

Abstract
Cerebral amyloid angiopathy (CAA), a prevalent cerebral small vessel disease in the elderly and a common comorbidity of Alzheimer’s disease, is characterized by cerebral vascular amyloid accumulation, cerebral infarction, microbleeds, intracerebral hemorrhages and is a prominent contributor to vascular cognitive impairment and dementia. Here, we investigate proteome changes associated with specific pathological features in several brain regions of rTg-DI rats, a preclinical model of CAA. Whereas varying degrees of microvascular amyloid and associated neuroinflammation are found in several brain regions the presence of microbleeds and occluded small vessels is largely restricted to the thalamic region of rTg-DI rats, indicating different levels of CAA and associated pathologies occur in distinct brain regions in this model. Here, using SWATHLC-MS/MS, we report specific proteomic analysis of isolated brain regions and employ pathway analysis to correlate regionally specific proteomic changes with uniquely implicated molecular pathways. Pathway analysis suggested common activation of tumor necrosis factor α (TNFα), abnormal nervous system morphology and neutrophil degranulation in all three regions. Activation of transforming growth factor-β1 (TGF-β1) was common to the hippocampus and thalamus, which share high CAA loads, while the thalamus which uniquely exhibits thrombotic events additionally displayed activation of thrombin and aggregation of blood cells. Thus, we present significant and new insight into the cerebral proteome changes found in distinct brain regions with differential CAA-related pathologies of rTg-DI rats and provide new information on potential pathogenic mechanisms associated with these regional disease processes.
Keywords: Cerebral amyloid angiopathy, amyloid beta (Aβ), animal model, proteomics, transgenic rat
Graphical Abstract
JNC-2021-0187.R1_InThisIssue
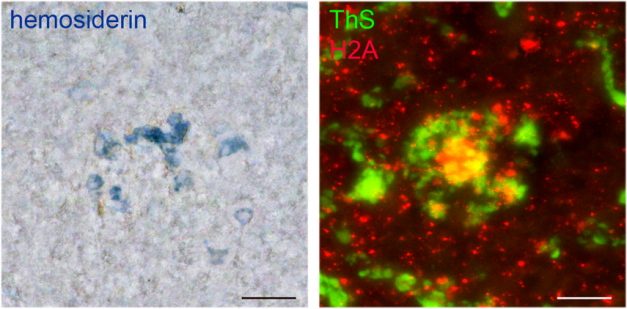
We report regional SWATHLC-MS (Sequential Window Acquisition of all Theoretical Mass Spectra-Mass spectrometry) proteomics data, and pathway analysis from the cortex, hippocampus, and thalamus of rTg-DI rats, a model of cerebral amyloid angiopathy (CAA). Each region develops significant vascular Aβ deposition and neuroinflammation, while only the thalamus develops thrombosis, vessel occlusion and cerebral microbleeds. Proteomic analysis revealed the presence of neutrophil extracellular traps (NETs) uniquely in the thalamus. In areas of thalamic microbleeds (blue, left panel) numerous vascular amyloid deposits (green, right panel) and NETs marker histone 2A (red, right panel) are found. The present findings provide new insight into the underlying mechanisms of vascular pathologies in CAA.
1. INTRODUCTION
Cerebral amyloid angiopathy (CAA) is a prevalent cerebral small vessel disease, arising from the deposition of fibrillar amyloid beta peptide (Aβ) within the meningeal and cortical arteries and arterioles, and brain capillaries (Attems et al. 2011; Rensink et al. 2003). CAA pathology is prominent in Alzheimer’s disease (AD) patients, with reported prevalence in >80% of cases (Attems et al. 2011; Rensink et al. 2003; Arvanitakis et al. 2011; Viswanathan and Greenberg 2011). Sporadic CAA, in the absence of AD, is also widespread among the elderly, occurring in >50% of individuals over the age of 80 years (Arvanitakis et al. 2011). Characterized clinically by cerebral infarction, microbleeds and intracerebral hemorrhages (ICH), CAA is a prominent contributor to vascular cognitive impairment and dementia (VCID) (Attems et al. 2011; Rensink et al. 2003; Boyle et al. 2015; Greenberg et al. 2009).
CAA typically presents as two types. CAA type-2 is restricted to larger vessels, found primarily in the meningeal and intracortical cerebral arterioles whereas in CAA type-1 the Aβ deposition is largely observed in the cortical capillaries, and is distinctly marked by fibrillar amyloid protruding into the adjacent brain parenchyma (Thal et al. 2002). This ‘dyshoric’ amyloid triggers significant perivascular neuroinflammation (Attems et al. 2011; Thal et al. 2002; Eikelenboom et al. 2008; Richard et al. 2010). It is postulated that perivascular drainage is impaired by pericapillary Aβ, disrupting its clearance and further adding to its deposition in capillary walls (Attems et al. 2011). The severity of CAA type-1 shows strong correlations with cognitive decline and dementia (Attems and Jellinger 2004; Bailey et al. 2004; Thal et al. 2003). Despite its obvious clinical relevance, the precise mechanisms linking capillary CAA to neuropathic outcomes are still largely unknown. Thus, a deeper understanding of the molecular pathways involved in CAA pathologies is necessary to develop effective therapeutic strategies.
There is increased use of proteomic analysis to identify potential biomarkers and disease pathways, and coupling liquid chromatography with tandem mass spectrometry (LC-MS/MS) has become the standard for reliable and sensitive high-throughput proteome investigation (Ludwig et al. 2018; Aebersold and Mann 2016). Sequential Window Acquisition of all Theoretical Mass Spectra-MS (SWATH-MS), an innovate data-independent acquisition (DIA) method, delivers consistent and accurate quantitative analysis of all detected protein fragments within a sample across a defined mass range (Ludwig et al. 2018; López‐Sánchez et al. 2019; Gillet et al. 2012). This ability to quantitatively analyze thousands of proteins within a sample makes SWATH-MS particularly useful in cohort proteome studies (Ludwig et al. 2018; López‐Sánchez et al. 2019).
We recently reported a novel transgenic rat model (rTg-DI) that expresses low amounts of human chimeric Dutch (E22Q)/Iowa (D23N) familial CAA mutant Aβ in brain. The Dutch and Iowa familial CAA mutations do not appreciably affect AβPP processing but rather alter the biophysical properties of the Aβ peptide strongly enhancing its assembly into amyloid fibrils (Van Nostrand et al. 2001). Although rTg-DI rats develop consistent early-onset and extensive microvascular CAA, parenchymal fibrillar amyloid plaques and larger vessel CAA type-2 are largely absent. In this model, microvascular CAA is accompanied by extensive perivascular neuroinflammation, degeneration and loss of capillary pericytes, cerebral microbleeds, microvessel occlusions, loss of axonal integrity, and white matter degeneration (Davis et al. 2018; Zhu et al. 2020a; Lee et al. 2020; Zhu et al. 2020b). Further, rTg-DI rats develop a ‘cognitive slowing’ phenotype (Popescu et al. 2020) that is similar to the deteriorating executive function observed in human CAA (Case et al. 2016; Planton et al. 2017). Therefore, rTg-DI rats provide a valid and useful pre-clinical model for the investigation of CAA type-1.
Here, we show regionally distinct pathological features in the cortex, hippocampus and thalamus of rTg-DI rats, and using SWATH-MS report specific proteomic analysis of these isolated regions. Additionally, we employ bioinformatics analysis using Ingenuity Pathway Analysis (IPA) (Qiagen, Venlo, Netherlands), to correlate regionally specific proteomic changes with uniquely implicated molecular pathways. Thus, we provide mechanistic insight into the distinct regional pathological phenotypes in rTg-DI rats and report potential pathways involved in the progression of CAA type-1.
2. Materials and Methods
2.1. Animals
All work with animals was performed in accordance with the United States Public Health Service’s Policy on Humane Care and Use of Laboratory Animals and was approved by the University of Rhode Island Institutional Animal Care and Use Committee (AN1718–008). The generation of rTg-DI transgenic rats was previously described (Davis et al. 2018). This line expresses low levels of human Swedish/Dutch/Iowa mutant AβPP under the control of the neuronal-specific Thy1.2 promoter and produces chimeric Dutch/Iowa CAA mutant Aβ peptides in the brain. The accumulation of microvascular fibrillar amyloid emerges at ≈3 M and progresses with age. Three male and three female heterozygous rTg-DI rats (n = 6) and non-transgenic, wild-type (WT) rats (n = 6) at 12 M were used in the present study based on the consistency of the pathology of this model and our previous experience in similar studies (Davis et al. 2018; Zhu et al. 2020a; Zhu et al. 2020b). All rats were housed in a controlled room (22 ± 2°C and 40–60% humidity) on a standard 12 h light cycle. Rat chow and water were available ad libitum. Animals were assigned to each experimental group based on genotype and sex, and no randomization/blinding was performed. The study was not pre-registered and was exploratory. No animals were excluded in this study and no sample size calculations were performed.
2.2. Brain tissue collection and preparation
Rats were anesthetized by intraperitoneal injection with ketamine(75mg/kg)/xylazine(10mg/kg) and then transcardially perfused with phosphate-buffered saline (PBS, pH 7.4) for 15 min at a rate of 20 ml/min. The rat brain was surgically removed and bisected in the mid-sagittal plane. One hemisphere was fixed in 4% paraformaldehyde for immunohistochemical analysis and the other hemisphere was snap-frozen in optimal cutting temperature medium (OCT 4585, Fisher Healthcare) directly.
2.3. Measurement of CAA load
Paraformaldehdye-fixed brain sections were cut in the sagittal plane at 10 μm thickness using a microtome (Leica, Buffalo Grove, IL), placed on slides and then rehydrated by immersing in xylene with decreasing concentrations of ethanol. Antigen retrieval was conducted via 5 min incubation with proteinase K (IB05406, IBI Scientific, Peosta, IA) (0.2 mg/ml) at 22° C. Tissue sections were then blocked in Superblock blocking buffer (cat. #37518, ThermoFisher, Franklin, MA) containing 0.3% Triton X-100 at room temperature for 30 min and incubated with rabbit polyclonal antibody to collagen IV to detect cerebral blood vessels (1:250, SD2365885, Invitrogen, Waltham, MA). Primary antibody was detected with Alexa Fluorescent 594-conjugated secondary antibody (1:1000) (RRID: AB_141359, Molecular Probes, Leiden, Netherlands). Deposited fibrillar amyloid was detected with thioflavin S (123H0598, Sigma-Aldrich, St. Louis, MO). Stacked images were captured on a KEYENCE BZ-X710 fluorescence microscope and analyzed with BZ-X Analyzer software (Version 1.3.1.1, 2013, Keyence, Itasca, IL). The percent area of capillaries covered with amyloid was quantified using a series of non-overlapping images covering the cortex, hippocampus and thalamus were analyzed by Image J software. For each field, the sum area of vascular thioflavin-S was divided by that for the total collagen IV immunoreactive capillary area x 100 to yield the percent area of microvascular amyloid.
2.4. Immunohistochemical analyses
Paraformaldehdye-fixed brain sections were cut in the sagittal plane at 10 μm thickness using a microtome (Leica, Buffalo Grove, IL), placed on slides and then rehydrated by immersing in xylene with decreasing concentrations of ethanol. Antigen retrieval was conducted via 5 min incubation with proteinase K (IB05406, IBI Scientific, Peosta, IA) (0.2 mg/ml) at 22° C. Tissue sections were then blocked in Superblock blocking buffer (cat. #37518, ThermoFisher, Franklin, MA) containing 0.3% Triton X-100 at room temperature for 30 min and incubated with individual primary antibodies at the following dilutions overnight: rabbit monoclonal antibody to apolipoprotein E (APOE, 1:250, RRID: AB_2832971, Abcam, Cambridge, MA), rabbit polyclonal antibody to Annexin A3 (Anxa3, 1:250, PA5082483, Invitrogen, Waltham, MA), rabbit polyclonal antibody to elastase (1:250, RRID# AB_2746314, Invitrogen), or rabbit polyclonal antibody to histone 2A (H2A, 1:250, RRID# AB_2735313, Invitrogen). Primary antibodies were detected with Alexa Fluorescent 594 (1:1000, RRID: AB_141359, Molecular Probes, Leiden, Netherlands). Deposited fibrillar amyloid was detected with thioflavin S (123H0598, Sigma-Aldrich, St. Louis, MO). Immunohistological stacked images were captured on a KEYENCE BZ-X710 fluorescence microscope and analyzed with BZ-X Analyzer software (Version 1.3.1.1, 2013, Keyence, Itasca, IL).
2.5. Laser capture microdissection and protein digest
Twenty sagittal sections (25 μM thick) were prepared from hemispheres of fresh frozen 12 M rTg-DI and WT rat brain on laser microdissection framed membrane slides (Leica, Buffalo Grove, IL), 2 sections per slide, and the cortical, hippocampal and thalamic regions from each section were collected via laser capture microdissection (LCM). Collected tissue regions were lysed in radioimmunoprecipitation assay (RIPA) buffer via sonication (12 × 1 sec bursts) on ice, followed by 1 h incubation on ice, and samples were normalized to equal total protein concentrations. Lysed tissue samples (~100 μg protein in 100 μL RIPA buffer) were spiked with 2 μg bovine serum albumin. Protein was denatured by adding 25 μL DTT (100 mM) and incubation with shaking (300 rpm) at 95° C for 15 min in a plate mixer (Mixer HC, USA Scientific, Ocala, FL). Samples were then alkylated with 25 μL iodoacetamide (200 mM) and incubated for 30 min in the dark at room temperature. Protein was then precipitated and concentrated by the addition of cold water, methanol and chloroform (1:2:1) and subsequent centrifugation (15,300 × g, 5 min at 4° C). The protein pellet was washed once with ice cold methanol and resuspended in 125 μL sodium deoxycholate (3 % w/v in 50 mM ammonium bicarbonate). 6.25 μL TPCK-treated trypsin (1 μg/μL in water) (Sciex, Framingham, MA) was added and samples were transferred to digestion PCT microtubes (Pressure Biosciences Inc., Easton, MA). Protein samples were then digested in a barocycler (Pressure Biosciences Inc, Easton, MA) run at 50 °C, for 75 cycles with 60 sec pressure cycles (50-sec high pressure, 10-sec ambient pressure, 25 kpsi). Another 6.25 μL of trypsin was then added to each sample, and further digestion occurred over an additional 60 cycles in the barocycler. To precipitate the DOC, formic acid (5 % v/v, in 50 % v/v acetonitrile in water) was added to each sample for final a concentration of 0.5 % v/v. To remove precipitated DOC, samples were centrifuged (15,300 × g, 5 min, at RT) and supernatant was collected.
2.6. Analysis by LC-QTOF/MS
Proteomic experiments were all conducted on a SCIEX 5600 TripleTOF mass spectrometer, in positive ion mode using a DuoSpray™ ion source (AB Sciex, Concord, Canada), coupled to an Acquity UPLC HClass system (Waters Corp., Milford, MA, USA) for chromatographic separation. Peptide separation was achieved using an Acquity UPLC Peptide BEH C18 (2.1 X 150 mm, 300 Å, 1.7 μm) column preceded by an Acquity VanGuard pre-column (2.1 X 5 mm, 300 Å, 1.7 μm), employing a 180 min gradient method at flow rate of 10 μL/min. The gradient utilized mobile phase A (water containing 0.1 % w/v formic acid) and mobile phase B (acetonitrile containing 0.1 % w/v formic acid) with a solvent composition scheme as follows: 98% A from 0 to 5 min, 98% to 70% A from 5 to 155 min, 70% to 50% A from 155 to 160 min, 50% to 5% A from 160 to 170 min, 5% to 98% A held from 170 to 175 min. Gradient was returned to initial conditions from 170 to 175 min to equilibrate the column between samples. Samples were maintained at 10 °C in the autosampler, and column temperature was held at 50 °C. Trypsin-digested β-galactosidase (cat#4465938, AB Sciex, Concord, Canada) was injected every 4 samples to monitor TOF mass calibration.
Data was acquired using Analyst TF 1.7.1 software (2019, AB Sciex, Concord, Canada). Data-dependent acquisition (DDA) was performed to generate peptide ion libraries, followed by SWATH-MS analysis for protein quantification. For DDA experiments, positive ionization mode via the DuoSpray™ ion source was employed, with ion source gas 1 (GS1), ion source gas 2 (GS2), and curtain gas (CUR) set to 55, 60, and 25 psi, respectively. The source temperature (TEM) was set to 450 °C and ion spray voltage floating (ISVF) was 5500 V. Declustering potential (DP), collision energy (CE) and collision energy spread (CES) were 100, 10 and 15 respectively. Ions within a mass range of m/z 300–1250 and exceeding 25 cps were selected for MS/MS analysis during survey scans. Former target ions were excluded for 8 sec and the mass tolerance for TOF-MS was 50 mDa with a 100 milliseconds accumulation time. During the product scan, data were acquired from 100–1250 m/z with an accumulation time of 75 milliseconds and total cycle time of 3.5 sec, and product ion analysis occurred under dynamic accumulation and rolling collision energy. SWATH-MS analysis was performed with the same parameters except ion selection was based on a TOF mass range of 300–1500 m/z and SWATH data was acquired using 70 SWATH windows of 10 Da each over a mass range of 400–1100 m/z.
2.7. Data processing
DDA measurements were used to generate the spectral library, via spectral library generation in Spectronaut (release version 13.919110643655, 2019; Biognosys, Schlieren, Switzerland), and referencing the Rat Uniprot FASTA database (searched December 2019). Experiments included 6 biological replicates (3 males and 3 females of both rTg-DI and WT rats), and 1 technical replicate per sample. All Spectronaut settings were kept at factory default. There were 21465 database entries searched. Protease specificity was set to Trypsin-P, fixed modification considered was carbamidomethyl, and variable modifications considered were acetyl (protein n-term) and oxidation (M). MS1 and MS2 mass tolerances were set to dynamic with the optimal range determined by Spectronaut. Individual spectra were selected via threshold scoring based on relative ion intensity relative to the most intense (base) peak. Relative ion intensities < 5% were filtered. False discovery rate (FDR) was set to 0.01 and estimated by Pulsar at the peptide, protein, and peptide-spectrum match (PSM) levels. Proteins were then identified and quantified from the DIA data using Spectronaut and the spectral library. Again, all Spectronaut settings were factory default except “used Biognosys’ iRT kit” and “PTM localization” were deselected. Precursor ion detection was performed with extracted ion current chromatogram (XIC) RT, with mass tolerance set to dynamic, and optimally determined by Spectronaut. Precursor identification was based on a q-value (FDR) cutoff of 0.01, and only precursors satisfying this cutoff were accepted. XIC RT window was set to dynamic, with optimal tolerances determined by Spectronaut, and the correction factor set to system default (1, no correction). A maximum of three and a minimum of one peak were considered for identifying peptides, and were selected based on intensity, with selection method set to “maximum intensity”. All mass tolerances were set to dynamic and optimally determined by Spectronaut. False discovery rate (FDR) was set to 0.01 and estimated by Pulsar at the peptide, protein, and peptide-spectrum match (PSM) levels. FDR was estimated using decoy peptides generated in-silico, as the fraction of decoys in the total identified peptides. Decoy generation method was set to ‘mutated’. Protein intensities were normalized by Spectronaut with the normalization strategy set to “Local normalization” (factory default).
2.8. Data Analysis
Raw protein intensities from Spectronaut were then converted to molar concentrations (pmol/total protein) using the total protein approach (TPA) (Wiśniewski and Rakus 2014; Wiśniewski et al. 2012). Differential expression of individual proteins was determined by head to head comparison of average WT and rTg-DI total protein concentrations within a specific brain region. Significance of differentially expressed proteins was therefore determined via student’s t test (p < 0.5) in Excel (version 2102, 2020, Microsoft, Redmond, WA). For proteins quantities of zero in individual samples, we imputed a baseline protein concentration (0.013 pmol/mg), which was determined by the lowest mean protein concentration for a protein quantified in all samples. Effect threshold cutoffs were used as selection criteria for differentially expressed proteins, and only proteins with ≥ 50% increase or ≥ 34% decrease compared to WT were considered. The data was not subjected to a normality test. Throughout the study, no exclusion criteria were predetermined, no tests for outliers were performed, and no data points were excluded.
2.9. Bioinformatics Analysis
We conducted pathway analysis of differentially expressed proteins using Ingenuity Pathway Analysis (release version 62089861, 2021; Qiagen, Hilden Germany). Total protein concentrations of the characterized proteins in each brain region of the rTg-DI rats were converted to expression ratios relative to the WT concentration, and these values, along with corresponding p values for significance based on the WT comparison within that same region, were uploaded to the IPA database. Expression ratios were then converted to fold change within IPA. Analysis was restricted to pathways and proteins occurring in endothelial cells, epithelial cells, immune cells, and the nervous system. Proteins were filtered base on an expression fold change threshold of ± 1.5 (50% increase, 34% decrease) and a p value of ≤ 0.05. IPA calculates the statistical significance of overlap between differentially expressed proteins in the uploaded dataset and canonical pathways, disease functions, and upstream regulators using the Fisher’s Exact Test, and provides an ‘overlap p value’ for these pathways (Krämer et al. 2014a). Predictions of activation or inhibition of annotations (pathways, regulators, and disease functions) are determined by the comparison of directional expression changes in the dataset with expected expression patterns based on the literature for that particular annotation, and assigned z scores for statistical significance. A thorough description of the IPA statistical methods has been published (Krämer et al. 2014a).
2.10. Data Availability
Raw mass spectrometry data can be found in the MassIVE repository (massive.ucsd.edu/ProteoSAFe/static/massive.jsp), project ID#: MSV000086432, and password: rTgDIrgprt1219.
3. RESULTS
3.1. Distinct brain regional pathologies associated with microvascular amyloid accumulation in rTg-DI rats
The rTg-DI rat model was previously shown to develop early-onset and progressive accumulation of cerebral microvascular amyloid in different brain regions ( Davis et al. 2018; Zhu et al. 2020a). Here we show a similar pattern of CAA severity in 12 M rTg-DI rats with moderate levels in the cortex and higher levels in the hippocampus and thalamus (Fig. 1). Previously, we showed that ≈90% of accumulated cerebral microvascular amyloid in rTg-DI rats is composed of the shorter Aβ40 isoform (Davis et al. 2018).
Fig. 1. Regional accumulation of cerebral microvascular amyloid in 12 M rTg-DI rat brain.
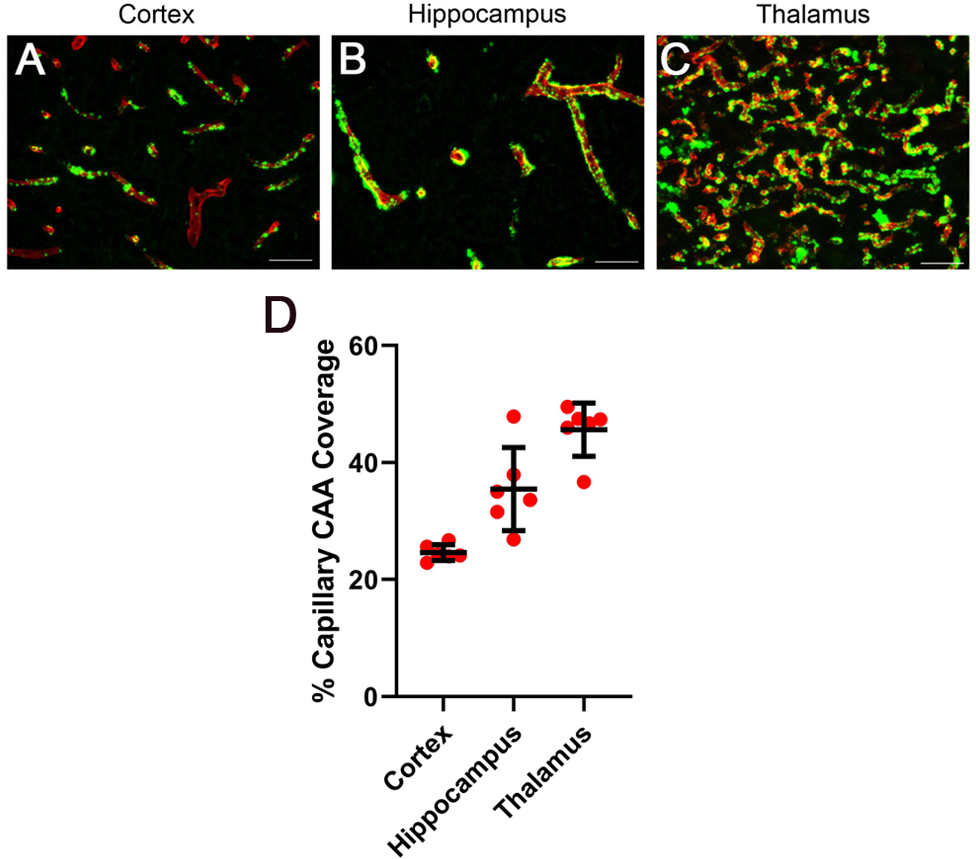
Brain sections from the present cohort of 12 M rTg-DI rats were immunolabeled with rabbit polyclonal antibody to collagen IV to specifically detect cerebral microvessels (red) and stained with thioflavin S to identify fibrillar amyloid (green). Representative images show the cerebral microvascular fibrillar amyloid deposits in the cortex (A), hippocampus (B), and thalamus (C). Scale bars = 50 μm. (D) Quantitation of cerebral microvascular amyloid load in different brain regions of 12 M rTg-DI rats. Data points show the results from each rat and the group mean ± SD of n=6 rTg-DI rats.
At 12 M of age, the presence of moderate to severe microvascular amyloid in rTg-DI rats promotes a well-documented neuroinflammatory response including markedly increased numbers of astrocytes in the cortex, hippocampus and thalamus (Zhu et al. 2020a). Supplemental Information (SI) Fig. S1 confirms this response in the present cohort of rTg-DI rats showing that the three brain regions have significantly increased numbers of astrocytes compared to similarly aged wild-type (WT) rats (p < 0.001) with the most dramatic increase in the thalamus. Likewise, the numbers of microglia were significantly increased (p < 0.001) four-fold in the cortex and hippocampus with even higher increases again in the thalamic region (SI Fig. S2).
Although moderate to severe levels of microvascular amyloid and elevated levels of neuroinflammatory cells are characteristic to all three brain regions the presence of thrombotic events is largely restricted to the thalamic region of rTg-DI rats (Davis et al. 2018; Zhu et al. 2020a; Lee et al. 2020; Zhu et al. 2020b). For example, at 12 M microbleeds are prominently observed bilaterally in the thalamic region based on magnetic resonance imaging and histological staining for hemosiderin (Davis et al. 2018; Zhu et al. 2020a; Zhu et al. 2020b; Lee et al. 2020). Consistent with these previous findings, thalamic hemosiderin deposits in the present cohort are shown in SI Fig. S3A,C. The thalamic region of rTg-DI rats also exclusively exhibit numerous calcified, occluded microvessels and capillaries (Davis et al. 2018; Zhu et al. 2020a; Lee et al. 2020). SI Fig. S3B,D show that these thalamic calcified, occluded vessels are similarly observed in the present cohort of rTg-DI rats.
Thus, the representative images and quantitative measures presented in Fig. 1 and SI Figs. S1–S3 indicate that at 12 M of age moderate to severe levels of CAA and associated pathologies are observed in all three brain regions of rTg-DI rats, with the exception of the thalamic region which distinctly presents with numerous thrombotic vessel events. Based on these findings, we focused our proteomic profiling on these three brain regions. Accordingly, the cortex, hippocampus and thalamus were collected from 12 M rTg-DI and WT rats using laser capture microdissection (LCM). SI Fig. S4 shows a representative image of the three distinct brain regions that were microdissected and collected from each rat.
3.2. Regional protein expression enhancements in rTg-DI rats
We employed a DIA LC-MS/MS proteomics approach to investigate regional protein expression changes in LCM captured tissue samples of rTg-DI and WT rat cortex, hippocampus and thalamus as described in “Materials and Methods”. Proteins were identified and quantified from DIA data using the software Spectronaut (Biognosys, Schlieren, Switzerland). Raw spectral protein intensities of the Spectronaut output were converted to molar concentrations according to the “total protein approach” (TPA) (Wiśniewski and Mann 2016) using the equation:
where c(i) is the concentration of protein i (in pmol/mg of total protein) and MW is its molar mass (in mg/mol). Regional protein concentrations of the rTg-DI rats were then compared with those from corresponding regions in WT rats for differential expression.
Of the 2814 total characterized proteins across all three regions, 487 proteins were found to have significant increases (p < 0.05) in expression as compared to the corresponding regions in the similarly aged WT rats. We observed the highest number of elevated proteins in the thalamus, with 273 significantly increased proteins, which was not surprising due to the extensive and distinct CAA-related pathology in this region. In comparison, the number of observed increased proteins in the hippocampus and cortex were 233 and 151, respectively.
Most likely due to the small sample size used in this study, we found multiple testing corrected false discovery rates (FDR) to be too blunt and restrictive, a difficulty commonly reported in small n proteomic studies (Hondius et al. 2018; Pascovici et al. 2016), and only a fraction of the differentially expressed proteins remained significant. Therefore, we decided to consider the uncorrected p values for the differentially expressed proteins, and manage the FDR by applying effect thresholds as has been suggested elsewhere (Pascovici et al. 2016). Thus, we compared all proteins with an observed increase of ≥50%, of which there were 264 proteins identified across the three brain regions. In total, the thalamus had 189 proteins with ≥50% increase, while the hippocampus and the cortex had 107 and 60 proteins, respectively (Fig. 2A). Lists of these increased proteins for each brain region can be found in SI Tables S1–S3. The thalamus, hippocampus and cortex had 129, 51, and 20 uniquely elevated proteins, respectively (Fig. 2A). We found 30 proteins were common to all three brain regions, which shared a moderate-high CAA load and neuroinflammation (SI Table S4). On the other hand, an additional 23 proteins were shared between the thalamus and hippocampus (Fig. 2A), which share a higher CAA burden. The cortex shared only an additional 7 elevated proteins with the thalamus, and an additional 3 with the hippocampus (Fig. 2A). Thus, in order of greatest number of changes the regions are ranked as follows: thalamus > hippocampus > cortex, with the largest commonality shared between the thalamus and the hippocampus, two regions that share a high CAA load (Fig. 1).
Figure 2. Significantly increased and decreased proteins in rTg-DI cortex, hippocampus and thalamus.
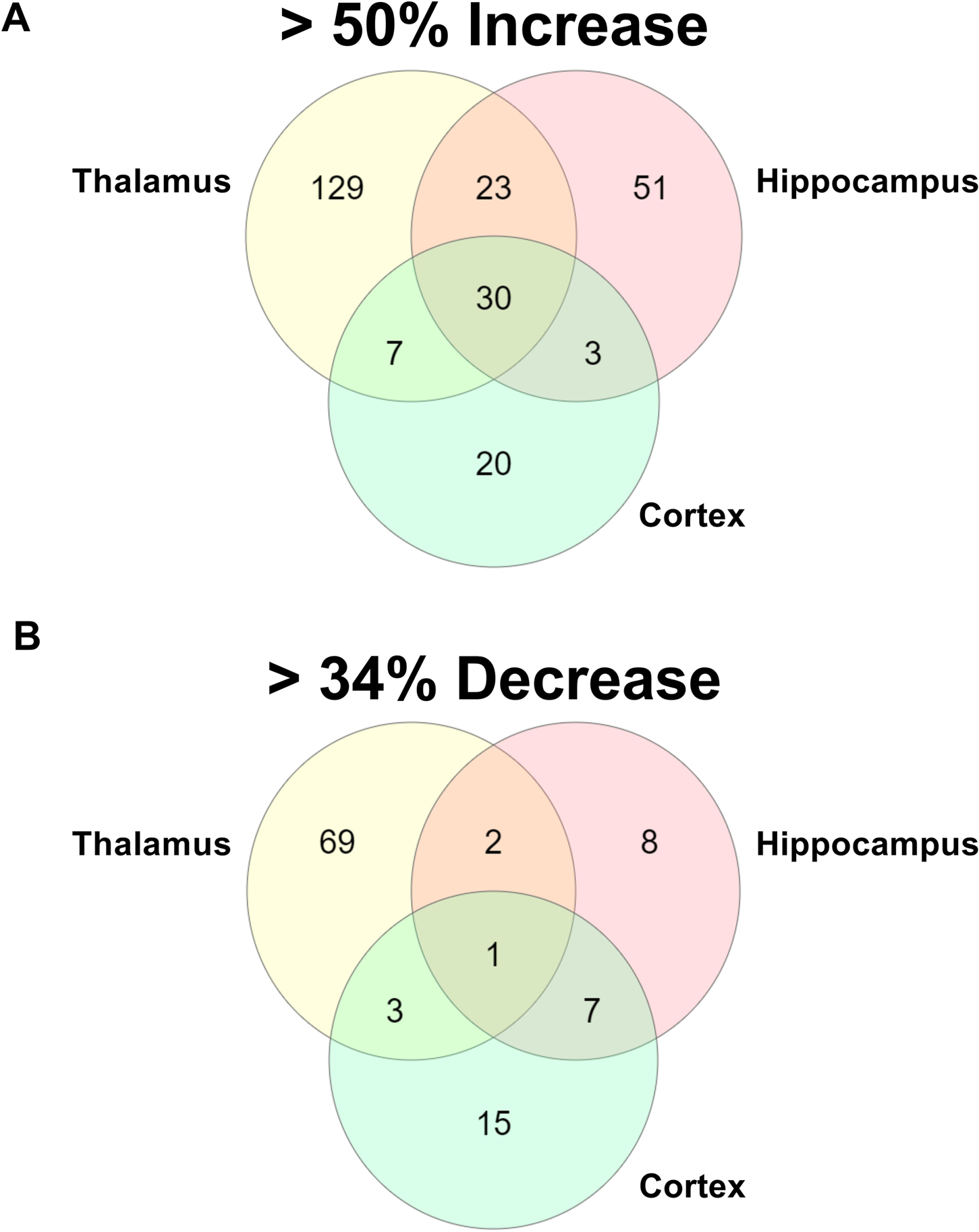
A. Venn diagram comparing significantly (p < 0.05) enhanced proteins by ≥ 50% of the WT concentration in cortex, hippocampus and thalamus of rTg-DI rats (n = 6). B. Venn diagram comparing significantly (p < 0.05) decreased proteins by ≥ 34% of the WT concentration in cortex, hippocampus and thalamus of rTg-DI rats (n = 6).
Of the 30 common proteins glial fibrillary acidic protein (GFAP), a widely used astrocyte marker (Eng et al. 1971; Eng et al. 2000; Guttenplan and Liddelow 2019), was the most abundant in all three regions (SI Table S4), consistent with the elevated numbers of astrocytes in rTg-DI rats (SI Fig. S1). Additionally, apolipoprotein E (ApoE), another predominantly astrocytic protein with reported roles in AD and suggested as a marker for CAA (Rannikmäe et al. 2014; Safieh et al. 2019; Hondius et al. 2018) was the second most abundant protein in each region, lending further confidence to the fidelity of the rTg-DI model and our proteomic analysis. While each of the 30 common proteins displayed an increase of >50%, the increases range from ~56–4500%, with the largest increases seen in GFAP and ApoE as expected, along with proteins S100 calcium binding protein A4 (S100A4), keratin 8 (Krt8), phosphatidate cytidylyltransferase 2 (Cds2), and annexin A3 (Anxa3).
Due to the high CAA load and neuroinflammation, and the degree of commonality in enhanced protein expression observed in the thalamus and hippocampus, elevated proteins common to these two specific regions are also of interest. Of the 23 elevated proteins shared in these regions, greatest increases were observed in heat shock protein beta-1 (Hspb1, also known as Hsp27), S100 calcium binding protein A6 (S100a6), histone H1.3 (Hist1h1d), keratin 75 (Krt75), and megalencephalic leukoencephalopathy with subcortical cysts 1 (Mlc1). Interestingly, it was reported that Hspb1 upregulation is not associated with CAA (Wilhelmus et al. 2009). Enhancement of S100a6 and Hist1h1d has been reported in AD (Manavalan et al. 2013; Boom et al. 2004), but not yet associated with CAA, and enhancement of Krt75 has not yet been linked to either AD or CAA.
The presence of thrombotic events and significant microbleeds observed in the thalamus led us to further examine the proteins elevated only in this region. Unexpectedly, the three most abundant proteins were Histone 4 (H4, Hist1h4b), Histone H2B (H2B), and H3 histone family member 3C (H3f3c), with increases of ~82, 70, and 55 percent respectively (SI Table S3). Additionally, two H1 Histone family proteins, Hist1h1c and H1f0, were among the most strongly elevated with ~150 and 120 percent increases, respectively (SI Table S3). All of these core histone proteins were previously reported as characteristic markers of neutrophil extracellular traps (NETs), an innate immune response reported to cause endothelial/epithelial disruption, loss of blood vessel integrity, bleeding, promotion of thrombosis and significant inflammation related tissue damage (Pietronigro et al. 2017; Hsieh et al.; Lim et al. 2018; Martinod and Wagner 2014; Kolaczkowska et al. 2015; Zhou et al. 2020).
3.3. Regional protein expression decreases in rTg-DI rats
We next investigated proteins with significantly reduced expression in each of the three brain regions in the rTg-DI rats compared to WT rats using an effect threshold as above. We observed a total of 105 proteins decreased in expression by at least 34% across the three brain regions with 75, 18 and 26 of those occurring in the thalamus, hippocampus and cortex, respectively (Fig. 2B). Lists of each of these proteins can be found in SI Tables S5–S7. In general, the three regions displayed much less commonality in reduced proteins, and shared just 1 reduced protein, phosphoglucomutase 2 like 1 (PGM2L1), with only an additional 2 shared between the thalamus and hippocampus, 3 between the thalamus and cortex, and 7 between the hippocampus and cortex (Fig. 2B). Of particular note, neurofilament medium length (Nefm) was commonly reduced in the cortex and thalamus. Dysregulation of Nefm has been used as a marker for neurodegeneration (Liu et al. 2011). Additionally, synaptotagmin-2 (Syt2), displayed reduced expression in the cortex and hippocampus, and is also a hallmark of neurodegenerative diseases (Bereczki et al. 2018).
We then validated our LC-MS/MS SWATH results for a subset of the proteins mentioned above, including GFAP, ApoE, Anxa3, Aqp4, and Mbp using quantitative immunoblotting. Quantification and representative images of these immunoblots are presented in SI Fig. S5 and these results are consistent with the changes observed in the LC-MS/MS SWATH data.
3.4. IPA reveals distinct upstream regulators and casual networks
We next conducted bioinformatics analysis of the differentially expressed proteins using Ingenuity Pathway Analysis (IPA) (Qiagen, Hilden, Germany), to identify key upstream regulators, disease functions and other pathways implicated by these proteomic changes. Proteins with ≥ 50% increase or ≥ 34% decrease compared to WT levels were included in this analysis. IPA predicts significantly activated (z score >2) or inhibited (z score < −2) upstream regulators based on the differential expression of downstream target molecules within the dataset (Krämer et al. 2014b). In this way, IPA can predict activation states of proteins not detected by LC-MS/MS based on the expression of multiple downstream molecular targets. The only commonly activated upstream regulator among the three brain regions was TNFα, and a heat map depicting implicated downstream target molecules identified in our proteomic analysis from each region is shown in Fig. 3A, including color shading indicating increased (red) and decreased (green) expression. Of particular interest to our study, increased release of TNFα within the cerebral microvessels of AD patients has been reported (Grammas and Ovase 2001). Thus, TNFα as a commonly activated upstream regulator among the three regions is not surprising, as there is a moderate/high level of fibrillar Aβ deposition in the microvasculature in each region. This is also consistent with our recent findings that TNFα mRNA levels are significantly elevated in 12 M rTg-DI rats (Zhu et al. 2020a). Commonly elevated target molecules among the three regions included cathepsin S, clusterin (Clu), hexosaminidase beta subunit (HexB), glutathione S-transferase A1 (Gsta1) and Annexin A11 (Anxa11). Of particular note, enhanced expression of Clu has been previously reported in human CAA patients (Manousopoulou et al. 2017).
Figure 3. IPA identified upstream regulators and causal networks.
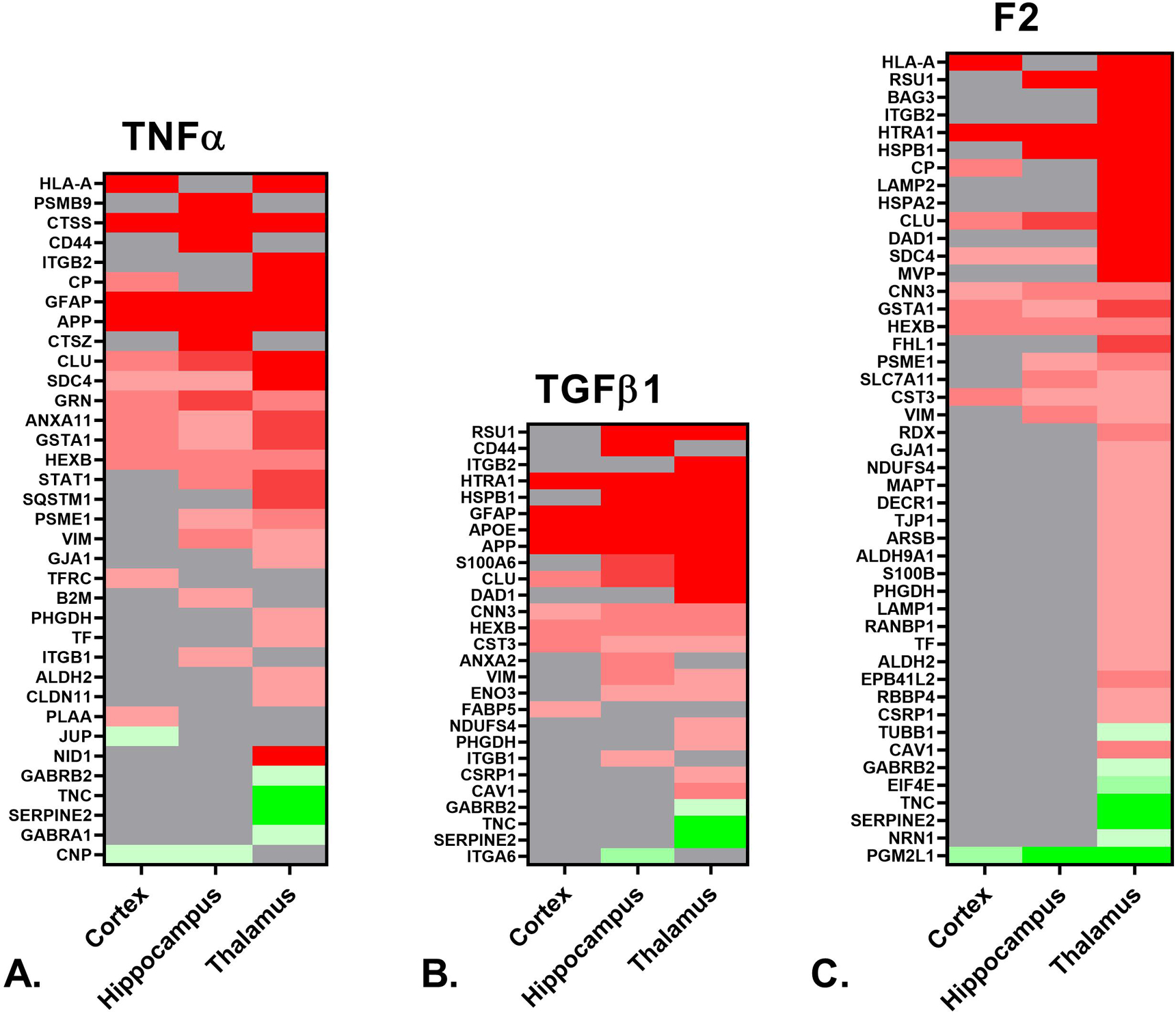
A. Heat map depicting the differentially expressed proteins (≥ 50% increase or ≥ 34% decrease, p < 0.05) in the cortex, hippocampus and thalamus of rTg-DI rats associated with upstream regulator TNFα. B. Heat map depicting differentially expressed proteins in the cortex, hippocampus and thalamus of rTg-DI rats associated with upstream regulator TGFβ1. C. Heat map depicting the differentially expressed proteins in the cortex, hippocampus, and thalamus of rTg-DI rats associated with the F2/Thrombin causal network. Red indicates increased, green decreased and grey not differentially expressed proteins, and color intensity correlates with degree of change.
Interestingly, the IPA analysis indicated activation for the anti-inflammatory marker TGF-β1 both in the hippocampus and thalamus, with z scores of 2.227 and 2.292, respectively. A heat map indicating relative expression of the downstream target molecules identified in our proteomic analysis for all three regions is depicted in Fig. 3B. Many of the mapped strongly elevated proteins common to the hippocampus and thalamus are specifically expressed in astrocytes including: Hspb1, S100a6, Clu, Htra1, and GFAP (Nafar et al. 2016; Dukay et al. 2021; Yamashita et al. 1999; Morgan et al. 1995; Launay et al. 2008), while other markers such as Vim and ApoE can be expressed in both astrocytes and microglia (O’Leary et al. 2020; Graeber et al. 1988). Activation of TGF-β1 is consistent with our recent findings that its mRNA expression is significantly elevated in 12 M rTg-DI rats (Zhu et al. 2020a).
IPA also conducts “causal network analysis”, where differential expression of downstream molecular targets are connected via direct and indirect connections with upstream regulators to predict activation states of these causal networks (Krämer et al. 2014b). Of interest to the present study is the thrombin/F2 causal network, indicated as activated (z-score = 3.539) in the thalamus but not the hippocampus or cortex. A heat map depicting the relative expression of the downstream targets in the F2 causal network for each region is shown in Fig. 3C, displaying numerous uniquely differentially expressed proteins in the thalamus. Notable was the down regulation of the thrombin inhibitor Serpine2, uniquely down regulated in the thalamus, along with the distinctive upregulation of lysosomal associated membrane protein 1 and 2 (LAMP1, LAMP2). Increased surface expression of LAMP1 and 2 is known to occur in platelets activated by thrombin (Nofer et al. 2004), thus their enhanced expression in the rTg-DI thalamus, which exhibits numerous microbleeds and thrombotic events is consistent with this distinct regional phenotype.
3.5. IPA reveals downstream effects analysis
Downstream Effects Analysis (DEA) was also employed to identify specific processes and functions causally affected by differentially expressed proteins within the dataset. There are similar and distinguishing functions, along with varying degrees of activation/inactivation, among the three investigated brain regions. Three functions of interest to the rTg-DI CAA model are depicted in Fig. 4, including observed proteins associated with each function, and color shading indicating increased (red) or decreased (green) expression as compared to WT animals. In some cases, reference information for causal predictions is unavailable, and activation z scores for a given function are indeterminant. Here, IPA provides an “overlap p-value” by comparing the number of differentially expressed proteins within a dataset that overlap with molecules known to be associated with a specific function (Krämer et al. 2014b). For example, activation z scores for abnormal morphology of the nervous system (Fig. 4A) are indeterminant. However, DEA indicates this function is significantly altered in all three regions, with overlap p-values of 9.78 × 10−5, 7.67 × 10−7 and 2.41 × 10−9, and 16, 24 and 43 differentially expressed target molecules in the cortex, hippocampus and thalamus, respectively. Fig. 5A depicts a heat map of relative regional differential expression (compared to WT) of these target proteins, and clearly indicates the increasing severity from the cortex to the thalamus. Additionally, neurofilament proteins are significantly reduced in both the cortex and thalamus (Fig. 5A). Commonly indicated abnormal morphology of the nervous system is consistent with our previous findings of swollen and fragmented axons and a cellular redistribution of neurofilament in the cortex, hippocampus and thalamus of rTg-DI rats (Zhu et al. 2020a).
Figure 4. IPA downstream effects analysis networks.
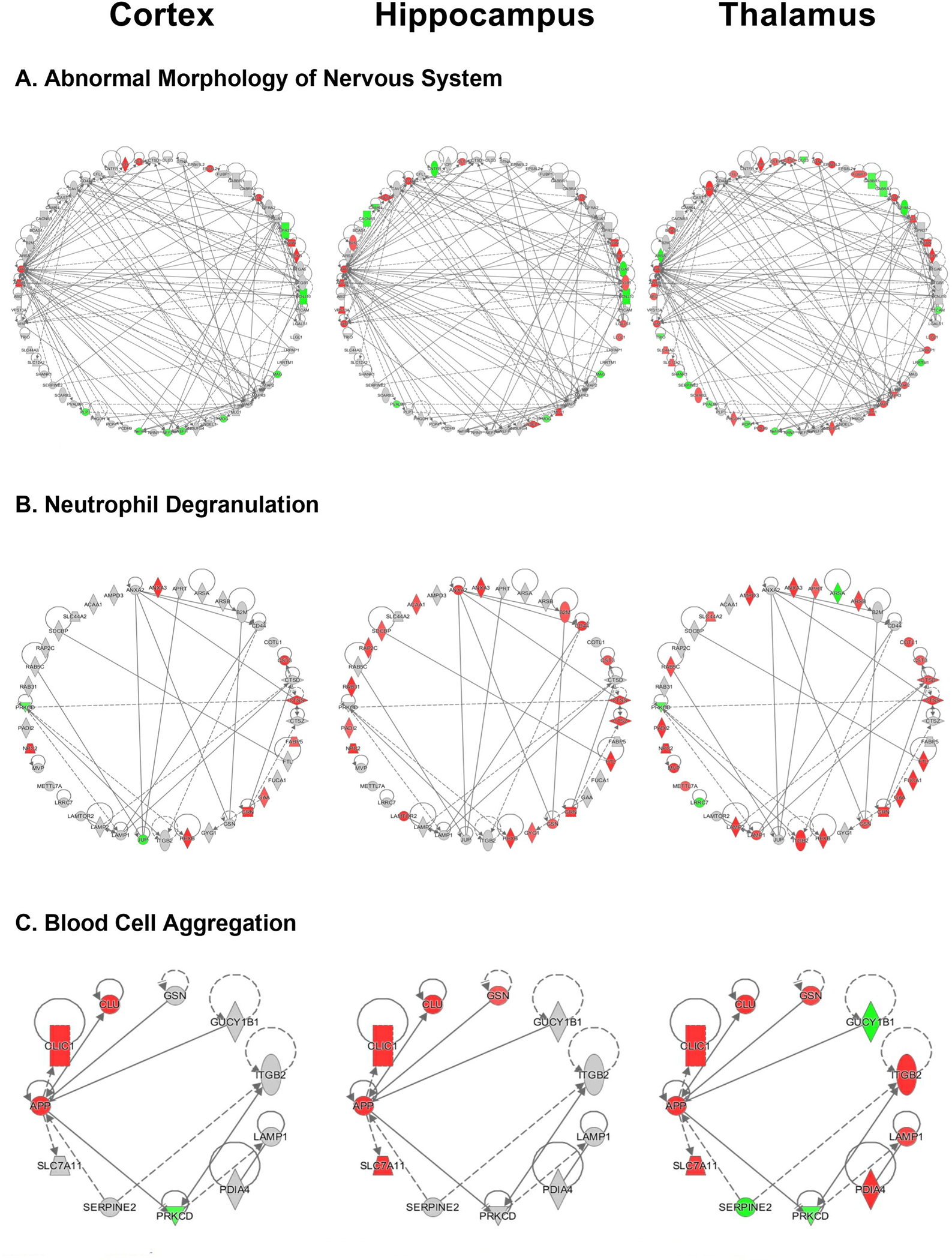
A. Abnormal morphology of nervous system network for the cortex, hippocampus and thalamus of rTg-DI rats including all associated differentially expressed proteins (≥ 50% increase or ≥ 34% decrease, p < 0.05). B. Neutrophil degranulation network depicted for the cortex, hippocampus and thalamus of rTg-DI rats. C. Aggregation of blood cells network created in IPA depicted for the cortex, hippocampus, and thalamus of rTg-DI rats. Red shading indicates increased, green decreased and grey not differentially expressed proteins in that region.
Figure 5. Downstream effects analysis networks heat maps.
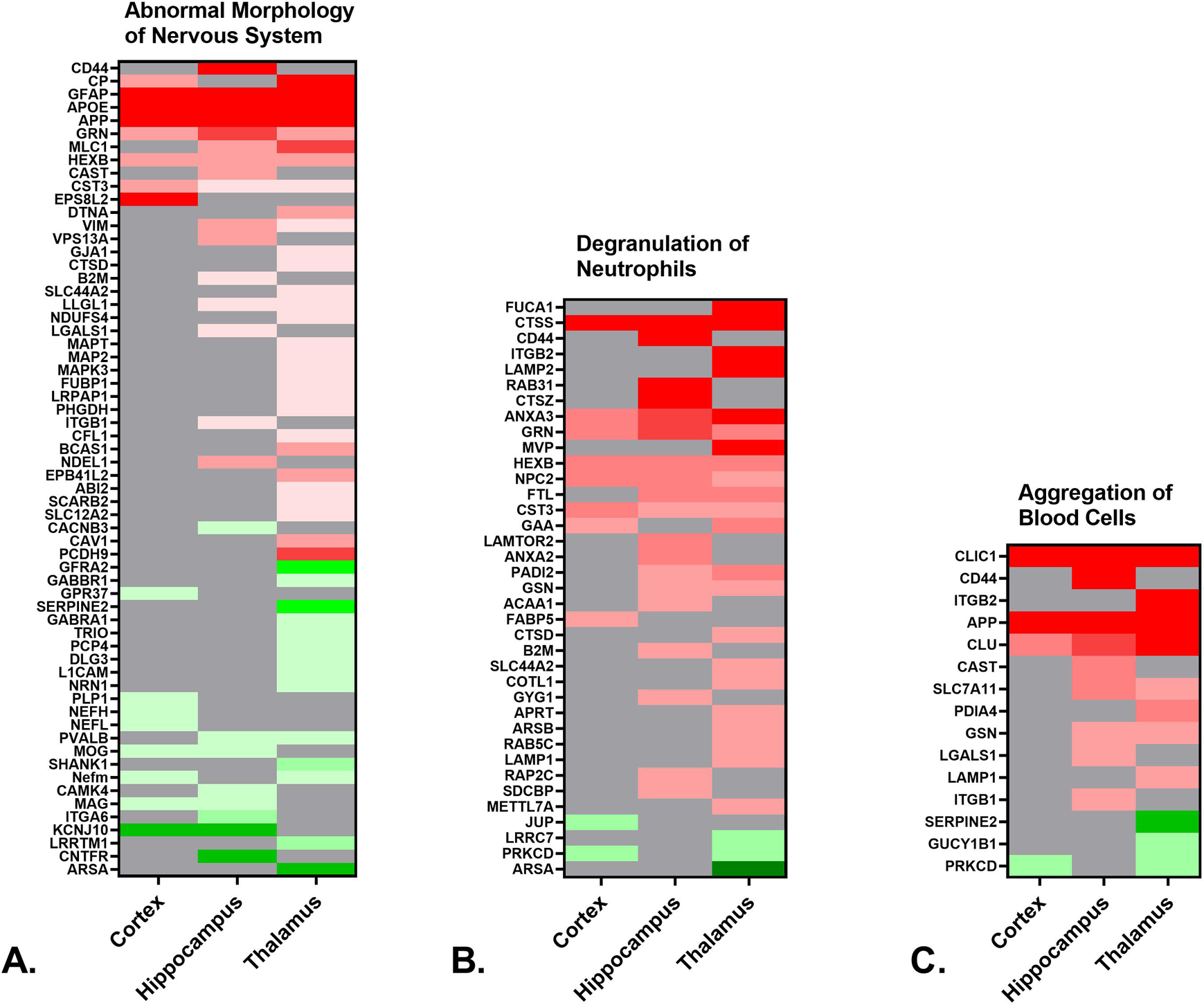
A. Heat map depicting the differentially expressed proteins (≥ 50% increase or ≥ 34% decrease, p < 0.05) in the cortex, hippocampus and thalamus of rTg-DI rats associated with abnormal morphology of the nervous system. B. Heat map depicting the differentially expressed proteins in the cortex, hippocampus and thalamus of rTg-DI rats associated with neutrophil degranulation. C. Heat map depicting the differentially expressed proteins in the cortex, hippocampus and thalamus of rTg-DI rats associated with aggregation of blood cells. Red indicates increased, green decreased, and grey not differentially expressed proteins, and color intensity correlates with degree of change.
DEA also identified neutrophil degranulation (Fig. 4B) as significantly altered in each region, and to greater extents in the hippocampus and thalamus, although, with indeterminant z-scores. Overlap p-values were 9.79 × 10−5, 6.44 × 10−10, and 1.59 × 10−9, and the number of differentially expressed proteins were 10, 19, and 25 in the cortex, hippocampus and thalamus, respectively. A heat map depicting the relative expression of these target molecules is depicted in Fig. 5B. Neutrophils are an important component of innate immunity, and influence the inflammatory response by acting as phagocytes, releasing pro- and anti-inflammatory granules, and proteinases involved in membrane degradation and matrix rearrangement (Selders et al. 2017). Additionally prolonged activity of released neutrophil granule matrix metalloproteinase 9 (MMP9) and neutrophil elastase (NE) have been reported to play a role in epithelial disruption and inflammation related tissue damage (Boxio et al. 2016; Fujie et al. 1999; Yang et al. 2003). Progranulin (Grn), commonly elevated among the three regions, is an indication of activated microglia, but can also inhibit neutrophil mediated inflammation (Cenik et al. 2012; Kessenbrock et al. 2008). However, progranulin is a substrate for NE, and is inactivated in prolonged neutrophil mediated inflammation (Kessenbrock et al. 2008). Similarly, Anxa3 mapped to this disease function and is an indicator of microglia activation (Junker et al. 2007; Smithson and Kawaja 2010). On the other hand, β2 integrin (ITGB2), uniquely elevated in the thalamus, mediates neutrophil adhesion and exocytosis (Pietronigro et al. 2017). Protein-arginine deiminase type-2 (PADI2), which catalyzes citrullination of many proteins and is elevated in the hippocampus and thalamus, is released upon neutrophil activation (O’Neil and Kaplan 2019). Thus, while these markers may indicate neutrophil degranulation, certain could also be markers of microgliosis which occurs in all three brain regions of rTg-DI rats.
DEA also identified aggregation of blood cells as uniquely activated in the thalamus (z-score = 2.381), the region that uniquely presents thrombotic events (SI Fig. S3). Heat maps for this function in all three regions is shown in Fig 5C.
3.6. Spatial imaging of enhanced markers
We next investigated the regional localization of some of the prominently enhanced proteins for mechanistic insight for common and regionally specific pathologies. Immunolabeling for the astrocytic protein ApoE in the cortex, hippocampus and thalamus of 12 M rTg-DI rats confirmed elevated levels and revealed strong accumulation with vascular amyloid deposits in each region (Fig 6D–F). The percent increase of ApoE protein measured by LC-MS/MS was revealed to be 448%, 395% and 795% in the cortex, hippocampus and thalamus, respectively which correlates with the relative astrocyte densities (SI Fig S1) and CAA load (Fig 1) in each region. Thus, increases of ApoE are relatively proportional to the extent of CAA load and number of astrocytes in each region.
Figure 6. Increased immunolabeling for ApoE and Anax3 in the cortex, hippocampus and thalamus of rTg-DI rats.
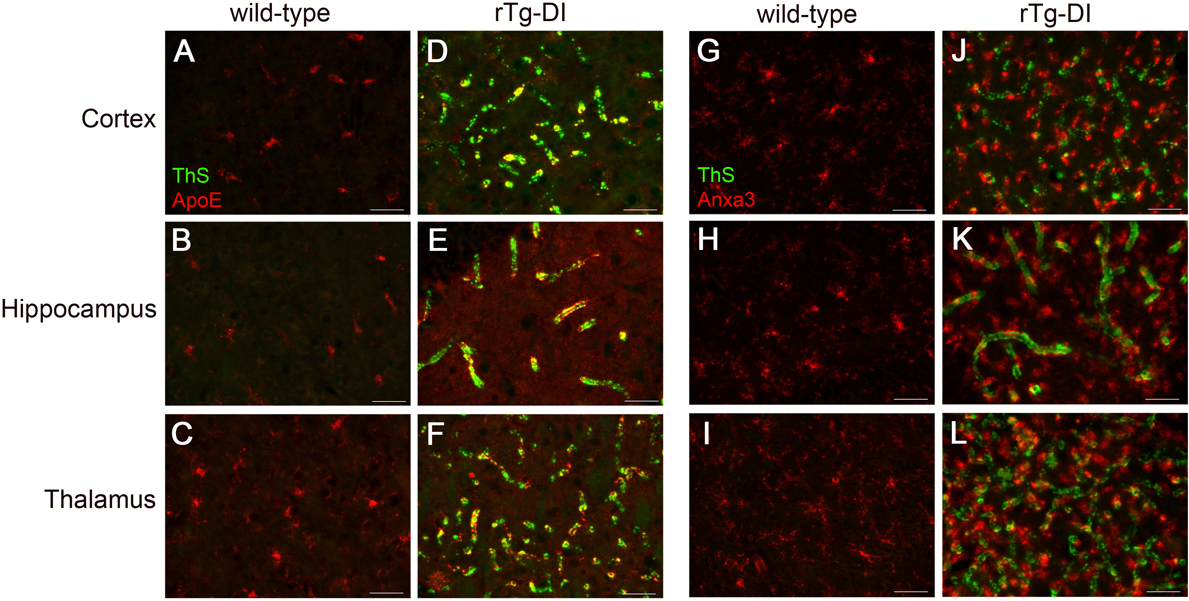
Brain sections from the present cohort of 12 M wild-type rats (A,B,C and G,H,I) and rTg-DI rats (D,E,F and J,K,L) were stained with thioflavin S to detect microvascular fibrillar amyloid (green) and mouse monoclonal antibody to ApoE (red) (A-F), or rabbit polyclonal antibody to Anax3 (red) (G-L). Scale bars = 50 μm. Representative images show increased ApoE and Anax3 in rTg-DI rats compared to wild-type rats in all brain regions.
Anxa3 has been previously suggested as a marker of activated microglia (Junker et al. 2007; Smithson and Kawaja 2010). Immunolabeling for Anxa3 in the cortex, hippocampus and thalamus of 12 M rTg-DI rats confirmed significant upregulation in each region (Fig 6J–L) compared to similarly aged WT rats (Fig. 6G–I). The elevated Anxa3 was co-localized with the numerous activated microglia surrounding the vascular amyloid deposits as confirmed by double labeling with the marker Iba-1 (SI Fig. S6). LC-MS/MS measured increases in Anxa3 were 163%, 223%, and 314% in the cortex, hippocampus and thalamus, respectively, which correlates with the relative microglia densities (SI Fig S2) and CAA load (Fig 1). Thus, the regional increases in Anxa3 are relatively proportional to the increases of microglia, and the cellular localization indicates these increases are indicative of an activated microglial response to vascular Aβ deposits in those regions.
IPA indicated Neutrophil Degranulation as an implicated disease function (Fig. 4). Interestingly, LC-MS/MS revealed many NET markers that were specifically enhanced in the thalamus, the region that uniquely develops progressive microbleeds and occluded vessels. Therefore, we conducted immunolabeling for the presence of NET-related proteins in the rTg-DI rats. NET imaging has been previously achieved using antibodies targeting neutrophil elastase (NE) and histone 2A (H2A) (Kolaczkowska et al. 2015). Immunolabeling in 12M rTg-DI rats revealed a dramatic and distinct presence of NE (Fig 7F) and H2A (Fig 7L) uniquely in the thalamus, confirming the presence of NET structures in this region of the CAA animals. NETs have been reported to promote thrombosis and microvessel breakdown in the cerebral vasculature, and cause subsequent inflammation related tissue damage (Kolaczkowska et al. 2015; Pietronigro et al. 2017; Zenaro et al. 2015). The unique presence of NET structures in the rTg-DI thalamus, the only region displaying microbleeds and occluded vessels, suggest that NETs likely contribute to these vasculopathies observed in that region.
Figure 7. Increased immunolabeling for neutrophil elastase and H2A exclusively in the thalamus of rTg-DI rats.
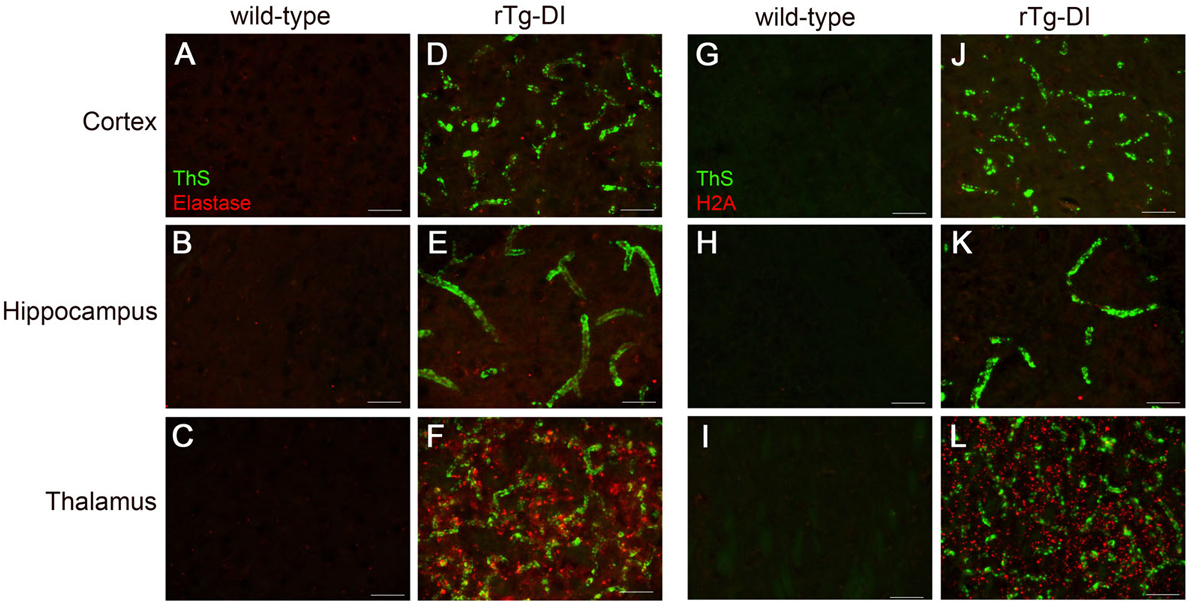
Brain sections from the present cohort of 12 M wild-type rats (A,B,C and G,H,I) and rTg-DI rats (D,E,F and J,K,L) were stained with thioflavin S to detect microvascular fibrillar amyloid (green) and rabbit polyclonal antibody to neutrophil elastase (red) (A-F), or rabbit polyclonal antibody to histone 2A (red) (G-L). Scale bars = 50 μm. Representative images show increased neutrophil elastase and histone 2A is increased only in the thalamus of rTg-DI rats.
4. DISCUSSION
The mechanistic link between the accumulation of fibrillar amyloid within the brain microvasculature and resultant pathological outcomes remains largely unknown. Thus, an important goal of this study is to connect differentially expressed proteins with the distinct regional pathologies associated with CAA, i.e. vascular amyloid load, neuroinflammation and thrombotic microbleeds/occluded microvessels. Here, consistent with our previous findings (Davis et al. 2018; Zhu et al. 2020a; Zhu et al. 2020b; Lee et al. 2020), we show in the present cohort of rTg-DI rats that the cortex, hippocampus and thalamus exhibit varying degrees of CAA, but a shared neuroinflammatory response, while thrombotic microbleeds and occluded vessels are restricted to the thalamus (Fig. 1, SI Figs. S1–S3). Therefore, by analyzing the proteome of each region individually, and then comparatively, including pathway/bioinformatics analysis and subsequent immunostaining, select differential proteins and pathways can be connected to specific pathological features.
There have been recent attempts to identify changes in the proteome of human CAA patients, either by isolation of leptomeningeal arteries (primarily CAA type-2) or brain tissue sections of CAA type-1 patients where affected vessels were isolated using laser microdissection (Hondius et al. 2018; Manousopoulou et al. 2017). In contrast, our study includes analysis of surrounding perivascular tissues which involves areas of induced neuroinflammation. Nevertheless, consistent with these studies involving human CAA we similarly identified ApoE, Htra1, Clu and markers for neuroinflammation as specifically enhanced in rTg-DI rats. However, there were notable differences. For example, in the study of leptomeningeal arteries, tissue inhibitor of metalloproteinase 3 (Timp3) was prominently identified (Manousopoulou et al. 2017), which was not seen in our results. Similarly, isolated vessels in human CAA type-1 highlighted serum amyloid P-component, norrin and collagen alpha-2 (VI) that were not found in our analysis (Hondius et al. 2018). On the other hand, notable proteins with increased expression in rTg-DI rats that were not prominent in the human CAA studies include Anxa3, Hspb1, S100a4, Aqp4 and Vim, as well as downregulated proteins in rTg-DI rats such as the neurofilament proteins and Mbp that were not reported by the human studies. There are several possibilities for the observed disparities between results obtained from the human vessels and our CAA rat model. One explanation may be the species differences between human patients and rodent pre-clinical models. Another noted difference is that the human studies involved CAA cases comprised of normal, non-mutated Aβ whereas the rTg-DI rats deposit Dutch/Iowa familial CAA mutant Aβ peptide in the cerebral vessels. Thus, structural differences between wild-type and CAA mutant Aβ peptides could elicit different responses. Lastly, differences could result from the distinct brain regions, surrounding tissues or vascular beds that are affected and were investigated in the different studies.
Another caveat performing proteomic analyses with individual human cases of CAA is that they exist on the background of a plethora of genetic, environmental, lifestyle and medication differences that vary from patient to patient thus potentially confounding the identification of specific CAA-associated changes. Advantages of using preclinical rodent models of CAA include their maintenance on a constant genetic background and consistent environmental and lifestyle conditions, which make population studies more uniform.
Approaches to proteomic investigation of rodent models have typically utilized whole brain homogenate samples or smaller manually dissected areas of localized disease progression (Njavro et al. 2020; Zheng et al. 2020; Deng et al. 2018). A strength of our approach includes the use of the novel rTg-DI rat model of microvascular CAA that largely reflects many of the pathological changes and cognitive deficits observed in humans with CAA and is largely devoid of any parenchymal fibrillar plaque deposition. This allowed us to investigate proteome changes that are specific for cerebral microvascular amyloid deposition without the confounds of parenchymal amyloid or tau pathologies. Furthermore, the rTg-DI line presents with a highly consistent onset and progression of cerebral microvascular amyloid deposition and associated pathologies. This permitted us to use relatively small cohorts of rats (n = 6) to obtain consistent measures lacking high degrees of variability between subjects. Lastly, the focus on specific brain regions (i.e. cortex, hippocampus and thalamus) enabled us to determine what proteome changes are associated with distinct CAA-related pathologies in each of these regions isolated from the same animals for comparative proteomic analysis. Thus, our experimental design was advantageously positioned to deliver regionally specified proteomic data concerning unique mechanisms that uniformly reflect CAA type-1 progression in this model.
ApoE was among the most abundantly expressed and most significantly increased proteins in all three regions with the greatest enhancement in the thalamus and least in the cortex, correlating with the astrocyte densities and CAA load. In the rTg-DI rats, the increased ApoE strongly accumulated with the microvascular amyloid deposits (Fig. 6D–F), consistent with earlier findings in the Tg-SwDI mouse model of CAA (Xu et al. 2008). ApoE has been proposed to mediate astrocytic degradation of Aβ (Koistinaho et al. 2004). More specific to CAA, ApoE has been shown to accumulate in the walls and perivascular spaces of microvessels impacted by small vessel disease, suggesting a disruption of the blood-brain barrier (BBB) (Rannikmäe et al. 2014; Grinberg and Thal 2010; Utter et al. 2008). Other astrocyte markers linked to CAA, such as Aqp4, were similarly elevated in each region. Aqp4 was previously reported as elevated in a model of mixed vascular dementia and may play an important role in reducing brain edema following intracerebral hemorrhage (Denver et al. 2019; Chu et al. 2013).
A novel finding of the present study was that Anxa3 was also among the most abundantly expressed and most significantly increased proteins in all three regions in rTg-DI rats. Anxa3 has been suggested as a marker of activated brain microglia/macrophages in models of cerebral ischemia, though has not yet been associated with CAA (Junker et al. 2007; Smithson and Kawaja 2010). While Anxa3 can be expressed in neutrophils, immunolabeling confirmed its expression in microglia in rTg-DI rats (Fig 6; SI Fig. S6). Anxa3 regional enhancements were greatest in the thalamus and least in the cortex, correlating with the relative microglial densities and CAA loads in each region. Thus, Anxa3 may be a novel marker for the microglial response to vascular amyloid and could play a role in cerebral small vessel pathology.
Other novel identified, abundant proteins that were commonly enhanced in both the hippocampus and thalamus included Vim, S100a6, and Hspb1. TGF-β1 stimulated expression of Vim upregulation has been reported to disrupt brain endothelial cell function (Derada Troletti et al. 2016), while S100a6 is reported to promote lymphocyte penetration through the BBB (Tsai et al. 2019). Conversely, Hspb1 may harbor protective functions, and is reported to reduce BBB permeability and promote microvascular integrity in models of cerebral ischemia (Leak et al. 2013). Therefore, the common enhancement of vimentin and S100a6 in the hippocampus and thalamus could be related to loss of microvascular integrity caused by a higher CAA load whereas the enhancement of Hspb1 could be compensatory in these regions.
To further investigate the functional significance of such a large number of altered proteins (547 total) in three distinct brain regions, we utilized IPA, as have many recent CNS and brain microvascular proteomic studies (Zhang et al. 2020; Gonzalez-Lozano et al. 2016; Ojo et al. 2020; Herland et al. 2020; Drummond et al. 2017). IPA identified multiple upstream regulators and causal networks, and disease related functions, both associating and distinguishing between the different regions. Although TNFα was not detected in our proteomic analysis, it was identified by IPA as the only commonly activated upstream regulator in all three brain regions and, notably, we recently showed its expression is increased in rTg-DI rats (Zhu et al. 2020a). Expression levels of TNFα were likely below the limits of detection for SWATH-MS analysis. However, IPA can use the differential expression of downstream target molecules to predict its activation state whether or not it has been detected (Krämer et al. 2014b). As a pro-inflammatory mediator, TNFα is reported to stimulate astrogliosis and microgliosis (Wang et al. 2015; Decourt et al. 2017), promote amyloidogensis (Blasko et al. 1999; Liao et al. 2004; Lahiri et al. 2003) and inhibit phagocytosis of Aβ plaques by macrophages (Koenigsknecht-Talboo and Landreth 2005). TNFα activation was common to all three brain regions, including the cortex where there is only a moderate CAA load. Therefore, TNFα may be responsible for some of the common neuroinflammatory features observed with moderate/severe CAA load in all three brain regions.
TGFβ1 was indicated as activated by IPA in the hippocampus and thalamus, which is consistent with our recent findings of increased expression in the brains of rTg-DI rats (Zhu et al. 2020a). Further, previous studies showed that TGFβ1 mRNA and that of its induced pro-fibrotic target genes is upregulated in Dutch-type CAA and is associated with increased CAA severity (Moursel et al. 2018; Zhang and Yang 2020). TGFβ1 has been reported to induce vascular fibrosis, mediate brain endothelial dysfunction, increased BBB permeability through downstream molecules such as FSP1 (S100A4) and Vim, and is associated with perivascular accumulation of ApoE (Moursel et al. 2018; Derada Troletti et al. 2016; Derada Troletti et al. 2019; Ueberham et al. 2005). FSP1, Vim and ApoE are all elevated in the rTg-DI rats (Table S1–3). Conversely, TGFβ1 released from pericytes and astrocytes promotes BBB integrity, and deficient signaling of TGFβ1 in the neurovascular unit increases BBB permeability (Derada Troletti et al. 2016; Garcia et al. 2004). Therefore, TGFβ1 signaling can have disparate roles on cerebral vascular integrity depending on where and when it is released.
Another unique finding was the activation of the causal network for thrombin (F2), the downstream function of aggregation of blood cells and the distinct down regulation of the thrombin inhibitor Serpine2 was observed in the thalamus, but not in the hippocampus or cortex. This is consistent with the presence of the microbleed and occluded vessel thrombotic phenotypes that are largely restricted to the thalamus (SI Fig. S3 and Davis et al. 2018; Zhu et al. 2020a; Lee et al. 2020). Thrombin has been reported as an important inflammatory mediator in AD related vasculopathies, and thrombin signaling is linked with neurovascular reorganization and loss of BBB function (Stanimirovic and Friedman 2012; Tripathy et al. 2013). Also, thrombin can promote neurodegeneration (Tripathy et al. 2013). Thus, connecting distinct thrombosis-related proteomic changes in the thalamus with these causal networks and functions provides valuable insight to these specific pathologies.
Neutrophil degranulation was indicated as impacted in each of the three regions by IPA. Neutrophils release pro- and anti-inflammatory granules and proteinases that mediate membrane degradation and matrix rearrangement (Selders et al. 2017). Additionally, neutrophil exocytosis leads to the formation of extracellular Neutrophil Extracellular Traps (NET), or web-like structures consisting of nucleic acid, histones and proteinases involved in membrane degradation, such as neutrophil elastase (NE) and MMP9 (Selders et al. 2017; Kolaczkowska et al. 2015). In the liver, vascular NET formation was shown to cause thrombosis, and is responsible for loss of vascular integrity, bleeding and up to 80% of infection related tissue damage (Kolaczkowska et al. 2015). Furthermore, NETs can serve as fibrin and thrombus scaffolds, enhance platelet aggregation and promote thrombosis in the cerebral vasculature (Martinod and Wagner 2014; Zhou et al. 2020; Peña-Martínez Carolina et al. 2019). The IPA indication of neutrophil degranulation as impacted, the upregulation of multiple NET markers (H1, 2, 3, and 4) in the thalamus and the striking similarities between vasculopathies observed in the rTg-DI thalamus and in NETosis prompted us to investigate the presence of the NET markers NE and H2A in rTg-DI rats (Fig. 7). The clear and specific presence of both markers in the rTg-DI thalamus, the only region exhibiting thrombotic events, is strong indication of the presence of NETs and linking them to the thalamic specific pathologies. Interestingly, Aβ fibrils have been reported to induce the release of NETs (Azevedo et al. 2012) and NET formation and neutrophil extravasation in areas of Aβ deposition in AD mouse models has been reported (Pietronigro et al. 2017; Zenaro et al. 2015). However, this is the first time the presence of NETs has been demonstrated in a model of CAA. As mentioned above, many of the proteins uniquely elevated in the thalamus (H1, 2, 3, and 4, β2 integrin) either mediate neutrophil adherence and exocytosis in the vasculature or have been identified in NET proteomes and used as NET markers (Lim et al. 2018; Urban et al. 2009; O’Donoghue et al. 2013; Pietronigro et al. 2017). Thus, regionally specific proteomic changes, coupled with IPA analysis and subsequent immunolabeling, now indicate NET formation as a thalamic-specific mechanism in the rTg-DI rats. This observation of NETs localized to areas of cerebral microbleeds provides a potentially important mechanistic link between vascular amyloid deposition and resultant vasculopathies present in CAA type-1.
Limitations
The present study focuses on proteome changes associated specifically with microvascular CAA and thus the absence of any appreciable parenchymal plaque or phospho-tau pathology limits our understanding of how these parenchymal pathologies would interact with microvascular CAA to influence regional proteome changes. Additionally, the small n = 3 female and male rats used in the present study limit our ability to identify sex-specific changes. Although, we have not observed significant differences in microvascular amyloid load nor incidence of cerebral microbleeds between male and female rTg-DI rats and there were no apparent significant differences in protein expression, a n = 3 is not sufficient to determine if relevant sex differences exist. Larger studies populated with more female and male rats will be required to better address this issue. Finally, our study focused on animals with later stage disease at 12 M. Future studies should explore proteome changes in younger rats at earlier stages of CAA.
Conclusion
The present study reveals important and new insight into the cerebral proteome changes found in distinct brain regions of rTg-DI rats that exhibit unique CAA-related pathological changes. These findings provide new information on potential pathogenic mechanisms of disease processes that can be further interrogated in future studies.
Supplementary Material
ACKNOWLEDGEMENTS
This research was supported by National Institutes of Health research grant RO1NS094201. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Technical assistance was provided by Dr. Xiaoyue Zhu, Dr. Benjamin Barlock and Ms. Judianne Davis.
ABBREVIATIONS
- Aβ
amyloid beta peptide
- AD
Alzheimer’s disease
- Anxa3
annexin A3
- ApoE
apolipoprotein E
- BBB
blood-brain barrier
- BDNF
brain derived neurotrophic factor
- CAA
cerebral amyloid angiopathy
- CDS2
phosphatidate cytidylyltransferase 2
- CNS
central nervous system
- DDA
data-dependent acquisition
- DEA
Downstream Effects Analysis
- DIA
data-independent acquisition
- DOC
sodium deoxycholate
- ECM
extracellular matrix
- EMT
epithelial to mesenchymal transition
- EndoMT
endothelial to mesenchymal transition
- FDR
false discovery rates
- Fsp1
fibroblast-specific protein 1
- F2
thrombin
- GFAP
glial fibrillary acidic protein
- H2A
histone H2A
- H2Ax
phosphorylated histone H2A
- Hspb1
heat shock protein beta-1
- Hsp27
heat shock protein 27
- Hist1h1d
histone H1.3
- IAA
iodoacetamide
- ICH
intracerebral hemorrhages
- IPA
Ingenuity Pathway Analysis
- Krt8
keratin 8
- Krt75
keratin 75
- LCM
laser capture microdissection
- LTM
long-term memory
- LTP
long-term potentiation
- MlC1
megalencephalic leukoencephalopathy with subcortical cysts 1
- MMP9
matrix metalloproteinase 9
- NE
neutrophil elastase
- Nefm
neurofilament medium length
- NET
Neutrophil Extracellular Traps
- RIPA
radioimmunoprecipitation assay
- SWATH-MS
Sequential Window Acquisition of all Theoretical Mass Spectra-MS
- SVD
small vessel disease
- S100A4
S100 calcium binding protein A4
- S100A6
S100 calcium binding protein A6
- Syt2
synaptotagmin-2
- TGF-β1
transforming growth factor β1
- TNFα
tumor necrosis factor α
- TOF-MS
time of flight MS
- TPA
total protein approach
- UPLC
ultra performance liquid chromatography
- VCID
vascular cognitive impairment and dementia
- Vim
vimentin
- XIC
extracted ion current chromatogram
Footnotes
CONFLICT OF INTEREST
All authors declare that they have no conflicts of interest with the contents of this article.
OPEN SCIENCE
This article has received a badge for Open Materials and Open Data because it provided all relevant information to reproduce the study in the manuscript and because it made the data publicly available. The data can be accessed at the MassIVE repository (massive.ucsd.edu/ProteoSAFe/static/massive.jsp), project ID#: MSV000086432
More information about the Open Science badges can be found at https://cos.io/our-services/open-science-badges/
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are openly available in the MassIVE repository (massive.ucsd.edu/ProteoSAFe/static/massive.jsp) under project ID#: MSV000086432
REFERENCES
- Aebersold R, Mann M (2016) Mass-spectrometric exploration of proteome structure and function. Nature 537, 347–355. [DOI] [PubMed] [Google Scholar]
- Arvanitakis Z, Leurgans SE, Wang Z, Wilson RS, Bennett DA, Schneider JA (2011) Cerebral amyloid angiopathy pathology and cognitive domains in older persons. Ann. Neurol. 69, 320–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attems J, Jellinger KA (2004) Only cerebral capillary amyloid angiopathy correlates with Alzheimer pathology—a pilot study. Acta Neuropathol. (Berl.) 107, 83–90. [DOI] [PubMed] [Google Scholar]
- Attems J, Jellinger K, Thal DR, Van Nostrand W (2011) Review: sporadic cerebral amyloid angiopathy. Neuropathol. Appl. Neurobiol. 37, 75–93. [DOI] [PubMed] [Google Scholar]
- Azevedo EPC, Guimarães-Costa AB, Torezani GS, Braga CA, Palhano FL, Kelly JW, Saraiva EM, Foguel D (2012) Amyloid Fibrils Trigger the Release of Neutrophil Extracellular Traps (NETs), Causing Fibril Fragmentation by NET-associated Elastase*,. J. Biol. Chem. 287, 37206–37218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Rivara CB, Rocher AB, Hof PR (2004) The nature and effects of cortical microvascular pathology in aging and Alzheimer’s disease. Neurol. Res. 26, 573–578. [DOI] [PubMed] [Google Scholar]
- Bereczki E, Branca RM, Francis PT, Pereira JB, Baek J-H, Hortobágyi T, Winblad B, Ballard C, Lehtiö J, Aarsland D (2018) Synaptic markers of cognitive decline in neurodegenerative diseases: a proteomic approach. Brain 141, 582–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasko I, Marx F, Steiner E, Hartmann T, Grubeck-Loebenstein B (1999) TNFalpha plus IFNgamma induce the production of Alzheimer beta-amyloid peptides and decrease the secretion of APPs. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 13, 63–68. [DOI] [PubMed] [Google Scholar]
- Boom A, Pochet R, Authelet M, Pradier L, Borghgraef P, Van Leuven F, Heizmann CW, Brion J-P (2004) Astrocytic calcium/zinc binding protein S100A6 over expression in Alzheimer’s disease and in PS1/APP transgenic mice models. Biochim. Biophys. Acta BBA - Mol. Cell Res. 1742, 161–168. [DOI] [PubMed] [Google Scholar]
- Boxio R, Wartelle J, Nawrocki-Raby B, Lagrange B, Malleret L, Hirche T, Taggart C, Pacheco Y, Devouassoux G, Bentaher A (2016) Neutrophil elastase cleaves epithelial cadherin in acutely injured lung epithelium. Respir. Res. 17, 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle PA, Yu L, Nag S, Leurgans S, Wilson RS, Bennett DA, Schneider JA (2015) Cerebral amyloid angiopathy and cognitive outcomes in community-based older persons. Neurology 85, 1930–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case NF, Charlton A, Zwiers A, Batool S, McCreary CR, Hogan DB, Ismail Z, et al. (2016) Cerebral Amyloid Angiopathy Is Associated With Executive Dysfunction and Mild Cognitive Impairment. Stroke 47, 2010–2016. [DOI] [PubMed] [Google Scholar]
- Cenik B, Sephton CF, Kutluk Cenik B, Herz J, Yu G (2012) Progranulin: A Proteolytically Processed Protein at the Crossroads of Inflammation and Neurodegeneration. J. Biol. Chem. 287, 32298–32306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu H, Tang Y, Dong Q (2013) Protection of Vascular Endothelial Growth Factor to Brain Edema Following Intracerebral Hemorrhage and Its Involved Mechanisms: Effect of Aquaporin-4. PloS One 8, e66051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis J, Xu F, Hatfield J, Lee H, Hoos MD, Popescu D, Crooks E, et al. (2018) A Novel Transgenic Rat Model of Robust Cerebral Microvascular Amyloid with Prominent Vasculopathy. Am. J. Pathol. 188, 2877–2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decourt B, Lahiri DK, Sabbagh MN (2017) Targeting Tumor Necrosis Factor Alpha for Alzheimer’s Disease. Curr. Alzheimer Res. 14, 412–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng S, Feng S, Wang W, Zhao F, Gong Y (2018) Biomarker and Drug Target Discovery Using Quantitative Proteomics Post-Intracerebral Hemorrhage Stroke in the Rat Brain. J. Mol. Neurosci. 66, 639–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denver P, D’Adamo H, Hu S, Zuo X, Zhu C, Okuma C, Kim P, et al. (2019) A Novel Model of Mixed Vascular Dementia Incorporating Hypertension in a Rat Model of Alzheimer’s Disease. Front. Physiol. 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derada Troletti C, Fontijn RD, Gowing E, Charabati M, van Het Hof B., Didouh I, van der Pol S. M. A., et al. (2019) Inflammation-induced endothelial to mesenchymal transition promotes brain endothelial cell dysfunction and occurs during multiple sclerosis pathophysiology. Cell Death Dis. 10, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derada Troletti C, Goede P. de, Kamermans A, Vries H. E. de (2016) Molecular alterations of the blood–brain barrier under inflammatory conditions: The role of endothelial to mesenchymal transition. Biochim. Biophys. Acta BBA - Mol. Basis Dis. 1862, 452–460. [DOI] [PubMed] [Google Scholar]
- Drummond E, Nayak S, Faustin A, Pires G, Hickman RA, Askenazi M, Cohen M, et al. (2017) Proteomic differences in amyloid plaques in rapidly progressive and sporadic Alzheimer’s disease. Acta Neuropathol. (Berl.) 133, 933–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dukay B, Walter FR, Vigh JP, Barabási B, Hajdu P, Balassa T, Migh E, et al. (2021) Neuroinflammatory processes are augmented in mice overexpressing human heat-shock protein B1 following ethanol-induced brain injury. J. Neuroinflammation 18, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eikelenboom P, Veerhuis R, Familian A, Hoozemans JJM, van Gool W. A., Rozemuller AJM. (2008) Neuroinflammation in plaque and vascular beta-amyloid disorders: clinical and therapeutic implications. Neurodegener. Dis. 5, 190–193. [DOI] [PubMed] [Google Scholar]
- Eng LF, Ghirnikar RS, Lee YL (2000) Glial fibrillary acidic protein: GFAP-thirty-one years (1969–2000). Neurochem. Res. 25, 1439–1451. [DOI] [PubMed] [Google Scholar]
- Eng LF, Vanderhaeghen JJ, Bignami A, Gerstl B (1971) An acidic protein isolated from fibrous astrocytes. Brain Res. 28, 351–354. [DOI] [PubMed] [Google Scholar]
- Fujie K, Shinguh Y, Inamura N, Yasumitsu R, Okamoto M, Okuhara M (1999) Release of neutrophil elastase and its role in tissue injury in acute inflammation: effect of the elastase inhibitor, FR134043. Eur. J. Pharmacol. 374, 117–125. [DOI] [PubMed] [Google Scholar]
- Garcia CM, Darland DC, Massingham LJ, D’Amore PA (2004) Endothelial cell–astrocyte interactions and TGFβ are required for induction of blood–neural barrier properties. Dev. Brain Res. 152, 25–38. [DOI] [PubMed] [Google Scholar]
- Gillet LC, Navarro P, Tate S, Röst H, Selevsek N, Reiter L, Bonner R, Aebersold R (2012) Targeted Data Extraction of the MS/MS Spectra Generated by Data-independent Acquisition: A New Concept for Consistent and Accurate Proteome Analysis. Mol. Cell. Proteomics 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Lozano MA, Klemmer P, Gebuis T, Hassan C, van Nierop P., van Kesteren R. E., Smit AB, Li KW (2016) Dynamics of the mouse brain cortical synaptic proteome during postnatal brain development. Sci. Rep. 6, 35456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graeber MB, Streit WJ, Kreutzberg GW (1988) The microglial cytoskeleton: vimentin is localized within activated cells in situ. J. Neurocytol. 17, 573–580. [DOI] [PubMed] [Google Scholar]
- Grammas P, Ovase R (2001) Inflammatory factors are elevated in brain microvessels in Alzheimer’s disease. Neurobiol. Aging 22, 837–842. [DOI] [PubMed] [Google Scholar]
- Greenberg SM, Vernooij MW, Cordonnier C, Viswanathan A, Al-Shahi Salman R, Warach S, Launer LJ, Van Buchem MA, Breteler MM, Microbleed Study Group (2009) Cerebral microbleeds: a guide to detection and interpretation. Lancet Neurol. 8, 165–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grinberg LT, Thal DR (2010) Vascular pathology in the aged human brain. Acta Neuropathol. (Berl.) 119, 277–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttenplan KA, Liddelow SA (2019) Astrocytes and microglia: Models and tools. J. Exp. Med. 216, 71–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herland A, Maoz BM, FitzGerald EA, Grevesse T, Vidoudez C, Sheehy SP, Budnik N, et al. (2020) Proteomic and Metabolomic Characterization of Human Neurovascular Unit Cells in Response to Methamphetamine. Adv. Biosyst. 4, 1900230. [DOI] [PubMed] [Google Scholar]
- Hondius DC, Eigenhuis KN, Morrema THJ, van der Schors R. C., van Nierop P., Bugiani M, Li KW, Hoozemans JJM, Smit AB, Rozemuller AJM (2018) Proteomics analysis identifies new markers associated with capillary cerebral amyloid angiopathy in Alzheimer’s disease. Acta Neuropathol. Commun. 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh I-N, Deluna X, White MR, Hartshorn KL Histone H4 directly stimulates neutrophil activation through membrane permeabilization. J. Leukoc. Biol. n/a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junker H, Suofu Y, Venz S, Sascau M, Herndon JG, Kessler C, Walther R, Popa-Wagner A (2007) Proteomic identification of an upregulated isoform of annexin A3 in the rat brain following reversible cerebral ischemia. Glia 55, 1630–1637. [DOI] [PubMed] [Google Scholar]
- Kessenbrock K, Fröhlich L, Sixt M, Lämmermann T, Pfister H, Bateman A, Belaaouaj A, et al. (2008) Proteinase 3 and neutrophil elastase enhance inflammation in mice by inactivating antiinflammatory progranulin. J. Clin. Invest. 118, 2438–2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenigsknecht-Talboo J, Landreth GE (2005) Microglial Phagocytosis Induced by Fibrillar β-Amyloid and IgGs Are Differentially Regulated by Proinflammatory Cytokines. J. Neurosci. 25, 8240–8249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koistinaho M, Lin S, Wu X, Esterman M, Koger D, Hanson J, Higgs R, et al. (2004) Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-β peptides. Nat. Med. 10, 719–726. [DOI] [PubMed] [Google Scholar]
- Kolaczkowska E, Jenne CN, Surewaard BGJ, Thanabalasuriar A, Lee W-Y, Sanz M-J, Mowen K, Opdenakker G, Kubes P (2015) Molecular mechanisms of NET formation and degradation revealed by intravital imaging in the liver vasculature. Nat. Commun. 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krämer A, Green J, Pollard J, Tugendreich S (2014a) Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 30, 523–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krämer A, Green J, Pollard J, Tugendreich S (2014b) Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 30, 523–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahiri DK, Chen D, Vivien D, Ge Y-W, Greig NH, Rogers JT (2003) Role of cytokines in the gene expression of amyloid beta-protein precursor: identification of a 5’-UTR-binding nuclear factor and its implications in Alzheimer’s disease. J. Alzheimers Dis. JAD 5, 81–90. [DOI] [PubMed] [Google Scholar]
- Launay S, Maubert E, Lebeurrier N, Tennstaedt A, Campioni M, Docagne F, Gabriel C, et al. (2008) HtrA1-dependent proteolysis of TGF- β controls both neuronal maturation and developmental survival. Cell Death Differ. 15, 1408–1416. [DOI] [PubMed] [Google Scholar]
- Leak RK, Zhang L, Stetler RA, Weng Z, Li P, Atkins GB, Gao Y, Chen J (2013) HSP27 Protects the Blood-Brain Barrier Against Ischemia-Induced Loss of Integrity. CNS Neurol. Disord. Drug Targets 12, 325–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Xu F, Liu X, Koundal S, Zhu X, Davis J, Yanez D, et al. (2020) Diffuse white matter loss in a transgenic rat model of cerebral amyloid angiopathy: J. Cereb. Blood Flow Metab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y-F, Wang B-J, Cheng H-T, Kuo L-H, Wolfe MS (2004) Tumor necrosis factor-alpha, interleukin-1beta, and interferon-gamma stimulate gamma-secretase-mediated cleavage of amyloid precursor protein through a JNK-dependent MAPK pathway. J. Biol. Chem. 279, 49523–49532. [DOI] [PubMed] [Google Scholar]
- Lim CH, Adav SS, Sze SK, Choong YK, Saravanan R, Schmidtchen A (2018) Thrombin and Plasmin Alter the Proteome of Neutrophil Extracellular Traps. Front. Immunol. 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Xie F, Alvarado-Diaz A, Smith MA, Moreira PI, Zhu X, Perry G (2011) Neurofilamentopathy in Neurodegenerative Diseases. Open Neurol. J. 5, 58–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López‐Sánchez LM, Jiménez‐Izquierdo R, Peñarando J, Mena R, Guil‐Luna S, Toledano M, Conde F, et al. (2019) SWATH‐based proteomics reveals processes associated with immune evasion and metastasis in poor prognosis colorectal tumours. J. Cell. Mol. Med. 23, 8219–8232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig C, Gillet L, Rosenberger G, Amon S, Collins BC, Aebersold R (2018) Data-independent acquisition-based SWATH-MS for quantitative proteomics: a tutorial. Mol. Syst. Biol. 14, e8126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manavalan A, Mishra M, Feng L, Sze SK, Akatsu H, Heese K (2013) Brain site-specific proteome changes in aging-related dementia. Exp. Mol. Med. 45, e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manousopoulou A, Gatherer M, Smith C, Nicoll JAR, Woelk CH, Johnson M, Kalaria R, Attems J, Garbis SD, Carare RO (2017) Systems proteomic analysis reveals that clusterin and tissue inhibitor of metalloproteinases 3 increase in leptomeningeal arteries affected by cerebral amyloid angiopathy. Neuropathol. Appl. Neurobiol. 43, 492–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinod K, Wagner DD (2014) Thrombosis: tangled up in NETs. Blood 123, 2768–2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan TE, Laping NJ, Rozovsky I, Oda T, Hogan TH, Finch CE, Pasinetti GM (1995) Clusterin expression by astrocytes is influenced by transforming growth factor β1 and heterotypic cell interactions. J. Neuroimmunol. 58, 101–110. [DOI] [PubMed] [Google Scholar]
- Moursel LG, Munting LP, van der Graaf L. M., van Duinen S. G., Goumans M-JTH, Ueberham U, Natté R, van Buchem M. A., van Roon‐Mom W. M. C., van der Weerd L. (2018) TGFβ pathway deregulation and abnormal phospho-SMAD2/3 staining in hereditary cerebral hemorrhage with amyloidosis-Dutch type. Brain Pathol. 28, 495–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nafar F, Williams JB, Mearow KM (2016) Astrocytes Release HspB1 in Response to Amyloid-β Exposure in vitro. J. Alzheimers Dis. 49, 251–263. [DOI] [PubMed] [Google Scholar]
- Njavro JR, Klotz J, Dislich B, Wanngren J, Shmueli MD, Herber J, Kuhn P-H, et al. (2020) Mouse brain proteomics establishes MDGA1 and CACHD1 as in vivo substrates of the Alzheimer protease BACE1. FASEB J. 34, 2465–2482. [DOI] [PubMed] [Google Scholar]
- Nofer J-R, Herminghaus G, Brodde M, Morgenstern E, Rust S, Engel T, Seedorf U, Assmann G, Bluethmann H, Kehrel BE (2004) Impaired Platelet Activation in Familial High Density Lipoprotein Deficiency (Tangier Disease) *. J. Biol. Chem. 279, 34032–34037. [DOI] [PubMed] [Google Scholar]
- O’Donoghue AJ, Jin Y, Knudsen GM, Perera NC, Jenne DE, Murphy JE, Craik CS, Hermiston TW (2013) Global Substrate Profiling of Proteases in Human Neutrophil Extracellular Traps Reveals Consensus Motif Predominantly Contributed by Elastase. PLOS ONE 8, e75141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojo JO, Crynen G, Algamal M, Vallabhaneni P, Leary P, Mouzon B, Reed JM, Mullan M, Crawford F (2020) Unbiased Proteomic Approach Identifies Pathobiological Profiles in the Brains of Preclinical Models of Repetitive Mild Traumatic Brain Injury, Tauopathy, and Amyloidosis. ASN Neuro 12, 1759091420914768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Leary LA, Davoli MA, Belliveau C, Tanti A, Ma JC, Farmer WT, Turecki G, Murai KK, Mechawar N (2020) Characterization of Vimentin-Immunoreactive Astrocytes in the Human Brain. Front. Neuroanat. 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neil LJ, Kaplan MJ (2019) Neutrophils in Rheumatoid Arthritis: Breaking Immune Tolerance and Fueling Disease. Trends Mol. Med. 25, 215–227. [DOI] [PubMed] [Google Scholar]
- Pascovici D, Handler DCL, Wu JX, Haynes PA (2016) Multiple testing corrections in quantitative proteomics: A useful but blunt tool. PROTEOMICS 16, 2448–2453. [DOI] [PubMed] [Google Scholar]
- Carolina Peña-Martínez, Violeta Durán-Laforet, Alicia García-Culebras, Fernando Ostos, Macarena Hernández-Jiménez, Isabel Bravo-Ferrer, Alberto Pérez-Ruiz, et al. (2019) Pharmacological Modulation of Neutrophil Extracellular Traps Reverses Thrombotic Stroke tPA (Tissue-Type Plasminogen Activator) Resistance. Stroke 50, 3228–3237. [DOI] [PubMed] [Google Scholar]
- Pietronigro EC, Della Bianca V, Zenaro E, Constantin G (2017) NETosis in Alzheimer’s Disease. Front. Immunol. 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planton M, Raposo N, Albucher J-F, Pariente J (2017) Cerebral amyloid angiopathy-related cognitive impairment: The search for a specific neuropsychological pattern. Rev. Neurol. (Paris) 173, 562–565. [DOI] [PubMed] [Google Scholar]
- Popescu DL, Van Nostrand WE, Robinson JK (2020) Longitudinal Cognitive Decline in a Novel Rodent Model of Cerebral Amyloid Angiopathy Type-1. Int. J. Mol. Sci. 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rannikmäe K, Kalaria RN, Greenberg SM, Chui HC, Schmitt FA, Samarasekera N, Al-Shahi Salman R, Sudlow CLM (2014) APOE associations with severe CAA-associated vasculopathic changes: collaborative meta-analysis. J. Neurol. Neurosurg. Psychiatry 85, 300–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rensink AAM, Waal R. M. W. de, Kremer B, Verbeek MM (2003) Pathogenesis of cerebral amyloid angiopathy. Brain Res. Rev. 43, 207–223. [DOI] [PubMed] [Google Scholar]
- Richard E, Carrano A, Hoozemans JJ, van Horssen J., van Haastert E. S., Eurelings LS, de Vries H. E., et al. (2010) Characteristics of Dyshoric Capillary Cerebral Amyloid Angiopathy. J. Neuropathol. Exp. Neurol. 69, 1158–1167. [DOI] [PubMed] [Google Scholar]
- Safieh M, Korczyn AD, Michaelson DM (2019) ApoE4: an emerging therapeutic target for Alzheimer’s disease. BMC Med. 17, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selders GS, Fetz AE, Radic MZ, Bowlin GL (2017) An overview of the role of neutrophils in innate immunity, inflammation and host-biomaterial integration. Regen. Biomater. 4, 55–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smithson LJ, Kawaja MD (2010) Microglial/macrophage cells in mammalian olfactory nerve fascicles. J. Neurosci. Res. 88, 858–865. [DOI] [PubMed] [Google Scholar]
- Stanimirovic DB, Friedman A (2012) Pathophysiology of the Neurovascular Unit: Disease Cause or Consequence? J. Cereb. Blood Flow Metab. 32, 1207–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thal DR, Ghebremedhin E, Orantes M, Wiestler OD (2003) Vascular Pathology in Alzheimer Disease: Correlation of Cerebral Amyloid Angiopathy and Arteriosclerosis/Lipohyalinosis with Cognitive Decline. J. Neuropathol. Exp. Neurol. 62, 1287–1301. [DOI] [PubMed] [Google Scholar]
- Thal DR, Ghebremedhin E, Rüb U, Yamaguchi H, Del Tredici K, Braak H (2002) Two Types of Sporadic Cerebral Amyloid Angiopathy. J. Neuropathol. Exp. Neurol. 61, 282–293. [DOI] [PubMed] [Google Scholar]
- Tripathy D, Sanchez A, Yin X, Luo J, Martinez J, Grammas P (2013) Thrombin, a mediator of cerebrovascular inflammation in AD and hypoxia. Front. Aging Neurosci. 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai M-H, Lin C-H, Tsai K-W, Lin M-H, Ho C-J, Lu Y-T, Weng K-P, Lin Y, Lin P-H, Li S-C (2019) S100A6 Promotes B Lymphocyte Penetration Through the Blood–Brain Barrier in Autoimmune Encephalitis. Front. Genet. 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueberham U, Ueberham E, Brückner MK, Seeger G, Gärtner U, Gruschka H, Gebhardt R, Arendt T (2005) Inducible neuronal expression of transgenic TGF-beta1 in vivo: dissection of short-term and long-term effects. Eur. J. Neurosci. 22, 50–64. [DOI] [PubMed] [Google Scholar]
- Urban CF, Ermert D, Schmid M, Abu-Abed U, Goosmann C, Nacken W, Brinkmann V, Jungblut PR, Zychlinsky A (2009) Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PLoS Pathog. 5, e1000639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utter S, Tamboli IY, Walter J, Upadhaya AR, Birkenmeier G, Pietrzik CU, Ghebremedhin E, Thal DR (2008) Cerebral small vessel disease-induced apolipoprotein E leakage is associated with Alzheimer disease and the accumulation of amyloid beta-protein in perivascular astrocytes. J. Neuropathol. Exp. Neurol. 67, 842–856. [DOI] [PubMed] [Google Scholar]
- Van Nostrand WE, Melchor JP, Cho HS, Greenberg SM, Rebeck GW (2001) Pathogenic effects of D23N Iowa mutant amyloid beta -protein. J. Biol. Chem. 276, 32860–32866. [DOI] [PubMed] [Google Scholar]
- Viswanathan A, Greenberg SM (2011) Cerebral Amyloid Angiopathy in the Elderly. Ann. Neurol. 70, 871–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W-Y, Tan M-S, Yu J-T, Tan L (2015) Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann. Transl. Med. 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelmus MMM, Boelens WC, Kox M, Maat-Schieman MLC, Veerhuis R, Waal R. M. W. de, Verbeek MM (2009) Small heat shock proteins associated with cerebral amyloid angiopathy of hereditary cerebral hemorrhage with amyloidosis (Dutch type) induce interleukin-6 secretion. Neurobiol. Aging 30, 229–240. [DOI] [PubMed] [Google Scholar]
- Wiśniewski JR, Mann M (2016) A Proteomics Approach to the Protein Normalization Problem: Selection of Unvarying Proteins for MS-Based Proteomics and Western Blotting. J. Proteome Res. 15, 2321–2326. [DOI] [PubMed] [Google Scholar]
- Wiśniewski JR, Ostasiewicz P, Duś K, Zielińska DF, Gnad F, Mann M (2012) Extensive quantitative remodeling of the proteome between normal colon tissue and adenocarcinoma. Mol. Syst. Biol. 8, 611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiśniewski JR, Rakus D (2014) Multi-enzyme digestion FASP and the ‘Total Protein Approach’-based absolute quantification of the Escherichia coli proteome. J. Proteomics 109, 322–331. [DOI] [PubMed] [Google Scholar]
- Xu F, Vitek MP, Colton CA, Previti ML, Gharkholonarehe N, Davis J, Nostrand WEV (2008) Human Apolipoprotein E Redistributes Fibrillar Amyloid Deposition in Tg-SwDI Mice. J. Neurosci. 28, 5312–5320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita N, Ilg EC, Schäfer BW, Heizmann CW, Kosaka T (1999) Distribution of a specific calcium-binding protein of the S100 protein family, S100A6 (calcyclin), in subpopulations of neurons and glial cells of the adult rat nervous system. J. Comp. Neurol. 404, 235–257. [PubMed] [Google Scholar]
- Yang Y-N, Bauer D, Wasmuth S, Steuhl K-P, Heiligenhaus A (2003) Matrix metalloproteinases (MMP-2 and 9) and tissue inhibitors of matrix metalloproteinases (TIMP-1 and 2) during the course of experimental necrotizing herpetic keratitis. Exp. Eye Res. 77, 227–237. [DOI] [PubMed] [Google Scholar]
- Zenaro E, Pietronigro E, Della Bianca V, Piacentino G, Marongiu L, Budui S, Turano E, et al. (2015) Neutrophils promote Alzheimer’s disease-like pathology and cognitive decline via LFA-1 integrin. Nat. Med. 21, 880–886. [DOI] [PubMed] [Google Scholar]
- Zhang D, Dong X, Liu X, Ye L, Li S, Zhu R, Ye Y, Jiang Y (2020) Proteomic Analysis of Brain Regions Reveals Brain Regional Differences and the Involvement of Multiple Keratins in Chronic Alcohol Neurotoxicity. Alcohol Alcohol 55, 147–156. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Yang X (2020) The Roles of TGF-β Signaling in Cerebrovascular Diseases. Front. Cell Dev. Biol. 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng F, Zhou Y-T, Zeng Y-F, Liu T, Yang Z-Y, Tang T, Luo J-K, Wang Y (2020) Proteomics Analysis of Brain Tissue in a Rat Model of Ischemic Stroke in the Acute Phase. Front. Mol. Neurosci. 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P, Li T, Jin J, Liu Y, Li B, Sun Q, Tian J, et al. (2020) Interactions between neutrophil extracellular traps and activated platelets enhance procoagulant activity in acute stroke patients with ICA occlusion. EBioMedicine 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Hatfield J, Sullivan JK, Xu F, Van Nostrand WE (2020a) Robust neuroinflammation and perivascular pathology in rTg-DI rats, a novel model of microvascular cerebral amyloid angiopathy. J. Neuroinflammation 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Xu F, Hoos MD, Lee H, Benveniste H, Van Nostrand WE (2020b) Reduced Levels of Cerebrospinal Fluid/Plasma Aβ40 as an Early Biomarker for Cerebral Amyloid Angiopathy in RTg-DI Rats. Int. J. Mol. Sci. 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw mass spectrometry data can be found in the MassIVE repository (massive.ucsd.edu/ProteoSAFe/static/massive.jsp), project ID#: MSV000086432, and password: rTgDIrgprt1219.
The data that support the findings of this study are openly available in the MassIVE repository (massive.ucsd.edu/ProteoSAFe/static/massive.jsp) under project ID#: MSV000086432