

Abstract
NMR has long been instrumental in the characterization of intrinsically disordered proteins (IDPs) and intrinsically disordered regions (IDRs). This method continues to offer rich insights into the nature of IDPs in solution, especially in combination with other biophysical methods such as small-angle scattering, single-molecule fluorescence, EPR and mass spectrometry. Substantial advances have been made in recent years in studies of proteins containing both ordered and disordered domains and in characterization of problematic sequences containing repeated tracts of a single or a few amino acids. These sequences are relevant to disease states such as Alzheimer’s, Parkinson’s and Huntington’s diseases, where disordered proteins misfold into harmful amyloid. Innovative applications of NMR are providing novel insights into mechanisms of protein aggregation and the complexity of IDP interactions with their targets. As a basis for understanding the solution structural ensembles, dynamic behavior, and functional mechanisms of IDPs and IDRs, NMR continues to prove invaluable.
Keywords: intrinsically disordered proteins, intrinsically disordered regions, amyloid, protein dynamics, isotopic labeling
Graphical Abstract
Introduction
More than half of the proteins in the human proteome are entirely disordered (intrinsically disordered proteins; IDPs) or contain both structured and long disordered regions (IDRs). Molecular level characterization of proteins that contain both structured and disordered domains represents an enormous challenge to the traditional methods of structural biology. Most studies to date have relied upon a reductionist approach, in which the ordered and disordered regions are investigated in isolation. However, within the cell, the ordered and disordered domains act synergistically to allow a protein to perform its biological function; a full understanding of the underlying molecular mechanism can only be achieved through a holistic, rather than reductionist, approach.
The original experimental identification of disordered regions of functional importance in proteins was accomplished primarily by NMR spectroscopy [1]. The sequence signatures of disordered protein regions have been identified in numerous bioinformatic studies and disorder predictions are available in databases [2]. The past few years have seen extensive progress in the biophysical characterization of IDPs and IDRs; NMR continues to be a primary tool and continuing innovations have greatly advanced fundamental studies aimed at characterizing IDPs, IDRs and their functional interactions, and in exploring particular disordered systems of biomedical interest. Despite intense current interest in the role of IDPs in liquid-liquid phase separation and the direct application of NMR in many studies, this review will make only brief mention of the field as recent reviews have covered the subject thoroughly [3].
NMR Technical Advances
IDPs are inherently dynamic and sample an ensemble of conformations in solution. Characterization and validation of these ensembles is challenging and requires a hybrid approach in which experimental restraints from NMR, small angle X-ray scattering, and biophysical methods such as single molecule FRET are combined with computer modeling to generate structural models of the overall ensemble [4]. Although local elements of transient helical structure can be readily identified and the population of helix quantitated from secondary chemical shifts, characterization of transient β-sheet has proved elusive. Recently, an approach designated TSD-NMR (Transient Structure from chemical Denaturation by NMR) has been developed which, in combination with chemical shift-based structure determination, revealed 10% population of a small β-sheet near the C-terminus of the Alzheimer’s Aβ1-42 peptide [5*]. Chemical denaturant titrations have also been used to overcome a problem that is often encountered in NMR studies of larger IDPs – broadening of resonances due to formation of collapsed, molten globule-like states. For the oncoprotein Myc, this problem was resolved by titration with guanidinium hydrochloride to allow assignment of resonances and calculation of structural ensembles using restraints from chemical shifts and paramagnetic relaxation rates, extrapolated to zero denaturant concentration [6*].
One of the major hurdles to tackling the NMR characterization of full-length proteins containing both ordered and disordered domains is their structural heterogeneity. This poses major challenges, especially for higher molecular weight proteins where resonances arising from residues in the structured domain(s) experience substantial broadening while those associated with the disordered domain(s) have narrow linewidths but may be poorly resolved due to limited spectral dispersion and extensive resonance overlap. Methods used previously to elucidate the structure and dynamics of isolated fragments comprising the disordered or structured regions do not provide the vital insights into the synergistic interactions between the various domains of the full-length molecules. This problem has been central to a number of the studies described in the past few years.
Protein ligation and isotopic labeling.
Deconvoluting the NMR resonances of large disordered and mixed ordered-disordered proteins is a challenge that can be addressed in the first instance by clever applications of stable isotopic labeling. Uniform isotopic labeling with 15N and 13C is the standard method, but complications due to resonance overlap persist even with the amelioration of the relaxation problems of large molecular systems by transverse relaxation-optimized spectroscopy (TROSY) and deuteration. Protein ligation technology has come into its own in the last few years, as a means of producing proteins that are differentially labeled at the level of individual domains. Recent innovations have included traceless salt-inducible intein ligation[7]., and flexible ligation using variants of a sortase A enzyme from Staphylococcus aureus that can be employed to produce circularized proteins or lipidated proteins, as well as mixed ordered-disordered proteins, or to provide anchoring to nanodiscs [8]. Cell-free expression has proved to be a powerful method to obtain NMR-labeled samples of low-complexity proteins, particularly homorepeat proteins containing long stretches of single amino acids such as proline or glutamine: a compendium of useful cell-free expression methods was recently published [9]. By combining cell-free protein synthesis with nonsense suppression, site-specific labeling of residues within polyglutamine, polyproline or other homorepeat tracts can be accomplished, opening the way to structural and dynamic characterization of proteins containing these regions [10].
15N and 13C detection for IDPs
Traditionally, heteronuclear NMR experiments have focused on the detection of proton magnetization since the sensitivity of the proton is so much greater than those of other spin ½ nuclei (except for 19F). However, for the specific case of disordered proteins, direct detection of 15N and 13C offers the advantages of narrower linewidths, enhanced resolution, and the ability to acquire spectra at physiological pH, where exchange of amide protons with water typically results in broadening or disappearance of resonances in conventional amide 1HN-detected spectra [11,12]. Recent advances include development of a sensitivity enhanced 13C-detect experiment for monitoring phosphorylation of IDPs at physiological temperature and pH where kinases are active [13]. A suite of triple resonance experiments utilizing 1Hα-detection provides a 3-5-fold increase in sensitivity compared to their 13CO-detect analogs for samples at physiological or higher pH [14].
New ways to use NMR for IDPs
The community has produced a number of innovative new experimental techniques designed to give information beyond simple chemical shift assignments and relaxation measurements. These include NMR exchange spectroscopy for elucidating fuzzy interactions [15]., use of solvent paramagnetic relaxation enhancement (PRE)[16]., to identify low populations of locally structured states and to characterize the conformational ensemble [17]., the use of 15N chemical exchange saturation transfer (CEST) to obtain amide proton exchange rates [18]., and a combination of CEST and pulsed double electron-electron resonance (DEER) EPR to give information on interactions of huntingtin peptides with micellar nanoparticles [19]. In an innovative approach to overcome the problem of extreme IDP resonance broadening due to fast exchange of amide protons with solvent at physiological pH values, a weak 1H B1 field is swept through a three-dimensional 1Hα-detect spectrum to determine amide proton resonance frequencies [20*]. A novel method that combines transferred NOEs, 13C-methyl labeling, and isotope-edited/isotope-filtered NOESY has been reported for mapping interactions of intrinsically disordered short linear motifs (SLiMs) with target proteins [21]. This method should find general utility for mapping weak, fast-exchange interactions between IDPs and targets of up to ~80 kDa molecular weight. Weakly interacting, slowly-exchanging systems can be characterized by off-resonance R1ρ in cases such as liquid-liquid phase separation where there are large differences in relaxation rates but no differences in chemical shift between free and bound states [22].
Dynamics measurements
Since IDPs consist of ensembles of distinct structures rather than the single or closely-similar structures characteristic of globular proteins, the issue of the dynamics of the ensemble, and the kinetics of interconversion between members of the ensemble is a central one for disordered proteins. Considerable emphasis has been placed in the past few years on the characterization of IDP dynamics and its relevance to biological function. Extensive 15N NMR relaxation measurements have been reported for many IDPs in their unbound states. Distinct dynamic modes associated with fast (< 50 ps) librational motions, transitions between Ramachandran substates on an ~1 ns time scale, and slower (> 5 ns) segmental motions of the polypeptide chain have been identified [23,24]. Measurements of IDP relaxation data under conditions of molecular crowding show that the intermediate and slow time scale motions are strongly coupled to the friction of the solvent [25*]. These studies led to a unified model of IDP dynamics in complex environments such as phase separated states and the cellular milieu.
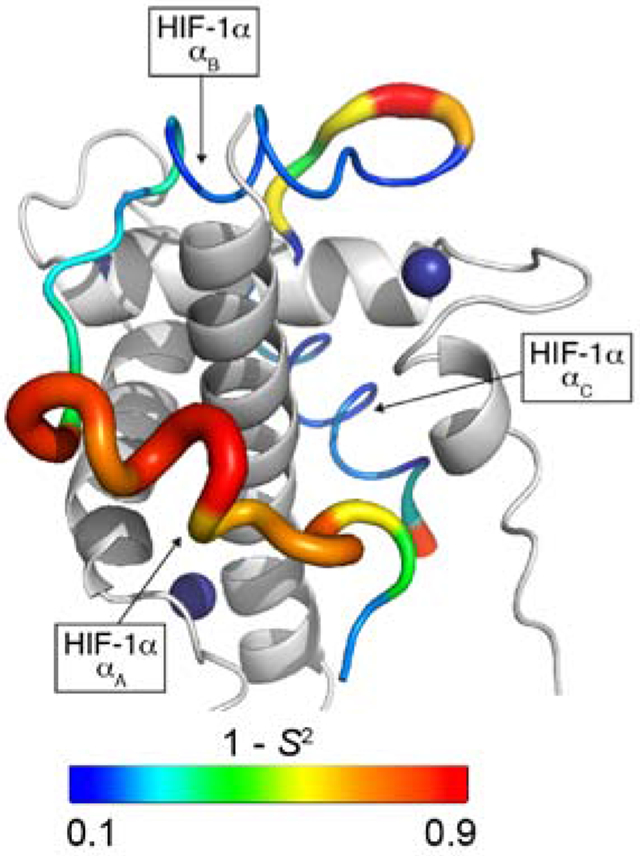
NMR relaxation measurements have been used to characterize fast (ps – ns) time scale dynamics in IDP complexes to provide insights into binding energetics, binding mechanisms, and allostery. Many IDPs bind their targets through multiple linear motifs separated by weakly interacting linkers; the overall affinity is determined by the binding free energy of each motif and the strength of thermodynamic coupling between them. NMR relaxation measurements provide a direct method for probing the heterogenous energy landscape of an IDP complex. Several studies have focused on complexes formed by the TAZ1 domain of the transcriptional coactivators CBP and p300, which binds the disordered activation domains of several cellular transcription factors. TAZ1 exhibits structural plasticity and both the backbone and side chain dynamics are differentially modulated by binding of different IDP ligands [26*,27]. The IDP ligands themselves exhibit heterogeneous dynamics; regions that do not interact with TAZ1 or make only fuzzy interactions undergo large amplitude motions while sequence motifs that dominate binding are motionally restricted, underscoring the complexity of IDP-target interactions [26*]. (Figure 1) Dynamics measurements provided important insights into the molecular mechanism by which the negative feedback regulator CITED2 promotes dissociation of the hypoxia inducible factor HIF-1α from TAZ1 to very efficiently down-regulate the hypoxic response. By binding to a region of TAZ1 where HIF-1α interactions are highly dynamic, CITED2 forms a transient ternary complex that drives a conformational change in TAZ1 and dissociation of HIF-1α [26*,28]. The plasticity of TAZ1 and the dynamic heterogeneity of the HIF-1α and CITED2 complexes are central to the mechanism of this molecular switch.
Figure 1.
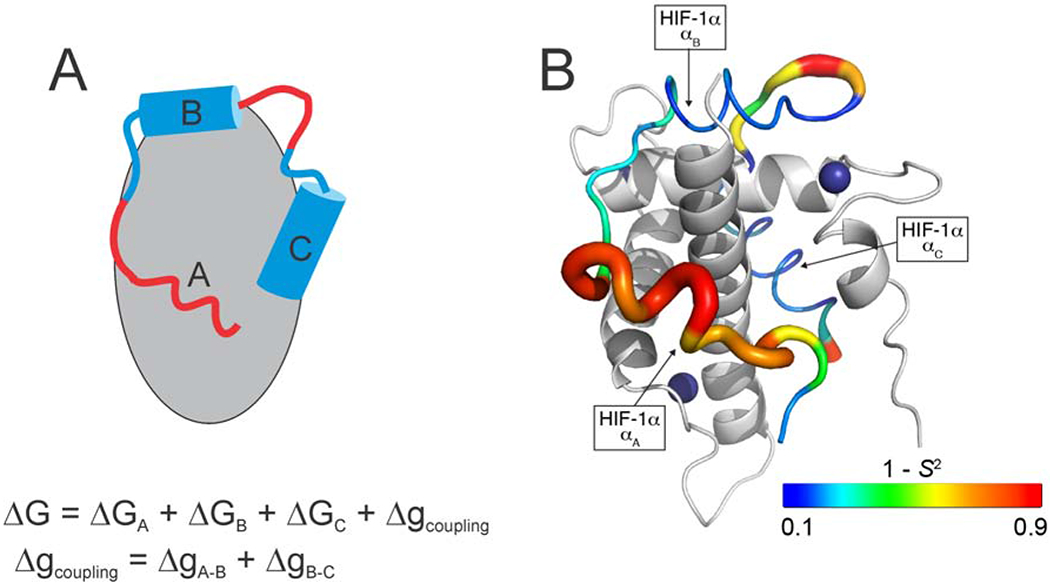
A. Schematic representation of heterogeneous interactions between an IDP and its target. Interactions with motif A are fuzzy and highly dynamic while motifs B and C interact strongly and specifically with the target such that their motions are restricted. The overall binding free energy is the sum of the free energies of interaction of each motif and the free energy associated with thermodynamic coupling (Δg) between the individual binding motifs. B. Backbone dynamics for the IDP HTF-1α in complex with TAZ1. The ribbon width of the HIF-1α transactivation domain peptide is scaled by 1-S2, where S2 is the order parameter describing the amplitude of N-H bond motions. The backbone color gradient ranges from blue (1-S2 = 0.1) in regions where motions are restricted by strong interactions with TAZ1 to red (1- S2 = 0.9) where intermolecular interactions are weak and there are large amplitude motions of the HIF-1α backbone. TAZ1 is shown in the cartoon representation in grey. Zinc atoms are shown as dark blue spheres. The secondary structural elements of HIF-1α are labeled. (Adapted from [26*] with permission).
A particularly fruitful approach appears to be the combination of NMR dynamics measurements with molecular dynamics calculations, which have been used, for example, in the delineation of solvent-dependent segmental dynamics [29]. A new strategy has been reported for generation of conformational ensembles for flexible multidomain proteins using residual dipolar coupling restraints and Langevin dynamics [30]. Solvent deuterium isotope effects were shown to have deleterious effects on the accuracy of 15N transverse relaxation measurements but this problem can be overcome by using a very low fraction of D2O in the buffer [31].
An IDP Problem: Repetitive low complexity tracts
Varying lengths of repeating sequences, sometimes of short peptide motifs and sometimes of just a single amino acid, are characteristic features of IDPs and IDRs. Such repetition is problematic for the residue-level characterization of IDPs by NMR, and several approaches have been successfully implemented to assign resonances in these regions. Proline-rich sequences are common in disordered regions, where they frequently participate in molecular recognition in signaling networks or may perform diverse roles such as a disorder promoter or compaction facilitator [32]. Proline is a problematic amino acid in the commonly-used 1HN-detected NMR experiments, as it lacks an amide proton. Proline-rich sequences can be assigned using 13C-detected CON-based strategies. Rather than requiring 4- and higher dimensional spectra, a simplified approach, of particular utility in systems with a very high proportion of prolines, uses a single 2D CON spectrum, with two 3D triple-resonance spectra to directly correlate adjacent CON units [33]. The CON spectrum provides a useful fingerprint for proline-rich IDPs, analogous to the 1H-15N HSQC spectrum [34]. A comprehensive characterization of the behavior of prolines in disordered proteins is essential for the description of a number of systems, such as a proline-rich domain (containing 30% proline residues) of the adaptor protein ALIX [35]. The CON strategy has also been used to identify the interactions between arginine-rich and aromatic-rich regions that drive phosphorylation-dependent, liquid-liquid phase separation in the FMRP-CAPRIN1 system at physiological pH (7.4) [36**].
Glutamine is another problematic amino acid that is frequently enriched in disordered regions of disease-related proteins. The problem here is not the absence of the amide proton, but the extreme resonance overlap caused by long tracts of polyglutamine. For huntingtin, where disease pathology in Huntington’s Disease is directly related to expansion of polyglutamine repeat sequences, characterization of these regions by NMR has required extensive design of specific labeling strategies [10]. which have led to the insight that the structural properties of the polyglutamine repeat regions are strongly influenced by the nature of the flanking regions [37**].
Fuzzy Complexes
Although early studies of the complexes formed by IDPs frequently showed the disordered domains in a folded state in the complex, we and others increasingly began to observe that in even quite tight complexes (Kd ~ nM), the bound IDP or regions of it can remain disordered. For example, the adenoviral E1A oncoprotein binds to the TAZ2 domain of the transcriptional activator CBP with two binding modes – part of the ligand folds to form a structured helix on the surface of TAZ2 [38]. but the binding affinity is increased 4-fold by the presence of an N-terminal region of E1A that makes only transient interactions with TAZ2 [39]. Complexes that display structural polymorphism and structural disorder have been termed fuzzy complexes [40].
The functional advantages of fuzziness have been described elsewhere [40]. and there are now numerous published examples in a number of biomedically important systems. Recent examples include a comprehensive study that identified an extremely high-affinity interaction between two disordered proteins, prothymosin-α and histone H1, that is mediated through electrostatics and shows all of the hallmarks of a fuzzy complex [41,42]. Other examples include the C-terminal domain of the splicing factor FBP21, which binds with high specificity to a large site on the Brr2 helicase but remains highly dynamic in the complex [43]. and binding of the transcription factor c-Jun to the ubiquitin ligase SCFFbw7 and the proline isomerase Pin1 through dynamic interactions with multiple phospho-degron motifs located in intrinsically disordered regions of c-Jun [44]. A particularly interesting and complex case involved an order-disorder transformation of a redox switch in the chloroplast protein CP12 upon interaction with glyceraldehyde-3-phosphate dehydrogenase [45]. which is interpreted as contributing to an entropy increase that amplifies the affinity of the complex.
Increasingly, NMR is combined with molecular dynamics simulations to provide a molecular level description of fuzzy complexes. By combining solution and solid-state NMR with computer simulations, a recent study provided novel insights into the highly dynamic interactions of a disordered protein, corresponding to the cytoplasmic region of the divisisome protein ChiZ, with an acidic membrane. Interactions occurred predominantly through arginine side chains and the membrane lipids were observed to redistribute to adopt arginine-proximal positions [46*]. In a very elegant study, methyl TROSY NMR, paramagnetic relaxation enhancement, small angle neutron scattering (SANS), and molecular modeling were used to determine the solution structure of the complex between the acetyltransferase Rtt109, the histone chaperones Asf1 and Vps75, and the histone H3:H4 dimer [47**]. Electrostatically-mediated, fuzzy interactions between the intrinsically disordered Vps75 C-terminal region and the disordered histone H3 tail promote binding of H3 lysine residues in the Rtt109 catalytic site and thereby enhance acetylation specificity with minimum entropic cost.
Chaperone interactions
Among the paradigmatic interactions of folded proteins with unfolded domains are those between molecular chaperones and nascent or unfolded proteins. One of the most prominent functions of chaperones is to sequester nascent protein chains as they emerge from ribosomes. NMR has been used with great success in the past to elucidate the disordered conformations of nascent chains [48]. More recently, an NMR and ESI-mass spectrometry study showed that the nascent polypeptide-associated complex, a ribosome-associated chaperone, exhibits conformational flexibility that allows it to bind weakly to nascent chains and IDPs with widely varying polypeptide sequences [49]. The chaperone HdeA becomes disordered when activated, exposing hydrophobic surfaces for binding clients [50]. CEST experiments were used to identify local acid-sensitive regions that regulate the structural change and exposure of the client binding sites [51]. NMR experiments also show that another small chaperone, HspB1, responds to phosphorylation of IDR clients by local unfolding and increased exposure of hydrophobic sites [52]. and that the same type of hydrophobic exposure mediates interactions of HspB1 with the disordered protein tau [53].
Besides their function as “holdases” in the stabilization of nascent transcripts emerging from ribosomes, and as “foldases” facilitating the correct folding of globular proteins in the cytoplasm, chaperones are also heavily utilized in secretion pathways. The requirement for secreted proteins to be unfolded before passing through the membrane in complex with chaperones has long been known. Differences in the mechanisms of these processes in the case of the Yersinia virulence factors YopE and YopH have recently been elucidated with methods combining NMR, SAXS and molecular modeling [54]. Bacterial virulence factors are secreted in complex with a specific chaperone. For YopE, the secretion complex was shown to be ordered, while YopH employed an entirely different mechanism with part of the C-terminus extruded from the surface of the complex.
Recent NMR studies give a definitive mechanistic picture of the interaction between the chaperone Hsp70 and client proteins, in this case a destabilized mutant domain of chicken brain α-spectrin termed R17* and an SH3 domain [55]. Both proteins exist in equilibrium between folded and unfolded states in solution. 1H-13C CEST and zz-exchange experiments were used to define the fluxes through induced-fit (IF) and conformational selection (CS) pathways of binding of R17* to Hsp70. The Hsp70 binds both clients by CS mechanism, preferentially binding to the unfolded state rather than the natively folded form. In an alternative approach, DEER EPR was recently used to characterize the interaction of a disordered protein, Tau, with the chaperone Hsp90 [56].
Progress in the Study of Aggregation Diseases
Protein aggregation diseases such as Alzheimer’s, Huntington’s and Parkinson’s diseases are among the most frustratingly resistant to treatment. Considerable effort has been expended in characterizing the mechanisms of aggregation and toxicity of the proteins involved. In-cell NMR gave extensive insights into the molecular state of the Parkinson’s disease protein α-synuclein, confirming that it remains disordered even in the crowded environment of the cell [57]. A more recent study employing in-cell NMR showed that a specific motif in α-synuclein interacts with Hsp70 and Hsp90 chaperones both in vitro and in the cell; inhibition of this interaction in the cell causes α-synuclein to re-localize to mitochondria and aggregate [58*].
New NMR methods have been introduced to probe transient oligomerization processes of intrinsically disordered proteins that lead to aggregation and amyloid formation. A novel pressure-jump NMR method has been applied to study Alzheimer’s Aβ oligomers at site-specific resolution [59*]. By cycling between low and high pressure, the structure and dynamics of the oligomeric state can be probed by monitoring NMR spectra of the monomer to provide new insights into the earliest aggregation events. A set of NMR relaxation experiments, including relaxation dispersion, rotating frame relaxation, and exchange-induced chemical shifts, recorded over a range of protein concentrations, has been used to monitor early oligomerization processes in a truncated region of the huntingtin protein [60**]. A branched kinetic scheme for oligomerization, involving on- and off-pathway processes, was derived and structural models of the productive intermediates were determined from PRE data and validated by DEER EPR measurements. The approach was later extended to the full huntingtin exon 1 region containing a tract of 7 glutamines and two polyproline tracts [61]. Human profilin I inhibits huntingtin aggregation by binding to one of the polyproline tracts and abrogating the productive oligomerization pathway.
NMR Approaches to Full-Length Proteins Containing IDRs
The experimental and computational methods outlined above (and other techniques that are beyond the scope of this review) form part of an arsenal that can be used to investigate full-length proteins that contain fully-folded and disordered domains. These efforts are clearly ongoing, but good progress has been made in recent years for a number of widely different systems.
Viral proteins
Protein disorder is particularly prevalent in transcriptional and cell cycle control [62]. and these processes are targeted by viruses that use virally encoded IDPs to hijack cellular pathways [63]. Disordered viral proteins target, for example, the retinoblastoma protein pRb and the transcriptional coactivator CBP/p300, through sequence-specific and high-affinity interactions that compete with disordered cellular factors such as the tumor suppressor p53. Recent NMR studies include characterization of the interaction of the disordered activation domain of the human T cell leukemia virus (HTLV-1) protein HBZ, which maintains chronic infection and promotes leukemogenesis, with the KIX domain of CBP [64]. The TAZ2 and NCBD domains of CBP/p300 interact with the adenoviral E1A protein [38,65]. as well as a previously-unrecognized sequence motif in the the N-terminal IDR of CBP [66].
Multi-domain viral proteins have been a major focus of several labs over a number of years. Early studies of the Measles virus capsid protein employed the reductionist approaches that were standard at the time, but recent work has captured many of the features of the full-length proteins, including liquid-liquid phase separation upon mixing the phosphoprotein and capsid protein to form droplets that facilitate encapsidation of RNA [67]. Two recent studies have focused on the Nipah virus phosphoprotein that contains both disordered and structured regions. Using 13C-detected and conventional 1HN detected experiments, ~90% of the residues in a disordered, 406 amino acid N-terminal domain were successfully assigned [68]. . Characterization of the structural ensemble of the full-length Nipah virus phosphoprotein tetramer exemplifies the challenges of characterizing the structure of large proteins and illustrates the power of a combined approach that applies NMR, X-ray crystallography, SAXS and other biophysical methods [69].
Tumor Suppressor p53
The voluminous literature on p53 has received a number of additions in the past two years. It contains both structured (the DNA-binding domain (DBD) and the tetramerization domain) and disordered domains (the N-terminal activation domain (NTAD), the proline-rich domain (PRD) and the C-terminal regulatory domain). p53 has been extensively studied for many years, but recent advances in NMR and isotopic labeling methods have allowed new, atomic-level insights into synergistic interactions between domains that modulate the behavior of the full-length protein. Two recent NMR studies using either full-length p53 or an NTAD-DBD fragment showed the presence of intramolecular interactions between the DBD and the NTAD that function to enhance discrimination between cognate and non-cognate DNA sequences [70,71]. Studies of the full-length protein were facilitated by the use of intein methods to segmentally 15N label the p53 tetramer so that only the NTAD resonances are observed in the NMR spectra [70]. These studies were subsequently extended to investigate the role of Thr55, located within the NTAD, which functions as a phosphorylation-dependent switch that regulates DNA binding [72].
Conclusion
NMR remains one of the most powerful techniques for characterization of the structure, dynamics, interactions, and mechanism of action of proteins, particularly those that contain regions of intrinsic disorder. Our current survey of the most recent literature identifies a few recurring themes. New advances in NMR methodology are constantly stretching the limits on the types of biological systems that can be studied. Innovative and elegant isotope labeling strategies are opening the way to NMR studies of large proteins containing both ordered and disordered domains and are beginning to provide unprecedented insights into complex systems. Combined approaches using multiple biophysical methods, including NMR, small angle scattering, single molecule FRET, DEER EPR, and X-ray crystallography, coupled with molecular dynamics simulations, promise to greatly advance our understanding of disordered proteins. We now have tools available to tackle some of the most challenging multi-domain proteins and can anticipate the emergence of a new, non-reductionist era of structural biology in which the functional synergies between ordered and disordered domains is fully revealed.
Figure 2.
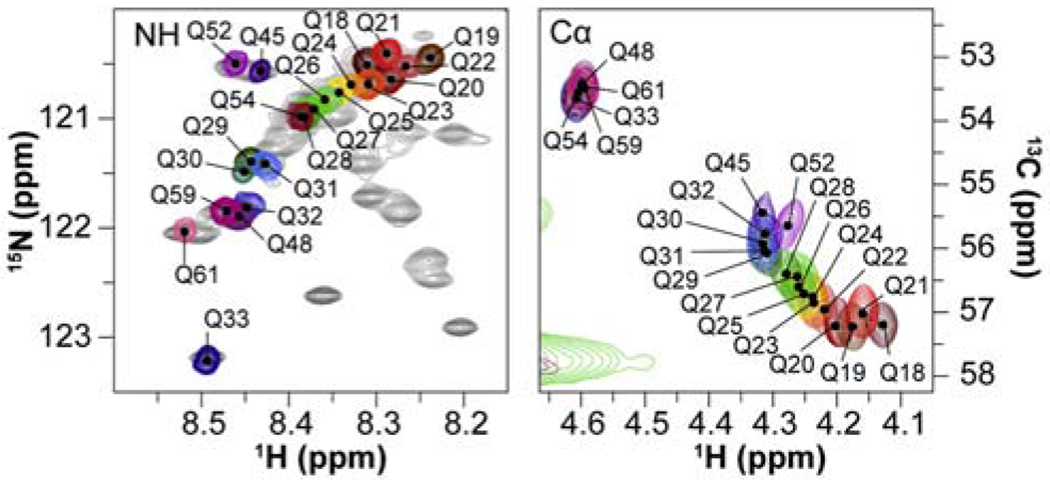
Portions of 1H-15N HSQC (left) and 1H-13C HSQC (right) spectra showing cross peaks belonging to members of a polyglutamine tract in huntingtin. The color-coded peaks were assigned by site-specific isotopic labeling [10,37**]. (adapted from reference [37**]. with permission).
Acknowledgements
This work was supported by grants GM131693, CA214054, CA229652, and GM075995 from the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interest
The authors declare no conflict of interest.
References
- 1.Dyson HJ, Wright PE: Perspective: the essential role of NMR in the discovery and characterization of intrinsically disordered proteins. J Biomol NMR 2019, 73:651–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Oates ME, Romero P, Ishida T, Ghalwash M, Mizianty MJ, Xue B, Dosztanyi Z, Uversky VN, Obradovic Z, Kurgan L, Dunker AK, Gough J: D2P2: database of disordered protein predictions. Nucl Acids Res 2013, 41:D508–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Murthy AC, Fawzi NL: The (un)structural biology of biomolecular liquid-liquid phase separation using NMR spectroscopy. J Biol Chem 2020, 295:2375–2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gomes GW, Krzeminski M, Namini A, Martin EW, Mittag T, Head-Gordon T, Forman-Kay JD, Gradinaru CC: Conformational ensembles of an intrinsically disordered protein consistent with NMR, SAXS, and single-molecule FRET. J Am Chem Soc 2020, 142:15697–15710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *5.Kakeshpour T, Ramanujam V, Barnes CA, Shen Y, Ying J, Bax A: A lowly populated, transient β-sheet structure in monomeric Aβ1-42 identified by multinuclear NMR of chemical denaturation. Biophys Chem 2021, 270:106531. [DOI] [PMC free article] [PubMed] [Google Scholar]; Application of chemical denaturant titrations and chemical shift-based structure determination to identify a transient, weakly populated β-sheet in the Alzheimer’s Aβ1-42 peptide.
- *6.Panova S, Cliff MJ, Macek P, Blackledge M, Jensen MR, Nissink JWM, Embrey KJ, Davies R, Waltho JP: Mapping hidden residual structure within the Myc bHLH-LZ domain using chemical denaturant titration. Structure 2019, 27:1537–1546.e4. [DOI] [PubMed] [Google Scholar]; Application of chemical denaturant titrations to overcome problems arising from broadening of resonances due to formation of a collapsed state of an IDP.
- 7.Ciragan A, Backlund SM, Mikula KM, Beyer HM, Samuli Ollila OH, Iwaï H: NMR structure and dynamics of TonB investigated by scar-less segmental isotopic labeling using a salt-inducible split intein. Front Chem 2020, 8:136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li J, Zhang Y, Soubias O, Khago D, Chao FA, Li Y, Shaw K, Byrd RA: Optimization of sortase A ligation for flexible engineering of complex protein systems. J Biol Chem 2020, 295:2664–2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morató A, Elena-Real CA, Popovic M, Fournet A, Zhang K, Allemand F, Sibille N, Urbanek A, Bernadó P: Robust cell-free expression of sub-pathological and pathological huntingtin exon-1 for NMR studies. General approaches for the isotopic labeling of low-complexity proteins. Biomolecules 2020, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Urbanek A, Morató A, Allemand F, Delaforge E, Fournet A, Popovic M, Delbecq S, Sibille N, Bernadó P: A general strategy to access structural information at atomic resolution in polyglutamine homorepeats. Angew Chem Int Ed Engl 2018, 57:3598–3601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gil S, Hošek T, Solyom Z, Kümmerle R, Brutscher B, Pierattelli R, Felli IC: NMR Spectroscopic studies of intrinsically disordered proteins at near-physiological conditions. Angew Chem Int Ed Engl 2013, 52:11808–11812. [DOI] [PubMed] [Google Scholar]
- 12.Chhabra S, Fischer P, Takeuchi K, Dubey A, Ziarek JJ, Boeszoermenyi A, Mathieu D, Bermel W, Davey NE, Wagner G, Arthanari H: 15N detection harnesses the slow relaxation property of nitrogen: Delivering enhanced resolution for intrinsically disordered proteins. Proc Natl Acad Sci USA 2018, 115:E1710–E1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alik A, Bouguechtouli C, Julien M, Bermel W, Ghouil R, Zinn-Justin S, Theillet FX: Sensitivity-enhanced 13C-NMR spectroscopy for monitoring multisite phosphorylation at physiological temperature and pH. Angew Chem Int Ed Engl 2020, 59:10411–10415. [DOI] [PubMed] [Google Scholar]
- 14.Wong LE, Kim TH, Muhandiram DR, Forman-Kay JD, Kay LE: NMR experiments for studies of dilute and condensed protein phases: application to the phase-separating protein CAPRIN1. J Am Chem Soc 2020, 142:2471–2489. [DOI] [PubMed] [Google Scholar]
- 15.Delaforge E, Kragelj J, Tengo L, Palencia A, Milles S, Bouvignies G, Salvi N, Blackledge M, Jensen MR: Deciphering the dynamic interaction profile of an intrinsically disordered protein by NMR exchange spectroscopy. J Am Chem Soc 2018, 140:1148–1158. [DOI] [PubMed] [Google Scholar]
- 16.Hartlmüller C, Spreitzer E, Göbl C, Falsone F, Madl T: NMR characterization of solvent accessibility and transient structure in intrinsically disordered proteins. J Biomol NMR 2019, 73:305–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kooshapur H, Schwieters C, Tjandra N: Conformational ensemble of disordered proteins probed by solvent Paramagnetic Relaxation Enhancement (sPRE). Angew Chem Int Ed Engl 2018, 57:13519–13522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yuwen T, Bah A, Brady JP, Ferrage F, Bouvignies G, Kay LE: Measuring solvent hydrogen exchange rates by multi-frequency excitation 15N CEST. J Phys Chem B 2018, 122:11206–11217. [DOI] [PubMed] [Google Scholar]
- 19.Ceccon A, Schmidt T, Tugarinov V, Kotler SA, Schwieters CD, Clore GM: Interaction of Huntingtin exon-1 peptides with lipid-based micellar nanoparticles probed by solution NMR and Q-band pulsed EPR. J Am Chem Soc 2018, 140:6199–6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *20.Wong LE, Kim TH, Rennella E, Vallurupalli P, Kay LE: Confronting the invisible: assignment of protein 1H15N chemical shifts in cases of extreme broadening. J Phys Chem Lett 2020, 11:3384–3389. [DOI] [PubMed] [Google Scholar]; A clever approach to measure accurate chemical shifts of amide protons whose resonances are rendered invisible due to rapid exchange with solvent at physiological or higher pH.
- 21.Kumar S, Akabayov SR, Kessler N, Cohen LS, Solanki J, Naider F, Kay LE, Anglister J: The methyl 13C-edited/13C-filtered transferred NOE for studying protein interactions with short linear motifs. J Biomol NMR 2020, 74:681–693. [DOI] [PubMed] [Google Scholar]
- 22.Yuwen T, Brady JP, Kay LE: Probing conformational exchange in weakly interacting, slowly exchanging protein systems via off-resonance R1ρ experiments: application to studies of protein phase separation. J Am Chem Soc 2018, 140:2115–2126. [DOI] [PubMed] [Google Scholar]
- 23.Abyzov A, Salvi N, Schneider R, Maurin D, Ruigrok RWH, Jensen MR, Blackledge M: Identification of dynamic modes in an intrinsically disordered protein using temperature-dependent NMR relaxation. J Am Chem Soc 2016, 138:6240–6251. [DOI] [PubMed] [Google Scholar]
- 24.Rezaei-Ghaleh N, Parigi G, Soranno A, Holla A, Becker S, Schuler B, Luchinat C, Zweckstetter M: Local and global dynamics in intrinsically disordered synuclein. Angew Chem Int Ed Engl 2018, 57:15262–15266. [DOI] [PubMed] [Google Scholar]
- *25.Adamski W, Salvi N, Maurin D, Magnat J, Milles S, Jensen MR, Abyzov A, Moreau CJ, Blackledge M: A unified description of intrinsically disordered protein dynamics under physiological conditions using NMR spectroscopy. J Am Chem Soc 2019, 141:17817–17829. [DOI] [PubMed] [Google Scholar]; Presents a unified model to describe the dynamics of IDPs in solution or in the complex environment of cells or phase separated states. Spin relaxation data acquired under conditions of molecular crowding show that the backbone dynamics are strongly coupled to the friction of the solvent.
- *26.Berlow RB, Martinez-Yamout MA, Dyson HJ, Wright PE: Role of backbone dynamics in modulating the interactions of disordered ligands with the TAZ1 domain of the CREB-binding protein. Biochemistry 2019, 58:1354–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]; Characterization of the heterogenous backbone dynamics of two IDPs, HIF-1α and CITED2, bound to the TAZ1 domain. Regions of the IDPs that make only transient interactions with TAZ1 undergo large amplitude motions in the bound state while local motifs that dominate binding are motionally restricted, underscoring the complexity of IDP-target interactions.
- 27.Nyqvist I, Dogan J: Characterization of the dynamics and the conformational entropy in the binding between TAZ1 and CTAD-HIF-1α. Sci Rep 2019, 9:16557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Berlow RB, Dyson HJ, Wright PE: Hypersensitive termination of the hypoxic response by a disordered protein switch. Nature 2017, 543:447–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Salvi N, Abyzov A, Blackledge M: Solvent-dependent segmental dynamics in intrinsically disordered proteins. Science advances 2019, 5:eaax2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bouchard JJ, Xia J, Case DA, Peng JW: Enhanced sampling of interdomain motion using map-restrained Langevin dynamics and NMR: application to Pin1. J Mol Biol 2018, 430:2164–2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kumari P, Frey L, Sobol A, Lakomek N-A, Riek R: 15N transverse relaxation measurements for the characterization of μs-ms dynamics are deteriorated by the deuterium isotope effect on 15N resulting from solvent exchange. J Biomol NMR 2018, 72:125–137. [DOI] [PubMed] [Google Scholar]
- 32.Mateos B, Conrad-Billroth C, Schiavina M, Beier A, Kontaxis G, Konrat R, Felli IC, Pierattelli R: The ambivalent role of proline residues in an intrinsically disordered protein: from disorder promoters to compaction facilitators. J Mol Biol 2020, 432:3093–3111. [DOI] [PubMed] [Google Scholar]
- 33.Chaves-Arquero B, Pantoja-Uceda D, Roque A, Ponte I, Suau P, Jiménez MA: A CON-based NMR assignment strategy for pro-rich intrinsically disordered proteins with low signal dispersion: the C-terminal domain of histone H1.0 as a case study. J Biomol NMR 2018, 72:139–148. [DOI] [PubMed] [Google Scholar]
- 34.Murrali MG, Piai A, Bermel W, Felli IC, Pierattelli R: Proline fingerprint in intrinsically disordered proteins. Chembiochem 2018, 19:1625–1629. [DOI] [PubMed] [Google Scholar]
- 35.Elias RD, Ma W, Ghirlando R, Schwieters CD, Reddy VS, Deshmukh L: Proline-rich domain of human ALIX contains multiple TSG101-UEV interaction sites and forms phosphorylation-mediated reversible amyloids. Proc Natl Acad Sci USA 2020, 117:24274–24284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **36.Kim TH, Tsang B, Vernon RM, Sonenberg N, Kay LE, Forman-Kay JD: Phospho-dependent phase separation of FMRP and CAPRIN1 recapitulates regulation of translation and deadenylation. Science 2019, 365:825–829. [DOI] [PubMed] [Google Scholar]; Elegant application of 13C-detect NMR to identify the molecular interactions that drive phase separation of the translational regulators FMRP and CAPRIN1. Use of the CON experiment circumvented the problem of fast amide proton exchange at the pH required for phase separation and yielded well-resolved spectra in the highly viscous condensed phase.
- **37.Urbanek A, Popovic M, Morató A, Estaña A, Elena-Real CA, Mier P, Fournet A, Allemand F, Delbecq S, Andrade-Navarro MA, Cortés J, Sibille N, Bernadó P: Flanking regions determine the structure of the poly-glutamine in huntingtin through mechanisms common among glutamine-rich human proteins. Structure 2020, 28:733–746. [DOI] [PubMed] [Google Scholar]; Application of site-specific labeling to characterize the conformational propensities of the polyglutamine tract in the N-terminal region of huntingtin and determine the mechanism by which the flanking regions promote or inhibit aggregation. By combining cell-free expression and nonsense suppression, site-specific assignments were made for the 16 consecutive glutamine residues in the polyglutamine tract.
- 38.Ferreon JC, Martinez-Yamout MA, Dyson HJ, Wright PE: Structural basis for subversion of cellular control mechanisms by the adenoviral E1A oncoprotein. Proc Natl Acad Sci USA 2009, 106:13260–13265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ferreon AC, Ferreon JC, Wright PE, Deniz AA: Modulation of allostery by protein intrinsic disorder. Nature 2013, 498:390–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tompa P, Fuxreiter M: Fuzzy complexes: polymorphism and structural disorder in protein-protein interactions. Trends Biochem Sci 2008, 33:2–8. [DOI] [PubMed] [Google Scholar]
- 41.Borgia A, Borgia MB, Bugge K, Kissling VM, Heidarsson PO, Fernandes CB, Sottini A, Soranno A, Buholzer KJ, Nettels D, Kragelund BB, Best RB, Schuler B: Extreme disorder in an ultrahigh-affinity protein complex. Nature 2018, 555:61–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sottini A, Borgia A, Borgia MB, Bugge K, Nettels D, Chowdhury A, Heidarsson PO, Zosel F, Best RB, Kragelund BB, Schuler B: Polyelectrolyte interactions enable rapid association and dissociation in high-affinity disordered protein complexes. Nat Commun 2020, 11:5736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sticht J, Bertazzon M, Henning LM, Licha JR, Abualrous ET, Freund C: FBP21’s C-terminal domain remains dynamic when wrapped around the c-Sec63 unit of Brr2 helicase. Biophys J 2019, 116:406–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Csizmok V, Montecchio M, Lin H, Tyers M, Sunnerhagen M, Forman-Kay JD: Multivalent interactions with Fbw7 and Pin1 facilitate recognition of c-Jun by the SCFFbw7 ubiquitin ligase. Structure 2018, 26:28–39.e2. [DOI] [PubMed] [Google Scholar]
- 45.Launay H, Barré P, Puppo C, Zhang Y, Maneville S, Gontero B, Receveur-Bréchot V: Cryptic disorder out of disorder: encounter between conditionally disordered CP12 and glyceraldehyde-3-phosphate dehydrogenase. J Mol Biol 2018, 430:1218–1234. [DOI] [PubMed] [Google Scholar]
- *46.Hicks A, Escobar CA, Cross TA, Zhou H-X: Fuzzy association of an intrinsically disordered protein with acidic membranes. J ACS Au 2021, 1:66–78. [DOI] [PMC free article] [PubMed] [Google Scholar]; Application of solid state and solution NMR together with molecular dynamics simulations to characterize the fuzzy interactions between an IDP and a lipid membrane. The acidic lipids were observed to dynamically redistribute to maximize association with arginine residues in the protein.
- **47.Danilenko N, Lercher L, Kirkpatrick J, Gabel F, Codutti L, Carlomagno T: Histone chaperone exploits intrinsic disorder to switch acetylation specificity. Nat Commun 2019, 10:3435. [DOI] [PMC free article] [PubMed] [Google Scholar]; Application of methyl TROSY NMR, small angle neutron scattering, and molecular modeling to determine the structure of a high molecular weight complex of an acetyltransferase, histone chaperones, and the histone H3:H4 dimer. Within this complex, the disordered histone H3 tail makes fuzzy interactions in the active site, dynamically localizing target lysine residues in the catalytic site with minimal entropic cost.
- 48.Hsu STD, Fucini P, Cabrita LD, Launay H, Dobson CM, Christodoulou J: Structure and dynamics of a ribosome-bound nascent chain by NMR spectroscopy. Proc Natl Acad Sci USA 2007, 104:16516–16521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Martin EM, Jackson MP, Gamerdinger M, Gense K, Karamanos TK, Humes JR, Deuerling E, Ashcroft AE, Radford SE: Conformational flexibility within the nascent polypeptide–associated complex enables its interactions with structurally diverse client proteins. J Biol Chem 2018, 293:8554–8568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Salmon L, Stull F, Sayle S, Cato C, Akgül Ş, Foit L, Ahlstrom LS, Eisenmesser EZ, Al-Hashimi HM, Bardwell JCA, Horowitz S: The mechanism of HdeA unfolding and chaperone activation. J Mol Biol 2018, 430:33–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yu X-C, Hu Y, Ding J, Li H, Jin C: Structural basis and mechanism of the unfolding-induced activation of HdeA, a bacterial acid response chaperone. J Biol Chem 2019, 294:3192–3206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Collier MP, Alderson TR, de Villiers CP, Nicholls D, Gastall HY, Allison TM, Degiacomi MT, Jiang H, Mlynek G, Fürst DO, van der Ven PFM, Djinovic-Carugo K, Baldwin AJ, Watkins H, Gehmlich K, Benesch JLP: HspB1 phosphorylation regulates its intramolecular dynamics and mechanosensitive molecular chaperone interaction with filamin C. Sci Adv 2019, 5:eaav8421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Baughman HER, Pham T-HT, Adams CS, Nath A, Klevit RE: Release of a disordered domain enhances HspB1 chaperone activity toward tau. Proc Natl Acad Sci USA 2020:201915099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gupta AA, Reinartz I, Karunanithy G, Spilotros A, Jonna VR, Hofer A, Svergun DI, Baldwin AJ, Schug A, Wolf-Watz M: Formation of a secretion-competent protein complex by a dynamic wrap-around binding mechanism. J Mol Biol 2018, 430:3157–3169. [DOI] [PubMed] [Google Scholar]
- 55.Sekhar A, Velyvis A, Zoltsman G, Rosenzweig R, Bouvignies G, Kay LE: Conserved conformational selection mechanism of Hsp70 chaperone-substrate interactions. Elife 2018, 7:e32764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Weickert S, Wawrzyniuk M, John LH, Rüdiger SGD, Drescher M: The mechanism of Hsp90-induced oligomerizaton of Tau. Sci Adv 2020, 6:eaax6999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Theillet F-X, Binolfi A, Bekei B, Martorana A, Rose HM, Stuiver M, Verzini S, Lorenz D, van Rossum M, Goldfarb D, Selenko P: Structural disorder of monomeric α-synuclein persists in mammalian cells. Nature 2016, 530:45–50. [DOI] [PubMed] [Google Scholar]
- *58.Burmann BM, Gerez JA, Matečko-Burmann I, Campioni S, Kumari P, Ghosh D, Mazur A, Aspholm EE, Šulskis D, Wawrzyniuk M, Bock T, Schmidt A, Rüdiger SGD, Riek R, Hiller S: Regulation of α-synuclein by chaperones in mammalian cells. Nature 2020, 577:127–132. [DOI] [PMC free article] [PubMed] [Google Scholar]; NMR was used to characterize interactions between α-synuclein and a series of chaperones both in vitro and in mammalian cells. Disruption of the chaperone interactions in the cell cause α-synuclein to relocalize to mitochondria and aggregate.
- *59.Barnes CA, Robertson AJ, Louis JM, Anfinrud P, Bax A: Observation of β-amyloid peptide oligomerization by pressure-jump NMR spectroscopy. J Am Chem Soc 2019, 141:13762–13766. [DOI] [PMC free article] [PubMed] [Google Scholar]; Pressure-jump NMR is used to study oligomerization of the Alzheimer’s Aβ peptide. The protein is cycled between the NMR-invisible oligomer, formed at low pressure, and dissociated monomeric states, for which NMR signals can be observed, that are formed at high pressure. In this way, information about the structure and dynamics of the oligomer can be read out through the spectra of the monomer.
- **60.Kotler SA, Tugarinov V, Schmidt T, Ceccon A, Libich DS, Ghirlando R, Schwieters CD, Clore GM: Probing initial transient oligomerization events facilitating Huntingtin fibril nucleation at atomic resolution by relaxation-based NMR. Proc Natl Acad Sci USA 2019, 116:3562–3571. [DOI] [PMC free article] [PubMed] [Google Scholar]; Application of relaxation dispersion, rotating frame relaxation, and exchange-induced chemical shift measurements to probe the initial events in oligomerization of a disordered huntingtin peptide. By measuring the concentration dependence of the relaxation parameters, a detailed kinetic model of the oligomerization mechanism was derived.
- 61.Ceccon A, Tugarinov V, Ghirlando R, Clore GM: Abrogation of prenucleation, transient oligomerization of the Huntingtin exon 1 protein by human profilin I. Proc Natl Acad Sci USA 2020, 117:5844–5852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wright PE, Dyson HJ: Intrinsically disordered proteins in cellular signalling and regulation. Nat Rev Mol Cell Biol 2015, 16:18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dyson HJ, Wright PE: How do intrinsically disordered viral proteins hijack the cell? Biochemistry 2018, 57:4045–4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yang K, Stanfield RL, Martinez-Yamout MA, Dyson HJ, Wilson IA, Wright PE: Structural basis for cooperative regulation of KIX-mediated transcription pathways by the HTLV-1 HBZ activation domain. Proc Natl Acad Sci USA 2018, 115:10040–10045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Haberz P, Arai M, Martinez-Yamout MA, Dyson HJ, Wright PE: Mapping the interactions of adenoviral E1A proteins with the p160 nuclear receptor coactivator binding domain of CBP. Protein Sci 2016, 25:2256–2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Murrali MG, Felli IC, Pierattelli R: Adenoviral E1A exploits flexibility and disorder to target cellular proteins. Biomolecules 2020, 10:1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Guseva S, Milles S, Jensen MR, Salvi N, Kleman J-P, Maurin D, Ruigrok RWH, Blackledge M: Measles virus nucleo- and phosphoproteins form liquid-like phase-separated compartments that promote nucleocapsid assembly. Sci Adv 2020, 6:eaaz7095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schiavina M, Salladini E, Murrali MG, Tria G, Felli IC, Pierattelli R, Longhi S: Ensemble description of the intrinsically disordered N-terminal domain of the Nipah virus P/V protein from combined NMR and SAXS. Sci Rep 2020, 10:19574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jensen MR, Yabukarski F, Communie G, Condamine E, Mas C, Volchkova V, Tarbouriech N, Bourhis J-M, Volchkov V, Blackledge M, Jamin M: Structural description of the Nipah virus phosphoprotein and its interaction with STAT1. Biophys J 2020, 118:2470–2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Krois AS, Dyson HJ, Wright PE: Long-range regulation of p53 DNA binding by its intrinsically disordered N-terminal transactivation domain. Proc Natl Acad Sci USA 2018, 115:E11302–E11310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.He F, Borcherds W, Song T, Wei X, Das M, Chen L, Daughdrill GW, Chen J: Interaction between p53 N terminus and core domain regulates specific and nonspecific DNA binding. Proc Natl Acad Sci USA 2019, 116:8859–8868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sun X, Dyson HJ, Wright PE: A phosphorylation-dependent switch in the disordered p53 transactivation domain regulates DNA binding. Proc Natl Acad Sci USA 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]