

An update of the etiology of Alzheimer’s disease (AD): The current theory of the etiology of AD and the guidelines for most of the wide-ranging treatments activities are built around amyloid and tau protein as causative agents of the disease (Atlante et al., 2020). At present, based on a comprehensive evaluation of existing and contemporary studies, important questions arise regarding the causal role of amyloid and tau protein in the pathogenesis of AD (Morris et al., 2018). Analyzes of the available evidence does not allow obvious conclusion that amyloid, and especially tau protein, plays a key role in the etiology of AD (Morris et al., 2018). Evaluation of new data shows that accumulation of amyloid and altered tau protein is not the main cause of AD and more research is needed (Morris et al., 2018). As for the two substances planted to contribute to the development of AD, recent data indicate that the changes of amyloid and tau protein level and structure is triggered by some as yet unspecified factors, and then the triggered amyloid and tau protein interact with each other, exerting synergistic toxic effects on neurons and damaged neurons initiate the development of AD (Morris et al., 2018). It is certain that the pathology of amyloid and tau protein is currently ruled out as the sole cause of the development of dementia, as it cannot explain why about half of the world’s population have accumulation of different kinds of amyloid plaques and neurofibrillary tangles in the absence of dementia (Atlante et al., 2020). In these people, it was observed that the accumulation of neurofibrillary tangles increased exponentially with age (Atlante et al., 2020). In addition, it was noted that the appearance of hippocampus atrophy in elderly people with normal cognitive performance was not dependent on the presence of level and structure of amyloid (Atlante et al., 2020). Moreover, a multicenter study found that in patients diagnosed with AD, about one-third of the cases had no brain amyloid (Atlante et al., 2020).
Thus, research clearly indicates that amyloid plaques and neurofibrillary tangles do not initiate AD and are only two of the many degenerative changes that occur in this disease. The above data suggest that continuing this approach will not solve the problem of the etiology of AD. These data clearly suggest that we should seek alternative views on the etiology of AD that are currently potentially under consideration, such as brain ischemia. We suggest that in order to thoroughly understand the etiology of AD, the amyloidocentric theory should be abandoned, without ruling out the role of amyloid. Experimental and clinical studies on brain ischemia provide ample evidence that ischemia is involved in the development of the phenotype and genotype of AD, suggesting that brain ischemia studies may be useful in elucidating the pathogenesis of AD (Figure 1) (Pluta et al., 2009; Kiryk et al., 2011; Gemmell et al., 2012; Lo et al., 2019).
Figure 1.
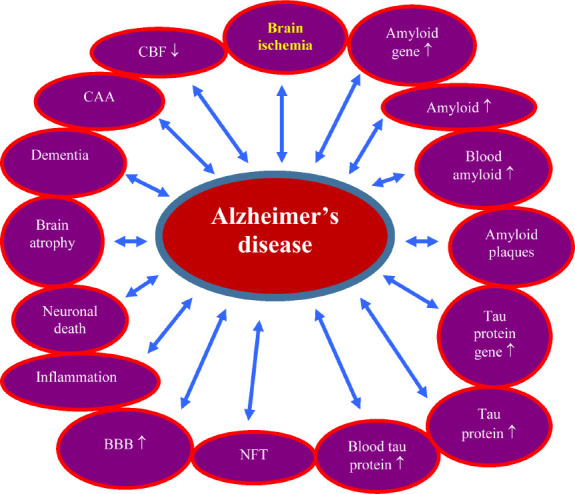
The role of ischemia in the development of the Alzheimer’s disease phenotype and genotype.
↑: Increase; ↓: decrease. BBB: Blood-brain barrier; CAA: cerebral amyloid angiopathy; CBF: cerebral blood flow; NFT: neurofibrillary tangles.
Post-ischemic dementia mimics dementia in AD: Currently, the number of post-ischemic cases worldwide is estimated at approximately 33 million (Bejot et al., 2016). If the trend of ischemic stroke continues worldwide, there will be 70 million stroke survivors in 2030, and more than 200 million disability-adjusted life years will be recorded annually (Bejot et al., 2016). A dangerous consequence for patients after brain ischemia is the progressive and irreversible development of dementia. The prevalence of dementia after the first ischemic stroke and recurrent stroke is estimated at 10% and 41%, respectively (Lo et al., 2019). Worldwide, post-ischemic dementia is estimated to occur in approximately 50% of patients (Lo et al., 2019). Experimental studies have also shown a synergistic link between cerebral ischemia and AD, which increases the risk of developing AD dementia (Figure 1) (Kiryk et al., 2011).
Post-ischemic neurodegeneration mimics neurodegeneration in AD: A reversible episode of cerebral ischemia in humans and animals causes massive neuronal death in the hippocampus and cerebral cortex. Death of neurons in the CA1 region of the ischemic hippocampus occurs within 7 days of survival (Pluta et al., 2009). Prolonged post-ischemic survival of up to 2 years results in additional neuronal loss in the CA3 region of the hippocampus (Pluta et al., 2009). The same kind of neuronal changes are seen in patients with ischemic brain injury (Gemmell et al., 2012). Ischemia opens the movement of cellular and non-cellular blood elements, e.g., platelets, lymphocytes, monocytes, neutrophils, and amyloid and tau protein, across the ischemic blood-brain barrier into brain. This consequently causes the development of vasoconstriction, amyloidosis, cerebral amyloid angiopathy, and inflammatory changes in the brain. Inflammatory factors are also triggered by microglia and astrocytes, which in turn causes a self-sustaining cycle that leads to progressive ischemic neurodegeneration of the brain. These processes trigger additional changes in neurons and their death, with permanent disruption of the neural network. Substances released by microglia and astrocytes, e.g. interleukin-1 and tumor necrosis factor α, further increase the permeability of the ischemic blood-brain barrier. This initiates secondary damage to already damaged brain. Changes in white matter with proliferation of neuroglial cells are observed in animals and humans post-ischemia. In the years following ischemia in humans and animals, ischemic neurodegenerative processes cause general brain atrophy, including the hippocampus.
Genes involved in the production of amyloid in post-ischemic brain: After experimental ischemic brain injury with a survival of up to 2 years, diffuse and senile amyloid plaques have been documented in the brain (Pluta et al., 2009). These data indicate that post-ischemia, the production of amyloid is additionally responsible for the progression of neurodegeneration that worsen the outcome of ischemia through progressive neuronal death. Also in humans after cerebral ischemia, both diffuse and senile amyloid plaques have been observed in the hippocampus and cerebral cortex.
In the CA1 area of the ischemic hippocampus with a 2-day survival, the expression of the amyloid protein precursor gene fell below the control value (Kocki et al., 2015). But 7–30 days post-ischemia, expression of this gene increased above the control value (Kocki et al., 2015). Expression of the β-secretase gene increased above the control value 2–7 days post-ischemia (Kocki et al., 2015). In contrast, at 30 days post-ischemia, the expression of the β-secretase gene fell below the control value (Kocki et al., 2015). But the expression of the presenilin 1 and 2 genes was above the control value at 2–7 days post-ischemia (Kocki et al., 2015). Thirty days post-ischemia, the expression of these genes fell below the control values (Kocki et al., 2015).
In the CA3 area, 2, 7 and 30 days post-ischemia, the expression of the amyloid protein precursor gene increased above the control values (Pluta et al., 2020). In contrast, expression of the α-secretase gene fell below the control values 2, 7 and 30 days post-ischemia (Pluta et al., 2020). Expression of the β-secretase gene also fell below the control value but only for 2–7 days post-ischemia. However, at 30 days post-ischemia, β-secretase gene expression increased above the control value (Pluta et al., 2020). Presenilin 1 gene expression was above the control value for 2–7 days post-ischemia. In contrast, at 30 days post-ischemia, the expression of this gene fell below the control value (Pluta et al., 2020). Presenilin 2 gene expression decreased at 2–7 days post-ischemia, but at 30 days post-ischemia it increased above the control value (Pluta et al., 2020).
The tau protein gene in the post-ischemic brain: Post-ischemia, intense staining of the tau protein was observed in neurons in the hippocampus and the cerebral cortex. Neurofibrillary tangles were also found in post-ischemic brain (Hatsuta et al., 2019). In the CA1 region of the ischemic hippocampus, tau protein gene expression was above the control value for 2 days, but at 7–30 days post-ischemia, gene expression dropped below the control value (Pluta et al., 2018). In the CA3 region, the expression of the tau protein gene decreased for 2 days post-ischemia, but within 7–30 days post-ischemia, gene expression increased above the control value (Pluta et al., 2020).
Conclusion: The combination of amyloid and tau protein does not lead to the development of AD, and the fact that ischemia may play a role in AD has led a handful of scientists to believe that research should be directed at understanding the interactions between these three factors. Increased levels of amyloid in the brain following ischemia cause amyloid accumulation in the vessel wall with primary atherosclerosis, leading to the development of cerebral amyloid angiopathy (Pluta et al., 2009). The development of cerebral amyloid angiopathy (Pluta et al., 2009) limits the transport of energy substrates to the brain across the ischemic blood-brain barrier, and also reduces clearance of amyloid from the brain. Additionally, factors released during neuroinflammation increase the leakage of the blood-brain barrier. An additional increase in the permeability of the blood-brain barrier allows inflammatory cells, amyloid and tau protein to travel from the circulatory system to the brain, leading to a vicious cycle. It is clear from the available evidence that much remains to be understood about the role of ischemia in the etiology of AD and to aid in the development of preventive and therapeutic strategies.
Footnotes
P-Reviewers: Radenovic L, Lehotsky J; C-Editors: Zhao M, Liu WJ, Qiu Y; T-Editor: Jia Y
Copyright license agreement: The Copyright License Agreement has been signed by the author before publication.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open peer reviewers: Lidija Radenovic, University of Belgrade, Serbia; Ján Lehotsky, Comenius University, Slovakia.
References
- 1.Atlante A, Amadoro G, Bobba A, Latina V. Functional foods: an approach to modulate molecular mechanisms of Alzheimer’s disease. Cells. 2020;9:2347. doi: 10.3390/cells9112347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bejot Y, Daubail B, Giroud M. Epidemiology of stroke and transient ischemic attacks: Current knowledge and perspectives. Rev Neurol. 2016;172:59–68. doi: 10.1016/j.neurol.2015.07.013. [DOI] [PubMed] [Google Scholar]
- 3.Gemmell E, Bosomworth H, Allan L, Hall R, Khundakar A, Oakley AE, Deramecourt V, Polvikoski TM, O’Brien JT, Kalaria RN. Hippocampal neuronal atrophy and cognitive function in delayed post-stroke and aging-related dementias. Stroke. 2012;43:808–814. doi: 10.1161/STROKEAHA.111.636498. [DOI] [PubMed] [Google Scholar]
- 4.Hatsuta H, Takao M, Nogami A, Uchino A, Sumikura H, Takata T, Morimoto S, Kanemaru K, Adachi T, Arai T, Hasegawa M, Murayama S. Tau and TDP-43 accumulation of the basal nucleus of Meynert in individuals with cerebral lobar infarcts or hemorrhage. Acta Neuropathol Commu. 2019;7:49. doi: 10.1186/s40478-019-0700-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kiryk A, Pluta R, Figiel I, Mikosz M, Ułamek M, Niewiadomska G, Jabłoński M, Kaczmarek L. Transient brain ischemia due to cardiac arrest causes irreversible long-lasting cognitive injury. Behav Brain Res. 2011;219:1–7. doi: 10.1016/j.bbr.2010.12.004. [DOI] [PubMed] [Google Scholar]
- 6.Kocki J, Ułamek-Kozioł M, Bogucka-Kocka A, Januszewski S, Jabłoński M, Gil-Kulik P, Brzozowska J, Petniak A, Furmaga-Jabłońska W, Bogucki J, Czuczwar SJ, Pluta R. Dysregulation of amyloid precursor protein, β-secretase, presenilin 1 and 2 genes in the rat selectively vulnerable CA1 subfield of hippocampus following transient global brain ischemia. J Alzheimers Dis. 2015;47:1047–1056. doi: 10.3233/JAD-150299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lo JW, Crawford JD, Desmond DW, Godefroy O, Jokinen H, Mahinrad S, Bae HJ, Lim JS, Kohler S, Douven E, Staals J, Chen C, Xu X, Chong EJ, Akinyemi RO, Kalaria RN, Ogunniyi A, Barbay M, Roussel M, Lee BC, et al. Profile of and risk factors for post-stroke cognitive impairment in diverse ethno-regional groups. Neurology. 2019;93:e2257–2271. doi: 10.1212/WNL.0000000000008612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morris GP, Clark IA, Vissel B. Questions concerning the role of amyloid-β in the definition, aetiology and diagnosis of Alzheimer’s disease. Acta Neuropathol. 2018;136:663–689. doi: 10.1007/s00401-018-1918-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pluta R, Ułamek M, Jabłoński M. Alzheimer’s mechanisms in ischemic brain degeneration. Anat Rec. 2009;292:1863–1881. doi: 10.1002/ar.21018. [DOI] [PubMed] [Google Scholar]
- 10.Pluta R, Bogucka-Kocka A, Ułamek-Kozioł M, Bogucki J, Czuczwar SJ. Ischemic tau protein gene induction as an additional key factor driving development of Alzheimer’s phenotype changes in CA1 area of hippocampus in an ischemic model of Alzheimer’s disease. Pharmacol Rep. 2018;70:881–884. doi: 10.1016/j.pharep.2018.03.004. [DOI] [PubMed] [Google Scholar]
- 11.Pluta R, Ułamek-Kozioł M, Kocki J, Bogucki J, Januszewski S, Bogucka-Kocka A, Czuczwar SJ. Expression of the tau protein and amyloid protein precursor processing genes in the CA3 area of the hippocampus in the ischemic model of Alzheimer’s disease in the rat. Mol Neurobiol. 2020;57:1281–1290. doi: 10.1007/s12035-019-01799-z. [DOI] [PMC free article] [PubMed] [Google Scholar]