

Abstract
Purpose:
Copy number-high endometrial carcinomas (ECs) were described by The Cancer Genome Atlas as high-grade endometrioid and serous cancers showing frequent copy number alterations (CNAs), low mutational burden (i.e. non-hypermutant), near universal TP53 mutation and unfavorable clinical outcomes. We sought to investigate and compare the clinicopathologic and molecular characteristics of non-hypermutant TP53-altered ECs of four histologic types.
Design:
TP53-mutated ECs, defined as TP53-mutant tumors lacking microsatellite instability or pathogenic POLE mutations, were identified (n=238) in a cohort of 1,239 ECs subjected to clinical massively parallel sequencing of 410–468 cancer-related genes. Somatic mutations and CNAs (n=238), and clinicopathologic features were determined (n=185, initial treatment planning at our institution).
Results:
TP53-mutated ECs encompassed uterine serous (n=102, 55.1%), histologically ambiguous high-grade EC-NOS (n=44, 23.8%), endometrioid carcinomas of all tumor grades (n=28, 15.1%), and clear cell (n=11, 5.9%) carcinomas. PTEN mutations were significantly more frequent in endometrioid carcinomas, SPOP mutations in clear cell carcinomas, and CCNE1 amplification in serous carcinomas/EC-NOS; however, none of these genomic alterations were exclusive to any given histologic type. ERBB2 amplification was present at similar frequencies across TP53-mutated histologic types (7.7%−18.6%). Although overall survival was similar across histologic types, serous carcinomas presented more frequently at stage IV, had more persistent and/or recurrent disease, and reduced disease-free survival.
Conclusions:
TP53-mutated ECs display clinical and molecular similarities across histologic subtypes. Our data provide evidence to suggest performance of ERBB2 assessment in all TP53-mutated ECs. Given the distinct clinical features of serous carcinomas, histologic classification continues to be relevant.
Keywords: Endometrial cancer, copy-number high, TP53 mutation, histologic type, sequencing, molecular subtype
TRANSLATIONAL RELEVANCE
Endometrial cancers (ECs) of copy number-high molecular subtype, described initially by The Cancer Genome Atlas to encompass primarily serous and high-grade endometrioid ECs, harbor recurrent TP53 mutations, lack POLE mutations and are microsatellite stable (i.e. non-hypermutant), and have poor clinical outcomes. An analysis of >1,200 ECs subjected to targeted sequencing using an FDA-authorized assay revealed that non-hypermutant TP53-altered ECs irrespective of histologic type have considerable clinical and genomic similarities. We show that the overall landscape of targetable genetic alterations affecting cancer-related genes, including ERBB2, is similar between TP53-mutant ECs across histologic types, supporting the notion that ERBB2 assessment should be performed on all non-hypermutant TP53-altered ECs. As serous carcinomas showed distinct features, however, including advanced stage at diagnosis and more frequent persistent and/or recurrent disease, our findings further demonstrate that despite their similar molecular profiles, histologic subtyping of TP53-mutant ECs remains important.
INTRODUCTION
The Cancer Genome Atlas (TCGA) study of endometrioid and serous carcinomas of the endometrium posited the existence of four genomic subtypes of endometrial carcinoma (EC): ultramutated (hotspot POLE-mutated), microsatellite instability-high (MSI-H) hypermutated (DNA mismatch repair (MMR)-deficient), copy number-low (CN-low) and copy number-high (CN-H; serous-like) (1). CN-H ECs comprised all serous carcinomas and a subset of high-grade endometrioid and mixed epithelial ECs. They were characterized by high levels of CN alterations; almost all harbored deleterious TP53 mutations, lacked pathogenic POLE mutations, were microsatellite stable (i.e. non-hypermutant), and had a significantly more aggressive clinical course compared with tumors from the other molecular categories. There were insufficient data, however, to compare outcomes of CN-H serous ECs versus CN-H endometrioid ECs (1). A follow-up study, focusing on endometrioid ECs only, indicated that TP53-mutated microsatellite stable (MSS) endometrioid ECs lacking hotspot POLE mutation (i.e. non-hypermutated), roughly equivalent to CN-H, had the shortest overall survival and recurrence-free survival (2). Furthermore, it has been reported that both clear cell EC and uterine carcinosarcoma could be similarly stratified into meaningful genomic categories that retained comparable associations with clinical outcomes (3–5). Although non-hypermutant TP53-altered/ CN-H ECs have been shown to be clinically aggressive, it is currently unknown whether the histologic type is of significance in these TP53-mutated tumors. Therefore, we sought to define the clinicopathologic and molecular characteristics of a large group of histologically heterogeneous non-hypermutant TP53-altered ECs and to determine whether histologic type provides useful information in the setting of TP53-mutation status.
MATERIALS AND METHODS
Case selection
This study was approved by the Institutional Review Board at Memorial Sloan Kettering Cancer Center (MSK), and written informed consent was obtained from all patients. This study was conducted in accordance with the principles of the Declaration of Helsinki. All ECs that underwent clinical FDA-authorized tumor-normal targeted massively parallel sequencing analysis of 410–468 cancer-related genes (MSK-Integrated Mutation Profiling of Actionable Cancer Targets; MSK-IMPACT) (6,7), from 2014 to 2019 were evaluated (n=1,239; Figure 1). Non-hypermutant TP53-mutated ECs (referred to henceforth as TP53-mutated ECs) were identified using a surrogate model, modified from the one described by Talhouk et al (8): All ECs that harbored a somatic TP53 alteration (n=514) were selected; carcinosarcomas were not included in this study. From the 514 TP53-mutated ECs, those with DNA MMR deficiency (n=158), as defined by loss of expression of any MMR protein (i.e. MLH1, MSH2, MSH6 and PMS2) by immunohistochemistry (IHC), or MSI determined by PCR or massively parallel sequencing, and tumors harboring hotspot POLE exonuclease domain mutations (n=59) (9) were excluded, leaving 297 TP53-mutated ECs (Figure 1).
Figure 1. CONSORT diagram summarizing the selection process of non-hypermutant TP53-mutated endometrial cancers included in this study.
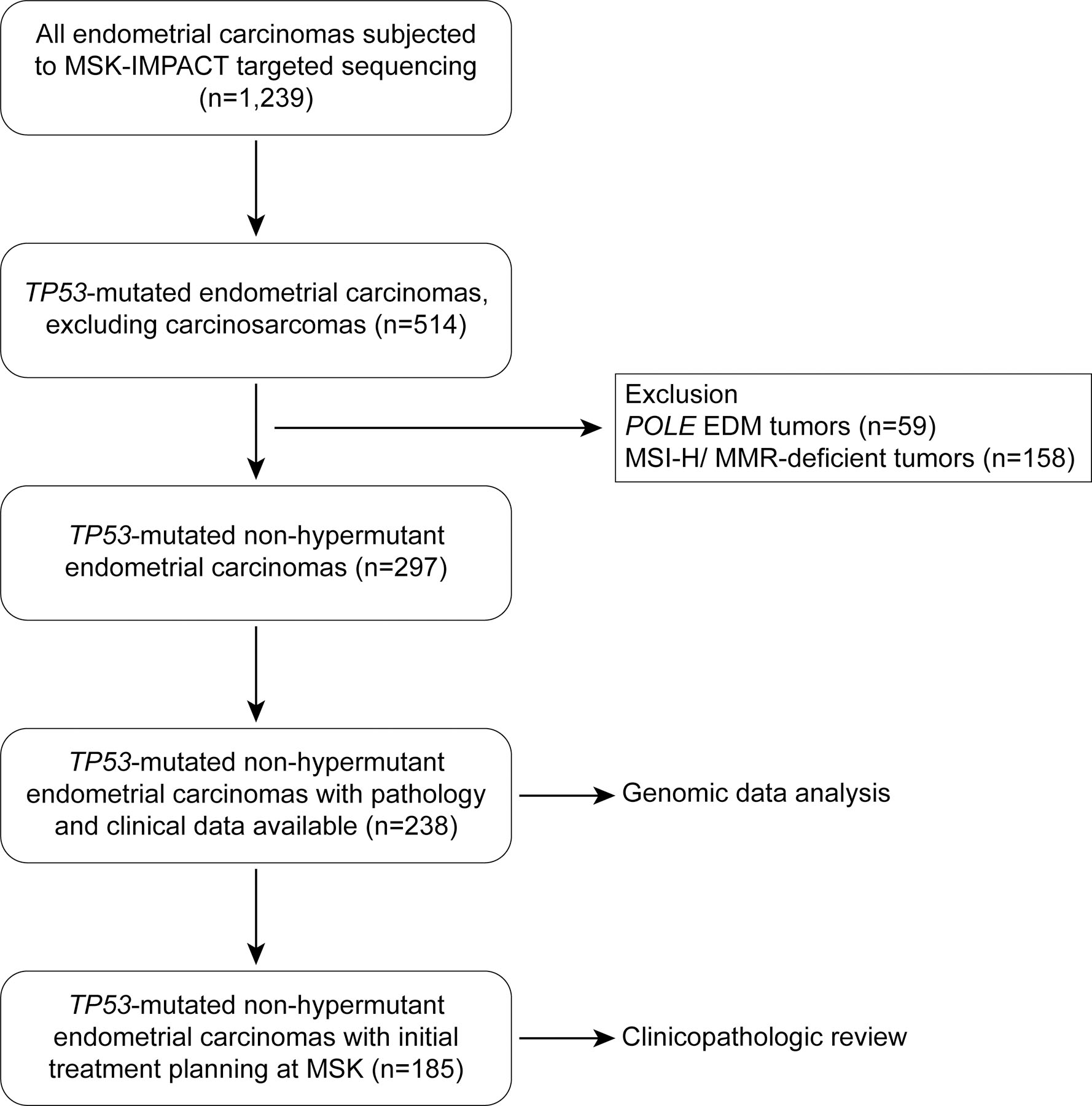
EDM, exonuclease domain mutant; MMR, DNA mismatch repair; MSI, microsatellite instability
For a case to be included, a gynecologic pathologist at MSK must have confirmed the histologic diagnosis of EC (as discussed below). Cases were eligible for inclusion in the analyses of clinical outcomes as long as patients had their initial treatment planning at MSK within three months of diagnosis, whether or not the primary surgery was done at MSK or elsewhere. Women who presented to MSK at time of recurrence, women with a concurrent advanced malignancy, and women with only pathology review or lacking sufficient follow-up were excluded from the analyses of clinical outcomes. Of the 297 TP53-mutated ECs, 238 had sufficient follow-up and pathology data. Of these 238 cases, 185 were seen at the time of their initial treatment planning, 142 of whom had their initial surgery at MSK. Fifty-three patients presented to MSK at the time of recurrence. For evaluation of the clinicopathologic factors, only the cases that were seen at the time of initial treatment planning were included (n=185). The CONSORT diagram summarizing case selection is shown in Figure 1. Patients’ demographic and clinical data were extracted by review of the electronic medical record (EMR), with information related to disease progression and survival captured. Disease status and disease progression were extracted from medical oncology notes, in combination with follow-up imaging studies according to the PRISSMM data model approach (10).
Pathology review
For each case, the histopathologic and morphologic data were extracted from the synoptic pathology report. For histologic typing, to mitigate the effect of suboptimal interobserver concordance (11,12), we performed a single-institution study with a group of experienced gynecologic pathologists. Biweekly diagnostic consensus conferences encouraged a uniform diagnostic approach within the group, as did frequent review of each other’s cases for tumor board and quality assurance. In general, a histologically high-grade EC was diagnosed as endometrioid when there was a component of low-grade endometrioid carcinoma or squamous, tubal, or mucinous differentiation. This approach was chosen based on data showing that high-grade endometrioid ECs, diagnosed according to these guidelines, were more likely than other high-grade ECs to harbor mutations associated with endometrioid tumorigenesis, such as PTEN and ARID1A (13,14). DNA MMR IHC was performed almost universally (215/238 cases), whereas PTEN and ARID1A IHC was performed in histologically ambiguous cases. Loss of DNA MMR protein, PTEN or ARID1A protein expression or heterogeneous (subclonal/geographic) expression favored endometrioid or clear cell EC, rather than serous EC. Clear cell carcinomas were diagnosed according to the guidelines of Fadare et al (15), which emphasize exclusionary features that are more commonly encountered in endometrioid and serous carcinomas. Tumors with variable morphologic features and absence of characteristics for confident histologic subclassification were assigned as unclassifiable high-grade EC (EC-NOS; histologically ambiguous), as previously described (16). These tumors had overlapping features of endometrioid, serous and clear cell carcinoma throughout the tumor with no predominant histologic features of one of the histologic subtypes present (16). The Gilks’ grading scheme (17) was used to determine “high grade” when it was uncertain whether to apply FIGO grading criteria. Slides from all FIGO grade 1 and 2 TP53-mutated endometrioid ECs were re-reviewed to ascertain grade.
Genomic data extraction
The genomic data were extracted from the MSK-IMPACT assay, including somatic mutations, CN alterations, and structural variants (6). Analysis of broad CN alterations was performed using the GISTIC 2.0 algorithm (18). Breakpoint instability index (BPII) was calculated using the modified Bayes Information Criterion, implemented in the R package WBS, using segmentation files containing focal copy number data (19).
Statistical analysis
Disease-free survival (DFS) and overall survival (OS) were evaluated by calculating survival curves using the Kaplan–Meier method, using the Log-rank test to compare subgroups, with the start date set as the date of initial diagnostic biopsy. Univariate and multivariate Cox Proportional Hazards analysis was performed to determine the hazard ratio (HR). Comparisons of quantitative data between the groups were performed using ANOVA with post-hoc Tukey test and comparison of qualitative data, including associations between clinicopathologic features and molecular data, were performed using the Chi-squared (X2) test. P-values of <0.05 were considered statistically significant.
RESULTS
Clinicopathologic landscape of non-hypermutant TP53-altered ECs
Of the 238 patients with TP53-mutated ECs included in this study, 185 (77.7%) were seen at the time of their initial treatment planning (Figure 1), with 142 having their initial surgery at MSK and 43 referred to our institution after disease recurrence. The evaluation of clinicopathologic data was limited to the 185 patients seen at the time of initial treatment planning (Figure 1).
The most common histologic subtype of TP53-mutated EC was uterine serous carcinoma (USC; 102/185, 55.1%), followed by unclassifiable high-grade EC (EC-NOS; histologically ambiguous; 44/185, 23.8%), uterine endometrioid carcinoma (UEC; 28/185, 15.1%) and uterine clear cell carcinoma (UCC; 11/185, 5.9%). FIGO grade information for the 28 UECs revealed that the majority were FIGO grade 3 (19/28, 67.9%), as expected; however, FIGO grade 2 (7/28, 25%) and FIGO grade 1 (2/28, 7.1%) TP53-mutated UECs were also identified (Figures 2A-F).
Figure 2. Histologic subtypes of non-hypermutant TP53-mutated endometrial carcinomas and p53 expression.
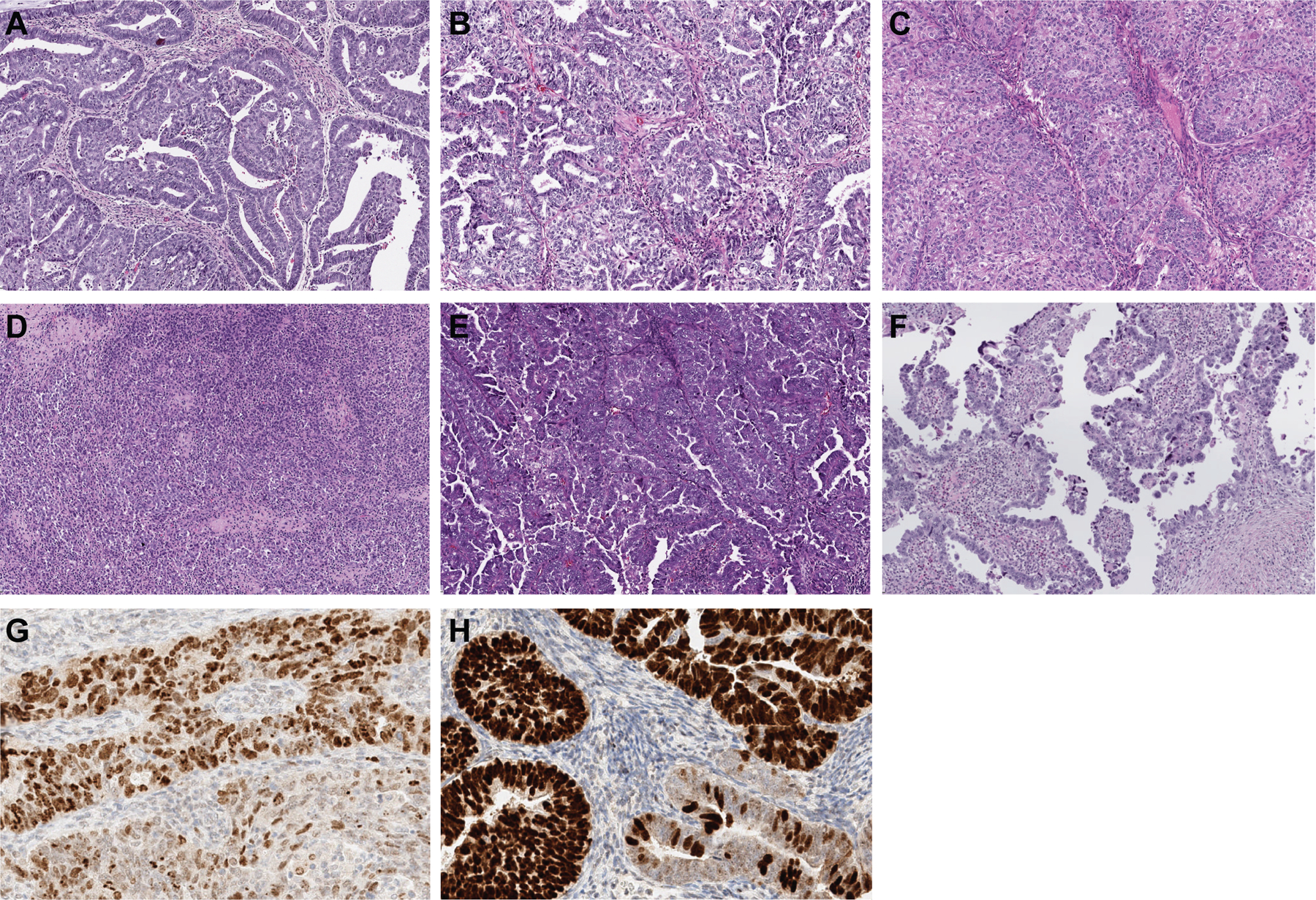
TP53-mutated endometrial carcinomas are of different histologic subtypes. TP53-mutated endometrioid endometrial carcinoma can present as (A) FIGO grade 1, (B) grade 2, or (C) grade 3. (D) TP53-mutated endometrial carcinomas can have ambiguous morphologic features. (E) The most common histologic subtype is endometrial serous carcinoma. (F) Endometrial clear cell carcinoma is the least common morphologic variant. In some TP53-mutated endometrial carcinomas, p53 aberrant expression can be subclonal. The subclonal aberrant expression in (G) shows variable overexpression of p53 in most cells of UEC (top), whereas the subclonal aberrant expression in (H), a high-grade endometrial carcinoma with ambiguous features/ not otherwise specified (EC-NOS), shows more uniform overexpression of p53 (top), the bottom glands in both (G) and (H) show wild-type (heterogenous) p53 staining pattern.
The median age at EC diagnosis was 69 years (range, 39–93 years; Table 1). Women with UEC were found to be younger (median 63 years, range 44–79 years) than those with other subtypes of TP53-mutated EC (median age 70, range 39–93; p <0.001, one-way ANOVA with post-test Tukey means comparison). No significant difference in age at EC diagnosis was found between the other histologic subtypes (Figure 3A).
Table 1.
Summary of clinicopathological findings in non-hypermutated TP53-altered endometrial carcinomas
| All | EC-NOS | UCC | UEC | USC | ||
|---|---|---|---|---|---|---|
| Samples (n) |
185 | 44 | 11 | 28 | 102 | |
| Median age at diagnosis (range) (years) |
69 (39–93) | 69 (39–93) | 73 (67–87) | 63 (44–79) | 70 (55–92) | |
| Tumors with myometrial invasion (%) |
62.2 | 61.4 | 63.6 | 60.7 | 62.7 | |
| Myometrial invasion depth (%) |
48.7 | 47.3 | 45.8 | 43.1 | 51.2 | |
| Tumors with cervical stromal invasion (%) |
22.2 | 20.5 | 25.0 | 18.5 | 30.9 | |
| Tumors with lymphovascular invasion (%) |
61.6 | 56.8 | 54.5 | 42.9 | 69.6 | |
| Tumors with adnexal involvement (%) |
29.7 | 38.6 | 18.2 | 21.4 | 29.4 | |
| FIGO Stage (%) | I | 30.3 | 25.0 | 45.4 | 53.6 | 24.5 |
| II | 5.4 | 4.5 | 9.1 | 0.0 | 6.9 | |
| III | 31.3 | 34.1 | 27.3 | 39.3 | 28.4 | |
| IV | 33.0 | 36.4 | 18.2 | 7.1 | 40.2 | |
|
Patients with disease progression (%) |
74.1 | 65.9 | 54.5 | 53.6 | 85.3 | |
| Median disease-free survival (months) |
16.6 | 19.3 | 31.3 | 25.7 | 14.5 | |
| Patients alive (%) |
64 | 61 | 100.0 | 71 | 59 | |
| Median overall survival (months) | 53.9 | 89.2 | N/A | 98.4 | 43.8 | |
EC-NOS: Histologically ambiguous high-grade endometrial carcinoma, UCC: Uterine clear cell carcinoma, UEC: Uterine endometrioid carcinoma, USC: Uterine serous carcinoma.
Figure 3. Clinicopathologic features of non-hypermutant TP53-mutated endometrial carcinomas and association with outcome.
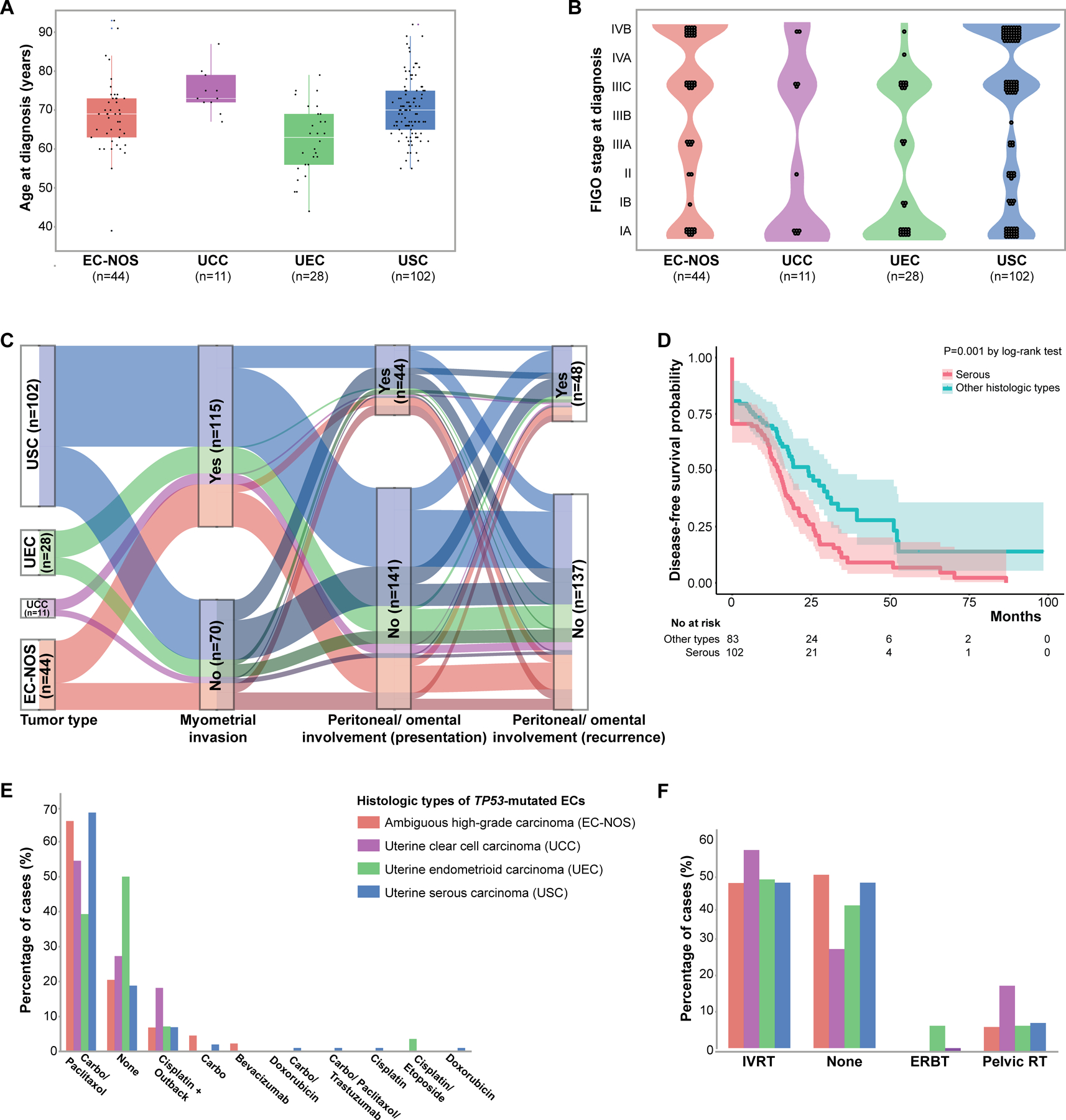
(A) Age at TP53-mutated endometrial cancer diagnosis. These tumors mostly occur in elderly patients. (B) FIGO stage at TP53-mutated endometrial cancer diagnosis. The majority of TP53-mutated tumors present with advanced FIGO stages. (C) Alluvial graph showing myometrial invasion, peritoneal/ omental involvement at presentation and recurrence in patients with TP53-mutated endometrial carcinomas. Note that TP53-mutated endometrial cancers have similar rates of peritoneal involvement irrespective of histologic subtype. Cases with non-myoinvasive disease are shown in darker shades. (D), Kaplan-Meier curve for disease-free survival of patients with TP53-mutated endometrial cancer of serous histology and those of other histologic subtypes (i.e. clear cell, endometrioid, and ambiguous high-grade). (E) Percentage of patients with TP53-mutated endometrial cancer who received adjuvant chemotherapy. Note that the choice of adjuvant therapy was different between UEC and USC. (Carbo: carboplatin). (F) Percentage of patients with TP53-mutated endometrial cancer who received adjuvant radiation therapy. IVRT, intravascular radiation therapy; ERBT, External beam radiation therapy; RT, radiotherapy. EC-NOS, high-grade endometrial carcinoma with ambiguous features/ not otherwise specified; UCC, uterine clear cell carcinoma; UEC, uterine endometrioid carcinoma; USC, uterine serous carcinoma.
The distribution between the FIGO stages at TP53-mutated EC diagnosis was similar between stage I (56/185, 30.3%), stage III (58/185, 31.3%) and stage IV (61/185, 33%), while FIGO stage II disease was less common (10/185, 5.4%). We noted, however, that TP53-mutated USC was more likely to present at stage IV compared with the other morphologic subtypes (41/102 vs. 20/83; X2 p=0.021; Figure 3B). No significant differences were observed between different histologic subtypes in the average depth of myometrial invasion, lymphovascular invasion, cervical stromal or adnexal involvement (Table 1).
Clinical behavior of TP53-mutated ECs
Of the 185 patients with TP53-mutated EC, 56.2% (n=104) presented with extrauterine disease at diagnosis (n=56 extrauterine sites, n=64 lymph nodes). The most common site of metastasis at presentation was peritoneum/omentum, with 44 patients having peritoneal disease at presentation (23.8%; Figure 3C); the majority of cases showed involvement of intrapelvic peritoneum (n=35) followed by omentum (n=9). Metastatic involvement of other sites at presentation were also found, including lung (n=8) and liver (n=2).
One hundred and thirty-seven patients (74.1%) with TP53-mutated EC had disease recurrence/progression. Among them, the most common site of disease spread at recurrence was peritoneum (48 patients, 35%), with intrapelvic peritoneal recurrence in 47 patients, and extra-pelvic/omental recurrence in 2 patients (Figure 3C). Lung metastasis was also common (22/137, 16.1%). Other metastatic sites included vagina (n=4) and liver (n=3). The location of the recurrence was not significantly different between histologic subtypes (data not shown).
Extrauterine disease and recurrences were not restricted to TP53-mutated EC with myoinvasive disease. Of the 70 patients whose tumors were non-myoinvasive, 37 (52.9%) had disease spread at the time of hysterectomy (n=25 (35.7%) distant metastasis; n=11 (15.7%) lymph node metastasis), 9 (12.9%) showed adnexal involvement, and 48 (68.6%) subsequently recurred.
Among the entire cohort, the overall all-cause mortality rate of patients with TP53-mutated EC was 36.2% (n=67), with a median OS of 54.7 months and a median DFS of 16.6 months (Table 1). While OS was not different between the histologic subtypes (p=0.103, Log-rank test), DFS was significantly different; compared to all other TP53-mutated histologic types, patients with USCs had a clearly reduced DFS (p=0.001, Log-rank; Figure 3D). Multivariate Cox Proportional Hazards analysis revealed that serous morphology was a predictor of DFS (adjusted HR: 1.65, p=0.007) but not OS (adjusted HR: 1.62, p=0.06) in this series of TP53-mutated EC (Supplementary Tables S1 and S2). Finally, we found that a larger proportion of patients with USC had disease persistence/recurrence (85.3% vs. 60.2% all other subtypes, p=0.0001), and all-cause mortality was higher in this group (41.2% vs. 30.1% all other subtypes); however this difference was not statistically significant (X2, p=0.12).
As expected, we found that, irrespective of histologic type, stage was a predictor of outcome in TP53-mutated ECs, with patients presenting at FIGO stages IIIC and IV having the worst outcomes (multivariate HR for combined stage IIIC/IV versus lower stages for OS: 2.46, p=0.001). No statistically significant difference in outcomes of TP53-mutated EC was observed when comparing FIGO stage I, stage II and stage III-A and III-B disease (Supplementary Tables S1 and S2, Supplementary Figure S1).
Treatment of TP53-mutated ECs
Of the 185 patients with TP53-mutated EC, 27 (14.6%) received neoadjuvant therapy (n=26 neoadjuvant chemotherapy, n=1 radiation therapy), with the majority of neoadjuvant chemotherapy regimens (23/27) including carboplatin and paclitaxel. The choice of neoadjuvant therapy was significantly different between the distinct histologic types: 19/102 (18.6%) patients with USC and 7/44 (15.9%) patients with EC-NOS received neoadjuvant therapy, compared with none of the 28 patients with UEC and only 1/11 with UCC (p=0.017). Furthermore, of the 185 patients with TP53-mutated EC, 160 (86.5%) received adjuvant therapy; of these, 82 patients (82/160=44.3%) had chemotherapy and radiation therapy, 57 (30.8%) chemotherapy alone, and 21 (11.3%) radiation therapy alone. The most common adjuvant chemotherapy regimen included carboplatin and paclitaxel (n=115; Figure 3E). As expected, the choice of adjuvant therapy regimen was significantly different between USC and UEC (p=0.001), with TP53-mutated UEC patients more likely to receive radiation therapy alone (32.1% vs. 5.9%, p=0.0001), and USC patients more likely to receive chemotherapy with/without radiation therapy (75.3% vs. 20.7%, p=0.001; Figures 3E and 3F). On univariate analysis, adjuvant therapy showed OS and DFS benefit (Log-rank p-value for OS and DFS: 0.04 and 0.04 respectively), with multivariate analysis only showing DFS benefit (multivariate HR for adjuvant therapy: 0.29, p=0.012). Finally, 12 TP53-mutated EC patients (6.5%) received no neoadjuvant or adjuvant therapy.
Genomic landscape of TP53-mutated ECs
Of the TP53-mutated ECs identified, 238 had clinical, pathology and molecular profiling data available for analysis (Figure 1). The median number of non-synonymous somatic mutations in these 238 TP53-mutated ECs was 5 (range, 1–10) and the median fraction of the genome altered was 23.8% (range, 0–75.5%), which were not significantly different between histologic types. TP53-mutated ECs exhibited a high degree of chromosomal instability, with the mean number of chromosomal breakpoints being 127.6 (range: 2–455) with no statistically significant difference observed between various TP53-mutated EC histologic types.
Based on our inclusion criteria, all ECs harbored TP53 somatic mutations (100%). Other recurrent genetic alterations found in the TP53-mutated ECs included PIK3CA (44%), PPP2R1A (30%), and FBXW7 (18%) somatic mutations as well as ERBB2 alterations (21%; 5.5% mutations, 17.2% amplification; Figure 4A). The majority of somatic mutations detected in these recurrently altered genes were either known to be oncogenic or were predicted/likely to be oncogenic (Supplementary Figure S2). The frequency of ERBB2 amplification did not differ between the histologic types, with 18.6% of USC, 12.2% of UEC, 7.7% of UCC, and 18.2% of EC-NOS tumors harboring ERBB2 amplification (p=0.57; p=0.065 for USC versus UEC). Of note, 13/238 (5.5%) of TP53-mutated ECs harbored ERBB2 point mutations, with 5 activating hotspot mutations in the kinase domain, which have shown association with sensitivity to anti-HER2 therapy in other cancer types (20) (Figure 4A, Supplementary Figure S3); all 5 hotspot kinase domain mutations were observed in USCs. The landscape of somatic mutations affecting the 468 cancer-related genes studied was similar across the various histologic types of TP53-mutated ECs, and no mutation was exclusive to a specific histologic type. However, the frequency of specific alterations was found to be distinct between histologic subtypes. Notable among these was PTEN, which was more frequently altered in TP53-mutated UECs (34% vs. 10% in all other morphologies, X2 p=0.001), and SPOP, which was more commonly altered in TP53-mutated UCCs (31% vs. 3% in all other morphologies, X2 p=0.01). Across histologic types, there was statistically significant mutual exclusivity between FBXW7 and PPP2R1A somatic mutations and between PIK3R1 and PIK3CA somatic mutations (p <0.01). On the other hand, alterations in ARID1A, PTEN, PIK3R1, ZFHX3, and FGFR2 showed statistically significant co-occurrence (p <0.05; Figure 4B). The proportion of TP53-mutated UECs harboring PTEN alterations was higher than other subtypes (35% vs. 10%) and this proportion was highly correlated with FIGO grade (100%, 66.6% and 31.2% of FIGO grade I, grade II, grade III tumors, respectively).
Figure 4. Somatic mutations and gene copy number alterations in non-hypermutant TP53-mutated endometrial carcinomas.
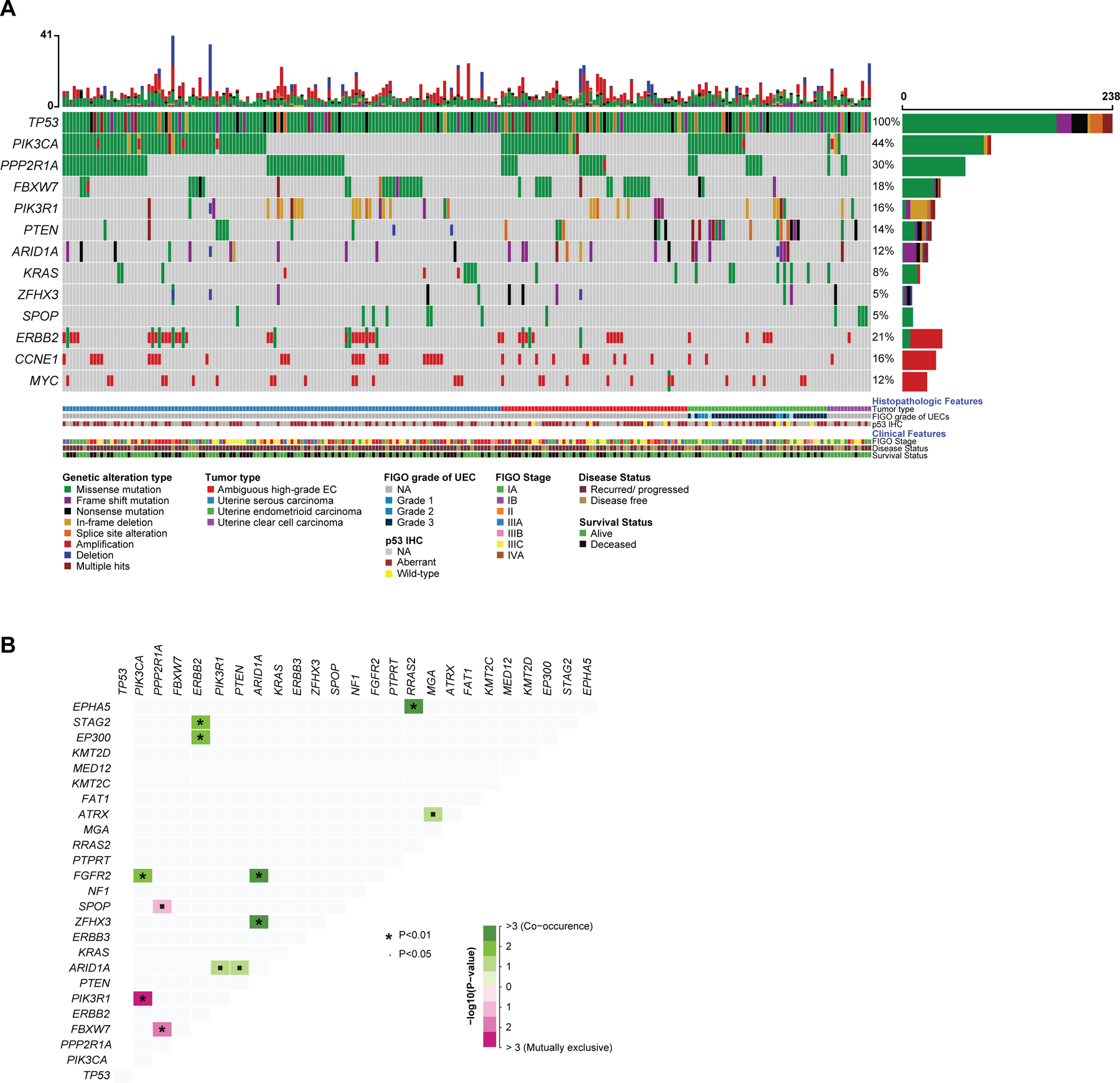
(A) Oncoprint depicting the most recurrent genomic alterations in TP53-mutated endometrial carcinomas. Each column represents a tumor with the bar graph at the top depicting the number/distribution of alterations per sample, and the Oncoprint rows showing alterations for each gene. The bottom part of the graph shows the summary of histopathologic and clinical information for each case. The bar graph on the right of the panel shows the number and distribution of alterations for each gene. Mutation types and clinicopathologic features are color-coded according to the legend. NA, not available. (B) Distance matrix showing the somatic interaction of commonly altered genes in TP53-mutated endometrial carcinomas. Significant co-occurrences are marked in shades of green and the significant mutual exclusivity marked in shades of purple. The size of the * depicting the p-value and the intensity of color shows the degree of association or exclusion.
In terms of copy number alterations, the most commonly amplified cancer-related genes in TP53-mutated ECs were ERBB2 (16.8%), as noted above, CCNE1 (16%, 37/238), and MYC (12%, 29/238). CCNE1 amplifications were mainly present in USC (25/131) and EC-NOS (9/55) and only rarely observed in UEC (2/41) and UCC (1/11; X2 p=0.009). In terms of broad level CN alterations, the most common high-level gains were of chromosomes 19 and 20, as well as chromosomal arms 1q, 3q, and 8q; the most common losses were chromosomal arms 17p, 16q, 22q, 9q, and 8p (Supplementary Figure S4).
As an exploratory, hypothesis-generating analysis we assessed whether specific genetic alterations would be associated with outcome in the 185 TP53-mutant EC patients who had initial treatment planning at MSK (for details, see Supplementary Methods, Supplementary Figure S5). These findings need to be confirmed in larger independent series of TP53-mutated ECs.
Correlation of TP53 mutation status with p53 immunohistochemistry (IHC)
Of the 238 TP53-mutated ECs included in this study, p53 IHC was performed and reported in 118 (49.6%). Of these, 110 tumors showed aberrant expression of p53 and 8 (6.8%) were reported to have heterogeneous (wild-type) expression patterns; however, based on our inclusion criteria, all TP53-mutated ECs harbored TP53 mutations. All 61 USCs with p53 IHC performed had concordant p53 IHC and TP53 mutation results, while 4 TP53-mutated UECs and 4 EC-NOSs had discordant p53 protein expression/TP53 mutation results (Figure 4A).
Of note, all TP53 somatic mutations in the p53 IHC wild-type expression group affected mutation hotspots, with seven missense and one splice-site mutation (p.C238R, p.R248Q, p.R181C, p.G245S, p.P278R, p.X126_splice, p.R273H and p.P152L). All truncating TP53 mutations displayed aberrant p53 protein expression patterns by IHC (data not shown). No differences in TP53 variant allele frequency were observed when comparing tumors with and without abnormal p53 expression in the TP53-mutated ECs. Furthermore, no particular protein change was shown to be exclusive to the p53 wild-type group. The p53 IHC was available for re-review in 6 of the tumors with a wild-type expression pattern (4 UEC, 2 EC-NOS). Three showed an aberrant subclonal staining pattern: i.e., aberrant staining in an area of the tumor with wild-type staining in other areas (Figures 2G and 2H); while the other 3 showed true wild-type staining patterns, an uncommon phenomenon that has also been observed in ovarian high-grade serous carcinomas (21).
DISCUSSION
ECs with aberrant p53 protein expression were traditionally thought to be composed of USCs (22) and small subsets of high-grade UECs (23,24) and UCCs (25). This was corroborated by the TCGA EC study, which showed that a subset of grade 3 UECs may harbor a so-called serous-like genotype as manifested by an abundance of CN alterations and TP53 mutations (1); these tumors also showed adverse clinical outcomes, with a mortality rate similar to that of USCs. These findings led to a proposal for molecular classification of ECs with CN-H as one of the categories (1,2), with further studies demonstrating that presence of deleterious TP53 mutations in absence of a hyper-/ultra-mutated setting can be used as a surrogate for CN-H tumors (2). Here we demonstrate that in a series of ECs subjected to clinical MSK-IMPACT sequencing, only about half (54%) of TP53-mutated ECs were of serous histology, whereas the other half were UECs, UCCs and histologically ambiguous high-grade EC-NOS, which exhibit a hybrid of serous, endometrioid and/or clear cell features (16,26). Furthermore, in the group of TP53-mutated ECs of endometrioid histology, not all were grade 3; even grade 2 and 1 tumors harbored TP53 mutations in the absence of MMR deficiency or POLE exonuclease domain mutations. While acknowledging an ascertainment bias, we show that the overall demographic and clinical pictures of TP53-mutated ECs were similar: they occurred in older women with disproportionately advanced stage disease and were frequently associated with adverse outcomes, such as disease progression (74.1%) and mortality (36.2%).
Akin to the serous and high-grade endometrioid CN-H ECs in the TCGA study (1), the TP53-mutated ECs of various histologic types studied here had frequent PI3K pathway alterations, with 44% harboring PIK3CA alterations and 16% PIK3R1 mutations; 14% harbored PTEN alterations, primarily affecting UECs. We confirmed the high frequency of PPP2R1A and FBXW7 somatic mutations in USC but also found that PPP2R1A and FBXW7 were recurrently altered in histologically ambiguous high-grade ECs, UECs and UCCs. We also noted that there was a tendency towards mutual exclusivity between FBXW7 and PPP2R1A alterations. Cyclin E is one of the proteins ubiquitinated by FBXW7, and prior studies have reported mutual exclusivity between CCNE1 amplification and FBXW7 (27); in our series, however, this failed to reach statistical significance (X2, p=0.6). The overall repertoire of somatic genetic alterations was similar across the histologic spectrum of TP53-mutated tumors, with the exception of PTEN alterations (more common in UEC), SPOP mutations (more common in UCC) and CCNE1 amplification, which was very rare in UEC and UCC, but none of the other genomic alterations were specific to any subtype. Theoretically, the temporal order of acquired mutations could underlie the morphologic differences between the subtypes. Epigenomic/transcriptomic differences might also contribute (28,29). Regardless of the initiating event, however, all the TP53-mutated ECs appear to follow a convergent genomic evolution leading to similarities in disease progression and clinical outcome.
Despite the global chromosomal instability observed in TP53-mutated tumors, the patterns of CN alterations were not entirely random, and many recurrent alterations were identified. Among these, ERBB2 amplification was the most common CN event observed in TP53-mutated ECs. ERBB2 amplification was originally described in USCs, with up to 20% reportedly having amplification of ERBB2, with both fluorescence in situ hybridization (FISH) and IHC for HER-2/neu supporting these findings (30). Following a clinical trial showing the effectiveness of anti-HER2 treatment in patients with advanced USC (31), guidelines now suggest the assessment of HER2 in advanced/recurrent USC and the addition of trastuzumab to chemotherapy. Here we used an FDA-approved sequencing assay showing that, while ERBB2 amplification was seen in 18.6% of USCs, the other histologic subtypes of TP53-mutated EC also harbored ERBB2 amplification in a smaller proportion of tumors. As such, there may be justification for determining the ERBB2 amplification status in all advanced TP53-mutated ECs with consideration given to targeted treatment regardless of histologic subtype. Furthermore, a subset of TP53-mutated ECs harbored activating ERBB2 mutations rather than ERBB2 amplification (5.5% of TP53-mutated ECs, all USC); this may also predict response to targeted HER2 therapy, emphasizing the importance of sequencing ECs (32).
The data on the prognostic importance of morphologic classification of high-grade ECs have been controversial. Studies have shown that histologic type is not associated with clinical outcomes in high-grade/p53abnormal ECs and, regardless of the histologic type, these tumors tend to have poor outcomes (14,33–35). This suggests that molecular classification can supplant morphologic classification (11,17,36), with a recent study showing near identical survival outcomes for USC and TP53-mutated MMR-proficient UEC (37). Although it is acknowledged that morphologic subtyping of high-grade ECs is associated with considerable inter-observer variability (12,33), a recent meta-analysis reported on a higher HR for non-endometrioid TP53-mutant ECs in comparison with endometrioid TP53-mutant ECs (35). With respect to this, our data demonstrate that, while a pan-morphologic comparison fails to identify significant differences in clinical outcome, a comparison between pure serous versus other TP53-mutated ECs (including histologically ambiguous tumors, clear cell and endometrioid) showed a statistically significant difference in DFS. It should be noted, however, that the clinical significance of this difference in DFS is not entirely clear, in light of the fact that a considerable number of patients who experience recurrence will ultimately succumb to the disease.
Regardless of histologic type, all TP53-mutated ECs had adverse outcomes overall: 69.7% of patients presented at FIGO stages II and above, with frequent disease progression (74.1%), and a high mortality rate (36.2%). The pattern of disease spread, including the frequency of peritoneal involvement at the time of hysterectomy and recurrence, was found to be similar among the various histologic types of TP53-mutated ECs. Current surgical practices recommend omental sampling for USC and UCC (38); however, our data suggest that patients with other TP53-mutated tumors may also benefit from omental sampling at the time of hysterectomy. High rates of disease progression and spread were not restricted to myoinvasive TP53-mutated ECs; 52.9% and 68.6% of patients in non-myoinvasive TP53-mutated carcinomas had disease beyond the uterine corpus at the time of hysterectomy and showed disease progression, respectively. This phenomenon has been previously described for USC and endometrial serous intraepithelial carcinoma (39,40); our data demonstrate that other non-myoinvasive TP53-mutated ECs can present with extrauterine and metastatic disease. This has important implications for the implementation of sentinel lymph node protocols for these tumors. Sentinel lymph node protocols have been limited to myoinvasive tumors (41), but our data indicate that ultra-staging may need to be extended to non-myoinvasive high-grade ECs (42). FIGO grade is part of the risk stratification algorithms for patients with UEC; low-grade UECs (FIGO grades 1 and 2) generally have a favorable clinical course (17). It should be noted, however, that each FIGO grade can be genomically heterogeneous (43,44). As our data show, even low-grade UECs can have aberrant p53 expression/ habor somatic TP53 mutations, portending aggressive disease when unassociated with POLE mutation or MSI (45).
Current guidelines for the treatment of UEC do not include molecular subtyping in clinical decision algorithms although it has been proposed by the NCCN (46) and is increasingly used to detect POLE mutation in an effort to de-escalate therapy and to determine eligibility for checkpoint inhibitor therapy. Our data show that molecular subtyping allowed detection of the rare TP53 mutated low-grade UECs, which may currently masquerade as low-risk tumors (47).
Almost all CN-H tumors harbor TP53 mutations (1), and p53 IHC analysis has been used as a surrogate (2,45). p53 IHC is a robust and reproducible predictor of adverse clinical outcomes, with numerous studies confirming a high degree of concordance between p53 expression by IHC and TP53 mutational status (48,49). We observed a high degree of concordance between TP53 mutational status and p53 protein expression by IHC; of the 118 tumors for which p53 IHC results were reported, 110 (93.2%) had concordant results and 8 (6.8%) were found to have a wild-type expression pattern. Re-review of the p53 IHC analysis in 6/8 cases led to reclassification of an additional 3 cases, as these were found to have subclonal aberrant p53 expression. Importantly, however, all 8 UECs/EC-NOS with discordant p53 IHC/TP53 mutation status harbored TP53 hotspot mutations; the detection of p53/TP53 alterations is importance in determining the correct molecular subtyping (i.e. these tumors would be classified as CN-low) and in treatment, as these may portend aggressive disease (45). As our results reaffirm the utility of p53 IHC as a surrogate marker for TP53-mutant/ CN-H endometrial tumors overall, we suggest that p53 IHC be performed on all malignant endometrial biopsies or curettings, and the immunophenotype of a carcinoma, represented in a biopsy or curetting, tends to be concordant with the matched resection specimen (50). This is especially useful for morphologically ambiguous tumors in which the combination of DNA MMR protein and p53 IHC can be used for risk stratification (16). One complicating factor is our current inability to distinguish between clinically significant driver mutations (in CN-H carcinomas) and clinically insignificant passenger TP53 mutations (such as in POLE mutated carcinomas) without sequencing.
In our cohort, despite the similarities in outcome between USCs and UECs, choice of neoadjuvant/adjuvant therapy differed; patients with UECs were less likely to receive neoadjuvant therapy and adjuvant chemotherapy. Additional work is warranted to understand the clinical behavior of non-hypermutant TP53-mutated low-grade UECs. Nevertheless, our data emphasize the importance of molecular risk stratification in addition to histologic grading in UECs.
Our study has several limitations. Although we studied a large series of TP53-mutated ECs, UCCs are rare and the sample size small; hence, the subgroup analyses on UCC should be interpreted as exploratory and warrant further independent validation. Furthermore, the mutational analyses we performed were restricted to cancer-related genes; whole-exome or whole-genome sequencing analyses are warranted to assess whether other genetic alterations, including structural rearrangements, may play roles in the different TP53-mutated histologic subtypes ECs studied, which could not be assessed here.
Taken together, this study demonstrates that TP53-mutated ECs across histologic subtypes harbor similar molecular profiles. Our findings further suggest that ERBB2 assessment should be performed on all TP53-mutated ECs. Although TP53-mutant ECs of distinct histologic types display genomic and clinical similarities, serous carcinoma were more frequently stage IV at presentation and had more persistent and/or recurrent disease, which provides justification for the continuing histologic classification of TP53-mutated ECs alongside molecular stratification/ characterization.
Supplementary Material
ACKNOWLEDGMENTS
BW was funded in part by Breast Cancer Research Foundation, Cycle for Survival and Stand Up To Cancer grants. Research reported in this publication was supported in part by a Cancer Center Support Grant of the NIH/NCI (Grant No. P30CA008748).
Footnotes
Conflicts of interest: None declared.
Disclosures: NR-A reports grants from Stryker/Novadaq, Olympus and GRAIL, outside the submitted work. CA reports personal fees from Tesaro, personal fees from Immunogen, grants and personal fees from Clovis, personal fees from Mateon Therapeutics, personal fees from Cerulean Pharma, grants from Genentech, grants from AbbVie, grants from Astra Zeneca, outside the submitted work. SC is a consultant for AstraZeneca, outside of the submitted work. LHE is Editor-in-Chief of the International Journal of Gynecological Pathology and is paid by both the publisher WWK and the International Society of Gynecological Pathologists, reports royalties from Springer as co-Editor of the textbook Blaustein’s Pathology of the Female Genital Tract, outside the submitted work. ML has received advisory board compensation from Merck, Lilly Oncology, AstraZeneca, Bristol-Myers Squibb, Takeda, Blueprint Medicines, Bayer, and Paige.AI, and research support from LOXO Oncology, Helsinn Healthcare, Elevation Oncology and Merus, outside the submitted work. RAS reports personal fees from Ebix/Oakstone, royalties from Cambridge University Press, royalties from Springer Publishers, personal fees from Roche, outside the submitted work. BW reports ad hoc membership of the scientific advisory board of REPARE Therapeutics, outside the submitted work. CV reports consulting fees from Paige AI and Docdoc Ltd. (Singapore), equity interest in Paige AI, outside the submitted work.
DATA AVAILABILITY
Targeted sequencing data that support the findings of this study will be available at cBioPortal (www.cbioportal.org) upon publication of the manuscript.
REFERENCES
- 1.Cancer Genome Atlas Research Network, Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013;497:67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kommoss S, McConechy MK, Kommoss F, Leung S, Bunz A, Magrill J, et al. Final validation of the ProMisE molecular classifier for endometrial carcinoma in a large population-based case series. Ann Oncol 2018;29:1180–8. [DOI] [PubMed] [Google Scholar]
- 3.DeLair DF, Burke KA, Selenica P, Lim RS, Scott SN, Middha S, et al. The genetic landscape of endometrial clear cell carcinomas. J Pathol 2017;243:230–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cherniack AD, Shen H, Walter V, Stewart C, Murray BA, Bowlby R, et al. Integrated Molecular Characterization of Uterine Carcinosarcoma. Cancer Cell 2017;31:411–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim SR, Cloutier BT, Leung S, Cochrane D, Britton H, Pina A, et al. Molecular subtypes of clear cell carcinoma of the endometrium: Opportunities for prognostic and predictive stratification. Gynecol Oncol 2020;158:3–11. [DOI] [PubMed] [Google Scholar]
- 6.Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nature Medicine 2017;23:703–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. J Mol Diagn 2015;17:251–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Talhouk A, McConechy MK, Leung S, Li-Chang HH, Kwon JS, Melnyk N, et al. A clinically applicable molecular-based classification for endometrial cancers. Br J Cancer 2015;113:299–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leon-Castillo A, Britton H, McConechy MK, McAlpine JN, Nout R, Kommoss S, et al. Interpretation of somatic POLE mutations in endometrial carcinoma. J Pathol 2020;250:323–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kehl KL, Xu W, Lepisto E, Elmarakeby H, Hassett MJ, Van Allen EM, et al. Natural Language Processing to Ascertain Cancer Outcomes From Medical Oncologist Notes. JCO Clin Cancer Inform 2020;4:680–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carlson J, McCluggage WG. Reclassifying endometrial carcinomas with a combined morphological and molecular approach. Curr Opin Oncol 2019;31:411–9. [DOI] [PubMed] [Google Scholar]
- 12.Hoang LN, Kinloch MA, Leo JM, Grondin K, Lee CH, Ewanowich C, et al. Interobserver Agreement in Endometrial Carcinoma Histotype Diagnosis Varies Depending on The Cancer Genome Atlas (TCGA)-based Molecular Subgroup. Am J Surg Pathol 2017;41:245–52. [DOI] [PubMed] [Google Scholar]
- 13.Wiegand KC, Lee AF, Al‐Agha OM, Chow C, Kalloger SE, Scott DW, et al. Loss of BAF250a (ARID1A) is frequent in high‐grade endometrial carcinomas. J Pathol 2011;224:328–33. [DOI] [PubMed] [Google Scholar]
- 14.Hoang LN, McConechy MK, Kobel M, Han G, Rouzbahman M, Davidson B, et al. Histotype-genotype correlation in 36 high-grade endometrial carcinomas. Am J Surg Pathol 2013;37:1421–32. [DOI] [PubMed] [Google Scholar]
- 15.Fadare O, Zheng W, Crispens MA, Jones HW, Khabele D, Gwin K, et al. Morphologic and other clinicopathologic features of endometrial clear cell carcinoma: a comprehensive analysis of 50 rigorously classified cases. Am J Cancer Res 2013;3:70–95. [PMC free article] [PubMed] [Google Scholar]
- 16.Garg K, Leitao MM Jr., Wynveen CA, Sica GL, Shia J, Shi W, et al. p53 overexpression in morphologically ambiguous endometrial carcinomas correlates with adverse clinical outcomes. Mod Pathol 2010;23:80–92. [DOI] [PubMed] [Google Scholar]
- 17.Soslow RA, Tornos C, Park KJ, Malpica A, Matias-Guiu X, Oliva E, et al. Endometrial Carcinoma Diagnosis: Use of FIGO Grading and Genomic Subcategories in Clinical Practice: Recommendations of the International Society of Gynecological Pathologists. Int J Gynecol Pathol 2019;38 Suppl 1:S64–S74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, Getz G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol 2011;12:R41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang NR, Siegmund DO. A modified Bayes information criterion with applications to the analysis of comparative genomic hybridization data. Biometrics 2007;63:22–32. [DOI] [PubMed] [Google Scholar]
- 20.Hyman DM, Piha-Paul SA, Won H, Rodon J, Saura C, Shapiro GI, et al. HER kinase inhibition in patients with HER2- and HER3-mutant cancers. Nature 2018;554:189–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee S, Piskorz AM, Le Page C, Mes Masson AM, Provencher D, Huntsman D, et al. Calibration and Optimization of p53, WT1, and Napsin A Immunohistochemistry Ancillary Tests for Histotyping of Ovarian Carcinoma: Canadian Immunohistochemistry Quality Control (CIQC) Experience. Int J Gynecol Pathol 2016;35:209–21. [DOI] [PubMed] [Google Scholar]
- 22.Pere H, Tapper J, Wahlstrom T, Knuutila S, Butzow R. Distinct chromosomal imbalances in uterine serous and endometrioid carcinomas. Cancer Res 1998;58:892–5. [PubMed] [Google Scholar]
- 23.Zheng W, Cao P, Zheng M, Kramer EE, Godwin TA. p53 overexpression and bcl-2 persistence in endometrial carcinoma: comparison of papillary serous and endometrioid subtypes. Gynecol Oncol 1996;61:167–74. [DOI] [PubMed] [Google Scholar]
- 24.Lax SF, Pizer ES, Ronnett BM, Kurman RJ. Comparison of estrogen and progesterone receptor, Ki-67, and p53 immunoreactivity in uterine endometrioid carcinoma and endometrioid carcinoma with squamous, mucinous, secretory, and ciliated cell differentiation. Hum Pathol 1998;29:924–31. [DOI] [PubMed] [Google Scholar]
- 25.Fadare O, Gwin K, Desouki MM, Crispens MA, Jones HW 3rd, Khabele D, et al. The clinicopathologic significance of p53 and BAF-250a (ARID1A) expression in clear cell carcinoma of the endometrium. Mod Pathol 2013;26:1101–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Soslow RA. Endometrial carcinomas with ambiguous features 2010. Elsevier. p 261–73. [DOI] [PubMed] [Google Scholar]
- 27.Kuhn E, Wu RC, Guan B, Wu G, Zhang J, Wang Y, et al. Identification of molecular pathway aberrations in uterine serous carcinoma by genome-wide analyses. J Natl Cancer Inst 2012;104:1503–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vicente-Duenas C, Hauer J, Cobaleda C, Borkhardt A, Sanchez-Garcia I. Epigenetic Priming in Cancer Initiation. Trends Cancer 2018;4:408–17. [DOI] [PubMed] [Google Scholar]
- 29.Zhong Q, Fan J, Chu H, Pang M, Li J, Fan Y, et al. Integrative analysis of genomic and epigenetic regulation of endometrial cancer. Aging 2020;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saffari B, Jones LA, Elnaggar A, Felix JC, George J, Press MF. Amplification and Overexpression of Her-2/Neu (C-Erbb2) in Endometrial Cancers - Correlation with Overall Survival. Cancer Res 1995;55:5693–8. [PubMed] [Google Scholar]
- 31.Fader AN, Roque DM, Siegel E, Buza N, Hui P, Abdelghany O, et al. Randomized Phase II Trial of Carboplatin-Paclitaxel Versus Carboplatin-Paclitaxel-Trastuzumab in Uterine Serous Carcinomas That Overexpress Human Epidermal Growth Factor Receptor 2/neu. J Clin Oncol 2018;36:2044–51. [DOI] [PubMed] [Google Scholar]
- 32.Connell CM, Doherty GJ. Activating HER2 mutations as emerging targets in multiple solid cancers. ESMO Open 2017;2:e000279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gilks CB, Oliva E, Soslow RA. Poor interobserver reproducibility in the diagnosis of high-grade endometrial carcinoma. Am J Surg Pathol 2013;37:874–81. [DOI] [PubMed] [Google Scholar]
- 34.Li WH, Li L, Wu M, Lang JH, Bi YL. The Prognosis of Stage IA Mixed Endometrial Carcinoma A Retrospective Cohort Study. Am J Surg Pathol 2019;152:616–24. [DOI] [PubMed] [Google Scholar]
- 35.Travaglino A, Raffone A, Stradella C, Esposito R, Moretta P, Gallo C, et al. Impact of endometrial carcinoma histotype on the prognostic value of the TCGA molecular subgroups. Arch Gynecol Obstet 2020;301:1355–63. [DOI] [PubMed] [Google Scholar]
- 36.Ayeni TA, Bakkum-Gamez JN, Mariani A, McGree ME, Weaver AL, Haddock MG, et al. Comparative outcomes assessment of uterine grade 3 endometrioid, serous, and clear cell carcinomas. Gynecol Oncol 2013;129:478–85. [DOI] [PubMed] [Google Scholar]
- 37.Brett MA, Atenafu EG, Singh N, Ghatage P, Clarke BA, Nelson GS, et al. Equivalent Survival of p53 Mutated Endometrial Endometrioid Carcinoma Grade 3 and Endometrial Serous Carcinoma. Int J Gynecol Pathol 2020. Online ahead of print. [DOI] [PubMed] [Google Scholar]
- 38.Luz R, MacDonald N, Mould T. Omental Biopsy for Surgical Staging of Uterine Serous Carcinoma. Int J Gynecol Cancer 2016;26:1448–54. [DOI] [PubMed] [Google Scholar]
- 39.Baergen RN, Warren CD, Isacson C, Ellenson LH. Early uterine serous carcinoma: clonal origin of extrauterine disease. Int J Gynecol Pathol 2001;20:214–9. [DOI] [PubMed] [Google Scholar]
- 40.Soslow RA, Pirog E, Isacson C. Endometrial intraepithelial carcinoma with associated peritoneal carcinomatosis. Am J Surg Pathol 2000;24:726–32. [DOI] [PubMed] [Google Scholar]
- 41.Euscher E, Sui DW, Soliman P, Westin S, Ramalingam P, Bassett R, et al. Ultrastaging of Sentinel Lymph Nodes in Endometrial Carcinoma According to Use of 2 Different Methods. International Journal of Gynecological Pathology 2018;37:242–51. [DOI] [PubMed] [Google Scholar]
- 42.Mueller JJ, Pedra Nobre S, Braxton K, Alektiar KM, Leitao MM Jr., Aghajanian C, et al. Incidence of pelvic lymph node metastasis using modern FIGO staging and sentinel lymph node mapping with ultrastaging in surgically staged patients with endometrioid and serous endometrial carcinoma. Gynecol Oncol 2020;157:619–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hussein YR, Broaddus R, Weigelt B, Levine DA, Soslow RA. The Genomic Heterogeneity of FIGO Grade 3 Endometrioid Carcinoma Impacts Diagnostic Accuracy and Reproducibility. Int J Gynecol Pathol 2016;35:16–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Travaglino A, Raffone A, Mollo A, Borrelli G, Alfano P, Zannoni GF, et al. TCGA molecular subgroups and FIGO grade in endometrial endometrioid carcinoma. Arch Gynecol Obstet 2020;301:1117–25. [DOI] [PubMed] [Google Scholar]
- 45.Yano M, Ito K, Yabuno A, Ogane N, Katoh T, Miyazawa M, et al. Impact of TP53 immunohistochemistry on the histological grading system for endometrial endometrioid carcinoma. Mod Pathol 2019;32:1023. [DOI] [PubMed] [Google Scholar]
- 46.Koh WJ, Abu-Rustum NR, Bean S, Bradley K, Campos SM, Cho KR, et al. Uterine Neoplasms, Version 1.2018, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw 2018;16:170–99. [DOI] [PubMed] [Google Scholar]
- 47.Vermij L, Smit V, Nout R, Bosse T. Incorporation of molecular characteristics into endometrial cancer management. Histopathol 2020;76:52–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kobel M, Ronnett BM, Singh N, Soslow RA, Gilks CB, McCluggage WG. Interpretation of P53 Immunohistochemistry in Endometrial Carcinomas: Toward Increased Reproducibility. Int J Gynecol Pathol 2019;38 Suppl 1:S123–S31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Singh N, Piskorz AM, Bosse T, Jimenez-Linan M, Rous B, Brenton JD, et al. p53 immunohistochemistry is an accurate surrogate for TP53 mutational analysis in endometrial carcinoma biopsies. J Pathol 2020;250:336–45. [DOI] [PubMed] [Google Scholar]
- 50.Talhouk A, Hoang LN, McConechy MK, Nakonechny Q, Leo J, Cheng A, et al. Molecular classification of endometrial carcinoma on diagnostic specimens is highly concordant with final hysterectomy: Earlier prognostic information to guide treatment. Gynecol Oncol 2016;143:46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.