

Abstract
Depression is a common condition but current treatments for depression are only effective in a subset of individuals. To identify novel treatment targets, we integrated depression GWAS results (N=500,199) with human brain proteomes (N=376) to perform a proteome-wide association study (PWAS) of depression, followed by Mendelian randomization. We identified 19 genes consistent with being causal in depression, acting via their cis-regulated brain protein abundance. We replicated 9 of these genes using an independent depression GWAS (N=307,353) and human brain proteomic dataset (N=152). Eleven of these 19 genes also had their cis-regulated mRNA levels associated with depression based on integration of the depression GWAS with human brain transcriptomes (N=888). Meta-analysis of the discovery and replication PWAS identified 25 brain proteins consistent with being causal in depression, and 20 were not previously implicated in depression by GWAS. Together, these findings provide novel promising brain protein targets for further mechanistic and therapeutic studies.
Depression is a common mental illness and a leading cause of disability worldwide1,2. Current treatments for depression, however, are ineffective in a large subset of the patients3–5. Hence, we sought to identify potential causal brain proteins in depression pathogenesis to support the development of novel therapeutics.
We investigated brain proteins for potential drug targets for several reasons. First, proteins are the final products of gene expression and the main functional components of cells and biological processes. Second, most drug targets and biomarkers are proteins6,7. Third, depression studies have examined genetic, epigenetic, and transcriptomic factors, but far fewer studies have examined brain proteins directly5,8–10. Indeed, genes undergo regulation at the post-transcriptional, translational, and post-translational levels11,12, which highlights the need to examine brain proteins directly.
To identify potentially causal brain proteins in depression, we hypothesized that specific genetic variants influence depression by altering brain protein expression levels. Two recent advances made testing this hypothesis feasible. The first is the advent of high throughput proteomic sequencing of complex tissues that has enabled large-scale quantification of human brain proteins13–15. The second are new frameworks that integrate gene expression and results of genome-wide association studies (GWAS) as implemented in functional summary-based imputation (FUSION)16,17 and summary data-based Mendelian randomization (SMR)18. Simply put, FUSION identifies genes whose cis-regulated brain protein abundance is associated with depression, and SMR tests whether these brain proteins mediate the association between genetic variants and depression. The inference is that the identified genes contribute to depression pathogenesis through modulating brain protein abundance16,18. The causal inference of this integrative analytical approach has been experimentally tested and found to be robust17,19,20.
We applied these analytic approaches to a discovery dataset consisting of human brain proteomic and genetic data from 376 individuals13 and the latest depression GWAS by Howard et al. (N=500,199)10. Additionally, we performed a replication analysis in an independent set of human brain proteomic and genetic data (N=152) and depression GWAS in 23andMe participants from Hyde et al21 (N=307,353). Together, we presented here the first proteome-wide association study (PWAS) of depression to identify specific brain proteins as potential treatment targets for depression.
Results
Discovery PWAS of depression
We integrated reference human brain proteomes with the latest depression GWAS results10 to perform a PWAS of depression using the FUSION pipeline16. The human brain proteomes were generated from the dorsolateral prefrontal cortex (dPFC) of 376 participants of European descent of the Religious Order Study and Rush Memory and Aging Project (ROS/MAP)13,22. They included 8356 proteins after quality control and 1468 of these had significant SNP-based heritability (p-value <0.01) and were included in the PWAS. The depression GWAS summary statistics were from 500,199 participants of European descent that were not 23andMe,Inc. participants from the latest Howard et al depression GWAS10. The discovery PWAS identified 24 genes whose cis-regulated brain protein abundances were associated with depression at FDR q<0.05 (Figure 1, Table 1, Supplementary Table 1). Each of these 24 genes is located >500 kb apart.
Figure 1.
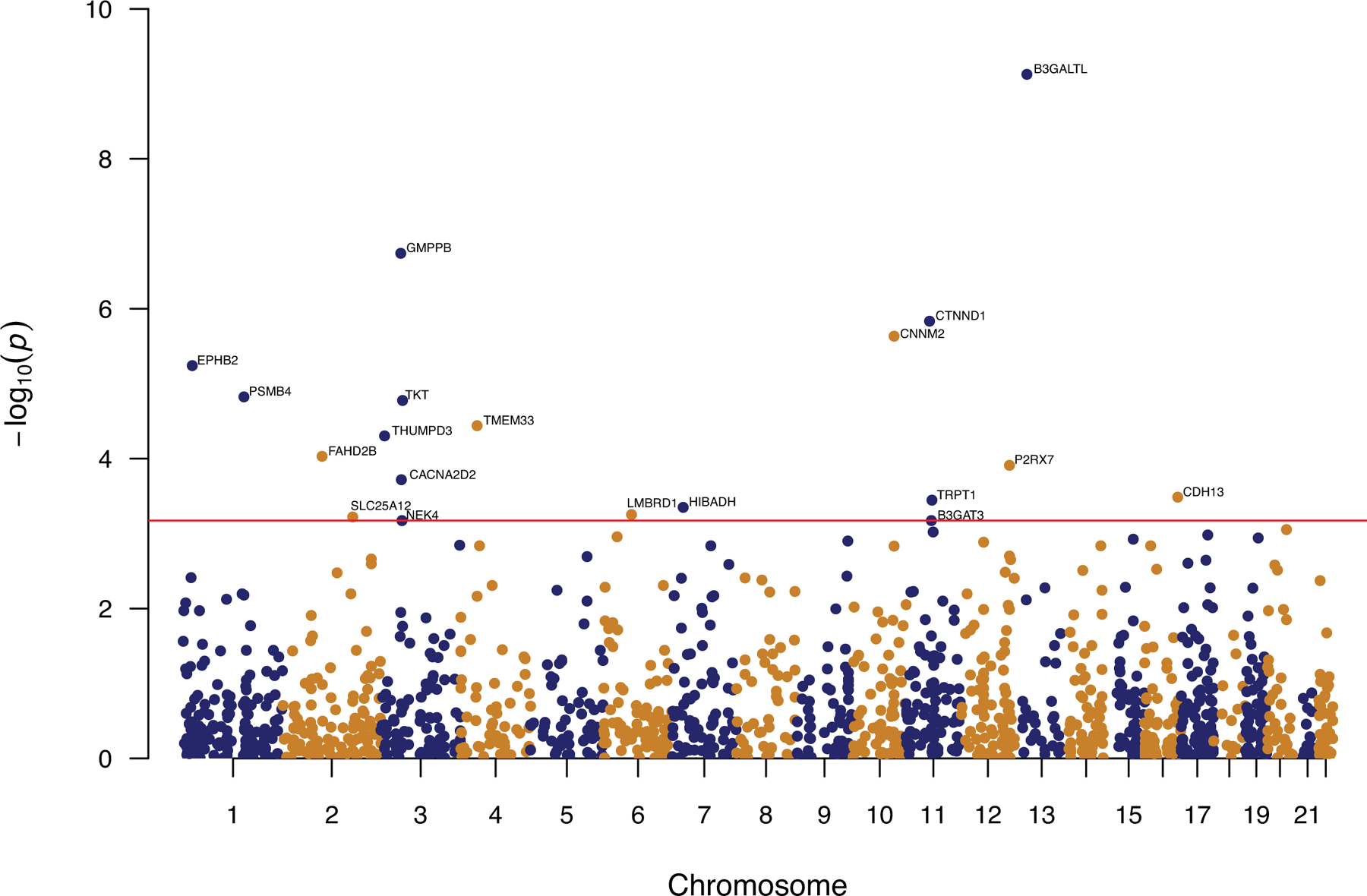
The discovery PWAS identified 19 proteins consistent with being causal in depression.
This figure shows the Manhattan plot of the 19 proteins identified in the discovery PWAS of depression using FUSION, followed by SMR and HEIDI. These 19 genes likely contribute to depression pathogenesis via their cis-regulated brain protein abundances.
Table 1.
Results of the discovery PWAS of depression in which the depression GWAS (N=500,199) summary statistics were integrated with the ROS/MAP human brain proteomic and genetic data (N = 376) using the FUSION pipeline, followed by SMR and HEIDI tests. Together, these analyses identified 19 genes that are consistent with being causal in depression. These genes contribute to depression pathogenesis via their cis-regulated brain protein abundance.
| Gene | Chr | PWAS Z | PWAS p |
PWAS FDR q |
SMR p |
HEIDI p |
Causal gene | |
|---|---|---|---|---|---|---|---|---|
| 1 | B3GALTL* | 13 | −6.16 | 7.4E-10 | 1.1E-06 | 6.1E-06 | 0.13 | consistent |
| 2 | GMPPB* | 3 | 5.22 | 1.8E-07 | 8.9E-05 | 4.9E-06 | 0.28 | consistent |
| 3 | CTNND1 | 11 | −4.82 | 1.5E-06 | 5.3E-04 | 3.7E-03 | 0.69 | consistent |
| 4 | CNNM2* | 10 | −4.72 | 2.3E-06 | 6.8E-04 | 2.2E-04 | 0.21 | consistent |
| 5 | EPHB2* | 1 | −4.54 | 5.8E-06 | 1.4E-03 | 2.3E-03 | 0.40 | consistent |
| 6 | PSMB4 | 1 | 4.33 | 1.5E-05 | 3.0E-03 | 2.7E-05 | 0.27 | consistent |
| 7 | TKT* | 3 | 4.31 | 1.7E-05 | 3.0E-03 | 2.3E-03 | 0.37 | consistent |
| 8 | TMEM33 | 4 | 4.13 | 3.6E-05 | 5.9E-03 | 2.3E-02 | 0.18 | consistent |
| 9 | THUMPD3* | 3 | 4.06 | 5.0E-05 | 7.3E-03 | 7.2E-03 | 0.21 | consistent |
| 10 | FAHD2B* | 2 | −3.91 | 9.4E-05 | 1.1E-02 | 2.1E-03 | 0.32 | consistent |
| 11 | P2RX7 | 12 | −3.84 | 1.2E-04 | 1.4E-02 | 9.1E-05 | 0.06 | consistent |
| 12 | CACNA2D2 | 3 | −3.73 | 1.9E-04 | 2.0E-02 | 1.4E-02 | 0.55 | consistent |
| 13 | CDH13* | 16 | −3.59 | 3.3E-04 | 3.1E-02 | 3.5E-03 | 0.13 | consistent |
| 14 | TRPT1 | 11 | −3.57 | 3.6E-04 | 3.1E-02 | 2.8E-02 | 0.77 | consistent |
| 15 | HIBADH* | 7 | 3.51 | 4.5E-04 | 3.6E-02 | 9.8E-03 | 0.93 | consistent |
| 16 | LMBRD1 | 6 | −3.45 | 5.6E-04 | 4.0E-02 | 1.6E-02 | 0.69 | consistent |
| 17 | SLC25A12* | 2 | 3.43 | 6.0E-04 | 4.1E-02 | 4.2E-03 | 1.00 | consistent |
| 18 | B3GAT3 | 11 | −3.40 | 6.7E-04 | 4.1E-02 | 5.4E-03 | 0.92 | consistent |
| 19 | NEK4* | 3 | 3.40 | 6.7E-04 | 4.1E-02 | 4.2E-03 | 0.11 | consistent |
| 20 | TMEM25 | 11 | −3.48 | 5.0E-04 | 3.8E-02 | 2.8E-03 | NA | possible |
| 21 | RAB27B | 18 | 5.74 | 9.7E-09 | 7.1E-06 | 4.5E-06 | 0.05 | no |
| 22 | TMEM106B | 7 | 4.02 | 5.9E-05 | 7.9E-03 | 5.8E-04 | 0.01 | no |
| 23 | FAM172A | 5 | −3.56 | 3.7E-04 | 3.1E-02 | 1.4E-02 | 0.02 | no |
| 24 | RGS6 | 14 | −3.41 | 6.5E-04 | 4.1E-02 | 1.4E-02 | 0.03 | no |
Asterisk indicates genes whose cis-regulated brain mRNA levels were associated with depression based on a TWAS of depression that integrated the depression GWAS (N=500,199) with brain transcriptomic and genetic data (N=888). Chr: chromosome. SMR: summary data-level Mendelian randomization. HEIDI: Heterogeneity in dependent instrument. NA means missing value since there were not sufficient SNPs to run the HEIDI test.
To further evaluate whether the cis-regulated brain protein expression mediates the association between the genetic variants and depression for each of these 24 genes, we applied summary data-based Mendelian randomization (SMR)18 on the same discovery dataset. Results from SMR suggest that was the case for all 24 genes, at least at nominally significant level, and for 5 genes at FDR q<0.05 (Table 1; Supplementary Table 2). Next, we performed the Heterogeneity in Dependent Instrument (HEIDI)18 test to distinguish pleiotropy/causality from linkage for these 24 genes. HEIDI results suggest that 4 of the 24 genes were likely significant due to linkage disequilibrium, 1 was undetermined, and 19 were consistent with either pleiotropy or causality (Table 1, Supplementary Table 2). Taken together, results from FUSION, SMR, and HEIDI suggest that 19 genes likely contribute to depression pathogenesis via their cis-regulated brain protein abundance (Table 1).
Replication PWAS of depression
To increase confidence in our findings, we performed a replication PWAS of depression using proteomes and GWAS not included in our discovery analysis. The replication human brain proteomes were generated from the dPFC of 152 participants of European descent recruited by Banner Sun Health Research Institute23. They included 8,168 proteins after quality control and 1,139 of these had significant SNP-based heritability (p-value <0.01) and were included in the replication PWAS. Notably, there was a high degree of reproducibility of the protein weights estimated by FUSION between the discovery and replication proteomic datasets with a mean correlation of 0.79 and median of 0.85 (Supplementary Table 3). We integrated the replication proteomes with an independent depression GWAS of 23andMe participants of European descent (N=307,354) from Hyde et al21 to perform a replication PWAS of depression using FUSION (Supplementary Table 4). Due to the stochastic nature of high throughput proteomic sequencing, the replication proteomes profiled 17 of the 19 proteins identified in the discovery PWAS. However, only 13 of these 17 proteins were heritable, with SNP-based heritability p<0.01, and were part of the replication PWAS (Table 2). A hypothesis-driven meta-analysis for these 13 genes using data from the discovery and replication PWAS revealed evidence for replication for 9 of the 13 genes (69% replication rate; Table 2, Supplementary Table 4). Here, replication was declared when the meta-analysis p-value was smaller than both of the p-values from the discovery and replication PWAS.
gTable 2:
Results of the replication PWAS and meta-analysis of the discovery and replication PWAS. A replication PWAS was performed using an independent brain proteomic dataset (n=152) and an independent depression GWAS (N=307,353 23andMe participants; Supplementary Table 4). Focusing on the 19 FDR-significant genes identified by the discovery PWAS and consistent with being causal by SMR, 13 were found in the replication PWAS and a meta-analysis was performed. Replication was declared if the unadjusted meta-analysis p-value was smaller than both the unadjusted discovery and replication PWAS p-values and direction of effect was consistent between the discovery and replication PWAS results. Unadjusted p-values for the discovery, replication, and meta-analysis PWAS results are given. The direction of effect for the discovery and replication interpretation are given. Based on the replication criteria, 9 of the 13 genes (69%) showed evidence for replication.
| Gene | Chr | Discovery PWAS p |
Replication PWAS p |
meta-analysis p-value | Direction | Replicated | |
|---|---|---|---|---|---|---|---|
| 1 | CTNND1 | 11 | 1.5E-06 | 7.5E-04 | 4.1E-09 | −− | yes |
| 2 | CNNM2* | 10 | 2.3E-06 | 1.1E-02 | 1.0E-07 | −− | yes |
| 3 | P2RX7 | 12 | 1.2E-04 | 5.4E-04 | 2.8E-07 | −− | yes |
| 4 | PSMB4 | 1 | 1.5E-05 | 4.8E-02 | 2.9E-06 | ++ | yes |
| 5 | FAHD2B* | 2 | 9.4E-05 | 2.6E-02 | 7.6E-06 | −− | yes |
| 6 | HIBADH* | 7 | 4.5E-04 | 5.4E-02 | 6.8E-05 | ++ | yes |
| 7 | CACNA2D2 | 3 | 1.9E-04 | 1.1E-01 | 6.9E-05 | −− | yes |
| 8 | SLC25A12* | 2 | 6.0E-04 | 4.6E-02 | 7.7E-05 | ++ | yes |
| 9 | CDH13* | 16 | 3.3E-04 | 8.0E-02 | 8.0E-05 | −− | yes |
| 10 | B3GALTL* | 13 | 7.4E-10 | 4.1E-01 | 3.7E-08 | −− | no |
| 11 | GMPPB* | 3 | 1.8E-07 | 7.0E-01 | 7.2E-06 | ++ | no |
| 12 | THUMPD3* | 3 | 5.0E-05 | 9.0E-01 | 7.1E-04 | ++ | no |
| 13 | B3GAT3 | 11 | 6.7E-04 | 6.3E-01 | 1.2E-02 | −+ | no |
Asterisk indicates genes whose cis-regulated brain mRNA levels were associated with depression from a TWAS of depression integrating the depression GWAS (N=500,199) with transcriptomic and genetic data (N=888) using FUSION. Direction refers to the direction of association between the cis-regulated protein level and depression. Replicated refers to whether the gene fulfills criteria for replication, which are an unadjusted meta-analysis p-value smaller than both the unadjusted discovery and replication PWAS p-values and direction of effect was consistent between the discovery and replication PWAS results.
Meta-analysis of the discovery and replication PWAS of depression
A meta-analysis of the discovery and replication PWAS yielded 38 proteins associated with depression at FDR q <0.05 among the 798 proteins that were detected and heritable in both datasets (Supplementary Table 5). Among these 38, 25 were consistent with being causal based on SMR and HEIDI results (Table 3, Supplementary Table 6), and 12 of these proteins were part of the 19 potential causal proteins from the discovery PWAS.
Table 3:
Meta-analysis of the discovery and replication PWAS of depression yielded 38 proteins associated with depression at FDR q < 0.05 among the 798 proteins common between the two PWAS. Among these 38 proteins, 25 were consistent with being causal based on SMR/HEIDI tests and are shown here.
| Discovery PWAS p | Replication PWAS p | Z score | Meta-analysis p | Direction | Meta-analysis FDR q | SMR p | HEIDI p | Causal | |
|---|---|---|---|---|---|---|---|---|---|
| CTNND1 | 1.5E-06 | 7.5E-04 | −5.9 | 4.1E-09 | −− | 7.4E-06 | 2.6E-04 | 0.89 | yes |
| RAB27B | 9.7E-09 | 7.0E-02 | 5.7 | 1.1E-08 | ++ | 9.4E-06 | 9.0E-07 | 0.14 | yes |
| B3GALTL | 7.4E-10 | 4.1E-01 | −5.5 | 3.9E-08 | −− | 2.3E-05 | 1.6E-06 | 0.19 | yes |
| CNNM2 | 2.3E-06 | 1.1E-02 | −5.3 | 1.0E-07 | −− | 4.6E-05 | 1.8E-05 | 0.59 | yes |
| P2RX7 | 1.2E-04 | 5.4E-04 | −5.1 | 2.8E-07 | −− | 8.4E-05 | 2.5E-06 | 0.31 | yes |
| PSMB4 | 1.5E-05 | 4.8E-02 | 4.7 | 3.0E-06 | ++ | 7.6E-04 | 2.9E-06 | 0.63 | yes |
| GMPPB | 1.8E-07 | 7.0E-01 | 4.5 | 7.6E-06 | ++ | 1.4E-03 | 3.8E-05 | 0.37 | yes |
| FAHD2B | 9.4E-05 | 2.6E-02 | −4.5 | 7.6E-06 | −− | 1.4E-03 | 1.7E-03 | 0.08 | yes |
| PPP3CC | 3.9E-03 | 1.3E-03 | 4.2 | 2.5E-05 | ++ | 3.8E-03 | 6.5E-05 | 0.38 | yes |
| ABCA5 | 1.0E-03 | 1.2E-02 | 4.1 | 3.7E-05 | ++ | 4.7E-03 | 1.7E-04 | 0.93 | yes |
| CSE1L | 1.4E-02 | 4.0E-04 | −4.1 | 5.0E-05 | −− | 5.9E-03 | 6.3E-03 | 0.43 | yes |
| HIBADH | 4.5E-04 | 5.4E-02 | 4.0 | 6.9E-05 | ++ | 6.9E-03 | 3.3E-03 | 0.94 | yes |
| CACNA2D2 | 1.9E-04 | 1.1E-01 | −4.0 | 7.0E-05 | −− | 6.9E-03 | 3.9E-03 | 0.90 | yes |
| SLC25A12 | 6.0E-04 | 4.6E-02 | 4.0 | 7.7E-05 | ++ | 7.2E-03 | 7.5E-04 | 0.99 | yes |
| LYRM4 | 5.2E-03 | 7.8E-03 | 3.8 | 1.3E-04 | ++ | 1.0E-02 | 2.2E-04 | 0.69 | yes |
| CCDC92 | 2.2E-03 | 2.6E-02 | −3.8 | 1.6E-04 | −− | 1.2E-02 | 8.7E-04 | 0.16 | yes |
| LRP4 | 1.4E-02 | 9.8E-03 | −3.5 | 4.7E-04 | −− | 2.8E-02 | 1.5E-03 | 0.29 | yes |
| MKRN1 | 2.6E-03 | 8.3E-02 | 3.5 | 5.4E-04 | ++ | 3.0E-02 | 1.4E-03 | 0.72 | yes |
| THUMPD3 | 5.0E-05 | 9.0E-01 | 3.4 | 7.4E-04 | ++ | 3.4E-02 | 1.5E-03 | 0.78 | yes |
| CCS | 9.5E-04 | 2.8E-01 | 3.3 | 9.1E-04 | ++ | 3.9E-02 | 1.8E-02 | 0.31 | yes |
| ADCY3 | 3.7E-02 | 5.5E-03 | −3.3 | 9.3E-04 | −− | 3.9E-02 | 6.0E-03 | 0.77 | yes |
| NFXL1 | 1.5E-03 | 2.6E-01 | −3.2 | 1.2E-03 | −− | 4.4E-02 | 1.2E-03 | 0.65 | yes |
| GDI2 | 9.5E-03 | 5.7E-02 | −3.2 | 1.3E-03 | −− | 4.6E-02 | 1.9E-02 | 0.90 | yes |
| PDXDC1 | 1.5E-03 | 2.9E-01 | −3.2 | 1.4E-03 | −− | 4.8E-02 | 1.2E-02 | 0.49 | yes |
| ATG7 | 3.6E-01 | 3.0E-05 | 3.2 | 1.5E-03 | ++ | 4.9E-02 | 4.7E-03 | 0.88 | yes |
To investigate the connectivity among the 25 depression causal proteins from the meta-analysis, we used GeNets, a web platform for network-based genomic analyses24. We found two protein communities based on protein-protein interaction (PPI; Figure 2). A community is a set of proteins that are more connected to one another than they are to other groups of proteins24. The first community included CSE1L, CTNND1, SLC25A12, and PSMB4, and the second community LYRM4 and GMPPB proteins (Figure 2). GeNets also enables gene set enrichment analysis on genes within a PPI network using the canonical pathways from gene sets in the MSigDB25. We found that these 25 causal genes were enriched for genes involved in amyotrophic lateral sclerosis (CCS, PPP3CC), calcium signaling (PPP3CC, P2RX7, ADCY3), dilated cardiomyopathy (CACNA2D2, ADCY3), oocyte meiosis (ADCY3, PPP3CC) and metabolism of amino acids and derivatives (HIBADH, PSMB4; Figure 2).
Figure 2:
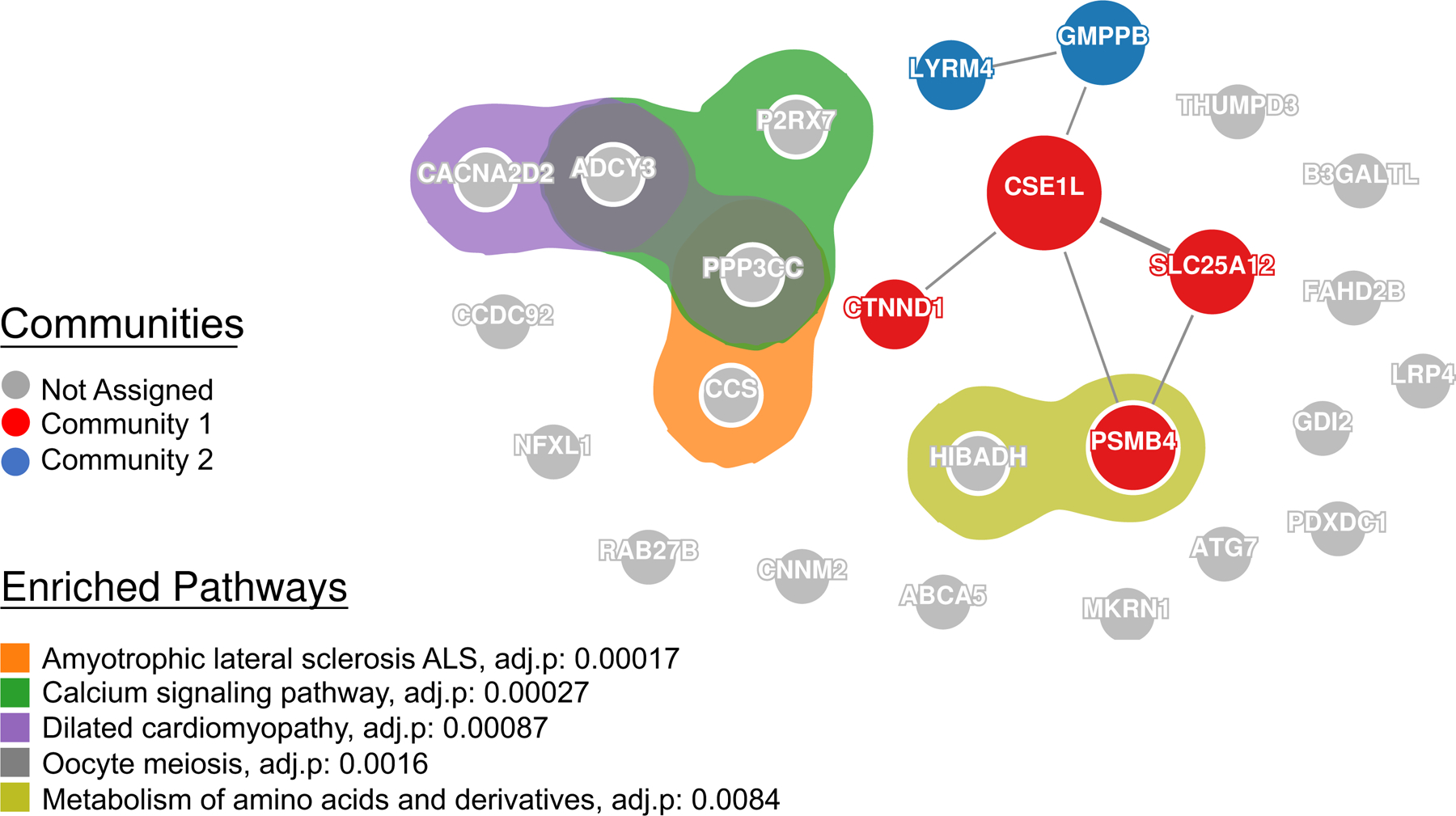
Protein-protein interaction (PPI) network and pathways among the 25 potentially causal proteins in depression from the meta-analysis of the discovery and replication PWAS of depression. The lines represent physical PPI. The thickness of the lines is proportional to the evidence for the PPI. Community 1 includes CTNND1, CSE1L, SLC25A12, and PSMB4. Community 2 includes LYRM4 and GMPPB. Enrichment of pathways was determined using a hypergenometric test with bonforroni adjustment for for multiple testing correction.
Specificity of the depression PWAS results
To understand the specificity of the depression PWAS results, we performed a PWAS for neuroticism, body mass index (BMI), and waist-to-hip ratio adjusting for BMI (WHRadjBMI). These traits were chosen because they have a range of estimated genetic correlations with depression – 0.7 for neuroticism, 0.09 for BMI, and 0.12 for WHR26. Thus, we expected that traits with evidence of higher genetic correlation would have more PWAS results in common. We used the GWAS results for neuroticism (N=390,278)27, BMI (N=681,275)28, and WHRadjBMI (N=694,649)29 from studies examining individuals of European descent and the discovery proteomic profiles (n=376) to perform a PWAS for each trait. Using FUSION, the PWAS of neuroticism identified 72 genes, of BMI identified 395 genes, and of WHRadjBMI identified 244 genes at FDR q<0.05 (Supplementary Tables 7–9). Next, as was done for the analysis of depression, we applied SMR and HEIDI on the PWAS significant genes to remove genes with SMR p ≥ 0.05, HEIDI p ≤ 0.05, or in cases where HEIDI p was unable to be determined. The goal of these additional tests is to focus on genes with evidence that their genetically regulated protein abundance mediates their association with the trait of interest and to remove genes likely to be the result of linkage disequilibrium. After considering findings from FUSION, SMR, and HEIDI, we identified 46 genes in neuroticism, 216 genes in BMI, and 117 genes in WHRadjBMI that likely contribute to these traits by modulating their brain protein abundance (Supplementary Tables 10–12). As expected, 11 of 46 (24%) of the neuroticism genes were also identified by the depression PWAS, which reflects their high degree of genetic correlation. By contrast, 4 of 216 (2%) of the BMI genes and 4 of 117 (3%) of the WHRadjBMI genes overlapped with the 19 depression PWAS-significant genes.
Examination of the potential depression-causal proteins at the mRNA level
To provide another layer of insight into the results of the 19 proteins identified by PWAS, we asked whether the cis-regulated mRNA levels of these genes were also associated with depression in a transcriptome-wide association study (TWAS) of depression using 888 reference human brain transcriptomes and the discovery depression GWAS (N=500,199). The brain transcriptomes were predominantly profiled from the frontal cortex of post-mortem brain samples of 783 participants of European descent from ROS/MAP, Mayo, and Mount Sinai Brain Bank studies30. Of the 13,650 mRNAs that passed quality control, 6735 had significant SNP-based heritability and were included in the TWAS. The TWAS of depression using FUSION identified 73 genes whose cis-regulated brain mRNA expression was associated with depression at FDR q<0.05 (Supplementary Table 13). Among these, 47 genes passed both the SMR (i.e., SMR p<0.05) and HEIDI test (i.e., HEIDI p ≥ 0.05; Supplementary Table 14). These 47 genes contribute to depression pathogenesis by modulating their genetically controlled brain mRNA expression.
All the 19 proteins identified in the discovery depression PWAS were profiled at the mRNA-level but only 14 had significant SNP-based heritability estimates for mRNAs (Supplementary Table 13). The TWAS using FUSION found that 13 of these 14 genes had their cis-regulated mRNA levels associated with depression at nominal significance level, and 11 of these 13 had consistent directions of effect for the mRNA and protein (Table 1; Supplementary Table 13a). All 11 genes passed SMR test but only 10 passed HEIDI test. CNNM2 did not pass the HEIDI test (HEIDI p=0.033). For the 11 genes with evidence for association with depression at both the mRNA and protein levels, SMR for two molecular traits31 (i.e., mRNA and protein) suggests that 9 of these 11 genes regulate brain protein abundance via their regulation of brain mRNA expression level (Supplementary Table 15).
Furthermore, among these 19 depression genes, 7 did not have evidence for association with depression at the mRNA-level in the TWAS, including those not heritable and thus not included in the TWAS (Table 1, genes without asterisk). Interestingly, 4 of the these 7 were significant in the discovery PWAS and were replicated (CTNND1, PSMB4, P2RX7, and CACNA2D2, Table 2) suggesting that the PWAS contribute new insights into the pathogenesis of depression beyond the TWAS.
Cell-type enrichment of expression of potential depression-causal genes
We asked whether the 19 genes identified by PWAS were enriched in a particular brain cell-type. Using human single-cell RNA sequencing data profiled from the dPFC32, we found that 10 of these 19 genes showed enrichment in specific cell types at FDR q <0.05 (adjusted for all 17,775 genes; Figure 2a; Supplementary Table 16a). Five genes were highly expressed in inhibitory neurons (CACNA2D2, CHD13, CNNM2, NEK4, SLC25A12) and three had enriched expression in excitatory neurons, albeit to a lesser degree than inhibitory neurons (CHD13, CNNM2, SLC25A12). To determine the specificity of this finding, we compared the enrichment of genes identified for BMI, WHR, and neuroticism for enrichment in inhibitory neurons. We found that depression genes were significantly enriched for inhibitory neurons compared to those for BMI (odds ratio (OR) = 5.6, p-value = 0.013) and WHR (OR = 9.5, p-value 0.004) but not to those for neuroticism (OR = 2.1, p-value = 0.450). Additionally, three genes were highly expressed in astrocytes (CTNND1, TKT, and TRPT1), one gene in oligodendrocytes (P2RX7), and one gene in microglia (LMBRD1; Figure 3a).
Figure 3:
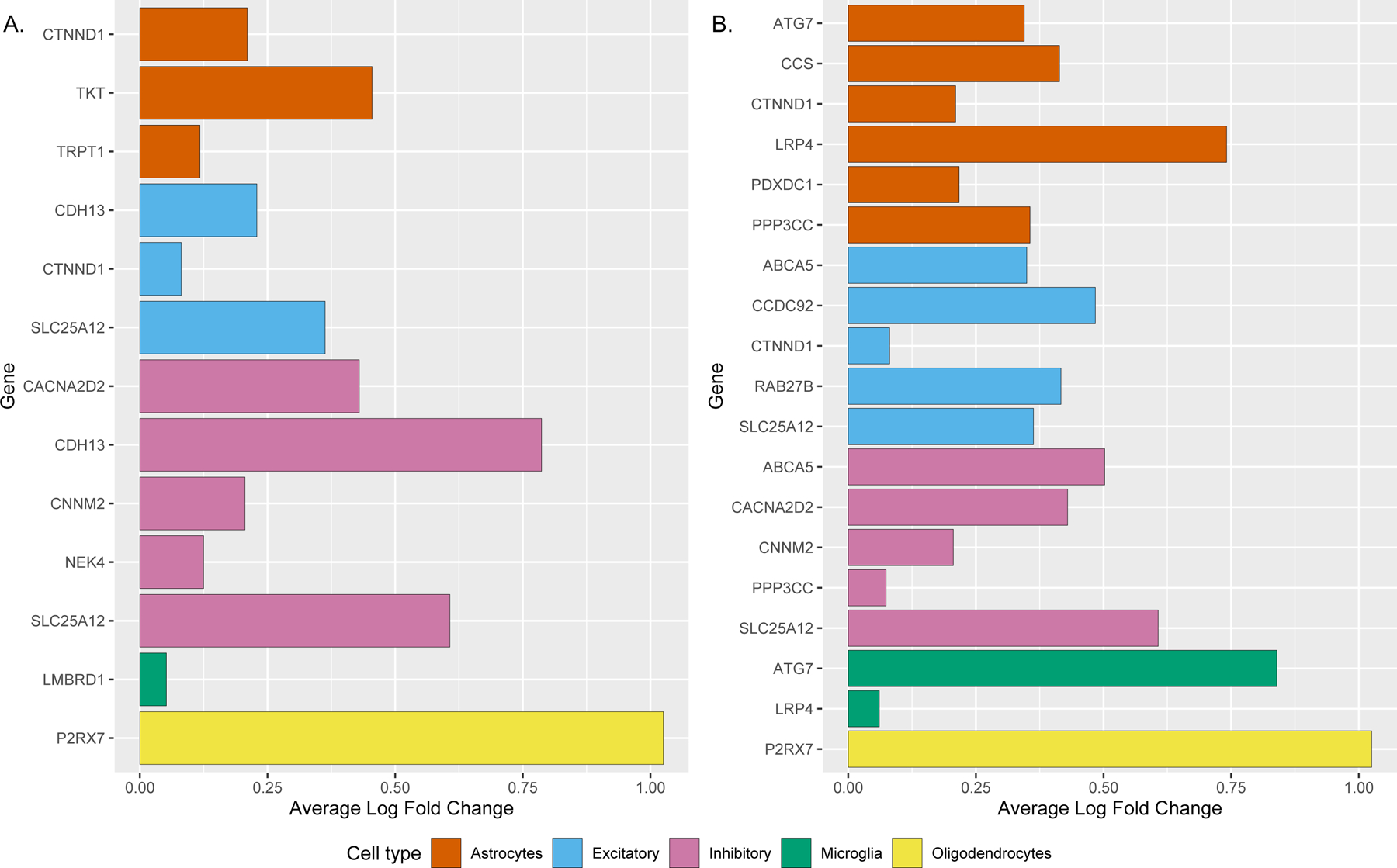
Bar graph of single-cell type enrichment for the A. Causal genes in depression from the discovery PWAS (n=19), and B. Causal genes in depression from the meta-analysis of the discovery and replication PWAS (n=25). The plot shows the average log fold change (x-axis) for each gene (y-axis) with evidence of significant enrichment within a particular brain cell-type (color of bar). A gene can be enriched in more than one cell type and only positive log fold change is plotted for simplicity. Enrichment is tested based on expression of the gene in a particular cell type versus in all other cell types using Wilcoxon rank sum test adjusted for 17,775 genes. The underlying data were from human brain single nuclei RNA-sequencing from the dPFC, and the full statistics are presented in Supplementary Table 16.
In addition, among the 25 potentially causal genes from the depression PWAS meta-analysis, 13 were enriched in one or more cell types – five in excitatory neurons, five in inhibitory neurons, six in astrocytes, two in microglia, and one in oligodendrocytes (Figure 3b; Supplementary Table 16b).
Novelty of the protein findings
To determine the novelty of the 25 potentially causal genes identified from the meta-analysis of the discovery and replication PWAS of depression, we identified the lowest p-values for the SNPs within 1 Mb of each of these 25 genes using the summary statistics from the largest depression GWAS (N=807,553)10. For five genes (RAB27B, P2RX7, B3GALTL, GMPPB, and CTNND1), the lowest p-values were less than 5×10−08, while the remaining 20 genes had SNPs with p-values ranging from 5.4×10−04 to 2.6×10−07 (Supplementary Table 17), implying genes not implicated in depression by GWAS. These findings are consistent with observations from other TWAS studies that found the novel genes to be from regions below genome-wide significant p-values17,33,34. Furthermore, the PWAS findings point to specific brain proteins that likely contribute to the pathogenesis of depression.
Discussion
In this study we sought to identify brain proteins that contribute to the pathogenesis of depression in order to find potential novel treatment targets for depression. We identified 19 potentially causal genes that act via modulating their brain protein abundances. Nine of these 19 genes were replicated in an independent PWAS of depression, providing a higher level of confidence in them. Among these 9 replicated genes, five (CNNM2, FAHD2B, HIBADH, SLC25A12, and CDH13) also had their cis-regulated mRNA levels associated with depression by TWAS, highlighting the consistent findings at both the mRNA and protein levels. Notably, 4 of these 9 replicated genes (CTNND1, P2RX7, PSMB4, and CACNA2D2) were only identified by the PWAS but not by the TWAS, underscoring the additional insights provided by studying brain proteins directly. Lastly, through a meta-analysis of the discovery and replication PWAS, we identified 25 brain proteins that are consistent with being causal in depression pathogenesis.
Three genes (EPHB2, TKT, and NEK4) were identified by the discovery PWAS but their replicability was undetermined due to technical reasons related to the stochastic nature of proteomic sequencing. Nevertheless, they were identified by the TWAS (Table 1, genes with asterisk), and the TWAS results offers a degree of validation. In sum, we have a high level of confidence for the 9 genes that replicated in the replication PWAS and moderate level of confidence for the 3 genes that were associated with depression at both the mRNA- and protein-levels (Table 1 and 2). Together, these 12 genes are likely important in the pathogenesis of depression, acting at the brain protein level, and serving as promising targets for further mechanistic and therapeutic studies.
Among the 19 causal genes identified by the discovery PWAS of depression, some play a role at the synapse. These include synaptogenesis (B3GALTL)35,36, synaptic signaling (CTNND1)37,38, and synaptic plasticity (EPHB2 and P2RX7)39,40. Specifically, B3GALTL, also known as B3GLCT, is a glycosyltransferase that adds glycans to proteins in the process of glycosylation, an abundant post-translational modification41. Via glycosylation, B3GALTL stabilizes thrombospondin 1 (TSP-1), a protein that induces synaptogenesis35,36, implicating B3GALTL in synaptogenesis. Our depression PWAS findings suggest that participants with depression had lower abundance of B3GALTL protein in the brain, which may negatively affect the stability of TSP-1 and subsequently synaptogenesis. Consistent with our finding, TSP-1 was upregulated in rat hippocampi following treatment of depression using electroconvulsive therapy, which quickly and effectively reverses the symptoms of depression in the short-term42. Taken together, decreased B3GALTL protein may negatively affect synaptogenesis, which may contribute to the pathogenesis of depression. Regarding synaptic signaling, CTNND1 functions in the glutamatergic excitatory synaptic signaling pathway during neuronal development38. With regard to synaptic plasticity, EPHB2 is an essential component of hippocampal synaptic plasticity39, and a number of studies have found that altering expression of EPHB2 in mouse models led to depression-like behavior, memory impairment, and defects of hippocampal neurogenesis and synaptic plasticity39,43. Likewise, P2RX7 drives synapse plasticity in the learned helplessness model of depression40 and has been previously implicated in depressive disorders40.
Other notable molecular roles for the 19 causal genes in depression are magnesium homeostasis, glycosylation, neuronal apoptosis, and cell adhesion. CNNM2 plays an important role in magnesium homeostasis44, and magnesium is a key component in neuronal maturation and neuropathology via its role in cell proliferation, differentiation, survival, and neuronal network formation45. CNNM2 has also been implicated in schizophrenia and impaired brain development44,46, and our finding suggests it contributes to the pathogenesis of depression. Both B3GALTL and GMPPB implicate a role for glycosylation, a common post-translational modification. B3GALTL, as mentioned above, is a glycosyltransferase that adds glycans to proteins in glycosylation41, and GMPPB, a GDP-mannose pyrophosphorylase B, catalyzes the formation of GDP-mannose, which is required for the glycosylation of lipids and proteins47. PSMB4 is a proteasome that is responsible for protein degradation and is involved in neuronal apoptosis in neuroinflammation48. Finally, CDH13 encodes a calcium-dependent cell adhesion protein that has been implicated in major depression, bipolar disorder, schizophrenia, ADHD, and autism spectrum disorder49. Taken together, these 19 genes implicate a number of molecular pathways involved in depression, and, in particular, emphasize the role of the synapse in depression.
Drug compounds targeting 4 of these 19 genes have been in phase II or III clinical trials and include PSMB4 (rated as high confidence level in our findings) for glioblastoma multiforme (2 drugs, phase III), P2RX7 (high confidence level) for rheumatoid arthritis and osteoarthritis (3 drugs, phase II), and CACNA2D2 (high confidence level) for generalized anxiety disorder, fibromyalgia, and neuropathic pain (3 drugs, phase III), and EPHB2 (moderate confidence level) for thyroid carcinoma (phase IV)50 (Supplementary Table 18). Secondary analyses of those and future compounds undergoing clinical investigations would likely prove valuable to test the hypothesis that these proteins play a causal role in development of depression.
Our findings should be interpreted in the context of its limitations. First, we are likely limited by the number of reference brain proteomes since about 18% (1468 / 8356) of the cis-regulated protein abundances could be estimated using 376 reference proteomes whereas 49% (6,735 /13,650) of the cis-regulated brain transcripts could be estimated from 888 reference transcriptomes. Nevertheless, we found 7 genes by TWAS and 1 gene by PWAS that were consistent with prior work using independent and smaller TWAS datasets26 (Supplementary Table 19). Second, we were limited by the stochastic nature of the discovery-based approach used to generate the human brain proteomes, which limited our ability to replicate 6 of the 19 proteins identified in the discovery PWAS. Both issues can be alleviated by larger brain proteomic datasets to estimate the genetic effect on protein abundance for more brain proteins. Despite these limitations, we presented results from the largest and deepest set of human brain proteomes available to our knowledge.
Our study has several strengths. First, this is the first PWAS of depression using the largest and deepest reference human proteomes and summary statistics from the latest GWAS of depression. Second, this is the first study to our knowledge that examined both the mRNA and protein levels in depression through both a PWAS and TWAS. Finally, we have a replication PWAS using an independent GWAS and independent reference human brain proteomic and genetic data.
In summary, we identified 19 genes that contribute to depression pathogenesis through modulating their brain protein abundance for future mechanistic and therapeutic studies to find effective treatments for depression.
Methods
Discovery Human Brain Proteomic and Genetic Data
The reference human brain proteomes for the discovery PWAS were profiled from the dorsolateral prefrontal cortex (dPFC) of post-mortem brain samples donated by 400 participants of European descent of the Religious Orders Study and Rush Memory and Aging Project (ROS/MAP)22. ROS/MAP participants provided informed consent, signed an Anatomic Gift Act, and a repository consent to allow their data and biospecimens to be repurposed. The studies were approved by an Institutional Review Board of Rush University Medical Center.
Proteomic profiling was performed using isobaric tandem mass tag (TMT) peptide labeling and analyzed by liquid chromatography coupled to mass spectrometry as described in detail previously13. Prior to TMT labeling, samples were randomized by co-variates (age, sex, post-mortem interval, cognitive diagnosis, and pathologies) into 50 total batches (8 samples per batch). Peptides from each individual sample (N=400) and the global internal standard (GIS; N=100) were labeled using the TMT 10-plex kit (ThermoFisher). High pH fractionation was performed as previously described with slight modifications51. Database searches and protein quantification have been described in detail here13. Briefly, all raw files were analyzed using the Proteome Discoverer suite (version 2.3 ThermoFisher Scientific) and MS2 spectra were searched against the canonical UniProtKB Human proteome database downloaded in February 2019 with 20,338 total sequences. Percolator was used to filter peptide spectral matches (PSM) and peptides to a false discovery rate (FDR) of less than 1%. Following spectral assignment, peptides were assembled into proteins, which were further filtered based on the combined probabilities of their constituent peptides to a final FDR of 1%. In cases of redundancy, shared peptides were assigned to the protein sequence based on parsimony. Reporter ions were quantified from MS2 or MS3 scans using an integration tolerance of 20 ppm with the most confident centroid setting.
The quality control of the proteomes has been described in detail previously13. Briefly, for each batch, the GIS were used to check for proteins outside of the 95% confidence interval and set to missing. Proteomic analysis in 400 subjects identified 12,691 proteins. Proteins with missing values in more than 50% of the 400 subjects were excluded. Each protein abundance was then scaled by a sample-specific total protein abundance to remove effects of protein loading differences, and then log2 transformed. Outlier samples were identified and removed through iterative principal component analysis (PCA). In each iteration, samples more than four standard deviations from the mean of either the first or second principal component were removed, and principal components were recalculated for the next iteration. A total of 9 outlier samples were removed. We used regression to remove effects of protein batch, MS2 versus MS3 reporter quantitation mode, sex, age at death, postmortem interval, study (ROS vs. MAP), and the final clinical diagnosis of cognitive status from the proteomic profile before estimating the protein weights.
Genotyping of the discovery dataset was generated by either whole genome sequencing (WGS) and/or genome-wide genotyping by either Illumina OmniQuad Express or Affymetrix GeneChip 6.0 platforms, as described52,53. We prioritized using WGS over genotyping where available and performed quality control of WGS and genotyping data separately using Plink54. We excluded individuals with genotyping missing rate >5%, variants with Hardy Weinberg equilibrium p-value < 1E-08, variants with missing genotype rate >5%, variants with minor allele frequency <1%, and variants that are not single nucleotide polymorphisms (SNPs). We used KING to remove individuals who were estimated to be closer than second degree kinship55. Genotyping for each individual was imputed to the 1000 Genome Project Phase 356 using the Michigan Imputation Server57 and SNPs with imputation R2 > 0.3 were retained. Finally, genotyping was filtered to include 1,190,321 HapMap SNPs from the 489 individuals of European descent from the 1000 Genomes Project16, which is provided by FUSION and often referred to as the linkage disequilibrium (LD) reference panel. After quality control, 376 subjects with both proteomic and genetic data were included in the discovery analyses. Of these, 262 subjects were female and mean age at death was 89 years old.
Replication Human Brain Proteomic and Genetic Data
The replication human brain proteomes were profiled from the dPFC of post-mortem brain samples donated by 198 European participants of the Banner Sun Health Research Institute (Banner). Most subjects were enrolled as cognitively unimpaired volunteers from the retirement communities of the greater Phoenix, Arizona, USA. Subjects received standardized general medical, neurological, and neuropsychological tests annually during life. A final clinicopathological diagnosis of normal cognition or Alzheimer’s disease or other dementias was rendered after review of all standardized clinical data, the most recent medical records, and neuropathological examination findings23. All enrolled subjects or their legal representatives signed an informed consent and the study was approved by the Banner Sun Health Research Institute Institutional Review Board. Only subjects with a final clinical diagnosis of normal cognition or AD were included in the proteomic analysis. Proteomic profiling was performed using the same approach as described for the discovery proteomes with two differences: only MS2 scans were obtained and MS2 spectra were searched against the UniProtKB human brain proteome database downloaded in April 2015. A total of 11,518 proteins were quantified. We used the same quality control procedures as the discovery proteomic data to remove proteins with more than 50% missing data, and identify and remove outliers, and remove the effects of clinical covariates (i.e., age, sex, the final clinical diagnosis of cognitive status) from the proteomic profile before estimating the protein weights.
Individuals from Banner were genotyped using Affymetrix Precision Medicine Array following the manufacturer’s protocol using DNA extracted from brain using Qiagen GenePure kit. We followed the same approach to quality control for the replication genotyping as was used for the discovery genotyping, including filtering based on data completeness, Hardy Weinberg equilibrium, MAF, and relatedness. Genotyping was also imputed to the 1000 Genome Project Phase 356 using the Michigan Imputation Server57 and SNPs with imputation R2 > 0.3 were retained. Finally, genotyping was filtered to only include sites on the LD reference panel provided by FUSION pipeline. After quality control, 152 subjects with proteomic and genetic data were included in our replication analyses. Among these, 87 were female and mean age at death was 85.
Brain Transcriptomic and Genetic Data
The 888 reference brain transcriptomes were profiled from post-mortem brain samples of 783 individuals of European descent recruited by the ROS/MAP, Mayo, and Mount Sinai Brain Bank studies22,58,59. The transcriptomes were mainly profiled from the dPFC and also from frontal cortex, temporal cortex, inferior frontal gyrus, superior temporal gyrus, and perirhinal gyrus. Alignment, quality control, and normalization of RNA-sequencing data have been described in detail before30. Briefly, BAM files were converted to FASTQ format using Picard v.2.2.4, followed by alignment of reads to GRCh38 reference genome using STAR v.2.460. Gene-level counts were computed using STAR. Genes with < 1 count per million in at least 50% of the samples and with missing gene length and percent GC content were removed. Outlier samples were removed. Effects of batch, sex, post-mortem interval, age at death, final diagnosis of cognitive status, and brain region were regressed from these transcriptomes. Normalized gene expression values derived from alignment to GRCh38 were assigned the lift-over to GRCh37 gene coordinates for purposes of defining 1MB cis-variant windows used to acquire variants passing quality control and filtered for those sites within the LD reference panel. This GRCh37 genome build matched variants to gene locus expression for all gene features and was used to estimate mRNA weights. After quality control, 13,650 mRNAs remained and were considered for the TWAS. Genome-wide genotypes were generated as previously described22,58,59, and quality control of the genotypes was the same as was done for the discovery dataset. Among the 783 subjects included in the analysis, 480 were female and mean age at death was 88 years old.
Depression GWAS Data
For the discovery analysis, we used summary statistics from 500,199 participants in the latest GWAS of depression by Howard and colleagues10 that did not include 23andMe participants and were of European descent. Of these, 361,315 were from the UK Biobank and 138,884 from the Psychiatric Genetics Consortium26. Approximately 34% of the participants had depression.
For the replication analysis, we used the summary statistics of the depression GWAS of 307,354 23andMe participants of European descent from Hyde et al21 provided by 23andMe research team for the replication PWAS of depression. These participants are independent from the participants of the depression GWAS10 used in the discovery analysis. About 48% of the 23andme participants were female and 24.6% had depression.
Statistical Approach
We used FUSION (downloaded from http://gusevlab.org/projects/fusion on May 2, 2019)16 to estimate protein weights in the discovery and replication datasets, separately. Briefly, we estimated SNP-based heritability for each gene using protein data. For proteins with significant SNP-based heritability (p-value <0.01), we used FUSION to compute the effect of SNPs on protein abundance using multiple predictive models (top1, blup, lasso, enet, bslmm)16, and the most predictive model was selected. For mRNA with significant SNP-based heritability (p-value <0.01), we modified how FUSION estimates mRNA weights to accommodate individuals with more than one brain region with transcriptomic data. First, the flag -scale 1 was added to handle pre-scaled expression values, as expression was scaled across individual tissues before filtering for matched genotype and combining across all tissues. Second, the family ID (FID) in the plink FAM file was used to ensure that within cross validation, all samples from the same individual were always in the same fold, and that no fold differed in size by more than 5% from any other fold. Similar to protein weights, mRNA weights were estimated using all models to train weights and the most predictive model was used. Next, FUSION combined the genetic effect of depression (i.e., depression GWAS Z-score) with the mRNA or protein expression weights by calculating the linear sum of Zscorre × weight for the independent SNPs at the locus to perform a PWAS or TWAS16.
Meta-analysis of the discovery and replication PWAS was performed using METAL61. For the hypothesis driven meta-analysis, i.e. meta-analysis of the genes found to be significant in the discovery PWAS, we declared as replicated for genes with meta-analysis p-values lower than the p-values of the discovery dataset and had the same direction of association in both datasets. For the proteome-wide meta-analysis for all genes present in both the discovery and replication PWAS, genes with FDR q <0.05 after the meta-analysis and with the same directions of association in both the discovery and replication datasets were declared replicated. The effective sample size for METAL was calculated using this formula: Neff = 2 / (1/Ncases + 1/Ncontrols)61.
Summary data-based Mendelian Randomization18 (SMR, downloaded from https://cnsgenomics.com/software/smr on May 22, 2019) was used to test whether the depression PWAS-significant genes (from the FUSION approach) were associated with depression via their cis-regulated brain protein expression. We used Plink54 to estimate protein quantitative trait loci (pQTL) in the discovery proteomic dataset by linear regression. Next, we used the pQTL results and the discovery depression GWAS summary statistics to perform SMR18. We used the conservative unadjusted p-value ≤ 0.05 from Heterogeneity in Dependent Instrument (HEIDI) to suggest that presence of linkage likely influences the main SMR findings. In addition, we performed SMR using the depression GWAS summary statistics from all 807,553 participants from Howard et al10 and the pQTL results from joining both the ROS/MAP and Banner proteomic profiles to determine mediation and pleiotropy/causality for the significant genes from the proteome-wide meta-analysis of the discovery and replication PWAS.
For genes with both mRNA and protein abundance associated with depression, we applied SMR for two molecular traits31 to the eQTL summary statistics from Siebert et al62 and pQTL summary statistics from the discovery proteomic profile to determine if cis-regulated mRNA level mediates association between SNP and cis-regulated protein levels for these genes.
Similarly, we performed SMR and HEIDI on the significant FUSION PWAS genes identified for neuroticism, BMI, and WHRadjBMI. For neuroticism, the meta-analysis GWAS summary statistics27 did not include beta and standard error, for which we calculated using the formula z / sqrt(2p(1− p)(n + z2)) and se=1/ sqrt(2p(1− p)(n + z2)), where p is allele frequency and n is sample size, as provided by SMR method paper18.
Protein-protein interaction network and pathway analysis
To investigate networks based on protein-protein interaction (PPI) among the 25 causal proteins from the meta-analysis of the depression PWAS, we used GeNets, a web platform for network-based genomic analyses24. GeNets used the PPI information from the InWeb database, a curated and computationally derived PPI network of 420,000 PPIs of high and lower probability interactions63. GeNets implements an algorithm originally presented in Clauset et al64 that identifies so-called communities in a set of genes. A community is a set of genes that are more connected to one another than they are to other groups of genes. Additionally, GeNets enables gene set enrichment analysis on genes within a PPI network using the canonical pathways from 2199 gene sets in the MSigDB25. GeNets applied a hypergeometric test to obtain p-value for the gene set enrichment and used Bonferroni correction to adjust for multiple hypothesis testing.
Brain cell-type specificity
Using human brain single-cell RNA-sequencing data profiled from the dPFC from Mathys et al32, we examined the cell-type specific expression of the 19 significant genes from the PWAS of depression. First, we performed data preprocessing and transformation using the Seurat package version 3.1.265. Genes were removed if they had fewer than 3 counts in a cell, and cells were removed if they had unique feature counts over 2,500 or less than 200. The RNA counts were normalized and scaled using the NormalizeData and ScaleData functions. The data had 17,926 genes in 70,634 cells before and 17,775 genes in 53,083 cells after quality control and normalization. We then focused on the 5 main cell types provided by Mathys et al 2019 using FindMarkers function: excitatory neuron, inhibitory neuron, astrocyte, microglia, and oligodendrocyte. For the 19 depression causal genes, we performed differential expression analysis to compare their expression levels in one cell type versus the rest of the other cell types using Wilcoxon Rank Sum test to determine if they are highly expressed in a particular cell type. The data met the assumptions of the statistical test used. For multiple testing correction, we used false discovery rate (FDR) adjusting for all 17,775 genes. We presented in Figure 2a only genes that had FDR p <0.05. Following the same approach, we determined if the 25 potentially causal genes from the meta-analysis of the discovery and replication depression PWAS are highly expressed in a particular cell type.
Reporting Summary.
Further information on research design is available in the Life Sciences Reporting Summary linked to this article.
Supplementary Material
Acknowledgements
We are grateful to the participants of the ROS, MAP, Mayo, Mount Sinai Brain Bank, and Banner Sun Health Research Institute Brain and Body Donation Program for their time and participation. The following NIH grants supported this work: P30 AG066511 (A.I.L.), P30 AG10161 (D.A.B.), P30 NS055077 (A.I.L.), P50 AG025688 (A.I.L.), R01 AG015819 (D.A.B.), R01 AG017917 (D.A.B.), R01 AG053960 (N.T.S.), R01 AG056533 (T.S.W., A.P.W.) R01 AG057911 (N.T.S.), R01 AG061800 (N.T.S.), R56 AG060757 (T.S.W.), R56 AG062256 (T.S.W.), RC2 AG036547 (D.A.B.), RF1 AG057470 (T.S.W.), U01 AG046152 (P.L.D.), U01 AG046161 (A.I.L.), U01 AG061356 (P.L.D.), U01 AG061357 (A.I.L.), and U01 MH115484 (A.P.W.). NIH grants include those that supported the Accelerating Medicine Partnership for AD, the NINDS Emory Neuroscience Core, and Goizueta Alzheimer’s Disease Research Center (ADRC) at Emory University, the Rush University ADRC, and Arizona State University ADRC that made this work possible. The following Veterans Administration grants supported this work: I01 BX003853 (A.P.W.) and IK4 BX005219 (A.P.W.). The Brain and Body Donation Program has been supported by NIH, the Arizona Department of Health Services, the Arizona Biomedical Research Commission and the Michael J. Fox Foundation for Parkinson’s Research. Additional support includes grants from the Alzheimer’s Association (N.T.S.), Alzheimer’s Research UK (N.T.S.), The Michael J. Fox Foundation for Parkinson’s Research (N.T.S.), and the Weston Brain Institute Biomarkers Across Neurodegenerative Diseases Grant 11060 (N.T.S.). The views expressed in this work do not necessarily represent the views of the Veterans Administration or the United States Government.
Footnotes
Competing interests
The authors declare no competing interest.
Data availability
Phenotypic, proteomic, and transcriptomic data used in this manuscript are available via the AD Knowledge Portal (https://adknowledgeportal.org). The AD Knowledge Portal is a platform for accessing data, analyses, and tools generated by the Accelerating Medicines Partnership (AMP-AD) Target Discovery Program and other National Institute on Aging (NIA)-supported programs to enable open-science practices and accelerate translational learning. The data, analyses and tools are shared early in the research cycle without a publication embargo on secondary use. Data is available for general research use according to the following requirements for data access and data attribution (https://adknowledgeportal.org/DataAccess/Instructions). For access to results of the pQTL analysis, protein weights, and transcript weights described in this manuscript see https://doi.org/10.7303/syn24872746
References
- 1.Lim GY, et al. Prevalence of Depression in the Community from 30 Countries between 1994 and 2014. Scientific reports 8, 2861 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Friedrich MJ Depression Is the Leading Cause of Disability Around the World. Jama 317, 1517 (2017). [DOI] [PubMed] [Google Scholar]
- 3.Rush AJ STAR*D: what have we learned? The American journal of psychiatry 164, 201–204 (2007). [DOI] [PubMed] [Google Scholar]
- 4.Thase ME & Schwartz TL Choosing medications for treatment-resistant depression based on mechanism of action. The Journal of clinical psychiatry 76, 720–727; quiz 727 (2015). [DOI] [PubMed] [Google Scholar]
- 5.Akil H, et al. Treatment resistant depression: A multi-scale, systems biology approach. Neuroscience and biobehavioral reviews 84, 272–288 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moya-García A, et al. Structural and Functional View of Polypharmacology. Scientific reports 7, 10102 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zheng J, et al. Phenome-wide Mendelian randomization mapping the influence of the plasma proteome on complex diseases. Nature genetics 52, 1122–1131 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pena CJ & Nestler EJ Progress in Epigenetics of Depression. Progress in molecular biology and translational science 157, 41–66 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arloth J, et al. Genetic Differences in the Immediate Transcriptome Response to Stress Predict Risk-Related Brain Function and Psychiatric Disorders. Neuron 86, 1189–1202 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Howard DM, et al. Genome-wide meta-analysis of depression identifies 102 independent variants and highlights the importance of the prefrontal brain regions. Nature neuroscience 22, 343–352 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sharma K, et al. Cell type- and brain region-resolved mouse brain proteome. Nature neuroscience 18, 1819–1831 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vogel C & Marcotte EM Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nature reviews. Genetics 13, 227–232 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wingo AP, et al. Shared proteomic effects of cerebral atherosclerosis and Alzheimer’s disease on the human brain. Nature neuroscience 23, 696–700 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wingo AP, et al. Large-scale proteomic analysis of human brain identifies proteins associated with cognitive trajectory in advanced age. Nat Commun 10, 1619 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Johnson ECB, et al. Large-scale proteomic analysis of Alzheimer’s disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nature medicine 26, 769–780 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gusev A, et al. Integrative approaches for large-scale transcriptome-wide association studies. Nature genetics 48, 245–252 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gusev A, et al. Transcriptome-wide association study of schizophrenia and chromatin activity yields mechanistic disease insights. Nature genetics 50, 538–548 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhu Z, et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nature genetics 48, 481–487 (2016). [DOI] [PubMed] [Google Scholar]
- 19.Wu L, et al. A transcriptome-wide association study of 229,000 women identifies new candidate susceptibility genes for breast cancer. Nature genetics 50, 968–978 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gusev A, et al. A transcriptome-wide association study of high-grade serous epithelial ovarian cancer identifies new susceptibility genes and splice variants. Nature genetics 51, 815–823 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hyde CL, et al. Identification of 15 genetic loci associated with risk of major depression in individuals of European descent. Nature genetics 48, 1031–1036 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bennett DA, et al. Religious Orders Study and Rush Memory and Aging Project. Journal of Alzheimer’s disease : JAD 64, S161–s189 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beach TG, et al. Arizona Study of Aging and Neurodegenerative Disorders and Brain and Body Donation Program. Neuropathology 35, 354–389 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li T, et al. GeNets: a unified web platform for network-based genomic analyses. Nature methods 15, 543–546 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Subramanian A, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wray NR, et al. Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nature genetics 50, 668–681 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nagel M, et al. Meta-analysis of genome-wide association studies for neuroticism in 449,484 individuals identifies novel genetic loci and pathways. Nature genetics 50, 920–927 (2018). [DOI] [PubMed] [Google Scholar]
- 28.Yengo L, et al. Meta-analysis of genome-wide association studies for height and body mass index in ∼700000 individuals of European ancestry. Human molecular genetics 27, 3641–3649 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pulit SL, et al. Meta-analysis of genome-wide association studies for body fat distribution in 694 649 individuals of European ancestry. Human molecular genetics 28, 166–174 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Logsdon BA, et al. Meta-analysis of the human brain transcriptome identifies heterogeneity across human AD coexpression modules robust to sample collection and methodological approach. bioRxiv, 510420 (2019).
- 31.Wu Y, et al. Integrative analysis of omics summary data reveals putative mechanisms underlying complex traits. Nat Commun 9, 918 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mathys H, et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 570, 332–337 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huckins LM, et al. Gene expression imputation across multiple brain regions provides insights into schizophrenia risk. Nature genetics 51, 659–674 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Raj T, et al. Integrative transcriptome analyses of the aging brain implicate altered splicing in Alzheimer’s disease susceptibility. Nature genetics 50, 1584–1592 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vasudevan D, Takeuchi H, Johar SS, Majerus E & Haltiwanger RS Peters plus syndrome mutations disrupt a noncanonical ER quality-control mechanism. Current biology : CB 25, 286–295 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Barres BA The mystery and magic of glia: a perspective on their roles in health and disease. Neuron 60, 430–440 (2008). [DOI] [PubMed] [Google Scholar]
- 37.Aho S, et al. Specific sequences in p120ctn determine subcellular distribution of its multiple isoforms involved in cellular adhesion of normal and malignant epithelial cells. Journal of cell science 115, 1391–1402 (2002). [DOI] [PubMed] [Google Scholar]
- 38.Jones SB, et al. Glutamate-induced delta-catenin redistribution and dissociation from postsynaptic receptor complexes. Neuroscience 115, 1009–1021 (2002). [DOI] [PubMed] [Google Scholar]
- 39.Grunwald IC, et al. Kinase-independent requirement of EphB2 receptors in hippocampal synaptic plasticity. Neuron 32, 1027–1040 (2001). [DOI] [PubMed] [Google Scholar]
- 40.Otrokocsi L, Kittel A & Sperlagh B P2X7 Receptors Drive Spine Synapse Plasticity in the Learned Helplessness Model of Depression. The international journal of neuropsychopharmacology 20, 813–822 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kurcon T, et al. miRNA proxy approach reveals hidden functions of glycosylation. Proceedings of the National Academy of Sciences of the United States of America 112, 7327–7332 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Okada-Tsuchioka M, et al. Electroconvulsive seizure induces thrombospondin-1 in the adult rat hippocampus. Progress in neuro-psychopharmacology & biological psychiatry 48, 236–244 (2014). [DOI] [PubMed] [Google Scholar]
- 43.Zhen L, et al. EphB2 Deficiency Induces Depression-Like Behaviors and Memory Impairment: Involvement of NMDA 2B Receptor Dependent Signaling. Frontiers in pharmacology 9, 862 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Arjona FJ, et al. CNNM2 mutations cause impaired brain development and seizures in patients with hypomagnesemia. PLoS genetics 10, e1004267 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yamanaka R, Shindo Y & Oka K Magnesium Is a Key Player in Neuronal Maturation and Neuropathology. International journal of molecular sciences 20(2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thyme SB, et al. Phenotypic Landscape of Schizophrenia-Associated Genes Defines Candidates and Their Shared Functions. Cell 177, 478–491.e420 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Maeda Y & Kinoshita T Dolichol-phosphate mannose synthase: structure, function and regulation. Biochimica et biophysica acta 1780, 861–868 (2008). [DOI] [PubMed] [Google Scholar]
- 48.Shi J, et al. Up-regulation of PSMB4 is associated with neuronal apoptosis after neuroinflammation induced by lipopolysaccharide. Journal of molecular histology 46, 457–466 (2015). [DOI] [PubMed] [Google Scholar]
- 49.Hawi Z, et al. The role of cadherin genes in five major psychiatric disorders: A literature update. American journal of medical genetics. Part B, Neuropsychiatric genetics : the official publication of the International Society of Psychiatric Genetics 177, 168–180 (2018). [DOI] [PubMed] [Google Scholar]
- 50.Carvalho-Silva D, et al. Open Targets Platform: new developments and updates two years on. Nucleic acids research 47, D1056–d1065 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
References
- 51.Mertins P, et al. Reproducible workflow for multiplexed deep-scale proteome and phosphoproteome analysis of tumor tissues by liquid chromatography-mass spectrometry. Nature protocols 13, 1632–1661 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.De Jager PL, et al. A genome-wide scan for common variants affecting the rate of age-related cognitive decline. Neurobiology of aging 33, 1017.e1011–1015 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.De Jager PL, et al. A multi-omic atlas of the human frontal cortex for aging and Alzheimer’s disease research. Scientific data 5, 180142 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Purcell S, et al. PLINK: a toolset for whole-genome association and population-based linkage analysis. American journal of human genetics 81, 559–575 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Manichaikul A, et al. Robust relationship inference in genome-wide association studies. Bioinformatics (Oxford, England) 26, 2867–2873 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Abecasis GR, et al. An integrated map of genetic variation from 1,092 human genomes. Nature 491, 56–65 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Das S, et al. Next-generation genotype imputation service and methods. Nature genetics 48, 1284–1287 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Allen M, et al. Human whole genome genotype and transcriptome data for Alzheimer’s and other neurodegenerative diseases. Scientific data 3, 160089 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang M, et al. The Mount Sinai cohort of large-scale genomic, transcriptomic and proteomic data in Alzheimer’s disease. Scientific data 5, 180185 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dobin A, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics (Oxford, England) 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Willer CJ, Li Y & Abecasis GR METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics (Oxford, England) 26, 2190–2191 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sieberts SK, et al. Large eQTL meta-analysis reveals differing patterns between cerebral cortical and cerebellar brain regions. Scientific data 7, 340 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lage K, et al. A human phenome-interactome network of protein complexes implicated in genetic disorders. Nature biotechnology 25, 309–316 (2007). [DOI] [PubMed] [Google Scholar]
- 64.Clauset A, Newman ME & Moore C Finding community structure in very large networks. Physical review. E, Statistical, nonlinear, and soft matter physics 70, 066111 (2004). [DOI] [PubMed] [Google Scholar]
- 65.Butler A, Hoffman P, Smibert P, Papalexi E & Satija R Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nature biotechnology 36, 411–420 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Phenotypic, proteomic, and transcriptomic data used in this manuscript are available via the AD Knowledge Portal (https://adknowledgeportal.org). The AD Knowledge Portal is a platform for accessing data, analyses, and tools generated by the Accelerating Medicines Partnership (AMP-AD) Target Discovery Program and other National Institute on Aging (NIA)-supported programs to enable open-science practices and accelerate translational learning. The data, analyses and tools are shared early in the research cycle without a publication embargo on secondary use. Data is available for general research use according to the following requirements for data access and data attribution (https://adknowledgeportal.org/DataAccess/Instructions). For access to results of the pQTL analysis, protein weights, and transcript weights described in this manuscript see https://doi.org/10.7303/syn24872746