470-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med. 2016;8:595–608. doi: 10.15252/emmm.201606210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Seo BJ, Yoon SH, Do JT. Mitochondrial dynamics in stem cells and differentiation. Int J Mol Sci. 2018;19:3893. doi: 10.3390/ijms19123893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Shen YC, Ou DL, Hsu C, Lin KL, Chang CY, Lin CY, Liu SH, Cheng AL. Activating oxidative phosphorylation by a pyruvate dehydrogenase kinase inhibitor overcomes sorafenib resistance of hepatocellular carcinoma. Br J Cancer. 2013;108:72–81. doi: 10.1038/bjc.2012.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Spalding KL, Bergmann O, Alkass K, Bernard S, Salehpour M, Huttner HB, Bostrom E, Westerlund I, Vial C, Buchholz BA, Possnert G, Mash DC, Druid H, Frisen J. Dynamics of hippocampal neurogenesis in adult humans. Cell. 2013;153:1219–1227. doi: 10.1016/j.cell.2013.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M. alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with lewy bodies. Proc Natl Acad Sci U S A. 1998;95:6469–6473. doi: 10.1073/pnas.95.11.6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Stefanis L. alpha-Synuclein in Parkinson’s disease. Cold Spring Harb Perspect Med. 2012;2:a009399. doi: 10.1101/cshperspect.a009399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Szaruga M, Munteanu B, Lismont S, Veugelen S, Horre K, Mercken M, Saido TC, Ryan NS, De Vos T, Savvides SN, Gallardo R, Schymkowitz J, Rousseau F, Fox NC, Hopf C, De Strooper B, Chavez-Gutierrez L. Alzheimer’s-causing mutations shift Abeta length by destabilizing gamma-secretase-abetan interactions. Cell. 2017;170:443–456. doi: 10.1016/j.cell.2017.07.004. [DOI] [PubMed] [Google Scholar]
- 104.Toda T, Gage FH. Review: adult neurogenesis contributes to hippocampal plasticity. Cell Tissue Res. 2018;373:693–709. doi: 10.1007/s00441-017-2735-4. [DOI] [PubMed] [Google Scholar]
- 105.Toglia P, Cheung KH, Mak DO, Ullah G. Impaired mitochondrial function due to familial Alzheimer’s disease-causing presenilins mutants via Ca(2+) disruptions. Cell Calcium. 2016;59:240–250. doi: 10.1016/j.ceca.2016.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Truban D, Hou X, Caulfield TR, Fiesel FC, Springer W. PINK1, Parkin, and mitochondrial quality control: what can we learn about Parkinson’s disease pathobiology? J Parkinsons Dis. 2017;7:13–29. doi: 10.3233/JPD-160989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Tu H, Nelson O, Bezprozvanny A, Wang Z, Lee SF, Hao YH, Serneels L, De Strooper B, Yu G, Bezprozvanny I. Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer’s disease-linked mutations. Cell. 2006;126:981–993. doi: 10.1016/j.cell.2006.06.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG, Albanese A, Nussbaum R, González-Maldonado R, Deller T, Salvi S, Cortelli P, Gilks WP, Latchman DS, Harvey RJ, Dallapiccola B, et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science. 2004;304:1158–1160. doi: 10.1126/science.1096284. [DOI] [PubMed] [Google Scholar]
- 109.Van Bergen NJ, Crowston JG, Craig JE, Burdon KP, Kearns LS, Sharma S, Hewitt AW, Mackey DA, Trounce IA. Measurement of systemic mitochondrial function in advanced primary open-angle glaucoma and leber hereditary optic neuropathy. PLoS One. 2015;10:e0140919. doi: 10.1371/journal.pone.0140919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wanet A, Arnould T, Najimi M, Renard P. Connecting mitochondria, metabolism, and stem cell fate. Stem Cells Dev. 2015;24:1957–1971. doi: 10.1089/scd.2015.0117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Weil R, Laplantine E, Curic S, Genin P. Role of optineurin in the mitochondrial dysfunction: potential implications in neurodegenerative diseases and cancer. Front Immunol. 2018;9:1243. doi: 10.3389/fimmu.2018.01243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Weinreb RN, Aung T, Medeiros FA. The pathophysiology and treatment of glaucoma: a review. JAMA. 2014;311:1901–1911. doi: 10.1001/jama.2014.3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Wiggs JL, Pasquale LR. Genetics of glaucoma. Hum Mol Genet. 2017;26:R21–27. doi: 10.1093/hmg/ddx184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Winner B, Winkler J. Adult neurogenesis in neurodegenerative diseases. Cold Spring Harb Perspect Biol. 2015;7:a021287. doi: 10.1101/cshperspect.a021287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wong CW, Quaranta V, Glenner GG. Neuritic plaques and cerebrovascular amyloid in Alzheimer disease are antigenically related. Proc Natl Acad Sci U S A. 1985;82:8729–8732. doi: 10.1073/pnas.82.24.8729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Wu JH, Zhang SH, Nickerson JM, Gao FJ, Sun Z, Chen XY, Zhang SJ, Gao F, Chen JY, Luo Y, Wang Y, Sun XH. Cumulative mtDNA damage and mutations contribute to the progressive loss of RGCs in a rat model of glaucoma. Neurobiol Dis. 2015;74:167–179. doi: 10.1016/j.nbd.2014.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Xavier JM, Morgado AL, Sola S, Rodrigues CM. Mitochondrial translocation of p53 modulates neuronal fate by preventing differentiation-induced mitochondrial stress. Antioxid Redox Signal. 2014;21:1009–1024. doi: 10.1089/ars.2013.5417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Yang L, Youngblood H, Wu C, Zhang Q. Mitochondria as a target for neuroprotection: role of methylene blue and photobiomodulation. Transl Neurodegener. 2020;9:19. doi: 10.1186/s40035-020-00197-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ying H, Yue BY. Cellular and molecular biology of optineurin. Int Rev Cell Mol Biol. 2012;294:223–258. doi: 10.1016/B978-0-12-394305-7.00005-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zheng X, Boyer L, Jin M, Mertens J, Kim Y, Ma L, Ma L, Hamm M, Gage FH, Hunter T. Metabolic reprogramming during neuronal differentiation from aerobic glycolysis to neuronal oxidative phosphorylation. Elife. 2016;5:e13374. doi: 10.7554/eLife.13374. [DOI] [PMC free article] [PubMed] [Google Scholar]