

Abstract
Objective -
Our hypothesis is that Secreted Frizzled Related Protein 2 (sFPR2) is an important mechanism mediating ischemic cardioprotection, since it is the most upregulated gene in the third window of ischemic preconditioning.
Approach and Results –
One week after permanent coronary artery occlusion (CAO), sFRP2 TG mice exhibited a 49% higher LV ejection fraction and a 36% reduction in infarct size, p<0.05, and reduced fibrosis in both adjacent and remote zones, along with an increase in collagen type III and a decrease in the collagen type I/III ratio compared with WTL. The ischemic cardioprotection was associated with increased angiogenesis and arteriogenesis, reflected by increased capillary and arteriolar proliferation in the ischemic zone, thereby preserving blood flow after CAO. The angiogenesis and arteriogenesis were mediated by cross talk between myocytes and endothelial cells. The mechanism for cardioprotection and angiogenesis/arteriogenesis did not involve a traditional vascular growth hormone, e.g., VEGF or FGF, but rather cTGF, and ATF6 through the stress signaling pathway. The ATF6 inhibitor, AEBSF, blocked the upregulation of cTGF and both the angiogenesis and arteriogenesis, resulting in abolition of the reduced infarct size and protection of cardiac function in the sFRP2 TG mouse following permanent CAO.
Conclusion -
sFRP2 is a novel mechanism to induce angiogenesis/arteriogenesis, mediated through the endoplasmatic reticulum (ER) stress signaling pathway, ATF6 and cTGF, which protects the heart from myocardial ischemia.
Keywords: secreted frizzled proteins, cardioprotection, angiogenesis, arteriogenesis
Subject Codes: Angiogenesis, Ischemia, Physiology
INTRODUCTION:
Most studies on myocardial ischemic cardioprotection utilize relatively acute models of myocardial ischemia. The “Third Window of Ischemic Preconditioning” is novel, as it involves ischemic preconditioning induced by 6 episodes of 90 min coronary stenosis, 12 hours apart [10, 39]. The goal of the current investigation was to determine whether novel upregulated genes in the third window, which differed from those in the first or second windows, induced cardioprotection. Microarray analysis identified the potential cardioprotective upregulated genes in the 3rd window and found that Secreted Frizzled Related Protein 2 (sFRP2) was the most up-regulated gene (250 fold increase) in the 3rd window, but was not upregulated in the 2nd window [10]. Secreted Frizzled-related proteins (sFRPs) are a family of secreted proteins, which are involved in embryonic development as well as pathological conditions including bone and myocardial disorders and cancer [1, 5, 21, 24, 32], with prior studies finding divergent results related to cardioprotection by sFRP2 [15, 25].
Accordingly, the hypothesis for the current investigation was that sFRP2 is an important mechanism mediating cardioprotection. To address this hypothesis, we developed an sFRP2 transgenic (TG) mouse and induced permanent coronary artery occlusion to examine infarct size, recovery of myocardial function, and protection against myocardial fibrosis. Once it was determined that the sFRP2 TG mouse was protected against myocardial ischemia, the next goal was to determine if the mechanism involved angiogenesis or arteriogenesis, since myocardial tissue cannot be rescued after permanent coronary occlusion in the absence of blood flow in the ischemic myocardium. The next goal was to identify the molecular mechanisms mediating this cardioprotection. The endoplasmic reticulum (ER) stress signaling pathway was examined, since it was found upregulated in the third window of preconditioning [10], and since this pathway has been linked to angiogenesis [3]. The stress signaling protein, ATF6, was specifically examined as the mechanism for the angiogenesis [3] and cardioprotection [4, 19], since it was found to be upregulated in the third window of preconditioning. Next, potential vascular growth factors were examined and cTGF was selected for further study, as it was upregulated the most of all growth factors studied in the sFRP2 TG heart. Finally, to confirm the essential role of ATF6 in mediating the cardioprotection, the ATF6 inhibitor, 4-benzenesulfonyl fluoride hydrochloride (AEBSF) was used to block the role of ATF6 in mediating the angiogenesis and cardioprotection.
MATERIALS AND METHODS
Animals:
3–5 month old male FVB background transgenic (TG) mice with cardiac-specific sFRP2 overexpression were generated, using the cardiac-specific promoter, α-myosin heavy chain. SFRP2 expression in the cardiac specific sFRP2 TG mouse heart increased by 15 fold compared to their WTL. sFRP2 TG mice and their respective WTLs were housed on a 12-hour light-dark cycle with standard chow and water. Development, breeding of the transgenic mice and the experiments were performed at Rutgers-New Jersey Medical School. All experiments were approved by the IACUC and all animals were maintained in accordance with the guidelines for the Guide for the Care and Use of Laboratory Animals (National Research Council, 8th Edition 2011). The in vivo and histology studies were performed in TG mice and their WTL, whereas cardiac myocytes isolated from neonatal rat hearts were used for in vitro cell culture experiments.
Permanent Coronary Artery Occlusion:
Coronary artery ligation was performed as previously described [41]. Briefly, sFRP2 TG mice and their WTLs, were treated with cefazolin (100 mg/kg i.m.), and were then anesthetized with sodium pentobarbital (60mg/kg i.p.). Rectal temperature was monitored and body temperature maintained at 37°C with an automatic heating lamp and a water-circulating pad. After intubation of the trachea, mice were ventilated with a tidal volume of 0.25 ml/kg at a rate of 150 strokes/min, a left thoracotomy was performed at the fourth intercostal space and left anterior descending coronary artery (LAD) was permanently ligated at the same site in all animals to induce similar amounts of ischemia in all animals. After completion of the ligation, the chest was closed, inside air was removed from the chest by 20 G IV catheter and syringe and buprenorphine (0.05mg/kg s.c.) was given as post-operative medication. The extent of infarction, LV function and angiogenesis/arteriogenesis were measured at baseline, after 1 week coronary artery occlusion and after 1week coronary artery occlusion in mice treated for 1 week with an ATF6 inhibitor.
Administration of the ATF6 Inhibitor:
The ATF6 inhibitor, 4-benzenesulfonyl fluoride hydrochloride (AEBSF) (catalog # 5175/100, R&D systems, MN), was administered at a dose of 2μg/kg/day via osmotic pumps for 1 week.
Echocardiography:
At baseline, or 1 week after permanent coronary artery occlusion, mice were anesthetized by injection of Avertin (12.5mg/ml). Transthoracic echocardiography was performed as previously described [13]. The animal body temperature was carefully monitored and maintained as close to 37°C as possible during the entire procedure. Using a VisualSonics Vevo 3100 ultrasound system, LV internal dimensions were measured in systole and diastole using leading-edge methods and guidelines of the American Society of Echocardiography [38]. LV ejection fraction was calculated using the Vevo 3100 software. Fractional area change (FAC) % was calculated as (LV end-diastolic area – LV end-systolic area) / LV end-diastolic area × 100% and diastolic index % was calculated as (LV 30% diastolic area – LV end-systolic area) / (LV end-diastolic area – LV end-systolic area) × 100% as previously described [33].
Myocardial Blood Flow Assessment:
At baseline or 1 week after coronary artery occlusion, myocardial blood flow was measured and quantified using the Vevo 3100 ultrasound system (VisualSonics Inc.). Animals were anesthetized using sodium pentobarbital (60mg/kg i.p.). An external jugular vein catheter was then inserted into each mouse. The mice were then placed in a supine position and their limbs were taped over the metal ECG leads. Their body temperature, heart rate, and respiratory rate were carefully monitored and were maintained within normal ranges. Following proper mounting of the animal, a MS-250 transducer was lowered on the animal and positioned in the Parasternal Long-Axis View (PLAX). This view allowed for proper visualization of the infarction as well as border and remote zones of the myocardium. To monitor relative blood flow, nonlinear contrast agent imaging was conducted. Micro Marker non-targeted contrast agents (VS-11913) were injected into the external jugular vein catheter of each mouse (microbubble concentration = 1 × 108/50 μl, 50μl injection per mouse) using an infusion pump at a standardized rate of 600μl/min. ECG and respiratory double-gating was implemented during the procedure. Following data acquisition, analysis of the perfused myocardium was completed by using the VevoCQ software (Visual Sonics Inc.). The region of interest (ROI) was positioned in both the LV cavity and the myocardium. The peak enhancement of the myocardium was standardized to the peak enhancement in the LV cavity, as previously described [23]. Data are presented as [dB]. The more negative value indicates less blood flow and the higher intensity of the red color reflects increases in blood flow.
Histology staining
Hearts were collected from TG and WTL mice with or without AEBSF treatment and fixed in 10% formalin. After embedding in paraffin, LV rings were sectioned at 5μm thickness and processed for the following:
Interstitial fibrosis - Tissue sections were stained with Picro-Sirius Red (PSR) and Masson Trichrome staining to identify interstitial fibrosis deposition. The percentage of total fibrosis was quantified from Masson Trichrome staining using Image Pro-Plus software, as previously described [35].
Collagen type I and III – Tissue sections were assessed by using immunofluorescence (collagen type I antibody: ab34710; collagen type III antibody: ab7778; 2nd antibody: Alexa Fluor 568 anti-rabbit antibody). Samples were visualized with an Olympus BX51 microscope using conventional wide-field fluorescence microscopy, and the pictures were taken under 20x magnification at the infarct zone. The percentage of collagen type I and type III was calculated using Image Pro-Plus software.
Infarct size - Using PSR staining, infarct size was assessed by quantification of the fibrotic area and calculated as a percentage of the whole myocardial area. Viable tissue was identified as the non-fibrotic area, which was quantified and expressed as the percentage of the ischemic zone.
Capillary density - Using isolectin staining (isolectin GS-IB4-Alexa448 conjugate, catalog # 1780254, Invitrogen), capillaries were identified as single endothelial cell layer with a diameter less than 25µm. Capillary density was quantified at 40x magnification as absolute number per unit in the ischemic area.
Capillary Proliferation, Ki67 positive capillary - Using Ki67 (Ki 67-SP6, catalog # RM-9106-S1, Thermo Scientific) and isolectin double immunostaining, Ki67 positive capillaries were quantified and expressed as absolute numbers per field in the ischemic area including the infarct zone.
Arteriole Proliferation, Ki67 positive arteriole - Using Ki67 (Ki 67-SP6, catalog # RM-9106-S1, Thermo Scientific) and smooth muscle actin (catalog # 9106S1102A, Dako) double immunostaining, Ki67 positive arterioles were quantified and expressed as absolute number per field in the ischemic area including the infarct zone.
Tubule formation assay
Cardiomyocytes isolated from neonatal rat hearts were cultured in the presence or absence of the sFRP2 agonist (25 nM, catalog # 6838-FR, R&D), and/or the ATF6 inhibitor, AEBSF (100 μM) for 4 hours, the medium was changed to remove the protein. After 48 hours, the conditioned medium from cardiomyocytes was collected and centrifuged at 1000 rpm for 10 minutes followed by filter sterilization using 100 μm syringe filters. The conditioned medium was then aliquoted and stored at −80°C until further use.
Direct Treatment:
For the tubule formation assay from direct contact with sFRP2 protein, human umbilical vein endothelial cells (HUVECs) (catalog# CRL-1730™, ATCC, Manassas, VA) were used. Matrigel (BD Bioscience) was placed in 96-well tissue culture plates and allowed to polymerize at 37⁰C for 30 min. HUVECs at a density of 5000 cells/well were then plated on Matrigel in the presence of either sFRP2 protein or vehicle.
Indirect Treatment:
For the tubule formation assay from indirect exposure to sFRP2, the conditioned medium obtained after exposure to sFRP2 was applied to HUVEC cells, HUVECs at a density of 5000 cells/well were plated on Matrigel in the presence of either unconditioned medium or the cardiomyocyte conditioned medium by sFRP2. For cTGF and VEGF-A inhibition, the cardiomyocyte conditioned medium was pre-treated for 1 hour with either a control IgG, anti-cTGF antibody (catalog # ab6992, Abcam) or anti-VEGF-A neutralizing antibody (catalog # sc-152, Santa Cruz) before being added to HUVECs. After 6 hours, the tubules were imaged at 10x magnification followed by quantification. Data were expressed as the number of tubules in the conditioned medium normalized to that of unconditioned medium.
Real-time PCR (qPCR)
LV tissue was homogenized, extracted and qPCR was performed as previously described [11, 37]. Briefly, RNA was extracted and VEGF-A, VEGF-B, TGF-1, FGF-2, PDGF, HIF-A, and cTGF levels were quantified relative to an internal control, HPRT (Hypoxanthine-guanine phosphoribosyltransferase) by qPCR. LV from WTL and sFRP2 mice were harvested in lysis buffer treated with DNase I and resulting RNA was reverse transcribed according to manufacturer’s instructions using TaqMan gene expression Cells-to-Ct kit. (catalog# 4399002, Life Technologies). The mRNA was quantified using real-time qPCR (40 cycles at 15s step at 95°C and a 1 min step at 60°C) on a 7500 ABI-Prizm sequence detector (Applied Biosystems) using standard TaqMan probes (Applied Biosystems) for VEGF-A, VEGF-B, TGF-1, FGF-2, PDGF, HIF-A, cTGF and HPRT.
Immunoblotting:
Proteins separated by SDS-PAGE were transferred to nitrocellulose membranes. The membranes were probed with primary antibody at 4 °C overnight. The bands were visualized using Chemiluminescence reagents. The linear range of detection for different proteins and band intensities were determined by densitometry. Blots were re-probed with GAPDH to equalize sample loading. The antibodies used were Wnt 3A (MAB9025, R&D, 1:1000), β-catenin (8480, Cell Signalling, 1:1000), GSK3β (ab131356, Abcam, 1:1000), pGSK3β (ab75745, Abcam, 1:1000), BMP1 (AF1927, R&D, 1:1000), Smad1 (9743, Cell Signalling, 1:1000), p42/44 (9102, Cell Signalling, 1:1000), sFRP2 (MAB1169, R&D, 1:1000), H11K (MAB4987, Abcam, 1:1000), HSP70 (ab2787, Abcam, 1:1000), HSP27 (ab2790, Abcam, 1:1000), ATF6 (65880, Cell Signalling, 1:1000), GRP78 (sc-376768, Santa Cruz, 1:1000), PKc zeta (ab59364, Abcam, 1:1000), cTGF (ab6992, Abcam, 1: 1000).
Statistical Analysis:
Data are expressed as mean ± SEM. Statistical significance was determined using Student’s t-test or one-way ANOVA plus Bonferroni post hoc test evaluations, where appropriate. p<0.05 was taken as a minimal level of significance
RESULTS:
Cardiac Protection in sFRP2 TG Mice After 1 Week Permanent Coronary Artery Occlusion.
At baseline, there were no differences in LV ejection fraction (Figure 1B), myocardial blood flow (Figures 2A, 2B), capillary density (Figure 3A) and arteriole density (Figure 3B).
Figure 1: Ischemic Protection in sFRP2 TG Mice with 1 week Coronary Artery Occlusion:
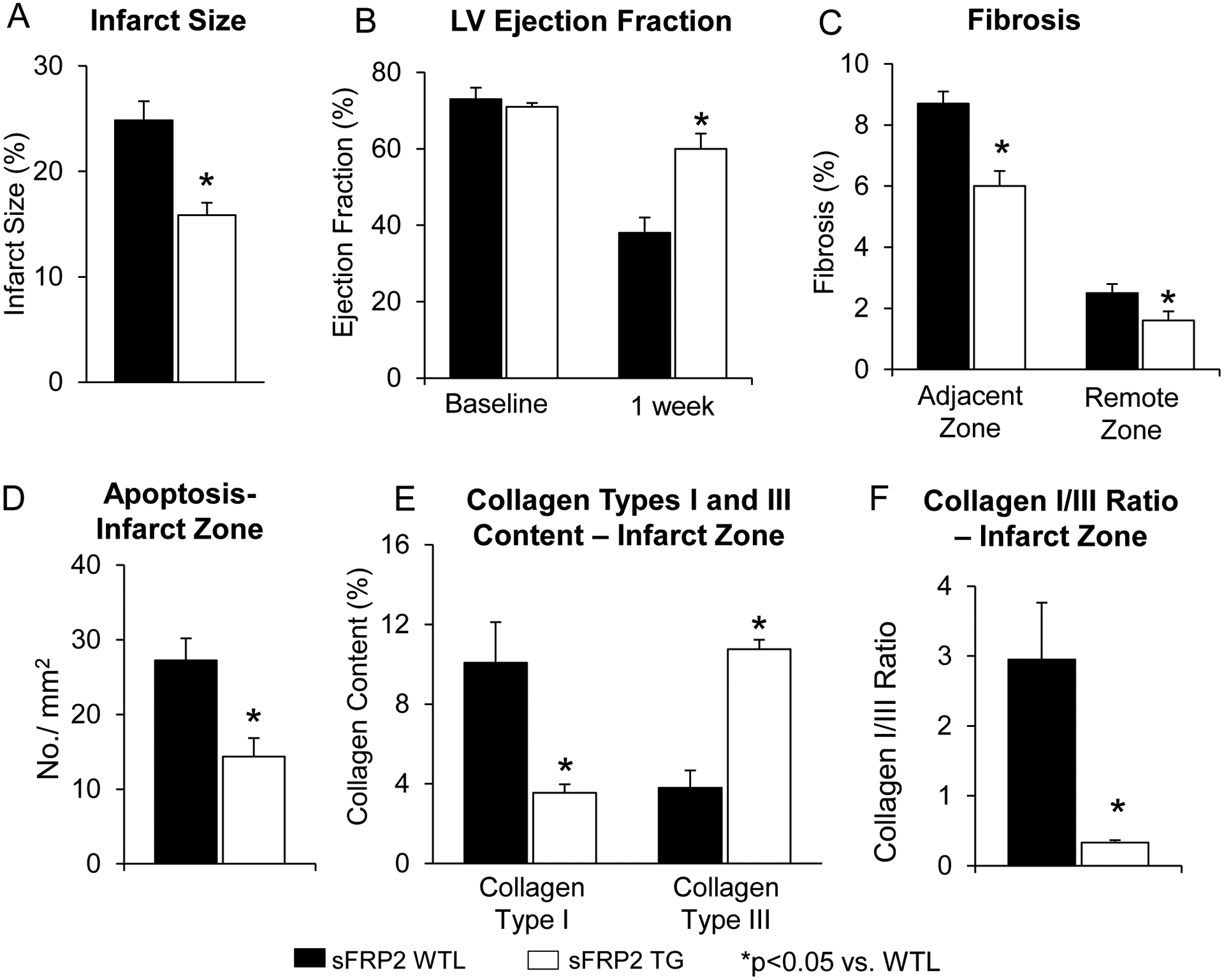
(A) Infarct size was significantly reduced in sFRP2 TG mice compared with infarct size in WTL after 1 week permanent coronary artery occlusion. (B) LV ejection fraction was preserved better in sFRP2 TG after 1 week permanent coronary artery occlusion. In the sfrp2 TG mice, there was less fibrosis in the adjacent and remote zones (C), and less apoptosis in the infarct zone (D). Collagen type III was increased (E) and the collagen type I/III ratio was significantly reduced in sFRP2 TG when compared to WTL (F). Results are expressed as mean ±SEM. *P<0.05 sFRP2 TG vs. WTL.
Figure 2: Myocardial Blood Flow is Increased in sFRP2 TG following 1 Week Coronary Artery Occlusion:
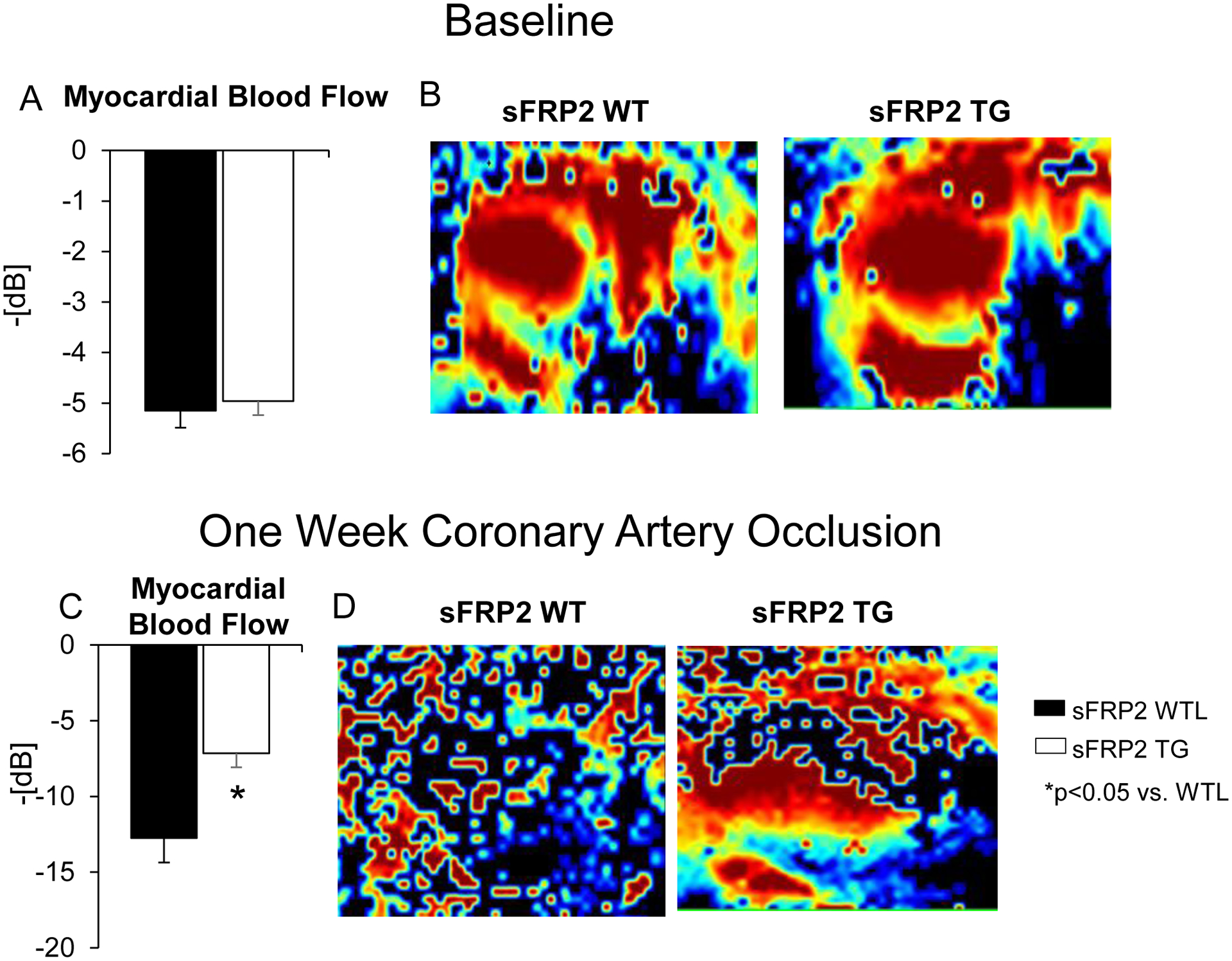
Myocardial blood flow measured by the Vevo 3100 (Visual Sonics Inc.). (A)No difference in myocardial blood flow was observed at baseline. (C) Myocardial blood flow was reduced less in the infarct zone in sFRP2 TG mice, than in WTL after 1 week coronary artery occlusion. The reduced negative numbers on the Y axis reflect higher blood flows. Representative figures showing similar levels of blood flow measured ultrasonically in the WTL andsFRP2 TG at baseline (B) and increased blood flow in sFRP2 TG after 1 week coronary artery occlusion (D). Results are expressed as mean ±SEM. *P<0.05 sFRP2 TG vs. WTL.
Figure 3: sFRP2 TG Mice Exhibit Enhanced Angiogeneis/Arteriogeneis:
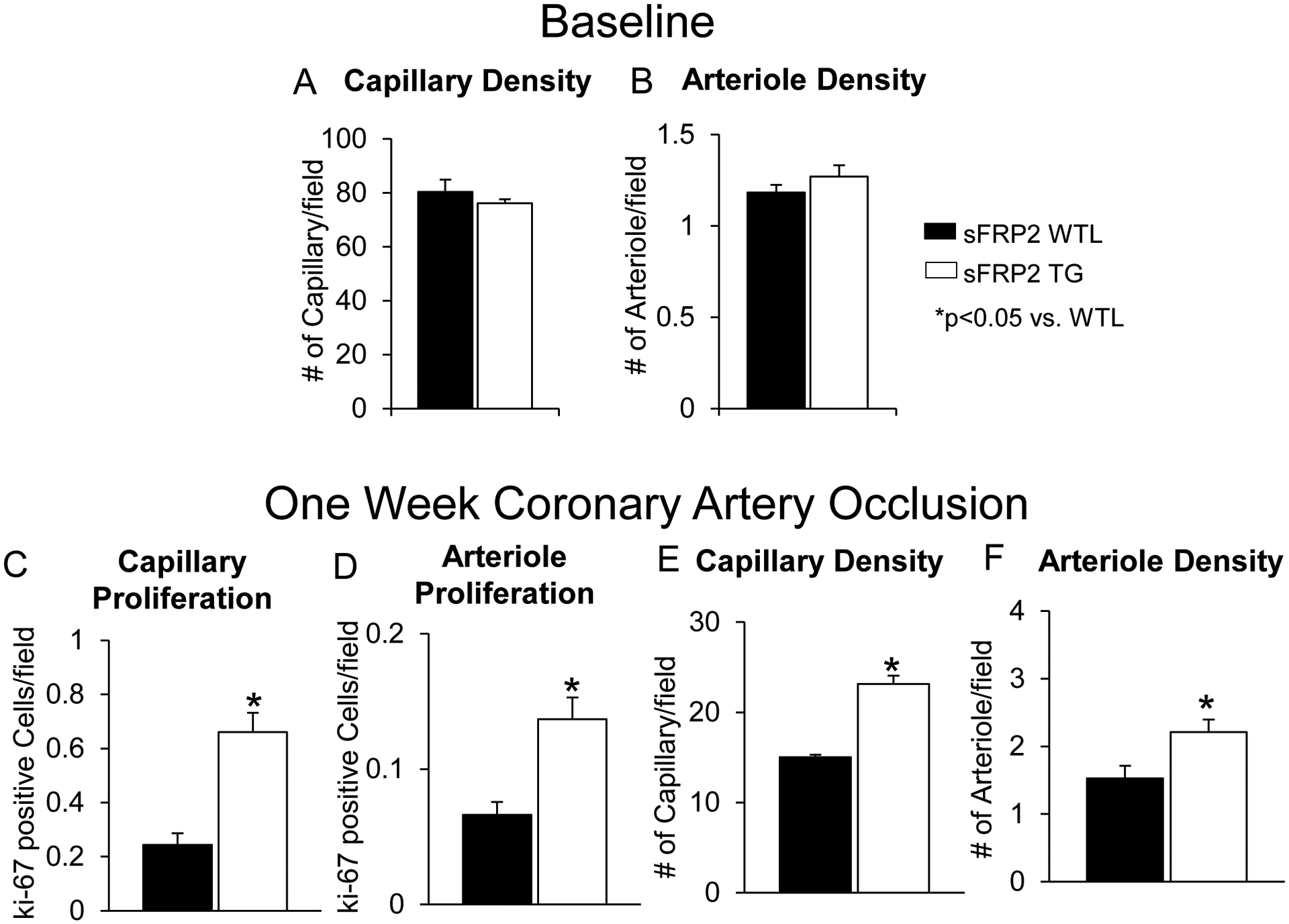
Prior to coronary artery occlusion, there is no difference in capillary (A) or arteriole density (B) was observed between sFRP2 TG mice and WTL. After 1 week permanent coronary artery occlusion, in the infarct zone, (C) capillary proliferation, measured by Ki-67 and isolectin, reflecting angiogenesis, was significantly increased in sFRP2 TG compared with their WTL. (D) Arteriole proliferation, measured by Ki-67 and α smooth muscle actin, reflecting arteriogenesis, was also significantly increased in sFRP2 TG compared with their WTL after 1 week permanent coronary artery occlusionin the infarct zone. (E) Capillary density and (F) arteriole density were significantly higher in sFRP2 TG compared with WTL after 1 week permanent coronary artery occlusion in the infarct zone. Results are expressed as mean ±SEM. *P<0.05 sFRP2 TG vs. WTL.
After 1 week coronary artery occlusion, there was improved LV ejection fraction (Figure 1B), and significant reductions in infarct size (Figure 1A), fibrosis in the adjacent and remote zones (Figure 1C), and apoptosis (Figure 1D) in sFRP2 TG mice compared with WTL. Collagen type III was increased (Figure 1E) and the collagen type I/III ratio was decreased in sFRP2 TG mice when compared to WTL (Figure 1F). Myocardial blood flow was lower in WTL mice blood flow in the ischemic zone, when compared to sFRP2 TG mice (Figure. 2C). Representative images of myocardial blood flow are shown in Figure 2D. This was associated with increased angiogenesis, reflected by increased capillary proliferation (Figure 3C) and increased capillary density (Figure 3E), as well as increased arteriogenesis (Figure 3D), reflected by increased arteriolar proliferation and increased arteriolar density (Figure 3F) in the ischemic zone.
Further analysis of systolic and diastolic function in sFRP2 TG mice and their WTL was conducted (Table 1). Following coronary artery occlusion, LV FAC and LV diastolic index were higher in sFRP2 TG mice when compared to WTL. Time to reach 50% of diastolic filling was preserved in sFRP2 TG mice following coronary artery occlusion while it increased in WTL. No differences were observed between sFRP2 TG and WTL at baseline.
Table 1:
Cardiac Function of sFRP2 TG at Baseline and After 1 Week Permanent Myocardial Ischemia (MI)
| Baseline | 1wk MI | |||
|---|---|---|---|---|
| WT | TG | WT | TG | |
| Heart Rate (bpm) | 432 ±27 | 429 ±45 | 456 ±27 | 424 ±25 |
| LV End Diastolic Diameter (mm) | 2.92 ±0.2 | 3.09 ±0.13 | 3.62 ±0.34 ƚ | 3.14 ±0.11 |
| LV End Systolic Diameter (mm) | 1.74 ±0.18 | 1.94 ±0.12 | 2.88 ±0.35 ƚ | 2.02 ±0.16 * |
| LV End Diastolic Volume (ul) | 40.5 ±4.0 | 41.9 ±3.7 | 56.8 ±3.0 ƚ | 40.8±4.0 |
| LV End Systolic Volume (ul) | 13.2 ±2.0 | 13.9 ±1.6 | 37.3 ±2.6 ƚ | 18.1±2.8 * |
| LV End Diastolic Area (mm2) | 10.2 ±0.4 | 10.2 ±0.4 | 13.7 ±0.9 ƚ | 12.2 ±1.2 |
| LV End Systolic Area (mm2) | 4.3 ±0.2 | 4.4 ±0.4 | 9.9 ±0.9 ƚ | 7.3 ±1.0 ƚ |
| LV FAC-Long Axis (%) | 57.1 ±1.7 | 56.9 ±4.5 | 28.4 ±3.1 ƚ | 42.2 ±3.7 ƚ* |
| LV Diastolic Index (%) | 61.1 ±3.7 | 60.9 ±4.3 | 29.0 ±3.4 ƚ | 55.7 ±3.3 * |
| Time to Reach 50% Diastolic Filling (s) | 0.04 ± 0.003 | 0.04 ± 0.001 | 0.06 ± 0.008 ƚ | 0.03 ± 0.002 * |
p< 0.05 WT vs TG;
p<0.05 baseline vs 1wk MI
Up-regulation of ER Stress Signaling Pathway and Vascular Growth Factors in sFRP2 TG Mouse Heart
Using microarray analysis to determine the signaling pathways of the 3rd window in the pig heart [10] and sFRP2 in the mouse heart, gene ontology (GO) analysis of microarray data showed that the most significantly up-regulated GO categories were the ER stress pathway and responses to unfolded protein in the sFRP2 TG mouse heart. These findings were further confirmed by western blotting showing that key ER stress response proteins: activating transcription factor 6 (ATF6), PKC zeta, GFR78 significantly increased in the sFRP2 TG mouse heart compared to that of WTL, as was the sFRP2 protein level (Figure 4A) at baseline. In addition, a group of heat shock proteins, e.g., H11 Kinase, HSP 70, significantly increased in the hearts from sFRP2 TG compared to WTL, suggesting that sFRP2 activates a novel Wnt-independent cell survival signaling pathway (Figure 4A) at baseline. Importantly, ATF6 was also upregulated in the pig heart with 3rd window, but not 2nd window preconditioning (Figure 4B).
Figure 4: Up-regulation of ER Stress Signaling Pathway and Vascular Growth Factors in sFRP2 TG mouse heart.
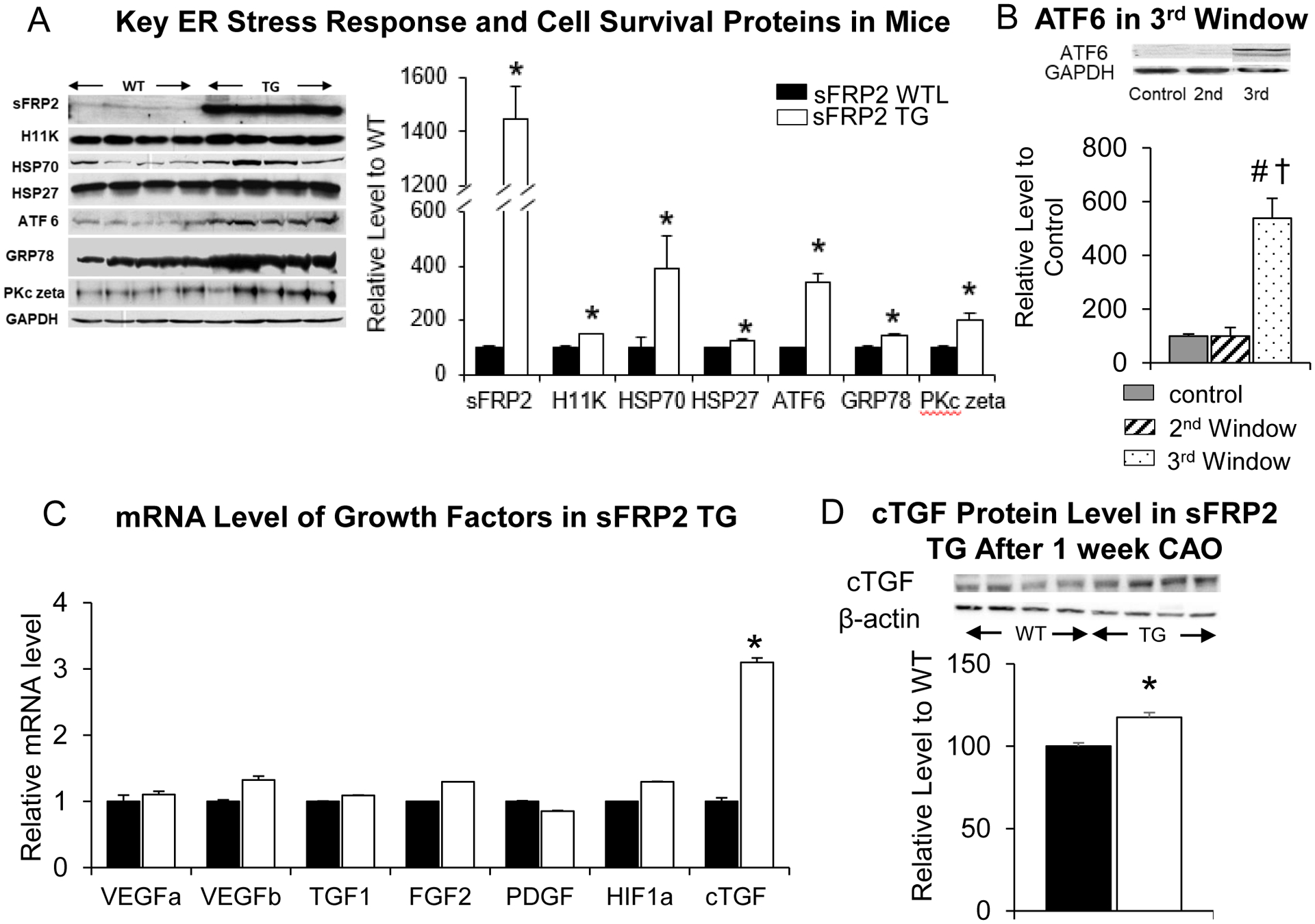
(A) ER stress response and cell survival proteins were upregulated in sFRP2 TG mice, identified by western blotting, indicating that stimulation of the ER stress signaling pathway is a unique mechanism of sFRP2 cardiac protection. ATF6, downstream in the signaling pathway from sFRP2 and critical for angiogenesis was upregulated in sFRP2 TG at baseline. (B) ATF6 was also up-regulated by six-fold in pigs in the third window of preconditioning, but not in the 2nd window of preconditioning. (C) mRNA level of the most common vascular growth factors were not increased in the sFRP2 TG mouse heart at baseline. However, cTGF was significantly increased in the sFRP2 TG compared with WTL confirmed by qPCR. (D) cTGF protein level was also found increased at 1 week after coronary artery occlusion in sFRP2 TG heart. Results are expressed as mean ±SEM. *p<0.05 vs. WTL. †P<0.05 vs. control (no preconditioning); #P<0.05 vs. 2nd window.
Since ATF6 has been reported to link to angiogenesis and cardioprotection [3, 4, 19], the major vascular growth factors were examined. VEGF, HIF1α, TGF1, FGF2 and PDGF, were not increased in the sFRP2 TG mouse heart. However, another growth factor, cTGF, was significantly increased in TG compared to WTL at baseline at the mRNA level (Figure 4C). Additionally, cTGF was increased in sFRP2 TG mice at the protein level one week after coronary artery occlusion (Figure 4D).
sFRP2 Promotes Angiogenesis in vitro Through Cross Talk Between Myocytes and Endothelial Cells, Mediated by cTGF.
Since angiogenesis and arteriogenesis were upregulated in sFRP2 TG following permanent coronary artery occlusion and since the sFRP2 TG mouse is cardiac specific, the next goal was to determine how sFRP2 in myocytes promotes cardiac angiogenesis in coronary vessels. To accomplish this, human umbilical vein endothelial cells (HUVEC) were directly treated with sFRP2 protein or vehicle, and cell proliferation and tubule formation were measured, in vitro. No differences were observed in sFRP2 protein treated HUVEC compared with vehicle control, indicating that sFRP2 does not induce angiogenesis by a direct action on vascular cells (Figure 5A, left panel). However, when rat neonatal myocytes were isolated and infected with sFRP2 protein or vehicle and the myocyte culture medium was collected 48 hours later and added to HUVEC, the conditioned medium from sFRP2 treated myocytes induced a significant increase in tubule formation in HUVECs by 27% compared to vehicle control (Figure 5A, B), indicating that sFRP2 increases the angiogenesis through cross talk between myocytes and endothelial cells via a paracrine mechanism. To confirm the key role of cTGF in mediating sFRP2 angiogenesis, we examined whether inhibition of cTGF, which was upregulated, or VEGF which was not, mediates the induction of angiogenesis by sFRP2 cross talk in vitro. The cTGF or VEGF antibody in sFRP2 conditioned medium was added to HUVECs cultured medium. The enhanced tubule formation was abolished by the cTGF antibody, but not by the VEGF antibody (Figures 5C, D), supporting cTGF as the mechanism for angiogenesis.
Figure 5: sFRP2 Promotes Angiogenesis in vitro Through Cross Talk Between Myocytes and Endothelial Cells, Mediated by cTGF.
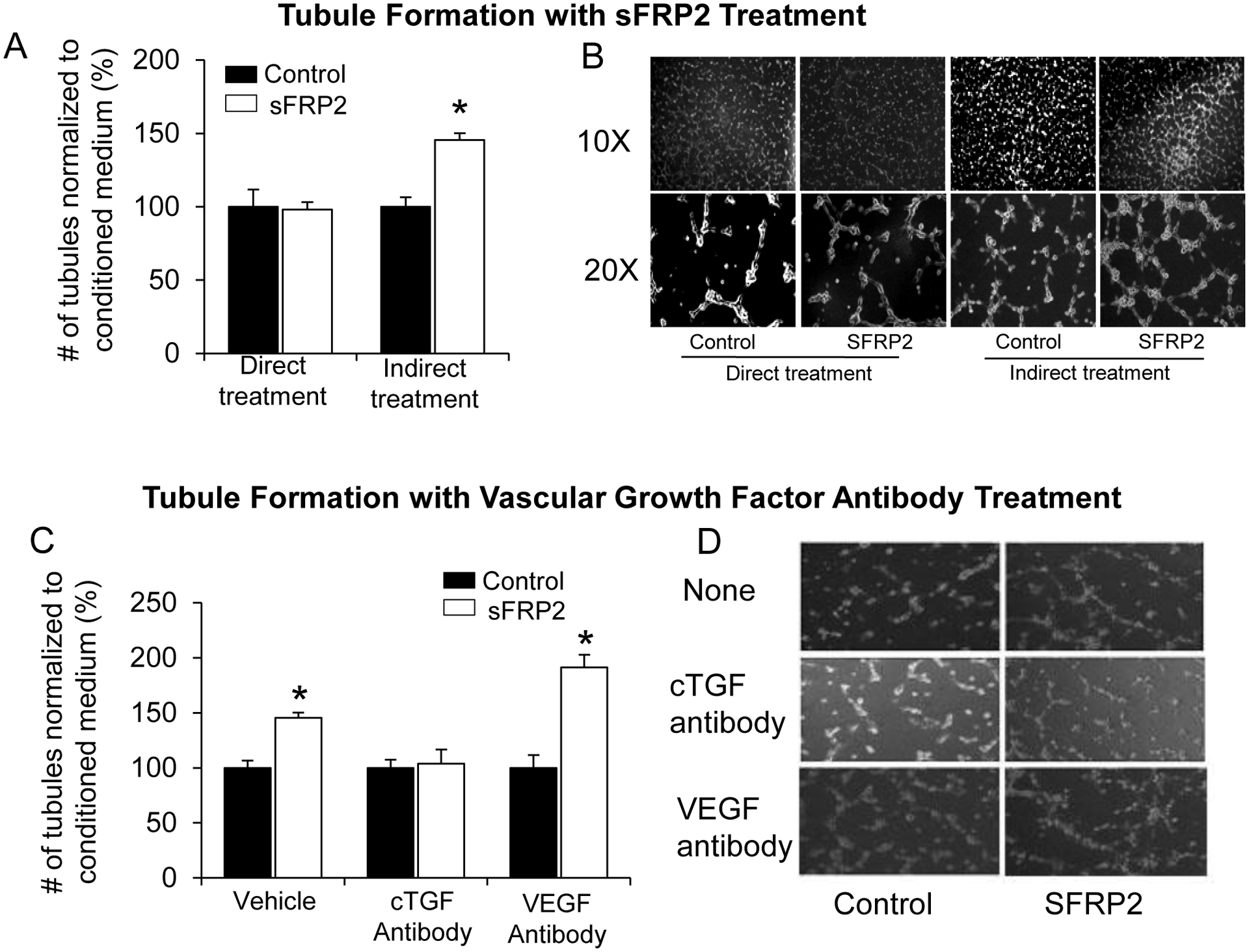
(A) Increased tubule formation of HUVEC was observed with conditioned medium treatment prior to treatment with sFRP2 protein, noted as indirect treatment, but not with direct application of sFRP2 protein to HUVEC. (B) Representative pictures of HUVEC tubule formation showing a significant increase in tubule formation in sFRP2 indirectly treated HUVECs. (C, D) Tubule formation, induced by indirect treatment of conditioned medium with sFRP2, was blocked with cTGF antibody, but not by VEGF antibody treatment. Results are expressed as mean ±SEM.*p<0.05 vs. control (no sFRP2 protein). Results are expressed as mean ± SEM.*p<0.05 vs. WTL.
Inhibiting ATF6 with AEBSF Blocked the sFRP2 Mediated Cardiac Protection and Angiogenesis and Arteriogenesis
We next examined the effects of inhibiting ATF6 with AEBSF. With AEBSF treatment, the reduced infarct size (Figure 6A), the preserved LV ejection fraction (Figure 6B), the increased capillary proliferation (Figure 6D) and arteriole proliferation (Figure 6E), and cTGF level (Figure 6F), and decreased apoptosis (Figure 6C) were no longer observed in the sFRP2 TG mice after 1 week coronary artery occlusion.
Figure 6: sFRP2 TG Cardioprotection with 1 week Coronary Artery Occlusion is Mediated via the ATF6 Pathway.
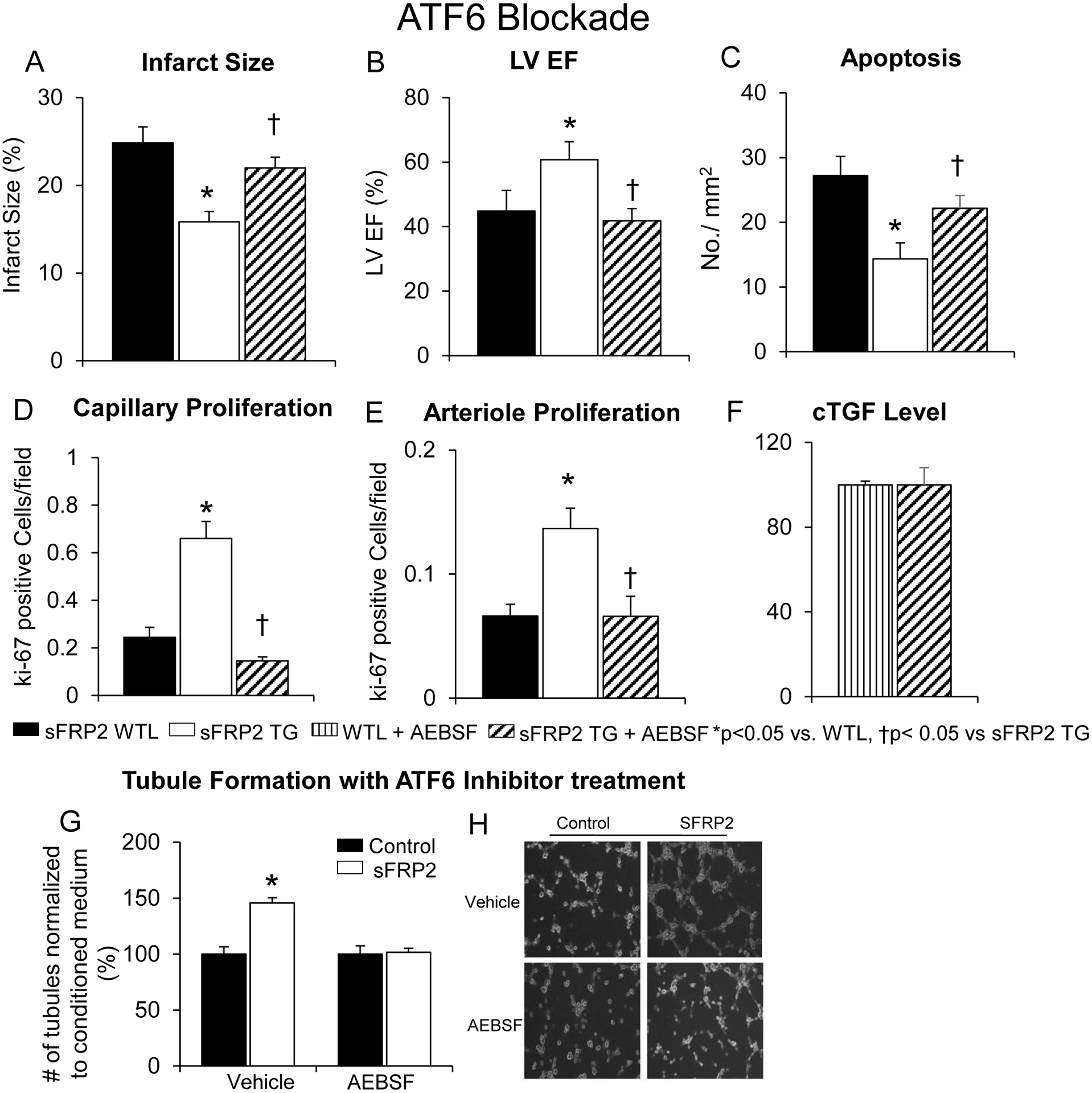
Following 1 week of permanent coronary artery occlusion, infarct size was reduced (A), LV ejection fraction was preserved (B), capillary proliferation (D), and arteriole proliferation (E) were all up-regulated, and apoptosis (C) was reduced in sFRP2 TG vs. WTL. Inhibition of ATF6 by AEBSF abolished all the cardioprotective features observed in sFRP2 TG with 1 week of permanent coronary artery occlusion. (F) cTGF levels were no different with treatment of AEBSF between sFRP2 TG and WTL mice. (G, H) Inhibition of ATF6 with AEBSF also blocked the increased tubule formation induced by sFRP2. Results are expressed as mean ±SEM.*p<0.05 vs. WTL; †P<0.05 vs. sFRP2 TG.
To confirm that ATF6 was involved in the mechanism of angiogenesis induced by sFRP2, AEBSF also blocked the increased tubule formation in HUVECs by sFRP2 cross talk (Figures 6G, 6H), indicating that sFRP2 activates angiogenesis through ATF6 signaling.
DISCUSSION
When examining upregulated genes in the heart of the third window model of ischemic preconditioning, sFRP2 emerged as the most highly upregulated gene of the 20,201 genes examined [10]. This finding led to the hypothesis that sFRP2 is a novel mechanism mediating ischemic cardioprotection. Accordingly, the first goal of this investigation was to determine the extent to which sFRP2 mediated cardioprotection following 1 week of permanent coronary artery occlusion. To address this goal we developed a cardiac specific sFRP2 TG mouse. After one week of permanent coronary occlusion, infarct size was reduced in the sFRP2 TG compared to WTL by 36% and LV ejection fraction was 49% higher in the sFRP2 TG compared with WTL, while myocardial fibrosis was reduced by 31% in the adjacent zone and by 37% in the remote zone. Further analysis of fibrosis revealed increased collagen type III and a reduced collagen I/III ratio in sFRP2 TG mice, supporting an improved healing process, when compared to WTL [7, 26, 30]. The next goal was to determine if there was angiogenesis or arteriogenesis in the ischemic sFRP2 TG heart. We found both angiogenesis (increased capillaries) and arteriogenesis (increased arterioles) in the sFRP2 TG heart at one week after permanent coronary artery occlusion, along with increased blood flow to the ischemic myocardium, resulting in protection of the sFRP2 TG heart, and mediating the reduced infarct size and preservation of cardiac function. Since angiogenesis and arteriogenesis were upregulated in sFRP2 TG following permanent coronary artery occlusion and since the sFRP2 TG mouse is cardiac specific, the next goal was to determine how sFRP2 in myocytes promotes cardiac angiogenesis in coronary vessels. This was accomplished by showing that sFRP2 increases the angiogenesis through cross talk between myocytes and endothelial cells via a paracrine mechanism. We determined this by demonstrating increased tubule formation, in vitro, by indirect exposure of the tubules to sFRP2 protein, as opposed to direct exposure.
Prior studies on cardioprotection induced by sFRP2 have been controversial [15, 25]. One study examined exogenously administered sFRP2 [15], whereas another used a sFRP2 knockout [25]. These studies should have had opposite results with similar conclusions, but actually had almost opposite conclusions [15, 25]. One key measurement lacking in these studies [15, 25] is whether sFRP2 induced angiogenesis, since an increase in blood flow to the ischemic tissue would be necessary for salvation of ischemic myocardium in the presence of permanent coronary artery occlusion. No prior study has found that sFRP2 induces angiogenesis in the heart, although some studies have found increased angiogenesis in other organs [8, 12, 40]. Other prior literature on this topic, but not assessing myocardial infarction, has also been controversial, with some studies finding that sFRP2 does induce angiogenesis [8, 40], but other studies found the opposite, i.e., that sFRP2 blockade induces angiogenesis [31].
The final goal of this study was to determine the mechanism of the angiogenesis/arteriogenesis. Since the ER stress signaling pathway was upregulated in the third window [10], and ATF6, a key protein in this pathway was also upregulated in the third window (Figure 4), this mechanism mediating the cardioprotection was examined, using the ATF6 blocker, AEBSF. Blocking ATF6 eliminated the reduction in infarct size and improvement of cardiac performance in the sFRP2 heart one week after permanent coronary artery occlusion. Blocking ATF6 also eliminated the angiogenesis and arteriogenesis, which mediated the cardioprotection. These data suggest that sFRP2 activates angiogenesis through ATF6 signaling. It is important to note that this is the first study to show ATF6 induces angiogenesis in the heart, which can then protect against myocardial infarction.
In order to further elucidate the mechanism of the angiogenesis/arteriogenesis in the sFRP2 heart, vascular growth factors were examined. VEGF, HIF1α, TGF1, FGF2 and PDGF, the major vascular growth factors, were not increased in the sFRP2 TG mouse heart. Rather, another growth factor, cTGF, was significantly increased in sFRP2 TG compared to WTL, and has been shown to regulate angiogenesis and endothelial cell function [6, 9, 28]. ATF6 inhibition by AEBSF also blocked the increased expression of cTGF in the sFRP2 TG heart, indicating that the mechanism of angiogenesis induced by sFRP2 involved cTGF, downstream in the ATF6 pathway. It is important to note that this is the first study to show cTGF induces angiogenesis in the heart, which can then protect against myocardial infarction.
Prior studies have emphasized the role of the Wnt signaling pathway in mediating the effects of sFRP2 [20, 27, 36]. Our data show that there is no difference in traditional Wnt signaling and its down-stream targets, e.g., β-catenin and GSK-3β between sFRP2 TG and WTL mouse hearts (Figure 7). We also did not find differences in BMP1 signaling in sFRP2 TG compared to WTL, although it has been reported as the target of sFRP2 protein [15]. Our data indicate that the mechanisms mediating the sFRP2 cardioprotection and angiogenesis/arteriogenesis in response to permanent coronary artery occlusion differ from traditional Wnt signaling. Rather, we found that the ER stress signaling pathways, ATF6 and cTGF, are involved in mediating the cardioprotection, reducing infarct size, and improving cardiac function through angiogenesis and arteriogenesis in the sFRP2 TG mouse following permanent coronary artery occlusion (Figure 8).
Figure 7:
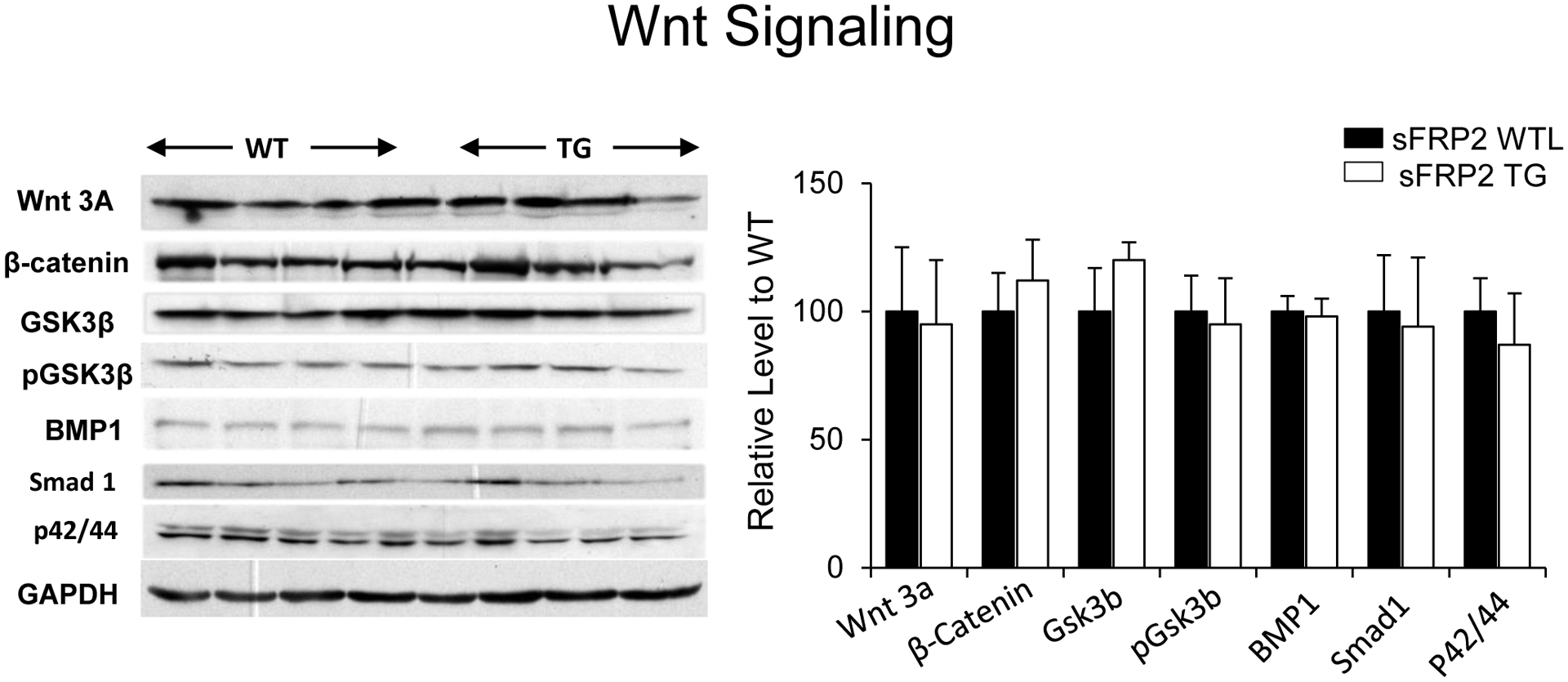
There were no differences in results from western blotting of traditional Wnt/β-catenin signaling and its downstream targets including GSK3β, BMP1, Smad1 and p42/44 proteins between sFRP2 TG and WTL at baseline. Results are expressed as mean ±SEM.*p<0.05 vs. WTL; †P<0.05 vs. sFRP2 TG.
Figure 8:
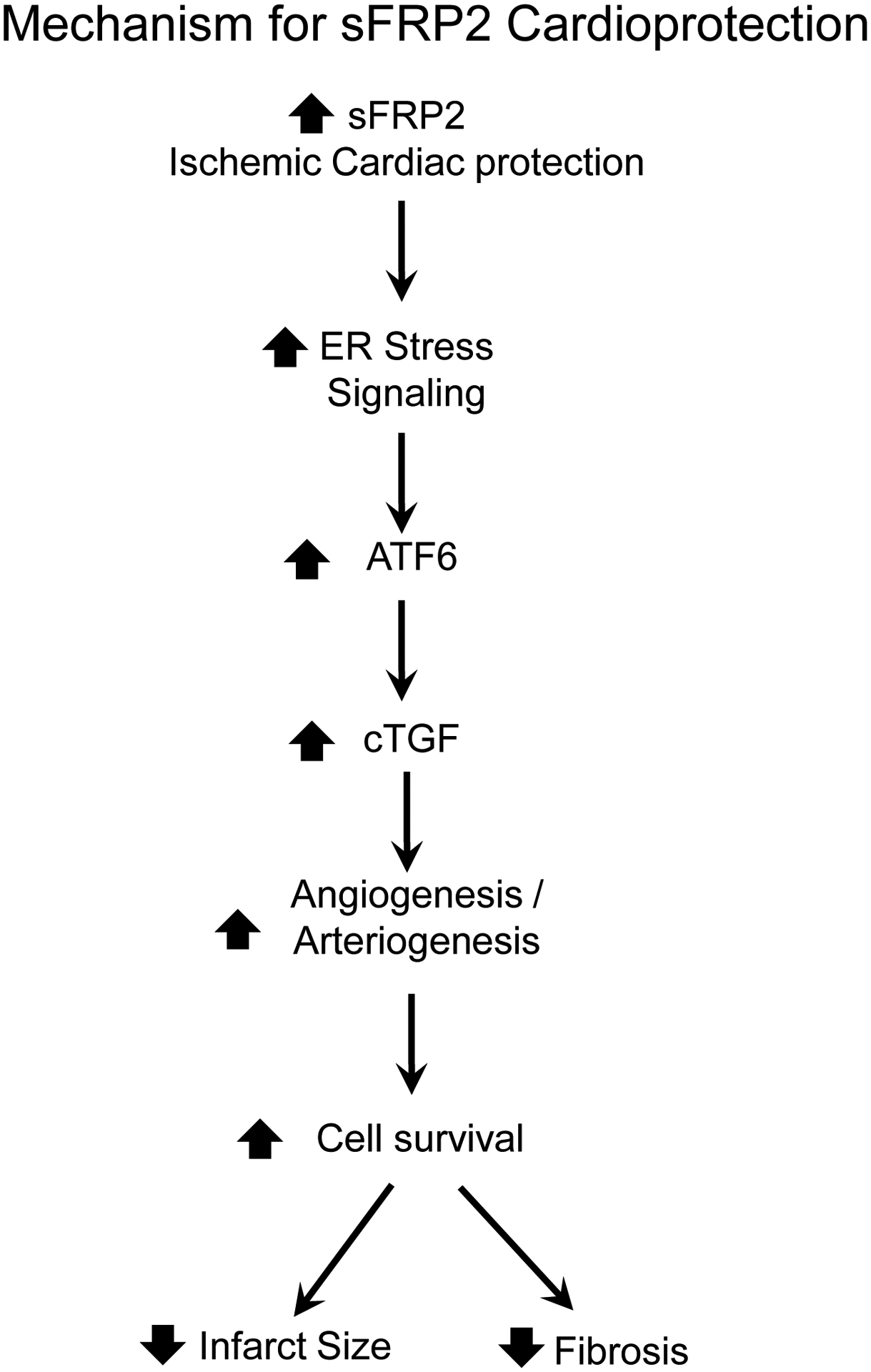
sFRP2 represents a novel mechanism of ischemic cardiac protection by increasing both angiogenesis and arteriogenesis mediated by cTGF and ATF6 signaling.
The angiogenesis and arteriogenesis findings may have translational importance. Despite promising results on VEGF inducing angiogenesis in animal models [2, 14, 29, 34] and recent positive results for lymphangiongenesis [42], recruitment of cardioprotective signaling, in general, has been disappointing [18], and clinical trials have failed [16, 17, 22]. It is conceivable that another growth factor may be more effective clinically to induce angiogenesis. cTGF is a novel growth factor for angiogenesis and it is conceivable it could be successful clinically, where VEGF failed. Furthermore, this finding offers further support for the use of exogenous sFRP2 as a potential therapeutic, given its initial success in reducing fibrosis following myocardial infarction in rats [15]. Angiogenesis and arteriogenesis are critical mechanisms to maintain tissue viability in the presence of chronic ischemia. With the elucidation of sFRP2’s role in vascular proliferation via cTGF, sFRP2 can now be explored in other models of ischemia, e.g. acute mesenteric ischemia, ischemic stroke, and peripheral artery disease, to determine further therapeutic value of this protein and its associated signaling mechanisms. In support of this hypothesis, it is important to keep in mind that the genesis for this study was the discovery of these molecular pathways in a clinically relevant model of ischemic preconditioning, i.e., the third window [10, 39].
ACKNOWLEDGMENTS
We appreciate Dr. Xiangzhen Sui, Dr. Yimin Tian, Dr. Seonghun Yoon and Dr. Xin Zhao for technical support.
SOURCES OF FUNDING
This study was supported by National Institutes of Health grants R01HL102472, R01HL106511, R01HL119464, R01HL124282, R01HL130848, R01HL137368, R01HL137405, R21AG053514, and S10 OD025238.
Footnotes
DISCLOSURES
None
REFERENCES:
- 1.Alfaro MP, Vincent A, Saraswati S, Thorne CA, Hong CC, Lee E, Young PP (2010) sFRP2 suppression of bone morphogenic protein (BMP) and Wnt signaling mediates mesenchymal stem cell (MSC) self-renewal promoting engraftment and myocardial repair. J Biol Chem 285:35645–35653 doi: 10.1074/jbc.M110.135335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Banai S, Jaklitsch MT, Shou M, Lazarous DF, Scheinowitz M, Biro S, Epstein SE, Unger EF (1994) Angiogenic-induced enhancement of collateral blood flow to ischemic myocardium by vascular endothelial growth factor in dogs. Circulation 89:2183–2189 [DOI] [PubMed] [Google Scholar]
- 3.Binet F, Sapieha P (2015) ER Stress and Angiogenesis. Cell Metab 22:560–575 doi: 10.1016/j.cmet.2015.07.010 [DOI] [PubMed] [Google Scholar]
- 4.Blackwood EA, Hofmann C, Santo Domingo M, Bilal AS, Sarakki A, Stauffer W, Arrieta A, Thuerauf DJ, Kolkhorst FW, Muller OJ, Jakobi T, Dieterich C, Katus HA, Doroudgar S, Glembotski CC (2019) ATF6 Regulates Cardiac Hypertrophy by Transcriptional Induction of the mTORC1 Activator, Rheb. Circ Res 124:79–93 doi: 10.1161/CIRCRESAHA.118.313854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bovolenta P, Esteve P, Ruiz JM, Cisneros E, Lopez-Rios J (2008) Beyond Wnt inhibition: new functions of secreted Frizzled-related proteins in development and disease. J Cell Sci 121:737–746 doi: 10.1242/jcs.026096 [DOI] [PubMed] [Google Scholar]
- 6.Brigstock DR (2002) Regulation of angiogenesis and endothelial cell function by connective tissue growth factor (CTGF) and cysteine-rich 61 (CYR61). Angiogenesis 5:153–165. doi: 10.1023/a:1023823803510. [DOI] [PubMed] [Google Scholar]
- 7.Clore JN, Cohen IK, Diegelmann RF (1979) Quantitation of collagen types I and III during wound healing in rat skin. Proc Soc Exp Biol Med 161:337–340 doi: 10.3181/00379727-161-40548 [DOI] [PubMed] [Google Scholar]
- 8.Courtwright A, Siamakpour-Reihani S, Arbiser JL, Banet N, Hilliard E, Fried L, Livasy C, Ketelsen D, Nepal DB, Perou CM, Patterson C, Klauber-Demore N (2009) Secreted frizzle-related protein 2 stimulates angiogenesis via a calcineurin/NFAT signaling pathway. Cancer Res 69:4621–4628 doi: 10.1158/0008-5472.CAN-08-3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Das MK, Basak S, Ahmed MS, Attramadal H, Duttaroy AK (2014) Connective tissue growth factor induces tube formation and IL-8 production in first trimester human placental trophoblast cells. Eur J Obstet Gynecol Reprod Biol 181:183–188 doi: 10.1016/j.ejogrb.2014.07.045 [DOI] [PubMed] [Google Scholar]
- 10.Depre C, Park JY, Shen YT, Zhao X, Qiu H, Yan L, Tian B, Vatner SF, Vatner DE (2010) Molecular mechanisms mediating preconditioning following chronic ischemia differ from those in classical second window. Am J Physiol Heart Circ Physiol 299:H752–762. doi: 10.1152/ajpheart.00147.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Depre C, Tomlinson JE, Kudej RK, Gaussin V, Thompson E, Kim SJ, Vatner DE, Topper JN, Vatner SF (2001) Gene program for cardiac cell survival induced by transient ischemia in conscious pigs. Proc Natl Acad Sci U S A 98:9336–9341. doi: 10.1073/pnas.171297498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fontenot E, Rossi E, Mumper R, Snyder S, Siamakpour-Reihani S, Ma P, Hilliard E, Bone B, Ketelsen D, Santos C, Patterson C, Klauber-DeMore N (2013) A novel monoclonal antibody to secreted frizzled-related protein 2 inhibits tumor growth. Mol Cancer Ther 12:685–695 doi: 10.1158/1535-7163.MCT-12-1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gao S, Ho D, Vatner DE, Vatner SF (2011) Echocardiography in Mice. Curr Protoc Mouse Biol 1:71–83 DOI: 10.1002/9780470942390.mo100130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harada K, Friedman M, Lopez JJ, Wang SY, Li J, Prasad PV, Pearlman JD, Edelman ER, Sellke FW, Simons M (1996) Vascular endothelial growth factor administration in chronic myocardial ischemia. Am J Physiol 270:H1791–1802 doi: 10.1152/ajpheart.1996.270.5.H1791 [DOI] [PubMed] [Google Scholar]
- 15.He W, Zhang L, Ni A, Zhang Z, Mirotsou M, Mao L, Pratt RE, Dzau VJ (2010) Exogenously administered secreted frizzled related protein 2 (Sfrp2) reduces fibrosis and improves cardiac function in a rat model of myocardial infarction. Proc Natl Acad Sci U S A 107:21110–21115 doi: 10.1073/pnas.1004708107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hedman M, Hartikainen J, Syvanne M, Stjernvall J, Hedman A, Kivela A, Vanninen E, Mussalo H, Kauppila E, Simula S, Narvanen O, Rantala A, Peuhkurinen K, Nieminen MS, Laakso M, Yla-Herttuala S (2003) Safety and feasibility of catheter-based local intracoronary vascular endothelial growth factor gene transfer in the prevention of postangioplasty and in-stent restenosis and in the treatment of chronic myocardial ischemia: phase II results of the Kuopio Angiogenesis Trial (KAT). Circulation 107:2677–2683 doi: 10.1161/01.CIR.0000070540.80780.92 [DOI] [PubMed] [Google Scholar]
- 17.Henry TD, Annex BH, McKendall GR, Azrin MA, Lopez JJ, Giordano FJ, Shah PK, Willerson JT, Benza RL, Berman DS, Gibson CM, Bajamonde A, Rundle AC, Fine J, McCluskey ER, Investigators V (2003) The VIVA trial: Vascular endothelial growth factor in Ischemia for Vascular Angiogenesis. Circulation 107:1359–1365. DOI: 10.1161/01.cir.0000061911.47710.8a [DOI] [PubMed] [Google Scholar]
- 18.Heusch G (2015) Molecular basis of cardioprotection: signal transduction in ischemic pre-, post-, and remote conditioning. Circ Res 116:674–699 doi: 10.1161/CIRCRESAHA.116.305348 [DOI] [PubMed] [Google Scholar]
- 19.Jin JK, Blackwood EA, Azizi K, Thuerauf DJ, Fahem AG, Hofmann C, Kaufman RJ, Doroudgar S, Glembotski CC (2017) ATF6 Decreases Myocardial Ischemia/Reperfusion Damage and Links ER Stress and Oxidative Stress Signaling Pathways in the Heart. Circ Res 120:862–875 doi: 10.1161/CIRCRESAHA.116.310266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jin L, Cao Y, Yu G, Wang J, Lin X, Ge L, Du J, Wang L, Diao S, Lian X, Wang S, Dong R, Shan Z (2017) SFRP2 enhances the osteogenic differentiation of apical papilla stem cells by antagonizing the canonical WNT pathway. Cell Mol Biol Lett 22:14 doi: 10.1186/s11658-017-0044-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jones SE, Jomary C (2002) Secreted Frizzled-related proteins: searching for relationships and patterns. Bioessays 24:811–820 doi: 10.1002/bies.10136 [DOI] [PubMed] [Google Scholar]
- 22.Kastrup J, Jorgensen E, Ruck A, Tagil K, Glogar D, Ruzyllo W, Botker HE, Dudek D, Drvota V, Hesse B, Thuesen L, Blomberg P, Gyongyosi M, Sylven C, Euroinject One G (2005) Direct intramyocardial plasmid vascular endothelial growth factor-A165 gene therapy in patients with stable severe angina pectoris A randomized double-blind placebo-controlled study: the Euroinject One trial. J Am Coll Cardiol 45:982–988 doi: 10.1016/j.jacc.2004.12.068 [DOI] [PubMed] [Google Scholar]
- 23.Kawamura M, Paulsen MJ, Goldstone AB, Shudo Y, Wang H, Steele AN, Stapleton LM, Edwards BB, Eskandari A, Truong VN, Jaatinen KJ, Ingason AB, Miyagawa S, Sawa Y, Woo YJ (2017) Tissue-engineered smooth muscle cell and endothelial progenitor cell bi-level cell sheets prevent progression of cardiac dysfunction, microvascular dysfunction, and interstitial fibrosis in a rodent model of type 1 diabetes-induced cardiomyopathy. Cardiovasc Diabetol 16:142 doi: 10.1186/s12933-017-0625-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kele J, Andersson ER, Villaescusa JC, Cajanek L, Parish CL, Bonilla S, Toledo EM, Bryja V, Rubin JS, Shimono A, Arenas E (2012) SFRP1 and SFRP2 dose-dependently regulate midbrain dopamine neuron development in vivo and in embryonic stem cells. Stem Cells 30:865–875 doi: 10.1002/stem.1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kobayashi K, Luo M, Zhang Y, Wilkes DC, Ge G, Grieskamp T, Yamada C, Liu TC, Huang G, Basson CT, Kispert A, Greenspan DS, Sato TN (2009) Secreted Frizzled-related protein 2 is a procollagen C proteinase enhancer with a role in fibrosis associated with myocardial infarction. Nat Cell Biol 11:46–55 doi: 10.1038/ncb1811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li Z, Masumoto H, Jo JI, Yamazaki K, Ikeda T, Tabata Y, Minatoya K (2018) Sustained release of basic fibroblast growth factor using gelatin hydrogel improved left ventricular function through the alteration of collagen subtype in a rat chronic myocardial infarction model. Gen Thorac Cardiovasc Surg 66:641–647 doi: 10.1007/s11748-018-0969-z [DOI] [PubMed] [Google Scholar]
- 27.Lin H, Angeli M, Chung KJ, Ejimadu C, Rosa AR, Lee T (2016) sFRP2 activates Wnt/beta-catenin signaling in cardiac fibroblasts: differential roles in cell growth, energy metabolism, and extracellular matrix remodeling. Am J Physiol Cell Physiol 311:C710–C719 doi: 10.1152/ajpcell.00137.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu SC, Chuang SM, Hsu CJ, Tsai CH, Wang SW, Tang CH (2014) CTGF increases vascular endothelial growth factor-dependent angiogenesis in human synovial fibroblasts by increasing miR-210 expression. Cell Death Dis 5:e1485 doi: 10.1038/cddis.2014.453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lopez JJ, Laham RJ, Stamler A, Pearlman JD, Bunting S, Kaplan A, Carrozza JP, Sellke FW, Simons M (1998) VEGF administration in chronic myocardial ischemia in pigs. Cardiovasc Res 40:272–281 do: 10.1016/s0008-6363(98)00136-9 [DOI] [PubMed] [Google Scholar]
- 30.Marijianowski MM, Teeling P, Becker AE (1997) Remodeling after myocardial infarction in humans is not associated with interstitial fibrosis of noninfarcted myocardium. J Am Coll Cardiol 30:76–82 doi: 10.1016/s0735-1097(97)00100-9 [DOI] [PubMed] [Google Scholar]
- 31.Mastri M, Shah Z, Hsieh K, Wang X, Wooldridge B, Martin S, Suzuki G, Lee T (2014) Secreted Frizzled-related protein 2 as a target in antifibrotic therapeutic intervention. Am J Physiol Cell Physiol 306:C531–539 doi: 10.1152/ajpcell.00238.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mii Y, Taira M (2011) Secreted Wnt “inhibitors” are not just inhibitors: regulation of extracellular Wnt by secreted Frizzled-related proteins. Dev Growth Differ 53:911–923 doi: 10.1111/j.1440-169X.2011.01299.x [DOI] [PubMed] [Google Scholar]
- 33.Okayama S, Nakano T, Uemura S, Fujimoto S, Somekawa S, Watanabe M, Nakajima T, Saito Y (2013) Evaluation of left ventricular diastolic function by fractional area change using cine cardiovascular magnetic resonance: a feasibility study. J Cardiovasc Magn Reson 15:87 doi: 10.1186/1532-429X-15-87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pearlman JD, Hibberd MG, Chuang ML, Harada K, Lopez JJ, Gladstone SR, Friedman M, Sellke FW, Simons M (1995) Magnetic resonance mapping demonstrates benefits of VEGF-induced myocardial angiogenesis. Nat Med 1:1085–1089 DOI: 10.1038/nm1095-1085 [DOI] [PubMed] [Google Scholar]
- 35.Peter PS, Brady JE, Yan L, Chen W, Engelhardt S, Wang Y, Sadoshima J, Vatner SF, Vatner DE (2007) Inhibition of p38 alpha MAPK rescues cardiomyopathy induced by overexpressed beta 2-adrenergic receptor, but not beta 1-adrenergic receptor. J Clin Invest 117:1335–1343 DOI: 10.1172/JCI29576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pomduk K, Kheolamai P, Y UP, Wattanapanitch M, Klincumhom N, Issaragrisil S (2015) Enhanced human mesenchymal stem cell survival under oxidative stress by overexpression of secreted frizzled-related protein 2 gene. Ann Hematol 94:319–327 doi: 10.1007/s00277-014-2210-1 [DOI] [PubMed] [Google Scholar]
- 37.Qiu H, Tian B, Resuello RG, Natividad FF, Peppas A, Shen YT, Vatner DE, Vatner SF, Depre C (2007) Sex-specific regulation of gene expression in the aging monkey aorta. Physiol Genomics 29:169–180. DOI: 10.1152/physiolgenomics.00229.2006 [DOI] [PubMed] [Google Scholar]
- 38.Sahn DJ, DeMaria A, Kisslo J, Weyman A (1978) Recommendations regarding quantitation in M-mode echocardiography: results of a survey of echocardiographic measurements. Circulation 58:1072–1083. DOI: 10.1161/01.cir.58.6.1072 [DOI] [PubMed] [Google Scholar]
- 39.Shen YT, Depre C, Yan L, Park JY, Tian B, Jain K, Chen L, Zhang Y, Kudej RK, Zhao X, Sadoshima J, Vatner DE, Vatner SF (2008) Repetitive ischemia by coronary stenosis induces a novel window of ischemic preconditioning. Circulation 118:1961–1969 doi: 10.1161/CIRCULATIONAHA.108.788240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Siamakpour-Reihani S, Caster J, Bandhu Nepal D, Courtwright A, Hilliard E, Usary J, Ketelsen D, Darr D, Shen XJ, Patterson C, Klauber-Demore N (2011) The role of calcineurin/NFAT in SFRP2 induced angiogenesis--a rationale for breast cancer treatment with the calcineurin inhibitor tacrolimus. PloS one 6:e20412 doi: 10.1371/journal.pone.0020412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yuan C, Yan L, Solanki P, Vatner SF, Vatner DE, Schwarz MA (2015) Blockade of EMAP II protects cardiac function after chronic myocardial infarction by inducing angiogenesis. J Mol Cell Cardiol 79:224–231 doi: 10.1016/j.yjmcc.2014.11.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang HF, Wang YL, Tan YZ, Wang HJ, Tao P, Zhou P (2019) Enhancement of cardiac lymphangiogenesis by transplantation of CD34(+)VEGFR-3(+) endothelial progenitor cells and sustained release of VEGF-C. Basic Res Cardiol 114:43 doi: 10.1007/s00395-019-0752-z [DOI] [PMC free article] [PubMed] [Google Scholar]