

Key Words: ATG7, autophagy, cerebral infarction, cerebral ischemia/reperfusion injury, long non-coding RNA, miR-181c-5p, neuron, oxygen and glucose deprivation/reoxygenation
Abstract
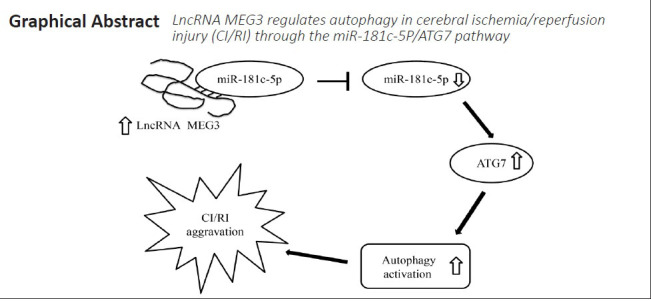
Severe cerebral ischemia/reperfusion injury has been shown to induce high-level autophagy and neuronal death. Therefore, it is extremely important to search for a target that inhibits autophagy activation. Long non-coding RNA MEG3 participates in autophagy. However, it remains unclear whether it can be targeted to regulate cerebral ischemia/reperfusion injury. Our results revealed that in oxygen and glucose deprivation/reoxygenation-treated HT22 cells, MEG3 expression was obviously upregulated, and autophagy was increased, while knockdown of MEG3 expression greatly reduced autophagy. Furthermore, MEG3 bound miR-181c-5p and inhibited its expression, while miR-181c-5p bound to autophagy-related gene ATG7 and inhibited its expression. Further experiments revealed that mir-181c-5p overexpression reversed the effect of MEG3 on autophagy and ATG7 expression in HT22 cells subjected to oxygen and glucose deprivation/reoxygenation. In vivo experiments revealed that MEG3 knockdown suppressed autophagy, infarct volume and behavioral deficits in cerebral ischemia/reperfusion mice. These findings suggest that MEG3 knockdown inhibited autophagy and alleviated cerebral ischemia/reperfusion injury through the miR-181c-5p/ATG7 signaling pathway. Therefore, MEG3 can be considered as an intervention target for the treatment of cerebral ischemia/reperfusion injury. This study was approved by the Animal Ethics Committee of the First Affiliated Hospital of Zhengzhou University, China (approval No. XF20190538) on January 4, 2019.
Chinese Library Classification No. R456; R743; Q522
Introduction
Cerebral ischemia-reperfusion injury (CI/RI) refers to the resumption of blood after a period of cerebral ischemia and usually occurs in the treatment stage of ischemic disease, which can aggravate the damage of brain tissue and even cause irreversible damage (Bai and Lyden, 2015). The pathogenesis of CI/RI is complex and includes apoptosis, oxidative stress, inflammation, and mitochondrial dysfunction (Bai and Lyden, 2015; Wu et al., 2018). Despite the significant amount of research on CI/RI, treating reperfusion injury remains a challenge for clinicians.
Recent studies have found that autophagy is activated during CI/RI (Xu and Zhang, 2011). Autophagy is a biological process that degrades damaged organelles and macromolecules in the cytoplasm through the lysosomal system and is widely involved in physiological and pathological processes (Parzych and Klionsky, 2014). Additionally, autophagy plays an important role in CI/RI. In addition to removing harmful substances and maintaining the stability of nerve cells it leads to nerve cell death (Xu and Zhang, 2011). Autophagy is modulated by evolutionarily conserved autophagy-related genes (ATG), and ATG7 is essential for the formation and closure of autophagosomes (Yu et al., 2018a; Levine and Kroemer, 2019). Knockdown of ATG7 improved the therapeutic effect of drugs by inhibiting autophagy in some diseases, like ovarian cancer and prolonged hypokalemia (Yu et al., 2018b; Kim et al., 2019). Furthermore, He et al. (2016) found that the ATG7 level was increased and autophagy was activated in cell models of CI/RI. However, the exact mechanism of ATG7 in CI/RI is unclear.
Long non-coding RNAs (lncRNAs) have been shown to participate in biological processes through a variety of mechanisms, and are involved in modulating the occurrence and progression of many diseases (Schmitz et al., 2016). In recent years, the abnormal expression of some lncRNAs including maternally expressed gene 3 (MEG3) in CI/RI models has been revealed by RNA-sequencing, deep sequencing, and microarrays techniques (Bao et al., 2018). Yan et al. (2017) found that MEG3 knockdown protected against ischemic damage and ameliorated neurological functions in CI/RI models. Moreover, MEG3 promoted morphine-induced autophagy in mouse hippocampal neuronal cell line HT22 (Gao et al., 2019). Moreover, several studies have demonstrated that MEG3 promoted CI/RI by increasing pyroptosis and that inhibition of MEG3 restored nerve growth and alleviated neurological impairment in CI/RI (You and You, 2019; Liang et al., 2020). Nevertheless, whether MEG3 affects CI/RI by regulating autophagy in neurons has not been reported.
The biological function of lncRNAs is usually relevant to microRNAs, and the interaction between lncRNAs and microRNAs plays important roles in many types of diseases. It has been reported that some lncRNAs can be used as competitive endogenous RNAs to suppress microRNA silencing to target genes during CI/RI. For instance, Cai et al. (2019) demonstrated that knockdown of lncRNA Gm11974 protected against cerebral ischemic reperfusion through NR3C2 by acting as a competitive endogenous RNA of miR-766-3p. Additionally, Guo et al. (2017) reported that inhibition of MALAT1 alleviated neuronal cell death by restraining autophagy via modulating miR-30a in cerebral ischemic stroke. In the current study, we explored the role and mechanism of MEG3 in autophagy in CI/RI.
Materials and Methods
Cell culture and oxygen and glucose deprivation/reoxygenation treatment
HT22 cell line, which had been authenticated by short tandem repeat (STR) markers, was obtained from American Type Culture Collection (Cat# AC337709, Gaithersburg, MD, USA). HT22 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum (Gibco, Grand Island, NY, USA) and 1% penicillin-streptomycin (Solarbio, Beijing, China). The cells were cultured at 37°C with 5% CO2. To mimic CI/RI in vitro, the cells were exposed to oxygen and glucose deprivation/reoxygenation (OGD/R) injury, the medium was changed into glucose-free DMEM and the cells were cultured at 37°C in a humidified incubator with 5% CO2 for 2 hours. Then, the glucose-free DMEM was replaced with normal medium. Cells in control group (without any treatment) and OGD/R treatment group (at 12, 24, and 48 hours) were harvested for analyses.
Cell transfection
Si-MEG3, si-NC (control), miR-181c-5p mimic, NC mimic, miR-181c-5p inhibitor, NC inhibitor, pcDNA-3.1, and pcDNA-3.1-MEG3 were synthesized by GenePharma (Shanghai, China). Rapamycin (RAPA; a mammalian target of rapamycin inhibitor) was purchased from Selleck (Cat# 53123-88-9, Houston, TX, USA). HT22 cells were seeded in 24-well plates; when they reached more than 80% confluence, they were transfected with pcDNA3.1/si-NC (100 μM) or co-transfected with MEG3-WT/MEG3-MUT (100 μM) and miR-181c-5p mimic/NC mimic (100 μM) for 48 hours. Lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA) was used as the transfection reagent according to standard protocols. RAPA (200 nM) was added to the cells and cultured for 48 hours. After transfection or incubation with RAPA, cells were harvested for further analysis. This procedure was performed before OGD/reoxygenation (OGD/R) treatment.
CI/RI mouse model
Forty C57BL/6 mice (male, 6–7 weeks old, 20–22 g weight) in specific pathogen-free conditions were purchased from Laboratory Animal Center of Zhengzhou University [SYXK (Yu) 2011-0001] and housed at 22–25°C with a 12-hour light/dark cycle with adequate food and water. The study was approved by the Animal Care Committee of the First Affiliated Hospital of Zhengzhou University, China (approval No. XF20190538) on January 4, 2019.
Mice were randomly assigned into four groups (n = 10): sham, middle cerebral artery occlusion/reperfusion (MCAO), MCAO + si-MEG3, and MCAO + si-NC.
The CI/RI mouse model was established by performing MCAO (Hu et al., 2012; Guo et al., 2017). The mice were anesthetized with pentobarbital sodium (35 mg/kg, CAS# 57-33-0, Hubei XinRunde Chemical Co., Ltd., Wuhan, China) by intraperitoneal injection in the right lower abdomen with a syringe. MCAO was performed with a 4-0 surgical nylon filament. After 60 minutes, the suture was removed and the blood flow was restored (reperfusion). Anesthesia and surgery procedures in the Sham group were the same, except for the MCAO.
Si-NC or si-MEG3 (both 100 μM, GenePharma) and Lipofectamine RNAiMAX Transfection Reagent (Invitrogen) were mixed according to the manufacturer’s instructions and incubated for 20 minutes. This solution (7 μL) was injected into the right cerebral ventricle immediately before the MCAO (Wang et al., 2017). Seven days later, the mice were euthanized by 100% CO2 inhalation at 25% cubage per minute for 3–4 minutes, and then the brain tissue was collected. Three mice of each group were used for 2,3,5-triphenyltetrazolium chloride (TTC) staining, four mice were used for quantitative real-time PCR (qRT-PCR) and western blotting, and the remaining three mice were used for terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) assay.
Cell Counting Kit-8 assay
HT22 cells were seeded in 96-well plates (3 × 103 cells/well). Ten microliters of Cell Counting Kit-8 (CCK8; Beyotime, Shanghai, China) was added to each well and incubated for 60 minutes. The optical density was measured and recorded at 450 nm. Five replicate wells were set for each sample. At least three independent experiments were performed on each group.
Quantitative real-time PCR
Total RNA of HT22 cells or brain tissue was extracted with Trizol (Invitrogen). The concentration and purity of the RNA extracts were determined by Nanodrop (Thermo Scientific, Cleveland, OH, USA); the acceptable range of the A260/A280 ratio was 1.9 to 2.1. Complementary DNAs were obtained and then quantified by an ABI 7500 Real-Time PCR system (Applied Biosystems, Waltham, MA, USA). The internal controls used were glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and U6. The primer sequences are listed in Additional Table 1.
Additional Table 1.
Primer sequences for quantitative real-time polymerase chain reaction
| Gene | Primer sequence |
|---|---|
| MEG3 | Forward: 5´-AGC GCTTCT GAAGAC CAAAC-3´ |
| Reverse: 5´-GAACACAAAAAAGACACC CAG CA-3´ | |
| miR-181c-5p | Forward: 5´-AACATT CAACCT GTC GGT GAG T-3´ |
| Reverse: 5´-CATGTCAGC CAG TGT TGAATG TC-3´ | |
| U6 | Forward: 5´-CTC GCTTCG GCAGCACA-3´ |
| Reverse: 5´-AAC GCTTCACGAATTTGC GT-3´ | |
| GAPDH | Forward:5´-GAC CTGACC TGC CGT CTA-3´ |
| Reverse: 5´-AGGAGT GGG TGT CGC TGT-3´ |
GAPDH: Glyceraldehyde 3-phosphate dehydrogenase; MEG3: maternally expressed gene 3.
Western blot assay
HT22 cells or brain tissue were lysed and the protein concentration was determined with a bicinchoninic acid kit (Beyotime). The proteins were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and then transferred to polyvinylidene difluoride membranes (Millipore, Temecula, CA, USA). The membranes were then incubated in blocking solution (Cat# SW3015BSA; Solarbio, Beijing, China), followed by incubation at 4°C overnight with antibodies for anti-mouse ATG7 (Cat# ab133528; 1:5000; Abcam, Cambridge, UK), anti-mouse Beclin1 (Cat# ab210498; 1:1000; Abcam), anti-mouse light chain 3 (LC3) (Cat# ab128025; 1:500; Abcam), or anti-mouse β-actin (Cat# ab227387; 1:5000; Abcam). Next, the membranes were incubated with goat anti-rabbit IgG secondary antibodies (Cat# ab97048; 1:5000; Abcam) for 3–4 hours at room temperature. The proteins were visualized with a chemiluminescence system (Beyotime) and imaged using the ECL Plus Western Blotting Substrate (Thermo Fisher). The quantitative analysis of the blots was conducted with ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Monodansylcadaverine staining
HT22 cells were seeded in 6-well plates with a coverslip in each well and different treatments were applied. Cells were collected, washed, and fixed. Then, the slides were exposed to 0.05 mM monodansylcadaverine (MDC; MilliporeSigma, St. Louis, MO, USA) at 37°C in the dark for 20 minutes. Slides were observed under an inverted fluorescence microscope (Olympus, Tokyo, Japan). Five fields were randomly selected for each group to quantify the number of MDC stained cells.
RNA fluorescence in situ hybridization assay
This assay was performed according to the guidelines of the Ribo™ lncRNA fluorescence in situ hybridization Probe Mix (RiboBio, Guangzhou, China). The cells were fixed in 4% paraformaldehyde (Solarbio) for 15 minutes at room temperature, and then incubated with 0.2% Triton X-100 for 10 minutes. Next, the cells were treated with proteinase K, glycine, and acetamidine reagent, followed by addition of 250 μL of prehybridization solution, which was then removed and replaced with hybridization solution containing the lncRNA MEG3 probe that was incubated overnight in the dark at 42°C. The mixture was washed three times with PBT solution and 4′,6-diamidino-2-phenylindole (MilliporeSigma) was added for 5 minutes. Finally, the samples were sealed with an anti-fluorescence quencher and the images were obtained using an inverted fluorescence microscope.
RNA immunoprecipitation
Argonaute 2 (Ago2)-RNA-immunoprecipitation (IP) assay was performed using the Magna RNA immunoprecipitation (RIP) RNA-Binding Protein Immunoprecipitation Kit (Millipore Corporation, Billerica, MA, USA) to immunoprecipitate miR-181c-5p and examine the interaction between miR-181c-5p and MEG3. Briefly, the miR-181c-5p mimic or NC mimic were transfected into HT22 cells, which were then collected and lysed with RIP lysis buffer. The cell lysate was incubated with RIP buffer with magnetic beads conjugated to anti-Ago2 or anti-IgG antibodies (negative control). Proteinase K was added to digest the protein, and the immunoprecipitates (Ago2-miR-181c-5p complex) were isolated. qRT-PCR analysis was used to detect MEG3 expression in the immunoprecipitates to determine the interaction of miR-181c-5p and MEG3.
Dual-luciferase activity assay
MEG3 and ATG7 3′-untranslated region were predicted by RNA Association Interaction Database 2.0 (http://www.rna-society.org/raid2/search.html) and TargetScan Human 7.2 (http://www.targetscan.org/vert_72/) to have binding sites for miR-181c-5p. These regions were cloned into pRL-TK vector (Promega, Madison, WI, USA) as the wild type (WT) MEG3 and ATG7 reporter vectors. The mutated (MUT) MEG3 and ATG7 reporter vectors were obtained from Invitrogen and served as negative controls. Next, the vectors and miR-181c-5p mimic were co-transfected into HT22 cells and cultured for 48 hours. The luciferase activity was determined by the dual-luciferase reporter system (Promega).
Mouse behavior assessment
Three mouse behaviors were assessed in this study. In the elevated plus maze test, two open arms (16 cm × 5 cm), two closed arms (16 cm × 5 cm × 12 cm), and one connecting central platform (5 cm × 5 cm) formed the maze (Wuhan Yihong Co., Ltd., Wuhan, China). Mice were put on the end of an open arm. The time it took the mice to enter the closed arm from the open arm with all the four legs was measured as transfer latency time (TLT). The test was performed on 3 consecutive days. The TLT record on day 3 was used as an indicator of normal memory, and the TLT record on day 4 was used as an indicator of the memory state after MCAO. After day 3, TLT was recorded, mice were subjected to CI/RI and this test was performed again (Itoh et al., 1990; Gupta et al., 2003).
The inclined beam-walking test was conducted to estimate the coordination of fore and hind limb movement. Mice were put on a metal bar (55 cm long and 1.5 cm wide) at 60° from the floor. The motor ability scores ranged from 0 to 4. A score of 0 indicated that the mouse easily went through the beam, and a score 4 meant that the mouse could hardly walk on the beam. This test was conducted before and 12 and 24 hours after CI/RI (Feeney et al., 1981; Kaur et al., 2017).
For the lateral push test, mice were put on a rough surface (plastic coated paper, counter protection paper, Kimberly-Clark, Irving, TX, USA) to enhance grip, and then we assessed the resistance to shoulder pushed on both sides. This test was conducted before and 12 and 24 hours after CI/RI. The resistance to a lateral push before MCAO was regarded as the primary resistance (100 score). Mice with decreased resistance to a lateral push after MCAO were assigned corresponding scores and the percentage relative to the primary resistance was calculated (Gupta et al., 2003).
TTC staining
The infarct volume was measured by TTC staining. The incubation of brain slices with 2% TTC (MilliporeSigma) was performed at 37°C for 25 minutes, and then the slices were fixed. The white areas are the cerebral infarction. Stained slices were observed and then the images were analyzed by ImageJ software. As previously reported (Gao et al., 2016; Han et al., 2018), the infarct volume was obtained by integrating the infarct areas in all sections, and it is expressed as a percentage of infarct area.
TUNEL assay
TUNEL was performed to evaluate cell death in brain tissues. Paraffin sections were dewaxed using xylene for 10 minutes. The xylene was replaced with fresh xylene and sections were dewaxed for another 10 minutes. Proteinase K (20 μg/mL; Beyotime) was added dropwise, and specimens were incubated for 25 minutes at room temperature after adding Enhanced Peroxidase Blocking Buffer (Beyotime). Cell death was detected with the Colorimetric TUNEL Kit (Beyotime) following the manufacturer’s guidelines. The staining results were observed and counted under a fluorescence microscope (BX60, Olympus, Tokyo, Japan). Six high power fields were selected for each sample and the average percentage of TUNEL-positive cells was calculated for statistical analysis.
Statistical analysis
Data were analyzed by SPSS (version 13.0, SPSS, Chicago, IL, USA) and are presented as mean ± standard deviation (SD). Unpaired t-test was used for comparison between two groups, and the difference among multiple groups was compared by one-way analysis of variance followed by the least significant difference post hoc test. P < 0.05 was considered statistically significant.
Results
Expression levels of MEG3 and ATG7 protein are increased and autophagy is elevated in OGD/R-induced HT22 cells
OGD/R-induced HT22 cells for 12, 24, and 48 hours were used to mimic CI/RI in vitro. The results showed that the cell viability of the reoxygenated cells gradually decreased for at least 48 hours (P < 0.001; Figure 1A). Additionally, the expression levels of MEG3 and ATG7 protein gradually increased for at least 48 hours (P < 0.001; Figure 1B–D). Furthermore, MDC staining showed that the autophagy in HT22 cells gradually increased until 48 hours (Figure 1E). Therefore, we reoxygenated OGD-induced cells for 48 hours in the following experiments.
Figure 1.

OGD/R elevates the expression of MEG3 and ATG7 protein and the autophagy level in HT22 cells.
HT22 cells were induced by OGD for 6 hours, followed by reoxygenation for different durations (12, 24, and 48 hours). The control group was normal HT22 cells without any treatment. (A) Cell viability was measured by the CCK8 assay. (B) The MEG3 expression was quantified by qRT-PCR and normalized to the control group. (C, D) The ATG7 protein level was evaluated by western blotting and normalized to the control group. (E) MDC staining was conducted to determine autophagy (arrows) in HT22 cells (original magnification 200×, scale bar: 100 μm). Autophagy in HT22 cells gradually increased until 48 hours. Data are representative of three independent experiments (mean ± SD). ***P < 0.001, vs. control group (one-way analysis of variance followed by the least significant difference post hoc test). ATG7: Autophagy-related gene 7; CCK8: Cell Counting Kit-8; CI/RI: cerebral ischemia-reperfusion injury; MDC: monodansylcadaverine; MEG3: maternally expressed gene 3; OD: optical density; OGD/R: oxygen and glucose deprivation/reoxygenation; qRT-PCR: quantitative real-time PCR.
Inhibition of MEG3 suppresses OGD/R-induced autophagy in HT22 cells
It has been has demonstrated that autophagy is activated in a CI/RI model (Xu and Zhang, 2011). To investigate whether MEG3 affects OGD/R-induced autophagy, we transfected si-MEG3 to knockdown MEG3 in HT22 cells subjected to OGD/R. We discovered that the expression of MEG3 was decreased after transfection of si-MEG3, implying the si-MEG3 transfection was effective (P < 0.001; Figure 2A). Compared with the control cells, the protein levels of ATG7, Beclin1, and LC3II/I were significantly elevated (P < 0.001; Figure 2B and C), cell autophagy (detected by MDC staining) was increased (P < 0.001; Figure 2D and E), and cell viability was decreased (P < 0.001; Figure 2F) in OGD/R-induced cells. These results implied that autophagy was activated after OGD/R treatment. We further found that MEG3 knockdown reduced the levels of ATG7, Beclin1, and LC3II/I (P < 0.01 or P < 0.001; Figure 2B and C), decreased cell autophagy (P < 0.001; Figure 2D and E), and increased cell viability (P < 0.001; Figure 2F) in OGD/R-induced cells. Next, we used RAPA to increase autophagy in OGD/R- induced HT22 cells and transfected si-MEG3 to examine the effect on autophagy. The results indicated that the levels of ATG7, LC3II/I, and Beclin1 were markedly elevated (P < 0.05 or P < 0.01; Figure 2B and C) and cell autophagy was increased (P < 0.01; Figure 2D and E), but the cell viability decreased in RAPA-treated OGD/R-induced cells (P < 0.01; Figure 2F). However, MEG3 knockdown reversed the effects of RAPA (P < 0.05, P < 0.01, or P < 0.001; Figure 2B–F). These data suggest that MEG3 knockdown inhibited OGD/R-induced autophagy in HT22 cells.
Figure 2.

Inhibition of MEG3 suppresses OGD/R-induced HT22 autophagy.
Control group: Normal cultured HT22 cells; OGD/R group: HT22 cells were reoxygenated for 48 hours after OGD treatment for 6 hours; OGD/R + si-NC group: HT22 cells were transfected with si-NC before OGD/R treatment; OGD/R + si-MEG3 group: HT22 cells were transfected with si-MEG3 before OGD/R treatment; OGD/R + RAPA group: HT22 cells were exposed to RAPA before OGD/R treatment; OGD/R + si-MEG3 + RAPA group: HT22 cells were transfected with si-MEG3 and then exposed to RAPA before treatment with OGD/R. (A) MEG3 expression was quantified by qRT-PCR and normalized to the control group. (B, C) ATG7, Beclin1, and LC3II/I protein levels were evaluated by western blotting. The target protein expression was normalized to the control group. (D, E) MDC staining was used to assess cell autophagy (arrows) in HT22 cells (scale bar: 100 μm; original magnification 200×). si-MEG3 transfection increased cell autophagy, and MEG3 knockdown decreased cell autophagy. (F) Cell viability was examined by the CCK8 assay. Data are representative of three independent experiments (mean ± SD). ***P < 0.001, vs. control group; &P < 0.05, &&P < 0.01, vs. OGD/R group; ##P < 0.01, ###P < 0.001, vs. OGD/R + si-NC group; $P < 0.05, $$P < 0.01, $$$P < 0.001, vs. OGD/R + si-MEG3 group (one-way analysis of variance followed by the least significant difference post hoc test). ATG7: Autophagy-related gene 7; CCK8: Cell Counting Kit-8; CI/RI: cerebral ischemia-reperfusion injury; LC3: light chain 3; MDC: monodansylcadaverine; MEG3: maternally expressed gene 3; OGD/R: oxygen and glucose deprivation/reoxygenation; qRT-PCR: quantitative real-time PCR; RAPA: rapamycin.
MiR-181c-5p interacts with MEG3 and ATG7 in OGD/R- induced HT22 cells
A previous study has revealed that miR-181c-5p expression was reduced in OGD-induced neuronal cells (Zhang et al., 2019). To elucidate whether miR-181c-5p interacts with MEG3 and ATG7, RNA fluorescence in situ hybridization assay was performed to identify the localization of MEG3, and then bioinformatics software (RNA Association Interaction Database 2.0 and TargetScan Human 7.2) were used to predict its interactions. The fluorescence in situ hybridization assay results showed that MEG3 was expressed in the cytoplasm of neuronal cells (Figure 3A), and miR-181c-5p had binding sites for MEG3 and ATG7 sequences (Figure 3B and I, respectively). Then, the dual-luciferase reporter assay was performed to verify the interactions in HT22 cells. We found that miR-181c-5p overexpression reduced the relative luciferase activity of MEG3-WT (P < 0.01), but it had no significant effect on MEG3-MUT (P > 0.05; Figure 3C). The RIP assay indicated that the MEG3 level was higher with Ago2 antibodies than it was with IgG antibodies (P < 0.001; Figure 3D). We then transfected the MEG3 overexpression plasmid or si-MEG3 into HT22 cells and found that MEG3 overexpression significantly decreased the miR-181c-5p level (P < 0.01; Figure 3E and F). Conversely, MEG3 knockdown significantly increased the miR-181c-5p expression (P < 0.001; Figure 3G and H). The above data suggest that MEG3 bound to miR-181c-5p and inhibited its expression in HT22 cells.
Figure 3.

MiR-181c-5p interacts with MEG3 and ATG7.
(A) The fluorescence in situ hybridization assay was performed to identify the cellular localization of MEG (red; original magnification 200×; scale bar: 10 μm). (B, C) The luciferase plasmid containing MEG3-WT or MEG3-MUT and miR-181c-5p mimic or control (NC mimic) were co-transfected into HT22 cells to examine luciferase activity. Red bases represent the complementary sequences and binding sites. Relative luciferase activity = firefly luciferase activity/renilla luciferase activity. (D) RIP assay was performed to verify the interaction of miR-181c-5p and MEG3. %Input = MEG3 enrichment/input (1) × 100. (E–H) MEG3 overexpression plasmid (MEG3), si-MEG3, or their control (pcDNA3.1 or si-NC, respectively) was transfected into HT22 cells. The levels of MEG3 and miR-181c-5p were quantified by qRT-PCR. The expression was normalized to the pcDNA3.1 or si-NC group. (I, J) The luciferase plasmid containing ATG7-WT or ATG7-MUT and miR-181c-5p mimic or control (NC mimic) were co-transfected into HT22 cells. Red bases represent the complementary sequences and binding sites. Luciferase activity was measured. (K–N) miR-181c-5p mimic, miR-181c-5p inhibitor, or their control (NC mimic or NC inhibitor, respectively) was transfected into HT22 cells. The levels of miR-181c-5p and ATG7 were measured by qRT-PCR and western blotting, respectively. The target miRNA/protein expression was normalized to the pcDNA3.1 or si-NC group. All data are representative of three independent experiments (mean ± SD). **P < 0.01, ***P < 0.001, vs. NC mimic, pcDNA3.1, or si-NC group; ###P < 0.001, vs. NC inhibitor (unpaired t-test in C–H and J–L; one-way analysis of variance followed by the least significant difference post hoc test in M). ATG7: Autophagy-related gene 7; CCK8: Cell Counting Kit-8; LC3: light chain 3; MDC: monodansylcadaverine; MEG3: maternally expressed gene 3; OGD/R: oxygen and glucose deprivation/reoxygenation.
Furthermore, we observed that miR-181c-5p overexpression remarkably reduced the relative luciferase activity of ATG7-WT (P < 0.01), but it had no significant effect on ATG7-MUT (P > 0.05 (Figure 3I and J). Moreover, transfection of miR-181c-5p mimic or miR-181c-5p inhibitor markedly promoted or decreased miR-181c-5p expression, respectively (P < 0.001 (Figure 3K and L). Additionally, miR-181c-5p knockdown increased the ATG7 protein level and miR-181c-5p overexpression had the opposite effect (P < 0.001 (Figure 3M and N). These findings indicated that miR-181c-5p bound to ATG7 and negatively regulated its expression in HT22 cells.
MEG3 regulates cell autophagy through the miR-181c-5p/ATG7 pathway
To further explore whether MEG3 regulates cell autophagy through the miR-181c-5p/ATG7 pathway, we transfected or co-transfected the MEG3 overexpression plasmid and miR-181c-5p mimic into HT22 cells prior to subjecting them to OGD/R, and then examined the effects on ATG7 expression and cell autophagy. We found that after transfection of the MEG3 overexpression plasmid, the protein levels of ATG7, Beclin1, and LC3II/I were significantly elevated (P < 0.001; Figure 4A and B), the cell autophagy analyzed by MDC staining was remarkably increased (P < 0.01; Figure 4C and D), but the cell viability decreased (P < 0.01; Figure 4E). Conversely, transfection of miR-181c-5p mimic into HT22 cells significantly downregulated the expression of ATG7, Beclin1, and LC3II/I (P < 0.05 or P < 0.001; Figure 4A and B), significantly decreased cell autophagy (P < 0.01; Figure 4C and D), and significantly increased the cell viability (P < 0.01; Figure 4E). Moreover, overexpression of miR-181c-5p reversed the above-mentioned effects of MEG3 overexpression (P < 0.05 or P < 0.001; Figure 4A–E). These results suggest that MEG3 regulated autophagy in OGD/R-induced HT22 cells through the miR-181c-5p/ATG7 pathway.
Figure 4.

MEG3 regulates cell autophagy during OGD/R through miR-181c-5p/ATG7.
HT22 cells were transfected or co-transfected with MEG3 overexpression plasmid (MEG3), miR-181c-5p mimic, and control (pcDNA3.1 + NC mimic). Then, the cells were reoxygenated for 48 hours after OGD treatment for 6 hours. pcDNA3.1 + NC mimic group: cells were co-transfected with empty plasmid (pcDNA3.1) and NC mimic; MEG3: cells were transfected with MEG3 overexpression plasmid; miR-181c-5p mimic group: cells were transfected with miR-181c-5p mimic; MEG3+ miR-181c-5p group: cells were co-transfected with MEG3 overexpression plasmid and miR-181c-5p mimic. (A, B) The levels of autophagy-specific proteins ATG7, Beclin1, and LC3II/I were examined by western blotting. The target protein expression was normalized to the pcDNA3.1 + mimic group. (C, D) MDC staining was used to measure cell autophagy (arrows) (scale bar: 100 μm; original magnification 200×). After transfection with miR-181c-5p mimic, cell autophagy was significantly decreased. Overexpression of miR-181c-5p reversed the effect of MEG3 overexpression on autophagy. (E) Cell viability was measured by the CCK8 assay. All data are representative of three independent experiments (mean ± SD). *P < 0.05, **P < 0.01, ***P < 0.001, vs. pcDNA3.1 + mimic group; #P < 0.05, ###P < 0.001, vs. MEG3 group (one-way analysis of variance followed by the least significant difference post hoc test). ATG7: Autophagy-related gene 7; CCK8: Cell Counting Kit-8; LC3: light chain 3; MDC: monodansylcadaverine; MEG3: maternally expressed gene 3; OD: optical density; OGD/R: oxygen and glucose deprivation/reoxygenation.
MEG3 knockdown inhibits autophagy and alleviates CI/RI in vivo
To verify the effects of MEG3 in vivo, the right ventricle of the mice was injected with si-MEG3 or si-NC, and then the CI/RI model was established. The results showed that compared with the Sham group, after CI/RI, there were many cerebral infarction areas in the mice (P < 0.001 (Figure 5A and B), MEG3 expression was markedly upregulated (P < 0.01; Figure 5C), miR-181c-5p was downregulated (P < 0.01; Figure 5D), the protein levels of ATG7, Beclin1, and LC3II/I were significantly increased (P < 0.01 or P < 0.001 (Figure 5E and F), and the cell death in brain tissue was significantly increased (P < 0.001 (Figure 5G and H). However, the si-MEG3 injection reversed these effects (P < 0.05 or P < 0.01; Figure 5A–H). The above results indicated that MEG3 knockdown inhibited cell autophagy in CI/RI model mouse brain tissue, thereby alleviating CI/RI.
Figure 5.

MEG3 knockdown inhibits autophagy and alleviates CI/RI.
The right ventricle of mice was transfected with si-MEG3 or si-NC, and then the CI/RI mouse model was established. Sham group: mice did not undergo MCAO procedure, but the other surgery was the same as that performed on the MCAO group; MCAO group: mice underwent MCAO surgery; MCAO+si-NC group: mice were injected with a mixture of si-NC and Lipofectamine RNAiMAX through the right cerebral ventricle, and then underwent the MCAO procedure; MCAO+si-MEG3 group: mice were injected with a mixture of MEG3 and Lipofectamine RNAiMAX through the right cerebral ventricle, and then underwent the MCAO procedure. (A, B) Brain tissue was stained with TTC, and the infarct volume (marked with yellow lines) was measured (n = 3 mice/group). (C, D) The expression of MEG3 and miR-181c-5p was quantified by qRT-PCR (n = 4 mice/group) and normalized to the Sham group. (E, F) The levels of autophagy-specific proteins ATG7, Beclin1, and LC3II/I were examined by western blotting (n = 4 mice/group). The target protein expression was normalized to the Sham group. (G, H) TUNEL assay was used to evaluate cell death in mouse brain tissues (scale bar: 3 μm; original magnification 400×). After CI/RI, cell death in the brain tissue was significantly increased and si-MEG3 injection reversed the cell death. Arrows indicate dead cells. Data are expressed as mean ± SD. **P < 0.01, ***P < 0.001, vs. Sham group; #P < 0.05, ##P < 0.01, vs. MCAO + si-NC group (one-way analysis of variance followed by the least significant difference post hoc test). ATG7: Autophagy-related gene 7; CI/RI: cerebral ischemia-reperfusion injury; LC3: light chain 3; MEG3: maternally expressed gene 3; qRT-PCR: quantitative real-time PCR; TTC: 2,3,5-triphenyltetrazolium chloride; TUNEL: terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling.
MEG3 knockdown ameliorates CI/RI-induced behavioral deficits
To further explore the effects of MEG3 on the behaviors of CI/RI-induced mice, we conducted the elevated plus maze test, the inclined beam-walking test, and the lateral push test. We found no significant changes in the TLT among the groups immediately after treatment, however, at 4 days after CI/RI, the TLT was significantly upregulated in the MCAO group (P < 0.001; Figure 6A). Furthermore, this result was partly reversed by si-MEG3 injection (P < 0.001; Figure 6A). The motor ability scores in the inclined beam-walking test increased 12 and 24 hours after CI/RI (P < 0.001), but si-MEG3 injection partly reversed this CI/RI effect (P < 0.001; Figure 6B). Furthermore, the resistance to lateral push was significantly declined in MCAO-induced mice at 12 and 24 hours (P < 0.001), but si-MEG3 injection reversed this effect (P < 0.001; Figure 6C). Collectively, these data suggest that MEG3 knockdown ameliorates the brain damage induced by CI/RI.
Figure 6.

MEG3 knockdown ameliorates CI/RI-induced behavioral deficits.
Si-MEG3 or si-NC was injected into the right ventricle of the mice, and then MCAO was performed for 1 hour, followed by reperfusion. (A) The TLT in the elevated plus maze test was measured immediately and 4 days after reperfusion. (B) The motor ability score in the inclined beam-walking test was determined at 0, 12, and 24 hours after reperfusion. (C) The lateral push resistance in the lateral push test was evaluated at 0, 12, and 24 hours after reperfusion. Data are expressed as mean ± SD (n = 4). ††P < 0.01, vs. MCAO group (one-way analysis of variance followed by the least significant difference post hoc test). CI/RI: Cerebral ischemia-reperfusion injury; MCAO: middle cerebral artery occlusion; MEG3: maternally expressed gene 3; TLT: transfer latency time.
Discussion
Previous studies have suggested that MEG3 is involved in the CI/RI process through different mechanisms. For instance, Liang et al. (2020) showed that MEG3 activated caspase 1 via the miR-485/AIM2 axis to promote pyroptosis during CI/R, thereby affecting CI/RI. You and You (2019) demonstrated that MEG3 knockdown promoted nerve growth and alleviated nerve injury by Wnt/β-catenin in CI/RI rat models. However, it has not been reported whether MEG3 influences CI/RI by regulating neuronal autophagy. Our study demonstrated that MEG3 knockdown inhibited autophagy in CI/RI models and alleviated CI/RI. These findings provide novel insights into the mechanisms by which MEG3 regulates CI/RI.
In recent years, it has been demonstrated in various CI/RI animal models that autophagy is activated in damaged brain tissue (Zhang et al., 2019; Sun et al., 2020). Further studies have shown that autophagy is activated in neurons, astrocytes, and endothelial cells in the ischemic region of the brain (Wen et al., 2008; Xu and Zhang, 2011; Sun et al., 2018). Autophagy is a double-edged sword in the CI/RI process. During the first few hours of reperfusion, activation of autophagy clears damaged organelles and promotes the recycling of energy and substances to protect nerve cells (Xu and Zhang, 2011; Chen et al., 2014). However, in the middle and later periods of the CI/RI, the excessive activation of autophagy can cause excessive degradation of unimpaired organelles and proteins, which ultimately leads to cell self-digestion and induces neural cell death (Gao et al., 2012). Therefore, autophagy regulation is considered as a potential therapeutic target for CI/RI. Our study demonstrated that MEG3 knockdown inhibited autophagy, reduced the protein levels of ATG7, Beclin1, and LC3II/I, and improved cell viability in the CI/RI model. These results indicate that inhibition of MEG3 can protect neurons from death by suppressing autophagy during reperfusion, thereby alleviating cerebral infarction and behavioral deficits in CI/RI. Although cell viability depends on multiple factors, we discovered it negatively correlated with cell autophagy in the MCAO model, which is consistent with a previous report (Pei et al., 2019). Autophagy activation during different periods of the CI/RI has different effects on neural cells and neurological deficits, and further exploration is clearly warranted to identify these effects.
The miR-181 family plays an important role in cerebrovascular diseases including CI/RI (Di et al., 2014; Volný et al., 2015). It has been suggested that miR-181c-5p expression is significantly reduced and is involved in neuronal apoptosis and inflammation during CI/RI (Zhang et al., 2019; Cao et al., 2020a). The prediction result of the Targetscan database suggested that miR-181c-5p binds the ATG7 3′-untranslated region. Our study identified this interaction and confirmed that miR-181c-5p inhibited ATG7 expression and suppressed autophagy in the CI/RI model. Similar to our results, Cao et al. (2020b) demonstrated that miR-485 targeted ATG7 and inhibited its expression in CI/RI. Zhang et al. (2020) indicated that miR-143 targeted ATG7 to regulate autophagy in acute myeloid leukemia cells. We also revealed that MEG3 bound miR-181c-5p and inhibited its expression. Furthermore, miR-181c-5p overexpression reversed the effects of MEG3 on autophagy and ATG7 in the in vitro CI/RI model. These results suggest that MEG3 can be used as a competitive endogenous RNA to inhibit the expression of miR-181c-5p and enhance the ATG7 level, thereby promoting autophagy and ameliorating brain tissue damage in CI/RI. Additionally, this study illustrated for the first time that MEG3 had an effect of ameliorating the behavioral deficits induced by CI/RI, thereby contributing to the behavioral field of CI/RI. However, miR-181c-5p may have other target genes, and ATG7 may be regulated by other miRNAs, hence further research is required.
The present study has a few limitations. We only demonstrated the autophagy regulation and potential mechanism of MEG3 in CI/RI in cells and mouse models; therefore, the clinical role of MEG3 should be further clarified in future studies. Moreover, our study explored the regulatory effect of MEG3 on ATG7, but it did not explore whether MEG3 also affects other ATG family members that co-regulate autophagy with ATG7. Other limitations of our study include the absence of morphological analysis by transmission electron microscopy, insufficient generalizability of the model used, and the limited age range of the animals. Thus, additional more comprehensive studies are needed to verify these limitations.
In summary, lncRNA MEG3 regulates autophagy in neurons and brain tissues in CI/RI models. Specifically, it regulates autophagy during CI/RI through the miR-181c-5p/ATG7 signaling pathway. This study improved our understanding of the role and potential mechanism of MEG3 in the pathogenesis and progression of CI/RI.
Additional file:
Additional Table 1: Primer sequences for quantitative real-time polymerase chain reaction.
Footnotes
C-Editor: Zhao M; S-Editors: Yu J, Li CH; L-Editors: Yu J, Song LP; T-Editor: Jia Y
Conflicts of interest: The authors declare no conflict of interest.
Financial support: None.
Institutional review board statement: The animal experimental procedures were approved by the Animal Care Committee of the First Affiliated Hospital of Zhengzhou University, China (approval No. XF20190538) on January 4, 2019.
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Data sharing statement: Datasets analyzed during the current study are available from the corresponding author on reasonable request.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
References
- 1.Bai J, Lyden PD. Revisiting cerebral postischemic reperfusion injury: new insights in understanding reperfusion failure, hemorrhage, and edema. Int J Stroke. 2015;10:143–152. doi: 10.1111/ijs.12434. [DOI] [PubMed] [Google Scholar]
- 2.Bao MH, Szeto V, Yang BB, Zhu SZ, Sun HS, Feng ZP. Long non-coding RNAs in ischemic stroke. Cell Death Dis. 2018;9:281. doi: 10.1038/s41419-018-0282-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cai J, Shangguan S, Li G, Cai Y, Chen Y, Ma G, Miao Z, Liu L, Deng Y. Knockdown of lncRNA Gm11974 protect against cerebral ischemic reperfusion through miR-766-3p/NR3C2 axis. Artif Cells Nanomed Biotechnol. 2019;47:3847–3853. doi: 10.1080/21691401.2019.1666859. [DOI] [PubMed] [Google Scholar]
- 4.Cao DW, Liu MM, Duan R, Tao YF, Zhou JS, Fang WR, Zhu JR, Niu L, Sun JG. The lncRNA Malat1 functions as a ceRNA to contribute to berberine-mediated inhibition of HMGB1 by sponging miR-181c-5p in poststroke inflammation. Acta Pharmacol Sin. 2020a;41:22–33. doi: 10.1038/s41401-019-0284-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cao Y, Pan L, Zhang X, Guo W, Huang D. LncRNA SNHG3 promotes autophagy-induced neuronal cell apoptosis by acting as a ceRNA for miR-485 to up-regulate ATG7 expression. Metab Brain Dis. 2020b;35:1361–1369. doi: 10.1007/s11011-020-00607-1. [DOI] [PubMed] [Google Scholar]
- 6.Chen W, Sun Y, Liu K, Sun X. Autophagy: a double-edged sword for neuronal survival after cerebral ischemia. Neural Regen Res. 2014;9:1210–1216. doi: 10.4103/1673-5374.135329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Di Y, Lei Y, Yu F, Changfeng F, Song W, Xuming M. MicroRNAs expression and function in cerebral ischemia reperfusion injury. J Mol Neurosci. 2014;53:242–250. doi: 10.1007/s12031-014-0293-8. [DOI] [PubMed] [Google Scholar]
- 8.Feeney DM, Boyeson MG, Linn RT, Murray HM, Dail WG. Responses to cortical injury: I. Methodology and local effects of contusions in the rat. Brain Res. 1981;211:67–77. doi: 10.1016/0006-8993(81)90067-6. [DOI] [PubMed] [Google Scholar]
- 9.Gao L, Jiang T, Guo J, Liu Y, Cui G, Gu L, Su L, Zhang Y. Inhibition of autophagy contributes to ischemic postconditioning-induced neuroprotection against focal cerebral ischemia in rats. PLoS One. 2012;7:e46092. doi: 10.1371/journal.pone.0046092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gao S, Li E, Gao H. Long non-coding RNA MEG3 attends to morphine-mediated autophagy of HT22 cells through modulating ERK pathway. Pharm Biol. 2019;57:536–542. doi: 10.1080/13880209.2019.1651343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gao Y, Chen T, Lei X, Li Y, Dai X, Cao Y, Ding Q, Lei X, Li T, Lin X. Neuroprotective effects of polydatin against mitochondrial-dependent apoptosis in the rat cerebral cortex following ischemia/reperfusion injury. Mol Med Rep. 2016;14:5481–5488. doi: 10.3892/mmr.2016.5936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guo D, Ma J, Yan L, Li T, Li Z, Han X, Shui S. Down-regulation of Lncrna MALAT1 attenuates neuronal cell death through suppressing Beclin1-dependent autophagy by regulating Mir-30a in cerebral ischemic stroke. Cell Physiol Biochem. 2017;43:182–194. doi: 10.1159/000480337. [DOI] [PubMed] [Google Scholar]
- 13.Gupta R, Singh M, Sharma A. Neuroprotective effect of antioxidants on ischaemia and reperfusion-induced cerebral injury. Pharmacol Res. 2003;48:209–215. doi: 10.1016/s1043-6618(03)00102-6. [DOI] [PubMed] [Google Scholar]
- 14.Han B, Zhang Y, Zhang Y, Bai Y, Chen X, Huang R, Wu F, Leng S, Chao J, Zhang JH, Hu G, Yao H. Novel insight into circular RNA HECTD1 in astrocyte activation via autophagy by targeting MIR142-TIPARP: implications for cerebral ischemic stroke. Autophagy. 2018;14:1164–1184. doi: 10.1080/15548627.2018.1458173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.He G, Xu W, Tong L, Li S, Su S, Tan X, Li C. Gadd45b prevents autophagy and apoptosis against rat cerebral neuron oxygen-glucose deprivation/reperfusion injury. Apoptosis. 2016;21:390–403. doi: 10.1007/s10495-016-1213-x. [DOI] [PubMed] [Google Scholar]
- 16.Hu X, Li P, Guo Y, Wang H, Leak RK, Chen S, Gao Y, Chen J. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke. 2012;43:3063–3070. doi: 10.1161/STROKEAHA.112.659656. [DOI] [PubMed] [Google Scholar]
- 17.Itoh J, Nabeshima T, Kameyama T. Utility of an elevated plus-maze for the evaluation of memory in mice: effects of nootropics, scopolamine and electroconvulsive shock. Psychopharmacology (Berl) 1990;101:27–33. doi: 10.1007/BF02253713. [DOI] [PubMed] [Google Scholar]
- 18.Kaur I, Kumar A, Jaggi AS, Singh N. Evidence for the role of histaminergic pathways in neuroprotective mechanism of ischemic postconditioning in mice. Fundam Clin Pharmacol. 2017;31:456–470. doi: 10.1111/fcp.12275. [DOI] [PubMed] [Google Scholar]
- 19.Kim WY, Nam SA, Choi A, Kim YM, Park SH, Kim HL, Kim H, Han KH, Yang CW, Lee MS, Kim YK, Kim J. Atg7-dependent canonical autophagy regulates the degradation of aquaporin 2 in prolonged hypokalemia. Sci Rep. 2019;9:3021. doi: 10.1038/s41598-019-39702-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Levine B, Kroemer G. Biological functions of autophagy genes: a disease perspective. Cell. 2019;176:11–42. doi: 10.1016/j.cell.2018.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liang J, Wang Q, Li JQ, Guo T, Yu D. Long non-coding RNA MEG3 promotes cerebral ischemia-reperfusion injury through increasing pyroptosis by targeting miR-485/AIM2 axis. Exp Neurol. 2020;325:113139. doi: 10.1016/j.expneurol.2019.113139. [DOI] [PubMed] [Google Scholar]
- 22.Parzych KR, Klionsky DJ. An overview of autophagy: morphology, mechanism, and regulation. Antioxid Redox Signal. 2014;20:460–473. doi: 10.1089/ars.2013.5371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pei X, Li Y, Zhu L, Zhou Z. Astrocyte-derived exosomes suppress autophagy and ameliorate neuronal damage in experimental ischemic stroke. Exp Cell Res. 2019;382:111474. doi: 10.1016/j.yexcr.2019.06.019. [DOI] [PubMed] [Google Scholar]
- 24.Schmitz SU, Grote P, Herrmann BG. Mechanisms of long noncoding RNA function in development and disease. Cell Mol Life Sci. 2016;73:2491–2509. doi: 10.1007/s00018-016-2174-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sun X, Wang D, Zhang T, Lu X, Duan F, Ju L, Zhuang X, Jiang X. Eugenol attenuates cerebral ischemia-reperfusion injury by enhancing autophagy via AMPK-mTOR-P70S6K pathway. Front Pharmacol. 2020;11:84. doi: 10.3389/fphar.2020.00084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sun Y, Zhang T, Zhang Y, Li J, Jin L, Sun Y, Shi N, Liu K, Sun X. Ischemic postconditioning alleviates cerebral ischemia-reperfusion injury through activating autophagy during early reperfusion in rats. Neurochem Res. 2018;43:1826–1840. doi: 10.1007/s11064-018-2599-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Volný O, Kašičková L, Coufalová D, Cimflová P, Novák J. microRNAs in cerebrovascular disease. Adv Exp Med Biol. 2015;888:155–195. doi: 10.1007/978-3-319-22671-2_9. [DOI] [PubMed] [Google Scholar]
- 28.Wang J, Zhao H, Fan Z, Li G, Ma Q, Tao Z, Wang R, Feng J, Luo Y. Long noncoding RNA H19 promotes neuroinflammation in ischemic stroke by driving histone deacetylase 1-dependent M1 microglial polarization. Stroke. 2017;48:2211–2221. doi: 10.1161/STROKEAHA.117.017387. [DOI] [PubMed] [Google Scholar]
- 29.Wen YD, Sheng R, Zhang LS, Han R, Zhang X, Zhang XD, Han F, Fukunaga K, Qin ZH. Neuronal injury in rat model of permanent focal cerebral ischemia is associated with activation of autophagic and lysosomal pathways. Autophagy. 2008;4:762–769. doi: 10.4161/auto.6412. [DOI] [PubMed] [Google Scholar]
- 30.Wu MY, Yiang GT, Liao WT, Tsai AP, Cheng YL, Cheng PW, Li CY, Li CJ. Current mechanistic concepts in ischemia and reperfusion injury. Cell Physiol Biochem. 2018;46:1650–1667. doi: 10.1159/000489241. [DOI] [PubMed] [Google Scholar]
- 31.Xu M, Zhang HL. Death and survival of neuronal and astrocytic cells in ischemic brain injury: a role of autophagy. Acta Pharmacol Sin. 2011;32:1089–1099. doi: 10.1038/aps.2011.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yan H, Rao J, Yuan J, Gao L, Huang W, Zhao L, Ren J. Long non-coding RNA MEG3 functions as a competing endogenous RNA to regulate ischemic neuronal death by targeting miR-21/PDCD4 signaling pathway. Cell Death Dis. 2017;8:3211. doi: 10.1038/s41419-017-0047-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.You D, You H. Repression of long non-coding RNA MEG3 restores nerve growth and alleviates neurological impairment after cerebral ischemia-reperfusion injury in a rat model. Biomed Pharmacother. 2019;111:1447–1457. doi: 10.1016/j.biopha.2018.12.067. [DOI] [PubMed] [Google Scholar]
- 34.Yu L, Chen Y, Tooze SA. Autophagy pathway: Cellular and molecular mechanisms. Autophagy. 2018a;14:207–215. doi: 10.1080/15548627.2017.1378838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yu Y, Zhang X, Tian H, Zhang Z, Tian Y. Knockdown of long non-coding RNA HOTAIR increases cisplatin sensitivity in ovarian cancer by inhibiting cisplatin-induced autophagy. J BUON. 2018b;23:1396–1401. [PubMed] [Google Scholar]
- 36.Zhang H, Kang J, Liu L, Chen L, Ren S, Tao Y. MicroRNA-143 sensitizes acute myeloid leukemia cells to cytarabine via targeting ATG7- and ATG2B-dependent autophagy. Aging (Albany NY) 2020;12:20111–20126. doi: 10.18632/aging.103614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang X, Liu Z, Shu Q, Yuan S, Xing Z, Song J. LncRNA SNHG6 functions as a ceRNA to regulate neuronal cell apoptosis by modulating miR-181c-5p/BIM signalling in ischaemic stroke. J Cell Mol Med. 2019;23:6120–6130. doi: 10.1111/jcmm.14480. [DOI] [PMC free article] [PubMed] [Google Scholar]