

Abstract
Chromatin modulation provides a key checkpoint for controlling cell cycle regulated gene networks. The replicative canonical histone genes are one such gene family under tight regulation during cell division. These genes are most highly expressed during S phase when histones are needed to chromatinize the new DNA template. While this fact has been known for a while, limited knowledge exists about the specific chromatin regulators controlling their temporal expression during cell cycle. Since histones and their associated mutations are emerging as major players in diseases such as cancer, identifying the chromatin factors modulating their expression is critical. The histone lysine tri-demethylase KDM4A is regulated over cell cycle and plays a direct role in DNA replication timing, site-specific rereplication, and DNA amplifications during S phase. Here, we establish an unappreciated role for the catalytically active KDM4A in directly regulating canonical replicative histone gene networks during cell cycle. Of interest, we further demonstrate that KDM4A interacts with proteins controlling histone expression and RNA processing (i.e., hnRNPUL1 and FUS/TLS). Together, this study provides a new function for KDM4A in modulating canonical histone gene expression.
Keywords: KDM4A, histones, hnRNPUL1, FUS/TLS, cell cycle, ATAC, transcription, demethylation, epigenetics
INTRODUCTION
Replicative canonical histone gene expression is a highly regulated process that is still not fully understood [1, 2]. Increased levels of histones are needed during S phase, when DNA replication occurs, in order to chromatinize the new DNA. However, while insufficient histones levels will alter S phase progression [3–5], the excess of histones, specifically in other phases of the cell cycle, is toxic for the cells [2]. Therefore, their gene expression is highly regulated in a timely manner. The replicative canonical histone genes are organized in three clusters: HIST1 is on chromosome 6 (50 genes), HIST2 (10 genes) and HIST3 (3 genes) are on chromosome 1 [2]. These clusters are in syntenic loci, highlighting a likely conserved regulation of expression [6].
Transcription and processing of replicative histones occur within histone locus bodies, and while clusters reside within different topologically associated domains (TADs), these TADs interact together [2]. In addition to their differential expression across the cell cycle, replicative histones mRNAs differ from other histone variants. They lack introns, have short UTRs (untranslated regions), and harbor a 3’ stem-loop structure in place of the polyA tail. Furthermore, several genes code for the same histone proteins, which likely reflects their cellular importance and high demand during S phase [1, 2].
KDM4A is a histone 3 lysine 9 and 36 tri-demethylase that is regulated at the protein level during cell cycle by ubiquitination and proteasomal degradation [7–9]. KDM4A protein levels peak in G1/S and are the lowest in G2/M [8, 10]. This suggests that KDM4A protein levels are modulated so that cell cycle progression or associated gene mechanisms are properly controlled. In the context of DNA replication, KDM4A stabilization or overexpression displaced heterochromatin and promoted replication timing changes, rereplication, and DNA copy gains of specific genomic regions [8, 10–14]. KDM4A interacts with the replication machinery and promotes site-specific DNA rereplication and transient site-specific copy gains of regions affiliated with drug resistance [12, 15]. While KDM4A has been linked to DNA replication directly, little knowledge exists to date about how KDM4A impacts gene networks in a cell cycle-specific manner. In this study, we set out to determine whether KDM4A modulates gene networks during specific cell cycle phases. We discovered that replicative canonical histones are a direct target of KDM4A and that KDM4A interacts with proteins involved in their expression and processing. Therefore, this study uncovers an unappreciated role for KDM4A in properly regulating the histone genes expression and processing during cell cycle progression.
MATERIALS AND METHODS
Cell Culture, Sorting, and Transfections
Stable cell lines have been generated as in [12]. Cells were incubated with Hoechst 33342 (ThermoFisher Scientific H3570) at 1/1000 directly into the media for 1h at 37 degrees. Cells were then trypsinized and resuspended in media containing Hoechst at 1/1000. Cells were sorted with a BD FACS Fusion using the laser BV421-A into Qiazol, based on DNA content. The gates for the sorting were set such as the G1 phase was collected from the bottom left of the first peak to the middle of the right slop, the G2 phase was collected from the bottom left of the second peak to the bottom right of that same peak. The ES and LS phase were equally divided in between G1 and G2 phases. Figure 1A presents the gating.
Figure 1.
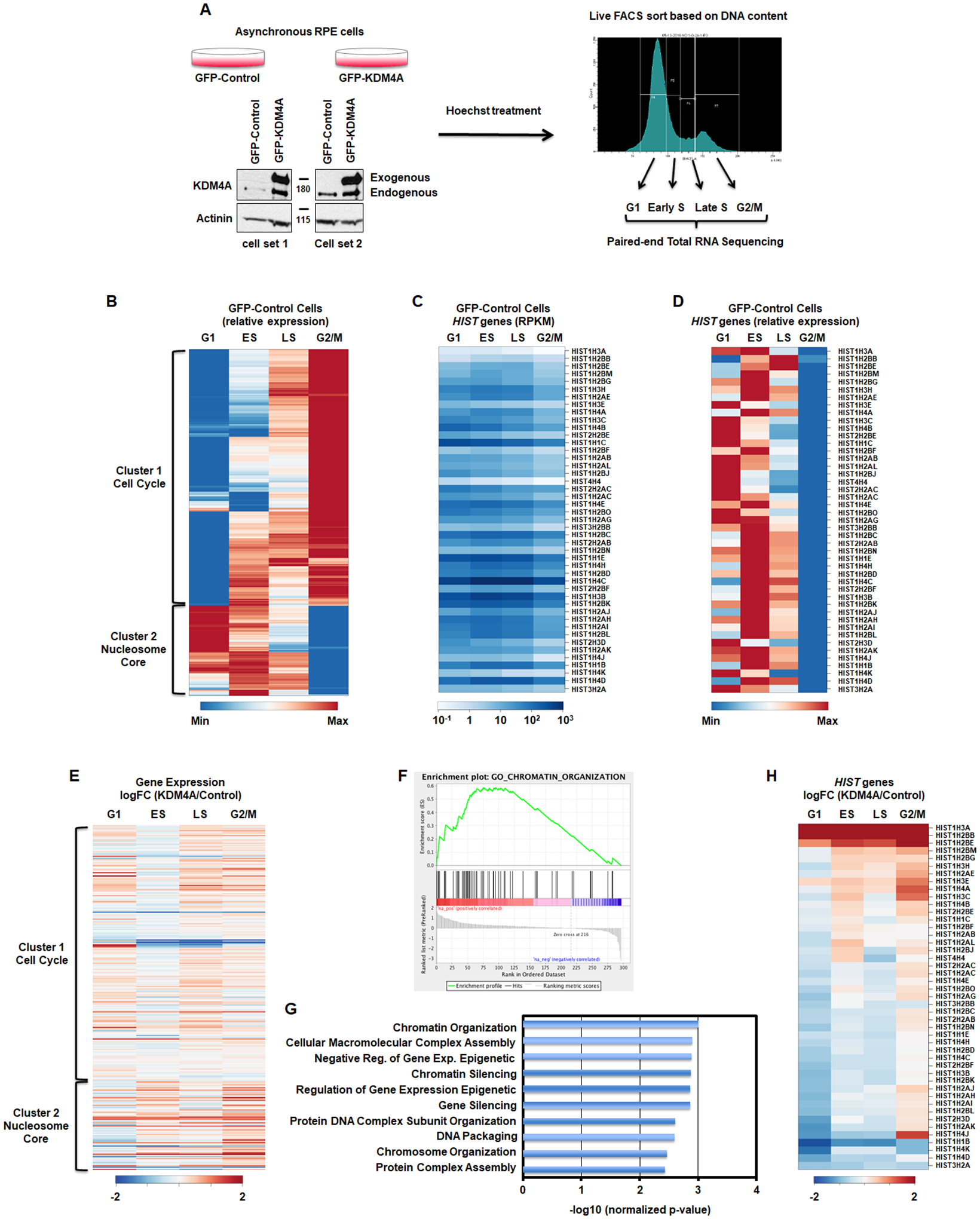
KDM4A regulates gene expression over cell cycle. A. Description of the method. B. Differential gene expression patterns across cell cycle in control RPE cells. The heatmap represents the levels of expression at four phases of cell cycle for the genes that show more than two-fold change in expression between any two phases. Colors indicate relative expression values scaled between minimum and maximum levels for a given gene. Clusters of expression patterns are annotated by highly overrepresented gene categories (Cell Cycle: p-value = 4.5e-55; Nucleosome Core: p-value = 1.1e-32). C,D. Absolute gene expression values (RPKM) and relative expression values of replicative histone genes across cell cycle. E. Differential gene expression between KDM4A and control cells across cell cycle (log fold change). Gene order is the same as in B. All results represent the average over two biological replicates. F. The cell cycle regulated genes whose expression was changed upon KDM4A overexpression were enriched in chromatin organization related genes. GSEA enrichment plot for the GO category of Chromatin Organization. G. Enrichment p-value for top 10 functional categories detected by GSEA among the cell cycle regulated genes that were misregulated upon KDM4A overexpression. H. Differential expression of replicative histone genes between KDM4A and control cells (log fold change of RPKM). All results represent the average over two biological replicates.
The siRNA were transfected using Lipofectamine 3000 (ThermoFisher Scientific) according to the supplier recommendations. The siRNAs were purchased from ThermoFisher Scientific (Silencer Select): KDM4A (s18635, s18636, s18637), FUS/TLS (s5401, s5402), hnRNPUL1 (s21883, s21884).
RNA Purification, cDNA preparation and Sequencing
RNA was purified using the Qiagen miRNeasy kit including a DNAse treatment. cDNAs were prepared using the SuperScript IV Reverse Transcriptase (ThermoFisher Scientific) using random hexamer. Quantitative PCR was performed with the following primers:
HIST1H3A: For – tttccagagctccgctgt, Rev – tgtcctcaaatagccctacca
HIST1H4A: For – tgttgcgtgacaacatcca, Rev – cagagatccgcttcacacc
HIST1H3C: For – gaaatccgtcgctaccagaa, Rev – ttagcgtgaatagcgcacag
TBP: For – gaacatcatggatcagaacaaca, Rev – atagggattccgggagtcat
Total RNA sequencing libraries were prepped using the TruSeq Stranded Total RNA Sample Preparation with Ribo-Zero kit (Illumina). Libraries were sequenced as paired-end 150 bp reads using a NextSeq500 instrument (Illumina).
ATAC Sequencing
Four 15cm plates of 2 million RPE were seeded for 48h. Cells were incubated with media containing Hoechst 33342 at 1/1000 for 1h at 37C. Cells were trypsinized and resuspended in Hoechst-containing media. Cells were sorted with a BD FACS Fusion BV421-H laser based on DNA content as reported in Figure 1A and in the Sorting section. 50,000 or 100,000 cells were collected per phase.
The library preparation protocol was adapted from [16]. Sorted cells were pelleted and resuspended in 250 μl cold RBS buffer (10 mM Tris-HCl pH 7.4 – 10 mM NaCl – 3 mM MgCl2) before being pelleted by centrifugation. Pellet was resuspended in 250 μl cold RBS buffer – 0.1% NP-40, incubated 5 minutes on ice and pelleted by centrifugation. Pellet was resuspended in 50 μl Tagmentation mix (22.5 μl H2O – 25 μl 2X Tagment DNA buffer – 2.5 μl Tagment DNA enzyme, Nextera Illumina FC-121–1030) and incubated 30 minutes at 37 degrees before being placed ion ice. DNA was purified with MinElute PCR Purification kit (Qiagen) following supplier’s instructions and eluted in 10 μl. Purified DNA was amplified by PCR by adding 2.5 μl 25 μM Primer Ad2 (barcode, use different primers for each samples) and 37.5 μl PCR mix (10 μl H2O – 2.5 μl 25 μM Primer Ad1 – 25 μl 2X NEBnext master mix, NEB E6040S) and incubated as follows: 72 degrees 5 minutes, 98 degrees 30 seconds, [98 degrees 10 seconds, 63 degrees 30 seconds, 72 degrees 1 minute] × 12 cycles, 4 degrees. Amplified DNA was purified using a Promega Gel Purification Kit following supplier’s instructions and eluted in 25 μl H2O. Library length distribution were assessed by TapeStation (Agilent) and quantified with Qubit (ThermoFisher). Libraries were paired-end sequenced (43 cycles each way) using a NextSeq500 (Illumina).
Primers sequences [16]:
Ad2 primers contain the barcodes (Ad2.1_TAAGGC: TAAGGC is the barcode). Ad1: AATGATACGGCGACCACCGAGATCTACACTCGTCGGCAGCGTCAGATGTG
Ad2.1_TAAGGC:
CAAGCAGAAGACGGCATACGAGATTCGCCTTAGTCTCGTGGGCTCGGAGATGT
Ad2.2_CGTACT:
CAAGCAGAAGACGGCATACGAGATCTAGTACGGTCTCGTGGGCTCGGAGATGT
Ad2.3_AGGCAG:
CAAGCAGAAGACGGCATACGAGATTTCTGCCTGTCTCGTGGGCTCGGAGATGT
Ad2.4_TCCTGA:
CAAGCAGAAGACGGCATACGAGATGCTCAGGAGTCTCGTGGGCTCGGAGATGT
Ad2.5_GGACTC:
CAAGCAGAAGACGGCATACGAGATAGGAGTCCGTCTCGTGGGCTCGGAGATGT
Ad2.6_TAGGCA:
CAAGCAGAAGACGGCATACGAGATCATGCCTAGTCTCGTGGGCTCGGAGATGT
Ad2.7_CTCTCT:
CAAGCAGAAGACGGCATACGAGATGTAGAGAGGTCTCGTGGGCTCGGAGATGT
Ad2.8_CAGAGA:
CAAGCAGAAGACGGCATACGAGATCCTCTCTGGTCTCGTGGGCTCGGAGATGT
Ad2.9_GCTACG:
CAAGCAGAAGACGGCATACGAGATAGCGTAGCGTCTCGTGGGCTCGGAGATGT
Ad2.10_CGAGGC:
CAAGCAGAAGACGGCATACGAGATCAGCCTCGGTCTCGTGGGCTCGGAGATGT
Ad2.11_AAGAGG:
CAAGCAGAAGACGGCATACGAGATTGCCTCTTGTCTCGTGGGCTCGGAGATGT
Ad2.12_GTAGAG:
CAAGCAGAAGACGGCATACGAGATTCCTCTACGTCTCGTGGGCTCGGAGATGT
Ad2.13_GTCGTG:
CAAGCAGAAGACGGCATACGAGATATCACGACGTCTCGTGGGCTCGGAGATGT
Ad2.14_ACCACT:
CAAGCAGAAGACGGCATACGAGATACAGTGGTGTCTCGTGGGCTCGGAGATGT
Ad2.15_TGGATC:
CAAGCAGAAGACGGCATACGAGATCAGATCCAGTCTCGTGGGCTCGGAGATGT
Ad2.16_CCGTTT:
CAAGCAGAAGACGGCATACGAGATACAAACGGGTCTCGTGGGCTCGGAGATGT
Ad2.17_TGCTGG:
CAAGCAGAAGACGGCATACGAGATACCCAGCAGTCTCGTGGGCTCGGAGATGT
Ad2.18_GAGGGG:
CAAGCAGAAGACGGCATACGAGATAACCCCTCGTCTCGTGGGCTCGGAGATGT
Ad2.19_AGGTTG:
CAAGCAGAAGACGGCATACGAGATCCCAACCTGTCTCGTGGGCTCGGAGATGT
Ad2.20_GTGTGG:
CAAGCAGAAGACGGCATACGAGATCACCACACGTCTCGTGGGCTCGGAGATGT
Ad2.21_TGGGTT:
CAAGCAGAAGACGGCATACGAGATGAAACCCAGTCTCGTGGGCTCGGAGATGT
Ad2.22_TGGTCA:
CAAGCAGAAGACGGCATACGAGATTGTGACCAGTCTCGTGGGCTCGGAGATGT
Ad2.23_TTGACC:
CAAGCAGAAGACGGCATACGAGATAGGGTCAAGTCTCGTGGGCTCGGAGATGT
Ad2.24_CCACTC:
CAAGCAGAAGACGGCATACGAGATAGGAGTGGGTCTCGTGGGCTCGGAGATGT
Bioinformatics Analyses of RNA-, ATAC-, ChIP-Seq Data
For RNA-seq analyses, STAR [17] aligner was used to map sequencing reads to transcripts in human hg19 reference genome. Read counts for individual transcripts were produced with HTseq-count [18], followed by the estimation of expression values and detection of differentially expressed transcripts using edgeR [19]. Differentially expressed genes were defined based on the criteria of >2-fold change in expression value and false discovery rate (FDR) < 0.05. For functional enrichment analysis of differentially expressed genes, we used the DAVID tool [20] and GSEA [21] applied against the MSigDB set module.
ATAC-seq and ChIP-seq reads were aligned against the hg19 reference genome using BWA [22]. Alignments were filtered for uniquely mapped none-mitochondrial reads and duplicates were removed. RPE Chip-seq data were previously published [15]. Public data were download from GEO database for KDM4A ChIP (GSE95190) and H3.3 (GSM2265640).
GM12878 K36me3 and K9me3 (GSM733679) ChIP-seq BAM files were downloaded from ENCODE [23]. Enrichment over gene body for each transcript was calculated as the log ratio of ChIP and input read counts.
Other public ChIP-seq datasets [24] were downloaded as fastq files from GEO [25], followed by mapping to the hg19 reference genome using BWA [22] and the generation of input-normalized coverage tracks using Deeptools [26]. All sequencing data presented in this study are available from NCBI Gene Expression Omnibus (GEO) with the accession number GSE154684.
Coimmunoprecipitation
Coimmunoprecipitations experiments were performed as described previously [8]. Westerns were auto-contrasted and cropped. If the proteins of interest have comparable molecular weights, the immunoprecipitation was divided and run on separate gels.
Fractionations
Fractionations experiments were performed as described in [27]. For western blot an equal volume of each fraction was analyzed (data not shown).
Mass Spectrometry Analysis
Mass spectrometry analyzes are the same as in [28]. Briefly, a duplicate set of immunoprecipitations were performed and analyzed as follows. For Ingenuity Pathway Analysis (IPA) analyses, KDM4A and IgG IPs were compared and any proteins with more peptides counts within KDM4A IPs than within IgG IPs were used in IPA. IPA was conducted on both independent list of proteins from each IP. Normalized spectral abundance factors (NSAF) were calculated and compared between the KDM4A and IgG IP protein sets and plotted in Figure 4B. The RNA post-transcriptional modification category from IPA analyses are outlined in red in Figure 4B.
Figure 4.
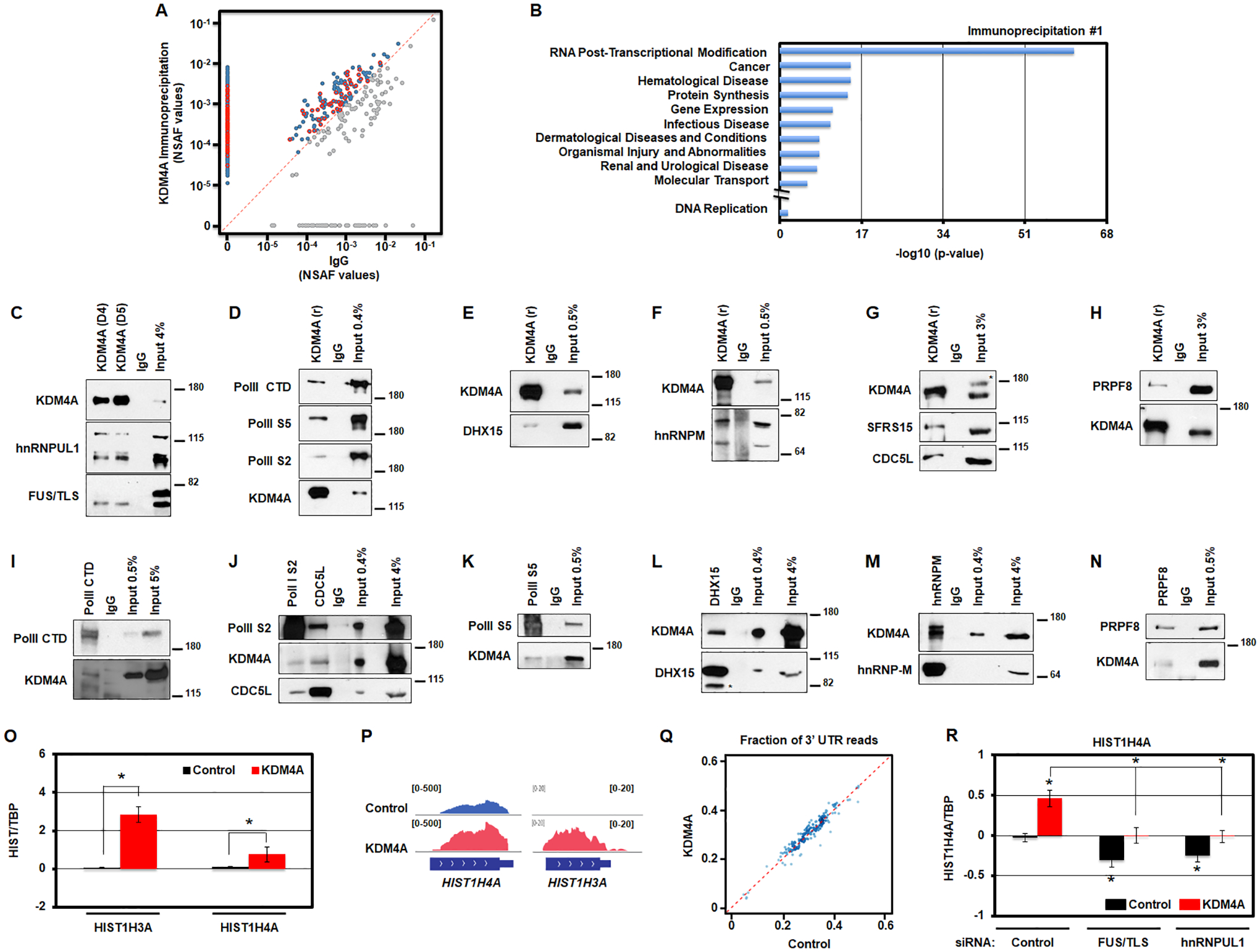
KDM4A interacts with the transcription/processing machinery controlling replicative canonical histone gene expression. A. KDM4A interacting proteins are enriched in the functional category associated with RNA post-transcriptional modifications. Scatter plot of normalized spectral abundance factors (NSAF) from mass spectrometry experiments for immunoprecipitation (IP) with KDM4A (y axis) vs IgG as a control (x axis). Blue points, proteins with stronger presence in KDM4A IP. Red points, proteins from “RNA Post-Transcriptional Modification” category according to Ingenuity Pathway Analysis (IPA). B. Enrichment p-values for functional categories detected by Ingenuity Pathway Analysis (IPA) among protein sets from mass spectrometry analyses of endogenous KDM4A complexes. In addition to top 10 categories represented, p-value for DNA replication category studied in [12] is shown. C. Endogenous KDM4A was immunoprecipitated from 293T cells with two antibodies (D4 and D5) followed by an immunoblot against hnRNPUL1 and FUS/TLS. 4% of input material was loaded. D. Endogenous KDM4A was immunoprecipitated from 293T cells followed by an immunoblot against Polymerase II (PolII CTD), Polymerase II phosphorylated on Serine 5 (PolII S5) and Serine 2 (PolII S2). 0.4% of input material was loaded. E. Endogenous KDM4A was immunoprecipitated from 293T cells followed by an immunoblot against DHX15. 0.5% of input material was loaded. F. Endogenous KDM4A was immunoprecipitated from 293T cells followed by an immunoblot against hnRNP-M. 0.5% of input material was loaded. G. Endogenous KDM4A was immunoprecipitated from 293T cells followed by an immunoblot against SFRS15 and CDC5L. 3% of input material was loaded. * indicates non-specific band on western. H. Endogenous KDM4A was immunoprecipitated from 293T cells followed by an immunoblot against PRPF8. 3% of input material was loaded. I-N. Endogenous members of the transcription/splicing machinery were immunoprecipitated from 293T cells followed by an immunoblot against KDM4A and other annotated proteins. * indicates non-specific band on western (L). O. Log2 ratios between the amount of RNAs from chromatin fraction for specific histone transcripts and TBP, relative to control cells, for control (black) and KDM4A overexpressing cells (red). Averages of five replicates for two biologically independent cells lines are shown. P. Examples of RNA sequencing tracks at replicative histone genes in control and KDM4A overexpressing cells. Q. Scatter plot of RNA sequencing coverage at 3’ UTR as a fraction of the coverage over the entire gene for all replicative histone genes in KDM4A overexpressing cells versus control cells. Averages of two biologically independent cell lines are shown. * T-test p-value < 0.05 relative to control cells. R. Log2 ratios between expression values of HIST1H4A and TBP, relative to control cells siRNA control, for control (black) and KDM4A overexpressing cells (red). The results represent the average of three replicates for two biologically independent cell lines transfected with two independent siRNAs. * T-test p-value < 0.05 relative to control cells or to control cells siRNA control, or between the two samples indicated with brackets when * is above a bracket.
Antibodies
KDM4A: immunoprecipitation D4 (KDM4A-P006), D5 (KDM4A-P014)[28], rabbit polyclonal antibody [referred as KDM4A(r) in the figures [10]; western blot NeuromAB 75–189. Actinin (Santa-Cruz sc-17829), hnRNPUL1 (Abcam ab68420), FUS/TLS (Abcam ab124923), PolII CTD (Abcam ab5408), PolII phosphor Ser5 (Abcam ab5131), PolII phosphor Ser2 (Abcam ab5095), SFRS15 (Abcam ab110522), CDC5L (Abcam ab31779), PRPF8 (Abcam ab51366), hnRNPM (Santa-Cruz sc-134360), DHX15 (Abcam ab70454), snRNP70 (Abcam ab51266), hnRNPUL1 (Abcam ab68420), FUS/TLS (Abcam ab124923).
RESULTS
KDM4A regulates replicative histone genes expression during cell cycle
In order to study the impact KDM4A has on cell cycle gene expression and avoid drug induced cell cycle profiles and interference with KDM4A function, we developed a fluorescence-activated cell sorting (FACS) approach to analyze cell cycle genes regulated by KDM4A at G1/S, early S (ES), late S (LS) and G2/M phases (Fig. 1A). We used the karyotypically stable and immortalized, but not transformed RPE-1-hTERT (RPE) cells that stably overexpressed GFP-KDM4A or GFP as a control (Fig. 1A). Specifically, these cells were Hoechst treated and sorted based on DNA content in four phases: G1, early S (ES), late S (LS) and G2/M. Total RNA was then extracted and processed for paired-end total RNA sequencing (Fig. 1A).
First, we assessed the gene expression profiles for the control RPE cells across cell cycle phases. Figure 1B and Table S1 represent the differentially expressed genes with at least a two-fold difference and FDR<0.05 between any two time points across the four cell cycle phases. These 297 genes include 220 “cell cycle” genes annotated by GO database (Table S1). Figure S1A represents canonical cell cycle-regulated genes that validate our FACS sort method for evaluation of cell cycle regulated genes [29, 30]. The heatmap represented in Figure 1B presents two major clusters. According to the analysis of functional gene set enrichment using the DAVID tool [20] (Table S1), cluster 1 was enriched in cell cycle genes (p-value = 4.53e-55); while, cluster 2 was enriched in nucleosome-associated genes, which mainly represented replicative canonical histones (HIST) genes (p-value = 1.05e-32).
We observed a range of canonical histone gene isoform expression across each cell cycle phases in the RPE cells (Figure 1C–D and Table S2). For example, replicative histone H2B isoforms HIST1H2BB and HIST1H2BE are expressed the highest in late S phase, while HIST1H2BJ and HIST1H2BO levels peak in G1 (Figure 1C–D and Table S2). As another example, replicative histone isoforms HIST1H4C and HIST1H4D are expressed the highest in early S phase, while HIST1H4B peaks in G1 (Figure 1C–D and Table S2). While the differential expression pattern between histones variants has been well defined (for example between H3.1/H3.2 and H3.3), the reason behind differential expression patterns of canonical histone genes has not been well documented. As expected, all replicative histone mRNAs are the lowest in G2/M, once replication is complete (Figure 1C–D and Table S2).
We then evaluated the impact that KDM4A overexpression has on differentially expressed genes irrespective of cell cycle regulation, and then focused separately on the differentially regulated cell-cycle genes (Table S3). KDM4A overexpression resulted in differential expression for 200 genes with at least a two-fold difference and FDR<0.05 at any one of four cell cycles phases (Table S3). Gene set enrichment analysis (GSEA) [21] of genome-wide expression changes revealed multiple enriched functional gene categories (Fig. S1B, Table S3). Of the top eight enriched categories, four were RNA binding categories and one the RNA polymerase activity category (Fig. S1B).
We then focused on the effect of KDM4A overexpression on genes that were differentially expressed during cell cycle in control cells. We observed that 11% of these cell cycle-regulated genes (Fig. 1B, 297 genes with >2-fold change of expression, FDR < 0.05) were mis-regulated when KDM4A was overexpressed, mostly in the G2/M phase (Fig. 1E and Table S1). To analyze the functional enrichment associated with specific effects of KDM4A overexpression on these cell-cycle regulated genes, GSEA [21] was applied to the cell-cycle regulated genes (Fig. 1B) sorted by the fold change of their expression in G2/M between KDM4A overexpressing and control cells. This analysis revealed enrichment for genes associated with chromatin organization, which primarily consisted of replicative canonical histone genes (Fig. 1F–G).
Consistent with this category being altered, KDM4A overexpression mis-regulated genes were significantly enriched in cluster 2 in Figure 1E (p-value = 7.1e-10, hypergeometric test). Therefore, we evaluated the expression patterns for all replicative canonical histone genes in KDM4A overexpressing cells compared to control cells (Fig. 1H). The heatmap represented in Figure 1H reveals that KDM4A regulates the proper expression of most replicative histone genes across cell cycle (25 of 45 over 1.5-fold change; p-value = 1.6e-37, hypergeometric test). For example, control RPE cells have reduced replicative histone gene expression as they enter the G2/M phase of the cell cycle (Table S2). However, upon KDM4A overexpression, most replicative histones are upregulated in the G2/M phase (e.g., HIST1H3A, HIST1H4A and HIST1H3C; Fig. 1H and Fig. 2A–C). We also observed that a third of the replicative histone genes are upregulated in early S phase [e.g., HIST1H3A, HIST1H4A and HIST1H3C; Fig. 1H (top of the heatmap) and Fig. 2A–C]. Only a few histone genes were overexpressed throughout each cell cycle phase (Fig. 1H). In fact, their expression went from silent/minimal expression to moderate/detectable (i.e., HIST1H2BB, HIST1H3A; Fig. 1C, 1H, 2A, and Table S2). These observations were validated through qRT-PCR (i.e., HIST1H3A, HIST1H4A and HIST1H3C; Fig. 2D–F). Consistent with KDM4A protein levels regulating the replicative histones during cell cycle, siRNA depletion of KDM4A lead to inversed expression of these genes when compared to KDM4A overexpression (i.e., HIST1H3A, HIST1H4A, and HIST1H3C; Fig. 2G–I and Fig. S2).
Figure 2.
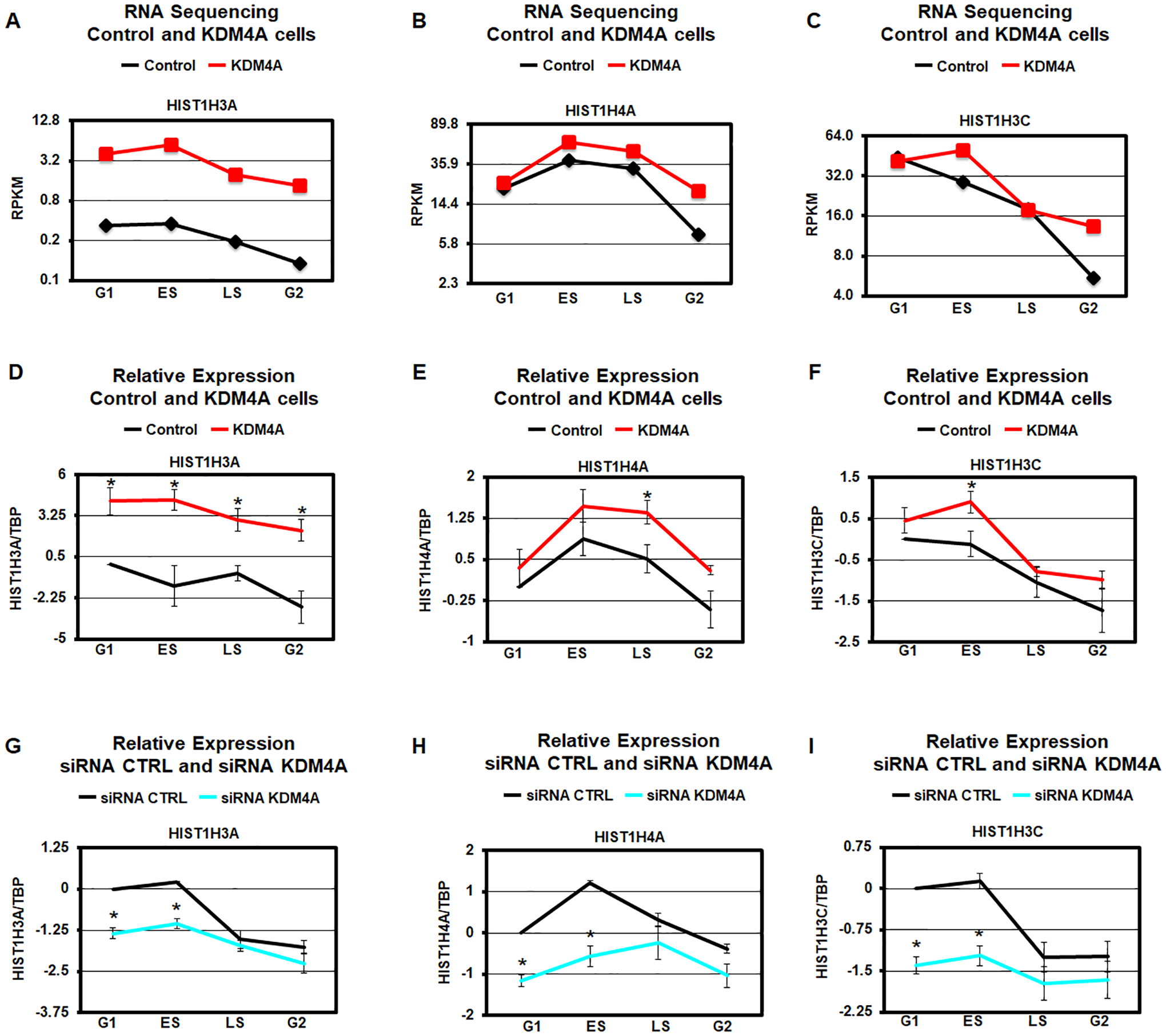
KDM4A regulates replicative histone gene expression over cell cycle. A-C. Expression levels (average RPKM over two biological replicates) of specific histone genes in control (black) and KDM4A overexpressing cells (red). D-F. Log2 ratios between expression values of specific histone genes and TBP, relative to control cells in G1, for control (black) and KDM4A overexpressing cells (red). Averages over two biological replicates are shown. G-I. Log2 ratios between expression values of specific histone genes and TBP, relative to control siRNA in G1, for control siRNA (black) and siRNA against KDM4A (blue). The results represent the average of three independent siRNAs. * T-test p-value < 0.05 for the comparison between KDM4A and control cells or siRNA KDM4A relative to siRNA control for the same phase of cycle.
KDM4A catalytic activity is required for histone gene regulation
We then assessed whether the increased expression of replicative histones required KDM4A enzymatic activity. We made RPE stable cell lines overexpressing either an empty vector, full length KDM4A, or KDM4A catalytic mutant (H188A) and demonstrated that altered expression was catalytically-dependent (HIST1H3A and HIST1H4A; Fig. 3A–B). Consistent with this observation, we observed enriched genome accessibility at the replicative canonical histones upon KDM4A overexpression (Fig. 3C; p=5.3×10−4). We further demonstrated that KDM4A occupied replicative canonical histone genes upon analyzing publicly available KDM4A ChIP sequencing data [24] (Fig. 3D and Fig. S3; p=7.7×10−4). These data are consistent with catalytically active KDM4A directly regulating replicative canonical histone gene expression.
Figure 3.
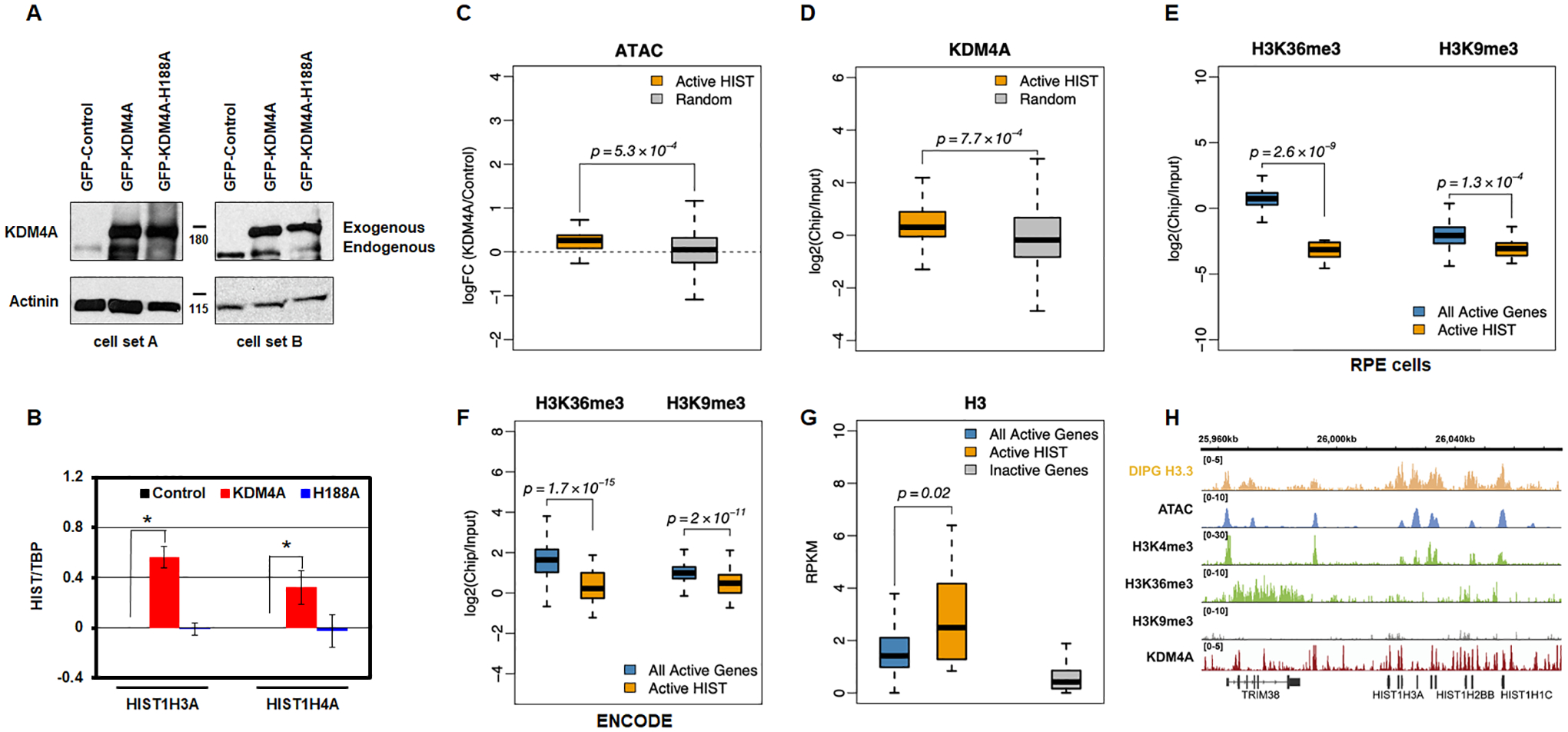
KDM4A regulation of replicative histone genes requires its catalytic activity. A. Western blot representing two biologically independent cell lines used to assess KDM4A catalytic activity. B. Log2 ratios between expression values of specific histone genes and TBP, relative to control cells, for control (black), KDM4A (red), and KDM4A-H188A overexpressing cells (catalytic mutant, blue). Averages over two biological replicates are shown. C. Promoters of active replicative histone genes increase their accessibility upon KDM4A overexpression. Boxplots of the fold change in ATAC-seq read density upon KDM4A overexpression, compared to control, at TSS-proximal regions (TSS +/− 1Kbp) of active histone genes, compared to a random sample of other transcriptionally active genes. D. KDM4A presents an increased occupancy at the promoters of active replicative histone genes. Boxplots of the ChIP-seq enrichment of KDM4A (log ratio of ChIP over input) at TSS-proximal regions (TSS +/− 1Kbp) of active histone genes compared to a random sample of other transcriptionally active genes (defined based on RNA-seq expression values, RPKM>1). E. Unlike other transcriptionally active genes (defined based on RNA-seq expression values, RPKM>1), the bodies of histone genes are depleted in H3K36me3. They are also depleted in H3K9me3. ChIP-seq enrichment of H3K36me3 and H3K9me3 at histone genes and other transcriptionally active genes. F. H3K36me3 and H3K9me3 enrichment at all active genes and active replicative histone genes from ENCODE human GM12878 human cells. G. Similar to other transcriptionally active genes, histone genes are enriched in histone H3. ChIP-seq enrichment of histone H3.3 at histone genes and other transcriptionally active genes based on public ChIP-seq data from DIPG cells [31]. H. Open chromatin and enrichment of active promoter histone marks and histone H3 at histone genes. Genomic tracks show the peaks of ATAC-seq signal and ChIP-seq enrichment of active histone mark H3K4me3, but no strong enrichment of H3K36me3 or H3K9me3 marks. The ChIP-seq enrichment of histone H3.3 [public data from DIPG cells (DIPG H3 ChIP-seq; [31]) is shown as a separate track on top. The KDM4A ChIP-seq enrichment [24] is shown as a separate track on the bottom.
Even though KDM4A appears to directly regulate replicative canonical histone genes, we observed an absence of the profiled histone modifications at the gene bodies of modulated histone genes (i.e., H3K9me3 and H3K36me3 [15]; Fig 3E, RPE cells). Consistent with this observation, analysis of public ChIP sequencing data from ENCODE [23] suggests that histone gene bodies have unusually low levels of active histone modification H3K36me3 given their active transcriptional state (Fig. 3F). While these modifications were unusually low at the replicative canonical histones, we observed histone 3 enrichment across the whole gene (H3; Fig. 3G) [31] as well as the presence of other active modifications that are specific to promoter regions (i.e., H3K4me3; Fig. 3H). These data suggest that KDM4A could be modulating another histone modification at these loci or an unidentified non-histone substrate involved in histone gene regulation since catalytic activity was required and KDM4A directly binds these target genes.
KDM4A interacts with the replicative canonical histone transcription and processing machinery
In order to further understand the biochemical role that KDM4A could have in modulating replicative canonical histone gene expression, we immunoprecipitated endogenous KDM4A and performed mass spectrometry analysis on two independent replicates so that interacting partners could be identified (Fig 4A; 518 proteins, Table S4). Ingenuity Pathway Analysis (IPA) of KDM4A interactors from the independent experiments revealed that KDM4A most significantly interacted with proteins in the RNA post-transcriptional modification category (Fig. 4A–B, Fig. S4A and Table S4). Specifically, these complexes were enriched for factors involved in transcription and RNA processing (e.g., splicing factors and RNA Polymerase II; Table S4). These interactions were confirmed by endogenous KDM4A co-immunoprecipitations (Fig. 4C–H) and reciprocal co-immunoprecipitations (Fig. 4I–N). These data are also consistent with the GSEA categories we observed upon KDM4A overexpression (Fig. S1B).
Next, we tested whether KDM4A overexpression resulted in increased histone RNA transcription by evaluating the RNA levels in the chromatin fraction. RNAs associated with the chromatin fractions were purified and then analyzed by qRT-PCR (Fig. 4O). We observed a significant enrichment for histone gene RNAs in the chromatin fraction upon KDM4A overexpression (Fig. 4O). Therefore, KDM4A appears to regulate histone gene expression directly on the chromatin.
Of interest, these experiments identified interactions with key members of the replicative histone transcription hnRNPUL1 and FUS/TLS as well as the active RNA Polymerase II (Fig. 4C–D, 4I–K) [1]. hnRNPUL1 and FUS/TLS are also implicated in replicative histone 3’-UTR processing [1, 32]. Therefore, we assessed whether the observed expression differences were associated with altered 3’-UTR processing. Upon analyzing the ratios of 3’-UTR coverage to the gene body coverage in our RNA-sequencing data, we observed very similar ratios for all replicative histones in control and KDM4A overexpressing cells (Fig. 4P–Q). Strong differences were not observed across any of these histone genes (Fig. 4Q; r2 = 0.92). The upregulated histones had universally increased RNA coverage across the entire mRNA (Fig. 4P). These results are consistent with KDM4A overexpression equally regulating transcription and processing of canonical histone genes.
In order to assess whether KDM4A modulates histone gene expression in conjunction with hnRNPUL1 and FUS/TLS, we tested whether FUS/TLS or hnRNPUL1 depletion could prevent the increased histone expression upon KDM4A overexpression. Either FUS/TLS or hnRNPUL1 siRNA depletion significantly reduced the HIST1H4A expression observed in KDM4A overexpressing cells (Fig. 4R and Fig. S4B). In fact, KDM4A overexpression prevented the reduced expression caused by FUS/TLS or hnRNPUL1 depletion in control cells. Taken together, our data suggest a role for KDM4A in modulating the transcription and processing of histone genes through FUS/TLS and hnRNPUL1.
DISCUSSION
In this manuscript, we report a new role for KDM4A in modulating replicative canonical histone gene expression throughout the cell cycle. These canonical histones are cleaved at the 3’-end and typically do not contain polyA tails, which requires specific transcriptional and processing factors [1, 2]. We demonstrate that catalytically active KDM4A is required for the upregulation of histone gene expression. Furthermore, KDM4A interacts with members of the transcription/processing machinery controlling histone gene expression. For example, KDM4A interacts with hnRNPUL1 and FUS/TLS, which are key proteins controlling histone transcription and RNA processing. These proteins are also required for the increased expression caused by KDM4A overexpression. These data suggest that these regulatory proteins are interacting with KDM4A in order to modulate histone expression. Future studies need to further resolve whether they are recruited by KDM4A and working directly with catalytically active KDM4A or whether they are functioning in a parallel pathway to catalytically active KDM4A in order to control the expression of histone genes during cell cycle.
Our data is consistent with KDM4A directly binding and regulating canonical histones genes. Even though catalytic activity is required for the increased expression of these genes, we observed an absence of known KDM4A histone methylated substrates (H3K9/36me3) at their locus. The lack of the modifications is observed in our cell model and in the ENCODE datasets. Since KDM4A occupies canonical histone genes, H3K9/36me3 levels could be maintained at a low level, which explains the lack of their presence at these genes. However, some histone genes are not occupied by KDM4A and also have low/no occupancy of H3K9/36me3. Nevertheless, future studies should assess the impact KDM4A depletion or inhibition have on histone methylation states. Future studies should also address whether modest alterations in regions with low levels of methylation could have a profound impact on expression or whether there could be unappreciated KDM4A histone substrates at the histone genes.
Since KDM4A catalytic activity was required for the increased histone expression, studies should also test whether the regulation of histone genes is through an unappreciated non-histone substrate within the transcription/processing complex associated with KDM4A. For example, while arginine methylation has been known to regulate transcription and mRNA processing factors [33], lysine methylation has only recently been shown to occur on these types of proteins [e.g., snRNP70 [34]], which highlights a new mode of regulation for these protein complexes. Since KDM4A overexpression also mis-regulated the RNA binding and transcription gene categories, there is a possibility that their mis-regulation could have an impact on transcriptional changes downstream of KDM4A overexpression. Further studies are required to resolve the exact mechanism by which KDM4A is directly or indirectly controlling both canonical replicative histones and other modulated genes.
KDM4A protein levels are tightly regulated across the cell cycle, and overexpression of this enzyme leads to altered replication timing and DNA copy gains during S phase [8, 10–15]. The regulation of replicative canonical histone genes by KDM4A adds another layer to its general roles in S phase. Taken together, this study highlights KDM4A as a novel master regulator for replicative histones during the cell cycle and describes a new role for KDM4A during cell cycle.
Supplementary Material
Table S2. Related to Figure 1. Fold-change values of histone gene expression differences shown in heatmap in Figure 1D. The colors used in the table are the same as used in the heatmap.
Table S1. Related to Figure 1. Fold-change values of gene expression differences shown in heatmap in Figure 1B. The colors used in the table are the same as used in the heatmap.
Table S4. Related to Figure 4. Proteins from the most enriched functional categories among protein sets detected by mass spectrometry analysis after endogenous KDM4A immunoprecipitation in 293T cells. IPA results for two replicate experiments.
Table S3. Related to Figure 1 and Figure S1. Differentially expressed genes (>2-fold change, FDR<0.05) in KDM4A overexpressing cells at any phase of the cell cycle, and their GSEA enriched functional categories.
Figure S1. Related to Figure 1. A. Canonical cell cycle-regulated genes. Differential gene expression patterns across cell cycle in control RPE cells. The heatmap represents the levels of expression at four phases of cell cycle for the genes that show more than two-fold change in expression between any two phases. Colors indicate relative expression values scaled between minimum and maximum levels for a given gene. B. Enrichment p-values for top 10 functional categories detected by GSEA that were misregulated upon KDM4A overexpression.
Figure S2. Related to Figure 2. Western blot representing the siRNA used in Figure 2G–I.
Figure S3. Related to Figure 3. KDM4A occupancy at the promoters of active replicative histone genes. The heatmap represents the levels of ChIP-seq enrichment of KDM4A (log ratio of ChIP over input) at TSS-proximal regions (TSS +/− 1Kbp) of annotated histone genes.
Figure S4. Related to Figure 4. A. Enrichment p-values for functional categories detected by Ingenuity Pathway Analysis (IPA) among protein sets from an independent mass spectrometry analyses of endogenous KDM4A complexes. In addition to top 10 categories represented, p-value for DNA replication category studied in [12] is shown. B. Western blots representing the knock-down used in Figure 4R.
HIGHLIGHTS.
Catalytically active KDM4A regulates canonical replicative histone genes during cell cycle
KDM4A binds and regulates histone gene accessibility
KDM4A interacts with transcriptional and RNA processing factors regulating histone genes
KDM4A overexpression promotes histone gene expression and 3’-UTR processing
ACKNOWLEDGEMENTS
We are grateful to Ravi Mylvaganam, Christina Luo, Hiranmayi Ravichandran, Rachel Servis, Jeremy Hon and the Massachusetts General Hospital Flow Cytometry Core for assistance with flow cytometry. We thank Dr. Sangita Pal for technical contributions. We would like to thank Drs. Alfonso Bellacosa, Jon Chernoff, Thomas Clarke and Vasily Studitsky for comments on the manuscript. Work related to this study is supported by funding to Johnathan R. Whetstine from R01GM097360 and the NIH/NCI Cancer Center Support Grant P30 CA006927. J.R.W. is a recipient of an American Lung Association Lung Cancer Discovery Award. S.M. was partially funded by the Senior Research Training Fellowship from the American Lung Association. R.I.S. was supported by NIH NIDDK P30 DK040561.
CONFLICT OF INTEREST
J.R. Whetstine has received funding from AstraZeneca, is a consultant at Qsonica, previously consulted for Celgene, and served on the Salarius Pharmaceuticals advisory board and served as a consultant. The other authors do not declare any conflict of interest.1
Footnotes
“The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.”
REFERENCES
- [1].Romeo V, Schumperli D, Cycling in the nucleus: regulation of RNA 3’ processing and nuclear organization of replication-dependent histone genes, Curr Opin Cell Biol, 40 (2016) 23–31. [DOI] [PubMed] [Google Scholar]
- [2].Mendiratta S, Gatto A, Almouzni G, Histone supply: Multitiered regulation ensures chromatin dynamics throughout the cell cycle, J Cell Biol, 218 (2019) 39–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Groth A, Ray-Gallet D, Quivy JP, Lukas J, Bartek J, Almouzni G, Human Asf1 regulates the flow of S phase histones during replicational stress, Mol Cell, 17 (2005) 301–311. [DOI] [PubMed] [Google Scholar]
- [4].Groth A, Corpet A, Cook AJ, Roche D, Bartek J, Lukas J, Almouzni G, Regulation of replication fork progression through histone supply and demand, Science, 318 (2007) 1928–1931. [DOI] [PubMed] [Google Scholar]
- [5].Zhao X, McKillop-Smith S, Muller B, The human histone gene expression regulator HBP/SLBP is required for histone and DNA synthesis, cell cycle progression and cell proliferation in mitotic cells, J Cell Sci, 117 (2004) 6043–6051. [DOI] [PubMed] [Google Scholar]
- [6].Marzluff WF, Gongidi P, Woods KR, Jin J, Maltais LJ, The human and mouse replication-dependent histone genes, Genomics, 80 (2002) 487–498. [PubMed] [Google Scholar]
- [7].Black JC, Van Rechem C, Whetstine JR, Histone lysine methylation dynamics: establishment, regulation, and biological impact, Mol Cell, 48 (2012) 491–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Van Rechem C, Black JC, Abbas T, Allen A, Rinehart CA, Yuan GC, Dutta A, Whetstine JR, The SKP1-Cul1-F-box and leucine-rich repeat protein 4 (SCF-FbxL4) ubiquitin ligase regulates lysine demethylase 4A (KDM4A)/Jumonji domain-containing 2A (JMJD2A) protein, J Biol Chem, 286 (2011) 30462–30470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Whetstine JR, Nottke A, Lan F, Huarte M, Smolikov S, Chen Z, Spooner E, Li E, Zhang G, Colaiacovo M, Shi Y, Reversal of histone lysine trimethylation by the JMJD2 family of histone demethylases, Cell, 125 (2006) 467–481. [DOI] [PubMed] [Google Scholar]
- [10].Black JC, Allen A, Van Rechem C, Forbes E, Longworth M, Tschop K, Rinehart C, Quiton J, Walsh R, Smallwood A, Dyson NJ, Whetstine JR, Conserved antagonism between JMJD2A/KDM4A and HP1gamma during cell cycle progression, Molecular cell, 40 (2010) 736–748. [DOI] [PubMed] [Google Scholar]
- [11].Black JC, Atabakhsh E, Kim J, Biette KM, Van Rechem C, Ladd B, Burrowes PD, Donado C, Mattoo H, Kleinstiver BP, Song B, Andriani G, Joung JK, Iliopoulos O, Montagna C, Pillai S, Getz G, Whetstine JR, Hypoxia drives transient site-specific copy gain and drug-resistant gene expression, Genes & development, 29 (2015) 1018–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Black JC, Manning AL, Van Rechem C, Kim J, Ladd B, Cho J, Pineda CM, Murphy N, Daniels DL, Montagna C, Lewis PW, Glass K, Allis CD, Dyson NJ, Getz G, Whetstine JR, KDM4A lysine demethylase induces site-specific copy gain and rereplication of regions amplified in tumors, Cell, 154 (2013) 541–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Black JC, Zhang H, Kim J, Getz G, Whetstine JR, Regulation of Transient Site-specific Copy Gain by MicroRNA, J Biol Chem, 291 (2016) 4862–4871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Mishra S, Van Rechem C, Pal S, Clarke TL, Chakraborty D, Mahan SD, Black JC, Murphy SE, Lawrence MS, Daniels DL, Whetstine JR, Cross-talk between Lysine-Modifying Enzymes Controls Site-Specific DNA Amplifications, Cell, 174 (2018) 803–817 e816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Clarke TL, Tang R, Chakraborty D, Van Rechem C, Ji F, Mishra S, Ma A, Kaniskan HU, Jin J, Lawrence MS, Sadreyev RI, Whetstine JR, Histone Lysine Methylation Dynamics Control EGFR DNA Copy-Number Amplification, Cancer Discov, 10 (2020) 306–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ, Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position, Nat Methods, 10 (2013) 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR, STAR: ultrafast universal RNA-seq aligner, Bioinformatics, 29 (2013) 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Anders S, Pyl PT, Huber W, HTSeq--a Python framework to work with high-throughput sequencing data, Bioinformatics, 31 (2015) 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Robinson MD, McCarthy DJ, Smyth GK, edgeR: a Bioconductor package for differential expression analysis of digital gene expression data, Bioinformatics, 26 (2010) 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Huang da W, Sherman BT, Lempicki RA, Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources, Nat Protoc, 4 (2009) 44–57. [DOI] [PubMed] [Google Scholar]
- [21].Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP, Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles, Proc Natl Acad Sci U S A, 102 (2005) 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Li H, Durbin R, Fast and accurate short read alignment with Burrows-Wheeler transform, Bioinformatics, 25 (2009) 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Consortium EP, An integrated encyclopedia of DNA elements in the human genome, Nature, 489 (2012) 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Metzger E, Stepputtis SS, Strietz J, Preca BT, Urban S, Willmann D, Allen A, Zenk F, Iovino N, Bronsert P, Proske A, Follo M, Boerries M, Stickeler E, Xu J, Wallace MB, Stafford JA, Kanouni T, Maurer J, Schule R, KDM4 Inhibition Targets Breast Cancer Stem-like Cells, Cancer Res, 77 (2017) 5900–5912. [DOI] [PubMed] [Google Scholar]
- [25].Edgar R, Domrachev M, Lash AE, Gene Expression Omnibus: NCBI gene expression and hybridization array data repository, Nucleic Acids Res, 30 (2002) 207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Ramirez F, Ryan DP, Gruning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dundar F, Manke T, deepTools2: a next generation web server for deep-sequencing data analysis, Nucleic Acids Res, 44 (2016) W160–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Henriques T, Gilchrist DA, Nechaev S, Bern M, Muse GW, Burkholder A, Fargo DC, Adelman K, Stable pausing by RNA polymerase II provides an opportunity to target and integrate regulatory signals, Mol Cell, 52 (2013) 517–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Van Rechem C, Black JC, Boukhali M, Aryee MJ, Graslund S, Haas W, Benes CH, Whetstine JR, Lysine Demethylase KDM4A Associates with Translation Machinery and Regulates Protein Synthesis, Cancer Discov, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Bertoli C, Skotheim JM, de Bruin RA, Control of cell cycle transcription during G1 and S phases, Nat Rev Mol Cell Biol, 14 (2013) 518–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Goldenson B, Crispino JD, The aurora kinases in cell cycle and leukemia, Oncogene, 34 (2015) 537–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Piunti A, Hashizume R, Morgan MA, Bartom ET, Horbinski CM, Marshall SA, Rendleman EJ, Ma Q, Takahashi YH, Woodfin AR, Misharin AV, Abshiru NA, Lulla RR, Saratsis AM, Kelleher NL, James CD, Shilatifard A, Therapeutic targeting of polycomb and BET bromodomain proteins in diffuse intrinsic pontine gliomas, Nat Med, 23 (2017) 493–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Raczynska KD, Ruepp MD, Brzek A, Reber S, Romeo V, Rindlisbacher B, Heller M, Szweykowska-Kulinska Z, Jarmolowski A, Schumperli D, FUS/TLS contributes to replication-dependent histone gene expression by interaction with U7 snRNPs and histone-specific transcription factors, Nucleic Acids Res, 43 (2015) 9711–9728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Bedford MT, Clarke SG, Protein arginine methylation in mammals: who, what, and why, Mol Cell, 33 (2009) 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Carlson SM, Moore KE, Sankaran SM, Reynoird N, Elias JE, Gozani O, A Proteomic Strategy Identifies Lysine Methylation of Splicing Factor snRNP70 by the SETMAR Enzyme, J Biol Chem, 290 (2015) 12040–12047. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S2. Related to Figure 1. Fold-change values of histone gene expression differences shown in heatmap in Figure 1D. The colors used in the table are the same as used in the heatmap.
Table S1. Related to Figure 1. Fold-change values of gene expression differences shown in heatmap in Figure 1B. The colors used in the table are the same as used in the heatmap.
Table S4. Related to Figure 4. Proteins from the most enriched functional categories among protein sets detected by mass spectrometry analysis after endogenous KDM4A immunoprecipitation in 293T cells. IPA results for two replicate experiments.
Table S3. Related to Figure 1 and Figure S1. Differentially expressed genes (>2-fold change, FDR<0.05) in KDM4A overexpressing cells at any phase of the cell cycle, and their GSEA enriched functional categories.
Figure S1. Related to Figure 1. A. Canonical cell cycle-regulated genes. Differential gene expression patterns across cell cycle in control RPE cells. The heatmap represents the levels of expression at four phases of cell cycle for the genes that show more than two-fold change in expression between any two phases. Colors indicate relative expression values scaled between minimum and maximum levels for a given gene. B. Enrichment p-values for top 10 functional categories detected by GSEA that were misregulated upon KDM4A overexpression.
Figure S2. Related to Figure 2. Western blot representing the siRNA used in Figure 2G–I.
Figure S3. Related to Figure 3. KDM4A occupancy at the promoters of active replicative histone genes. The heatmap represents the levels of ChIP-seq enrichment of KDM4A (log ratio of ChIP over input) at TSS-proximal regions (TSS +/− 1Kbp) of annotated histone genes.
Figure S4. Related to Figure 4. A. Enrichment p-values for functional categories detected by Ingenuity Pathway Analysis (IPA) among protein sets from an independent mass spectrometry analyses of endogenous KDM4A complexes. In addition to top 10 categories represented, p-value for DNA replication category studied in [12] is shown. B. Western blots representing the knock-down used in Figure 4R.