

ABSTRACT
MiR-25 is a well-documented oncogenic miRNA implicated in esophageal squamous cell carcinoma (ESCC) development, progression and metastasis. However, whether and how miR-25 is involved in the development and metastasis of ESCC remain un-addressed. By using qRT-PCR analysis to compare levels of miR-25 in ESCC tissues with or without lymph node metastasis (LNM), it showed that ESCC tissues with LNM had increased levels of miR-25, which was correlated with tumor metastasis and poor prognosis. Gain- and loss-of-function assays indicated that targeting miR-25 could reverse EMT, and reduce in vitro cell migration and invasion, but not apoptosis and proliferation of ESCC. Furthermore, targeting miR-25 inhibited in vivo lung metastasis, and vice versa. And E-cadherin was a direct target of miR-25 through which affected EMT process and metastasis of ESCC. It is therefore indicated that miR-25 promotes metastasis of ESCC through E-cadherin and EMT events, thus may serves as a negative prognostic factor and possible target for treatment of ESCC patients.
KEYWORDS: Esophageal squamous cell carcinoma, lymph node metastasis, epithelial–mesenchymal transition, miR-25, E-cadherin
Introduction
Esophageal squamous cell carcinoma (ESCC) accounts for about 90% of the 456,000 incident esophageal cancers each year [1]. It is also the most common type of esophageal cancer in China, which accounts for more than 90% of cases [2]. The majority of esophageal cancer patients are diagnosed with advanced disease due to unclear early symptoms. The prognosis is very poor, with 5-year survival of only 10%, due to early invasion and metastasis via the well-developed network of submucosal lymphatic vessels [3]. Although previous studies have demonstrated that alterations of numerous oncogenes and tumor-suppressor genes are involved in ESCC, the underlying molecular and genetic mechanism of esophageal carcinogenesis remains largely unknown.
Numerous studies have indicated that microRNAs (miRNAs) are involved in multiple cellular processes as post-transcriptional regulators and particularly in cancer development and progression [4,5]. miRNAs are aberrantly expressed in various cancers and function as a novel class of oncogenes or tumor-suppressor genes depending on their targets. MiR-25 was reported to be associated with tumor carcinogenesis and metastasis, including ESCC [6–8]. But its exact mechanisms and roles in vivo are unclear.
Epithelial–mesenchymal transition (EMT) is considered to play an important role in the initial parts of the metastatic cascade-providing cancer cells with invasive, migratory, and cancer stem cell properties, and the induction of EMT leads to the downregulation of E-cadherin, expression of distinct mesenchymal markers such as vimentin, fibronectin, and N-cadherin, and morphological changes [9,10]. Several miRNAs, such as miR-200 family members, have been found to regulate EMT by targeting the E-cadherin repressors ZEB1 and ZEB2. MiR-124, a novel tumor-suppressor miRNA that is epigenetically silenced in EC, can reverse EMT and the invasive properties, by attenuating the expression of the IQGAP1 oncogene [11]. In HK-2 cells, enhanced expression of miR-106b attenuated EMT by retaining the epithelial morphology of HK-2 cells, reducing the levels of α-smooth muscle actin (α-SMA), and increasing the levels of E-cadherin [12]. In cervical cancer cells, miR‐25‐3p is an important regulator of cervical cancer EMT and chemoresistance [13].
In the present study, we investigated whether the relative expression of miR-25 between tumor and normal tissues is correlated with lymph node metastasis in ESCC patients and the mechanism by which miR-25 promotes ESCC metastasis and the same in turn. The results showed that high miR-25 expression is correlated with tumor metastasis and poor prognosis in ESCC patients. Targeting miR-25 could reverse EMT and reduce in vitro cell migration, invasion and in vivo lung metastasis, the mechanisms of which were by miR-25-dependent E-cadherin regulation. Thus, targeting miR-25 could be a novel approach to inhibit metastasis in patients with ESCC.
Material and methods
Tissue specimens
Seventy-nine cancer tissues and adjacent normal tissues were obtained from ESCC patients who underwent surgery at the central hospital of Linyi, Shandong, China. The protocol was approved by the Ethics Committee of the hospital, and all experiments were conducted after acquiring informed consent. All samples were separated into two parts. One part was collected, snap-frozen immediately, and stored at −80°C for RNA isolation using TRIzol reagent (Invitrogen, Thermo Fisher Scientific). Another part was detected by immunohistochemistry probed with antibodies against E-cadherin (Signaling Technology Inc, Danvers, MA, USA).
Cell culture
The ESCC cell lines KYSE-410, Ec-109, CaEs-17 and KYSE-510 were obtained from DSMZ, the German Resource Center for Biological Materials and maintained in RPMI-1640 (Life Technologies, Shanghai, China) supplemented with 5% fetal calf serum at 37°C in a humidified atmosphere containing 5% CO2 and 95% air. All cell lines have been DNA fingerprinted using the PowerPlex 1.2 kit (Promega, Beijing Biotech Co., Ltd., Beijing, China) and are mycoplasma free using the e-Myco kit (Boca Scientific, Florida, USA).
Stable cell line selection
A pre-miR‐25 expression vector was constructed using a BLOCK‐iT™Pol II miR RNAi Expression Vector Kit (Invitrogen, Shanghai, China) according to the manufacturer’s protocol and transfected into the Ec-109 cells for selection of stable miR‐25‐expressing cells. After 48 h of transfection, cells were incubated with 10 mg/mL blasticidin for 2 weeks. To construct stably expressing anti-miR‐25 cells, an anti-miR‐25 expression vector was transfected into the KYSE-510 cells. After 48 h of transfection, cells were incubated with 2 mg/mL puromycin for 2 weeks. Then, cells were frozen in aliquots for later use.
Transfection–transduction
Transient transfection of Ec-109 or KYSE-510 cells using Lipofectamine 2000 was performed with pre-miR-25 or anti-miR-25 or negative control pre-miR (anti-miR) according to the manufacturer’s instructions. At 72 h after transfection, cells were counted and the cellular lysates were collected for analysis of the protein expression of the selected putative miR-25 targets. To study the effects of miR-25 on E-cadherin expression, the anti-miR-25 transfected KYSE-510 cells were seeded at 2 × 105 cells per well in a 6-well plate and transfected with E-cadherin siRNA. After 48 h, cells were counted and lysed for RNA and protein extraction.
Quantitative RT-PCR
RNA was isolated from human tissue and cells using Trizol (Life Technologies, Shanghai, China) according to manufacturer’s protocol. miR qPCR was performed using Express SYBR GreenER Supermix with premixed ROX (Invitrogen, Shanghai, China). U6 was used as housekeeping gene for miR to normalize the Ct values (ΔCt), and relative expression was calculated using the 2−ΔΔCt method. The 75th percentiles of 2ΔΔCt were used as the cutoff point for patients with high and low levels of miR-25. Primers for miR-25 were purchased from Qiagen (Shanghai, China) and relative expression was calculated using the 2−ΔΔCt method.
Western blot analysis
Cells were harvested and washed with PBS twice, disrupted in IP buffer (Thermo) and centrifuged at 12,000 × g for 20 min. Protein (50 µg) from the supernatant fraction (quantified by the BCA Protein Assay Kit, Thermo) was subjected to SDS-PAGE, and transferred to a PVDF membrane (Millipore, Guangzhou, China). Membranes were blocked with 5% nonfat milk for 1 h at room temperature and then incubated with the anti-cadherin (Abcam, Shanghai, China), followed by the corresponding HRP-conjugated anti-mouse or anti-rabbit secondary antibody (Abcam, Shanghai, China). Protein bands were visualized by the Western lightening plus-ECL kit (Pierce, Wuhan, China).
Immunofluorescence
Cells grown on glass coverslips were fixed in 4% paraformaldehyde for 10 min at room temperature. Cells were washed twice with PBS. Blocking buffer (DakoCytomation, Glostrup, Denmark) was added for 30 min, and samples were then stained with primary E-cadherin antibodies (Santa Cruz Biotechnology, Shanghai, China) and double-stained with DAPI to reveal the nuclei. Images were taken using a fluorescence Nikon Ti-E inverted microscope (MP110, Molson Pavillion).
Cell migration and invasion assays
Cell migration and invasion assays were analyzed using the Transwell chambers assay (Costar, Corning Inc., Corning, NY, USA), with or without coated Matrigel (BD Biosciences, San Jose, CA, USA). Cells were plated at a density of 5 × 104 per well in the upper chamber without serum. The lower chamber of the Transwell device was filled with 500 µl RPMI 1640 supplemented with 10% fetal bovine serum. After incubation for 24 h, noninvading cells were removed from the top well with a cotton swab, whereas the bottom cells were fixed with 3% paraformaldehyde, stained with 0.1% crystal violet, and photographed in three independent fields for each well. They were finally extracted with 33% acetic acid and detected quantitatively using a standard microplate reader (at 570 nm). Three independent experiments were conducted in triplicate.
Wound healing assays
A total of 100,000 Ec-109 or KYSE-510 cells were grown to 70% confluency in a 6-well plate (Falcon Becton Dickinson). The cells were transfected with pre-miR-25/pre-miR (Ec-109) and anti-miR-25/anti-miR (KYSE-510). After 5 h of transfection, 10% DMEM was added to cells. The cells were then serum-starved accordingly and three separate scratches were made per well after 24 h of transfection. The cells were left in serum-free or complete media for 48 h and the wounds were imaged at 200× magnification using Evos FL Digital Fluorescence microscope. The data is representative of two independent experiments done in triplicate.
Cell proliferation and apoptosis assay
To measure cell proliferation, cells were plated at a density of 1 × 103 cells per well into 96-well plates at day 0 (24 h after pre-miR-25 or anti-miR-25 transfection). At days 1, 3 and 5, cell viability was measured using MTT as the manufacture’s instruction.
Immunohistochemical staining
Tissue sections were dewaxed with xylene and rehydrated through gradient ethanol into water. Antigen retrieval, lockage of endogenous peroxidase, and nonspecific binding were conducted before primary antibody incubation. Negative controls were obtained by omitting the primary antibody. Expression E-cadherin were evaluated as described previously. The percentage of positive tumor cells was determined semi-quantitatively by assessing the entire tumor section. Each sample was assigned to one of the following categories: 0 (0–4%), 1 (5–24%), 2 (25–49%), 3 (50–74%), or 4 (75–100%). The intensity of immunostaining was determined as 0 (negative), 1+ (weak), 2+ (moderate), or 3+ (strong). A final immunoreactive score between 0 and 12 was calculated by multiplying the percentage of positive cells with the staining intensity score. The two scores were then multiplied to produce a weighted score for each sample, the expression was considered positive when the score was >2. All slides were blindly evaluated for immunostaining without any knowledge of the clinical outcome of other clinical or pathological data.
Metastasis assays in nude mice
For lung metastasis model, 2 × 106 cells (stable Ec-109/pre-miR-25, KYSE-510/anti-miR-25 and their controls) of each group were washed in PBS and injected intravenously into 4- to 6-week-old age-matched female. Animals were sacrificed 4 weeks after injection and the lung surface tumor foci were counted. Lung tissues were fixed in formalin and sectioned for H&E staining.
Statistical analysis
All data were expressed as mean ± standard deviation, and SPSS 22.0 software was used for statistical analyses. All in vitro experiments were repeated at least three times unless stated otherwise. Differences among the groups and treatments were determined by Student’s t-test or one-way ANOVA unless stated otherwise. Survival was estimated using the Kaplan–Meier method. Significance between survival functions was identified by the log-rank test. P< 0.05 was considered statistically significant.
Results
Expression of miR-25 is closely associated with lymph node metastasis and poor survival
Seventy-nine ESCC tissues and neighboring non-cancerous tissues were assessed for miR-25 expression level by qRT-PCR. Quantitative data indicated that the levels of miR-25 in ESCC tissues were notably elevated compared to the matched non-cancerous tissues (t = 4.673, P = 0.0026, Figure 1(a)a). Furthermore, the levels of miR-25 were significantly higher in metastatic tumor tissues than that in the primary tumor tissues without detectable metastasis (t = 4.156, P = 0.0163, Figure 1(a,b) (Table 1). The 75th percentiles of 2ΔΔCt was used as the cutoff point for patients with high and low levels of miR-25. Kaplan–Meier survival estimate showed that the patients with high miR-25 expression (n = 34) had a shorter survival compared with the patients with low miR-25 expression (n = 45). The difference in the prognosis of these two groups was statistically significant (p = 0.006, log-rank test, Figure 1(b)).
Figure 1.
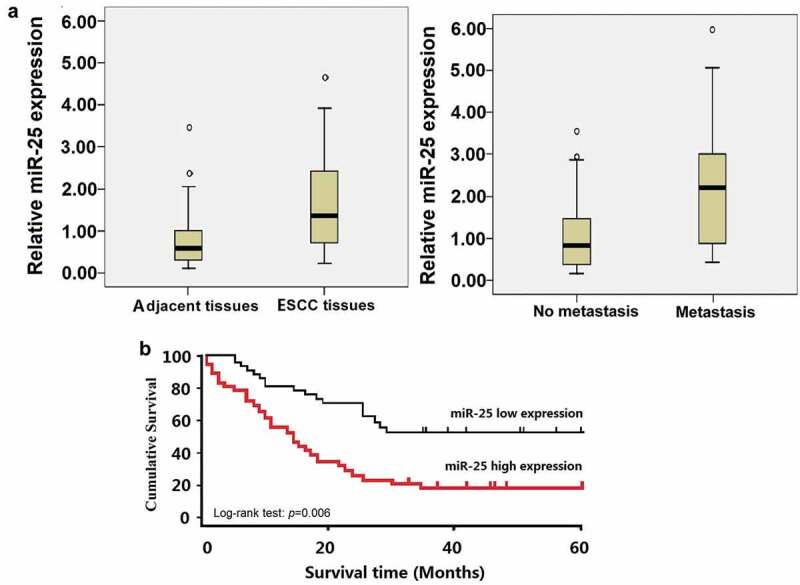
Expression of miR-25 in ESCC tissues and its relation with survival.
Table 1.
Correlation between E-cadherin, lymph node metastasis and miR-25 expression.
| miR-25 expression |
|||||
|---|---|---|---|---|---|
| Features | Number | Low | High | p-value | |
| E-cadherin | Negative | 60 | 33 | 27 | 0.0016 |
| Positive | 19 | 12 | 7 | ||
| Lymph node metastasis | No | 35 | 21 | 14 | 0.028 |
| Yes | 44 | 24 | 20 | ||
The industry composition follows the CSRC’s industry coding system in 2001. Manufacturing (labeled C) is divided into ten categories because more than 60% of the sample firms are manufacturers. We omit the banking and insurance industry (labeled I) in our sample.
miR-25 modulates migration and invasion but not proliferation in ESCC cells
In the pre-experiment, miR-25 expression was detected by qRT-PCR in KYSE-410, Ec-109, CaEs-17 and KYSE-510 cells, showing that Ec-109 cells have the lowest miR-25 expression, and KYSE-510 has the highest miR-25 expression (Figure 2(a)). So the Ec-109 and KYSE-510 cells were used for further study. To further investigate the biological mechanism of miR-25 on the invasiveness of ESCC, in vitro gain- and loss-of-function analysis was carried out through transient transfection in ESCC cells. miR-25 overexpression experiments were first performed by miR-25 precursor (pre-miR-25) transfection in the Ec-109 cell line. Overexpression of miR-25 was confirmed by qRT-PCR, as shown in Figure 2(b) (P = 0.004). In the wound healing assay, high miR-25 expression significantly promotes the ability of cells to migrate (Figure 2(c)). In the transwell invasion and migration assay, cells transfected with pre-miR-25 displayed an increase in invasion and migration ability when compared with the control groups (Figure 2(d–e)). Collectively, these results indicated that ectopic miR-25 significantly promoted cell migration and invasion in vitro.
Figure 2.
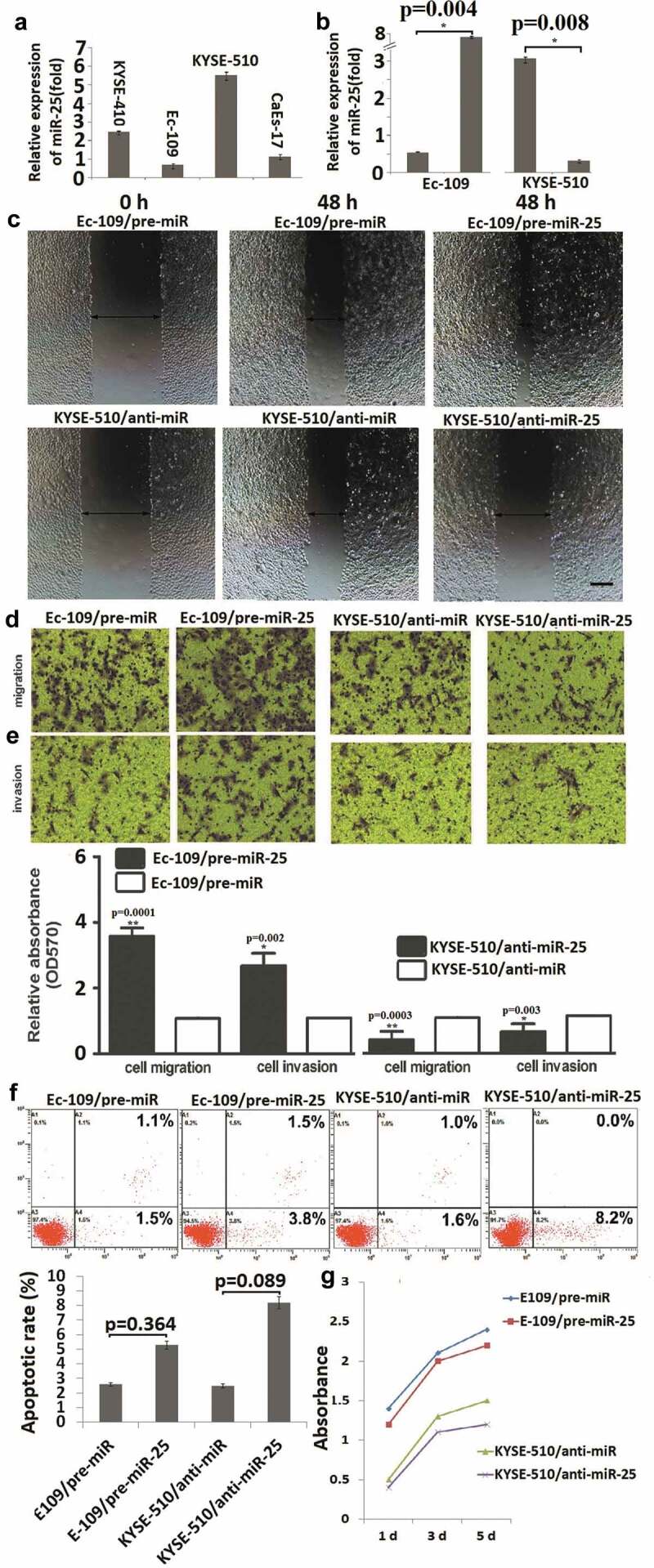
MiR-25 regulated the migration, invasion but not apoptosis in ESCC cells.
To be logical and direct demonstration of the consequences of the low expression levels of miR-25 in ESCC, miR-25 inhibition experiments were performed by miR-25 inhibitor (anti-miR-25) transfection in the KYSE-510 cell line. miR-25 was transiently inhibited in KYSE-510 cells with anti-miR-25 (P = 0.008, Figure 2(b)), and the effect of inhibition of miR-25 levels on cellular invasion and migration potential of KYSE-510 cells was analyzed. The results showed that the suppression of miR-25 decreased invasion and migration in KYSE-510 cells (Figure 2(c–e)). However, overexpression of miR-25 in Ec-109 cells (P = 0.364) and knock-down of miR-25 in KYSE-510 cells (P = 0.089) had no significant effect on cell apoptosis (Figure 2(g)) and proliferation (Figure 2(h)). These results demonstrated that miR-25 promotes ESCC cell migration and invasion but has no effect on cell apoptosis and proliferation.
Expression of miR-25 is closely associated with E-cadherin
Immunohistochemical staining showed that E-cadherin was strongly expressed in the membrane in non-cancerous tissues (100%, 79/79), while the positive expression rate was significantly lower in ESCC (19/79). Representative immunostaining showed that ESCC tissues with high expression of miR-25 had\weak staining of E-cadherin, and ESCC tissues with low expression of miR-25 had strong staining for E-cadherin (Figure 3). Statistical analysis showed that the expression of miR-25 was closely related with E-cadherin (P = 0.0016) (Table 1). The results taken together revealed that miR-25 expression was inversely correlated with E-cadherin expression.
Figure 3.
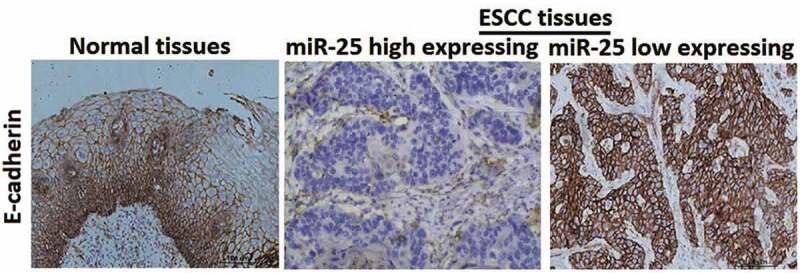
Immunohistochemical staining of E-cadherin.
miR-25 regulates E-cadherin expression in ESCC cells
To determine whether miR-25 regulates E-cadherin -induced EMT in ESCC cells, the effect of miR-25 on E-cadherin expression regulation was analyzed. Ec-109 cells were transfected with 100 nM pre-miR-25 or pre-miR control for 48 h, and KYSE-510 cells were transfected with 100 nM anti-miR-25 and anti-miR control for 48 h. The results showed that pre-miR-25 treatment inhibits E-cadherin expression in Ec-109 cells (Figure 4(a)). And anti-miR-25 treatment promotes E-cadherin expression in KYSE-510 cells (Figure 4(b)). Consistent with the western blot assay, immunofluorescence staining by confocal laser scanning microscope for E-cadherin showed the same results (Figure 4(c,d)).
Figure 4.
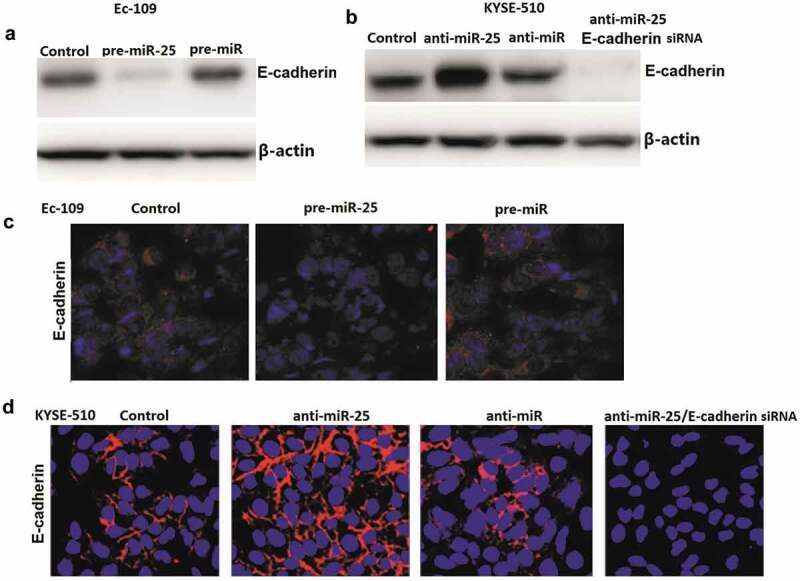
Effect of miR-25 on E-cadherin genes in Ec-109 and KYSE-510 cells.
E-cadherin is crucial for miR-25-mediated EMT
Untreated Ec-109 and KYSE-510 cells had an almost round shape under the phase-contrast microscopy (Figure 5). In Ec-109 cells, miR-25 transfection replaced cobblestone-like appearance with spindle-shaped cells like the mesenchymal cells. In KYSE-510 cells, anti-miR-25 transfection makes cobblestone-like appearance with more dense cell-to-cell adhesions and assembled closely, whereas anti-miR-25 + E-cadherin siRNA treated KYSE-510 cells developed a spindle-shaped morphology, the cell-to-cell adhesions became weak and the cells were scattered compared to the anti-miR-25 alone treated KYSE-510 cells (Figure 5).
Figure 5.
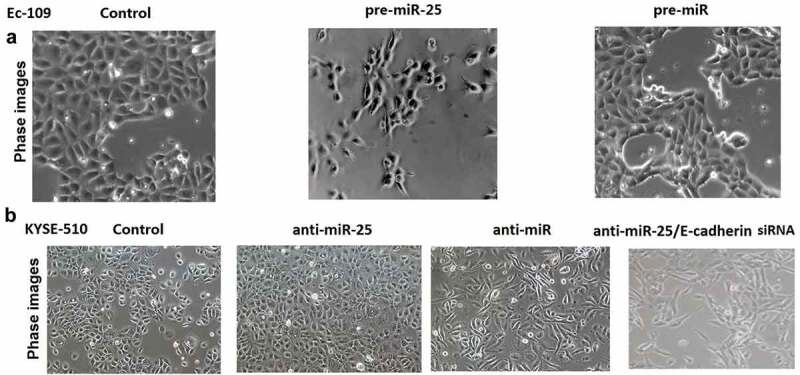
Down-regulation of miR-25 or up-regulation of miR-25 in cells having undergone EMT.
Upregulation of miR-25 induced lung metastasis, and targeting miR-25 inhibited lung metastasis in mice
In Ec-109 cells and KYSE-510 cells, the vector treated cells (Ec-109/pre-miR and KYSE-510/anti-miR) formed multiple and large-sized metastatic nodules in the lung (Figure 6(a-b)). In Ec-109 cells, more lung metastatic nodules were observed in the lungs of the pre-miR-25 expression groups compared with the control group (Figure 6(a)). However, in KYSE-510 cells, fewer lung metastatic nodules were observed in the lungs of the anti-miR-25 expression group compared with the control group (Figure 6(b)).
Figure 6.
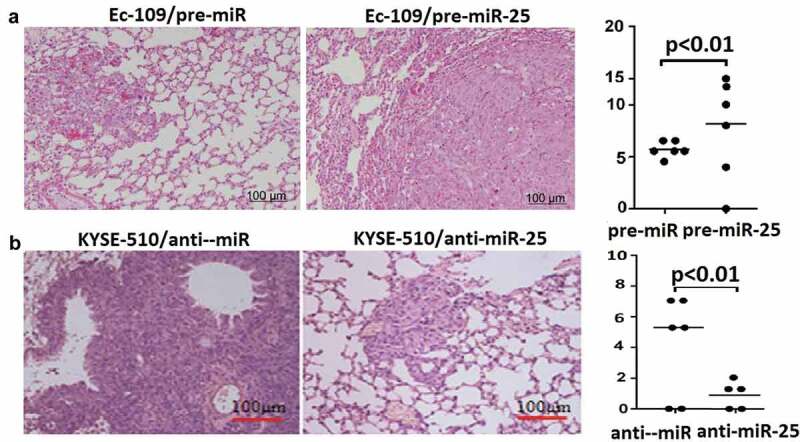
Effect of miR-25 lung metastasis in mice.
Discussion
In the present study, miR-25 is upregulated in the ESCC tissues than the matched non-cancerous tissues. In addition, miR-25 is more strongly upregulated in the metastatic ESCC tissues than that in primary tumor tissues without detectable metastasis. High miR-25 expression was significantly associated with lymph node metastasis and poor overall survival and was confirmed as an independent prognostic factor in multivariate analysis. miR-25 is independent of traditional clinicopathological factors, including patient sex, age, clinical stage, tumor differentiation, and tumor grade. Therefore, miR-25 showed correlated mRNA over-expression in ESCC development and progression, and could be a diagnostic biomarker and gene target. Our results are agreed with the previous study in ESCC tissues [8], but contrary to Wang’s report that overexpression of miR-25 showed a positive correlation with overall survival in ESCC patients [14]. Besides, it was also revealed that miR-25 is a critical positive regulator of EMT program mainly through affecting the E-cadherin, and it promotes ESCC cells migration and invasion in vitro and in vivo, and targeting miR-25 inhibits ESCC cells migration and invasion in vitro and in vivo.
Metastasis is the primary cause of mortality in most cancer patients [15]. Thus, to understand the molecular mechanisms of metastasis is one of the most important issues in cancer research. Epithelial–mesenchymal transition (EMT), which enables epithelial cells to acquire invasive mesenchymal phenotype, is attracting increasing attention as an important mechanism for the initial step of metastasis [16,17]. It has been proven that loss of expression and/or abnormal function of E-cadherin leads to loss of cell polarity and derangement of normal tissue architecture. In most cancers with epithelial origins, E-cadherin -mediated cell-cell adhesion is lost concomitantly with the acquisition of an invasive phenotype, high tumor grade, and low patient survival [18–20]. Loss or reduction of E-cadherin expression can be caused by a variety of mechanisms, such as somatic mutations, chromosomal deletions, proteolytic cleavage, and silencing of the E-cadherin promoter [21]. Our study revealed a novel mechanism by which E-cadherin is regulated. Our study showed that E-cadherin is directly targeted by miR-25 in ESCC cell lines, and an inverse correlation between miR-25 and E-cadherin in ESCC tumor tissues and cell lines was uncovered. Furthermore, restoration of E-cadherin expression inhibited cell invasion and migration induced by miR-25 overexpression, and targeting E-cadherin reversed cell invasion and migration induced by targeting miR-25, indicating that E-cadherin functions as a mediator of miR-25 in cell migration and ESCC progression.
EMT is a key factor to embryonic development and tumor metastasis. Our data demonstrated that miR-25 overexpression inhibited E-cadherin expression and induced EMT. However, targeting miR-25 induced E-cadherin expression and suppressed EMT, and targeting of E-cadherin in cancer cells could be sufficient to reverse EMT in anti-miR-25 treated cells, which was accompanied by a more invasive property. Our findings showed that miR-25 is required for EMT initiation and maintenance, which was related to E-cadherin regulation, and a common regulatory mechanism of tumor cell invasion.
Conclusions
Our study not only revealed the important role of miR-25/E-cadherin signaling pathway in ESCC pathogenesis, but also implied a potential role for miR-25 in the clinical diagnosis and treatment of ESCC. Our study also suggests that miR-25 is required for EMT initiation and maintenance. miR-25 plays an important role in EMT through direct targeting of E-cadherin. Therefore, miR-25 could be used as effect target for ESCC treatment.
Funding Statement
This work was supported by the grants from the National Natural Science Foundation of China [grant numbers: 81872417].
Highlight
High miR-25 expression is correlated with metastasis in patients with esophageal squamous cell carcinoma.
High miR-25 expression is correlated with poor prognosis in patients with esophageal squamous cell carcinoma.
Targeting miR-25 could reverse epithelial–mesenchymal transition in esophageal squamous cell carcinoma cells in vitro.
Targeting miR-25 reduces in vitro cell migration, invasion and in vivo lung metastasis.
miR-25 inhibits invasion and metastasis of esophageal squamous cell carcinoma cells through E-cadherin upregulation.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Abnet CC, Arnold M, Wei WQ. Epidemiology of esophageal squamous cell carcinoma. Gastroenterology 2018; 154:360–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Chen W, Zheng R, Baade PD. Cancer statistics in China, 2015. CA Cancer J Clin 2016; 66: 115–132. [DOI] [PubMed] [Google Scholar]
- [3].Enzinger PC, Mayer RJ.. Esophageal cancer. N Engl J Med 2003; 349:2241–2252. [DOI] [PubMed] [Google Scholar]
- [4].He L, Hannon GJ.. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet. 2004; 5(7):522–531. [DOI] [PubMed] [Google Scholar]
- [5].Calin GA, Croce CM.. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006; 6:857–866. [DOI] [PubMed] [Google Scholar]
- [6].Jia Y, Lu H, Wang C, et al. miR-25 is upregulated before the occurrence of esophageal squamous cell carcinoma. Am J Transl Res 2017;9:4458–4469. [PMC free article] [PubMed] [Google Scholar]
- [7].Wang M, Yang YO, Jin Q, et al. Function of miR-25 in the invasion and metastasis of esophageal squamous carcinoma cells and bioinformatical analysis of the miR-106b-25 cluster. Exp Ther Med 2018;15:440–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Xu X, Chen Z, Zhao X, et al. MicroRNA-25 promotes cell migration and invasion in esophageal squamous cell carcinoma.Biochem Biophys Res Commun 2012;421(4):640–645. [DOI] [PubMed] [Google Scholar]
- [9].Lu W, Kang Y. Epithelial- mesenchymal plasticity in cancer progression and metastasis. Dev Cell 2019;49:361–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Gloushankova NA, Zhitnyak IY, Rubtsova SN. Role of epithelial-mesenchymal transition in tumor progression.Biochem (Mosc) 2018;83:1469–1476. [DOI] [PubMed] [Google Scholar]
- [11].Dong P, Ihira K, Xiong Y, et al. Reactivation of epigenetically silenced miR-124 reverses the epithelial-to-mesenchymal transition and inhibits invasion in endometrial cancer cells via the direct repression of IQGAP1 expression. Oncotarget. 2016;7(15):20260–20270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Tang Q, Zhong H, Xie F, et al. Expression of miR-106b-25 induced by salvianolic acid B inhibits epithelial-to-mesenchymal transition in HK-2 cells. Eur J Pharmacol. 2014;741:97–103. [DOI] [PubMed] [Google Scholar]
- [13].Song J, Li Y. miR-25-3p reverses epithelial-mesenchymal transition via targeting Sema4C in cisplatin-resistance cervical cancer cells. Cancer Sci. 2017;108(1):23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Wu C, Wang C, Guan X, et al. Diagnostic and prognostic implications of a serum miRNA panel in oesophageal squamous cell carcinoma. PLoS One. 2014;9:e92292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Riihimäki M, Thomsen H, Hemminki A, et al.: Comparison of survival of patients with metastases from known versus unknown primaries: survival in metastatic cancer. BMC Cancer 2013; 13:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Pecina-Slaus N, Cicvara-Pecina T, Kafka A: Epithelial-to-mesenchymal transition: possible role in meningiomas. Front Biosci (Elite Ed) 2012; 4:889–896. [DOI] [PubMed] [Google Scholar]
- [17].Guarino M, Rubino B, Ballabio G: The role of epithelial-mesenchymal transition in cancer pathology. Pathology 2007; 39:305–318. [DOI] [PubMed] [Google Scholar]
- [18].Cavallaro U, Christofori G. Cell adhesion and signalling by cadherins and Ig-CAMs in cancer. Nat Rev Cancer. 2014;4(2):118–132. [DOI] [PubMed] [Google Scholar]
- [19].Karatzas G, Karayiannakis AJ, Syrigos KN, et al. E-cadherin expression correlates with tumor differentiation in colorectal cancer. Hepatogastroenterology. 1999. Jan-Feb;46(25):232–235. [PubMed] [Google Scholar]
- [20].Syrigos KN, Krausz T, Waxman J, et al. E-cadherin expression in bladder cancer using formalin-fixed, paraffin-embedded tissues: correlation with histopathological grade, tumour stage and survival. Int J Cancer. 1995;64(6):367–370. [DOI] [PubMed] [Google Scholar]
- [21].Maretzky T, Reiss K, Ludwig A, et al. ADAM10 mediates E-cadherin shedding and regulates epithelial cell-cell adhesion, migration, and beta-catenin translocation. Proc Natl Acad Sci U S A. 2015;102(26):9182–9187. [DOI] [PMC free article] [PubMed] [Google Scholar]