

Abstract
Objective:
Despite considerable research, the goal of finding non-surgical remedies against thoracic aortic aneurysm and acute aortic dissection (TAAD) remains elusive. We sought to identify a novel aortic protein kinase that can be pharmacologically targeted to mitigate aneurysmal disease in a well-established mouse model of early onset progressively severe Marfan syndrome (MFS).
Approach and Results:
Computational analyses of transcriptomic data derived from the ascending aorta of MFS mice predicted a probable association between TAAD development and the multifunctional, stress-activated homeodomain-interacting protein kinase 2 (HIPK2). Consistent with this prediction, Hipk2 gene inactivation significantly extended the survival of MFS mice by slowing aneurysm growth and delaying transmural rupture. HIPK2 also ranked among the top predicted protein kinases in computational analyses of genes differentially expressed in the dilated aorta of three MFS patients, which strengthened the clinical relevance of the experimental finding. Additional in silico analyses of the human and mouse data sets identified the TGFβ/Smad3 signaling pathway as a potential target of HIPK2 in the MFS aorta. Chronic treatment of MFS mice with an allosteric inhibitor of HIPK2-mediated stimulation of Smad3 signaling validated this prediction by mitigating TAAD pathology and partially improving aortic material stiffness.
Conclusions:
HIPK2 is a previously unrecognized determinant of aneurysmal disease and an attractive new target for anti-TAAD multidrug therapy.
Keywords: Acute aortic dissection, fibrillin-1, HIPK2, Marfan syndrome, thoracic aortic aneurysm
Subject Terms: Aneurysm, animal models of human diseases, computational biology, pharmacology, translational studies
Introduction
Aneurysms involving the aortic root and the proximal segment of the ascending aorta are life-threatening pathologies that can predispose the dilated vessel to acute medial dissection and transmural rupture (ruptured TAAD).1 Histopathological findings of aneurysmal disease include progressive degeneration of the medial layer associated with de-differentiation and loss of smooth muscle cells (SMCs), fragmentation of elastic fibers, and excessive accumulation of collagen (vascular fibrosis) and proteoglycans.1,2 Marfan syndrome (MFS) is the most common syndromic presentation of ruptured TAAD.3–5 Standard care currently includes chronic treatment with anti-hypertensive drugs to slow the rate of aneurysm growth and prophylactic surgery to prevent premature death from aortic dissection and rupture. However, more effective drug-based therapies are needed because TAAD progression is highly heterogeneous and surgical repair carries significant morbidity/mortality risk.1,3–5
MFS is a connective tissue disorder caused by mutations in the large extracellular matrix (ECM) glycoprotein, fibrillin-1.4,5 Studies using mouse models of MFS have demonstrated that, aside from impairing ECM integrity, fibrillin-1 deficiency also perturbs several signaling pathways that regulate aortic function and homeostasis.4,5 Components of these disease-related pathways are potential targets for drug interventions that, once validated in MFS mice, could be translated into more effective treatments for MFS patients. Unfortunately, development of such evidence-based therapies is severely hampered by our limited understanding of how disease-driving signaling pathways interact with one another during aneurysm development. A case in point is the controversial role of TGFβ signaling during thoracic aneurysm progression. Early pharmacological findings with MFS mice that rarely dissect (Fbn1C1039G/+ mice) suggested a primary pathogenic role of TGFβ signaling.6,7 However, subsequent studies of both Fbn1C1039G/+ mice and MFS mice that invariably die from ruptured TAAD (Fbn1mgR/mgR mice) demonstrated a protective role of baseline TGFβ signaling during early stage of aneurysm formation, and a pathogenic role of enhanced TGFβ signaling at later stages of arterial disease.10 Hence, there is promise for temporally separated combinational drug treatments that differentially target early and late determinants of TAAD onset and progression without interfering with protective or compensatory pathways.10
To overcome the predictive limitations of examining individual genes and pathways in isolation, we recently integrated transcriptomic data derived from aortic tissue of MFS mice and aortic SMCs of MFS patients to construct a network of pathways involved in whole cell function. This integration enabled us to computationally predict and experimentally demonstrate that GABAB receptor agonist baclofen is a promising new drug treatment against arterial disease.11 Using a similarly unbiased systems pharmacology approach, here we identified the multi-functional, stress-activated homeodomain-interacting protein kinase 2 (HIPK2) as a previously unrecognized cellular component involved in TAAD pathogenesis. We also present preclinical evidence supporting the notion that pharmacological inhibition of HIPK2 activity may represent a novel therapy to restrict TAAD development in MFS patients.
Materials and Methods
Mice:
Hypomorphic Fbn1 mice12 (herein referred to as MFS mice), wild type (WT) and Hipk2 conditional null (Hipk2Lox/Lox) mice13 were maintained on the C57BL/6J genetic background, whereas R26CreERT2 mice (Jackson mouse repository; stock no. 004847) were maintained on the mixed C57BL/6J;129T2/SvEmsJ background. A total of 399 mice were analyzed to generate the data presented in this study. This number of animals does not include MFS mice that either died of non-TAAD (i.e: pulmonary insufficiency, ~5%) or were sacrificed for humane end-points (i.e.: anal and/or uterine prolapse; ~15%)10–12. Of the 197 animals enrolled in the gene inactivation experiments, 112 were used to generate the survival curves (30 WT, 35 MFS, 16 Hipk2R−/R−, 31 MFS;Hipk2R−/R−), and examine aneurysm growth and histopathology in animals still alive at P90. Separate groups of mice were sacrificed at 4 different time points to estimate Hipk2 transcript levels (5 WT and 5 MFS per time point), at 3 different time points for RNASeq experiments (3 WT and 3 MFS per time point), at P60 to asses HIPK2 protein levels (9 WT, 9 MFS) or at P90 to monitor tamoxifen effect on TAAD progression (5 MFS untreated and 4 MFS treated). Of the 192 animals enrolled in the pharmacological experiments, 27 WT mice were used for pharmacokinetics and 119 to generate the survival curves (36 WT untreated, 24 WT treated, 25 MFS untreated, 34 MFS treated) and to examine aneurysm growth and histopathology in animals still alive at P90. Two other separate groups of P60 mice were used for biomechanics (8 WT untreated, 8 WT treated, 6 MFS untreated, 4 MFS treated) and to estimate p-Smad3 protein levels (5 WT untreated, 5 WT untreated, 5 MFS untreated, 5 MFS treated). Kaplan-Meier survival curves were generated from birth to P90, when surviving mice were sacrificed for tissue collection. All experiments employed male mice to ensure consistency with previous work using the same MFS model by avoiding possible sex-related differences in TAAD pathology. The Institutional Animal Care and Use Committees of the Icahn School of Medicine at Mount Sinai in New York City reviewed and approved the animal studies.
Genetic and pharmacological gene inactivation:
Hipk2Lox recombination was induced by intraperitoneal tamoxifen (Sigma) injections for five consecutive days (P1 to P5) at a daily dose of 100 mg/kg of body weight; control mice were injected with vehicle (corn oil).13 Gene recombination was monitored by DNA amplification using primers flanking exon 3 (5’-CCCCACATTTGTGACATTCC-3’ and 5’-CAGTGAAGCATGTCTGTGGG-3’), whereas expression of the recombined gene was estimated by RT-PCR (forward and reverse primers: 5’-CCGATGAAGAAGAGGAGCAG-3’ and 5’-TTGCCAGTGCAGTGATTCTC-3’, respectively).13 BT173 or vehicle (DMSO) was administered daily to mice by gavage from P16 until P90 at the concentration of 20 mg/kg of body weight in saline supplemented with 5% DMSO.13 BT173 was synthesized by Richman Chemical Inc. at 99.1% purity, as evidenced by HPLC analysis; NMR data confirmed the chemical structure and absence of any residual solvent. For pharmacokinetic analyses, plasma was collected from 30-day-old WT mice treated with BT173 at 0, 0.25hr, 0.5hr, 1hr, 1.5hr, 2hr, 4hr, 6hr, 8hr and 12hr and sacrificed by cardiac puncture at the indicated time points (n=3 per time point). Sample preparation including calibration curves, quality controls, LC-MS/MS, and data analysis were performed using verapamil as an internal standard.11 Aneurysm progression was evaluated using the VisualSonics Vevo 2100 imaging system equipped with a 40-MHz transducer, using long-axis parasternal views of the aorta in mice under isoflurane anesthesia.11 Two-dimensional videos captured in the B-mode were used to measure aortic root and ascending aorta diameter and analyzed with VisualSonics software. Two individuals blinded to genotype and treatment performed measurements in systole from 3 images per mouse and averaged values to obtain corresponding diameters per mouse. Tail-cuff blood pressure measurements were performed during 3 consecutive days on 3-month-old conscious mice acclimated to the procedure for 2 prior days using the CODA noninvasive blood pressure system (Kent Scientific).11 Average systolic, diastolic, and pulse pressure values were measured for each animal.
Patients:
Three male MFS patients who underwent aortic root surgery (patients FB490, FB1334 and FB2271) were recruited through the National Reference Center for MFS and Related Disorders (Hôpital Bichat) and diagnosed according to established clinical criteria at the age of 29 (FB490), 31 (FB1334) and 30 years (FB2271). Genetic and clinical features of patient FB490 have already been described.11 Patient FB1334 [FBN1 mutation: c.6661T>C – p.(Cys2241Arg)] underwent surgery at the age of 31 when aortic dilatation (50mm) was discovered without mitral valve prolapse (valve sparing aortic surgery). He presented scoliosis, wrist and thumb signs, dolichostenomelia, high arched palate, pes planus, striae and ectopia lentis associated with myopia. Patient FB2271 [FBN1 mutation: c.6038-?_6163+? (exon4 deletion)] underwent surgery at the age of 30 when aortic dilatation (53mm) was discovered. He presented wrist and thumb signs, dolichostenomelia, high arched palate, striae, and he had surgery for bilateral ectopia lentis at 2 years of age. MFS aortic specimens were collected during elective aortic surgery. Normal ascending aortas were obtained from 3 male organ transplant donors (AS1066, AS1088, and AS1207) aged 24, 21, and 30 years, respectively. Mean age at the time of aortic tissue collection was 30 years for MFS patients and 25 years for non-MFS individuals. The clinical research protocol was approved by the local ethical committee (CPP 05 04 32, Ambroise Paré, Boulogne, France, April 2005; updated in March 2008). Aortic specimens were collected from MFS patients during elective aortic surgery (Hôpital Bichat) and used in compliance with French regulations (Declaration of Conservation DC-2008–283). Ascending aortas from organ transplant donors were obtained with the authorization of the French Biomedicine Agency (PFS 09–007) and in accordance with the Declaration of Helsinki. Informed consent was obtained from all patients in agreement with the requirements of French bioethics laws.
RNA sequencing:
Human samples were collected from the outer curvature of the most dilated part of the proximal ascending aorta, stripped off of the adventitia layer and stored at −80°C immediately after surgery. Samples were cryogenically pulverized in liquid nitrogen with a freezer mill (model 6750 SPEX SamplePrep) and homogenized in RNAse-free distilled water. Total RNA was purified using RNeasy Plus Mini kit (Qiagen S.A.) according to the manufacturer’s instructions followed by DNAse digestion and estimates of RNA concentration and quality by measuring 260 nm absorbance and by agarose gel electrophoresis, respectively. Mouse RNA was extracted from frozen samples of the proximal ascending aorta spanning from immediately above the aortic root to immediately before the brachiocephalic artery. Libraries were prepared from 1 μg of total RNA using NEBNext® Ultra™ II Directional RNA Library Prep Kit (human libraries) or TruSeq Stranded mRNA kit protocol (mouse libraries) according to the supplier’s recommendations. Sequencing of random paired-end 75-bp or single ended 100-bp molecules was carried out on an Illumina HiSeq 4000 for human and mouse samples, respectively.
Computational analyses:
Sequencing reads were aligned to the human reference genome ‘hg19’ or mouse reference genome ‘mm9’ using Tophat 2.0.8., samtools-0.1.7, and bowtie 2.1.0. Differentially expressed genes (DEGs) were identified with cufflinks 1.3.0.11 All human and mouse samples were considered as separate sets and DEGs were identified between experimental (MFS) and control samples in each of them following our published protocol.14 DEGs were identified based on an FDR of 5% and a minimum absolute fold change (log2 [(FPKMcondition1 + 1)/(FPKMcondition2 + 1)]) of log2(1.3). Each of the four DEG lists was uploaded on the eXpression2Kinases (X2KWeb) computational pipeline to sequentially identify enriched transcription factor (TF)-DNA binding sites, sub-networks of TF-related protein–protein interactions and ranked lists of enriched TF-interacting protein kinases (PKs).15,16 For each dataset, top five predicted PKs were considered for further analysis. To predict pathways that regulate gene expression, we subjected the proteins of the sub-networks to enrichment analysis using Gene Ontology (GO) Biological Pathways 2018 that we downloaded from the Enrichr website.17,18. To focus on signaling pathways we only considered children of the GO term “signaling”, based on the “is_a” and “part_of” relationships obtained from www.geneontology.org.
Histological analyses:
Aortic specimens spanning from immediately above the aortic root to immediately before the brachiocephalic artery were harvested from control and experimental mice and fixed overnight in 10% formalin followed by paraffin embedding and sectioning at 5 μm.10,11 Sections were stained with hematoxylin and eosin (H&E), Masson’s trichrome, or Verhoeff–van Gieson stain. Images were acquired using a Leica microscope (Model DM2500) with a ×40 objective and analyzed using ImageJ (NIH). For tissue cellularity (H&E staining), nuclei were counted and normalized to area. The density of elastic (Verhoeff–van Gieson staining) and collagen fibers (Masson’s trichrome staining) were quantified using the ImageJ analysis software color threshold tool to define the area fraction of elastic fibers or collagen, and the ratio of their respective densities to the surface of the tunica media area was calculated. For each histological assessment, fifteen 5μm-thick consecutive aortic sections were imaged and independently analyzed by two individuals blinded to genotype and treatment.10,11
RNA and protein analyses:
In addition to the aforementioned Hipk2 primers, the q-PCR assay utilized Hprt forward (5’-GCAGTACAGCCCCAAAATGG-3’) and reverse (5’-GGTCCTTTTCACCAGCAAGCT-3’) primers; all amplified products were analyzed using the ΔΔCT method.10,11 For protein analyses, total protein extracts were processed for immunoblots using antibodies against Smad3 (Cell Signaling Technology; catalog # 9513) and against p-Smad3 (Abcam; catalog # ab52903). All antibodies were diluted 1:1000 in Tris-buffered saline, pH 7.4, and 0.1% (v/v) Tween 20 supplemented with 5% nonfat milk and incubated with the transfer membrane for 16 hours at 4°C.10,11 In all cases, Ponceau S solution (Sigma-Aldrich; catalog # P7170) staining of the transfer membrane was used as a protein loading control. Immunoreactive products were visualized by chemiluminescence using Amersham ECL Select Western Blotting Detection Reagent (GE Healthcare) and their relative intensity was evaluated using Photoshop (Adobe Systems Inc.).10,11
Biomechanics:
For ex vivo assessment of passive biaxial mechanical properties, following euthanasia the ascending aorta (from the root to brachiocephalic) was excised, cleaned of excess perivascular tissue, cannulated on glass micropipets, and placed within a custom computer-controlled testing device in a Hanks buffered physiologic solution maintained at 37°C. Material behaviors were quantified using a validated constitutive relation based on cyclic pressure-diameter and axial force-length tests.11
Statistics:
A 2 × 2 Factorial Analysis of Variance design (ANOVA) was used to compare the mean differences among groups that consist of two independent factors. If significant interactions were present, the comparison of different treatments were based on establishing Contrast tests - i.e. a linear combination of 2 or more factor level means with coefficients that sum to zero. When appropriate, differences among means where three or more groups were compared were analyzed using a one-way analysis of variance (ANOVA), with post hoc Tukey’s testing following Bartlett’s test for equal variances. If data were not assumed to be normal, the Kruskal Wallis test was employed followed by the Dwass, Steel, Critchlow-Fligner method for multiple comparison testing. All values were expressed as the mean ± SD. In the biochemical experiments, statistically significant differences were highlighted by asterisks signifying *p≤0.05, **p≤0.01,***p≤0.001. For experiments involving only two independent groups, data were tested to satisfy the assumption of normality. If this was not achieved, the nonparametric version of the Student’s t test, i.e. the Wilcoxon Rank Sum test was used instead.The Kaplan Meier product limit estimate was used to analyze survival distributions. Group comparisons were compared by the log rank test. P values were not adjusted for the multiple tests that were performed, protecting against the inflation of the Type I error. Analyses were performed using SAS System Software, Version 9.4 and graphs were constructed using GraphPad Prism version 7.0 (GraphPad Software Inc.).
Data and materials availability:
RNA-Seq data can be downloaded from the NCBI Gene Expression Omnibus GEO GSE128101 and GSE145903.
Results
Hipk2 is a computationally predicted contributor to TAAD pathogenesis
We sought to predict PKs related to TAAD pathogenesis from computational analyses of DEGs identified in the proximal ascending aortas of MFS mice vs. WT littermates harvested 16, 30 and 60 days after birth. PKs were chosen because they often regulate the activity of TFs, and changes in PK activities often lead to disease by dysregulating gene expression. Indeed, PKs are widely accepted drug targets for many types of diseases.
RNA sequencing (RNA-Seq) experiments revealed the number and identity of abnormally upregulated and down-regulated genes in the ascending aorta of MFS mice relative to their WT littermates (Fig. 1A and Supplemental Table I). DEGs in each of the three experimental settings were analyzed using the online application X2KWeb to identify and rank putative TAAD-related PKs and associated molecular pathways.14,15 X2K uses a three step approach to identify regulatory PKs as potential drug targets. First, it predicts regulatory TFs from DEGs; second, it connects TFs by a protein subnetwork predicted to regulate TF activities and thereby gene expression; and third, enrichment analysis of subnetwork proteins (including TFs) leads to prediction of regulatory PKs.
Figure 1. Computational analyses of DEGs in the aortas of MFS mice and patients.
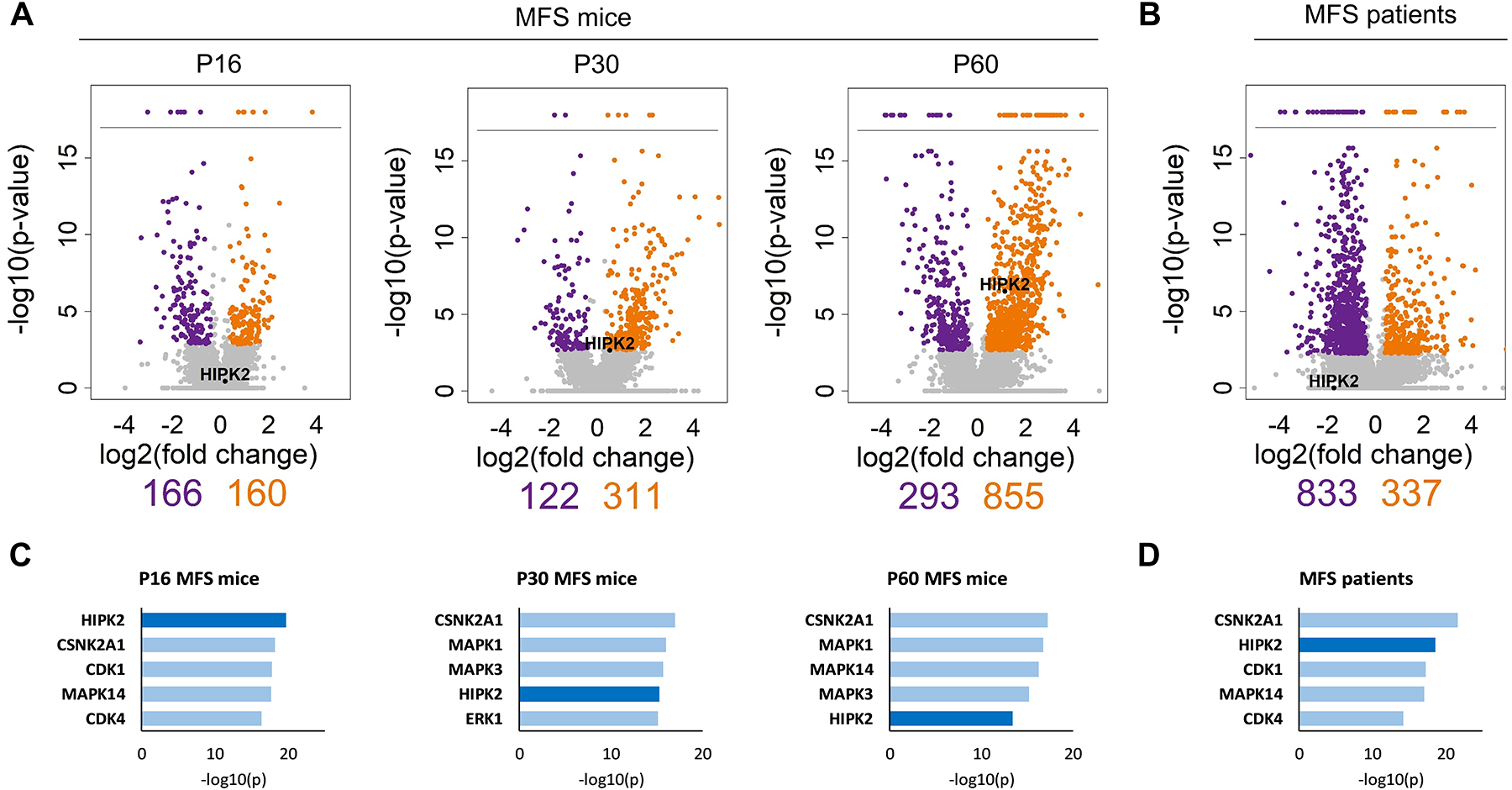
Volcano plots of DEGs in (A) aortas of MFS mice sacrificed at P16, P30 and P60 (n = 3 per genotype) and (B) aortas of MFS patients aged between 22 and 32 years (n = 3 per genotype). The log2 of fold change is plotted against the negative log10 of the p-value; orange dots indicate significantly upregulated genes, and purple dots indicate significantly downregulated genes. The number of significantly down- or upregulated genes in mouse and human samples are shown immediately below the Volcano plots; relative positions of Hipk2 expression in the mouse plots are also shown. Below are the top five PKs predicted through X2KWeb based on each list of DEGs obtained from MFS mice (C) and MFS patients (D)
HIPK2, p38 MAPK (MAPK14) and casein kinase II (CSNK2A) consistently scored at the top of the three mouse PK lists (Fig. 1C and Supplemental Table II). Ranking MAPK14 among the top PKs in 2 of the 3 data sets provide evidence for the potential validity of our computational predictions, as different experimental reports have demonstrated that MAPK14 is a major TAAD determinant in both fibrillin-1 deficient mice and MFS patient-derived iPSC differentiated into vascular SMCs.19,20 Although CSNK2A is an important developmental PK that regulates multiple signaling pathways21 and merits further study, this project focused on HIPK2 because of the PK’s established contribution to TGFβ-driven tissue fibrosis,13,22 which is a major histopathological finding in TAAD.5
Postnatal inactivation of the Hipk2 gene mitigates TAAD progression
To test our computational prediction, TAAD progression was compared in MFS mice with or without inactivation of the Hipk2 gene. Given the impaired fitness of germline Hipk2−/− mice,23 the PK was inactivated in all postnatal tissues of either WT or MFS mice by using Hipk2 conditional null (Hipk2Lox/Lox) mice and tamoxifen-inducible R26CreERT2 mice so as to produce Hipk2R−/R− and MFS;Hipk2R−/R− mice, respectively. DNA genotyping and q-PCR assays of mice injected with tamoxifen soon after birth (P1–P5) documented Cre-induced recombination of floxed Hipk2 alleles in the ascending aortas of Hipk2R−/R− and MFS;Hipk2R−/R− mice (Supplemental Fig. I). Like WT littermates, HIPK2-deficient mice showed no aortic phenotype or changed survival over 90 days (Fig. 2). HIPK2 deficiency in MFS mice significantly extended survival by slowing aneurysm growth and delaying death from acute dissection and rupture of the vessel wall (Fig. 2A and B). Consistent with these findings, elastic fiber fragmentation and vascular fibrosis were substantially attenuated in MFS;Hipk2R−/R− mice relative to MFS littermates (Fig. 2C). Postnatal loss of HIPK2 activity in MFS mice had no effects on blood pressure, thus excluding that reduced hemodynamic load might contribute to the protection (Supplemental Table III). Likewise, no major TAAD modifications were observed in a small cohort of MFS mice injected at P16 with tamoxifen once a day for 5 consecutive days, and sacrificed at P90 when aneurysm size and elastic fiber fragmentation are most evident (Supplemental Fig. II).This last finding is in contrast with the reported attenuation of experimental abdominal aortic aneurysm in rats harboring subcutaneous pellets that delivered tamoxifen continuously for 14 to 21 days.24 Differences in tamoxifen dosing and disease models may account for this apparent discrepancy.
Figure 2. HIPK2 contribution to TAAD progression in MFS mice.
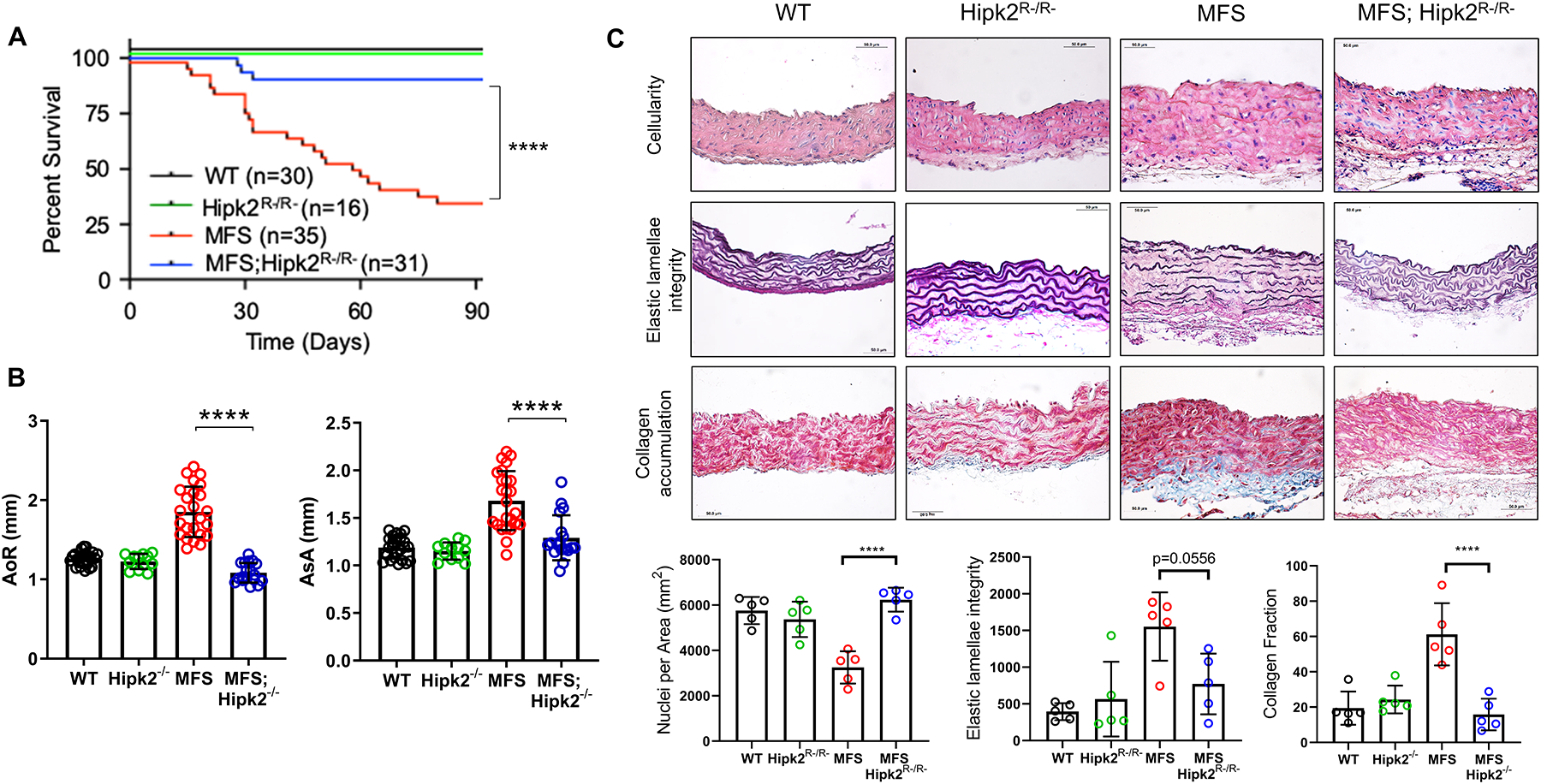
(A) Kaplan-Meier survival curves of mice of the indicated phenotype along with the number of animals examined in each group (n=16–35). The log rank test determined significance between the two groups of mice (p <0.0001). (B) Diameters of the aortic root (AoR; left) and proximal ascending aorta (AsA; right) in 3-month-old mice of the indicated genotypes (n = 16–35 mice per genotype). (C) Representative images of cross-sections of proximal ascending aortas of the indicated genotypes stained with Hematoxylin and Eosin (H&E) for nuclei count (upper panels; p <0.0001), Verhoeff–van Gieson for elastic fiber architecture (middle panels; p =0.0556), and Masson’s trichrome for collagen accumulation (lower panels; p <0.0001). Quantification of the data is shown in the plots below the panels (n= 5 mice per genotype). 2 × 2 Factorial ANOVA to test for interaction followed by main effect comparisons by CONTRAST statements were employed to determine statistically significant differences which are indicated by asterisks (****p<0.0001). Error bars indicate means ± SD
Computational analyses of DEGs identified in the medial layer of the dilated aortas harvested from three MFS patients ranked HIPK2 among the top PKs in the human specimens as well (Fig. 1B and D, and Supplemental Tables II and IV). Based on this evidence, we monitored Hipk2 expression during aneurysm development in MFS mice. Q-PCR amplification assays revealed abnormal accumulation of Hipk2 transcripts in the ascending aorta of MFS mice from early (P16) to advanced (P90) stages of TAAD development (Fig. 3A). Furthermore, immunoblots of total proteins isolated from the ascending aortas of 2-month-old MFS mice showed substantially more HIPK2 accumulation than in the WT counterparts (Fig. 3B). Absence of upregulated Hipk2 transcripts in some of the DEG data sets (Fig. 1A and Supplemental Tables I and IV) conceivably reflects the documented problem that RNA-Seq experiments, even with sufficient depth of sequencing (20 million reads), do not always capture changes in levels of all mRNAs that are observed by q–PCR assays.25 This point notwithstanding, our experiments revealed a previously unrecognized involvement of HIPK2 in TAAD pathogenesis.
Figure 3. Hipk2 expression in the ascending aorta of MFS mice.
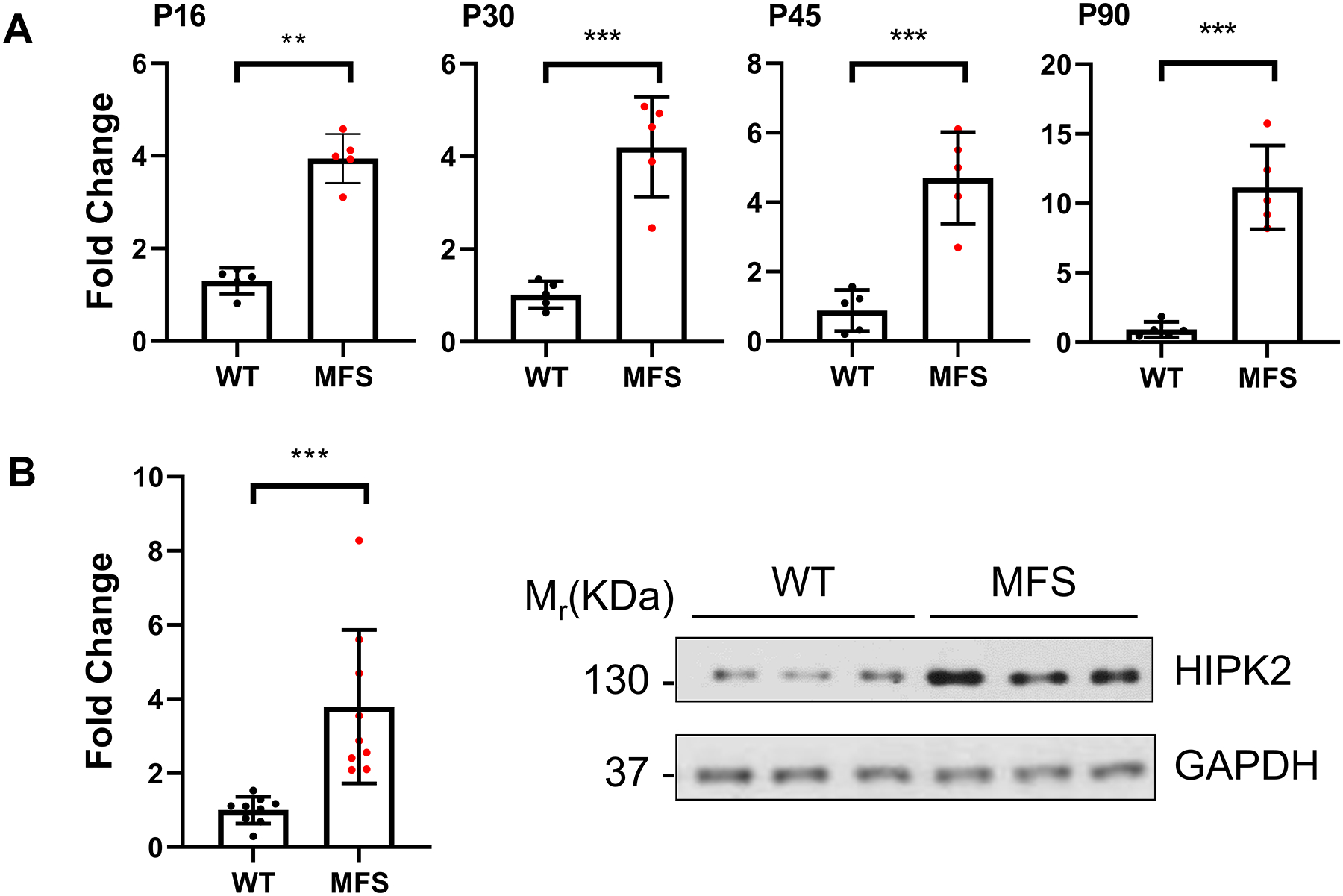
(A) Real-time q-PCR measurements of Hipk2 expression levels on total RNAs collected at the indicated time points from the proximal ascending aorta of MFS mice and WT littermates (n=5 per genotype and time point). (B) Representative immunoblots of HIPK2 detected in the proximal ascending aorta of P60 mice of the indicated genotypes (n=9 per genotype). Bar graphs summarize the relative levels of Hipk2 transcripts and HIPK2 protein, respectively; error bars indicate means with SD (p <0.05). The nonparametric Wilcoxon Rank sum test was used to compare the two groups. Statistically significant differences are indicated by asterisks (**p<0.01, ***p<0.001, ****p<0.0001).
Inhibition of HIPK2-dependent Smad3 signaling attenuates TAAD progression
Previous work has demonstrated that HIPK2 promotes TGFβ-mediated kidney fibrosis in part through interaction with Smad3.13,22 As vascular fibrosis is an histopathological hallmark of TAAD,1–5 we searched for correlative evidence of a possible connection between HIPK2 and TGFβ by focusing on differentially activated signaling pathways that may induce dysregulated gene expression in the MFS aorta. Accordingly, the human and mouse protein subnetworks (including TFs) used to predict HIPK2 dysregulation in the MFS aorta were subjected to pathway enrichment analysis. This step is the same as the last one performed by the X2Kweb pipeline, except that we replaced the KEA database by Gene Ontology and focused only on signaling pathways. Proteins associated with TGFβ signaling were significantly enriched in all subnetworks, suggesting a role of this pathway in driving differential gene expression in the MFS aorta (Supplemental Table V and Supplemental Fig. III). Investigation of potential HIPK2 protein targets in the mice and human subnetworks identified Smad3 in all data sets (Supplemental Tables II and V, Supplemental Fig. IV). Based on these findings, we hypothesized that HIPK2 dysregulation in the MFS aorta may contribute to pathogenic TGFβ/Smad3 pathway signaling and that pharmacological inhibition of this HIPK2 activity could be therapeutic.
To experimentally test the above hypothesis, WT and MFS mice were treated with a small molecule compound (BT173) that allosterically inhibits HIPK2-mediated stimulation of Smad3 signaling.13,26BT173 was administered at a daily dose of 20 mg/kg of body weight for 74 days starting at P16.13,26 Pharmacokinetic analyses were first employed to determine BT173 concentration in the plasma of treated mice.11 Plasma samples were collected at nine discrete time points (n= 3 per time point) following the dose until 12hr after administration. Quantitation of BT173 levels in plasma samples was achieved by LC/MS/MS and yielded the following pharmacokinetic parameters: an observed Cmax of 24.5 ng/mL at a Tmax of 1hr and elimination half-life of 9.8hr (Supplemental Table VI). Like genetic Hipk2 inactivation, BT173 administration had no effect on body weight or blood pressure of experimental and control animals, or on fitness and survival of WT mice (data not shown).
Consistent with BT173 mode of action,26 aortic p-Smad3 levels were reduced in BT173-treated compared to vehicle-treated MFS mice (Fig. 4A). Importantly, systemic administration of the inhibitor of HIPK2-dependent Smad3 signaling extended significantly the survival of MFS mice as result of slowing aneurysmal growth and delaying ruptured TAAD (Fig 4B and C). Compared with vehicle-treated MFS mice, BT173 treatment also improved medial cellularity and elastic fiber morphology, and mitigated excessive collagen accumulation (Fig. 5). TAAD mitigation in BT173-treated mice suggested a causal relationship among HIPK2 upregulation, increased Smad3-mediated signaling and aneurysm progression. Comparable degrees of TAAD mitigation by either Hipk2 gene inactivation or HIPK2 protein inhibition provided additional evidence excluding a beneficial effect of tamoxifen on aneurysmal disease in MFS mice.
Figure 4. TAAD progression in MFS mice systemically treated with BT173.
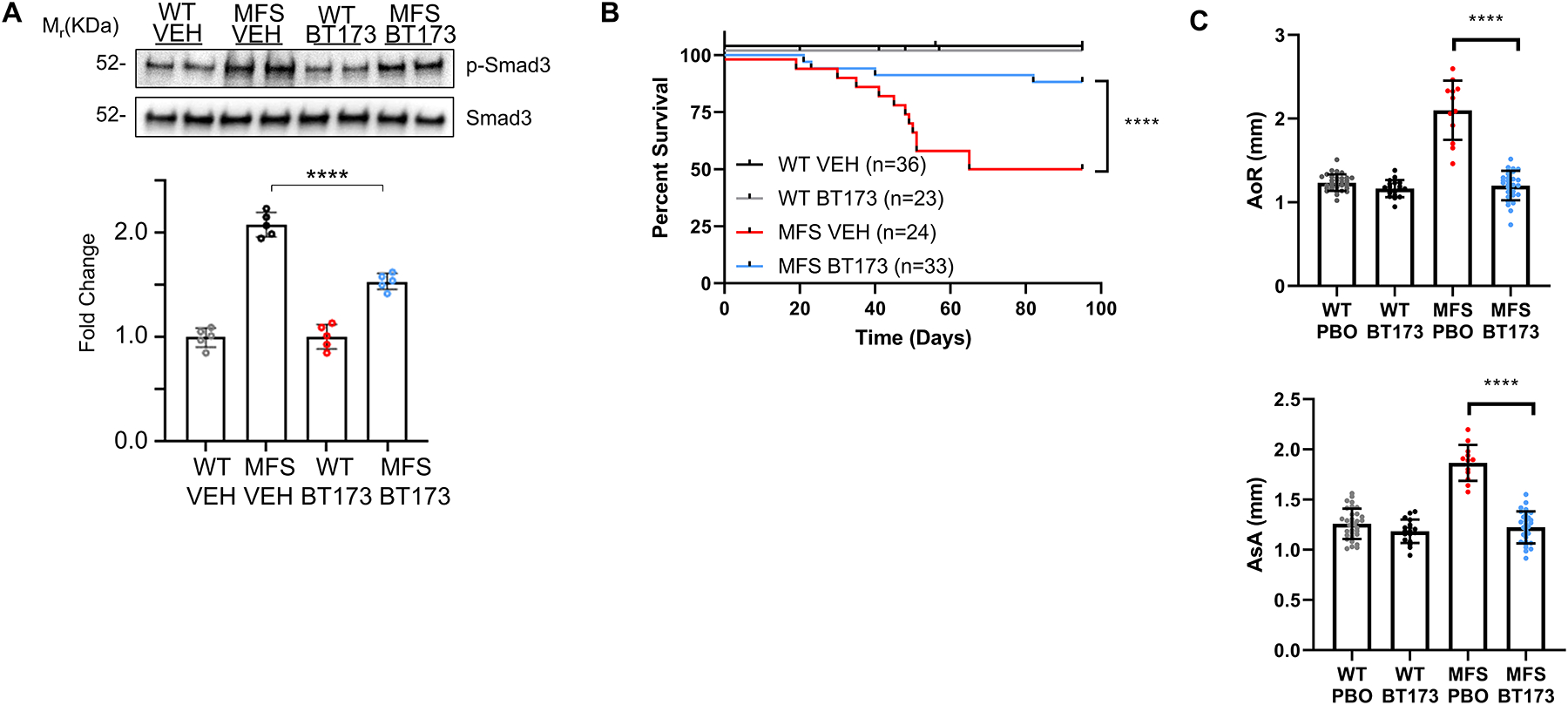
(A) Representative immunoblots of p-Smad3 and total Smad3 proteins detected in the proximal ascending aortas of P60 mice of the indicated phenotypes with quantitative plots shown below (n= 5 per genotype and treatment). 2 × 2 Factorial ANOVA to test for interaction followed by main effect comparisons by CONTRAST statements were employed to determine statistically significant differences which are indicated by asterisks (***p<0.001, ****p<0.0001). Error bars indicate means ± SD. Scale bar: 50 μm. (B) Kaplan-Meier survival curves of MFS mice treated with either BT173 or vehicle (VEH) shown along with the number of animals examined in each group (n= 24–36). The log rank test determined significance between the two groups of mice (p <0.0001). (C) Diameters of the aortic root (AoR, upper panel) and proximal ascending aorta (AsA, lower panel) in three-month old WT and MFS mice treated with either vehicle or BT173 (n=36).
Figure 5. TAAD pathology in MFS mice systemically treated with BT173.
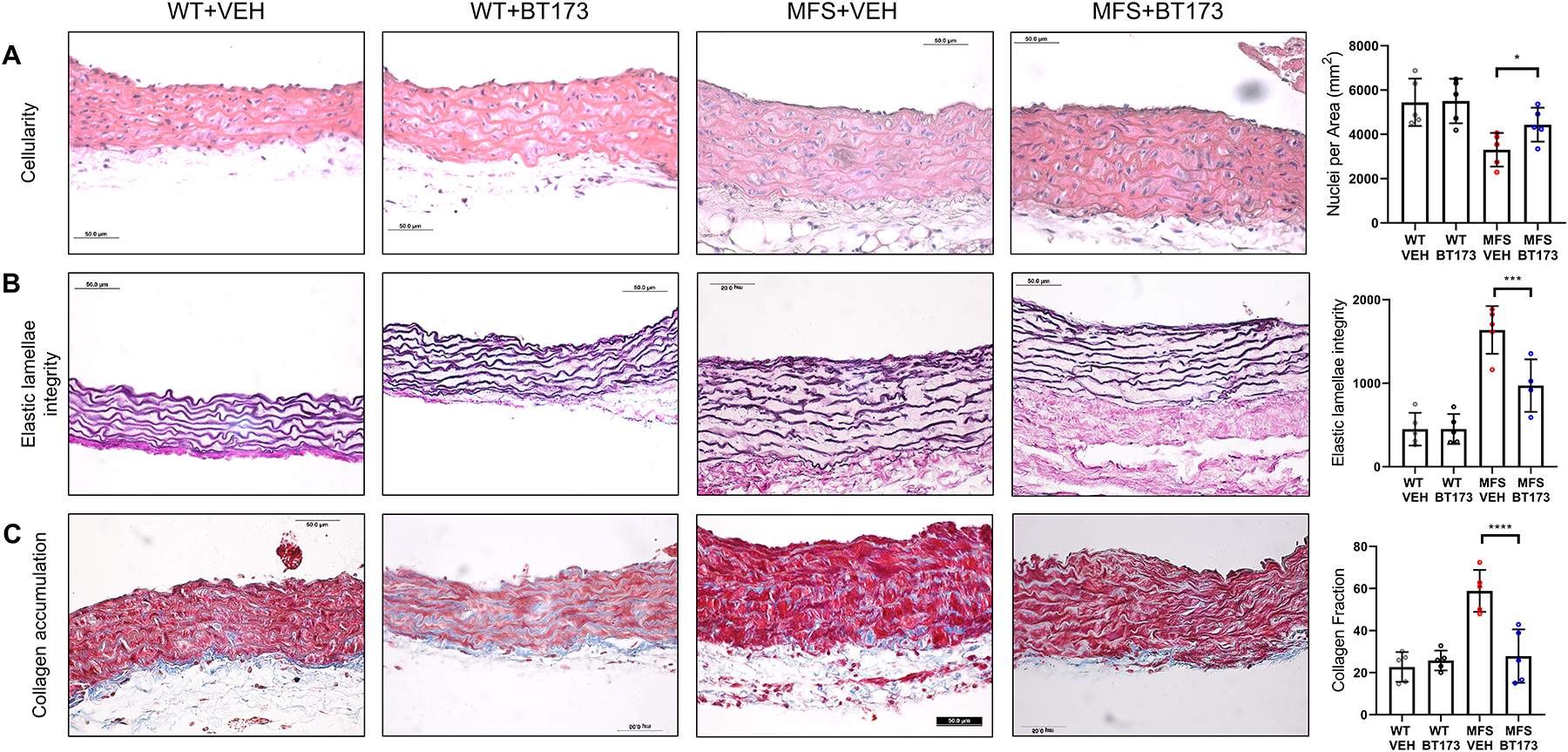
Representative images of cross-sections of ascending aortic walls of the indicated genotypes treated with vehicle (VEH) or BT173 and stained with (A) H&E to estimate nuclei count (p = 0.0470), (B) Verhoeff–van Gieson stain to estimate elastic fiber architecture (p = 0.0011), and (C) Masson’s trichrome to estimate collagen deposition (p <0.0001). Quantification of each dataset is shown in the dot plots next to each panel (n= 5 mice per genotype and treatment). 2 × 2 Factorial ANOVA to test for interaction followed by main effect comparisons by CONTRAST statements were employed to determine statistically significant differences which are indicated by asterisks (***p<0.001, ****p<0.0001). Error bars indicate means ± SD. Data presented as mean ± SD. Scale bars: 50 μm.
Tissue level manifestations of the histopathological findings were assessed via biaxial mechanical tests as a function of BT173 treatment of the fibrilin-1 deficient aorta. Among the multiple findings (Supplemental Table VII), treatment normalized circumferential stretch and improved circumferential stiffness, two parameters reflective of aneurysmal vulnerability,27 but did not modify stored elastic energy or distensibility, two parameters reflective of aortic conduit function (Fig. 6).28 We therefore speculate that pharmacological inhibition of HIPK2-mediated stimulation of the TGFβ/Smad3 signaling pathway contributes to delayed TAAD progression in mice with severe MFS, in part, by improving some of the biomechanical properties of the diseased aorta.
Figure 6. Biomechanical function of aortas of MFS mice systemically treated with BT173.
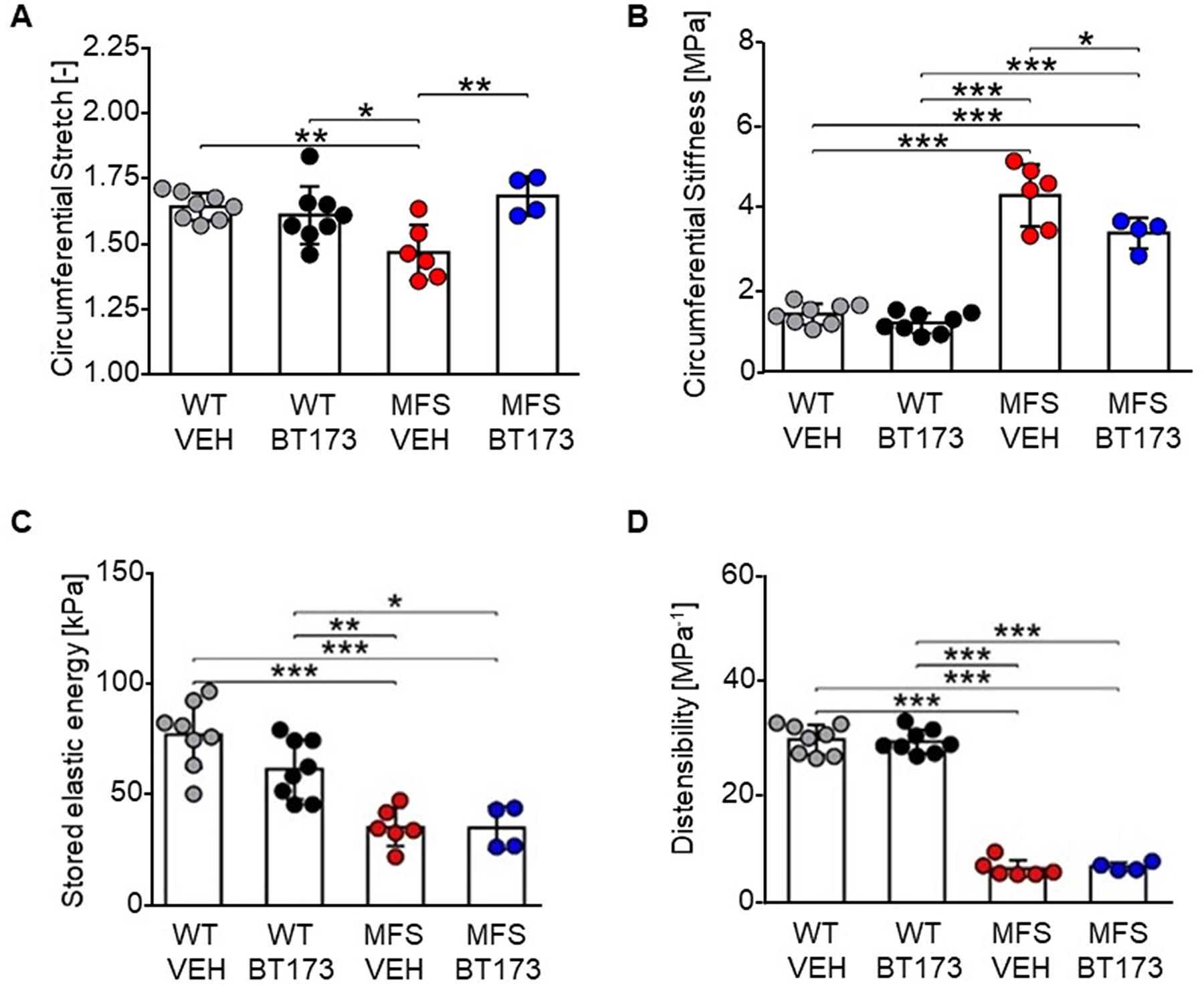
Results of ex vivo biaxial mechanical testing. Dots represent individual animals; bars represent mean ± SD. Treatment increased circumferential stretch (A) and decreased circumferential material stiffness (B) in MFS. Elastic energy storage capacity (C) and distensibility (D) were not affected by BT173 treatment. All metrics (n= 4–8) were evaluated at the individual in vivo values of the axial stretch. Metrics in panels A–C were calculated at the group-specific systolic pressure; distensibility (D) was calculated as(Ds − Dd)/(Dd × (Ps − Pd)), with P and D indicating pressure and inner diameter, and subscripts s and d systolic and diastolic, respectively. Additional biomechanical data are included in Supplemental Table VII. One-way analysis of variance (ANOVA) followed by Tukey’s post-hoc tests were employed to determine statistically significant differences (p≤0.05), which are indicated by asterisks (*p≤0.05, **p≤0.01, ***p≤0.001).
Discussion
Experimental and clinical evidence suggest that an effective TAAD pharmacotherapy in MFS is likely to rely on a multi-drug treatment strategy.5 We recently used an unbiased systems therapeutics approach that integrates patient- and mouse-derived transcriptomic data to identify an FDA-approved drug (baclofen) that could be repurposed to treat arterial disease in MFS.11 Here, we similarly integrated human and mouse gene expression data to computationally predict and experimentally validate a previously unrecognized association between a PK (HIPK2) and aneurysmal disease in MFS. Additional findings indicate that HIPK2 is a potential new drug target for combination anti-TAAD therapy.
Originally identified as a transcriptional co-repressors of NK homeoproteins,29 HIPK2 was subsequently implicated in several physiological processes and pathological conditions.30,31 HIPK2 functional diversity in embryonic development, adult tissue homeostasis and disease states reflects the PK’s ability to modulate a large plethora of signaling pathways and in turn, to be regulated by several post-translational modifications.29,30 Using a systems pharmacology approach, we identified HIPK2 among the highly ranked PKs predicted to dysregulate gene expression in the diseased aorta of both MFS patients and MFS mice, thus ensuring potential clinical relevance of the experimental data. Although computational identification of a druggable PK target was based on relatively small sample sizes (n=3 per each mouse time point and human aortic specimens), the pattern of top ranked PKs and HIPK2-interacting proteins are similar between mouse and human samples, suggesting a conservation of regulatory pathways involved in mechanisms of aneurysm progression in both species.
We experimentally validated our computational prediction by showing that (a) fibrillin-1 deficiency is associated with greater HIPK2 expression in the ascending aorta, and (b) postnatal Hipk2 inactivation attenuates TAAD pathology. A comparable TAAD mitigation was also observed by treating MFS mice with a selective allosteric inhibitor of HIPK2 that targets the TGFβ1/Smad3 signaling pathway.26 We have previously reported a dimorphic role of TGFβ signaling during TAAD development, namely a protective role during postnatal vessel growth and a pathogenic one at later stages of arterial disease.10 Diversification of TGFβ signaling outcome has been observed in several physiological and disease processes as result of stage-specific (contextual) mechanisms.32 The finding that neither genetic Hipk2 inactivation at P1–5 nor pharmacological inhibition of HIPK2/Smad3 interaction from P16 onward led to an earlier demise of MFS mice implicitly excludes that this PK may interfere with the protective function of TGFβ signaling. While currently lacking mechanistic insights into TGFβ dimorphism during TAAD, our findings nonetheless make BT173 an attractive small molecule compound that could be used to blunt pathogenic TGFβ/Smad3 signaling in a combination drug therapy.
An important consideration in selecting a pharmacological treatment is to expand the traditional surrogate markers of TAAD severity (morphological and histological abnormalities and mouse survival) to include impacts on the biomechanical function of the aorta, which affects hemodynamic loading.33 Four important biomechanical metrics are deformability (i.e., biaxial stretch), elastic energy storage (whereby aortic recoil augments blood flow), distensibility (which relates to pulse wave velocity and thus central pulse pressure), and circumferential material stiffness (which reflects ECM mechano-regulation by cells). In a comparison of diverse mouse models, decreased deformability and increased circumferential stiffness correlated strongly with aneurysmal propensity and presence.27 BT173 treatment of the MFS mice improved both of these physiological metrics, consistent with the reduced aortic diameter (morphology) and reduced elastic fiber fragmentation (histology). Neither energy storage nor distensibility improved, however, suggesting that the compound may not normalize hemodynamics by itself, again pointing to the potential importance of considering combinational therapies.
While our translational study identified an aortic PK that could be targeted pharmacologically to alleviate TAAD progression, additional work is needed to fully delineate the molecular mechanisms underlying HIPK2 pathogenic role in arterial disease. Ongoing investigations are aimed at characterizing how HIPK2 is upregulated in the MFS aorta, how this abnormality influences the TGFβ pathway and if additional pathways are involved. These and additional other mechanistic studies will greatly benefit from continued progress in fully resolving the 3D structure of HIPK2,34 an important pre-requisite to develop highly selective small molecule inhibitors of this multifunctional PK involved in several human diseases.
Supplementary Material
Highlights.
Integration of transcriptomic data from aortic specimens of patients and mice similarly afflicted with early onset progressively severe Marfan syndrome (MFS) predicted a probable involvement of the multifunctional homeodomain-interacting protein kinase 2 (HIPK2) in driving thoracic aortic aneurysm and acute dissection (TAAD).
Experimental validation of our computational prediction revealed abnormally high HIPK2 expression in the diseased aortas of MFS mice and delayed premature death from ruptured TAAD of mutant mice with genetically or pharmacologically disrupted HIPK2 activity.
Stress-activated HIPK2 therefore represents a previously unrecognized determinant of aneurysmal disease and an attractive new target for anti-TAAD multidrug therapy.
Acknowledgments
We thank Dr. B. Das for advices on BT173 usage, I. Miramontes for technical assistance, and K. Johnson for organizing the manuscript.
Sources of Funding
This work was supported by NIH grants HL126173 (to FR), HL134605 (to FR, RI and JDH) and DK088541 (to JCH); additional support was by the Netherlands Organisation for Scientific Research (Rubicon 452172006, to BS) and the European Union (Horizon 2020 grant 793805, to BS) and by the Elster Research Endowment (to FR).
Non-standard abbreviations and acronyms
- AT1r
angiotensin II type I receptor
- DEGs
differentially expressed genes
- ECM
extracellular matrix
- FBN1
fibrillin-1
- FDR
the false discovery rate
- iPSC
induced pluripotent stem cells
- LC-MS/MS
liquid chromatography with tandem mass spectrometry
- MFS
Marfan syndrome
- NMR
nuclear magnetic resonance
- PKs
protein kinases
- SMCs
smooth muscle cells
- TAAD
thoracic aortic aneurysms and dissection
- TF
transcription factor
- X2KWeb
eXpression2Kinases application
Footnotes
Disclosures
None.
The authors declare that all supporting data are available within the article [and its online supplementary files]
References
- 1.Milewicz DM, Prakash SK, Ramirez F. Therapeutics targeting drivers of thoracic aortic aneurysms and acute aortic dissections: insights from predisposing genes and mouse models. Annu Rev Med. 2017; 68:51–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu D, Shen YH, Russell L, Coselli JS, LeMaire SA. Molecular mechanisms of thoracic aortic dissection. J Surg Res. 2013; 184:907–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pinard A, Jones GT, Milewicz DM. Genetics of thoracic and abdominal aortic diseases. Circ Res. 2019; 124:588–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ramirez F, Caescu C, Wondimu E, Galatioto J. Marfan syndrome; A connective tissue disease at the crossroads of mechanotransduction, TGFβ signaling and cell stemness. Matrix Biol; 2018; 71–72:82–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Milewicz DM, Ramirez F. Therapies for thoracic aortic aneurysms and acute aortic dissections. Arterioscler Thromb Vasc Biol 2019; 39:126–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Habashi JP, Judge DP, Holm TM, Cohn RD, Loeys BL, Cooper TK, Myers L, Klein EC, Liu G, Calvi C, et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science 2006; 312:117–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Holm TM, Habashi JP, Doyle JJ, Bedja D, Chen Y, van Erp C, Lindsay ME, Kim D, Schoenhoff F, Cohn RD, et al. Noncanonical TGFβ signaling contributes to aortic aneurysm progression in Marfan syndrome mice. Science 2011; 332:358–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li W, Li Q, Jiao Y, Qin L, Ali R, Zhou J, Ferruzzi J, Kim RW, Geirsson A, Dietz HC, et al. Tgfbr2 disruption in postnatal smooth muscle impairs aortic wall homeostasis. J Clin Invest 2014; 124:755–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wei H, Hu J, Angelov SN, Fox K, Yan J, Enstrom R, Smith A, Dichek DA. Aortopathy in a mouse model of Marfan syndrome is not mediated by altered Transforming Growth Factor beta signaling. J Am Heart Assoc 2017; 6:e004968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cook JR, Clayton NP, Carta L, Galatioto J, Chiu E, Smaldone S, Nelson CA, Cheng SH, Wentworth BM, Ramirez F. Dimorphic effects of transforming growth factor-beta signaling during aortic aneurysm progression in mice suggest a combinatorial therapy for Marfan syndrome. Arterioscler Thromb Vasc Biol 2015; 35:911–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hansen J, Galatioto J, Caescu CI, Arnaud P, Calizo RC, Spronck B, Murtada SI, Borkar R, Weinberg A, Azeloglu EU et al. Systems pharmacology-based integration of human and mouse data for drug repurposing to treat thoracic aneurysms. JCI Insight 2019; 4:e127652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pereira L, Lee SY, Gayraud B, Andrikopoulos K, Shapiro SD, Bunton T, Biery NJ, Dietz HC, Sakai LY, Ramirez F. Pathogenetic sequence for aneurysm revealed in mice underexpressing fibrillin-1. Proc. Natl. Acad. Sci. USA 1999; 96:3819–3823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xiao W, E J, Bao L, Fan Y, Jin Y, Wang A, Bauman D, Li Z, Ya-Li Z, Liu R, Lee K, He JC. Tubular HIPK2 is a key contributor to renal fibrosis. JCI Insight 2021; 5: e136004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stillitano F, Hansen J, Kong CW, Karakikes I, Funck-Brentano C, Geng L, Scott S, Reynier S, Wu M, Valogne Y, Desseaux C, Salem JE, Jeziorowska D, Zahr N, Li R, Iyengar R, Hajjar RJ, Hulot JS. Modeling susceptibility to drug-induced long QT with a panel of subject-specific induced pluripotent stem cells. E Life 2017; 6:e19406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen EY, Xu H, Gordonov S, Lim MP, Perkins MH, Ma’ayan A. Expression2Kinases: mRNA profiling linked to multiple upstream regulatory layers. Bioinformatics 2012; 28:105–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Clarke DJB, Kuleshov MV, Schilder BM, Torre D, Duffy ME, Keenan AB, Lachmann A, Feldmann AS, Gundersen GW, Silverstein MC, et al. (eXpression2Kinases (X2K) Web: linking expression signatures to upstream cell signaling networks. Nucleic Acids Res 2018; 46: W171–W179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, Ma’ayan A. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics. 2013; 14: 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, McDermott MG, Monteiro CD, Gundersen GW, Ma’ayan A. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016; 44: W90–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carta L, Smaldone S, Zilberberg L, Loch D, Dietz HC, Rifkin DB, Ramirez F. p38 MAPK is an early determinant of promiscuous Smad2/3 signaling in the aortas of fibrillin-1 (Fbn1)-null mice. J Biol Chem 2009; 284:5630–5636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Granata A, Serrano F, Bernard WG, McNamara M, Low L, Sastry P, Sinha S. An iPSC-derived vascular model of Marfan syndrome identifies key mediators of smooth muscle cell death. Nat Genet 2017; 49:97–109. [DOI] [PubMed] [Google Scholar]
- 21.Dominguez I, Sonenshein GE, Seldi DC, Protein kinase CK2 in health and disease: CK2 and its role in Wnt and NF-kappaB signaling: linking development and cancer. Cell Mol Life Sci 2009; 66:1850–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jin Y, Ratnam K, Chuang PY, Fan Y, Zhong Y, Dai Y, Mazloom AR, Chen EY, D’Agati V, Xiong H, Ross MJ, Chen N, Ma’ayan A, He JC. A systems approach identifies HIPK2 as a key regulator of kidney fibrosis. Nat Med 2012; 18:580–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang J, Pho V, Bonasera SJ, Holtzman J, Tang AT, Hellmuth J, Tang S, Janak PH, Tecott LH, Huang EJ. Essential function of HIPK2 in TGFβ-dependent survival of midbrain dopamine neurons. Nat Neurosci 2007; 10:77–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grigoryants V, Hannawa KK, Pearce CG, Sinha I, Roelofs KJ, Ailawadi G, Deatrick KB, Woodrum DT, Cho BS, Henke PK, Stanley JC, Eagleton MJ, Upchurch GR. Tamoxifen up-regulates catalase production, inhibits vessel wall neutrophil infiltration, and attenuates development of experimental abdominal aortic aneurysm. J. Vasc. Surg 2005; 41:108–114. [DOI] [PubMed] [Google Scholar]
- 25.Zhang ZH, Jhaveri DJ, Marshall VM, Bauer DC, Edson J, Narayanan RK, Robinson GJ, Lundberg AE, Bartlett PF, Wray NR, Zhao QY. A comparative study of techniques for differential expression analysis on RNA-Seq data. PLoS One. 2014; 9: e103207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu R, Das B, Xiao W, Li Z, Li H, Lee K, He JC A Novel Inhibitor of Homeodomain Interacting Protein Kinase 2 Mitigates Kidney Fibrosis through Inhibition of the TGF-β1/Smad3 Pathway. J Am Soc Nephrol 2017; 28:2133–2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bellini C, Bersi MR, Caulk AW, Ferruzzi J, Milewicz DM, Ramirez F, Rifkin DB, Tellides G, Yanagisawa H, Humphrey JD. Comparison of 10 murine models reveals a distinct biomechanical phenotype in thoracic aortic aneurysms. J R Soc Interface 2017; 14:20161036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wagenseil JE, Mecham RP. Vascular extracellular matrix and arterial mechanics. Physiol Rev 2009; 957–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim YH, Choi CY, Lee SJ, Conti MA, Kim Y. Homeodomain-interacting protein kinases, a novel family of co-repressors for homeodomain transcription factors. J Biol Chem 1998; 273: 25875–25879. [DOI] [PubMed] [Google Scholar]
- 30.Blaquiere JA, Verheyen EM. Homeodomain-interacting protein kinases: diverse and complex roles in development and disease. Curr Top Dev Biol 2017; 123:73–103. [DOI] [PubMed] [Google Scholar]
- 31.Fan Y, Wang N, Chuang P, He JC. Role of HIPK2 in kidney fibrosis. Kidney Int Suppl 2014; 4:97–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Massague’ J. TGFβ signaling in context. Nat Rev Mol Cell Biol. 2012;13:616–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Teixido-Tura G, Redheuil A, Rodriguez-Palomares J, Gutierrez L, Sanchez V, Forteza A, Lima JA, Garcia-Dorado D, Evangelista A. Aortic biomechanics by magnetic resonance: early markers of aortic disease in Marfan syndrome regardless of aortic dilatation? Int J Cardiol 2014; 171:56–61. [DOI] [PubMed] [Google Scholar]
- 34.Agnew C, Liu L, Liu S, Xu W, You L, Yeung W, Kannan N, Jablons D, Jura N. The crystal structure of the protein kinase HIPK2 reveals a unique architecture of its CMGC-insert region. J Biol Chem 2019; 294:13545–13559. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.