

Abstract
Normal human cells exhibit a limited replicative life span in culture, eventually arresting growth by a process termed senescence. Progressive telomere shortening appears to trigger senescence in normal human fibroblasts and retinal pigment epithelial cells, as ectopic expression of the telomerase catalytic subunit, hTERT, immortalizes these cell types directly. Telomerase expression alone is insufficient to enable certain other cell types to evade senescence, however. Such cells, including keratinocytes and mammary epithelial cells, appear to require loss of the pRB/p16INK4a cell cycle control mechanism in addition to hTERT expression to achieve immortality. To investigate the relationships among telomerase activity, cell cycle control, senescence, and differentiation, we expressed hTERT in two epithelial cell types, keratinocytes and mesothelial cells, and determined the effect on proliferation potential and on the function of cell-type-specific growth control and differentiation systems. Ectopic hTERT expression immortalized normal mesothelial cells and a premalignant, p16INK4a-negative keratinocyte line. In contrast, when four keratinocyte strains cultured from normal tissue were transduced to express hTERT, they were incompletely rescued from senescence. After reaching the population doubling limit of their parent cell strains, hTERT+ keratinocytes entered a slow growth phase of indefinite length, from which rare, rapidly dividing immortal cells emerged. These immortal cell lines frequently had sustained deletions of the CDK2NA/INK4A locus or otherwise were deficient in p16INK4a expression. They nevertheless typically retained other keratinocyte growth controls and differentiated normally in culture and in xenografts. Thus, keratinocyte replicative potential is limited by a p16INK4a-dependent mechanism, the activation of which can occur independent of telomere length. Abrogation of this mechanism together with telomerase expression immortalizes keratinocytes without affecting other major growth control or differentiation systems.
Normal human somatic cells have a limited capacity to replicate in culture, even under conditions that appear to satisfy their nutritional and mitogen requirements (53, 56). These cells proliferate initially but eventually enter a state of permanent growth arrest termed senescence, clearly distinct from differentiation, in which they can remain metabolically active indefinitely. Progressive shortening of the telomeres, DNA-protein structures located at the ends of linear eukaryotic chromosomes, occurs during the 50- to 100-population-doubling (PD) life span of human fibroblasts in culture (19). The erosion of telomeric DNA with successive cell replications has led to the proposal that telomeres not only function to protect the chromosomes from end-to-end fusions but, when disrupted by shortening, also signal the onset of senescence (2).
Unlike most normal human somatic cell types, most advanced-stage cancer cells are replicatively immortal and express the enzyme telomerase. Telomerase is a multimeric ribonucleoprotein containing an RNA component that includes in its sequence the template for telomere synthesis (14) and a catalytic protein subunit that is a reverse transcriptase (34, 38). The expression of telomerase in immortal cancer cells apparently is responsible for their maintenance of a stable telomere length through an indefinite number of cell divisions (11). Although the telomerase RNA component is expressed constitutively (19), the catalytic subunit, hTERT, is expressed only in germ cells and in immortal cancer cells (34, 38), suggesting that hTERT is the activity-limiting component of the telomerase holoenzyme. Introduction of hTERT into presenescent human fibroblasts and retinal pigment epithelial cells was found to confer telomere maintenance and unlimited replicative potential to these cell types (5), giving strong support to the model that telomere shortening determines the onset of senescence. This simple interpretation, however, may not apply to all cell types, as it was reported recently that ectopic expression of hTERT is not sufficient to immortalize normal human keratinocytes and mammary epithelial cells (25).
We have sought to investigate the role of telomerase in cellular senescence, to identify potential ancillary genetic alterations necessary for immortalization of epithelial cells, and to determine the effects of immortalization on cell-type-specific growth control and differentiation mechanisms. We have expressed hTERT in two different types of epithelial cells, mesothelial cells and keratinocytes, both of which exhibit a finite life span in vitro and have well-characterized growth control systems and differentiation programs (10, 16, 47, 52). Our experiments indicate that these two epithelial cell types behave very differently in response to ectopically expressed hTERT and that such expression is not sufficient to immortalize keratinocytes. We have identified a complex pattern of p16INK4a expression in keratinocytes associated with senescence which functions independent of telomere shortening. Keratinocytes that express hTERT and also acquire a defect in triggering p16INK4a expression become immortalized but otherwise display normal growth characteristics and differentiation potential, indicating that the process of senescence in this cell type is complex but separate from mechanisms that regulate growth and differentiation.
MATERIALS AND METHODS
Cell lines and culture media.
Most of the cells studied (Table 1) were cell strains (sometimes termed primary cells) cultured from clinically and presumably genetically normal tissues (28, 45, 61). POE9n was cultured from a premalignant dysplastic oral epithelial lesion and was found to have a homozygous deletion at the CDKN2A/INK4A locus, to lack expression of p53, and to have an extended but finite life span (J. Rheinwald, J. Benwood, A. Palanisamy, Y. Ino, D. Louis, R. Feldman, and E. Sauter, unpublished data). LiF-Ep was cultured from phenotypically normal epidermis from an individual (National Cancer Institute repository identifier no. 1010 from kindred no. 1 [27]) with pancreatic cancer and Li-Fraumeni syndrome (31). The inherited mutation in this individual is a heterozygous, one-base-pair deletion at the exon 9-intron 9 junction of the TP53 gene. Somatic cells sampled from this individual previously were found by reverse transcription-PCR analysis not to express detectable levels of p53 mRNA encoded by the mutant allele (S. Verselis, personal communication).
TABLE 1.
Human cell lines
| Cell type | Cell line designation | Donor age, sexa | Replicative life span | Known abnormalities |
|---|---|---|---|---|
| Epidermal keratinocyte | N | Newborn, M | ∼52 PD | None |
| N/E7 | Newborn | ∼85 PD (extended) | HPV16E7+, pRB deficient | |
| LiF-Ep | 60, F | ∼35 PD | p53+/− | |
| Oral mucosal keratinocyte | OKF6 | 57, M | ∼45 PD | None |
| OKF4 | 28, M | ∼32 PD | None | |
| POE-19 | 65, M | ∼22 PD | None | |
| Premalignant oral mucosal keratinocyte | POE9n | 65, M | ∼85 PD (extended) | p16−/−, p53 deficient |
| Dermal fibroblast | S1F | Newborn, M | ∼90 PD | None |
| Peritoneal mesothelial cell | LP9 | 26, F | ∼60 PD | None |
| Epidermal SCC | SCC-13 | 56, F | Immortal | p16 deficient, p53 deficient |
M, male; F, female
Keratinocyte cultures were initiated and expanded through third passage by cocultivating with mitomycin-treated Swiss 3T3J2 cells in FAD medium (consisting of Dulbecco modified Eagle medium–F-12 medium [1:1, vol/vol] plus 5% calf serum [CS; HyClone], 10 ng of epidermal growth factor [EGF] per ml, 0.4 μg of hydrocortisone [HC] per ml, 5 μg of insulin per ml, 10 × 10−10 M cholera toxin, 2 × 10−11 M triiodothyronine, and 1.8 × 10−4 M adenine [44, 46]). Keratinocytes and derived transductants were subsequently cultured without feeder cells in keratinocyte serum-free medium (K-sfm) (GIBCO/BRL) plus 30 μg of bovine pituitary extract per ml, 0.1 ng of EGF per ml and additional CaCl2 to raise the [Ca2+] to 0.4 mM (52).
Dermal fibroblasts were cultured in medium 199 (M199) plus 15% CS plus 10 ng of EGF per ml. Mesothelial cells were cultured in M199 plus 15% CS plus 10 ng of EGF per ml plus 0.4 μg of HC per ml (10). 3T3J2 cells and PT67 retroviral vector producer cells (17) were cultured in pyruvate-free Dulbecco modified Eagle medium (GIBCO/BRL)–10% CS. A multiple-drug-resistant fibroblast feeder cell line, 3T3J2NHP, was generated by sequentially transducing 3T3J2 with the retroviral vectors LXSN (35), BABE/hygro, and BABE/puro (36) to confer resistance to G418, hygromycin, and puromycin, respectively. This line was used as feeder cells to support transduced keratinocyte populations during selection with any of these drugs.
Retroviral vectors and transduction.
PT67 amphotropic packaging cells producing BABE-hygro-hTERT and BABE-puro-hTERT (17) or control BABE-hygro and BABE-puro (36) retroviral vectors were used to generate retroviral supernatants in K-sfm, which was passed through a 0.45-μm-pore-size filter and stored at −80°C until use. Human cells plated 1 day earlier at ∼105 cells per 9-cm2 well were transduced by refeeding them for 5 to 7 h with retroviral supernatant plus Polybrene (4 μg/ml; Sigma). The treated cells were subcultured the next day into 75-cm2 flasks. Drug selection (5 to 10 μg of hygromycin per ml or 1 μg of puromycin per ml) was started 2 days after transduction and continued for 7 to 14 days. Some hTERT transductants were generated by coplating keratinocytes with mitomycin-treated retroviral producer cells in FAD medium. Four days later, the producer cells were selectively removed by brief incubation with EDTA and vigorous pipetting, mitomycin-treated 3T3J2NHP cells were added back to the cultures, and drug selection was started the next day.
Replicative life span determination.
Cells were plated at 1 × 105 to 3 × 105 cells per T75 or T175 flask or p100 dish in their appropriate growth medium, refed every 2 to 3 days, and subcultured 5 to 8 days later, before high cell density slowed their growth. PD per passage was calculated as log2 (number of cells at time of subculture/number of cells plated). No correction was made for cells that failed to reinitiate growth at subculture. Cumulative PD was plotted against total time in culture to assess replicative life span, senescence, slow growth, or crisis, and immortalization, the latter judged to have occurred if cells grew for at least 50 PD beyond the life span of the parent cell line.
Telomerase assays and telomere length determination.
Telomerase activity was detected using the PCR-based, telomerase repeat amplification protocol (TRAP) assay (24). Telomere length was measured by hybridizing a 32P-labeled telomeric (CCCTAA)3 probe to HinfI- and RsaI-digested genomic DNA separated on agarose gels (12).
CDKN2A/INK4A genomic analysis.
For genomic analysis of keratinocyte cell lines (Table 2), DNA was extracted from cell pellets Puregene DNA isolation kit (Gentra Systems). Allelic loss on the chromosome 9p21 region was assessed by comparing parent cell line-experimental cell line sets using microsatellite markers D9S126, D9S741, and D9S1748 (3) as described previously (30) (primer sequences are available from the genome database [gdbwww.gdb.org]). Exons 1 through 3 of the CDKN2A/INK4A gene were screened for mutations by single-strand conformation polymorphism (SSCP) analysis, followed by direct DNA sequencing of samples with shifted bands, as described previously (58). CDKN2A/INK4A homozygous deletions were detected by comparative multiplex PCR as described previously (39). Hypermethylation of CpG islands of the CDKN2A/INK4A promoter was assessed by methylation-specific PCR (20), with minor modifications as described previously (8).
TABLE 2.
p16INK4a genomic analysis of keratinocyte cell lines expressing hTERT
| Cell line | Deletion analysisa
|
SSCP mutation analysisb
|
Sequencing resultc
|
Promoter methylationd | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| D9S126 | D9S741 | D9S1748 | Multiplex PCR | Exon 1 | Exon 2(A) | Exon 2(B) | Exon 3 | Exon | Codon | Mutation (or polymorphism) | Amino acid change | ||
| LP9 (23 PD, mid-life span) | 12 | 12 | 12 | N | N | N | N | N | U | ||||
| LP9/TERT-1 (90 PD, RDI) | 12 | 12 | 12 | N | N | N | N | N | U | ||||
| N (42 PD, late life span) | 12 | NI | 12 | N | N | N | N | N | U | ||||
| N/TERT-1 (100 PD, RDI) | 12 | NI | 12 | N | N | N | N | N | U | ||||
| LiF-Ep (25 PD, mid-life span) | NI | 12 | 12 | N | N | N | S+N | Se | 2 | 148 | (GCG→ACG) | Ala→Thr | U |
| 3 | nt 499 C→G | 3′ UTR | |||||||||||
| LiF-Ep/TERT-1 (49 PD, RDI) | NI | LOH | LOH | N | N | N | Se | 2 | 148 | (GCG→ACG) | Ala→Thr | U | |
| 3 | nt 499 C→G | 3′ UTR | |||||||||||
| OKF6 (42 PD, late life span) | 12 | 12 | 12 | N | N | N | N | N | U | ||||
| OKF6/TERT-1 (42 PD, early SGP) | 12 | 12 | 12 | ND | ND | ND | ND | ND | ND | ||||
| OKF6/TERT-1 (63 PD, RDI) | LOH | LOH | LOH | N | N | N | N | N | U | ||||
| OKF6/TERT-1R (65 PD, RDI) | 12 | 12 | 12 | ND | ND | ND | ND | ND | ND | ||||
| OKF6/TERT-2 (52 PD, RDI) | 12 | 12 | (HD) | HD | (HD) | (HD) | (HD) | (HD) | (HD) | ||||
Changes from the parent cell line are noted in boldface. PCR primer sets were used to amplify the polymorphic microsatellite sequences D9S126, D9S741, and D9S1748 to detect loss of heterozygosity (LOH) in the 9p21 region. 12, two alleles detected; NI, not informative; LOH, allelic loss compared with parent cell line or normal cells from same donor. Multiplex PCR was performed with exon 2 primers and primers that amplified sequences from a control locus as described in Materials and Methods. N, exon 2 sequences detected; HD, homozygous deletion; ND, not determined; (HD), no PCR products observed because of HD.
Note that two overlapping regions of exon 2 (designated A and B) were amplified separately. N, PCR product migrating as expected for wild-type allele; S, shift in migration compared with that of the wild-type allele. The codon 148 missense G-to-A alteration in LiF-Ep, identified as shifted in the SSCP analysis, is a polymorphism that does not result in altered biological activity of p16INK4a (reference 42; note that the old codon numbering system in this paper designated codon 148 as 140).
nt, nucleotide; UTR, untranslated region.
Determined by methylation-sensitive PCR to detect methylation of the CpG island in the p16INK4a promoter most commonly found to be hypermethylated in human cancer cells, as described in Materials and Methods. U, unmethylated; M, only methylated sequences detected.
C→G substitution in nucleotide 494 of the 3′ untranslated region was identified in the allele of LiF-Ep that was retained by LiF-Ep/TERT-1.
Growth control assays.
Cells were plated at 103 to 3 × 103 cells per 9-cm2 well in various medium formulations, refed every 3 to 4 days, and counted after 9 to 11 days of growth, when cells cultured in the control, permissive medium were still actively proliferating.
Keratinocytes.
To assess mitogen dependence, cells were plated in K-sfm supplemented with the reduced bovine pituitary extract concentration of 10 μg/ml with or without EGF (0.1 ng/ml). To assess sensitivity to growth inhibitors, cells were cultured in K-sfm to which 10−9 M tetradecanoyl phorbol acetate (TPA; Sigma) or 0.3 ng of transforming growth factor β1 (R&D Systems) was added. Cells were cultured in control and experimental conditions for 7 to 9 days, and their growth rates were calculated as [log2 (number of cells at harvest/number of cells plated)]/number of days = PD/day.
Mesothelial cells.
To assess mitogen dependence, cells were plated in M199 plus 15% CS with or without EGF and HC and in M199 medium plus EGF plus HC plus 1% CS (10, 57).
Immunochemical analysis of cell cycle regulatory protein expression.
Cultured cells were trypsinized, rinsed in phosphate-buffered saline (PBS), pelleted, and lysed in 20 mM Tris buffer (pH 7.3)–2% sodium dodecyl sulfate–1 mM dithiothreitol. Twenty to 100 μg of protein was separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis using a polyacrylamide concentration of 14% for p16INK4a, 10 or 12% for cyclin D1, cdk4, and cdk6, or 7% for pRB. Gels were electrotransferred to nitrocellulose paper, and proteins were detected with antibodies specific for p16INK4a (JC1; a gift from E. Harlow, Massachusetts General Hospital, Boston), pRB (G3-245; PharMingen), p53 (DO-1), cdk4 (C-22), cdk6 (C21), and cyclin D1 (HD11) (the latter four antibodies all from Santa Cruz Biotechnologies), followed by peroxidase-labeled secondary antibody (Southern Biotechnologies) and enhanced chemiluminescence reagent (Amersham Corp.).
Cells growing on culture dishes, some which received 0.5 nM actinomycin D for the final 2 days to elicit a DNA damage-stimulated increase in p53 levels (23), were fixed in 4% paraformaldehyde in PBS for 30 min, permeabilized with 0.1% Triton X-100 in PBS for 5 min, and incubated for 30 min with antibodies specific for p16INK4a (JC8 or JC2 [8]; a gift from E. Harlow) or p53 (BP53.12; Zymed). Antibody binding was detected using ABC peroxidase (Vector Laboratories) with NovaRed colorimetric substrate.
Cell differentiation and histogenesis assays.
Keratinocyte differentiation-related proteins were detected immunocytochemically by ABC peroxidase staining (Vector Laboratories) as described elsewhere (52). Cultures were fixed in cold methanol and immunostained for involucrin (antibody SY5 [21]; from F. Watt, ICRF Laboratories, London, England), for keratin K10 (antibody AE20 [32]; from C. A. Loomis, New York University School of Medicine, New York, N.Y.), and for keratin K13 (antibody AE8 [13]; from T.-T. Sun, New York University School of Medicine).
The ability of hTERT-immortalized keratinocytes to form a differentiated, stratified squamous epithelium was assessed in organotypic culture (40, 52). Keratinocytes were seeded at 2 × 105 cells per ∼1-cm2 surface area onto collagen gels containing human foreskin fibroblasts (strain B256), cultured submerged for 4 days, and then at the air-liquid interface for 10 days. Cell lines of oral epithelial origin (i.e., OKF6/TERT-1, OKF6/TERT-2, and POE9n/TERT-1) received 10−8 M retinoic acid, which provides for more accurate recapitulation of in vivo histology by oral mucosal keratinocytes (52). Cultures were fixed in formalin and embedded in paraffin or were frozen in OCT compound. Sections were stained with hematoxylin and eosin (H&E).
For grafting, athymic NIH Swiss (nu/nu) mice were anesthetized with ketamine-xylazine, and a 1-cm2 area of full-thickness skin was excised from the middle of the back. An organotypic culture was transferred to the site and held in place with Vaseline-impregnated gauze covered by a Band-Aid. Grafts were kept covered for 1 week, after which they were exposed to the air. Mice were sacrificed 24 or 48 days postgrafting; the grafts were excised, fixed in formalin, paraffin embedded, sectioned, and stained with H&E or with a human involucrin-specific antiserum (BTI-601 [48]; Biomedical Technologies, Inc.) to distinguish human from mouse epithelium.
RESULTS
Effect of hTERT expression on human mesothelial cells, a simple squamous epithelial cell type.
To examine the effects of hTERT expression on the growth of epithelial cells, we used amphotropic retroviral vectors that transduced hTERT and a drug resistance selection marker, or a control vector expressing only the marker gene, into mid-life-span cultures of the normal mesothelial cell strain LP9. We also transduced these genes into the normal dermal fibroblast strain S1F to serve as a control for the function of our hTERT vector, since hTERT has previously been shown to immortalize normal human fibroblasts (5, 25). The resulting hTERT-expressing transductants exhibited readily detectable telomerase activity, whereas the control cells did not (data not shown). Later-passage LP9 and S1F control cells had short (∼3-kb) telomeres (Fig. 1b), whereas the respective hTERT transductants acquired and maintained average telomere lengths of ∼10 kb.
FIG. 1.

Telomerase activity and telomere maintenance in hTERT transductants. (a) TRAP assays of telomerase activity in control (vector) and hTERT-transduced cell lines tested at indicated PD levels. The group of three lanes designated by each number show assay results for 2 μg of extract preincubated with RNase as a control and for 1 and 2 μg of extract assayed. Groups: 1, N/BABE-2F, 50 PD; 2, N/TERT-2F, 48 PD (pre-SGP); 3, N/TERT-1, 57 PD (early SGP); 4, LiF-Ep/BABE-1, 35 PD; 5, LiF-Ep/BABE-2, 31 PD; 6, LiF-Ep/TERT-2, 39 PD (late SGP); 7, LiF-Ep/TERT-1, 45 PD (RDI); 8, OKF6, 18 PD; 9, OKF6/BABE-1, 39 PD; 10, OKF6/TERT-1, 42 PD (beginning of SGP); 11, OKF6/TERT-1, 60 PD (early RDI); 12, OKF6/TERT-1, 107 PD; 13, POE9n/BABE-1, 32 PD; 14, POE9n/BABE-1, 83 PD; 15, POE9n/TERT-1, 38 PD; 16, POE9n/TERT-1, 127 PD. HT, reaction mixture heat treated before thermocycling; IC, internal (positive) control amplification product showing activity of PCR reagents.
Control LP9 mesothelial cells (Fig. 2a) and S1F fibroblasts (data not shown) ultimately ceased dividing after 55 and 91 PD, respectively, which corresponded approximately to the replicative life spans previously determined for the parent cell strains (T. O'Connell-Willstaedt, J. Benwood, and J. Rheinwald, unpublished data). In contrast, the hTERT-transduced LP9 cells, designated LP9/TERT-1, and the hTERT-transduced S1F cells, designated S1F/TERT-1, continued to divide rapidly and indefinitely for at least 50 PD beyond their normal limit (Fig. 2a; Table 3). We concluded that these cells had become immortal, similar to the results reported for human fibroblasts transfected with hTERT (5).
FIG. 2.

Long-term replication characteristics of control and hTERT-expressing cell lines. Early- to mid-life-span cultures of LP9 (a), N (b and d), LiF-Ep (c), OKF6 (e), and POE9n (f) were transduced with control or hTERT expression vectors (time of transduction indicated by large arrow), were drug selected, and then were serially passaged. Graphs show passage history of the parent cell line before transduction (○), of control vector transductants (▵), and of hTERT transductants (■). The label in each panel is the name of the immortal cell line that resulted from that hTERT transduction. In panel c, +, +,−, and − indicate the appearance of p53-negative cells in the LiF-Ep/TERT-1 population during serial passage, as described in the text (+, 100 of 100 colonies examined were p53-positive; +,−, 51 of 100 colonies examined were p53 positive and 49 of 100 were p53 negative; −, 99 of 100 colonies examined were p53 negative). In panel d, after transduction with control or hTERT vector, half of each drug-selected population was serially passaged in K-sfm (▴, N/BABE-2G; ■, N/TERT-2G) and half was serially passaged in the feeder cell-FAD system (▵, N/BABE-2F; □, N/TERT-2F). In panel e, some OKF6/TERT-1 cells cryopreserved at the onset of SGP (indicated by *) were thawed and the serial passage was repeated (□), yielding another RDI line designated OKF6/TERT-1R. Panel f includes the replication history of POE-19, a line of apparently normal oral keratinocytes cultured from the same donor as POE9n. The short life span (∼20 PD) of POE-19 indicates that the long (∼85-PD) life span of POE9n and POE9n/BABE is abnormally extended, as discussed in the text.
TABLE 3.
Proliferation characteristics of keratinocytes and other cell types stably transduced to express hTERT
| Transductant | Proliferation rate (PD/day)
|
Length of SGP (days) | ||
|---|---|---|---|---|
| Parent cell line | During SGP | RDI cells | ||
| S1F/TERT-1 | 0.65 | 0.58 | 0 | |
| LP9/TERT-1 | 0.71 | 0.65 | 0 | |
| POE9n/TERT-1 | 0.80 | 0.96 | 0 | |
| N/E7/TERT-1 | 0.76 | 1.05 | 0 | |
| N/TERT-1 | 0.77 | 0.16 | 0.92 | 52 |
| N/TERT-2G | 0.77 | 0.23 | 0.76 | 23 |
| N/TERT-2F | 0.77 | 0.29 | ND | 89 |
| LiF-Ep/TERT-1 | 0.68 | 0.03 | 0.61 | 27 |
| LiF-Ep/TERT-2 | 0.68 | 0.08 | 0.66 | 33 |
| OKF6/TERT-1 | 0.64 | 0.04 | 0.69 | 87 |
| OKF6/TERT-1R | 0.64 | 0.05 | 0.45 | 84 |
| OKF6/TERT-2 | 0.64 | 0.10 | 0.86 | 28 |
| OKF6/TERT-3 | 0.64 | 0.13 | 77 | |
| OKF4/TERT-1 | 0.73 | 0.07 | 0.66 | 34 |
After LP9/TERT-1 cells had divided 50 PD beyond the normal replicative limit of LP9, we tested them for in vitro growth and differentiation phenotypes characteristic of this cell type (10, 57). LP9/TERT-1 cells were indistinguishable from the control cells with respect to dependence on EGF, HC, and serum for growth. When control and hTERT-expressing cells were deprived of EGF, they both became reversibly growth arrested and exhibited induction of keratin K18, characteristic of normal mesothelial cells (data not shown). These results served to validate our retroviral vectors and enabled us to include mesothelial cells among the cell types that can be immortalized by expression of hTERT. Moreover, they demonstrated that hTERT-mediated acquisition of unlimited proliferative potential was not accompanied by loss of other growth or differentiation control mechanisms.
Effects of expressing hTERT in normal human keratinocytes.
We then extended these experiments to the study of a different epithelial cell type, the keratinocyte. We used cells cultured from specimens of normal human epidermis (strain N) and from normal oral mucosal epithelium (strains OKF6 and OKF4). Mid-life-span cultures of N and OKF6 were infected with amphotropic retroviral vectors encoding hTERT or a drug resistance marker alone, as before. In each case, cells transduced with a retrovirus encoding hTERT demonstrated clearly detectable telomerase activity, while control cells were telomerase negative (Fig. 1a). Corresponding to this acquisition of telomerase activity, hTERT-expressing cells maintained their telomeres at lengths longer than those of control cells (Fig. 1b).
In each case, cells transduced with the control vector senesced between 35 and 55 PD, as evidenced by a lack of net population increase during passage (Fig. 2b to e) and complete cessation of division accompanied by enlargement of all cells in the culture. This senescence occurred after approximately the same number of cumulative PD as we had observed previously for the untransduced parent cell strains (data not shown). Senescent keratinocytes did not reattach efficiently when subcultured, resulting in progressive loss of cells at subsequent passages.
A different outcome was observed for the hTERT-expressing keratinocytes. After the same cumulative PD at which control cells senesced, further expansion of the hTERT-transduced population was severely curtailed but not completely arrested. Instead, the cells entered a slow-growth phase (SGP) characterized by low colony-forming efficiencies (<1 to 5% of plated cells) and lower than normal colony growth rates, which resulted in minimal to modest net population increase during serial passage. The length of the SGP ranged from about 3 weeks to 3 months for the nine hTERT-transduced keratinocyte cultures that we examined, during which time the PD rate remained quite constant for each hTERT+ culture, ranging among cultures from 0.03 to 0.29 PD/day (Fig. 2; Table 3). Similar to normal keratinocyte cultures (4, 60), dividing cells in hTERT+ SGP cultures were relatively small, whereas nondividing cells became enlarged. hTERT-transduced populations examined before they had entered or during their SGP were found to have telomerase enzyme activity (Fig. 1, lanes 2, 3, 6, and 10) and to have an average telomere length substantially greater than that of control cells approaching senescence (Fig. 1b, lane 6; Fig. 1c, lanes 2, 7, and 9). We therefore concluded that the low PD rate exhibited by hTERT-transduced keratinocytes after they had reached their normal replicative life span limit was not the result of failure to reverse telomere erosion.
RDI cells arise from hTERT+ keratinocyte populations.
In eight of the nine hTERT-transduced keratinocyte populations that we examined, rapidly dividing cells eventually appeared in the SGP cultures and took over the population within several passages of their first visual detection. Their first appearance, typically seen as a single colony or small number of rapidly growing colonies within an otherwise slowly dividing population, suggested clonal origin as the progeny of a single cell that had undergone a permanent heritable alteration. The rapidly dividing variant cells were uniformly small and had a high colony-forming efficiency and population growth rate, typically similar to that of early-passage cells of the parent line. These rapidly dividing, immortalized (RDI) cells expressed telomerase activity (Fig. 1a) and maintained telomeres at lengths greater than or equal to that of control cells (Fig. 1c). They continued to divide for at least 50 PD beyond the normal life span of the parent line. We therefore concluded that they had undergone immortalization.
The emergence of RDI cells from hTERT+ SGP populations appeared to be stochastic. The duration of SGP varied greatly (Table 3) among three independent hTERT-transduced OKF6 cultures, with one SGP population ceasing growth before emergence of an RDI variant. Cryopreserved SGP cells of the same transduction from which the OKF6/TERT-1 RDI cell line had emerged were thawed and reanalyzed by serially passage again under the same conditions. We again observed eventual emergence of an RDI line after an SGP of long duration (Fig. 2e; Table 3). This second RDI line (OKF6/TERT-1R) had a lower proliferation rate than OKF6/TERT-1 (Table 3) and, as described below, carried different genomic alterations. We therefore concluded that the two RDI lines represented progeny of independent genetic events that occurred in this hTERT+ population during the SGP.
We entertained the possibility that the two-stage immortalization process that we observed, and that was reported by others recently (25), might have been the result of the specific conditions that we were using to culture keratinocytes. Specifically, we wished to determine whether the absence of fibroblast feeder cells, previously found to be beneficial for long-term, albeit limited, serial propagation of normal human keratinocytes (2, 46, 47), might have caused the cells to have a limited expansion potential in spite of hTERT expression and telomere stabilization. In the experiment shown in Fig. 2d, N cells were transduced with hTERT or control vectors and the drug-selected populations then were divided, with half of the cells propagated subsequently in the feeder cell-FAD system and half placed in K-sfm medium without feeder cells. The PD levels at which control cells senesced and hTERT transductants entered SGP were very nearly the same in both culture systems, and in this experiment, RDI cells arose within the population passaged in K-sfm (designated N/TERT-2G) before they arose in the population passaged in the feeder cell-FAD system (designated N/TERT-2F) (Fig. 2d; Table 3). In another experiment, we transduced the normal oral keratinocyte strain OKF4 with the BABE-puro-hTERT vector and, after drug selection, serially passaged the hTERT transductants in the feeder cell-FAD system. The cells went through an SGP, and RDI cells (OKF4/TERT-1) emerged (Table 3), which we found were able to divide rapidly and indefinitely also in K-sfm medium (data not shown). We concluded that in both culture systems a second event, in addition to hTERT expression, is required for keratinocyte immortalization and that the same type(s) of second event can serve to complement hTERT in conferring an RDI phenotype to keratinocytes growing in either culture system.
Loss of p16INK4a expression and deletions involving the CDKN2A/INK4A locus in hTERT-immortalized keratinocytes.
We expressed hTERT in keratinocytes cultured from a premalignant oral lesion, strain POE9n, which was found to have a homozygous deletion of the CDKN2A/INK4A locus and to exhibit an abnormally extended but limited, replicative life span (J. Rheinwald, J. Benwood, A. Palanisamy, Y. Ino, D. Louis, R. Feldman, and E. Sauter, unpublished data). As shown in Fig. 2f, hTERT-transduced POE9n cells divided continuously for at least 50 doublings beyond the limit of control vector-expressing cells without exhibiting any period of slowed growth. We obtained similar results (i.e., direct immortalization without an SGP) when we expressed hTERT in strain N keratinocytes that had been previously transduced to express the human papillomavirus type 16 (HPV 16) E7 protein (N/E7 cells) (Tables 1 and 3), confirming a prior report (25). Since both POE9n cells and N/E7 cells lack a functional pRB cell cycle regulatory pathway (loss of p16INK4a and expression of the HPV16 E7 protein, which sequester and inactivate pRB, respectively), we hypothesized that defects in the pRB pathway complement the expression of hTERT in keratinocytes to bypass senescence and yield RDI cells.
To test this hypothesis, we examined expression of pRB pathway proteins in the RDI lines that ultimately had arisen following hTERT expression in the normal keratinocyte strains. Exponentially growing cultures of hTERT+ RDI lines of strains N, LiF-Ep, and OKF6 were compared with mid-life-span cultures of their parent cell lines for expression of p16INK4a and other cell cycle regulatory proteins that act in the pRB-mediated mechanism controlling the G1 restriction point. As shown in Fig. 3a, levels of cyclin D1, cdk4, cdk6, and pRB expressed by hTERT-immortalized keratinocytes were similar to those of the parent cell lines. Importantly, no evidence of cyclin D1 or cdk4/6 overexpression or loss of pRB expression was detected in the hTERT-expressing cells.
FIG. 3.

Expression of cell cycle regulatory proteins by hTERT transductants. (a) Western blots comparing parent cell lines with hTERT-immortalized cell lines. “RDI+n” indicates number of population doublings after RDI cells emerged from the SGP population. Lanes: 1, N, 37 PD (mid-life span); 2, N/TERT-1, 135 PD (RDI+68); 3, LiF-Ep, 20 PD (mid-life span); 4, LiF-Ep/TERT-1, 90 PD (RDI+55); 5, OKF6, 19 PD (mid-life span); 6, OKF6/TERT-1, 63 PD (RDI+11); 7, OKF6/TERT-2, 52 PD (RDI+15). The mobility of the protein recognized by each antibody was as expected for the protein, compared with molecular mass markers separated on the same gel (data not shown). (b) Western blot analysis of p16INK4a expression by hTERT-immortalized mesothelial cells and fibroblasts. Life span designations as noted above. Lanes 1, strain N keratinocytes, 33 PD (mid-life span); 2, LP9 mesothelial cells, 41 PD (mid-life span); 3, LP9/TERT-1, 108 PD; 4, S1F/BABE-1, 70 PD (late life span); 5, S1F/TERT-1, 147 PD.
Examination of the levels of p16INK4a protein in the hTERT+ RDI lines revealed a different result. We were unable to detect p16 protein in N/TERT-1 and OKF6/TERT-2 cells and found lower levels in LiF-Ep/TERT-1 and OKF6/TERT-1 cells than in control cells (Fig. 3a). In contrast, the LP9/TERT-1 mesothelial line and the S1F/TERT-1 fibroblast line, which appeared to have undergone direct immortalization as the result of hTERT expression (Fig. 2; Table 3), exhibited no decrease in p16 expression compared with control cells (Fig. 3b). These observations suggested that acquisition of the RDI phenotype by hTERT-expressing keratinocytes might be connected mechanistically with loss of p16-imposed control of the pRB pathway.
We next sought to identify the molecular basis for the reduced p16 expression in these cell lines. DNA isolated from the hTERT-expressing cells and their parent cell lines was analyzed using PCR-based methods to detect point mutations in the coding exons for p16INK4a, hypermethylation of the promoter, and loss of heterozygosity or homozygous deletion involving the CDKN2A/INK4A locus. LiF-Ep/TERT-1 and OKF6/TERT-1 cells had acquired heterozygous deletions, and OKF6/TERT-2 cells had acquired a homozygous deletion involving all three exons of p16INK4a. SSCP analysis disclosed no base substitution or deletion mutations in the coding regions of the remaining p16 allele in LiFEp/TERT-1 and OKF6/TERT-1, consistent with the possibility that the residual protein is functionally active in these cells. While OKF6/TERT-1 cells were found to have a heterozygous deletion of the p16INK4a gene, OKF6/TERT-1R, the RDI line that emerged from a second serial passage of the same hTERT-transduced cell population that yielded OKF6/TERT-1, did not carry any detectable deletion. We were unable to identify a molecular event responsible for the complete loss of p16INK4a protein expression in the N/TERT-1 RDI line. There were no detectable deletions, nor could we detect any DNA sequence alterations of either INK4A allele. In addition, the promoter of this gene was not methylated at the CpG island commonly found to be methylated in many p16-deficient cancer cells (20, 33). These findings suggested the possibility that this line acquired a lesion in a trans-acting regulator of p16 expression.
To identify potential differences in p16 expression between normal keratinocytes, hTERT+ SGP cells, and hTERT+ RDI cells that continued to express p16 protein detectable by Western blotting, we examined cultures immunocytochemically. We found that p16 expression in all of these cultures was heterogeneous (Fig. 4). Small dividing cells in normal keratinocyte cultures at any passage, even near senescence, were p16 negative, whereas p16 was expressed at high levels in larger, nondividing cells. hTERT+ cells during the SGP had the same pattern of p16 expression as that seen in normal keratinocytes. As expected from the Western blot result, N/TERT-1 RDI cells contained no detectable p16, whereas LiF-Ep/TERT-1 RDI cells (Fig. 4) and OKF6/TERT-1 RDI cells (data not shown) displayed a heterogeneous pattern of p16 expression. We concluded that a marked elevation of p16 levels is triggered in normal keratinocytes with an increasing probability as they are serially passaged, associated with their senescence-related, irreversible loss of division potential. We also concluded that the mechanism responsible for this increase in p16 expression was not affected by telomere length stabilization.
FIG. 4.
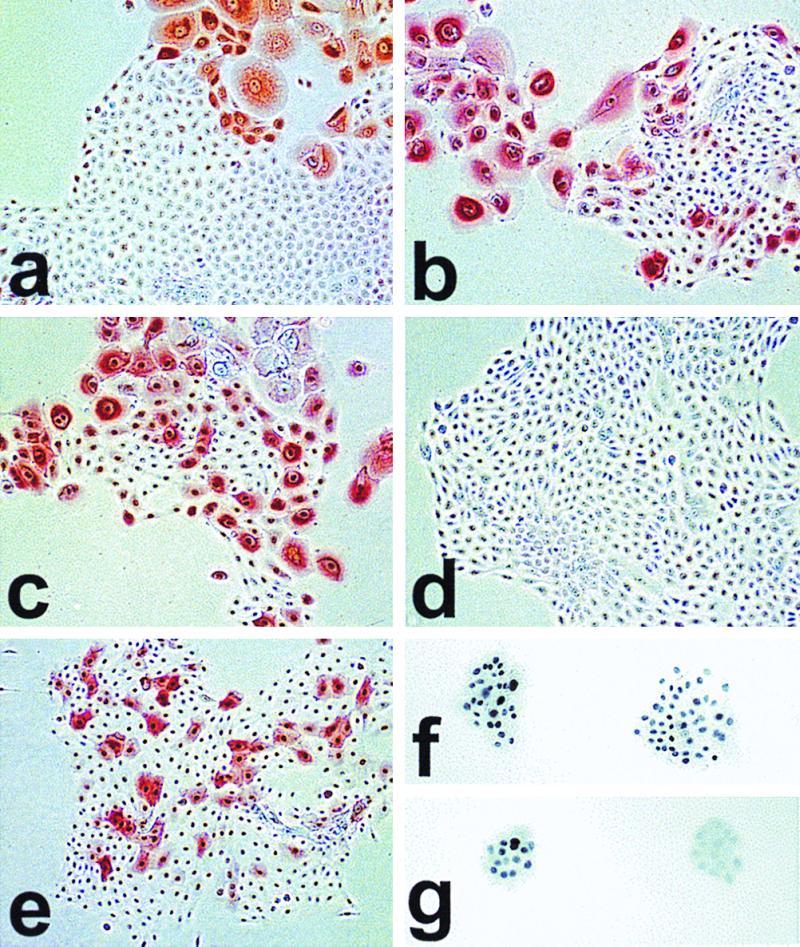
Immunocytochemical staining of cell lines for p16INK4a (a to e, phase contrast) and p53 (f and g, bright field). Cells were plated at clonal density in K-sfm and cultured for 4 to 7 days before staining. Life span designations are as for Fig. 3a. (a) N, 28 PD (mid-life span); (b) N/BABE-2G, 55 PD (near senescence); (c) N/TERT-2F, 80 PD (SGP); (d) N/TERT-1, 114 PD (RDI+43); (e) LiF-Ep/TERT-1, 67 PD (RDI+28); (f) LiF-Ep/TERT-1, 39 PD (early RDI); (g) LiF-Ep/TERT-1, 48 PD. Note the heterogeneity of p16 expression in normal N cells in panels a and b, confined to large nondividing cells, with small proliferating cells expressing no detectable p16 whether at mid-life span or in rare dividing cells in the nearly senescent population. As shown in panel c, hTERT-transduced keratinocytes continue the normal pattern of p16 expression during SGP. Some RDI lines that emerge from SGP retain the normal pattern of p16 expression, while some cease expressing p16. The p53+ colonies in panel f are representative of >99% of colonies in the early RDI LiF-Ep/TERT-1 population (as confirmed by the Western blot shown in Fig. 3b). In panel g, the p53+ colony at the left and the p53− colony at the right are representative of the mixed nature of the LiF-Ep/TERT-1 RDI population three passages (9 PD) later.
Lack of a complementary role for p53 mutations in keratinocyte immortalization by hTERT.
The TP53 gene product, p53, has been implicated as playing an important role in initiating senescence-associated growth arrest in fibroblasts and other cell types (53). It has been reported that cells cultured from individuals with Li-Fraumeni (inherited (p53+/−) syndrome may occasionally undergo spontaneous immortalization associated with loss of the wild-type p53 allele (50, 55). We therefore included in our transduction experiments LiF-Ep, a keratinocyte strain that we had cultured from a specimen of normal skin from an adult with Li-Fraumeni syndrome (see Materials and Methods). The pattern of senescence of control LiF-Ep cultures and the frequency with which RDI cells arose following transduction expression were indistinguishable from those of the normal, p53+/+ strains (Fig. 2; Table 3).
We also examined p53 expression in the RDI lines that had ultimately emerged following hTERT expression in LiF-Ep and in the other, p53+/+ keratinocyte strains. p53 protein levels expressed by the hTERT+ RDI lines during exponential growth were similar to those of the respective parent cell lines with the exception of LiF-Ep/TERT-1 (Fig. 3a). When examined at 90 PD (55 PD after emergence of RDI cells), LiF- Ep/TERT-1 cells had no p53 protein detectable by Western blotting. We therefore analyzed LiF-Ep/TERT-1 cells immunocytochemically for p53 expression and function at various times during the SGP and after emergence of the RDI line. Normal keratinocytes express low levels of p53 protein during exponential growth, but cells with functionally normal p53 expression and function greatly increase their levels of this protein in response to DNA damage, as results from exposure to low levels of actinomycin D (23). As shown in Fig. 4 and 2c, p53 protein-negative, presumably genetically p53−/− cells first appeared in the LiF-Ep/TERT-1 culture long after the event responsible for conversion of SGP cells to RDI status. The LiF-Ep/TERT-1 population consisted of at least 99% p53+ cells at the time rapidly dividing variants first emerged from the SGP (at 39 PD). Fifty percent of the cells in the RDI population were p53 negative three passages later (at 48 PD), and the culture consisted entirely of p53-negative cells by 66 PD (Fig. 4 and 2c).
A different hTERT transduction of LiF-Ep yielded an RDI line, LiF-Ep/TERT-2 (Table 3), in which apparently all of the RDI cells (i.e., 150 of 150 colonies examined immunocytochemically [data not shown]) remained phenotypically normal for p53 expression and function at least 20 PD after their emergence from SGP. We therefore concluded that p53-deficient keratinocytes do not have a selective advantage in hTERT+ populations during SGP and, therefore, that loss of p53 does not complement hTERT for keratinocyte immortalization.
Growth regulation and differentiation characteristics of hTERT-immortalized keratinocytes.
We wished to determine whether any aspect of immortalization, especially p16INK4a deficiency, which is a frequent characteristic of keratinocyte transformation in vivo (9) and of human squamous cell carcinoma (SCC) cell lines that can grow in culture (29, 37), impaired specific keratinocyte growth control and differentiation mechanisms. Four of the hTERT transductants were compared with their respective parent cell lines in several in vitro assays for function of several keratinocyte growth regulatory mechanisms that are frequently lost in SCC cells (41, 45, 51). In control conditions, the hTERT lines, tested at 45 to 68 PD after emergence as RDI cells, all had higher PD rates than mid- to late-life-span cells of their respective parent cell strains (Fig. 5). N/TERT-1, LiF-Ep/TERT-1, and OKF6/TERT-1 cells still retained dependence upon EGF for growth and sensitivity to growth inhibition by the phorbol ester TPA, exhibiting only minimal growth (less than one (<1-PD increase over the plating density during the course of the experiment) in the absence of EGF or presence of TPA. The exception was OKF6/TERT-2, which remained TPA sensitive but proliferated in the absence of EGF at 60% of the rate in the presence of EGF, approaching the degree of EGF independence of SCC-13. The four hTERT-transductants examined, including OKF6/TERT-2, were growth inhibited by transforming growth factor β (data not shown). We concluded that loss of normal keratinocyte growth regulatory mechanisms is not a necessary consequence of immortalization, even when the senescence-related pRB/p16INK4a growth arrest mechanism has been abrogated, as it is in N/TERT-1. However, our results could not eliminate the possibility that homozygous deletion of the p16INK4a locus was responsible for the EGF independence exhibited by OKF6/TERT-2 cells (Fig. 5a) and by POE9n and POE9n/TERT cells (data not shown).
FIG. 5.

Long-term retention of normal keratinocyte growth control mechanisms by many, but not all, hTERT-transductants. Cells were plated at low density in K-sfm medium, either in the presence (solid bar) or absence (stippled bar) of EGF (a) or in the presence (stippled bar) or absence (solid bar) of TPA (b), and counted 7 to 9 days later; growth under these conditions calculated as PD per day. Results shown are average of two experiments (standard error shown) except for LiF-Ep, LiF-Ep/TERT-1, and OKF6/TERT-2 in panel b, for which other experiments under slightly different conditions confirmed their TPA sensitivity (data not shown). PD levels of cells assayed: N, 50 PD (late life span); N/TERT-1, 135 P (RDI+68); LiF-Ep, 24 PD (mid-life span); LiF-Ep/TERT-1, 90 PD (RDI+55); OKF6, 37 PD (late life span); OKF6/TERT-1, 97 PD (RDI+45); OKF6/TERT-2, 115 PD (RDI+56); SCC-13 (∼50 PD).
We next examined the immortal hTERT transductants for their retention of normal keratinocyte differentiation and histogenesis functions. LiF-Ep/TERT-1 and OKF6/TERT-2 (Fig. 6a to d) and N/TERT-1 and OKF6/TERT-1 (data not shown) continued to express the terminal differentiation-related proteins involucrin and keratin K10 in suprabasal cells of stratified colonies, even >40 PD after emergence as RDI cells. The histogenic potential of the cells was examined in more detail using organotypic culture conditions, on a type I collagen gel at the air-liquid interface. Under these conditions, normal epidermal keratinocytes stratify and undergo an orthokeratinizing form of suprabasal differentiation (i.e., formation of a granular layer and outermost multilayer of flattened, stratum corneum cells), whereas oral mucosal keratinocytes undergo a nonkeratinizing form of suprabasal differentiation, similar to the behavior of these two keratinocyte subtypes in vivo (28, 40, 52).
FIG. 6.

Differentiation-related protein expression and epithelial tissue formation by hTERT-immortalized keratinocytes. Cells were cultured on conventional plastic dishes for 7 to 9 days in K-sfm (a and b) or in the feeder cell-FAD system (c and d) or were seeded on collagen gels to generate organotypic cultures (e to j), one of which was then grafted to the skin of an athymic mouse (h). Panels a (LiF-Ep, 28 PD) and b (LiF-Ep/TERT-1, 66 PD [RDI+43]) were immunostained for the suprabasal epidermal differentiation keratin K10. Panels c (OKF6, 30 PD) and d (OKF6/TERT-2, 122 PD [RDI+63]) were immunostained for the keratinocyte terminal differentiation protein involucrin. (e) N, 37 PD (mid-life span); f, N/TERT-1, 91 PD (RDI+24); g, LiF-Ep/TERT-1, 90 PD (RDI+55); h, a replicate organotypic culture of the one shown in panel g, 42 days after transplantation as a skin graft to an athymic mouse; i and j, OKF6/TERT-2, 86 PD (RDI+47). Panels e to i were stained with H&E (note that the intense blue of the cell layers below the stratum corneum in the epithelium shown in panel h is the result of a slightly different staining formulation and has no biological significance). Panel j was immunostained for keratin K13. Arrowheads point to the basal layer of the organotypic epithelium, showing that K13 expression begins several cell layers above the first suprabasal layer.
N/TERT-1 (Fig. 6f) and LiF-Ep/TERT-1 (Fig. 6g) cells formed a normally differentiating epidermis in organotypic culture. The hTERT-immortalized cells typically formed a more robust epithelium with a denser and more columnar basal cell layer and more layers of spinous cells than that formed by mid-life-span normal keratinocytes (52) (compare Fig. 6e and f). Organotypic cultures of N/TERT-1 (80 PD, 10 PD after emergence of RDI cells) and LiF-Ep/TERT-1 (90 PD, 55 PD after emergence of RDI cells) were transplanted as surface skin grafts onto athymic mice. N/TERT-1 and LiF-Ep/TERT-1 grafts recovered at 24 days (data not shown) and 48 days (Fig. 6h), respectively, were found to have formed a normally differentiating epidermis with no evidence of invasion of underlying connective tissue. OKF6/TERT-1 and -2 cells in organotypic culture underwent a nonkeratinizing form of stratified squamous epithelial differentiation, including suprabasal expression of keratin K13 (Fig. 6i and j), characteristic of floor-of-mouth mucosa (52), from which the parent line OKF6 was cultured. We concluded that no alteration necessary for immortalization, including loss of the ability to express p16INK4a, interferes with keratinocyte differentiation.
DISCUSSION
Several lines of evidence have implicated telomere erosion in limiting the proliferative potential of human cells, but the results presented here clearly indicate that a different mechanism is responsible for determining the replicative life span of human keratinocytes in culture. We have identified a precipitous increase in p16INK4a protein levels accompanying end-of-life-span growth arrest in normal keratinocytes, which appears to be triggered by a mechanism that is activated in cells with increasing probability as a cell population is serially propagated. Loss of this mechanism, whether by p16INK4a gene deletion, mutation, or altered regulation of expression, together with telomere stabilization effected by hTERT expression is necessary to enable a keratinocyte to become immortalized. Perhaps surprisingly, immortalization of keratinocytes by forced expression of telomerase and subsequent spontaneous events leading to loss of this p16INK4a-dependent mechanism generally does not disrupt other normal growth control mechanisms or affect the ability of the cells to form a differentiated epithelium. In contrast, senescence arrest is abrogated in cultured fibroblasts and retinal pigment epithelial cells by expression of hTERT (5), and our observations have added the mesothelial cell, a mesoderm-derived epithelial cell, to the group of cell types for which a telomere length-sensitive mechanism appears responsible for initiating senescence.
We have shown that expression of hTERT alone permits keratinocytes to escape complete growth arrest and to enter a phase of slow growth of variable length from which rapidly dividing immortal variants emerge. Such immortalized cells typically have identifiable defects in p16INK4a expression but retain functional p53. These results therefore confirm and extend those of Kiyono et al. (25), who demonstrated that expression of hTERT in combination with expression of the HPV16 E7 oncoprotein allowed human foreskin keratinocytes and mammary epithelial cells to bypass senescence. We have demonstrated that telomere length stabilization alone is unable to permit keratinocytes to bypass senescence, but that the subsequent slow, indefinite continued growth permitted by telomerase expression permits rare immortalized variants to arise.
Our observations clearly provide evidence supporting recent proposals (43, 62) that multiple “clocks” function to limit the proliferative capacity of human cells. The mechanism that triggers p16INK4a accumulation appears to sense the proliferative history of the keratinocyte, but preventing telomere erosion does not avoid its activation. In hTERT-expressing keratinocyte populations a small proportion of the cells indefinitely evade arrest, however. It is possible, therefore, that senescence-associated p16INK4a regulation is under two types of control, one that is tightly telomere length sensitive and another that is telomere length independent and stochastic. Whether the latter detects an aspect of cell aging related to number of cell divisions or to chronological time in culture remains to be determined because it is not possible to adjust culture conditions to slow or arrest keratinocyte growth without triggering irreversible commitment to terminal differentiation. The molecular mechanisms regulating p16INK4a gene expression are only beginning to be elucidated (18). It has been proposed that in vivo a subpopulation of keratinocytes serve as stem cells by possessing a very long or indefinite replicative potential (for example, see references 26 and 59). Whether stem cell status involves a special mechanism for avoiding senescence-related p16INK4a expression remains to be determined.
Mutations in the pRB/p16INK4a tumor suppressor pathway are found in the majority of human cancers (54). The observations presented here implicate this pathway as an essential control mechanism that must be subverted to create immortal cells. The role of p16INK4a in limiting epithelial cell proliferation is suggested by findings that variants of mammary epithelial cells that are unable to express p16 owing to promoter hypermethylation exhibit an extended replicative life span (6, 15); that p16 levels, but not p21 levels, progressively increase in keratinocyte cultures as they approach senescence (37); and that immortal SCC cell lines (29) and immortal variants of HPV16 E6-transfected prostate epithelial cell cultures (22) are found consistently to have lost p16 expression as a result of mutation or promoter hypermethylation.
It has been reported (25, 37) that p16INK4a levels, detected by Western blot analysis, increase in keratinocyte cultures as they are serially propagated. These data would support a model in which slowly increasing levels of p16INK4a in dividing keratinocytes eventually result in levels at which a subsequent G1-S transition can no longer occur. Our immunocytochemical analysis of normal and hTERT-transduced keratinocyte cultures revealed an unsuspected complexity in p16INK4a expression. We detected marked heterogeneity in p16INK4a levels within normal keratinocyte cultures at both early and late passage. p16INK4a protein was undetectable in small, rapidly dividing cells at any stage of the life span, at low to moderate levels in slightly larger cells in more slowly dividing colonies, and at very high levels in large, nondividing cells. This heterogeneous pattern of p16INK4a expression continued in hTERT+ keratinocyte populations during the SGP. These observations suggest that telomere-independent mechanisms signal senescence in keratinocytes by triggering a precipitous increase in p16INK4a levels that then causes cell cycle arrest. Moreover, they suggest that the incrementally increasing levels of p16INK4a observed in serially passaged keratinocyte cultures result from the increasing representation of cells that express high levels of p16INK4a amid a majority population of cells that remain p16INK4a negative. Since p21cip1 protein does not increase in senescent keratinocyte cultures (37), increased p16INK4a levels are likely to serve as the effector mechanism that enforces senescence-related growth arrest in keratinocytes.
In the majority of the rapidly dividing immortal variants that arose within the hTERT+ keratinocyte cultures examined here, we identified alterations compromising p16INK4a expression. Such lesions included homozygous deletion involving all p16INK4a exons, heterozygous deletion of the CDKN2A/INK4A locus with continued, heterogeneous expression of p16INK4a, and no detectable deletion, mutation, or promoter hypermethylation but complete loss of p16INK4a protein expression. The rapidly dividing immortal keratinocyte lines that continue to express p16INK4a may have mutations in other proteins involved in the pRB/p16INK4a pathway, or they may have acquired a lesion in a specific, senescence-related inducer of p16INK4a.
The deletions we detected at CDKN2A/INK4A also included loss of at least the second exon of p14ARF, another potential cell cycle inhibitor encoded in part by an alternate reading frame of the p16INK4a exon 2. p14ARF functions by preventing MDM2 from targeting p53 for degradation, thereby resulting in elevated levels of the latter and consequent cell cycle arrest (63). A recent study (37) has found that p14ARF levels do not increase during keratinocyte senescence and that all of 20 immortal human SCC lines examined had mutations compromising p16INK4a and none had mutations specifically affecting p14ARF. Our preliminary analyses also indicate that p14ARF is still expressed by hTERT-immortalized lines that still have at least one intact CDKN2A/INK4A allele (Z. Guo and J. Rheinwald, unpublished data), supporting the conclusion that it is loss of p16INK4a that is required to complement hTERT for keratinocyte immortalization.
We found no evidence that inactivation of any p53-dependent pathway is necessary for immortalization of keratinocytes that express hTERT. In neither of two hTERT transductions of the p53+/− LiF-Ep strain did we find RDI conversion associated with loss of the wild-type p53 allele. These observations are consistent with earlier studies suggesting that p53 is an essential component of the telomere length-sensitive growth arrest signal in fibroblasts (62), in that hTERT-expressing keratinocytes would be expected to permanently avoid activating such a signal.
Despite expression of hTERT and loss of p16INK4a function, the immortal keratinocytes described here are able to initiate their program of terminal differentiation, express suprabasal differentiation-specific proteins, and form differentiated epithelia in vitro and in vivo. In particular, the N/TERT-1 and LiF-Ep/TERT-1 lines continued to show a normal pattern of epidermal histogenesis in organotypic culture and in grafts to athymic mice. These results clearly indicate that p16 does not play an essential role in the irreversible growth arrest that precedes normal stratified squamous epithelial differentiation. Three of the four RDI TERT lines that we studied in detail also retained EGF dependence for growth at low density and sensitivity to irreversible growth arrest by TPA, which are normal keratinocyte growth control mechanisms consistently found to be lost in advanced SCCs (41, 45). Although OKF6/TERT-2, which had undergone homozygous deletion of the CDKN2A/INK4A locus, showed EGF-independent growth, our observations clearly demonstrate that the mechanisms involved in keratinocyte senescence and immortalization are distinct from those essential for other aspects of tissue growth control and differentiation.
Finally, the hTERT-immortalized keratinocyte lines that we have described here may have significant potential value for a wide variety of investigations into human epithelial biology, including characterization of the effects of expression or loss of specific gene products on acquisition of malignant phenotypes. Although we have demonstrated that hTERT-immortalized immortal cells can retain normal growth and differentiation control mechanisms, it is possible that the loss of the p16-mediated growth arrest mechanism and unlimited replicative potential predisposes such cells to further changes that may result in malignant transformation. Indeed, it has been shown recently that expression of hTERT cooperates with the simian virus 40 large T oncoprotein and oncogenic ras to transform human fibroblasts and kidney epithelial cells to tumorigenicity (17). For this reason, enthusiasm for potential clinical applications of hTERT-immortalized epithelial cell lines as therapeutic transplants should be tempered with caution.
ACKNOWLEDGMENTS
We thank J. Benwood, D. Long-Woodward, and K. O'Toole for technical assistance and S. Verselis for information about the mutant p53 allele in our Li-Fraumeni cell line. We thank D. Galloway for the HPV16 E7 retroviral vector, J. Koh, S. C, Ngwu, and E. Harlow for p16INK4a antibodies, and T.-T. Sun, C. A. Loomis, and F. M. Watt for keratin and involucrin antibodies.
This research was supported by Oral Cancer Program Project grant PO1 DE12467 from the NIDCR, Skin Disease Research Center grant P30 AR42689 from the NIAMS, and a research grant from Organogenesis, Inc. (J.G.R.). In addition, portions of this work were supported by a Daniel K. Ludwig and American Cancer Society Professorship (R.A.W.), a Culpeper Biomedical Initiative Pilot Grant (W.C.H. and R.A.W.), a Damon-Runyon/Walter Winchell Cancer Research Fund award, a Howard Hughes Medical Institute postdoctoral fellowship, and a Herman and Margaret Sokol postdoctoral fellowship (W.C.H.), and a Starr Foundation and American Cancer Society Clinical Research Professorship (F.P.L.).
REFERENCES
- 1.Alcorta D A, Xiong Y, Phelps D, Hannon G, Beach D, Barrett J C. Involvement of the cyclin-dependent kinase inhibitor p16 (INK4a) in replicative senescence of normal human fibroblasts. Proc Natl Acad Sci USA. 1996;93:13742–13747. doi: 10.1073/pnas.93.24.13742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Allsopp R C, Vazin H, Patterson C, Goldstein S, Younglai E V, Futcher A B, Greider C W, Harley C B. Telomere length predicts replicative capacity of human fibroblasts. Proc Natl Acad Sci USA. 1992;89:10114–10118. doi: 10.1073/pnas.89.21.10114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bahuau M, Vidaud D, Jenkins R B, Bieche I, Kimmel D W, Assouline B, Smith J S, Alderete B, Cayuela J M, Harpey J P, Caille B, Vidaud M. Germ-line deletion involving the INK4 locus in familial proneness to melanoma and nervous system tumors. Cancer Res. 1998;58:2298–2303. [PubMed] [Google Scholar]
- 4.Barrandon Y, Green H. Cell size as a determinant of the clone-forming ability of human keratinocytes. Proc Natl Acad Sci USA. 1985;82:5390–5394. doi: 10.1073/pnas.82.16.5390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bodnar A G, Ouelette M, Frolkis M, Holt A E, Chiu C-P, Morin G B, Harley C B, Shay J W, Lichsteiner S, Wright W E. Extension of lifespan by introduction of telomerase into normal human cells. Science. 1998;279:349–352. doi: 10.1126/science.279.5349.349. [DOI] [PubMed] [Google Scholar]
- 6.Brenner A J, Stampfer M R, Aldaz C M. Increased p16 expression with first senescence arrest in human mammary epithelial cells and extended growth capacity with p16 inactivation. Oncogene. 1998;17:199–205. doi: 10.1038/sj.onc.1201919. [DOI] [PubMed] [Google Scholar]
- 7.Brown J P, Wei W, Sedivy J M. Bypass of senescence after disruption of p21cip1/waf1 gene in normal diploid human fibroblasts. Science. 1997;277:831–834. doi: 10.1126/science.277.5327.831. [DOI] [PubMed] [Google Scholar]
- 8.Burns K L, Ueki K, Jhung S L, Koh J, Louis D N. Molecular genetic correlates of p16, cdk4, and pRb immunohistochemistry in glioblastomas. J Neuropathol Exp Neurol. 1998;57:122–130. doi: 10.1097/00005072-199802000-00003. [DOI] [PubMed] [Google Scholar]
- 9.Califano J, Van der Riet P, Westra W, Nawroz H, Clayman G, Piantadosi S, Corio R, Lee D, Greenberg B, Koch W, Sidransky D. Genetic progression model for head and neck cancer: implications for field cancerization. Cancer Res. 1996;56:2488–2492. [PubMed] [Google Scholar]
- 10.Connell N D, Rheinwald J G. Regulation of the cytoskeleton in mesothelial cells: reversible loss of keratin and increase in vimentin during rapid growth in culture. Cell. 1983;34:245–253. doi: 10.1016/0092-8674(83)90155-1. [DOI] [PubMed] [Google Scholar]
- 11.Counter C M, Avilion A A, LeFeuvre C E, Stewart N G, Greider C W, Harley C B, Bacchetti S. Telomere shortening associated with chromosome instability is arrested in immortal cells which express telomerase activity. EMBO J. 1992;11:1921–1929. doi: 10.1002/j.1460-2075.1992.tb05245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Counter C M, Botelho F M, Wang P, Harley C B, Bacchetti S. Stabilization of short telomeres and telomerase activity accompany immortalization of Epstein-Barr virus-transformed human B lymphocytes. J Virol. 1994;68:3410–3414. doi: 10.1128/jvi.68.5.3410-3414.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dhouailly D, Xu C, Manabe M, Schermer A, Sun T-T. Expression of hair-related keratins in a soft epithelium: subpopulations of human and mouse dorsal tongue keratinocytes express keratin markers for hair-, skin-, and esophageal-types of differentiation. Exp Cell Res. 1989;181:141–158. doi: 10.1016/0014-4827(89)90189-4. [DOI] [PubMed] [Google Scholar]
- 14.Feng J, Funk W D, Wang S S, Weinrich S L, Avilion A A, Chiu C P, Adams R R, Chang E, Allsopp R C, Yu J, Le S, West M D, Harley C B, Andrews W H, Greider C W, Villeponteau B. The RNA component of human telomerase. Science. 1995;269:1236–1241. doi: 10.1126/science.7544491. [DOI] [PubMed] [Google Scholar]
- 15.Foster S A, Wong D J, Barrett M T, Galloway D A. Inactivation of p16 in human mammary epithelial cells by CpG island methylation. Mol Cell Biol. 1998;18:1793–1801. doi: 10.1128/mcb.18.4.1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fuchs E. Epidermal differentiation: the bare essentials. J Cell Biol. 1990;111:2807–2814. doi: 10.1083/jcb.111.6.2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hahn W C, Counter C M, Lundberg A S, Beijersbergen R L, Brooks M W, Weinberg R A. Creation of human tumor cells with defined genetic elements. Nature. 1999;400:464–468. doi: 10.1038/22780. [DOI] [PubMed] [Google Scholar]
- 18.Hara E, Smith R, Parry D, Tahara H, Stone S, Peters G. Regulation of p16INK4a expression and its implications for cell immortalization and senescence. Mol Cell Biol. 1996;16:859–867. doi: 10.1128/mcb.16.3.859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harley C B, Futcher A B, Greider C W. Telomeres shorten during ageing of human fibroblasts. Nature. 1990;345:458–460. doi: 10.1038/345458a0. [DOI] [PubMed] [Google Scholar]
- 20.Herman J G, Graff J R, Myohanen S, Nelkin B D, Baylin S B. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci USA. 1996;93:9821–9826. doi: 10.1073/pnas.93.18.9821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hudson D L, Weiland K L, Dooley T P, Simon M, Watt F M. Characterisation of eight monoclonal antibodies to involucrin. Hybridoma. 1992;11:367–379. doi: 10.1089/hyb.1992.11.367. [DOI] [PubMed] [Google Scholar]
- 22.Jarrard D F, Sarkar S, Shi Y, Yeager T R, Magrane G, Kinoshita H, Nassif N, Meisner L, Newton M A, Waldman F M, Reznikoff C A. p16/pRb pathway alterations are required for bypassing senescence in human prostate epithelial cells. Cancer Res. 1999;59:2957–2964. [PubMed] [Google Scholar]
- 23.Kastan M B, Onyekwere O, Sidransky D, Vogelstein B, Craig R W. Participation of p53 protein in the cellular response to DNA damage. Cancer Res. 1991;51:6304–6311. [PubMed] [Google Scholar]
- 24.Kim N W, Wu F. Advances in quantification and characterization of telomerase activity by the telomeric repeat amplification protocol (TRAP) Nucleic Acids Res. 1997;25:2595–2597. doi: 10.1093/nar/25.13.2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kiyono T, Foster S A, Koop J I, McDougall J K, Galloway D A, Klingelhutz A J. Both Rb/p16ink4a inactivation and telomerase activity are required to immortalize human epithelial cells. Nature. 1998;396:84–88. doi: 10.1038/23962. [DOI] [PubMed] [Google Scholar]
- 26.Lavker R M, Sun T-T. Epidermal stem cells. J Investig Dermatol. 1983;81:1211s–127s. doi: 10.1111/1523-1747.ep12540880. [DOI] [PubMed] [Google Scholar]
- 27.Li F P, Fraumeni J F, Mulvihill J J, Blattner W A, Dreyfuss M G, Tucker M A, Miller R W. A cancer family syndrome in twenty-four kindreds. Cancer Res. 1988;48:5358–5362. [PubMed] [Google Scholar]
- 28.Lindberg K, Rheinwald J G. Three distinct keratinocyte subtypes identified in human oral epithelium by their patterns of keratin expression in culture and in xenografts. Differentiation. 1990;45:230–241. doi: 10.1111/j.1432-0436.1990.tb00477.x. [DOI] [PubMed] [Google Scholar]
- 29.Loughran O, Malliri A, Owens D, Gallimore P H, Stanley M A, Ozanne B, Frame M C, Parkinson E K. Association of CDKN2A/P16INK4a with human head and neck keratinocyte replicative senescence: relationship of dysfunction to immortality and neoplasia. Oncogene. 1996;13:561–568. [PubMed] [Google Scholar]
- 30.Louis D N, von Deimling A, Seizinger B R. A (CA)n dinucleotide repeat assay for evaluating loss of allelic heterozygosity in small and archival human brain tumor specimens. Am J Pathol. 1992;141:777–782. [PMC free article] [PubMed] [Google Scholar]
- 31.Malkin D, Li F P, Strong L C, Fraumeni J F, Nelson C E, Kim D H, Kassel J, Gryka M A, Bischoff F Z, Tainsky M A. Germline p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science. 1990;250:1233–1238. doi: 10.1126/science.1978757. [DOI] [PubMed] [Google Scholar]
- 32.Manabe M, Lim H W, Winzer M, Loomis C A. Architectural organization of filiform papillae in normal and black hairy tongue epithelium: dissection of differentiation pathways in a complex human epithelium according to their patterns of keratin expression. Arch Dermatol. 1999;135:177–181. doi: 10.1001/archderm.135.2.177. [DOI] [PubMed] [Google Scholar]
- 33.Merlo A, Herman J G, Mao L, Lee D J, Gabrielson E, Burger P C, Baylin S B, Sidransky D. 5′ CpG island methylation is associated with transcriptional silencing of the tumour suppressor p16/CDKN2/MTS1 in human cancers. Nat Med. 1995;1:686–692. doi: 10.1038/nm0795-686. [DOI] [PubMed] [Google Scholar]
- 34.Meyerson M, Counter C M, Eaton E N, Ellisen L W, Steiner P, Caddle S D, Ziaugru L, Beijersbergen R L, Davidoff M J, Liu Q, Bachetti S, Haber D A, Weinberg R A. hEST2, the putative human telomerase catalytic subunit gene, is up-regulated in tumor cells and during immortalization. Cell. 1997;90:785–795. doi: 10.1016/s0092-8674(00)80538-3. [DOI] [PubMed] [Google Scholar]
- 35.Miller A D, Rosman G J. Improved retroviral vectors for gene transfer and expression. BioTechniques. 1989;7:980–990. [PMC free article] [PubMed] [Google Scholar]
- 36.Morgenstern J P, Land H. Advanced mammalian gene transfer: high titer retroviral vectors with multiple drug selection markers and a complementary helper-free packaging cell line. Nucleic Acids Res. 1990;18:3585–3596. doi: 10.1093/nar/18.12.3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Munro J, Stott F J, Vousden K H, Peters G, Parkinson E K. Role of the alternative INK4A proteins in human keratinocyte senescence: evidence for the specific inactivation of p16INK4a upon immortalization. Cancer Res. 1999;59:2516–2521. [PubMed] [Google Scholar]
- 38.Nakamura T M, Morin G B, Chapman K B, Weinrich S L, Andrews W H, Lingner J, Harley C B, Cech T R. Telomerase catalytic subunit homologs from fission yeast and human. Science. 1997;277:955–959. doi: 10.1126/science.277.5328.955. [DOI] [PubMed] [Google Scholar]
- 39.Ono Y, Tamiya T, Ichikawa T, Kunishio K, Matsumoto K, Furuta T, Ohmoto T, Ueki K, Louis D N. Malignant astrocytomas with homozygous CDKN2/p16 gene deletions have higher Ki-67 proliferation indices. J Neuropathol Exp Neurol. 1996;55:1026–1031. [PubMed] [Google Scholar]
- 40.Parenteau N L, Nolte C M, Bilbo P, Rosenberg M, Wilkins L M, Johnson E W, Watson S, Mason V S, Bell E. Epidermis generated in vitro: practical considerations and applications. J Cell Biochem. 1991;45:245–251. doi: 10.1002/jcb.240450304. [DOI] [PubMed] [Google Scholar]
- 41.Parkinson E K, Grabham P, Emmerson A. A subpopulation of cultured human keratinocytes which is resistant to the induction of terminal differentiation-related changes by phorbol, 12-myristate, 13-acetate: evidence for an increase in the resistant population following transformation. Carcinogenesis. 1983;4:857–861. doi: 10.1093/carcin/4.7.857. [DOI] [PubMed] [Google Scholar]
- 42.Ranade K, Hussussian C J, Sikorski R S, Varmus H E, Goldstein A M, Tucker M A, Serrano M, Hannon G J, Beach D, Dracopoli N C. Mutations associated with familial melanoma impair p16ink4 function. Nat Genet. 1995;10:114–116. doi: 10.1038/ng0595-114. [DOI] [PubMed] [Google Scholar]
- 43.Reddel R R. A reassessment of the telomere hypothesis of senescence. Bioessays. 1998;20:977–984. doi: 10.1002/(SICI)1521-1878(199812)20:12<977::AID-BIES3>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 44.Rheinwald J G. Methods for clonal growth and serial cultivation of normal human epidermal keratinocytes and mesothelial cells. In: Baserga R, editor. Cell growth and division: a practical approach. Oxford, England: IRL Press; 1989. pp. 81–94. [Google Scholar]
- 45.Rheinwald J G, Beckett M A. Tumorigenic keratinocyte lines requiring anchorage and fibroblast support cultured from human squamous cell carcinomas. Cancer Res. 1981;41:1657–1663. [PubMed] [Google Scholar]
- 46.Rheinwald J G, Green H. Serial cultivation of strains of human epidermal keratinocytes: the formation of keratinizing colonies from single cells. Cell. 1975;6:331–344. doi: 10.1016/s0092-8674(75)80001-8. [DOI] [PubMed] [Google Scholar]
- 47.Rheinwald J G, Green H. Epidermal growth factor and the multiplication of cultured human epidermal keratinocytes. Nature. 1997;265:421–424. doi: 10.1038/265421a0. [DOI] [PubMed] [Google Scholar]
- 48.Rice R H, Green H. Presence in human epidermal cells of a soluble protein precursor of the cross-linked envelope: activation of the cross-linking by calcium ions. Cell. 1979;18:681–694. doi: 10.1016/0092-8674(79)90123-5. [DOI] [PubMed] [Google Scholar]
- 49.Robles S J, Adami G R. Agents that cause DNA double strand breaks lead to pi6INK4A enrichment and the premature senescence of normal fibroblasts. Oncogene. 1998;16:1113–1123. doi: 10.1038/sj.onc.1201862. [DOI] [PubMed] [Google Scholar]
- 50.Rogan E M, Bryan T M, Hukku B, Maclean K, Chang C-M, Moy E L, Englezou A, Warneford S G, Dalla-Pozza L, Reddel R R. Alterations in p53 and p16INK4a expression and telomere length during spontaneous immortalization of Li-Fraumeni syndrome fibroblasts. Mol Cell Biol. 1995;15:4745–4753. doi: 10.1128/mcb.15.9.4745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rollins B J, O'Connell T M, Bennett G, Burton L E, Stiles C D, Rheinwald J G. Environment-dependent growth inhibition of human epidermal keratinocytes by recombinant human transforming growth factor-β. J Cell Physiol. 1989;139:455–462. doi: 10.1002/jcp.1041390302. [DOI] [PubMed] [Google Scholar]
- 52.Schön M, Rheinwald J G. A limited role for retinoic acid and the RARα and RARβ receptors in regulating keratin 19 expression and keratinization in oral and epidermal keratinocytes. J Investig Dermatol. 1996;107:428–438. doi: 10.1111/1523-1747.ep12363411. [DOI] [PubMed] [Google Scholar]
- 53.Sedivy J M. Can ends justify the means?: telomeres and the mechanisms of replicative senescence and immortalization in mammalian cells. Proc Natl Acad Sci USA. 1998;95:9078–9081. doi: 10.1073/pnas.95.16.9078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sellers W R, Kaelin W G., Jr Role of the retinoblastoma protein in the pathogenesis of human cancer. J Clin Oncol. 1997;15:3301–3312. doi: 10.1200/JCO.1997.15.11.3301. [DOI] [PubMed] [Google Scholar]
- 55.Shay J W, Tomlinson G, Piatyszek W A, Gollahon L S. Spontaneous in vitro immortalization of breast epithelial cells from a patient with Li-Fraumeni syndrome. Mol Cell Biol. 1995;15:425–432. doi: 10.1128/mcb.15.1.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Smith J R, Pereira-Smith O M. Replicative senescence: implications for in vivo aging and tumor suppression. Science. 1996;273:63–67. doi: 10.1126/science.273.5271.63. [DOI] [PubMed] [Google Scholar]
- 57.Tubo R A, Rheinwald J G. Normal human mesothelial cells and fibroblasts transfected with the EJras oncogene become EGF-independent, but are not malignantly transformed. Oncogene Res. 1987;1:407–21. [PubMed] [Google Scholar]
- 58.Ueki K, Rubio M P, Ramesh V, Correa K M, Rutter J L, von Deimling A, Buckler A J, Gusella J F, Louis D N. MTS1/CDKN2 gene mutations are rare in primary human astrocytomas with allelic loss of chromosome 9p. Hum Mol Genet. 1994;3:1841–1845. doi: 10.1093/hmg/3.10.1841. [DOI] [PubMed] [Google Scholar]
- 59.Watt F M. Epidermal stem cells: markers, patterning and the control of stem cell fate. Philos Trans R Soc Lond Ser B. 1998;353:831–837. doi: 10.1098/rstb.1998.0247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Watt F M, Green H. Involucrin synthesis is correlated with cell size in human epidermal cultures. J Cell Biol. 1981;90:738–742. doi: 10.1083/jcb.90.3.738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wu Y-J, Parker L M, Binder N E, Beckett M A, Sinard J H, Griffiths C T, Rheinwald J G. The mesothelial keratins: a new family of cytoskeletal proteins identified in cultured mesothelial cells and nonkeratinizing epithelia. Cell. 1982;31:693–703. doi: 10.1016/0092-8674(82)90324-5. [DOI] [PubMed] [Google Scholar]
- 62.Wynford-Thomas D. Cellular senescence and cancer. J Pathol. 1999;187:100–111. doi: 10.1002/(SICI)1096-9896(199901)187:1<100::AID-PATH236>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 63.Zhang Y, Xiong Y, Yarbrough W G. ARF promotes MDM2 degradation and stabilizes p53: ARF-INK4a locus deletion impairs both the Rb and p53 tumor suppression pathways. Cell. 1998;92:725–734. doi: 10.1016/s0092-8674(00)81401-4. [DOI] [PubMed] [Google Scholar]