

1. Introduction
Selective intracellular protein degradation is involved in every aspect of cellular life. A unifying theme in eukaryotic protein degradation is found in the tagging and destruction mechanism called the ubiquitin proteasome system (UPS): a particular protein is targeted for degradation by the recursive covalent addition of the small protein ubiquitin (Ub), which leads to recognition of the resulting multiubiquitin chain by the 26S proteasome. The breadth of use of UPS in cellular life is allowed by the specificity of target protein recognition; in this way, drastic differences in protein half-life can be observed in the same cellular compartment, thus assuring the required specificity for a regulatory mechanism that is sufficiently flexible to destroy any cellular protein.
Ubiquitination of proteins occurs by the successive action of a cascade of three enzymes (Figure 1). This process is extensively detailed in numerous reviews 1 so a useful summary is described here. The E1 ubiquitin activating enzyme uses ATP to covalently activate and then add ubiquitin to an E2 ubiquitin conjugating (UBC) enzyme. Ubiquitin is then transferred from the ubiquitin-charged E2 to the substrate or the growing ubiquitin chain by the action of an E3 ubiquitin ligase, resulting in a substrate-attached multi-ubiquitin chain that is recognized by the proteasome, leading to degradation of the ubiquitinated substrate. The ubiquitination enzymes operate in a hierarchy: in yeast, there is one E1, 10 E2s and a few hundred E3 ubiquitin ligases. In the mammal the same organization exists, but with many more E2s and E3s. The E3 ligases are the most immediately involved in selective targeting of ubiquitination substrates, and thus often the focus of investigation when studying a degradation pathway. This three enzyme cascade presents a skeletal picture; in most cases ancillary factors participate in substrate recognition and transfer of the ubiquitinated product to the proteasome 2,3.
Figure 1. The enzymes of ubiquitination.
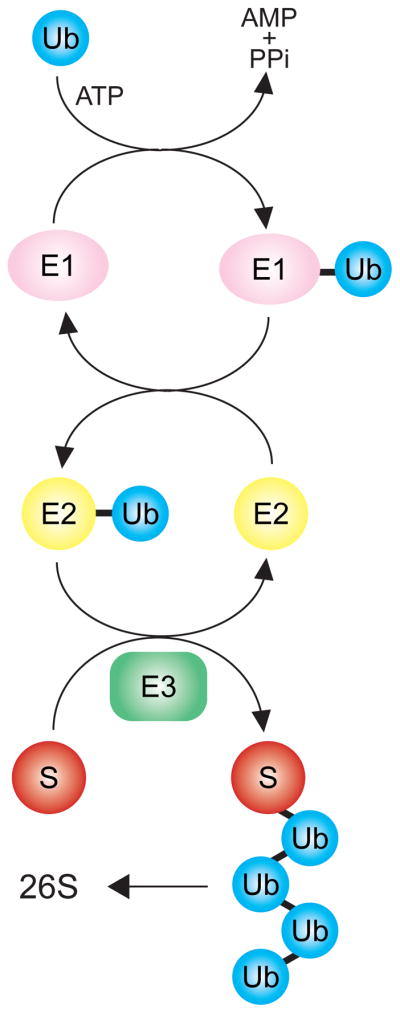
The covalent attachment of the small (7.6 kD) protein ubiquitin (Ub) to a target substrate (S) proceeds by a cascade of enzymes. The ubiquitin activating enzyme (UBE), or E1, adds Ub to itself in a thioester linkage on the E1. Ubiquitin conjugating enzymes (UBCs), E2s, are charged by transfer of ubiquitin from the E1 to a thioester linkage on the E2. Finally, a ubiquitin ligase, or E3, brokers the transfer of ubiquitin to the target substrate S, producing a ubiquitinated protein. This process is continued by continued addition of the next ubiquitin to the previously added one, producing a polyubiquitin chain. The polyubiquitin chain is recognized with high specificity by the 26S proteasome, causing degradation of the S target. The E3 is thus a critical determininat of specificity in this modification scheme. Typically, there is one E1, tens of E2s (10 in yeast, ~ 50 in mammals) and many E3s.
2. Degradation for regulation or protein quality control
Despite the wide range of uses and recognition mechanisms for UPS substrates, there are two general themes in protein degradation, referred to here as regulation and quality control. In regulation, ubiquitin-mediated degradation is used to effect regulatory changes in a specific proteins’s levels and activity. Regulated degradation is keyed to physiological or developmental signals to allow selective loss of a protein in conditions that demand its levels be altered. Examples include the regulated degradation of p53 4, temporally programmed destruction of cyclins and other cell cycle regulators 5, and the selective degradation of glucose-synthesizing enzymes after feeding 6. In all cases unique features of the targeted protein mediate destruction, ensuring high specificity. Thus, many E3 ligases have specific binding sites for a single or very few proteins; this specific ligase-substrate interaction is a principle determinant of selective ubiquitination by many E3s. E3-substrate binding is often sufficient to allow selective ubiquitination. This has been demonstrated in experiments where non-recognized proteins become substrates when made able to bind to an E3 subunit either by molecular biological alteration of the E3 7, or use of a small molecule to mediate binding to the heterologous target 8. Similarly, promoting binding of an E3 to a normally non-recognized protein to cause that proteins’ selective degradation is a tactic that viruses have employed in numerous circumstances 9,10.
Variations of regulated degradation abound, but all cause highly specific alterations of a target protein level by selective ubiquitination. Some of these include: phosphorylation of a targeted protein as a prerequisite for specific E3 association 11; association of the plant hormone auxin to an E3 subunit to create a binding site for an auxin-targeted transcriptional repressor 12, or E3 recognition of HIF1α that has undergone oxygen-dependent modification to couple HIF1α levels to oxygen demand 13. In even the most complex variations, high specificity between the E3 and its target is a lynchpin of the regulatory strategy.
A distinct, second theme in cellular proteolysis is protein degradation for purposes of cellular quality control. That is, the selective intracellular degradation of misfolded or inappropriately assembled proteins. Protein quality control is implicated in maintaining acceptably low levels of aberrant proteins, and is thought to be an important component in management of cellular stress. Separate pathways of protein quality control exist in numerous, and probably all, compartments of the cell 14,15, and apparently in all kingdoms of life 16,17.
It has been empirically observed in many instances that degradative quality control is, like regulation of individual proteins, highly specific and selective, but in a totally different sense. It is often found that a normal protein will be quite stable while a mutant that does not fold correctly or folds too slowly will be subject to selective degradation. A striking example is found in the single point mutant of the yeast vacuolar enzyme known as carboxypeptidase Y (CPY), called CPY*. While the normal protein is folded in the lumen of the ER and shipped off to the vacuole where it resides, the CPY* point mutant undergoes rapid quality control degradation. In fact, the CPY* variant was the prototype substrate of the now widely studied quality control pathway of ER-associated degradation (ERAD) 18, described in more detail below. Another example of this high specificity is found in the disease-causing CFTR-Δ508 variant of the CFTR transporter. Homozygosity of the Δ508 allele is responsible for the majority of cases of cystic fibrosis (CF). The CFTR-Δ508 protein is functional and abundantly produced, but ER-localized quality control degradation of this slowly folding form brings about the pathologically low levels responsible for the galaxy of CF symptoms 19. Similarly, uncomplexed subunits of multiprotein complexes, such as the T-cell receptor or the alpha 2 repressor proteins, are often subject to rapid degradation that ignores those proteins when part of their correct multimers 20,21. Because many of the diseases of ageing appear to have underpinnings in the improper management of misfolded proteins 22, it is clear that quality control degradation has important roles in the etiology of, and perhaps novel therapeutic strategies for, these various and very serious pathologies.
3. Possible functions of protein quality control
Much of our limited understanding of degradative quality control (QC) comes from the study of mutant proteins generated by nature or molecular biology, to gain insights about recognition mechanisms that presumably operate on natural substrates. Thus, the function of these degradation pathways is not inherently revealed by the use of these model substrates. The reasonable speculation is that QC pathways are present to limit levels of toxic and stress-inducing misfolded proteins in the cell. This appears to be the case in yeast ERAD, where loss of either pathway (HRD or DOA) leads to increased UPR signaling, indicating higher levels of naturally produced ER misfolded proteins, and increased sensitivity to drugs that promote misfolding in the ER23,24. Presumably there will be other examples of such “stress homeostasis” functions in degradative quality control.
There are other possibilities for the cellular functions of these degradative quality control pathways. Today’s QC pathways may have evolved when protein folding was less efficient, so that a given protein had a larger range of structures including those with poor, no, or even toxic functions. The proto-QC pathways could have operated to ensure optimal catalytic activity by lowering the number of less effective structures of a given protein. Today, these pathways may similarly function to effect the turnover of many proteins in the cell, and thus would be a critical part of natural cellular proteostasis. The proteome is as a dynamic entity; each “node” in the protein network has a steady-state level defined by synthesis and degradation. Perhaps the quality control pathways serve a broad function of allowing the turnover of normal proteins, recognizing the extreme members of the ensemble of structures that occur during a normal protein’s “breathing” within its folded state. By this speculative model, “quality control” mechanisms would promote the turnover of many normal proteins, degrading the naturally accessed conformations during its dynamic motions, and the more extreme cases of truly misfolded proteins caused by chemical or thermal damage, or clever molecular biologists.
The idea that normal proteins might be subjected to degradation by a quality control mechanism by recognition of naturally attainable extremes of the folding state allows for some interesting ideas about cellular employment of QC pathways. One notion from our and others’ work on regulation of sterol synthesis is that the high selectivity of protein quality control is harnessed to bring about physiological regulation of normal proteins. This “crossing” of the conceptual boundary between regulation and quality control will be described in detail for feedback regulation of the rate-limiting enzyme of sterol synthesis, HMG-CoA reductase (HMGR). Then some other potential examples of this idea from other sectors of biology will be described. In addition, the implications for understanding and harnessing these quality control or “proteostatic” degradative pathways will be discussed.
4. Regulated degradation of HMG-CoA reductase (HMGR)
HMG-CoA reductase (HMGR) is a key enzyme of the sterol pathway, by which sterols are synthesized from acetyl-CoA (Figure 2). The HMGR-catalyzed reaction produces mevalonic acid from HMG-CoA by reduction with NADPH. The resulting 6-carbon mevalonate is next converted into the isomeric, phosphorylated 5-carbon isoprene subunits dimethylallyl pyrophosphate (DMAPP) and isopentyl pyrophosphate (IPP), that are used as building blocks to construct myriad isoprenoids, including sterols such as cholesterol. This long biosynthetic pathway is feedback regulated at multiple levels, including transcription, translation, and regulated degradation 25,26. The basic theme in all modes is that increased demand for more sterol pathway molecules results in increased levels of the synthetic enzymes, while decreased demand for sterol pathway products results in decreased levels of those enzymes. The study of sterol pathway regulation has led to an astonishing amount of basic biology, and each branch of the regulation could (and often does) encompass a separate review. This work will necessarily focus on the regulated degradation of yeast HMGR, and the insights it has provided into possible functions and use of cellular quality control.
Figure 2. The sterol synthetic, or mevalonate pathway.

The synthesis of sterols and other isoprenes occurs by the sequential construction of the 5 carbon pyrophosphorylated isoprenes IPP and its isomer DMAPP (not shown), followed by iterated condensation to produce 10, 15 and higher carbon pyrophosphates. Twi 15 carbon farnesylpyrophosophate (FPP) molecules are the condensed by production of squalene (not show) to make the 30 carbon lanosterol, the first sterol in the pathway. This sterol is then used to make a variety of sterols, with cholesterol being the principle mammalian product, and ergosterol being that of yeast. The mevalonate pathway has many branches off the main pathway, delivering a wide variety of isroprene lipids for use in many corners of cell and organismal biology. One of these side branches included production of 20 carbon GGPP from 15 carbon FPP, discussed below. The key enzyme HMG-CoA reductase (HMGR) functions at the point indicated, catalyzing the reduction of the 6 carbon hydroxymethylglutaryl group on the CoA with 2NADPH to produce free mevalonic acid, as shown. To simplify the diagram, only the lipid structures are shown, while cofactors and non-lipids are absent.
Part of feedback regulation of the sterol pathway centers on regulated degradation of HMGR, which is a key and often rate-limiting enzyme for the sterol pathway. It was first observed in the mammalian cells that the stability of HMGR was decreased when early sterol precursors or sterols were added to cell culture medium, indicating that the stability of HMGR was subject to feedback regulation. This was borne out by a variety of studies showing that increased levels of pathway products led to increased degradation of HMGR, thus lowering enzyme levels, while decreased production of sterol pathway products led to increased stabilization and elevated enzyme levels. That is, feedback regulation in which sterol pathway activity is keyed to the degradation rate of the HMGR molecule. The mammalian HMGR molecule is an 8-spanning ER-resident integral membrane protein. A multispanning N-terminal domain keeps the protein anchored in the ER, and a linker connects the membrane anchor to the highly conserved, soluble catalytic C-terminal region responsible for HMGR’s essential enzyme activity in all eukaryotes and archebacteria 27,28. It is the N-terminal multispanning domain that mediates regulated degradation of the HMGR molecule, allowing replacement of the C-terminal catalytic region with enzymatic or optical reporters while still preserving the regulated degradation of the molecule.
Yeast has two isozymes of HMGR, Hmg1p and Hmg2p. Both have a similar “body plan” to the single mammalian HMGR: an N-terminal 8-spanning domain, a linker and the attached catalytic activity. The HMGR isozymes of yeast are about 50% identical in sequence over the transmembrane domain, and about 93% identical in the conserved catalytic regions (Figure 3). Furthermore, both Hmg1p and Hmg2p have the conserved motif known as a sterol sensing domain (SSD) that has been implicated in lipid sensing by a variety of proteins including HMGR and Hmg2p (discussed below). Nevertheless, only the Hmg2p isozyme undergoes regulated degradation, in a manner strikingly similar to the mammalian enzyme despite ~ 1 billion years of separate evolution: high flux through the sterol pathway promotes more degradation, while diminished production of sterol pathway products causes high stability. In this way the degradation of Hmg2p can be modulated between a half-life of 10 minutes up to 6 or more hours 29. By contrast, Hmg1p is highly stable in all conditions so far tested 30.
Figure 3. Organization of the HMG-CoA reductase protein.

The integral ER membrane protein HMGR in both mammals and yeast consists of an 8-spanning N-terminal membrane anchor, embedded in the ER membrane (grey) as shown, attached by a linker to the highly conserved catalytic domain that performs the essential enzyme action. Regulated degradation requires the N-terminal transmembrane domain. This region has the conserved sterol-sensing domain (SSD) motif distributed between spans 2 to 5, as discussed in the text.
To gain insight into the mechanisms of regulated HMGR degradation in yeast and in general, we have conducted numerous genetic analyses to find the HRD genes responsible for Hmg-Co A Reductase Degradation. Our initial work discovered the core ubiquitin ligase components called Hrd1p and Hrd3p 31. The ER-associated Hrd1p ligase is absolutely required for regulated degradation of Hmg2p; in the absence of Hrd1p, Hmg2p is completely stable no matter what the level of sterol pathway activity. The subsequent results of many genetic, biochemical, and proteomic studies have yielded much detail about the HRD pathway, which mediates one route of eukaryotic ER-associated degradation (ERAD).
5. The HRD pathway: ERAD and cellular quality control
The ER is the site of robust protein degradation. It has long been appreciated that both lumenal and membrane-bound ER-localized proteins can be subject to degradation 23,31,32. Collectively such degradation processes are now referred to as ERAD. The range of substrates for ERAD is quite large, and this process is best viewed as a quality control pathway that mediates the degradation of misfolded and misassembled lumenal and integral membrane ER-resident proteins. Thus, substrates as distinct as the misfolded, soluble point mutant CPY* and the mutant multispanning membrane protein Sec61-2 are subject to degradation by the HRD pathway, despite their complete lack of sequence similarity, while the normal proteins are completely stable. It appears that the HRD pathway functions in this capacity constitutively in eukaryotic cell. The expression of HRD pathway components is regulated by the Unfolded Protein Response (UPR), the monitoring system that measures unfolded proteins in the ER and regulates the expression of proteins that help remove or refold them 24,33. Cells that lack the HRD pathway by null mutation of the Hrd1p E3 ligase have higher levels of ER misfolded proteins in unstressed conditions and are more sensitive to perturbations that increase ER stress. In yeast, the HRD pathway functions alongside a distinct ERAD pathway mediated by the Doa10p ubiquitin ligase, which is involved in the degradation of a variety of misfolded ER proteins that generally are not HRD pathway substrates, as well as a number of proteins that do not reside in the ER 23. Like the HRD pathway, loss of the DOA pathway also causes increased ER stress and increased sensitivity to agents that trigger the UPR. Both pathways are conserved in mammals, and probably distinct ERAD mechanisms operate in addition 34,35. Considering that the ER is the site of continuous high-flux reception of nascent polypeptides that must be folded and assembled, it is not surprising that a multi-branched and high capacity system for degradation of misfolded proteins is associated with this organelle.
Although there are at least two distinct ERAD pathways in yeast, and more in mammals, there are features of the process that are common to all. All substrates are either present in the lumen of the ER as soluble proteins, or embedded in the ER membrane. Substrates appear to be selected for degradation based on structural criteria that can be shared, apparently, between totally distinct proteins. The recognition of lumenal substrates can involve specific glycosylations, features of the misfolded substrate, or both 36,37. Lumenal substrates must be partially moved to the cytosol, since the required E3 ligase active sites and E2 ubiquitin conjugating enzymes are all in the cytosol. As the substrate is ubiquitinated, this dislocation or retrotranslocation continues as ubiquitination proceeds, although the detailed order or interdependence of these processes has not been discerned. Finally, the ubiquitinated protein is delivered to the proteasome, which also resides in the cytosolic space, perhaps in loose association with the ER membrane. ER membrane protein degradation follows a similar path. Although ubiquitination of integral membrane substrates could be initiated on cytosolic lysines, membrane proteins similarly require the retrotranslocation machinery for ERAD, and these substrates are completely removed from the ER membrane in the course of degradation 38 (and R. Garza et al., manuscript submitted). Thus, ERAD is a ubiquitin-mediated pathway that is intimately involved with the cell biology of the ER where it is localized.
6. The machinery of the HRD pathway
This basic scheme of recognition, retrotranslocation, ubiquitination and proteasomal degradation occurs for all HRD substrates, and presumably in all ERAD pathways. The HRD machinery is fairly well characterized, although many questions remain about their functions. The HRD pathway is mediated by an ER-localized complex of proteins, including the E3 ligase Hrd1p, and a number of proteins that work in conjunction with Hrd1p to mediate recognition, ubiquitination and retrotranslocation of both membrane and lumenal substrates out of the ER (Figure 4). Hrd1p is an eight-spanning, ER-localized membrane protein with an N-terminal membrane anchor linked to the C-terminal cytosolic domain that bears a canonical RING-H2 ubiquitin ligase motif. Hrd1p is responsible for the ubiquitin ligase activity of the HRD complex. Hrd1p employs two E2 ubiquitin conjugating enzymes, primarily Ubc7p, and to a lesser extent, Ubc1p. The Ubc7p is held on the cytoplasmic face of the ER by Cue1p, which serves both as an docking site for this otherwise soluble E2, and as a potent activator of the E2 as well 39. No mechanism of docking for Ubc1p has been discerned, although it does appear to show localization to the ER surface (T. Sommer, personal communication).
Figure 4. Protein machinery of the HRD pathway.

The HRD complex functions in ER-associated degradation (ERAD) to recognize and ubiquitinate misfolded and misassembled ER proteins. The Hrd1p E3 ubiquitin ligase works in a complex that is involved in all aspects of ERAD substrate recognition, ubiquitination, and movement to the cytosolic face where the proteasome resides. Hrd1p/Hrd3p are the E3 complex, and Ubc7p is the principle E2. The Cdc48 AAA-ATPase hexamer functions to remove substrates from the ER. Hrd3p, Kar2p, and Yos9p all function to recognized luminal ERAD substrates. The Usa1p protein regulates Hrd1p stability and activity along with allowing optimal Hrd1p activity. The HRD pathways has a large set of substrates that are recognized by a variety of criteria that hallmark poor folding.
Together with Hrd1p and Hrd3p, a number of other proteins form a “HRD complex” that mediates the broad range of degradation events that hallmark this and other ERAD pathways (Figure 4). Hrd1p is found in stoichiometric complex with Hrd3p, another integral ER membrane protein with the majority of its sequence in the ER lumen. Hrd3p appears to participate in the recognition of some ER substrates and the recruitment of lumenal factors involved in detection and “HRDing” of substrates to Hrd1p-dependent ubiquitination. These lumenal factors include the classical Hsp70 chaperone Kar2p (BiP in mammals) and the lectin/chaperone Yos9p 40–42. How Kar2p, Yos9p, and Hrd3p divide the responsibility of lumenal substrate recognition is not clear, and probably depends on the structural features of a given substrate. In addition, there are several integral membrane proteins of the HRD complex, including the prototype “derlin” called Der1p, Ubx2p, and Usa1p. Der1p, and derlins in generals, have been proposed to be involved in movement of lumenal proteins across the ER membrane to the cytoplasmic ubuiquitiation machinery 43–45. Consistent with this, a der1Δ null mutation causes full stabilization of lumenal ERAD substrates such as CPY* 46. Ubx2p has a role in recruiting the cytosolic Cdc48p AAA-ATPase complex Cdc48p/Npl4p/Ufd1p to the ER membrane 47,48. The Cdc48p ATPase complex is required for retrotranslocation of both lumenal and membrane substrates 7,38, and probably functions to power retrotranslocation, or dislocation, by using its hexameric AAA-ATPase domains to derive ATP energy for this action (R. Garza et al., manuscript submitted). Although it has reasonably been proposed by numerous investigators that a protienacious channel mediates movement of ERAD substrate polypeptide segments across the ER membrane during retrotranslocation, no fully convincing candidate has emerged, although the derlins, the anterograde channel Sec61, and the large transmembrane domain of Hrd1p itself have all been put forth as reasonable candidates for this function. In addition, mechanisms that involve lipid intermediates rather than a canonical channel have been put forth, but so far with no experimental evidence for this channel-alternative view 49. Our recent work on Usa1p indicates that it has roles in both Hrd1p function and regulation. Usa1p is also absolutely required for Hrd1p-mediated self-degradation, which has been proposed to be a mechanism for self-regulation of this potentially toxic ligase 40. In addition, Usa1p also diminishes the toxicity of overexpressed Hrd1p by mechanisms independent of its degradation-mediating effects (S. Bartle and R. Hampton, manuscript submitted)
The broad range of damaged, unassembled, and misfolded proteins that are under the umbrella of HRD substrates begs the question of how such a panoply of proteins are recognized. Instead of there being a specific “degron” that allows recognition of each substrate, in all likelihood the substrates have shared structural features that allow entry into the HRD pathway. For Hrd1p substrates there is likely to be some division of recognition labor among the lumenal factors Yos9p, Kar2p, and Hrd3p itself to recognize misfolded lumenal proteins. Furthermore, both folding features and particular glycosylations can be involved the decision for glycoproteins to enter ERAD pathways. There are some substrates for which the appropriately modified N-linked glycan chain is required for ERAD, and some for which glycosylation is not a requirement for ERAD, including both membrane-bound proteins such as Hmg2p (our unpublished observations), and some lumenal substrates 36,37. Perhaps such a broad range of substrates requires a number of separate molecular devices dedicated to recognition of distinct criteria of misfolding. However, a genetic feature of the HRD pathway indicates that there may be multiple modes of recognition possible for a given substrate. Overexpression of the HRD1 gene from strong promoter can override a number of individual null mutants that show strong HRD defects when the ligase is at its normal genomic levels. The hrd3Δ null mutant shows degradation of both Hmg2p and CPY* when Hrd1p is sufficiently elevated, and overexpression of Hrd1p can similarly override the stabilizing effect of a usa1Δ null or a yos9Δ null 40 (and S. Bartle et al., manuscript submitted). Thus, no one factor appears to be required for recognition. One possibility is that Hrd1p has intrinsic ability to recognize substrates that is augmented by these lumenal factors. Alternatively, it may be that there are multiple routes for substrate recognition, so that removal of one factor by null mutation allows for lower efficiency recognition by the remaining factor(s) to be revealed by elevation of Hrd1p. It will be important to parse out the train of events and molecular components that bring about recognition, membrane localization, presentation to the cytosolic RING domain, and eventual retrotranslocation of HRD substrates.
7. Degradation of integral membrane proteins: ERAD-M
A defining feature of the HRD pathway is its ability to degrade both lumenal and membrane anchored proteins. The degradation of membrane-bound substrates is more poorly understood than the degradation of lumenal ones, but appears to involve fewer ERAD components. For example, degradation of Hmg2p does not require Der1p, nor Kar2p, nor Yos9p 50 (S. Bartle and R. Hampton, manuscript submitted; and unpublished observations). Furthermore, although Usa1p is strongly required for degradation of lumenal substrates, its plays only a small role in Hmg2p degradation. Such differences have led to separate designations of substrates as ERAD-L (for lumenal) and ERAD-M (for membrane). While these designations are made with a fairly small number of usually artificial substrates, they probably reflect differences between the mechanisms of degradation of each class that will be general.
How are misfolded membrane proteins detected and degraded? The most obvious difference between the two classes of substrates are the bilayer-crossing membrane spans present in the ERAD-M substrates. A misassembled or misfolded membrane protein might present structural hallmarks of these problems in the secluded environment of the bilayer. If so, the proteins used for detection of ERAD-L substrates, such as Kar2p or Yos9p, would not have access to this compartment, and so would not be useful for detecting intramembrane features of ERAD-M substrates. Furthermore the soluble detectors may use totally distinct biophysical criteria to do their job. In both cases the misfolded proteins would present residues that are normally buried due to the folding or the assembled state of the normal protein. A misfolded soluble protein would be expected to present hydrophobic residues that are normally sequestered away from water in a folded core, or a protein-protein interface. By contrast, a misfolded or misassembled membrane protein might reveal hydrophilic residues that are normally sequestered away from the hydrophobic environment of the bilayer. Thus, the detection apparatus would require access to the bilayer space, and might use aberrantly exposed hydrophilic residues as part of the recognition scheme. Biophysical studies show that this mechanism can mediate intramembrane protein interactions 51,52. Hrd1p has a large transmembrane domain, and is able to recognize membrane substrates without need for many of the recognition factors for lumenal, soluble substrates. An appealing idea is that the Hrd1p transmembrane region directly participates in the detection of intra-membrane determinants of misfolding present in ERAD-M substrates. Inspection of the Hrd1p transmembrane region reveals a surprising number of hydrophilic residues, distributed over numerous helices, which could be employed to engage exposed hyrophilic substrate residues in the bilayer by non-covalent binding, thus signaling the presence of an eligible substrate.
8. Hrd1p in ERAD-M substrate recognition
In recent work we tested the importance of the Hrd1p transmembrane region in ERAD substrate recognition, by systematic mutagenesis of this portion of the E3 ligase. Each of the numerous hydrophilic residues was individually changed to a more bilayer-friendly alanine. In addition, residues conserved between Hrd1p homolgues were modified, resulting in a collection of 78 site-directed point mutants, which were tested for degradation of ERAD-L or ERAD-M substrates, and Hrd1p self-degradation (B. Sato et al., manuscript submitted). The results strongly indicate that Hrd1p has a central role in the recognition of membrane-bound substrates. Within our collection of Hrd1p variants are mutants that only stabilize ERAD-M substrates, and have no effect on lumenal substrates such as CPY*, indicating that ERAD-M may involve distinct recognition mechanisms. Consistent with this, the mutant library also contained separate Hrd1p variants deficient for degradation of a single ERAD-M substrate, but normal for all others. Further analysis of a triple mutant 3A-Hrd1p indicate that the intramembrane residues altered in 3A-Hrd1p are involved in the decision to degrade Hmg2p independent of binding detected by cross linking or native co-IP. The model that emerges from these and earlier work is that Hrd1p “queries” a variety of polytopic membrane proteins by transient interactions that do not distinguish substrates from non-substrates, and that structural information gleaned by the transmembrane domain then activates the Hrd1p ligase for ubiquitination of only those proteins with hallmarks of misassemby or misfolding. Apparently our selective mutants can alter this “assessment phase” in a highly substrate-specific manner.
The 3A-Hrd1p variant that is selectively null for Hmg2p degradation has three bilayer-buried hydrophilic residues (two serines and one aspartate, see figure) that are converted to alanines, consistent with the idea that ERAD-M might depend on detection of exposed hydrophilic residues. However, other single ERAD-M substrate Hrd1p mutants we studied have changes in bulky hydrophobic residues, which could mean that the availability of hydrophilic residues near these changes are affected, or that other modes of substrate recognition are being employed. Until a detailed structure of Hrd1p is available, these conjectures will be hard to resolve. However, this demonstration that Hrd1p directly participates in recognition of membrane substrates will hopefully lend impetus to structural biologists accomplishing this important milestone.
9. Sterol pathway regulation of Hmg2p stability
This article starts by describing a basic conceptual distinction between protein degradation for regulation and degradation for quality control. However, Hmg2p regulation inconveniently crosses this boundary. Hmg2p degradation is under stringent regulation: its half life can vary from less than 10 minutes to over 6 hours depending on the availability of the sterol pathway signals that feedback on Hmg2p stability. But this regulated degradation of Hmg2p is mediated by the HRD quality control pathway that functions in the maintenance of ER proteostasis by degrading a wide variety of structurally aberrant proteins. So this is a case of regulated protein quality control, which smears the boundaries described (and still often very useful) in no uncertain terms. The regulation of Hmg2p by the HRD pathway will next be described, and how the HRD pathway is employed to bring this about.
The HRD-dependent degradation of Hmg2p is under the control the sterol pathway, in a manner consistent with feedback regulation. When pathway flux is high, degradation of Hmg2p tends to be high, resulting in lower levels of enzyme. Conversely, when flux is low, degradation is slowed and the steady state levels tend to be high 29,53. In this way, increasing demand for sterol pathway products is met with an increase in HMGR to increase their production. The sterol pathway in all eukaryotes has a uniform set of reactions whose products are used to form the sterol skeleton, which is then modified to form cholesterol in mammals, ergosterol in yeast and phytosterol in plants (Figure 2). Although the “linear pathway” depicted appears simple, the 5 carbon units defined by the isopentyl pyrophosphate (IPP) and dimethylallyl pyrophosphate (DMAPP) can be combined and modified to give thousands of distinct products. It has been estimated that there are approximately 30,000 distinct isoprenes generated by branches of this basic mevalonate pathway. Truly, the mevalonate pathway is “Darwin’s combinatoric library”, generating a staggering array of distinct lipid shapes from a simple chemical platform to perform tasks in all walks of organismal and cellular life. This potential diversity means that there are many possible structures for a low-abundance regulatory signal, and this caveat should be held in mind when analyzing sterol pathway sensing and regulation.
10. FPP as a positive regulator of Hmg2p
The degradation of Hmg2p is regulated by the production of the 15 carbon isoprenoid farnesyl pyrophosphate (FPP) also called farnesyl diphosphate (Figure 5). We choose the term pyrophosphate to avoid ambiguity about where the phosphates reside on the molecule. Our earlier work using a variety of in vivo techniques all point to FPP being a positive regulator of Hmg2p degradation 29. Inhibition of enzymes upstream of FPP, such as HMG-CoA reductase itself, or HMG-CoA synthase, stabilize Hmg2p. These manipulations would be expected to deplete FPP, and other molecules downstream of these enzymes. Conversely, inhibition of the enzyme squalene synthase with the highly specific natural product zaragozic acid (ZA) causes a drastic increase in Hmg2p degradation and ubiquitination. Since FPP is the substrate of squalene synthase, these drug studies indicate that FPP is a key positive regulator of Hmg2p degradation.
Figure 5. Isoprenes relevant to Hmg2p regulated degradation.

The structures of three isoprenes discussed in detail are shown, including 15 carbon farnesyl pyrophosphate (FPP), farnesol, and the 20 carbon geranylgeranyl pyrophosphate (GGPP). FPP levels in the cell are directly correlated with degradation rate of Hmg2p. Farnesol was employed in an the limited proteolysis assay to observe selective misfolding of the Hmg2p transmembrane domain. GGPP is featured in our most current studies on the signal that regulates Hmg2p degradation.
Consistent with this idea, overexpression of squalene synthase (SS; Figure 2), which depletes FPP, causes a striking increase in the stability of Hmg2p, and this effect is reversed by the squalene synthase inhibitor ZA. The facility of yeast genetic manipulation allowed further testing of the FPP model by directly manipulating the enzymes responsible for its synthesis or use in the sterol pathway. The prediction was that loss of FPP-synthase, responsible for FPP production, would stabilize Hmg2p, while loss of squalene synthase would, like inhibition with ZA, cause enhanced degradation of Hmg2p. The use of simple null mutations for this metabolic mapping is not possible because the pathway enzymes are all essential. Instead the individual promoters for the genes encoding relevant pathway enzymes were replaced with the methionine-regulated promoter from the MET3 gene, allowing acute shut-off of that enzyme by the addition of methionine to the culture medium. These studies bore out the prediction that FPP controls HRD-dependent Hmg2p degradation. More FPP production results in more Hmg2p degradation; lowered FPP production results in less Hmg2p degradation.
The linear isoprenoids like 15 carbon FPP and its 20 carbon derivative GGPP are used to covalently modify, or prenylate, a variety of proteins that have important functions in membrane dynamics. The observation of FPP as a key regulator of Hmg2p degradation leads one to wonder if this molecule affects degradation by farnesylating a protein involved in Hmg2p degradation. For example, perhaps a positive regulator of Hmg2p degradation becomes farnesylated to cause degradation, thus connecting FPP levels to enhanced degradation by increased farnesylation of a positive regulator. Although this is not ruled out, we know that regulation by increasing FPP occurs in the absence of protein synthesis. Since farnesylation (and other prenylations) usually occur co-translationally and irreversibly 54, the ability of FPP to control Hmg2p degradation when farnesylation of this putative regulator can not occur casts doubt on that mechanism, and we currently favor a direct mechanism of isoprene regulation of Hmg2p. Nevertheless, the sterol regulation field is full of interesting mechanistic surprises, so we can not rule out the possibility that a non-canonical prenylation could still be involved, or that a prenylated protein could work in concert with the direct signal.
How does FPP control Hmg2p entry into the HRD quality control pathway? Increasing cellular FPP production by a variety of means increases ubiquitination and degradation of Hmg2p, while lowering the levels of FPP decrease ubiquitination and degradation. FPP is a lipid, and many Hrd proteins including the Hrd1p E3 ligase have membrane-spanning domains. The simplest model that FPP somehow increases the general activity of the HRD pathway is not valid, since the degradation rates of many HRD substrates, including variants of Hmg2p itself, are unresponsive to changing levels of FPP 29,30. Thus, the sterol pathway regulation of Hmg2p is specific..
11. The Hmg2p transmembrane region
The Hmg2p transmembrane region is necessary and sufficient for FPP-regulated degradation; versions of Hmg2p with the C-terminal catalytic region replaced with reporter genes such as GFP are subject to regulation, while the free Hmg2p catalytic region is not. Extensive mutagenic analysis of Hmg2p has shown that loss of regulation can occur by many mutations, including gross changes like insertion of 6 tandem myc epitopes in a section of the transmembrane anchor 31, or very subtle mutations involving a few amino acids replaced by those with similar hydrophobicity 30,55. In many cases, the resulting mutants are constitutively degraded by the HRD pathway, and no longer have degradation rates that are dependent on FPP levels. This is not that surprising, since the HRD pathway functions to find and degraded misfolded proteins. Many of these constitutive mutants are not quite normal in structure and thus are HRD pathway substrates. A genetic version of this type of observation strengthens the idea that the wild-type, correctly made Hmg2p transmembrane domain functions to respond to the FPP-derived signal.
In a genetic screen for yeast genes required for regulation of Hmg2p, we isolated mutants that would not stabilize Hmg2p when the FPP-derived signal was lowered with an early blocker of the sterol pathway, incorporating counterscreens to cull those mutants that were simply unresponsive to the screening drug 56. This screen turned up 37 alleles of the same gene, COD1/SPF1, which encodes an ER-localized P-type ATPase transporter. The substrate that the Cod1p pump transports is not known 57, although we suspect it is a lipid flippase 58. In a cod1Δ null, Hmg2p undergoes constitutive HRD-dependent degradation, unaffected by levels of FPP in the cell. The encoded Cod1p protein is needed for a number of ER functions, including correct insertion of model ER membrane proteins 59. Consistent with this, in direct structural analyses of Hmg2p, it is clear that Hmg2p has an altered structure 55. Thus, the cod1Δ null mutant renders Hmg2p non-regulated by disallowing formation of its correct shape. Taken together, our studies indicate that the Hmg2p transmembrane domain has structural information required for regulated entry into the HRD pathway. Loss of that information by direct mutation or altered ability to fold Hmg2p results in Hmg2p that is constitutively degraded the HRD pathway, or in some cases, Hmg2p that is constitutively stable.
The Hmg2p transmembrane domain allows the degradative response to the FPP-derived signal. There are many scenarios that would allow such a regulatory loop, involving intermediary proteins. However, the studies on Cod1p imply that Hmg2p regulation might be fairly direct, since this high saturation screen (over 300,000 haploid colonies) only turned up only alleles of COD1 (37 of them) and no other genes. Although the absence of factors does not prove their absence in Hmg2p regulation, this outcome lent impetus to examining a direct role of the Hmg2p transmembrane domain in FPP-regulated Hmg2p degradation. The simplest model for the action of the FPP-derived signal is that it directly affects Hmg2p structure, allowing more efficient entry into the HRD pathway. We call this model the structural transition hypothesis.
12. Regulated misfolding of Hmg2p
The “structural transition hypothesis” predicts that normal Hmg2p acquires features of a misfolded protein when the FPP-derived signal is high (Figure 6). The degradative behavior of Hmg2p is consistent with it being recognized as a misfolded protein when the degradation signal is high. Hmg2p degradation is significantly temperature sensitive, showing marked slowing at room temperature, as is the degradation of a number of misfolded membrane proteins such as CFTR-Δ508. Many misfolded proteins undergo improved folding, both in vivo and in vitro, by treatment with a class of small molecules known as chemical chaperones, or cosmotrophs (order producers), the prototype molecule being glycerol, which acts as a chemical chaperone at 10–15 % by volume and is quite benign to most living cells 60. In cells where Hmg2p is undergoing rapid degradation due to high levels of signal, Hmg2p degradation is remarkably stabilized by the addition of glycerol at concentrations appropriate for chemical chaperoning 61,62. The effect is almost instantaneous (within five minutes) and completely reversible: removal of glycerol from the medium allows the immediate re-initiation of Hmg2p degradation. Importantly, grossly misfolded versions of Hmg2p, which are also HRD substrates, are not stabilized by glycerol. Furthermore, the stabilizing effects of glycerol and the stabilizing effect of FPP-lowering drugs are not additive. These studies indicate that in conditions of rapid degradation (high FPP) Hmg2p has features of a partially misfolded proteins, that is, more readily recognized by the HRD machinery, but remediable by chemical chaperones or low temperature.
Figure 6. The structural transition model for Hmg2p regulation.

Regulated degradation of Hmg2p is posited to occur by a regulated change in structure to one that is more susceptible to HRD pathway degradation. Three ways of altering the stability of Hmg2p are indicated, with the direction of arrow showing the effect of each item. The FPP-derived signal causes more degradation, and it thought to altere the structure to one with more features of a misfolded protein. Conversely, Nsg1p or glycerol (a chemical chaperone) both effectively stabilize Hmg2p. Similarly, lowering the FPP-derived signal causes improved folding and thus, increased stability. The lower panel shows the metabolic reaction that generates GGPP from FPP by addition of 5 carbons from IPP, as shown. This reaction is catalyzed by the BTS1 gene product.
The structural transition model was more directly evaluated with a limited proteolysis assay. Our extensive mutagenic analysis to find transmembrane determinants of Hmg2p regulated degradation also revealed regions of the Hmg2p transmembrane domain that could be altered without regulatory consequence 30. We chose such a “cold spot” to place a myc epitope tag in the first lumenal loop of the Hmg2p-GFP coding region, so that the myc tag was present in the ER lumen in the expressed protein. The resulting protein is called mycL-Hmg2p-GFP. Hmg2p-GFP is not catalytically active, but undergoes regulated degradation which can be quantitatively assessed by use of time dependent flow cytometery in living cells 61. As expected, mycL-Hmg2p-GFP was normally regulated, and so provided a way to evaluate the effect of altering degradation on the structure of the transmembrane domain. Microsomes prepared from strains expressing mycL-Hmg2p-GFP remain right-side out, leaving the myc epitope protected in the lumenal space. Treatment of the microsomes with proteases such as trypsin causes a time-dependent cleavage of the protein without loss of epitope signal, allowing an empirical gauge of the structure of the transmembrane region (Figure 7). When mycL-Hmg2p-GFP expressing microsomes are treated with trypsin and subject to myc immunoblotting after electrophoresis, a series of fragments are produced as the trypsin cuts at its sites. The order and timing of fragment appearance is very reproducible and this time-dependent pattern is a readout of Hmg2p structure. Alterations in the pattern reflect changes in the structure of Hmg2p caused by conditions of interest. For example, the presence of glycerol or other chemical chaperones in the limited proteolysis assay causes a drastic slowing of fragment generation, while trypsinolysis of the glycerol-unresponsive 6myc-Hmg2p was unaffected. Interestingly the fragments appear in the same temporal order but the rate of their production was about 10 times slower. Just as with its in vivo actions, the effect of glycerol on Hmg2p limited proteolysis was very rapid, and completely reversible: removal of the glycerol from the microsomes resulted in a subsequent trypsinolysis rate identical to untreated membranes 61. Thus, the structure of the Hmg2p transmembrane domain is highly sensitive to the presence of these folding-promoting substances.
Figure 7. Limited proteolysis assay for evaluating structural changes in Hmg2p.

A myc epitope tag (“tag”) was added to the first lumenal loop of Hmg2p-GFP to create the normally regulated mycL-Hmg2p-GFP, depicted on the right. The tag is protected in the ER lumen, allowing detection of fragments generated during limited proteolysis of intact ER microsomes. A typical proteolysis experiment is shown in the left, in which microsomes are incubated with trypsin for the indicated times, and then subjected to SDS-PAGE and myc tag immunoblotting. All perturbations described in the text cause changes in the rate of proteolysis without any change in the order or pattern of fragment appearance. Stabilizing treatments such as chemical chaperones or preincubation with early sterol pathway inhibitors cause the proteolysis pattern to be generated more slowly. Conversely, treatments that cause more rapid degradation promote more rapid generation of the trypsinolysis pattern.
These limited proteolysis studies show that the Hmg2p structure “tightens up” in the presence of the chemical chaperones, rendering the accessibility of the cut sites lower. This “structural flexibility” of Hmg2p could explain the stabilizing effect of glycerol on in vivo degradation of Hmg2p by the HRD pathway: presentation of misfolding determinants as the proteins goes through its range of steady-state conformations (its “breathing” as that process is sometimes called) occur less frequently in glycerol, and so HRD dependent degradation has slower kinetics. But does this pertain to the more physiological FPP-dependent regulation? We used the limited trypsinolysis assay to evaluate the action of the FPP-derived signal both in vivo and in vitro. In particular we wondered if the same simple “more open-less open” effect seen in the chemical chaperone studies is related to the action of the FPP-derived signal. If this were the case, increasing the FPP-derived signal would increase the rate of production of the trypsinolysis products, and decreasing this signal would slow the production of the assay products, and the temporal order of fragment production would be the same. To study the effects of the FPP-derived signal in vivo, strains expressing using mycL-Hmg2p-GFP were treated with drugs to raise or lower the FPP signal, and then the microsomes from each were isolated and subjected to the limited proteolysis assay 55. The results were surprisingly simple: increased exposure to FPP in vivo resulted in more rapid production of the product, and lowered FPP in vivo resulted in slower production of the same products. The pattern of band production was unaffected by changing intracellular FPP, but the rate of generation increased with increasing signal. Thus the physiological regulation of Hmg2p structure, at least at the resolution of the proteolysis assay, has similar structural features to the manipulation of stability with chemical chaperones. In both cases, conditions that promote degradation result in more access to the same trypsinolysis sites and conditions that promote stability result in less access to those sites.
A third mode of stability control in vivo also appears to use the same structural features of Hmg2p. We have found that the yeast homologues of the Insig proteins, so important for control of mammalian HMGR stability (see below), play a key role in regulated degradation of Hmg2p. Insigs are a conserved family of multispanning membrane proteins first discovered as centrally involved in mammalian sterol regulation, by their ability to bind mammalian HMGR and a protein with a related transmembrane domain called SCAP 26. The yeast Inisg, called Nsg1p, is able to stabilize Hmg2p, rendering it stable even in the presence of high degradation signals 63. Nsg1p binds directly to the Hmg2p transmembrane domain, and is required at similar levels to Hmg2p to effect stabilization. The limited proteolysis assay showed that the effect of Nsg1p on Hmg2p structure was similar to the other stabilizing influences of lowering the FPP-derived signal or increasing chemical chaperones: the presence of stabilizing levels of Nsg1p in the microsomes caused a slowing of Hmg2p trypsinolysis rate, again without any change in the pattern. Thus, we proposed that the Nsg proteins in yeast, and perhaps Insigs in general, are dedicated intramembrane chaperones that bind to Hmg2p and improve its folding. Taken together, it appears that chemical chaperones, FPP-mediated regulation and Nsg-dependent stabilization all operate by altering Hmg2p along the same structural spectrum (Figure 6), although rigorous testing of this idea must await more sophisticated methods.
13. Direct action of isoprenoid molecules on Hmg2p structure
The paucity of regulatory genes from our screens implied that the effects of the degradation signal on Hmg2p structure could be direct. We examined the action of candidate FPP-related lipids on Hmg2p structure by adding them directly to the isolated microsomes of the limited proteolysis assay. Of the lipids tested, we found that farnesol, the 15 carbon alcohol that results from removal of the FPP phosphates, caused the same opening of Hmg2p structure seen in the experiments where FPP levels were altered in vivo prior to the assay 55. The effect of farnesol was reversible, such that removal of the lipid from microsomes by sequestration restored Hmg2p proteolysis to that of untreated microsomes. Furthermore, a mutant form of Hmg2p that is unresponsive to the in vivo degradation signal, called NR1, was unaffected by farnesol in vivo. Finally, in vitro farnesol causes mycL-Hmg2p-GFP to adopt a more unfolded state, as measured by a thermal denaturation assay; this unfolding effect of farnesol is also not seen with the unregulated NR1 mutant, implying that the altered trypsinolysis and thermal stability are both indications of the same structural effects of the lipid. These effects of farnesol had some structural specificity. The 20 carbon geranylgeraniol has some effect. The 10 carbon analogous alcohol geraniol did nothing at very high concentrations, nor did fully hydrogenated farnesol with the same sigma bond structure but none of the hallmark isoprene double bonds.
Our model is that in vitro farnesol causes a structural change in Hmg2p that is the same as that caused by the FPP-derived signal in vivo, due to the numerous similarities between the two: in both cases the pattern of cleavage products produced by trypsin is not changed, but the time course is altered by the presence of the signal. The in vitro effect, like the degradation-enhancing effect of elevated in vivo signal, is reversed by chemical chaperone glycerol. Hmg2p variants that are degraded by the HRD pathway but unresponsive to the signal in vivo are also unresponsive to farnesol in vitro. Thus, it appears the in vitro structural assay is recapitulating aspects of the in vivo regulation that allows regulated degradation by the HRD quality control pathway.
We previously harbored the simple model that farnesol itself was the in vivo regulator. However, we have recently discovered that a more distal candidate molecule is most likely to be a more distal candidate for the FPP-derived regulator (R. Garza et al., manuscript submitted). Surprisingly, direct addition of the 20 carbon geranylgeranyl pyrophosphate (GGPP; Figure 5) to intact yeast cells causes rapid, high-potency enhancement of Hmg2p degradation. In striking contrast, addition of farnesol, while very toxic to yeast, does not have any effects on Hmg2p degradation, nor does added FPP, or the dephosphorylatin product of GGPP called GGOH. Added GGPP does not stimulate degradation by inhibiting squalene synthase, and this molecule can override the Hmg2p-stabilizing effects of sterol pathway inhibition, indicating a direct role in Hmg2p regulation. Elevation of the enzyme Bts1p that endogenously produces GGPP from FPP similarly accelerates degradation, while a bts1Δ null strain has a demonstrable slowing of Hmg2p degradation, indicating that GGPP production contributes to Hmg2p degradation in vivo. Like farnesol, GGPP causes the same in vitro structural transitions in the trypsinolysis assay described above, but unlike farnesol, has direct activity in the living cell as well. Because the in vitro trypisinolysis assay will not support intercoversion between added isoprenes, it appears that the ability to cause the in vitro effects on Hmg2p structure is a necessary condition of stimulated degradation shared by a number of related isoprenes, but only GGPP (or a molecule derived from it in vivo) has the other features needed to bring about regulated entry into the HRD pathway. These studies indicate that the FPP-derived molecule GGPP or a derivative is a, and perhaps the, bone fide regulator of Hmg2p. For now, we will continue to use the still-accurate term FPP-derived signal, with the understanding that it may well be GGPP or something derived from it. Interestingly, a geranylgeranyl signal has been implicated as participating in mammalian HMGR degradation, along with the more central sterol signal that clearly controls HMGR stability in mammals (see below). Whether this connection is a mechanistic bridge between the two regulatory processes will be an important future avenue to explore.
14. The sterol-sensing domain (SSD) in Hmg2p regulation
We have recently improved our understanding of how the FPP-derived signal alters Hmg2p degradation by exploring the role of a highly conserved motif called the sterol-sensing domain (SSD). The Hmg2p transmembrane domain contains detailed molecular information for responding to the FPP-derived signal. Even small perturbations in the structure or sequence cause loss of regulability, rendering versions of Hmg2p that are unresponsive to the degradation signal or lipids in the in limited proteolysis assay. This strong dependence on Hmg2p structure of Hmg2p led us to explore the role of a conserved sequence required for recognition of sterol pathway molecules in a variety of circumstances (T. Davis and R. Hampton, unpublished). The sterol-sensing domain (SSD) is a protein motif embedded in the lipid spanning helices of a variety of proteins involved in cholesterol homeostasis. In particular, the mammalian proteins SCAP and HMGR have SSDs that are intimately involved in feedback regulation of the sterol pathway. The SCAP protein is required for sterol regulation of SCAP-dependent activation of the SREBP transcription factor that causes transcription sterol-synthesizing and LDL receptor genes 26. Similarly, the SSD of HMGR mediates the sterol-stimulated degradation of HMGR, which occurs when sterol synthesis is abundant, in analogy to the regulation of Hmg2p by earlier pathway signals derived from FPP. It has been directly shown that SCAP binds cholesterol with high affinity in an SSD-dependent manner 64, and this is thought to be the mechanism by which all SSDs operate. Hmg2p has a conserved SSD and we wondered if this motif were similarly important for recognition and response to the FPP-derived signal. We made point mutants (usually alanine replacements) of each residue conserved between the SCAP mammalian SSD and the SSD found in yeast Hmg2p. The collection of mutants had several interesting features. Many of the mutants had no effect on regulation of Hmg2p, indicating that there they were not generally involved in the global structure of the transmembrane region. However, some of the mutations (e.g. S215A) had strong stabilizing effects on Hmg2p, consistent with the SSD having a role in recognition of the FPP-derived degradation signal, which although from the sterol pathway, is not a sterol. If this is the case, then the SSD would have a wider range of ligands, and perhaps in fact be a signal-sensing domain, keyed to the measurement of a variety of lipids depending of the details of the residues and the structural environment in which the SSD is embedded. Consistent with this idea, the Drosophila SCAP may be involved in recognition of the phospholipid phosphatidylethanolamine 65. However broad the role of this motif turns out the be, the Hmg2p SSD plays a critical role in the recognition of sterol pathway products that signal HRD pathway degradation.
The exact nature of the reversible structural transition that brings about Hmg2p entry into the HRD pathway is not known. One possibility is that individual Hmg2p transmembrane domains change in the presence of the signal, allowing increased probability of entry into the HRD pathway. Alternatively, it may be that the Hmg2p molecule exists as a multimer, and that the structural transition that allows entry into the HRD pathway is production of monomers that are quality control substrates. In this scenario, the FPP-derived signal would increase the production rate of that species, again increasing the chance for recognition by the HRD pathway. Although much remains to be done, the model that emerges from regulated degradation of Hmg2p is generally interesting and potentially useful both technically and clinically.
15. Regulated protein quality control in biology and medicine
As mentioned in the introduction regulation and quality control are usually considered to be separate arenas of intracellular protein degradation. To selectively degrade a protein for a desired regulatory outcome, it makes sense to capitalize on a highly specific E3-interaction with that protein. Since quality control is keyed to structural features shared by a wide variety of substrates, the mechanisms of recognition might not be seen as useful for controlling the degradation of a single protein. But the example of Hmg2p demonstrates a way to employ protein quality control for selective regulation of a particular target protein. Instead of the E3 determining the specificity, the interaction of the regulator (in this case the FPP-derived signal) with the target protein is highly specific, causing only Hmg2p to undergo a structural change that improves efficiency of HRD pathway recognition and destruction. So in the regulatory strategy, the high selectivity of protein quality control for misfolded (or misassembled) proteins is harnessed through selective misfolding of only the target protein. This idea has some interesting implications. First, since there appear to be multiple quality control pathways in each compartment of the cell, and all kingdoms of life, the idea of employing quality control pathways through selective misfolding of target proteins could be broadly used in biology; several possible cases will be described below. The diversity of examples indicates that the specificity of protein quality control pathways may be used in many circumstances.
Regulated quality control may also have applications in drug discovery. Hmg2p regulation provides a natural proof-of-concept that there could be a new class of drugs that would work by specifically driving a specific protein target down a quality control pathway to effect its clinically desirable elimination or diminution. Such “degradation angatonists” could be every bit as selective as enzyme inhibitors or receptor antagonists, but would work by selectively misfolding only the desired target. In a sense, this idea is the opposite of the very exciting “pharmaceutical chaperones” that Bouvier and others are pioneering 66. A pharmaceutical chaperone binds to a particular protein by virtue of a specific binding site on that protein, and promotes its folding or stabilization. Some clear examples of this effect come from studies of the multispanning G-protein coupled receptors (GPRCs) that undergo ER protein quality control during their biogenesis 67–69. Application of the correct receptor binding ligand will selectively spare the ligand-binding receptor from quality control due to improved binding-driven folding, leading to escape from ER quality control, and thus increased expression of the receptor on the cell surface. Pharmaceutical chaperones are being explored as a way correct a disease-causing mutants of GPCRs that undergo deleterious ER degradation due to their mutations, resulting in low cell surface levels and clinical symptoms as a result of low receptor levels. In this case, the specific binding of a ligand might spare disease variants from quality control, thus bringing about improved cell surface levels and alleviation of the clinical symptoms 70.
Our example of “degradation-based antagonists”-called this because they would lower or decrease (antagonize) the activity of a target by stimulating degradation-would be the opposite case: the specific binding of a molecule would cause selective misfolding rather than improved folding. Although it is often assumed that binding of a ligand to a protein would always stabilize the structure, that does not have to be the case. If the binding energy of the ligand can be harnessed to cause local or even global unfolding, or a change in multimer-monomer steady state, then this specific interaction could bring about selective enhancement of quality control degradation. Whether such compounds can be found, and exactly how to find them is still an open question, but hopefully one of sufficient interest and utility to promote creative attempts to explore these ideas.
16. Examples of regulated quality control
The example of Hmg2p undergoing regulated quality control degradation demonstrates a potentially useful and clearly interesting interface between distinct sectors of this field. But possibility and utility are two different features, and only the former is demonstrated by a single example. In this section examples from a range of biological processes will be described to show that nature appears to have capitalized on protein quality control to effect regulation of a specific protein in a number of circumstances. Thus, there is evolutionary utility in using this regulatory strategy; perhaps clinical utility will follow in the future.
Mammalian HMGR
In elegant work by Debose-Boyd and colleagues, mammalian HMG-CoA reductase (HMGR), like Hmg2p, was shown to undergo ubiquitin mediated regulated degradation in response to sterol pathway signals 71 HMGR degradation is mediated by gp78, an ER-localized multispanning transmembrane ubiquitin ligase with sequence homology to Hrd1p. In contrast to Hrd1p, gp78 has additional sections in its C-terminal RING domain that allows direct engagement of the principle ERAD E2 Ube2g2, which is the mammalian homolgue of Ubc7p 72. Furthermore, gp78 can also directly bind the retrotranslocation factor p97/Cdc48 73, while in yeast the ER association of this factor is mediated by the independent membrane protein Ubx2p 47,48, and probably other unknown anchors as well. Thus, gp78 can be viewed as “Hrd1p with a backpack” allowing more direct association with the necessary trans factors for ubiquitin transfer and substrate removal from the ER membrane. Regulation of HMGR degradation by gp78 is controlled by the sterol pathway molecule lanosterol, which is the first actual sterol produced in sterol biosynthesis (fig 2) and thus is a unique indicator of the level of sterol synthetic activity in the cell. When lanosterol levels are elevated, the HMGR transmembrane domain binds more efficiently to Insig, which in turns mediates the recruitment of gp78 74. Thus, Insig is part of the complex required for HMGR degradation. This is in curious contrast to the yeast Insig homolgue Nsg1p, that functions to block Hrd1p mediated degradation of Hmg2p, also by binding it, and appears to improve the folding of Hmg2p, lowering the efficiency of HRD mediated degradation 63. This “opposite” behavior of the Insigs led us to propose that they may be diverged chaperones of HMGR, since promoting folding of a partially folded substrate or recruitment of an E3 ligase to degrade such a substrate are two distinct functions of the more traditional chaperone Hsp70 in different biological contexts. In addition to the mechanistically well-understood sterol-mediated recruitment of gp78, there is a suspected role for the 20 carbon isoprenoid GGPP, due to the observed stimulation of HMGR degradation when cells are treated with GGOH, the non-phosphorylated precursor of this molecule 71. Whether this other axis is similar to the clear effects of GGPP in yeast is an intriguing open question. A more general question is whether controlled misfolding is part of regulated HMGR degradation, or alternatively if the QC ligase is prompted to degrade HMGR simply by brokering gp78 proximity to the normal protein in a lanosterol-dependent manner. Whatever the mechanism, it is clear that a quality control pathway is also employed to effect feedback regulation of HMGR in the mammal as well as yeast.
Mammalian Inositol triphosphate receptor (IP3R) regulation
The ER-localized IP3 receptor is an integral membrane protein of the sacroplasmic reticulum that releases Ca2+ stores into the cytoplasm in response to the soluble signaling molecule inositol triphosphate (IP3). Ongoing work by the Wojcikiewicz (Voy-chi-kay-vitch) laboratory has shown that IP3R is subject to feedback regulated degradation keyed to its signaling function. When cytoplasmic levels of both the IP3 signal and cytoplasmic calcium are elevated to the ranges that occur during IP3-gated calcium release, the receptor molecule is selectively ubiquitinated and degraded, thus providing a governor on the release pathway 75,76. Since the sarcoplasmic reticulum is a specialization of the ER, this is another example of an ERAD pathway being employed to program the selective degradation. The details of the ubiquitination mechanism are not yet clear, but two observations indicate that a quality control pathway is used for this purpose. First, the p97/Cdc48 AAA-ATPase is required for IP3R degradation 77, as would be expected for an ERAD substrate. Also, a novel ER-localized protein discovered in the analysis of this pathway, called SPFH2, is required for the stimulated ubiquitination of IP3R, and for the degradation of a variety of other misfolded ERAD substrates 78. The simplest model is that the combination of IP3 and Ca2+ ion causes a change in the large cytoplasmic domain of IP3R, presumably by binding, that promotes more efficient ubiquitination and degradation by one of the numerous ERAD E3 present in mammalian cells. The identity of the responsible ligase will be a most interesting part of this important example of regulated quality control.
Regulated cytochrome P450 degradation
The cytochrome P450 (CYP) family of oxidoreductases comprise a metabolic system for the detoxification and oxidation of a wide variety of endogenous and xenobiotic compounds. The human genome encodes 50 or more of these enzymes, which may have expanded to this number for enhanced protection from numerous toxins encountered in the hunter-gatherer period of hominid existence. These enzymes reside on the surface of the ER, anchored by a single membrane span, and are thus potentially susceptible to degradation by ERAD mechanisms. The levels of P450 enzymes vary greatly depending on the physiological and toxicological circumstances. For example, induction of CYP2E1, the cytochrome involved in ethanol toxicity, is induced through stabilization by acetone, one of its substrates 79. CPY2E1 undergoes degradation by several mechanisms, including chaperone-dependent ubiquitination by the quality control ligase CHIP 80. The mechanism of acetone-induced stabilization is not yet clear, but it would be consistent with a substrate-based version of pharmacological chaperoning.
The ER localization of CYPs leads one to wonder if ERAD pathways are involved in regulated degradation of this family of enzymes. Studies from the Corriera lab show that in a yeast model of CYP450 degradation and in cultured cells that naturally express the protein, CYP3A is subject to altered stability when encountering substrates, and in some cases inhibitors of the enzyme that alter the structure 81,82. It appears in both organisms, the ER-localized enzyme undergoes altered susceptibility to ER-associated degradation when certain inhibitors or substrates that specifically bind the cytchrome are present. The suicide inhibitor called DDEP, which specifically alters the mammalian CYP3A enzyme, causing increased ubiquitin-mediated degradation that appears to occur through ERAD mechanisms. In contrast, the use of a different inhibitor called TAO stabilizes CYP 3A, indicating that small molecules can specifically alter ERAD of a protein towards degradation or stability. This stabilization it thought to involve substrate-mediated protection of the enzyme from mechanism-based, self-inflicted oxidative damage that triggers ERAD due to the misfolding that this oxidation can cause. P450 regulation is complex, and varies widely between differing isozymes. It is not clear if these mechanisms of altering degradation by small molecules are part of natural induction or regulation of the enzyme. However, the observation of small molecules that alter ERAD of a particular P450 isoform bodes well for the idea that selective enhancers of quality control degradation can be found for a desired target protein.
Feedback regulated degradation of the LpxC in bacteria
The above examples involve proteins that are situated at the ER surface and thus accessible to known or novel ERAD pathways. In the future, as more pathways of quality control degradation are revealed it will be most interesting to evaluate if they too can be harnessed by biology or biotech for selective degradation of a target protein. The final example implies that this will be the case, since it takes us far away from the surface of the ER, to the eubacterial cytoplasm. Lipid A, the lipid core of lipopolysaccaride (LPS), is an essential phosphorylated lipid that resides on the outer leaflet of the Gram negative bacterium. It is the principle outer leaflet lipid of the bilayer and is thus required for the functions and structure of that membrane. Lipid A in LPS is also one of the principle “molecular patterns” recognized by the innate immune systems of mammals and other metazonans, and thus it plays key roles in both the physiology and medical biology of this class of prokaryotes. The lipid A biosynthetic pathway starts with N-acetyl glucosamine as the starting compound for the two sugar, six acyl chain structure of mature lipid A. One of the early reactions is catalyzed by a deacetylase called LpxC that removes the N-linked acetyl group from mono-acylated N-acetyl glucosamine to allow subsequent acylation of the denuded 2-nitrogen.
LpxC levels are regulated due to the need for the correct amount of this enzyme in the Gram negative; too much or too little activity each can have lethal ramifications 83. Studies with mutants in the lipid A pathway and inhibitors of LpxC itself show that the LpxC enzyme is under feedback control: lowering flux through the pathway by a hypomorph in an enzyme upstream of LpxC, or by application of the inhibitor at sub-lethal doses causes a dramatic increase in the amount of the LpxC protein new 84, with no change in the messenger RNA, indicating the the enzymes degradation may be under feedback control. Indeed, LpxC is degraded by the FstH hexameric ATPase, that is required for the proteolysis of some misfolded and misassembled membrane proteins 85. The FtsH quality control protease is the only essential ATP-dependent protease, and this lethality is suppressed by lowering LpxC levels. This indicates that the normal and essential function of FtsH is to keep the levels of LpxC low enough to avoid the toxic effects of overabundance. Thus a reasonable model is that a molecule from the lipid A pathway causes the degradation of LpxC by promoting its FtsH-mediated degradation, perhaps through exposure of a C-terminal degron caused by binding of the regulator 86. The direct connection between LpxC degradation rate and altered flux through the lipid A pathway has not yet been directly tested, although an impressive number of tools await application to this intriguing aspect of lipid regulation 87,88.
It would appear that there are many potential examples of regulated degradation of proteins that capitalize on machinery of proteostasis. Hopefully, the breadth of this naturally employed strategy bodes well for an equally wide application of these ideas to understanding regulation of cellular proteins, and to the development of new methods to selectively control the levels of clinical targets.
Acknowledgments
We thank all Hampton lab members for past and ongoing prodigious efforts to understand regulated and constitutive quality control degradation. RYH wishes in addition to thank Drs. Dan Finley and Chinfei Chen for recent, perexemplary generosity and hospitality. These studies were supported by NIH grant 5 R01 DK051996-15 to RYH, and a minority supplement to RG associated with the same grant.
Biographies

Randolph Hampton was born in New York State just before cars evolved fins. He grew up in Scarsdale NY, attended Ohio Wesleyan University, and by a circuitous route that included research at University of Michigan, running the office at Zingerman’s Delicatessen, and doing field work in music and comedy, finally earned a Ph. D. in Biochemistry in the laboratory of Christian Raetz, at University of Wisconsin-Madison and at Merck & Co. in Rahway NJ. He then headed west to do a postdoctoral fellowship with Jasper Rine at U. C. Berkeley, in one of the few laboratories in the world dedicated to both Yeast and Dog genetics, where the work on regulated degradation of HMG-CoA reductase (in yeast, not dog) was started. He next took a position in the Division of Biological Sciences at U. C. San Diego, back when it was the Department of Biology, where he so far remains. His scientific interests include cellular management of misfolded proteins, sterol regulation and the cholesterol pathway, metabolism, and ubiquitin in health and disease. His interests outside the laboratory include running, the banjo, avoidance of opera, and the complex synergies that govern the lives of us all.

Renee M. Garza grew up in Whittier, California, and received her B.A. in biology from California State University, Fullerton. There, she was exposed to the excitement of basic research in the laboratory of Dr. Rodrigo Lois. Renee joined Dr. Randolph Hampton’s research group in 2001 at University of California, San Diego, and studied the regulated degradation of yeast Hmg2p. She received her Ph.D. in 2008, and will begin a postdoctoral position exploring post-translational modifications and their effects on chromatin dynamics.
References
- 1.Ciechanover A, Iwai K. IUBMB Life. 2004;56:193. doi: 10.1080/1521654042000223616. [DOI] [PubMed] [Google Scholar]
- 2.Richly H, Rape M, Braun S, Rumpf S, Hoege C, Jentsch S. Cell. 2005;120:73. doi: 10.1016/j.cell.2004.11.013. [DOI] [PubMed] [Google Scholar]
- 3.Elsasser S, Chandler-Militello D, Muller B, Hanna J, Finley D. J Biol Chem. 2004;279:26817. doi: 10.1074/jbc.M404020200. [DOI] [PubMed] [Google Scholar]
- 4.Fang S, Jensen JP, Ludwig RL, Vousden KH, Weissman AM. J Biol Chem. 2000;275:8945. doi: 10.1074/jbc.275.12.8945. [DOI] [PubMed] [Google Scholar]
- 5.Pines J. Trends Cell Biol. 2006;16:55. doi: 10.1016/j.tcb.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 6.Regelmann J, Schule T, Josupeit FS, Horak J, Rose M, Entian KD, Thumm M, Wolf DH. Mol Biol Cell. 2003;14:1652. doi: 10.1091/mbc.E02-08-0456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bays NW, Gardner RG, Seelig LP, Joazeiro CA, Hampton RY. Nat Cell Biol. 2001;3:24. doi: 10.1038/35050524. [DOI] [PubMed] [Google Scholar]
- 8.Sakamoto KM, Kim KB, Verma R, Ransick A, Stein B, Crews CM, Deshaies RJ. Mol Cell Proteomics. 2003;2:1350. doi: 10.1074/mcp.T300009-MCP200. [DOI] [PubMed] [Google Scholar]
- 9.Meusser B, Sommer T. Mol Cell. 2004;14:247. doi: 10.1016/s1097-2765(04)00212-6. [DOI] [PubMed] [Google Scholar]
- 10.Beaudenon S, Huibregtse JM. BMC Biochem. 2008;9(Suppl 1):S4. doi: 10.1186/1471-2091-9-S1-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Orlicky S, Tang X, Willems A, Tyers M, Sicheri F. Cell. 2003;112:243. doi: 10.1016/s0092-8674(03)00034-5. [DOI] [PubMed] [Google Scholar]
- 12.Tan X, Calderon-Villalobos LI, Sharon M, Zheng C, Robinson CV, Estelle M, Zheng N. Nature. 2007;446:640. doi: 10.1038/nature05731. [DOI] [PubMed] [Google Scholar]
- 13.Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, Kriegsheim A, Hebestreit HF, Mukherji M, Schofield CJ, Maxwell PH, Pugh CW, Ratcliffe PJ. Science. 2001;292:468. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- 14.Gardner RG, Nelson ZW, Gottschling DE. Cell. 2005;120:803. doi: 10.1016/j.cell.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 15.McClellan AJ, Scott MD, Frydman J. Cell. 2005;121:739. doi: 10.1016/j.cell.2005.03.024. [DOI] [PubMed] [Google Scholar]
- 16.Hengge R, Bukau B. Mol Microbiol. 2003;49:1451. doi: 10.1046/j.1365-2958.2003.03693.x. [DOI] [PubMed] [Google Scholar]
- 17.Mogk A, Haslberger T, Tessarz P, Bukau B. Biochem Soc Trans. 2008;36:120. doi: 10.1042/BST0360120. [DOI] [PubMed] [Google Scholar]
- 18.Hiller MM, Finger A, Schweiger M, Wolf DH. Science. 1996;273:1725. doi: 10.1126/science.273.5282.1725. [DOI] [PubMed] [Google Scholar]
- 19.Ward CL, Omura S, Kopito RR. Cell. 1995;83:121. doi: 10.1016/0092-8674(95)90240-6. [DOI] [PubMed] [Google Scholar]
- 20.Bonifacino JS, Lippincott SJ. Curr Opin Cell Biol. 1991;3:592. doi: 10.1016/0955-0674(91)90028-w. [DOI] [PubMed] [Google Scholar]
- 21.Johnson PR, Swanson R, Rakhilina L, Hochstrasser M. Cell. 1998;94:217. doi: 10.1016/s0092-8674(00)81421-x. [DOI] [PubMed] [Google Scholar]
- 22.Cohen E, Bieschke J, Perciavalle RM, Kelly JW, Dillin A. Science. 2006;313:1604. doi: 10.1126/science.1124646. [DOI] [PubMed] [Google Scholar]
- 23.Swanson R, Locher M, Hochstrasser M. Genes Dev. 2001;15:2660. doi: 10.1101/gad.933301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Travers KJ, Patil CK, Wodicka L, Lockhart DJ, Weissman JS, Walter P. Cell. 2000;101:249. doi: 10.1016/s0092-8674(00)80835-1. [DOI] [PubMed] [Google Scholar]
- 25.Goldstein JL, Brown MS. Nature. 1990;343:425. doi: 10.1038/343425a0. [DOI] [PubMed] [Google Scholar]
- 26.Goldstein JL, DeBose-Boyd RA, Brown MS. Cell. 2006;124:35. doi: 10.1016/j.cell.2005.12.022. [DOI] [PubMed] [Google Scholar]
- 27.Basson ME, Thorsness M, Finer MJ, Stroud RM, Rine J. Mol Cell Biol. 1988;8:3797. doi: 10.1128/mcb.8.9.3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Friesen JA, Rodwell VW. Genome Biol. 2004;5:248. doi: 10.1186/gb-2004-5-11-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gardner RG, Hampton RY. J Biol Chem. 1999;274:31671. doi: 10.1074/jbc.274.44.31671. [DOI] [PubMed] [Google Scholar]
- 30.Gardner RG, Hampton RY. EMBO J. 1999;18:5994. doi: 10.1093/emboj/18.21.5994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hampton RY, Gardner RG, Rine J. Mol Biol Cell. 1996;7:2029. doi: 10.1091/mbc.7.12.2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bordallo J, Wolf DH. FEBS Lett. 1999;448:244. doi: 10.1016/s0014-5793(99)00362-2. [DOI] [PubMed] [Google Scholar]
- 33.Ng DT, Spear ED, Walter P. J Cell Biol. 2000;150:77. doi: 10.1083/jcb.150.1.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Younger JM, Chen L, Ren HY, Rosser MF, Turnbull EL, Fan CY, Patterson C, Cyr DM. Cell. 2006;126:571. doi: 10.1016/j.cell.2006.06.041. [DOI] [PubMed] [Google Scholar]
- 35.Meacham GC, Patterson C, Zhang W, Younger JM, Cyr DM. Nat Cell Biol. 2001;3:100. doi: 10.1038/35050509. [DOI] [PubMed] [Google Scholar]
- 36.Spear ED, Ng DT. J Cell Biol. 2005;169:73. doi: 10.1083/jcb.200411136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Okuda-Shimizu Y, Hendershot LM. Mol Cell. 2007;28:544. doi: 10.1016/j.molcel.2007.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ye Y, Meyer HH, Rapoport TA. Nature. 2001;414:652. doi: 10.1038/414652a. [DOI] [PubMed] [Google Scholar]
- 39.Bazirgan OA, Hampton RY. J Biol Chem. 2008;283:12797. doi: 10.1074/jbc.M801122200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bhamidipati A, Denic V, Quan EM, Weissman JS. Mol Cell. 2005;19:741. doi: 10.1016/j.molcel.2005.07.027. [DOI] [PubMed] [Google Scholar]
- 41.Kim W, Spear ED, Ng DT. Mol Cell. 2005;19:753. doi: 10.1016/j.molcel.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 42.Gauss R, Jarosch E, Sommer T, Hirsch C. Nat Cell Biol. 2006;8:849. doi: 10.1038/ncb1445. [DOI] [PubMed] [Google Scholar]
- 43.Wahlman J, DeMartino GN, Skach WR, Bulleid NJ, Brodsky JL, Johnson AE. Cell. 2007;129:943. doi: 10.1016/j.cell.2007.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lilley BN, Ploegh HL. Proc Natl Acad Sci U S A. 2005;102:14296. doi: 10.1073/pnas.0505014102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ye Y, Shibata Y, Yun C, Ron D, Rapoport TA. Nature. 2004;429:841–7. doi: 10.1038/nature02656. [DOI] [PubMed] [Google Scholar]
- 46.Knop M, Finger A, Braun T, Hellmuth K, Wolf DH. EMBO J. 1996;15:753. [PMC free article] [PubMed] [Google Scholar]
- 47.Neuber O, Jarosch E, Volkwein C, Walter J, Sommer T. Nat Cell Biol. 2005;7:993. doi: 10.1038/ncb1298. [DOI] [PubMed] [Google Scholar]
- 48.Schuberth C, Buchberger A. Nat Cell Biol. 2005;7:999. doi: 10.1038/ncb1299. [DOI] [PubMed] [Google Scholar]
- 49.Ploegh HL. Nature. 2007;448:435. doi: 10.1038/nature06004. [DOI] [PubMed] [Google Scholar]
- 50.Sato BK, Hampton RY. Yeast. 2006;23:1053–64. doi: 10.1002/yea.1407. [DOI] [PubMed] [Google Scholar]
- 51.Dawson JP, Weinger JS, Engelman DM. J Mol Biol. 2002;316:799–805. doi: 10.1006/jmbi.2001.5353. [DOI] [PubMed] [Google Scholar]
- 52.Zhou FX, Merianos HJ, Brunger AT, Engelman DM. Proc Natl Acad Sci U S A. 2001;98:2250. doi: 10.1073/pnas.041593698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hampton RY. Annu Rev Cell Dev Biol. 2002;18:345. doi: 10.1146/annurev.cellbio.18.032002.131219. [DOI] [PubMed] [Google Scholar]
- 54.Casey PJ, Seabra MC. J Biol Chem. 1996;271:5289. doi: 10.1074/jbc.271.10.5289. [DOI] [PubMed] [Google Scholar]
- 55.Shearer AG, Hampton RY. EMBO J. 2005;24:149. doi: 10.1038/sj.emboj.7600498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cronin SR, Khoury A, Ferry DK, Hampton RY. J Cell Biol. 2000;148:915. doi: 10.1083/jcb.148.5.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cronin SR, Rao R, Hampton RY. J Cell Biol. 2002;157:1017. doi: 10.1083/jcb.200203052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Saier MH, Ma CH, Rodgers L, Tamang DG, Yen MR. Adv Appl Microbiol. 2008;65:141. doi: 10.1016/S0065-2164(08)00606-0. [DOI] [PubMed] [Google Scholar]
- 59.Tipper DJ, Harley CA. Mol Biol Cell. 2002;13:1158. doi: 10.1091/mbc.01-10-0488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ward CL, Kopito RR. J Biol Chem. 1994;269:25710. [PubMed] [Google Scholar]
- 61.Shearer AG, Hampton RY. J Biol Chem. 2004;279:188. doi: 10.1074/jbc.M307734200. [DOI] [PubMed] [Google Scholar]
- 62.Gardner RG, Shearer AG, Hampton RY. Mol Cell Biol. 2001;21:4276. doi: 10.1128/MCB.21.13.4276-4291.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Flury I, Garza R, Shearer A, Rosen J, Cronin S, Hampton RY. EMBO J. 2005;24:3917. doi: 10.1038/sj.emboj.7600855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Radhakrishnan A, Sun LP, Kwon HJ, Brown MS, Goldstein JL. Mol Cell. 2004;15:259. doi: 10.1016/j.molcel.2004.06.019. [DOI] [PubMed] [Google Scholar]
- 65.Seegmiller AC, Dobrosotskaya I, Goldstein JL, Ho YK, Brown MS, Rawson RB. Dev Cell. 2002;2:229. doi: 10.1016/s1534-5807(01)00119-8. [DOI] [PubMed] [Google Scholar]
- 66.Bernier V, Lagace M, Bichet DG, Bouvier M. Trends Endocrinol Metab. 2004;15:222. doi: 10.1016/j.tem.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 67.Bernier V, Bichet DG, Bouvier M. Curr Opin Pharmacol. 2004;4:528. doi: 10.1016/j.coph.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 68.Bernier V, Lagace M, Lonergan M, Arthus MF, Bichet DG, Bouvier M. Mol Endocrinol. 2004;18:2074. doi: 10.1210/me.2004-0080. [DOI] [PubMed] [Google Scholar]
- 69.Petaja-Repo UE, Hogue M, Bhalla S, Laperriere A, Morello JP, Bouvier M. EMBO J. 2002;21:1628. doi: 10.1093/emboj/21.7.1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bernier V, Morello JP, Zarruk A, Debrand N, Salahpour A, Lonergan M, Arthus MF, Laperriere A, Brouard R, Bouvier M, Bichet DG. J Am Soc Nephrol. 2006;17:232. doi: 10.1681/ASN.2005080854. [DOI] [PubMed] [Google Scholar]
- 71.Sever N, Song BL, Yabe D, Goldstein JL, Brown MS, DeBose-Boyd RA. J Biol Chem. 2003;278:52479. doi: 10.1074/jbc.M310053200. [DOI] [PubMed] [Google Scholar]
- 72.Chen B, Mariano J, Tsai YC, Chan AH, Cohen M, Weissman AM. Proc Natl Acad Sci U S A. 2006;103:341. doi: 10.1073/pnas.0506618103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ballar P, Shen Y, Yang H, Fang S. J Biol Chem. 2006;281:35359. doi: 10.1074/jbc.M603355200. [DOI] [PubMed] [Google Scholar]
- 74.Song BL, Javitt NB, DeBose-Boyd RA. Cell Metabolism. 2005;1:179. doi: 10.1016/j.cmet.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 75.Sliter DA, Kubota K, Kirkpatrick DS, Alzayady KJ, Gygi SP, Wojcikiewicz RJ. J Biol Chem. 2008;283:35319. doi: 10.1074/jbc.M807288200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhu CC, Wojcikiewicz RJ. Biochem J. 2000;348(Pt 3):551. [PMC free article] [PubMed] [Google Scholar]
- 77.Alzayady KJ, Panning MM, Kelley GG, Wojcikiewicz RJ. J Biol Chem. 2005;280:34530. doi: 10.1074/jbc.M508890200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pearce MM, Wang Y, Kelley GG, Wojcikiewicz RJ. J Biol Chem. 2007;282:20104. doi: 10.1074/jbc.M701862200. [DOI] [PubMed] [Google Scholar]
- 79.Song BJ, Veech RL, Park SS, Gelboin HV, Gonzalez FJ. J Biol Chem. 1989;264:3568. [PubMed] [Google Scholar]
- 80.Morishima Y, Peng HM, Lin HL, Hollenberg PF, Sunahara RK, Osawa Y, Pratt WB. Biochemistry. 2005;44:16333. doi: 10.1021/bi0515570. [DOI] [PubMed] [Google Scholar]
- 81.Liao M, Faouzi S, Karyakin A, Correia MA. Mol Pharmacol. 2006;69:1897. doi: 10.1124/mol.105.021816. [DOI] [PubMed] [Google Scholar]
- 82.Faouzi S, Medzihradszky KF, Hefner C, Maher JJ, Correia MA. Biochemistry. 2007;46:7793. doi: 10.1021/bi700340n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ogura T, Inoue K, Tatsuta T, Suzaki T, Karata K, Young K, Su LH, Fierke CA, Jackman JE, Raetz CR, Coleman J, Tomoyasu T, Matsuzawa H. Mol Microbiol. 1999;31:833. doi: 10.1046/j.1365-2958.1999.01221.x. [DOI] [PubMed] [Google Scholar]
- 84.Sorensen PG, Lutkenhaus J, Young K, Eveland SS, Anderson MS, Raetz CR. J Biol Chem. 1996;271:25898. doi: 10.1074/jbc.271.42.25898. [DOI] [PubMed] [Google Scholar]
- 85.Ito K, Akiyama Y. Annu Rev Microbiol. 2005;59:211. doi: 10.1146/annurev.micro.59.030804.121316. [DOI] [PubMed] [Google Scholar]
- 86.Fuhrer F, Langklotz S, Narberhaus F. Mol Microbiol. 2006;59:1025. doi: 10.1111/j.1365-2958.2005.04994.x. [DOI] [PubMed] [Google Scholar]
- 87.Raetz CR, Whitfield C. Annu Rev Biochem. 2002;71:635. doi: 10.1146/annurev.biochem.71.110601.135414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Murphy RC, Raetz CR, Reynolds CM, Barkley RM. Prostaglandins Other Lipid Mediat. 2005;77:131. doi: 10.1016/j.prostaglandins.2004.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]