

Abstract
Background:
Correction of von Willebrand factor (VWF) deficiency with replacement products containing VWF can lead to the development of anti-VWF alloantibodies (i.e., VWF inhibitors) in patients with severe von Willebrand disease (VWD).
Objective:
Locate inhibitor-reactive regions within VWF using phage display.
Methods:
We screened a phage library displaying random, overlapping fragments covering the full-length VWF protein sequence for binding to a commercial anti-VWF antibody or to immunoglobulins from three type 3 VWD patients who developed VWF inhibitors in response to treatment with plasma-derived VWF. Immunoreactive phage clones were identified and quantified by next-generation DNA sequencing (NGS).
Results:
Next-generation DNA sequencing markedly increased the number of phages analyzed for locating immunoreactive regions within VWF following a single round of selection and identified regions not recognized in previous reports using standard phage display methods. Extending this approach to characterize VWF inhibitors from three type 3 VWD patients (including two siblings homozygous for the same VWF gene deletion) revealed patterns of immunoreactivity distinct from the commercial antibody and between unrelated patients, though with notable areas of overlap. Alloantibody reactivity against the VWF propeptide is consistent with incomplete removal of the propeptide from plasma-derived VWF replacement products.
Conclusion:
These results demonstrate the utility of phage display and NGS to characterize diverse anti-VWF antibody reactivities.
Keywords: anti-VWF alloantibodies, next-generation DNA sequencing, phage display, von Willebrand disease, von Willebrand factor
1 |. INTRODUCTION
Plasma von Willebrand factor (VWF) stabilizes coagulation factor VIII (FVIII) and directs platelets to sites of vascular injury.1,2 Abnormalities of VWF result in several types of von Willebrand disease (VWD).3 Intravenous infusion of plasma-derived VWF concentrate is the current standard of care for pediatric VWD patients with significant bleeding and poor response to desmopressin.3,4 However, the development of anti-VWF alloantibodies (i.e., VWF inhibitors), which primarily occurs in type 3 VWD patients (prevalence of ~1:1 000 000 of the general population), significantly complicates therapy.5,6 In contrast to the high rate of FVIII inhibitor development in severe hemophilia A patients (~30%), alloimmunization to exogenous VWF is less common (~5%–10% of type 3 VWD patients) and less well studied.5,7,8
Mature plasma VWF enters the blood after proteolytic removal of its propeptide (VWFpp) and circulates as large multimers, with specific functions localized to distinct domains within the monomer subunits.9 Previous reports using proteolytic and recombinant fragments of VWF have demonstrated the presence of alloantibodies that recognize the VWF platelet-binding domain and inhibit VWF binding to its platelet receptor, glycoprotein Ib.10–13 Heterogeneous immunoreactivity to other VWF domains between patients suggests that alloantibody epitopes and potential functional consequences may vary widely among VWD patients with inhibitors.10,12
Phage display is a high content method for studying protein interactions. In phage display, bacteriophage is engineered to fuse a coat protein with a library of amino acid variants or random protein fragments of an antigen, thereby linking the expressed peptide with its encoding DNA.14 These libraries typically contain up to ~106 independent clones that may express different parts or variants of a protein. Applying a selection pressure (e.g., antibody binding) to a phage display library separates clones with a selective advantage from clones that do not. In the example of antibody binding, advantageous clones are precipitated and collected whereas disadvantaged clones are removed, thereby enriching for antibody-binding clones. Following selection, clones are identified by DNA sequencing. This approach has been used to localize immunoreactive regions in VWF and FVIII for anti-VWF antibodies generated as research reagents and FVIII inhibitors found in patients with hemophilia A, respectively.15–18 However, the practical limits of standard Sanger DNA sequencing of individual, selected phage impede a comprehensive analysis of an antigenic landscape. Next-generation DNA sequencing (NGS) has extended the utility of display technologies (e.g., phage display) for epitope mapping with greater throughput and finer amino acid sequence resolution.19,20 By comparing each clone’s population proportion within the selected library to that of the unselected library, antigenic regions within a protein may be identified.
We previously reported the application of NGS combined with phage display to provide a comprehensive picture of ADAMTS13 interaction with its target sequence within the VWF A2 domain.21 We now extend this approach of combining phage display with NGS to identify the segments within VWF recognized by anti-VWF antibodies.
2 |. MATERIALS AND METHODS
2.1 |. DNA constructs
The sequences of all oligonucleotides (P1–P12, IDT DNA Technologies) are listed in Table 1. A FLAG tag (NH2-DYKDDDDK-COOH) was inserted into the phagemid pAY-E (GenBank KF384455) to facilitate specific elution of bound M13 with enterokinase.22 Oligonucleotides P1 and P2 were annealed (65.0°C) and ligated into the NotI and SgrAI sites of pAY-E to introduce the FLAG tag. Subsequently, the CcdB-CmR cassette (ThermoFisher) was PCR amplified with primers P3 and P4 and ligated into the SfiI and NotI restriction sites. The resulting phagemid, pAY-FE (GenBank MW464120), was maintained in in TOP10 CcdB Survival T1R cells (ThermoFisher). The sequence of pAY-FE verified by Sanger sequencing.
TABLE 1.
Oligonucleotides
| Primer | Sequence |
|---|---|
| P1 | GGCCGCAGGTGGTGGTGACTACAAGGACGATGACGATAAAGGAGGAGGAGGTGCG |
| P2 | CCGGCGCACCTCCTCCTCCTTTATCGTCATCGTCCTTGTAGTCACCACCACCTGC |
| P3 | TCGGCCTAGCCGGCCGGCGCGCCATTAGGCACCCCAGGCTTTACACTTTATGCTT |
| P4 | TGCGGCCGCGTCGACCTGCAGACTGGCTGTGTATAA |
| P5 | CGCGCCATCGGGAGGAGGG |
| P6 | /5PHOS/CCCTCCTCCCGATGG |
| P7 | /5PHOS/TGGCGGCGGTGC |
| P8 | GGCCGCACCGCCGCCA |
| P9 | TTCATGCTGCCGGCTTTCTCGG |
| P10 | TCGTCATCGTCCTTGTAGTCACCACCA |
| P11 | AATGATACGGCGACCACCGAGATCTACAC |
| P12 | CAAGCAGAAGACGGCATACGAGAT |
| P13 | TGTTCTGGGGAATATAATCTAGATGATCATAATCAGCCATACCACATTTGTAGAGGTTTT |
| P14 | CCATACCACATTTGTAGAGGTTTTACTTGCTTTAAAAAACCTCCCACACCTCCCCCTGAA |
| P15 | AAACAAGTTAACAACAACAATTGCATTCATTTTATGTTTCAGGTTCAGGGGGAGGTGTGG |
The expression plasmid for VWF has been previously described.23 To create the expression plasmid for the VWFpp, DNA generated by PCR amplification of primers P13–P15 (GoTaq Green, Promega) was subcloned by Gibson assembly (repliQa HiFi Assembly Mix, Quantabio) into the VWF expression plasmid that was digested with HindIII-HF (New England Biolabs). The resulting plasmid introduced a VWD variant, R763G, which inhibits furin-mediated proteolysis between the VWFpp and the tandem C-terminal myc epitope and His6 tags. The VWF and VWFpp expression plasmids were maintained and propagated in TOP10 cells (ThermoFisher). The sequence of the VWFpp expression plasmid was verified by Sanger sequencing.
2.2 |. Phage display VWF fragment library
The VWF cDNA subcloned into pBluescript was randomly fragmented by sonication. DNA ends were repaired and phosphorylated with the NEBNext End Repair kit (New England Biolabs). Phagemid adaptors were created by annealing oligonucleotides (P5/P6 and P7/P8, both at 50.0°C) and were ligated at ~20-fold molar excess onto the repaired VWF cDNA fragments. Adapted fragments were fractioned across a 2% (w/v) agarose gel, collected (~300 bp to ~1000 bp), purified by electroelution and phenol/chloroform extraction, and ligated into pAY-FE at the AscI and NotI sites. Ligation products were transformed into XL1-Blue MRF′ cells (Agilent) by electroporation in 0.1 cm cuvettes at 1.8 kV, 200 Ω, and 25 μF (Gene Pulser, Biorad). Transformed cells were recovered in SOC medium (ThermoFisher) at 37°C for 1 h and quantified for ampicillin resistance on LB-AG (Luria broth +100 μg/ml ampicillin +2% glucose) agar; the VWF fragment library depth was ~2.78 × 106 independent clones. This library (~12.5 ml) was expanded by inoculating 300 ml LB-AG and culturing at 37°C with aeration overnight. The expanded library was stored in fresh LB-AG +20% glycerol as 1-ml aliquots at −80°C.
Only a subset of the random VWF cDNA plasmid (in Bluescript) fragments cloned into the pAY-FE phagemid vector would be expected to express a VWF peptide fused to the N-terminus of phage coat protein, pIII (Figure S1A). Display of a VWF protein fragment requires that its subcloned, encoding cDNA be composed of VWF sequence (total plasmid content = ~3/4 VWF + ~1/4 pBluescript) whose orientation matched the phagemid orientation (1/2 of fragments) and whose 5′-end was the first nucleotide of a VWF codon (1/3 of fragments) and whose 3′-end was the second nucleotide of a VWF codon (1/3 of fragments). As a result, only ~1/24th (3/4 × 1/2 × 1/3 × 1/3) of the library is expected to display a VWF protein fragment. The remaining 23/24th of clones are expected to encode non-VWF peptides, premature stop codon(s), or frame-shift(s) that prevents expression of a C-terminal pIII fusion.
To generate phage virions displaying VWF fragments for screening, 100 ml LB-AG was inoculated with a 1-ml aliquot of the VWF fragment phage display library and cultured at 37°C with agitation until mid-log phase (OD600nm = 0.3–0.5). Cultures were then infected with M13KO7 helper phage (GE Healthcare Life Sciences) at a multiplicity of infection ≥10 at 37°C with agitation for 1 h. Cells were isolated by centrifugation, resuspended in 100 ml 2x yeast extract tryptone media containing 100 μg/ml ampicillin and 50 μg/ml kanamycin, and cultured at 30°C overnight with agitation. Cells were removed from the culture by centrifugation at 4000g and 4°C, and phage were precipitated from the supernatant with 2 ml PEG/NaCl (16.7% PEG 8000 + 19.5% NaCl) per 10 ml supernatant at 4°C. Precipitated phage were collected by centrifugation at 20 000g and 4°C, resuspended in TBS-T (50 mM Tris-HCl, pH = 7.5, +150 mM NaCl +0.05% Tween-20), and reprecipitated with PEG/NaCl (2 ml per 10 ml supernatant) at 4°C. Phage were finally collected by centrifugation at 20 000g at 4°C and resuspended in antibody selection buffer (TBS-T + 5% bovine serum albumin). Phage concentrations were quantified by infecting XL1-Blue MRF′ cells as previously described.22
2.3 |. Screening phage displayed VWF fragments for binding anti-VWF antibodies
Magnetic protein G beads (New England Biolabs) were blocked in antibody selection buffer and used to immobilize 10–15 μg polyclonal rabbit anti-VWF (Dako A0082, lots 00045319 and 00051141) or immunoglobulin G (IgG) in platelet-poor plasma (PPP) from patients diagnosed with an inhibitor against VWF. Total IgG in PPP was quantified with the Human IgG ELISA Quantitation Set (Bethyl Laboratories, E80–104). Following informed consent with protocols approved by institutional review boards of the University of Michigan and of Boston Children’s Hospital, PPP containing anti-VWF alloantibodies were collected from previously described pediatric patients diagnosed with type 3 VWD and inhibitors to plasma-derived VWF concentrate.24,25 PPP was prepared by centrifugation as previously described.1 IgG-bound beads were washed with antibody selection buffer and subsequently incubated with 1011–1012 phage from the VWF fragment library at room temperature for at least 1.5 h. Beads were then washed with antibody selection buffer followed by antibody selection elution buffer (5% bovine serum albumin +20 mM Tris-HCl, pH =7.4, +50 mM NaCl +2 mM CaCl2 + 0.005% Tween-20). Bound phage were eluted with 20 ng/ml enterokinase (New England Biolabs) at 4°C overnight. The supernatants containing the eluted phage were collected and stored at 4°C. The number of phage virions eluted was quantified as previously described.22
2.4 |. Phage identification
Single stranded DNA from 105–107 selected and unselected phage was liberated by proteinase K digestion in digestion buffer (100 mM Tris-HCl, pH = 8.5–8.8, +5 mM EDTA +200 mM NaCl +0.2% sodium dodecyl sulfate) overnight at 37°C and further purified with phenol/chloroform/isoamyl alcohol extraction.26 The region containing random fragments of the VWF plasmid was made double-stranded using primers P9/P10 (annealing temperature = 65.0°C) and Herculase II (Agilent) with 16–20 rounds of PCR amplification (Figure S1B). The PCR products were purified with AMPure XP beads (Beckman Coulter) and fragmented by five cycles of sonication on a Bioruptor (per cycle: power = high, interval = 30 s on/30 s off, 5 min; Diagenode) to facilitate efficient DNA clustering onto the Illumina sequencing platform. Fragmented PCR products were repaired and dA-tailed using the NEBNext End Repair/dA Tailing kit (New England Biolabs) according to the manufacturer’s instructions and subsequently barcoded with Illumina-compatible, barcoded NGS adaptors (NEXTflex DNA Barcodes, Bioo Scientific) using the NEBNext Ultra Ligation kit (New England Biolabs). The adapted PCR products were purified twice with AMPure XP beads and subsequently PCR-amplified for eight cycles with primers P11/P12 (annealing temperature = 60.0°C). The resulting sequencing libraries were quantified with PicoGreen (Thermo Fisher) and analyzed on a Bioanalyzer (Agilent) according to the manufacturers’ instructions. Sequencing was performed on the MiSeq platform (Illumina) using the 2 × 75 bp kit or on the HiSeq 2500 platform (100 base, paired end; Illumina) with the libraries clustered at 12–15 pM and mixed with 5% PhiX genomic DNA (Illumina).
2.5 |. Computational and statistical analyses
We used a custom Python script to process the sequencing data. Our script (1) combined paired reads into one sequence, (2) filtered sequences for junctions between the phagemid and the parental VWF plasmid, (3) mapped the adjoined VWF plasmid sequences, and (4) generated a summary of counts for the nucleotide locations within the parental VWF cDNA plasmid that were found immediately adjacent to the phagemid. The summary data were used as input for the subsequent statistical analyses. The bioinformatic, statistical, and graphing scripts are available on GitHub (https://github.com/yeea0 0/anti-VWF _Phage.git). Each phagemid/VWF nucleotide fragment was considered as an independent variable. Counts for the first and last VWF nucleotide within DNA fragments containing a phagemid/VWF fusion (Figure S1B) ranged from 0 to ~41 000 and were analyzed with DESeq2 to calculate the fold changes (selected vs. unselected), the mean nucleotide counts (between selected and unselected), and the p values adjusted for multiple comparisons (q values) by false discovery rate (FDR) correction.27 A q value < .1 was considered significant.
2.6 |. Clearance studies
Patients II-1 and II-2 were challenged with an intravenous infusion of plasma-derived VWF containing FVIII (Wilate, Octapharma) at 50 IU/kg. VWF antigen, VWF ristocetin cofactor activity, and FVIII activity (i.e., von Willebrand panel) were clinically evaluated at the indicated time points on a Siemens BCS coagulation analyzer by the Clinical Coagulation Laboratory at Boston Children’s Hospital. A similar clearance study of plasma-derived VWF for patient I-1 has been previously reported.25
2.7 |. Protein expression, immunoprecipitation, and analysis
Human embryonic kidney 293T cells were transiently transfected with VWF or VWFpp expression plasmids using polyethylenimine as previously described.1 Conditioned media were collected 2 days after transfection and clarified by centrifugation at 500g for 10 min at 22°C. The supernatant was adjusted to 20 mM Tris-HCl (pH = 7.5) and stored at 4°C until analysis.
Recombinant VWF or VWFpp were immunoprecipitated from the clarified conditioned media. Magnetic protein G beads (50 μl, New England Biolabs) or Ni-NTA beads (50 μl, Genesee Scientific) were blocked with antibody selection buffer. Polyclonal rabbit anti-VWF (15 μg; Dako A0082, lot 20057618) or IgG (15 μg) in patient PPP were immobilized onto blocked protein G beads at room temperature. Immunoglobulin-coupled protein G beads and blocked Ni-NTA beads were washed with antibody selection buffer and incubated with 1 ml of clarified conditioned media containing either multimeric VWF or VWFpp overnight at 4°C. Beads were then collected on a magnet (protein G) or by centrifugation at 800g for 1 min at room temperature (Ni-NTA) and washed with TBS-T. Proteins on washed beads were eluted and denatured with reducing Laemmli buffer (60 mM tris-HCl, pH = 8.0, +2% sodium dodecyl sulfate +5% β-mercaptoethanol +0.02% Orange G + 10% glycerol) at 95°C for 5 min.
Immunoprecipitated, denatured proteins were fractioned on a 6% tris-glycine gel (ThermoFisher) and transferred onto nitrocellulose as previously described.1 Blots were blocked in blocking buffer (Odyssey TBS Blocking Buffer, LI-COR) and probed first with 1 mg/ml rabbit-anti-myc antibody (Behtyl Laboratories, A190-105A) in immunoblotting buffer (blocking buffer +0.05% Tween-20) then with 0.133 μg/ml goat-anti-rabbit IgG conjugated to Alexa Fluor 680 (ThermoFisher, A21076) in immunoblotting buffer. Blots were imaged on an Odyssey CLx (LI-COR).
3 |. RESULTS AND DISCUSSION
Characterization of the unselected library with NGS showed a full representation of VWF nucleotides at the terminal positions (Figure S1C) with comparable frequencies (defined as the number of reads for a nucleotide per total number of reads for all nucleotides), demonstrating the absence of a strong selection for or against any particular VWF cDNA fragment in Escherichia coli (Figure S2A). Median frequencies were similar between terminal nucleotides of different VWF reading frames, though the distributions of nucleotide frequencies were different among the three frames (Anderson-Darling p < 1 × 10−130 for both replicates of sense nucleotides and p < .05 for both replicates of antisense nucleotides; Figure S2B). These nucleotide frequencies were well correlated between biological replicates of unselected phage (Pearson R = 0.85 and 0.93 for nucleotides from the sense and antisense strands of the parental VWF plasmid, respectively; Figure S2C), demonstrating comparability between different preparations of unselected phage.
In our previous analysis of 81 individual phage clones (32 unique) using Sanger sequencing, we identified eight distinct regions in VWF (E1–E8 in Figure 1) that bind to a commercial anti-VWF antibody.16 In this report, we expanded our analysis to 105–107 independent phage with NGS and identified a much more extensive immunoreactivity spanning the entire mature VWF sequence for the same commercial antibody. Following a single round of selection, 1758 N-terminal and 1466 C-terminal VWF residues were significantly enriched (fold change ≥1.5, FDR adjusted p value < .1, Figure S3). In the absence of anti-VWF antibodies, no specific VWF N- or C-terminal residues were strongly enriched (Figure S3). Comparison of fold-enrichment by two different lots of the same commercial anti-VWF antibody resulted in a high degree of agreement (Pearson R = 0.96 for N-terminal residues, Pearson R = 0.93 for C-terminal residues). The identified antibody-binding fragments localize exclusively to the mature VWF sequence, with no significantly enriched segments of the VWFpp (Figure 1), consistent with the use of plasma-derived VWF as the immunogen.28 The previously reported immunoreactive sites, E1–E8, encompassed multiple, significantly enriched terminal residues (Figure S4). Of note, the application of NGS required fragmentation of the ~400 bp to ~1100 bp phage insert amplicons to ~100 bp to ~600 bp segments (Figure S1B). As a result, we cannot define the specific N- and C-termini for any given fragment, limiting the ability to define minimal, contiguous immunoreactive sequences within VWF. Advances in NGS technologies that enable longer reads together with increased sequencing read depth should help circumvent this limitation in future experiments.
FIGURE 1.
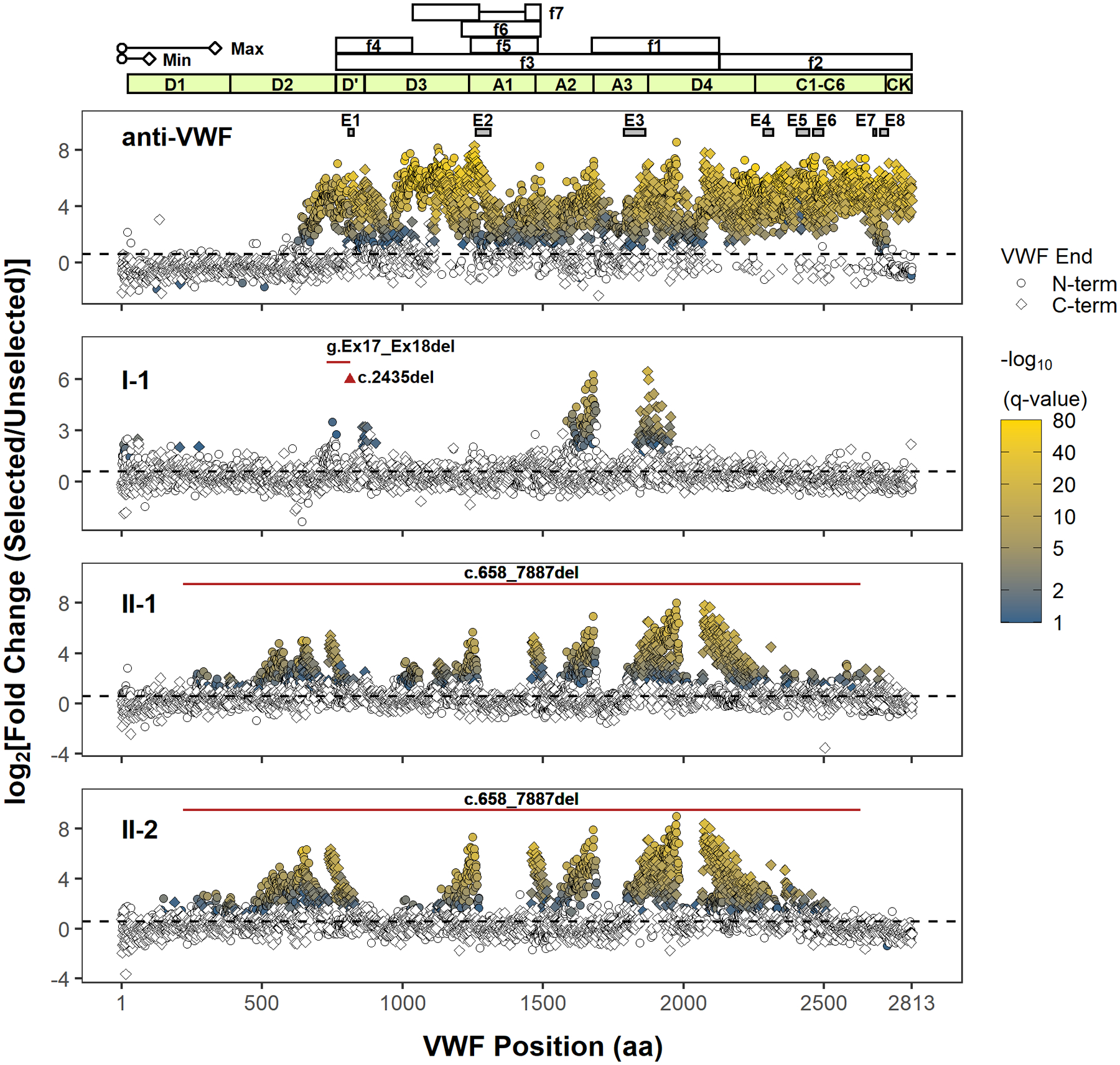
Next-generation DNA sequencing analysis of phage display identifies immunoreactive regions in VWF. Only the position of the first (N-terminal) or last (C-terminal) residue within the identified VWF fragment is shown. The approximate minimal (min, ~100 amino acids) and maximal (max, ~333 amino acids) lengths of phage displayed VWF peptides (depiction scaled according to the x-axis) are determined from the size range of VWF cDNA fragments subcloned into the phagemid. However, the phase of N- and C-termini of individual VWF fragments (i.e., which N-terminus is paired with which C-terminus) cannot be determined because of the fragmentation of PCR amplicons of phage DNA for NGS, limiting the delineation of unique epitopes. The coordinates of proteolytic VWF fragments, f1–f7, used to epitope map anti-VWF alloantibodies in prior reports are shown (f1: G1674-E2128; f2: E2129-K2813; f3: S764-E2128; f4: S764-R1035; f5: L1243-G1481; f6: V1212-K1491; f7: K1036-R1274, A1437-K1491).10–13 The VWF domain boundaries are annotated in light green. The black dashed line denotes a fold change of 1.5. Terminal residues of phage displayed VWF fragments are marked (VWF end) according to their location and enrichment. Only VWF fragment termini with a significant fold change (FDR adjusted p value < .1) are indicated in shading from blue to yellow, which scales with the significance of fold change (−log10(q value)). A polyclonal anti-VWF antibody (anti-VWF) recognizes fragments spanning the mature VWF sequence. Epitopes E1–E8 were previously identified by conventional phage display for the same antibody, and their coordinates are shown as gray boxes.16 The regions for E1–E8 are magnified in Figure S4. VWF inhibitors from type 3 VWD patients (I-1, II-1, and II-2) bind distinct regions in VWF. The location of the patients’ genetic deletion(s) is marked according to the corresponding VWF codon (red triangle for point deletion or red solid line for contiguous deletion). Note that the clustering of significantly enriched termini supports localization of a series of reactive VWF fragments whose internal sequences are graphically depicted as gaps between the clusters (e.g., the immunoreactive region in A3 for I-1 is bounded by clusters of significantly enriched N- and C-terminal residues on the left and right, respectively). Abbreviations: FDR, false discovery rate; NGS, next-generation sequencing; VWD, von Willebrand disease; VWF, von Willebrand factor
We next applied this approach to localize the VWF binding regions of anti-VWF alloantibodies that developed in three type 3 VWD patients who were treated with VWF replacement products (Figure 1 and Figure S3). Plasma IgG from patient I-1 (compound heterozygous for VWF c.2435del and for a deletion in VWF of at least 8.7 kb spanning exons 17 and 1825) selected VWF fragments that localized to the D′ and A3 domains, with marked enrichment for the A3 domain (Figure 1). Consistent with the lack of selection for VWF fragments spanning the platelet-binding A1 domain, patient I-1’s alloantibodies did not inhibit the ristocetin cofactor activity of infused VWF.25 Patients II-1 and II-2, brothers both homozygous for a large deletion in VWF (c.658_7887del),24 developed inhibitors with highly similar profiles in the selection of phage-displayed VWF fragments. Similar immunoreactivity to the D3, A1, A3, and D4 domains of mature VWF were found in both patients (Figure 1). Immunoreactivity to the D1 and D2 domains was also identified for both brothers (Figure 1), consistent with the presence of the VWFpp in their treatment (plasma-derived VWF derived from several different pharmaceutical sources). Immunoprecipitation of recombinant VWFpp confirmed the presence of anti-VWF pp alloantibodies in patients II-1 and II-2 (Figure 2). In comparison to previous studies that used proteolytic fragments of mature VWF, f1–f7 (Figure 1), to identify regions where anti-VWF alloantibodies bind,10–13 phage display unmasks distinct immunoreactive regions with greater detail for VWF inhibitors from II-1 and II-2. For example, fragment f1 contains the immunoreactive sequences found in A3 and D4 for II-1 and II-2—note the two gaps between the enriched N- and C-termini under A3 and D4 (Figure 1)—and cannot be expected to distinguish these two distinct regions. The diffuse immunoreactivity observed for II-1 and II-2 suggests the potential for broader inhibition of VWF function (e.g., platelet binding) than in patient I-1. However, the low levels of VWF antigen limits our ability to interpret the undetectable VWF ristocetin cofactor activity level in patient II-1 at 30 min postinfusion of VWF concentrate (Table 2). Rapid clearance of replacement VWF is most likely the primary cause of the failure to restore VWF-dependent hemostasis in these patients; infused VWF was completely cleared within 30 min for patients I-125 and II-2 (Table 2) and within 3 h for patient II-1 (Table 2).
FIGURE 2.
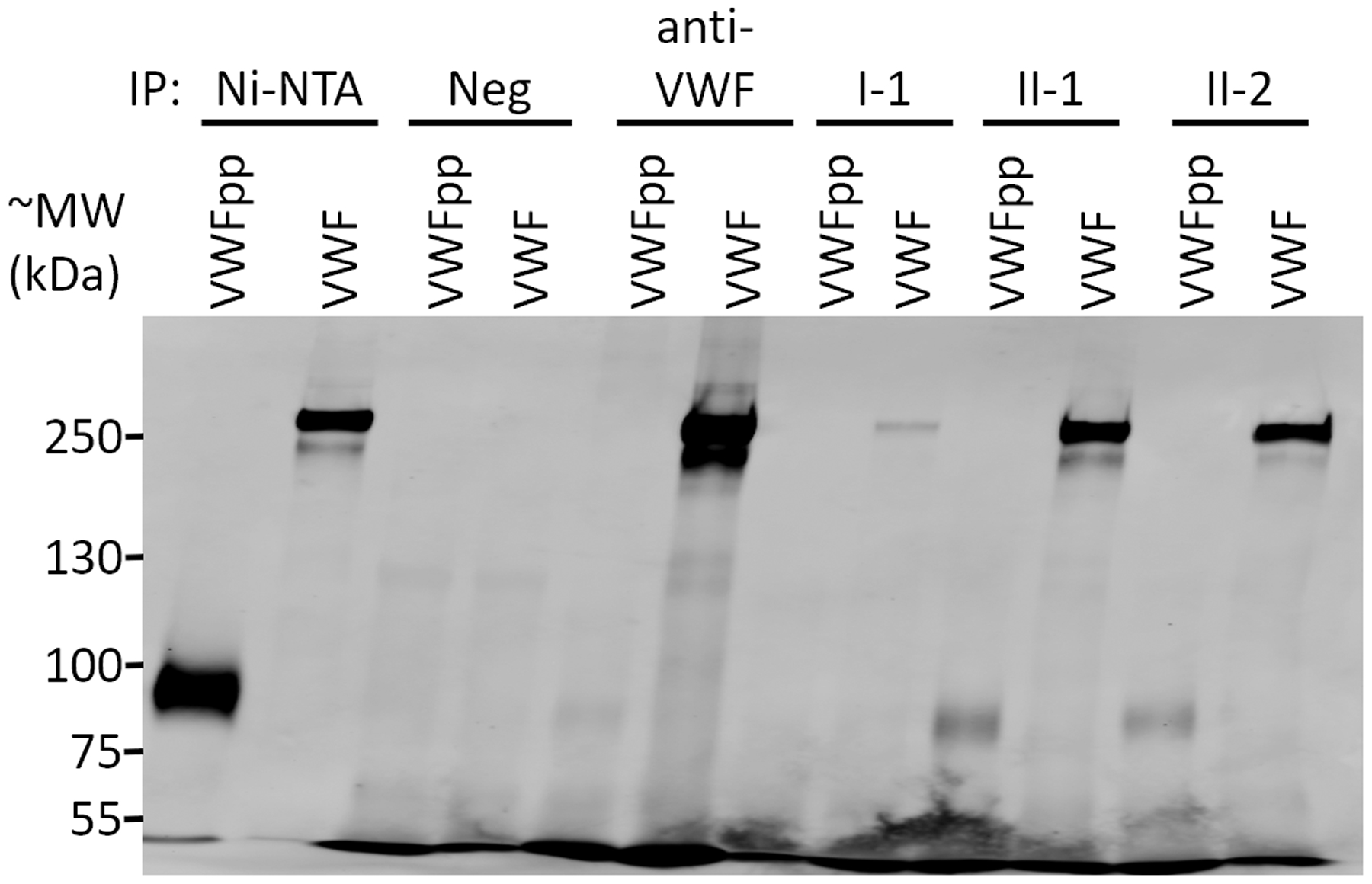
Anti-VWF alloantibodies react with VWFpp. Recombinant VWFpp or multimeric VWF (both with C-terminal myc and His6 tags) were immunoprecipitated (IP) with Ni-NTA beads, naïve protein G beads (Neg), or antibody-coupled protein G beads (anti-VWF , I-1, II-1, and II-2). Immunoprecipitation of VWFpp or VWF was determined by immunoblotting for the myc epitope tag. The expected molecular weight (MW) for VWFpp and VWF are ~90 kDa and ~260 kDa, respectively. Immunoreactivity to VWFpp was confirmed for patients II-1 and II-2. Although immunoreactivity to VWFpp was not detected for anti-VWF using phage display, a very faint band corresponding to VWFpp is noted here, suggesting a potential for a very low level of immunoreactivity to VWFpp for anti-VWF. We also cannot exclude the possibility that immunoreactivity to VWFpp may be different between lots of anti-VWF. Abbreviations: VWF, von Willebrand factor; VWFpp, von Willebrand factor propeptide
TABLE 2.
Clinical laboratory evaluation
| Patient | Test | Preinfusion | 30 min Postinfusion | 3 h Postinfusion |
|---|---|---|---|---|
| II-1 | VWF antigen (%) | 0 | 12 | 0 |
| VWF ristocetin cofactor activity (%) | 0 | 0 | 0 | |
| FVIII (%) | 3 | 59 | 7 | |
| II-2 | VWF antigen (%) | 0 | 0 | 0 |
| VWF ristocetin cofactor activity (%) | 0 | 0 | 0 | |
| FVIII (%) | 3 | 30 | 5 |
Abbreviations: FVIII, factor VIII; VWF, von Willebrand factor.
The immunoreactive profiles of the anti-VWF alloantibodies suggest the possibility for conformationally dependent epitopes. Although the size limit of our phage display library may exclude epitopes formed by distant residues that are greater than ~333 amino acids (aa) apart (VWF length = 2813 aa), several immunoreactive regions were identified to have lengths of contiguous aa sequences that are longer than the sequence of engineered epitope tags (e.g., FLAG) and within the size limits of our phage display approach. For example, the minimal gap width between the N- and C-terminal residues in the A1 region for patients II-1 and II-2 is 194 aa (P1266-D1459) and 187 aa (C1272-C1458), respectively (Figure S5). These minimal gaps are longer than the minimal VWF fragment that can be displayed (~100 aa) and encompass the cysteines responsible for the intramolecular disulfide bond of the A1 domain, which can be recapitulated with phage display as we previously reported.22 An epitope that depends only on the primary or secondary and not the tertiary or quaternary protein structures would be reflected by a narrower gap width. However, because immunoreactive phage clones were selected from a polyclonal mixture and because the sequencing limits precluded the identification of matching N- and C- termini, we cannot exclude the possibility of multiple antibody clones that target different or overlapping epitopes within A1.
Prior reports that used proteolytic VWF fragments to broadly map the immunoreactive regions of VWF inhibitors from a total of six unrelated patients concluded that target specificity is unique to each patient.10–13 We similarly conclude that unrelated patients may have different patterns of immunoreactivity. However, the mechanism(s) for the strikingly similar immunoreactive profiles between patients II-1 and II-2 is unclear. VWF inhibitor development is a complex trait that is unlikely to depend entirely on the VWD-causing variant(s). Not all patients with large genomic deletions in VWF develop VWF inhibitors.29 Furthermore, although patient I-1 and a previously reported patient with the same c.2435del variant in VWF both developed anti-VWF alloantibodies, other type 3 VWD patients with this variant (homozygous and heterozygous) do not have VWF inhibitors.29,30 Although patients with type 3 VWD have a higher risk for developing VWF inhibitors, alloantibodies have been reported in a patient with type 2B VWD (p.R1308C) who was treated with plasma-derived VWF, demonstrating that a complete lack of VWF expression is not the only risk factor.6
Notwithstanding the unknown mechanisms that drive anti-VWF alloantibody formation and epitope selection for the patients in this report, we demonstrate a high content approach to broadly survey their alloantibodies’ anti-VWF immunoreactivity. Future refinement of our approach with other molecular techniques may help delineate minimal epitopes in VWF and identify sequences in VWF that may be engineered to be less immunogenic.
Supplementary Material
Essentials.
Severe von Willebrand disease is treated with infused von Willebrand factor (VWF).
Infused VWF may lead to the development of VWF inhibitors.
Phage display with next generation DNA sequencing comprehensively maps immunoreactive regions.
von Willebrand factor inhibitors may target mature and propeptide sequences of VWF.
ACKNOWLEDGMENTS
We thank the patients for their contributions. NGS was performed by the University of Michigan DNA Sequencing Core. Funding was provided by the National Institutes of Health (HL039693 and HL135793, DG; HL150784 and HL125774, J.A.S.), American Society of Hematology Faculty Scholar Award (A.Y. and J.A.S.), Caroline Wiess Law Fund for Research in Molecular Medicine at Baylor College of Medicine (A.Y.), National Hemophilia Foundation Innovative Investigator Research Award (A.Y.), and Mary R. Gibson Foundation (A.Y.). D.G. is a Howard Hughes Medical Institute Investigator.
Funding information
Baylor College of Medicine, Grant/Award Number: Caroline Wiess Law Fund for Research in Molecular Medicine; Mary R. Gibson Foundation; National Institutes of Health, Grant/Award Number: HL039693, HL135793, HL150784 and HL125774; American Society of Hematology Faculty Scholar Award; National Hemophilia Foundation Innovative Investigator Research Award; Howard Hughes Medical Institute
Footnotes
CONFLICT OF INTEREST
David Ginsburg benefits from patent royalties from Takeda to Boston Children’s Hospital for recombinant VWF. Jordan A. Shavit has consulted for Takeda, Bayer, Sanofi, and HEMA Biologics. The remaining authors have no relevant disclosures.
SUPPORTING INFORMATION
Additional Supporting Information may be found online in the Supporting Information section.
REFERENCES
- 1.Yee A, Gildersleeve RD, Gu S, et al. A von Willebrand factor fragment containing the D’D3 domains is sufficient to stabilize coagulation factor VIII in mice. Blood. 2014;124:445–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yee A, Kretz CA. Von Willebrand factor: form for function. Semin Thromb Hemost. 2014;40:17–27. [DOI] [PubMed] [Google Scholar]
- 3.Johnsen JM, Ginsburg D. VON Willebrand disease. In: Kaushansky K, Prchal JT, Burns LJ, Lichtman MA, Levi M, Linch DC, Williams hematology, 10e. New York, NY: McGraw-Hill Education; 2021. [Google Scholar]
- 4.Connell NT, Flood VH, Brignardello-Petersen R, et al. ASH ISTH NHF WFH 2021 guidelines on the management of von Willebrand disease. Blood Adv. 2021;5:301–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.James PD, Lillicrap D, Mannucci PM. Alloantibodies in von Willebrand disease. Blood. 2013;122:636–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baaij M, van Galen KPM, Urbanus RT, Nigten J, Eikenboom JHC, Schutgens REG. First report of inhibitory von Willebrand factor alloantibodies in type 2B von Willebrand disease. Br J Haematol. 2015;171:424–427. [DOI] [PubMed] [Google Scholar]
- 7.Ehrenforth S, Kreuz W, Scharrer I, et al. Incidence of development of factor VIII and factor IX inhibitors in haemophiliacs. Lancet. 1992;339:594–598. [DOI] [PubMed] [Google Scholar]
- 8.Peyvandi F, Mannucci PM, Garagiola I, et al. A randomized trial of factor VIII and neutralizing antibodies in hemophilia A. N Engl J Med. 2016;374:2054–2064. [DOI] [PubMed] [Google Scholar]
- 9.Springer TA. von Willebrand factor, Jedi knight of the bloodstream. Blood. 2014;124:1412–1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lopez-Fernandez MF, Martin R, Lopez-Berges C, Ramos F, Bosch N, Batlle J. Further specificity characterization of von Willebrand factor inhibitors developed in two patients with severe von Willebrand disease. Blood. 1988;72:116–120. [PubMed] [Google Scholar]
- 11.Shibata M, Shima M, Fujimura Y, et al. Identification of the binding site for an alloantibody to von Willebrand factor which inhibits binding to glycoprotein Ib within the amino-terminal region flanking the A1 domain. Thromb Haemost. 1999;81:793–798. [PubMed] [Google Scholar]
- 12.Tout H, Obert B, Houllier A, et al. Mapping and functional studies of two alloantibodies developed in patients with type 3 von Willebrand disease. Thromb Haemost. 2000;83:274–281. [PubMed] [Google Scholar]
- 13.Mohri H, Yamazaki E, Suzuki Z, Takano T, Yokota S, Okubo T. Autoantibody selectively inhibits binding of von Willebrand factor to glycoprotein ib. Recognition site is located in the A1 loop of von Willebrand factor. Thromb Haemost. 1997;77:760–766. [PubMed] [Google Scholar]
- 14.Smith GP. Phage display: simple evolution in a petri dish (Nobel Lecture). Angew Chem Int Ed Engl. 2019;58:14428–14437. [DOI] [PubMed] [Google Scholar]
- 15.Bahou WF, Ginsburg D, Sikkink R, Litwiller R, Fass DN. A monoclonal antibody to von Willebrand factor (vWF) inhibits factor VIII binding. Localization of its antigenic determinant to a nonadecapeptide at the amino terminus of the mature vWF polypeptide. J Clin Invest. 1989;84:56–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tan FL, Ginsburg D. What a polyclonal antibody sees in von Willebrand factor. Thromb Res. 2008;121(4):519–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuwabara I, Maruyama H, Kamisue S, Shima M, Yoshioka A, Maruyama IN. Mapping of the minimal domain encoding a conformational epitope by lambda phage surface display: factor VIII inhibitor antibodies from haemophilia A patients. J Immunol Methods. 1999;224:89–99. [DOI] [PubMed] [Google Scholar]
- 18.Huang CC, Shen MC, Chen JY, Hung MH, Hsu TC, Lin SW. Epitope mapping of factor VIII inhibitor antibodies of Chinese origin. Br J Haematol. 2001;113:915–924. [DOI] [PubMed] [Google Scholar]
- 19.Van Blarcom T, Rossi A, Foletti D, Sundar P, et al. Precise and efficient antibody epitope determination through library design, yeast display and next-generation sequencing. J Mol Biol. 2015;427(6):1513–1534. [DOI] [PubMed] [Google Scholar]
- 20.Huang W, Soeung V, Boragine DM, et al. High-resolution mapping of human norovirus antigens via genomic phage display library selections and deep sequencing. J Virol. 2020;95(1):e01495–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kretz CA, Dai M, Soylemez O, et al. Massively parallel enzyme kinetics reveals the substrate recognition landscape of the metalloprotease ADAMTS13. Proc Natl Acad Sci USA. 2015;112(30):9328–9333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yee A, Tan FL, Ginsburg D. Functional display of platelet-binding VWF fragments on filamentous bacteriophage. PLoS One. 2013;8:e73518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mohlke KL, Purkayastha AA, Westrick RJ, et al. Mvwf, a dominant modifier of murine von Willebrand factor, results from altered lineage-specific expression of a glycosyltransferase. Cell. 1999;96:111–120. [DOI] [PubMed] [Google Scholar]
- 24.Berntorp E, Agren A, Aledort L, et al. Fifth Aland Island conference on von Willebrand disease. Haemophilia. 2018;24(Suppl 4):5–19. [DOI] [PubMed] [Google Scholar]
- 25.Weyand AC, Flood VH, Shavit JA, Pipe SW. Efficacy of emicizumab in a pediatric patient with type 3 von Willebrand disease and alloantibodies. Blood Adv. 2019;3:2748–2750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: a laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 27.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dako. Polyclonal rabbit anti-human von Willebrand factor code A0082. https://www.agilent.com/cs/library/packageinsert/public/SSA0082IVD-US_01.pdf. Accessed December 29, 2020.
- 29.Mohl A, Boda Z, Jager R, et al. Common large partial VWF gene deletion does not cause alloantibody formation in the Hungarian type 3 von Willebrand disease population. J Thromb Haemost. 2011;9:945–952. [DOI] [PubMed] [Google Scholar]
- 30.Faganel Kotnik B, Strandberg K, Debeljak M, et al. von Willebrand factor alloantibodies in type 3 von Willebrand disease. Blood Coagul Fibrinolysis. 2020;31:77–79. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.