

Abstract
Adoptive immunotherapy has emerged as a powerful approach to cure cancer and chronic infections. Currently, the generation of a massive number of T cells that provide long-lasting immunity is challenged by exhaustion and differentiation-associated senescence, which inevitably arise during in vitro cloning and expansion. To circumvent these problems, several studies have proposed an induced pluripotent stem cell (iPSC)-mediated rejuvenation strategy to revitalize the exhausted/senescent T cell clones. Because iPSC-derived cytotoxic T lymphocytes (iPSC-CTLs) generated via commonly used monolayer systems have unfavorable, innate-like features such as aberrant natural killer (NK) activity and limited replication potential, we modified the redifferentiation culture to generate CD8αβ+CD5+CCR7+CD45RA+CD56−-adaptive iPSC-CTLs. The modified iPSC-CTLs exhibited early memory phenotype, including high replicative capacity and the ability to give rise to potent effector cells. In expansion culture with an optimized cytokine cocktail, iPSC-CTLs proliferated more than 1015-fold in a feeder-free condition. Our redifferentiation and expansion package of early memory iPSC-CTLs could supply memory and effector T cells for both autologous and allogeneic immunotherapies.
Keywords: rejuvenated T cell, iPS cell, T cell differentiation, stemness
Graphical abstract
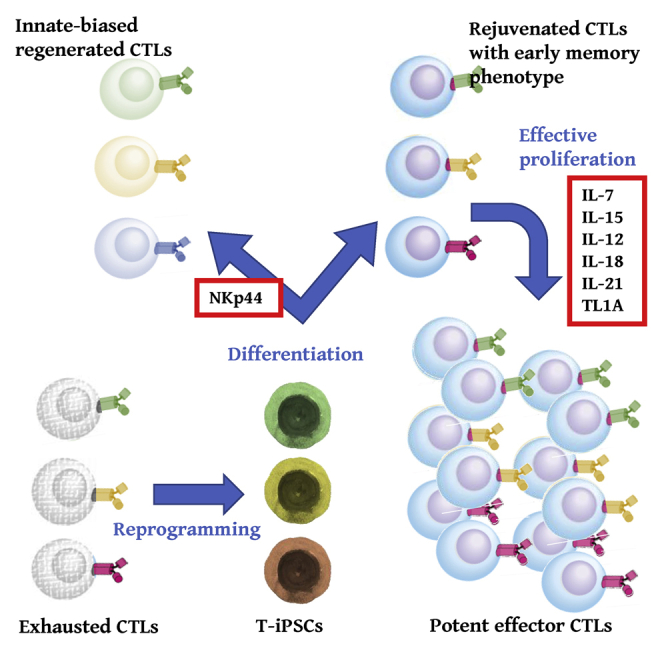
Kawai and Kaneko et al. reported regeneration of antigen-specific cytotoxic T cells with early memory phenotype including high replicative capacity and the ability to give rise to potent effector cells by a newly identified combination of cytokines, which would contribute to improved quality control of regenerated CTLs.
Introduction
Recent advances in immune checkpoint blockade1,2 and adoptive T cell transfer (ACT)3, 4, 5 for the treatment of various cancers and chronic infections have stimulated a new generation of immunotherapies. The first clinical example of ACT expanded the number of tumor-infiltrating lymphocytes (TILs), which are a naturally occurring cell source for tumor-reactive T cells.6 Another source is tumor-redirected T cells, in which patient-derived normal T cells are modified with a tumor-specific T cell receptor (TCR) or chimeric antigen receptor (CAR).7 Both TILs and tumor-redirected T cells have a striking ability to eliminate various cancers, especially melanoma and hematologic malignancies, with a high response rate and durable complete remission.7 At present, however, the preparation of a massive number of T cells retaining effector function as well as high avidity to diseased cells remains a challenge. This difficulty is mainly attributed to the cell exhaustion/senescence phenotype, which inevitably arises during the in vitro cell-expansion procedure and hampers not only the cell expansion but also the therapeutic efficacy.8 The differentiation state of infused T cells is another critical factor for the therapeutic efficacy of ACT. Mature T cells change their phenotype as they experience antigenic stimulation and differentiate from naive cells (TN; CCR7+CD45RA+) to memory stem cells (TSCM; CCR7+CD45RA+CD95+), central memory cells (TCM; CCR7+CD45RA−), effector memory cells (TEM; CCR7−CD45RA−), and effector cells (TEMRA; CCR7−CD45RA+). As differentiation progresses, T cells strengthen their effector function, such as cytotoxic activity and interferon (IFN)-γ production, and preferentially localize to non-lymphoid peripheral tissues to mount a quick immune response.9 Simultaneously, however, they lose their replication/survival potential as well as their systemic circulation capacity, leading to diminished immune-surveillance capability. Indeed, less-differentiated T cells such as TN and early memory T cells (TSCM and TCM) were shown to be superior cell sources for ACT compared with more-differentiated late memory (TEM) and TEMRA cells.10, 11, 12 Collectively, there is demand for a methodology that prepares target-reactive T cells while preserving stemness.
To meet this demand, several studies have proposed an induced pluripotent stem cell (iPSC)-mediated rejuvenation strategy, in which iPSCs of infinite proliferation capacity are established from T cell clones of finite proliferation capacity and then re-differentiated into functional T cells.13, 14, 15, 16, 17 iPSC-derived cytotoxic T lymphocytes (iPSC-CTLs) were the first T cell type generated using this strategy. These cells cannot only directly kill target cells efficiently and specifically but also provide long-lasting protection by generating memory cells. Moreover, iPSC-CTLs maintain the rearranged TCR sequence and antigen specificity of the parental CTLs.
Theoretically, reprogramming to iPSCs enables the generation of the full spectrum of CTL subsets including least-differentiated naive CTLs. However, to date, the most immature phenotype acquired by iPSC-CTLs in commonly used and clinically applicable OP9-DL1 co-culture is CCR7lo/−CD45RA− TEM-like cells.13,17 In line with the prematurely differentiated phenotype, the proliferative capacity of conventional iPSC-CTLs is considerably limited compared to fresh primary CTLs. Furthermore, conventional iPSC-CTLs exhibit the gene-expression profile of innate-like T cells such as γδ T cells,15 natural killer T cell (NKT),18 mucosa-associated invariant T cells (MAIT),19 or group 1 innate lymphoid cells (ILC1).20 They also exhibit the CD8αβlo/−CD5lo/−CD56+ phenotype and NK activity, which might aberrantly kill recipient normal cells and/or induce graft-versus-host disease (GVHD), especially in allogeneic transplantation settings.16,18,20 Moreover, the unstable expression of CD8β poses another problem, as the formation of the CD8αβ heterodimer is critical for co-receptor function and appropriate affinity between the peptide-human leukocyte antigen (HLA) complex and TCR.21 These properties contribute to the suboptimal antigen recognition of conventional iPSC-CTLs compared with parental CTLs.16,17
In the present study, we sought to correct these problems by re-differentiating iPSC-CTLs into a minimally differentiated early memory T cell phenotype that has high proliferative capacity and differentiation capacity to give rise to potent effector cells. The resulting iPSC-CTLs exhibited superior effector function both in vitro and in vivo.
Results
iPSC-CTLs acquired a CD8αβ+CD5+CCR7+CD45RA+CD56−-adaptive and naive-associated phenotype upon coordinated stimulation of TCR, Notch, and cytokine signals in the CD4/CD8-double-positive (DP)-to-CD8-single-positive (CD8SP) step
The T cell-derived iPSCs (T-iPSCs) used in this study came from two previously reported clones, TKT3V1-7 and H254SeV3,13 plus one new clone, H2531SeV3, generated from HIV group-specific antigen (gag) antigen-specific CD8 T cells. In the optimization process to generate minimally differentiated adaptive CD8αβ iPSC-CTLs, we focused on markers associated with the adaptive/innate and naive/memory axes including CCR7 (naive marker), CD5 (adaptive marker), and CD56 (innate marker) together with CD8αβ (2ST8.5H7 monoclonal antibody recognizes an epitope formed by the combination of CD8α and CD8β). The CD4/CD8-DP-to-CD8SP step was optimized by the combinational supplement of soluble anti-CD3ε antibody, plate-coated RetroNectin, and cytokines to preferentially generate CD8αβ iPSC-CTLs, which express more CCR7 and CD5 but less CD56 than conventional CD8αα cells (Figures 1A and S1). In detail, we found plate-coated RetroNectin enhanced cell viability overall, but the supportive effect was especially strong for CD8αβ+CD5+ cells, resulting in an increased proportion of CD8αβ+CD5+ cells (Figures 1B and S2). CCR7 expression was also maintained well on RetroNectin -stimulated CD8αβ+ iPSC-CTLs (Figures 1A and S1), as reported for primary T cells.22 In addition, the more and longer supplement of soluble anti-CD3ε antibody enhanced the yield of CD8αβ T cells compared to the starter DP cell number (maturation efficacy) with negligible effect on the ratio of CD8αβ-, CD5-, or CCR7-expressing cells (Figure S3). We found interleukin (IL)-15 increased the maturation efficacy of CD8αβ iPSC-CTLs (Figure 1C, top) but greatly dampened the expansion efficacy (Figure 1C, bottom). In parallel with the poor proliferative capacity, IL-15 lowered the expression of naive-associated molecules such as CD27 and CD28, but it upregulated the expression of NK-related molecules such as CD56 and NKp44 (Figures 1D and S4). These observations suggest that adding IL-15 preferentially induced the generation of more-differentiated innate CTLs over less-differentiated adaptive CTLs. IL-2 also dampened the expansion efficacy (Figure 1C, bottom) and decreased the expression of CD28 (Figure S4), albeit to a lesser degree compared with IL-15. IL-21 is also a common γ-chain cytokine but is secreted by helper T (Th) cells.23 In contrast to IL-15 and IL-2, IL-21 supplementation significantly improved both the maturation and expansion efficacies (Figure 1C). In addition, IL-21 augmented the expression of CD28, CCR7, and CD62L without inducing NK-related molecules (Figures 1D and S4). The expression levels of CD27 and CCR7, but not of CD28 or CD62L, on iPSC-CTLs reached those on primary CTLs when IL-21 was supplemented. Of note, the combination of IL-21 and Notch ligand impaired the yield of CD8αβ iPSC-CTLs for unknown reasons (Figure S5). Therefore, we separated the maturation step into two stages: a 2-day early stage with CD3 antibody + OP9-DL1 and a 5-day later stage with IL-21 + RetroNectin (Figure 1E). Consistent with the physiological positive selection of primary CTLs, iPSC-CTLs changed their CD45 type from CD45RO+CD45RA− at the DP stage to CD45RO−CD45RA+ at the CD8SP stage (Figure 1F). The downregulation of CD1a and upregulation of CD2724 and CCR7 were also observed (Figure 1F).
Figure 1.

DP-to-CD8SP maturation was optimized to guide iPSC-CTLs from innate to adaptive CD8αβ+CCR7+CD45RA+ cells
(A) Flow cytometric profile of CD8αβ and CD8αα iPSC-CTLs after modified maturation culture as described in Materials and methods. (B) Frequency of CD8αβ+CD5+ iPSC-CTLs after DP-to-CD8SP transition with or without RetroNectin coating in the presence of 1 μg/mL CD3ε antibody and 10 ng/ml IL-7 but not IL-21 (n = 3; mean ± SEM; ∗p < 0.05). (C and D) The effect of cytokine supplements on the maturation and subsequent expansion efficacy (C) and surface phenotype (D). (C) The CD8SP cell yield against the starting DP cell number was calculated as the maturation efficacy. The same CD8SP cells were subsequently expanded side by side in a CD3/CD28-based PBMC feeder-free condition, and the expansion efficacy was calculated at day 14 (top, n = 6; bottom, n = 10; mean ± SEM; one-way ANOVA comparing mean log10 of all groups with Tukey’s multiple comparisons test; ∗p < 0.05, ∗∗p < 0.005, ∗∗∗∗p < 0.0001). (D) The expression of innate-, adaptive-, and naive-associated markers in healthy donor-derived primary naive CTLs and iPSC-CTLs matured in the presence of the indicated cytokines at the DP-to-CD8SP transition step. (E) Schematic depiction of the modified maturation and expansion culture. (F) Flow cytometric profile of pre- and post-DP-to-CD8SP maturation iPSC-CTLs. Data are representative of three (A, B, D, and F) or more than six (C) independent experiments using TKT3V1-7-, H254SeV3-, and H2531SeV3-derived iPSC-CTLs.
NKp44− negative sorting can remove residual aberrant NK activity in the CD8αβ+ fraction and enrich adaptive iPSC-CTLs with an early memory phenotype
Although adaptive CD8αβ T cells become the dominant final product instead of innate-like CD8αα cells as a result of culture optimization, NK activity was still observed in the CD8αβ fraction with considerable inter-iPSC-clone and experimental-lot variation (Figure 2A). This observation prompted us to search for specific markers to monitor ectopic NK activity. We found that expression of the NK cell-activating receptor NKp4425 in iPSC-CTLs was highly correlated with NK activity (Figures 2A−2C). Of importance, the removal of NKp44+ cells neutralized the NK activity but kept the antigen-specific cytotoxic activity intact (Figure 2D), demonstrating that NKp44 expression could distinguish the two distinct cytotoxic activities and was effective for the quality control of aberrant NK activity. Furthermore, the initial NKp44 expression level influenced the durability of NKp44 negative sorting over subsequent cell expansions: NKp44lo iPSC-CTLs maintained the NKp44 null phenotype better than NKp44hi cells (Figure S6), again demonstrating the importance of checking NKp44 expression immediately after maturation. Although NKp44 may not be directly involved in NK activity, since the addition of NKp44 blocking antibody did not block NK activity (Figure S7), we performed NKp44 negative sorting together with CD8αβ and CD5 positive sorting for all of the following iPSC-CTLs to minimize batch variation.
Figure 2.

NKp44 negative sorting removed aberrant NK activity of CD8αβ iPSC-CTLs and enriched early memory phenotype
(A−C) NK activity (A) and NKp44 expression (B) of iPSC-CTLs re-differentiated from independent iPSC clones and healthy donor-derived primary CTLs. 51Cr release assay against peptide-unpulsed K562-HLA-A24 cells at E/T = 10 (A) and flow cytometry (B) was performed after one PHA-PBMC-feeder expansion. (C) The relationship between the NK activity (A) and NKp44+ ratio (B) of different iPSC-CTLs (n = 8). r is Pearson’s correlation coefficient. Representative of two independent experiments using TKT3V1-7 (#1, #2)-, H254SeV3 (#3, #4, #6)-, and H2531SeV3 (#5, #7, #8)-derived iPSC-CTLs. (D) Antigen-dependent and -independent cytotoxic activities of sorted NKp44+ and NKp44– iPSC-CTLs. NK-strong iPSC-CTLs (H2531SeV3) were expanded once with the PHA-PBMC feeder for 2 weeks, sorted for NKp44 expression, and underwent the 51Cr release assay against peptide-pulsed or -unpulsed K562-HLA-A24 cells. (E−H) RNA sequencing (E and F) and cytokine production (G and H) of iPSC-CTLs and the indicated subsets of healthy donor-derived primary CTLs. (E) The gene expressions were analyzed by RNA sequencing, and the correlation analysis was performed based on the 776 T cell-associated genes indicated in Table S1 (gene list from Muranski et al.26). (F) Messenger RNA expression levels (reads Per Kilobase of exon per Million mapped reads [RPKM]) of TCF7, LEF1, TAF4B, EOMES, TBX21, PRDM1, and GZMA in the indicated populations (iPSC-CTLs, n = 2; primary CTL subsets, n = 3 each; mean ± SEM). TKT3V1-7- and H254SeV3-derived iPSC-CTLs were used in (E) and (F). (G and H) Intracellular IL-2 and IFN-γ were analyzed with flow cytometry after 4 h of stimulation with 50 ng/mL PMA and 1 μg/mL ionomycin. Shown in (H) is the mean percent of the indicated IL-2- and/or IFN-γ-producing cells in three independent experiments using TKT3V1-7- and H254SeV3-derived iPSC-CTLs. (Data from one experiment of TKT3V1-7-derived iPSC-CTLs are shown in G. Data from other experiments are shown in Figure S12.)
Next, we characterized the surface phenotype of iPSC-CTLs in comparison with primary CTL subsets. We found iPSC-CTLs expressed memory markers such as CD95, CD218a (IL-18Rα), and CXCR3 (Figure S8) in addition to naive-associated markers, as mentioned above. Furthermore, we addressed the gene-expression profile of iPSC-CTLs using RNA sequencing. Cluster analysis based on the global gene-expression demonstrated that iPSC-CTLs are clearly distinguished from fully differentiated cytotoxic effector cells such as NK cells and TEMRA (Figure S9). NK-associated markers such as NKG2D, NKG2E, and NKG2F, whose expressions are observed in primary CTLs, were also expressed in iPSC-CTLs, but more NK-specific markers such as KIR, DAP12, and Fc-gamma receptor IIIb (CD16) were not expressed (Figure S10). To focus on the T cell memory profile, we performed a cluster analysis based on a reported dataset that includes 776 selected genes representing the molecular signatures of early memory cells and late memory T cells.26,27 We found that iPSC-CTLs were first clustered with TN and TSCM, then with TCM, and had low similarity with more differentiated TEM and TEMRA (Figures 2E and S11). More specifically, iPSC-CTLs expressed early memory genes such as TCF7, LEF1, and TRAF4B, whereas barely expressing late memory genes such as EOMES, TBX21, PRDM1, and GZMA (Figure 2F). The expression balance of early and late memory genes was similar to those of early memory T cells such as TSCM and TCM.
Functionally, we addressed the cytokine profile of iPSC-CTLs, since the cytokine profile of T cells changes depending on the differentiation stage: cytokine IL-2 production decreases, whereas effector cytokine IFN-γ increases as differentiation progresses.28 Modified iPSC-CTLs preferentially produced IL-2 rather than IFN-γ in response to polyclonal phorbol 12-myristate 13-acetate (PMA)/ionomycin stimulation (Figures 2G, 2H, and S12), and the percentages of IL-2 SP, IL-2/IFN-γ DP, and IFN-γ SP in iPSC-CTLs were equivalent to those of TCM (Figure 2H).
In summary, modified iPSC-CTLs have early an memory phenotype according to gene/protein expressions and cytokine profiles.
Modified iPSC-CTLs exhibit differentiation-related phenotypic changes like primary CTLs with vigorous proliferation
Early memory T cells are functionally characterized by high proliferative/survival potential and the capacity to generate late memory and effector cells. We examined the proliferative capacity of iPSC-CTLs in phytohemagglutinin (PHA)-based peripheral blood mononuclear cell (PBMC)-feeder expansion culture supplemented with IL-7 and IL-15. We found that iPSC-CTLs could be expanded over 1,000-fold consistently with one stimulation and estimated they expand over 1030-fold with 10 repeated stimulations (Figure 3A). In this condition, the growth curve followed that of primary naive CTLs but not the parental CTL clone, which committed apoptosis and endured just 3 or 4 repeated stimulations (Figure 3A). Repeatedly expanded iPSC-CTLs kept expressing CD8αβ stably, thus maintaining a capacity to bind specifically the peptide-HLA tetramer like the parental CTL clone (Figure 3B). On the other hand, conventional iPSC-CTLs expressed CD8αα instead of CD8αβ13,14 and exhibited much less binding to the peptide-HLA tetramer despite the equivalent expression of CD3 (Figure 3B). CD5 expression was also high in iPSC-CTLs over repeated expansions (Figure S13). Regarding other surface markers, iPSC-CTLs in expansion culture recapitulated the phenotypic change seen with primary CTL differentiation; they lost the expression of naive-associated markers such as CD28, CD27, CD62L, and CCR7 with multiple expansions and acquired CD45RO with a loss of CD45RA expression (Figure 3C). On the other hand, the expression of exhaustion and senescence markers such as programmed death-1 (PD-1), Lymphocyte activation gene 3 (LAG-3), cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), and CD5729 was not induced even after 10 repeated stimulations in this condition (Figures 3C and S14). CTLs change not only their surface phenotype but also their effector function as they differentiate into effector cells.28 We found the same was true for iPSC-CTLs in our expansion culture based on granzyme B expression (Figures 3D and S15), and cytotoxic activity increased up to the 4th∼6th expansion but decreased afterward (Figures 3E and S15). On the other hand, cytokine production, especially that of IL-2, decreased consistently from the early phase of multiple expansions, indicating the differentiation to TEM and TEMRA (Figures 3E and S15). Together, these results indicate that iPSC-CTLs are highly proliferative and have the potential to differentiate into fully functional TEMRA.
Figure 3.

Modified iPSC-CTLs exhibit high proliferation capacity while recapitulating the phenotypic changes of primary CTLs
(A−C) Growth curve (A) and flow cytometric profile (B and C) of iPSC-CTLs, the parental T cell clone, and healthy donor-derived primary naive CTLs in PHA-PBMC-feeder expansion culture. (A) The indicated cells were expanded and enumerated repeatedly at 2-week intervals in the presence of 5 ng/mL IL-7, 5 ng/mL IL-15, and other supplements described in Materials and methods. (B and C) The binding of the cognate peptide-HLA complex and expression of CD8 subunits, TCR (B), and T cell differentiation-related markers (C) after 4 (B) or 1 or 10 (C) rounds of expansion. (D and E) Intracellular granzyme B expression (D) and IL-2 and IFN-γ production and cytotoxic activity (E) of iPSC-CTLs after the indicated rounds of expansion. The frozen stocks of iPSC-CTLs were made at different times, but they were thawed, rested, and expanded once side by side in advance of the flow cytometry or the 51Cr release killing assay against 1 μM peptide-pulsed K562-A24 at an E/T = 1 (IL-2, IFN-γ) and 10 (cytotoxicity) (mean ± SD). (A−C) Data are representative of two independent experiments using TKT3V1-7- and H254SeV3-derived iPSC-CTLs, whereas data of (D) and (E) are the result of a single experiment using TKT3V1-7-derived iPSC-CTLs. (The results of H254SeV3- and H2531SeV3-derived iPSC-CTLs are shown in Figure S15.)
Supplementing IL-21, IL-12, IL-18, and TNF-like ligand 1A (TL1A) established a highly efficient feeder-free expansion culture with improved effector function
For the wide application of iPSC-CTLs in adoptive immunotherapy, feeder-free cell expansion is desirable especially in allogeneic transplantation settings. As previously reported, conventional feeder-free stimulation using CD3/CD28 microbeads and IL-2 cannot only transduce CAR and TCR genes but also expands primary T cells efficiently by a single stimulation. However, when CTLs are separated from other cells in PBMC culture, such as Th and myeloid cells, their expansion efficacy decreases dramatically after secondary and tertiary CD3/CD28 microbeads plus IL-2 stimulation. These observations suggest that improvement of the feeder-free expansion culture is needed to produce iPSC-CTLs massively and consistently with minimal batch effect. Therefore, we explored additional supplemental cytokines suitable for iPSC-CTL expansion without compromising effector function. As previously reported in primary CTLs,30, 31, 32, 33 IL-21 enhanced the expansion efficacy of iPSC-CTLs additively with IL-7 and IL-15 (Figure 4A). In addition, we found the combination of IL-12 and IL-18 potently induced IFN-γ production in iPSC-CTLs in a TCR-independent manner (data not shown). Although the expression of the IL-12 receptor was not very high in iPSC-CTLs, IL-12 enhanced IL-18 receptor expression on iPSC-CTLs (Figure S16) as reported on primary T cells.34 In fact, these two cytokines dramatically enhanced the TCR-dependent proliferation in an additive manner with IL-7, IL-15, and IL-21 (Figure 4A) but had negligible effect on TCR-independent proliferation (data not shown). We confirmed that IL-12/IL-18 enhanced the secondary expansion efficacy as well (Figure 4B), suggesting that these two cytokines enhanced the proliferation capacity rather than only enhanced the initial proliferation transiently. Since the tumor necrosis factor (TNF) family cytokine TL1A is known to induce IFN-γ production in a synergistic manner with IL-12 and IL-18,35 we measured the effect of TL1A and found that it enhanced the expansion efficacy in an additive manner with IL-21, IL-12, and IL-18 (Figures 4C and 4D). In accordance with the positive effect on proliferation, we found that none of IL-12, IL-18, or TL1A upregulated Eomesodermin (EOMES) or T-box expressed in T cells (T-bet) (Figure S17), although a previous report demonstrated that the IL-12/IL-18 supplement induced T-bet expression within a few days,36 an effect that promotes terminal differentiation.37 Therefore, we hypothesized that transiently supplementing the indicated cytokines only at the beginning of the stimulation is sufficient to enhanced proliferation without inducing T-bet expression. On the other hand, the production and accumulation of granzyme B were induced by these cytokines, especially IL-21, as previously reported in primary CTLs38,39 (Figure S17, bottom). Regarding effector function, IL-21, IL-12, IL-18, and TL1A exhibited little if any effect on cytotoxic activity in vitro, but they had an additive effect on effector cytokine production in iPSC-CTLs (Figure S18). Importantly, exhausted parental CTL clones also exhibited enhanced proliferation with sustained effector function when treated with IL-21, IL-12, IL-18, and TL1A (Figure S19). Furthermore, the cytokine cocktail partially enhanced the expansion efficacy of primary CTLs, especially that of purified CTLs separated from other cells in PBMC culture such as myeloid and Th cells (Figure 4E), demonstrating the cytokine cocktail is generally effective for the isolated expansion of CTLs. Altogether, supplementing the culture with the above cytokines markedly improved the expansion efficacy without compromising the effector function of CTLs. Furthermore, the estimated fold expansion was 1015 with 6 repeated stimulations, suggesting 300-fold∼3,000-fold per stimulation but with the highest fold change coming on the first expansion with PBMC-feeder cells (Figure 4F). We confirmed the expressions of CD57, killer cell lectin like receptor G1 (KLRG-1), PD-1, and T-cell immunoglobulin and mucin domain 3 (Tim-3) were hardly induced throughout the repeated expansions (Figure 4G), demonstrating that our modified feeder-free system could support iPSC-CTL expansion without excessive differentiation or exhaustion due to the initial early memory phenotype of the cells. Importantly, our protocol did not achieve multiple-round expansions of the parental T cell clone, presumably due to the permanent loss of the cells’ proliferation capacity. Overall, our findings demonstrate the effectiveness of iPSC-mediated rejuvenation to acquire a stemness phenotype, thus improving the proliferative capacity of the T cell clone dramatically.
Figure 4.

A highly efficient feeder-free expansion culture was developed with the supplementation of IL-21, IL-12, IL-18, and TL1A
(A−D) Fold expansion of iPSC-CTLs after 2 weeks of PBMC-feeder-free expansion supplemented with the indicated cytokines. Independent lots of iPSC-CTLs were once expanded in PHA-PBMC-feeder condition, then stimulated with immobilized anti-CD3 antibody in the presence of the indicated cytokines, and enumerated 2 weeks later to calculate the fold expansion (A, n = 8; C, n = 15; mean ± SEM; one-way ANOVA comparing mean log10 of all groups with Tukey’s multiple comparisons test (A and C); ∗p < 0.05, ∗∗p < 0.005, ∗∗∗∗p < 0.0005). (B and D) Fold expansion in sequential feeder-free stimulation with (+) or without (−) IL-12/IL-18 in addition to IL-7/IL-15/IL-21 (B) or with or without TL1A in addition to IL-7/IL-15/IL-21/IL-12/IL-18 (D). (E) Growth curve of purified or unpurified CTLs in healthy donor-derived PBMC in feeder-free expansion culture. Cells were stimulated first by CD3/CD28 microbeads and biweekly afterward by 1 μg/mL immobilized anti-CD3 antibody in the presence of IL-2 alone or IL-7, IL-15, IL-21, IL-12, IL-18, and TL1A. Plots show 4 (left) and 2 (right) independent experiments. (F) Growth curve of iPSC-CTLs, the parental T cell clone, and healthy donor-derived primary naive CTLs in feeder-free expansion culture supplemented with IL-7, IL-15, IL-21, IL-12, IL-18, and TL1A. iPSC-CTLs and primary naive CTLs experienced one-time PHA-PBMC-feeder expansion followed by repeated feeder-free expansions. The T cell clone experienced two feeder-free expansions after four repeated PBMC-feeder expansions (not shown). (G) The expression of exhaustion- and senescence-associated markers in eight repeated feeder-free expanded iPSC-CTLs. (F and G) Data are representative of two independent experiments using TKT3V1-7- and H254SeV3-derived iPSC-CTLs, whereas data of (A)−(D) are representative of more than eight independent experiments using TKT3V1-7-, H254SeV3-, and H2531SeV3-derived iPSC-CTLs.
Modified CD8αβ iPSC-CTLs can exert superior anti-viral and anti-tumor activity with superior proliferative capacity than the parental T cell clone
After 4 repeated stimulations, when iPSC-CTLs were expanded 1010-fold and estimated to have balanced effector function with maturity, we evaluated their effector function. At this time point, iPSC-CTLs or parental CTLs hardly expressed PD-1, KLRG-1, or CD57 (Figure S20). Based on the chromium release killing assay (51Cr release assay), we found similar antigen-specific cytotoxic activity against HIV peptide-pulsed target cells at all observed peptide doses and effector/target (E/T) ratios between iPSC-CTLs and the parental clone (Figures 5A and 5C, left). Neither iPSC-CTLs nor the parental clone exhibited TCR-independent cytotoxicity against NK-susceptible K562 cells (Figure 5A). Comparable TCR-dependent and -independent cytotoxicities were also observed against the CD4 T cell line GXR (Figure S21). Furthermore, we examined if these iPSC-CTLs have effector function to control pathogenic cells that present the target peptide after endogenous processing of a whole protein. When iPSC-CTLs were co-cultured with one of two HIV-1-infected human CD4+ T cell lines, GXR and ACH-2 (see Materials and methods), both of which were transduced with HLA-A∗24:02, showed strong HLA-dependent cytotoxicity against target cells on top of the baseline cell death caused by the cytopathic effect of HIV (Figure 5D). Regarding cytokine production, iPSC-CTLs were superior in IL-2 production but inferior in IFN-γ production compared with the parental clone (Figures 5B and 5C, middle and right), presumably reflecting their earlier memory phenotype. In line with this observation, the proliferation response to IL-15 without TCR stimulation, another feature of early memory T cells,11 was superior in iPSC-CTLs than in the parental T cell clone (Figure 5E). Next, to address antigen-dependent expansion and continuous cytotoxic activity in a clinical setting, we cocultured effector cells with target cells, HIV negative factor (nef) gene-expressing K562/HLA-A∗24:02, at an E/T = 0.1 for a prolonged period (2 weeks), and monitored their cytotoxic capacity (Figure 5F). When co-cultured with the outnumbering target cells that constitutively expressed the cognate peptide, iPSC-CTLs showed vigorous proliferation and complete target cell eradication. On the other hand, the parental T cell clone could not proliferate and was overwhelmed by the target cells. Despite the strong and direct cytotoxic activity observed in the 51Cr release assay, the parental T cell clone easily committed activation-induced apoptosis after killing only a few target cells in contrast to iPSC-CTLs, which exhibited frequent serial killing (Video S1). These observations could explain why the parental T cell clone disappeared eventually. Lastly, we addressed the in vivo property of iPSC-CTLs and the parental clone. We found iPSC-CTLs persisted in vivo for a longer period than the parental clone in non-obese diabetic (NOD)-severe combined immunodeficiency (SCID) IL-2Rγcnull (NSG) mice under a weekly supplement of 100 ng human IL-15 (hIL-15) (Figures 5G and S22). It is reported that the survival and proliferative capacity of T cells are critical for the cells’ in vivo functions.40 Therefore, we examined the in vivo cytotoxicity of iPSC-CTLs in a murine xenograft model, in which target cells were inoculated subcutaneously, and 4 and 7 days later, the iPSC-CTLs or the parental T cell clone were infused intravenously. For these experiments, we used HIV Nef antigen-transduced K562/HLA-A24 cells as the target cells to evaluate iPSC-CTL-mediated cytotoxicity for a long period separately from the virus-induced cytopathic effect on the target cells. We found that the efficacy of tumor growth inhibition and survival was significantly higher for iPSC-CTLs (Figures 5H, 5I, and S23). Altogether, these findings suggest that modified CD8αβ iPSC-CTLs are more therapeutically effective with equivalent cytotoxicity but superior proliferative capacity and persistency in vivo than the parental T cell clone.
Figure 5.

Modified CD8αβ iPSC-CTLs exert superior anti-viral and anti-tumor activity with superior proliferative capacity than parental T cell clone
Cytotoxicity and cytokine production against K562/A24 cells, cytotoxicity (A−C) against HIV-1-infected human CD4 T cell lines (D), in vitro proliferation (E), long-term coculture killing (F), in vivo persistence (G), and in vivo killing assay (H and I) of iPSC-CTLs and parental T cell clones. All T cells were thawed and expanded for 2 weeks once in feeder-free condition side by side before the functional assays. (A−C) 51Cr release assay (A) and cytometric bead array (CBA) cytokine assay for IL-2 and IFN-γ production (B) were performed after coculture for 5 h with K562-A24 cells pulsed with the indicated dose of peptide at an E/T = 1 (B), 9 (A, left), or as indicated (A, right). Shown in (C) is a summary of independent experiments when iPSC-CTLs or T cell clones were cocultured with 1 μM (IL-2 and cytotoxicity) or 10 nM (IFN-γ) peptide-pulsed K562-A24 cells (cytotoxicity, n = 5; IL-2 and IFN-γ, n = 4 per group; mean ± SEM; ∗p < 0.05; NS, not significant). (D) Cytotoxicity of T cells against HIV-1-infected human CD4 T cell lines based on the number of surviving target cells after co-culture with T cells at the indicated E/T ratio. p24 (ACH-2) or inducible-GFP (GXR) was used to calculate the number of HIV-1-infected cells. The relative percentages of p24-positive or GFP-positive cells in the target cells to that of the control, which was normalized to 100%. (E) Carboxyfluorescein succinimidyl ester (CFSE) dilution of iPSC-CTLs and the parental T cell clone during 1-week culture with 100 pg/mL or 10 ng/mL IL-15 without TCR stimulation. (F) Fold expansion of T cells and K562-A24-N138Rluc in 2-week coculture, calculated using flow cytometry. (G) IVIS images of NOD-SCID IL2Rγcnull (NSG) mice engrafted with 2 × 106 luciferase-transduced iPSC-CTLs or the parental T cell clone intraperitoneally. 100 ng/head recombinant hIL-15 was infused weekly. Representative of two independent experiments using H254SeV3-derived iPSC-CTLs (5 mice per group). (H and I) IVIS images (H) and Kaplan-Meier survival curves (I) of NSG mice with ∼2 cm3 large tumors after subcutaneous inoculation with 2 × 105 cognate-Nef-peptide expressing K562-A24-N138Rluc followed by the injection of PBS, iPSC-CTLs, or the parental clone 4 days and 7 days later (2 × 106 T cells at each time point). Mice were euthanized when the tumor volume exceeded 2 cm3 on either side. At day 4 immediately before the T cell infusion, top and bottom photos are identical but processed with different color scales. Combined results of two independent experiments using H254SeV3-derived iPSC-CTLs are shown (12 mice per group [5 and 7 mice for each experiment]; ∗p < 0.05 by the log-rank [Mantel-Cox] test; tumor volume = [length × width2]/241).
Discussion
In the present study, by optimizing a method that induces the antigen-specific early memory phenotype, we addressed ways to resolve several weaknesses of iPSC-CTLs including poor replication capacity, ectopic NK activity,16,17 and unstable CD8β expression.13,15,42 Conventional iPSC-CTLs have low CD5 expression,13,15 and their phenotype resembles those of ILCs or innate-like T cells.18,20 We sought to correct these unfavorable phenotypes altogether by modifying the developmental pathway of iPSC-CTLs from the innate to the adaptive lineage. In the maturation culture we report here, we added a high dose of anti-CD3ε antibody to generate proliferative CD8αβ CTLs efficiently, which should deliver agonistic signals and induce the maturation of innate-like CTLs.43, 44, 45, 46 To prevent diversion to the innate lineage, we optimized the cytokine combination and found that NK-activating cytokines such as IL-15 and IL-2 should be excluded from the maturation culture, because they profoundly diminished the intrinsic proliferative capacity of iPSC-CTLs in the expansion process. Previous reports demonstrated that IL-15 is indispensable for the maturation of innate-like T cells (NKT cells and a subset of intraepithelial lymphocytes [IELs]) but dispensable for the maturation of adaptive T cells.47 Even in HLA-restricted adaptive subsets, IL-15 preferentially induces the maturation of unconventional T cells, which show a unique memory phenotype and are hypo-responsive to antigenic stimulation.48, 49, 50 These properties resemble the innate and TCR-hyporesponsive phenotypes of conventional iPSC-CTLs when supplemented with IL-15 during the differentiation process. Although IL-2 did not elicit as strong a phenotypic change as IL-15, it also dampened the expansion efficacy of iPSC-CTLs significantly. This negative effect may be attributed to IL-2-induced differentiation,51 downregulation of the IL-2 common γ-chain receptor,52,53 and NK activity in double-negative (DN) iPSC-CTLs, which kills neighboring CD8αβ iPSC-CTLs.16 Unlike IL-2 and IL-15, IL-21 preferentially improved the viability of adaptive T cells over innate cells54 and maintained the naive phenotype of CTLs.39,51,55 Indeed, we found that IL-21 enhanced the expression of naive-associated markers such as CCR7 and CD28 in iPSC-CTLs and improved the efficacy of both the maturation and subsequent expansion cultures. IL-21 is a highly pleiotropic cytokine, but its physiological role in CTL maturation remains unclear, as no abnormalities have been observed in the thymic development of IL-21R knockout (KO) mice.56,57 The functions of IL-21 have been mainly reported on post-maturation CTLs in the periphery, such as the maintenance of antigen responsiveness and prevention of cell exhaustion.30, 31, 32 Thus, the favorable effects of IL-21 in the final maturation step might stem from improved cell maintenance rather than maturation of the desired cell types.
Modified iPSC-CTLs express important naive-associated markers such as CCR7 and CD27 equivalently to primary naive CTLs. Additionally, whereas the expression levels of CD28 and CD62L in iPSC-CTLs are improved, they are still lower than those in primary naive CTLs. This phenotype might be a functional bottleneck for iPSC-CTLs and should be overcome through further research, since a high expression of CD62L is known to be necessary from the early stage of positive selection to the late stage, in which mature SP T cells acquire naive T cell properties. The low expression of CD62L in our iPSC-CTLs may resemble DP cells that fail to pass correct physiological selection.58,59 However, we confirmed that iPSC-derived T cells had functional competence, such as cytokine production and proliferation, in response to agonistic stimulation. These are hallmark features of mature lymphoid cells and clearly contrast with immature DP cells, which commit apoptosis in response to agonistic stimulation, thus mirroring physiological negative selection.
Proliferative capacity and aberrant NK activity can cause batch variation. Regarding proliferative capacity, the expression of CD5 in iPSC-CTLs might be a good indicator, since CD5 is reportedly an adaptive marker and also a positive marker for in vivo persistence and antigen responsiveness in the periphery.60 We frequently observed stable CD5 expression in highly proliferative iPSC-CTLs. Regarding, NK activity, the variation in the quality of iPSC clones rather than experimental lots seems to be more responsible for the batch variation. Some previous reports have overlooked the NK activity of iPSC-CTLs, because NK-resistant target cells such as lymphoblastoid cell lines (LCLs) in the latent phase, which express HLA and NK-activating stress ligands weakly, were used.13,61 We monitored NK activity against NK-sensitive K562 cells in the 51Cr release assay and found that NKp44 was a good marker to specifically track NK activity in iPSC-CTLs. Because NKp44 is not directly involved in NK activity in our experimental settings, it is necessary to identify other NK receptors that are directly involved and molecular pathways that induce NKp44 and the causative NK receptors to truly control aberrant NK activity in iPSC-CTLs.
The stemness phenotype is functionally defined by high self-renewal potential and differentiation potential to memory and effector cells. Although the self-renewal capacity remains to be verified due to technical limitations in xenograft animal models, iPSC-CTLs exhibited remarkably high proliferative capacity consistently in in vitro expansion culture without signs of exhaustion. Also, iPSC-CTLs exhibited high precursor potential to generate potent effector cells over multiple expansions. These features suggested the usefulness of a rejuvenated iPSC-CTL-mediated immunotherapy against fatal chronic infections and cancer.
However, our iPSC-CTLs could not readily or consistently expand in conventional anti-CD3/CD28 bead-based PBMC feeder-free culture due to experimental and clonal variation. To maximize the potential of each iPSC-CTL clone, we supplemented the expansion culture with IL-12, IL-18, and TL1A in addition to IL-7, IL-15, and IL-21. IL-12 and IL-18 are reported to reciprocally upregulate the other’s receptor and deliver potent Th type 1 (Th1) co-stimulatory cytokine signals synergistically.34,62 Although potent Th1 signals reportedly induce differentiation, we found that IL-12/IL-18 potentiated the replication capacity of iPSC-CTLs durably over repeated expansions, which was reminiscent of the sensitization of primary naive CTLs to tonic TCR signals by IL-18/IL-7 treatment.63 Also, we found that TL1A enhanced the expansion efficacy in an additive manner with IL-21, IL-12, and IL-18, as previously reported in primary T cells.64 Although the costimulatory role of TL1A has been mainly reported for IFN-γ production,35,65 we confirmed that enhancement of the expansion efficacy by TL1A was substantial and durable especially for iPSC-CTLs, which otherwise did not expand well. The nuclear factor κB (NF-κB) pathway might be a responsible signaling mediator, since it is activated by death domain receptor 3 (DR3), a cognate receptor for TL1A,65 and is deeply involved in T cell differentiation.66 Altogether, a supplement combining the above cytokines made possible the repeated PBMC feeder-free isolated expansion of functional iPSC-CTLs beyond 1015-fold, a level that approaches the number required for clinical settings. Although the production efficacy of DP and CD8SP cells is still moderate, we suppose this problem could be solved with cell expansion, which could compensate for any inefficient maturation.
Accumulating lines of evidence have demonstrated that early memory T cells such as TSCM and TCM are an ideal cell source for ACT, as they have the optimal combination of naive and memory phenotypes.40,67 Early memory T cells can generate the wide spectrum of memory and effector cells while providing long-lasting immunity with high self-renewal capacity. However, the clinical use of early memory T cells, especially TSCM, has been hampered by their rarity in circulation, but the mass production of early memory iPSC-CTLs could solve this problem. Furthermore, early memory iPSC-CTLs would be powerful not only as infused cells but also as a cell source to generate various effector cells in vitro. Thus, early memory iPSC-CTLs might provide desired T cell types depending on the clinical setting: long-lasting early memory T cells immediately after maturation and highly cytotoxic short-lived effector cells after multiple rounds of expansion.
To conclude, we show evidence that an almost unlimited number of both memory and effector iPSC-CTLs could be generated in principle from one dish of iPSCs. Some reports have generated T cells expressing considerably stable transgenic TCR from primary T cells without mispairing between endogenous and transgenic TCR.68, 69, 70 Although these T cell products too have potential clinical application, an iPSC-based formulation would be superior in facilitating quality control for cell products that underwent genomic manipulations such as tumor-redirecting receptor transduction, HLA modification,71 and cytokine/chemokine signal modification,72 which are technically laborious for a peripheral T cell with current approaches. Thus, iPSC technology could offer a novel platform for effective and long-lasting adoptive immunotherapy.
Materials and methods
iPS cell lines
Human iPSC line TKT3V1-7 was induced from an antigen non-specified CD3 T cell of a healthy volunteer by using retroviral vector harboring OCT3/4, KLF-4, SOX-2, and C-MYC.13 Human iPSC lines H254SeV313 and H2531SeV3 were, respectively, induced from HIV Nef and Gag antigen-specific CD8 T cell clones expanded by multicycle peptide stimulation to peripheral blood CD8 T cells from HIV-infected patients by using Sendai virus vector harboring OCT3/4, KLF-4, SOX-2, and C-MYC.
T cell differentiation from iPSCs
To differentiate human iPSCs to DP cells, we followed a previously reported protocol13 with slight modification to the culture medium in OP9-DL1 coculture, which consisted of alpha Modified Eagle Minimum Essential Medium (αMEM) supplemented with 15% fetal bovine serum (FBS), 1% penicillin-streptomycin-L-glutamine (PSG; Sigma-Aldrich), 50 μg/mL ascorbic acid 2-phosphate (Nacalai, Kyoto, Japan), 1 ng/mL recombinant hIL-7 (PeproTech, Rocky Hill, NJ, USA), and 10 ng/mL human recombinant Fms-like tyrosine kinase 3 (Flt3L) (PeproTech). To differentiate from DP to CD8SP, iPSC-derived DP cells were first cultured for 2 days on OP9-DL1 in the presence of 1 μg/mL anti-CD3ε antibody, 10 ng/mL IL-7, 10 ng/mL Flt3L, 15% FBS, 1% PSG, insulin-transferrin-selenium (ITS), and ascorbic acid 2-phosphate, then the antibody was washed away, and the cells were transferred to a RetroNectin-coated culture dish and cultured for 5 days in the presence of 10 ng/mL IL-7, 10 ng/mL Flt3L, 20 ng/mL IL-21, 15% FBS, 1% PSG, ITS, and ascorbic acid 2-phosphate. After completion of the maturation (CD1a downregulation), CD8αβ+CD5+NKp44− iPSC-CTLs were sorted with a FACSAria II flow cytometer (BD Biosciences, San Jose, CA, USA) and subjected to cell expansion (see below).
T cell expansion
A previously described protocol was used13 with some modification to conduct the PHA-based PBMC-feeder expansion. T cells were mixed with 40 Gy-irradiated PBMCs at a responder/feeder ratio of ∼1/50 in αMEM supplemented with 2 μg/mL PHA, 10 μM pan caspase fmk inhibitor Z-VAD (Z-VAD-fmk; R&D Systems), 5 ng/mL IL-7, 5 ng/mL IL-15 (PeproTech), 15% FBS, 1% PSG, ITS, and ascorbic acid 2-phosphate. The culture was split and replenished with fresh medium every 2−3 days to keep the sub-confluent cell density constant. For feeder-free expansion, T cells were put on an anti-CD3ε antibody-coated plate (coated overnight at 4°C and 1 μg/mL concentration) and cultured in αMEM supplemented with 10 μM Z-VAD-fmk (R&D Systems), 5 ng/mL IL-7, 5 ng/mL IL-15 (PeproTech), 15% FBS, 1% PSG, 1× ITS, 50 μg/mL ascorbic acid 2-phosphate, 20 ng/mL IL-21, 50 ng/mL IL-12, 50 ng/mL IL-18, and 50 ng/mL TL1A where indicated. 16 h later, the T cells were transferred to an antibody-uncoated plate, then split, and replenished with fresh medium like the PHA-PBMC-feeder expansion. Cytokines other than IL-7 and IL-15 were not supplemented in the medium change.
Flow cytometry
Antibodies used in the cytometry and functional assays are as follows. CD3ε (UCHT1), CD3ε (OKT3; for maturation), CD4 (OKT4), CD8α (SK1), CCR7 (G043H7), CD56 (HCD56), NKp44 (P44-8; both for fluorescence-activated cell sorting [FACS] and functional blocking), CD27 (O323), CD28 (CD28.2), CD62L (DREG-56), CD45RA (HI100), CD45RO (UCHL1), CD1a (HI149), NKG2D (1D11), IL-2 (MQ1-17H12), CD95 (DX2), CXCR3 (G025H7), CCR5 (HEK/1/85a), IL-18R (H44), PD-1 (EH12.2H7), Tim-3 (F38-2E2), CD57 (HCD57), T-bet (4B10), KLRG-1 (SA231A2), and granzyme B (GB11) were all purchased from BioLegend (San Diego, CA, USA); CD5 (UCHT2) and EOMES (WD1928) from eBioscience (San Diego, CA, USA); CD8β (2ST8.5H7) from Beckman Coulter (Marseille, France); and IFN-γ (B27) from BD Biosciences. The HLA-A24/Nef134-8 tetramer was kindly provided by Dr. Kawana-Tachikawa (National Institute of Infectious Diseases, Japan). The stained cell samples were analyzed using FACSAria II or LSRFortessa (BD Biosciences), and the data were processed using FlowJo software (Tree Star, Ashland, OR, USA).
RNA sequencing
cDNA was synthesized using a SMARTer Ultra Low Input RNA and sequenced with an Illumina Sequencing-HV kit (Clontech, Mountain View, CA, USA), after which the Illumina library was prepared using a Low Input Library Prep kit (Clontech). The libraries were sequenced using HiSeq 2500 in 93 cycle Single-Read mode. All sequence reads were extracted in FASTQ format using BCL2FASTQ Conversion Software 1.8.4 in the CASAVA 1.8.2 pipeline. The sequence reads were mapped to an hg19 reference genome; downloaded on December 10, 2012, using TopHat v2.0.8b; and quantified using rpkmforgenes. Hierarchical clustering was conducted using the Euclidean distance and the ward method of hclust function in R3.2.1. The data have been deposited in NCBI Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/; NCBI: GSE173377). Gene-expression profiles of CD8β+CD5+NKp44− iPS-T cells were compared to those of NK cells and the indicated subsets of CTLs, which were separated from the PBMCs of healthy donors.
51Cr release assay
NK and antigen-specific cytotoxic activities were measured in the 51Cr release assay as previously described.73 K562 cells expressing HLA-A24 (K562-A24) were pulsed or unpulsed with the cognate Nef134-8 peptide and used as antigen-presenting cells.
In vitro killing assay against HIV-1-infected cells
A GFP reporter CD4+ T cell line, CEM-GXR25 (GXR),74 and HLA-A∗24:02-expressing GXR (A24-GXR) were used as target cells to measure the killing activity of iPSC-CTLs or T cell clones against HIV-1-infected cells. A24-GXR and GXR were infected with HIV-1 NL4-3 for 4 days, when the percentage of HIV-1-infected (GFP+) cells achieved 80%. HIV-1-infected A24-GXR and GXR were co-cultured with or without CTLs labeled with CellTrace Far Red (Thermo Fisher Scientific) for 4 h.
An HIV-1 latent T cell clone, ACH-2,75 and HLA-A∗24:02-expressing ACH-2 (A24-ACH-2) were also used as target cells for the in vitro killing assay. ACH-2 and A24-ACH-2 were treated with 10 ng/mL TNF-α to reactivate latently infected HIV-1. The reactivation of HIV-1 was detected by the intracellular staining of HIV-1 p24 protein using Alexa Fluor 700-conjugated HIV-1 p24-specific Nu24 monoclonal antibody, a kind gift from Dr. Yasuko Tsunetsugu-Yokota.76 The percentage of HIV-1-reactivated (p24+) cells was around 80%, 48 h after TNF-α reactivation. The reactivated A24-ACH-2 and ACH-2 cells were co-cultured with or without the labeled CTLs for 4 h.
The percentage of GFP+ or p24+ cells in the non-labeled cell population was measured by FACSCanto II (BD Biosciences). The relative percentage of GFP+ or p24+ cells to that of target cells without CTLs is shown in the figures.
In vivo killing assay
5- to 7-week-old female NSG mice (Oriental Bioservice, Japan) were subcutaneously inoculated at two sides of the dorsal skin with 2 × 105 K562-HLA-A24 cells expressing N138 (cognate peptide)-renilla luciferase fusion protein (K562-A24-N138Rluc). 4 and 7 days later, PBS, 2 × 106 iPSC-CTLs, or 2 × 106 cells from the parental clone were injected intravenously into randomly grouped mice. Tumor burden was measured by a caliper (tumor volume = [length × width2]/241) and in vivo bioluminescence imaging (IVIS 100 Imaging System) until the tumor volume exceeded 2 cm3 on either side, after which the mice were euthanized. All in vivo experiments were approved by the Kyoto University Animal Care Committee.
Statistics
All data are presented as mean ± SD unless otherwise indicated. All statistics were performed using Excel (Microsoft) and Prism (GraphPad software). The two-tailed Student’s t test was applied to compare two groups, and one-way ANOVA with Tukey’s multiple comparisons test was applied to compare more than three groups. Values of p < 0.05 were considered significant.
Acknowledgments
We thank Mr. Daisuke Seki (Kyoto Univ.) for technical assistance with the animal experiments; Dr. Yuta Mishima, Mr. Kohei Ohara, and Katsura Noda (Kyoto Univ.) for technical assistance; and Dr. Peter Karagiannis (Kyoto Univ.) for editing the manuscript. The entire study was conducted in accordance with the Declaration of Helsinki and permitted by the Institutional Ethical Board of Kyoto University. This work was partially supported by a JSPS Grant-in-Aid (18K15278, 25293226, and 23591413) from the Ministry of Education, Culture, Sports, Science and Technology (JST-MEXT), Japan; Research Program on Emerging and Re-emerging Infectious Diseases; and Core Center for iPS Cell Research of Research Center Network for Realization of Regenerative Medicine from the Japan Agency for Medical Research and Development (AMED).
Author contributions
.K. and S.K. designed the study; Y.K. and S.K. interpreted the data; Y.K., A.KT., S.Ki., T.U., S.M. and A.W. performed the experiments. Y.K., S.K. and A.KT. analysed the data. S.K. supervised the study; and Y. K. and S.K. wrote the manuscript.
Declaration of interests
S. Kaneko is a founder, shareholder, and chief scientific officer at Thyas Co., Ltd., and received research funding from Takeda Pharmaceutical Co., Ltd.; Kirin Co., Ltd.; Thyas Co., Ltd.; Astellas Co., Ltd.; Terumo Co., Ltd.; and Tosoh Co., Ltd.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2021.05.016.
Supplemental information
References
- 1.Topalian S.L., Drake C.G., Pardoll D.M. Targeting the PD-1/B7-H1(PD-L1) pathway to activate anti-tumor immunity. Curr. Opin. Immunol. 2012;24:207–212. doi: 10.1016/j.coi.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Callahan M.K., Postow M.A., Wolchok J.D. Targeting T Cell Co-receptors for Cancer Therapy. Immunity. 2016;44:1069–1078. doi: 10.1016/j.immuni.2016.04.023. [DOI] [PubMed] [Google Scholar]
- 3.Maus M.V., Fraietta J.A., Levine B.L., Kalos M., Zhao Y., June C.H. Adoptive immunotherapy for cancer or viruses. Annu. Rev. Immunol. 2014;32:189–225. doi: 10.1146/annurev-immunol-032713-120136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rosenberg S.A., Restifo N.P. Adoptive cell transfer as personalized immunotherapy for human cancer. Science. 2015;348:62–68. doi: 10.1126/science.aaa4967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fesnak A.D., June C.H., Levine B.L. Engineered T cells: the promise and challenges of cancer immunotherapy. Nat. Rev. Cancer. 2016;16:566–581. doi: 10.1038/nrc.2016.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Old L.J. Tumor immunology: the first century. Curr. Opin. Immunol. 1992;4:603–607. doi: 10.1016/0952-7915(92)90034-c. [DOI] [PubMed] [Google Scholar]
- 7.Sadelain M. Chimeric antigen receptors: driving immunology towards synthetic biology. Curr. Opin. Immunol. 2016;41:68–76. doi: 10.1016/j.coi.2016.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wherry E.J., Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015;15:486–499. doi: 10.1038/nri3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Farber D.L., Yudanin N.A., Restifo N.P. Human memory T cells: generation, compartmentalization and homeostasis. Nat. Rev. Immunol. 2014;14:24–35. doi: 10.1038/nri3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hinrichs C.S., Borman Z.A., Cassard L., Gattinoni L., Spolski R., Yu Z., Sanchez-Perez L., Muranski P., Kern S.J., Logun C. Adoptively transferred effector cells derived from naive rather than central memory CD8+ T cells mediate superior antitumor immunity. Proc. Natl. Acad. Sci. USA. 2009;106:17469–17474. doi: 10.1073/pnas.0907448106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gattinoni L., Lugli E., Ji Y., Pos Z., Paulos C.M., Quigley M.F., Almeida J.R., Gostick E., Yu Z., Carpenito C. A human memory T cell subset with stem cell-like properties. Nat. Med. 2011;17:1290–1297. doi: 10.1038/nm.2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hinrichs C.S., Borman Z.A., Gattinoni L., Yu Z., Burns W.R., Huang J., Klebanoff C.A., Johnson L.A., Kerkar S.P., Yang S. Human effector CD8+ T cells derived from naive rather than memory subsets possess superior traits for adoptive immunotherapy. Blood. 2011;117:808–814. doi: 10.1182/blood-2010-05-286286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nishimura T., Kaneko S., Kawana-Tachikawa A., Tajima Y., Goto H., Zhu D., Nakayama-Hosoya K., Iriguchi S., Uemura Y., Shimizu T. Generation of rejuvenated antigen-specific T cells by reprogramming to pluripotency and redifferentiation. Cell Stem Cell. 2013;12:114–126. doi: 10.1016/j.stem.2012.11.002. [DOI] [PubMed] [Google Scholar]
- 14.Vizcardo R., Masuda K., Yamada D., Ikawa T., Shimizu K., Fujii S., Koseki H., Kawamoto H. Regeneration of human tumor antigen-specific T cells from iPSCs derived from mature CD8(+) T cells. Cell Stem Cell. 2013;12:31–36. doi: 10.1016/j.stem.2012.12.006. [DOI] [PubMed] [Google Scholar]
- 15.Themeli M., Kloss C.C., Ciriello G., Fedorov V.D., Perna F., Gonen M., Sadelain M. Generation of tumor-targeted human T lymphocytes from induced pluripotent stem cells for cancer therapy. Nat. Biotechnol. 2013;31:928–933. doi: 10.1038/nbt.2678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maeda T., Nagano S., Ichise H., Kataoka K., Yamada D., Ogawa S., Koseki H., Kitawaki T., Kadowaki N., Takaori-Kondo A. Regeneration of CD8αβ T Cells from T-cell-Derived iPSC Imparts Potent Tumor Antigen-Specific Cytotoxicity. Cancer Res. 2016;76:6839–6850. doi: 10.1158/0008-5472.CAN-16-1149. [DOI] [PubMed] [Google Scholar]
- 17.Minagawa A., Yoshikawa T., Yasukawa M., Hotta A., Kunitomo M., Iriguchi S., Takiguchi M., Kassai Y., Imai E., Yasui Y. Enhancing T Cell Receptor Stability in Rejuvenated iPSC-Derived T Cells Improves Their Use in Cancer Immunotherapy. Cell Stem Cell. 2018;23:850–858.e4. doi: 10.1016/j.stem.2018.10.005. [DOI] [PubMed] [Google Scholar]
- 18.Kitayama S., Zhang R., Liu T.Y., Ueda N., Iriguchi S., Yasui Y., Kawai Y., Tatsumi M., Hirai N., Mizoro Y. Cellular Adjuvant Properties, Direct Cytotoxicity of Re-differentiated Vα24 Invariant NKT-like Cells from Human Induced Pluripotent Stem Cells. Stem Cell Reports. 2016;6:213–227. doi: 10.1016/j.stemcr.2016.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wakao H., Yoshikiyo K., Koshimizu U., Furukawa T., Enomoto K., Matsunaga T., Tanaka T., Yasutomi Y., Yamada T., Minakami H. Expansion of functional human mucosal-associated invariant T cells via reprogramming to pluripotency and redifferentiation. Cell Stem Cell. 2013;12:546–558. doi: 10.1016/j.stem.2013.03.001. [DOI] [PubMed] [Google Scholar]
- 20.Ueda N., Uemura Y., Zhang R., Kitayama S., Iriguchi S., Kawai Y., Yasui Y., Tatsumi M., Ueda T., Liu T.Y. Generation of TCR-Expressing Innate Lymphoid-like Helper Cells that Induce Cytotoxic T Cell-Mediated Anti-leukemic Cell Response. Stem Cell Reports. 2018;10:1935–1946. doi: 10.1016/j.stemcr.2018.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheroutre H., Lambolez F. Doubting the TCR coreceptor function of CD8alphaalpha. Immunity. 2008;28:149–159. doi: 10.1016/j.immuni.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 22.Stock S., Hoffmann J.M., Schubert M.L., Wang L., Wang S., Gong W., Neuber B., Gern U., Schmitt A., Müller-Tidow C. Influence of Retronectin-Mediated T-Cell Activation on Expansion and Phenotype of CD19-Specific Chimeric Antigen Receptor T Cells. Hum. Gene Ther. 2018;29:1167–1182. doi: 10.1089/hum.2017.237. [DOI] [PubMed] [Google Scholar]
- 23.Tangye S.G. Advances in IL-21 biology - enhancing our understanding of human disease. Curr. Opin. Immunol. 2015;34:107–115. doi: 10.1016/j.coi.2015.02.010. [DOI] [PubMed] [Google Scholar]
- 24.Res P., Blom B., Hori T., Weijer K., Spits H. Downregulation of CD1 marks acquisition of functional maturation of human thymocytes and defines a control point in late stages of human T cell development. J. Exp. Med. 1997;185:141–151. doi: 10.1084/jem.185.1.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Horton N.C., Mathew P.A. NKp44 and Natural Cytotoxicity Receptors as Damage-Associated Molecular Pattern Recognition Receptors. Front. Immunol. 2015;6:31. doi: 10.3389/fimmu.2015.00031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Muranski P., Borman Z.A., Kerkar S.P., Klebanoff C.A., Ji Y., Sanchez-Perez L., Sukumar M., Reger R.N., Yu Z., Kern S.J. Th17 cells are long lived and retain a stem cell-like molecular signature. Immunity. 2011;35:972–985. doi: 10.1016/j.immuni.2011.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wirth T.C., Xue H.H., Rai D., Sabel J.T., Bair T., Harty J.T., Badovinac V.P. Repetitive antigen stimulation induces stepwise transcriptome diversification but preserves a core signature of memory CD8(+) T cell differentiation. Immunity. 2010;33:128–140. doi: 10.1016/j.immuni.2010.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sallusto F., Lenig D., Förster R., Lipp M., Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999;401:708–712. doi: 10.1038/44385. [DOI] [PubMed] [Google Scholar]
- 29.Brenchley J.M., Karandikar N.J., Betts M.R., Ambrozak D.R., Hill B.J., Crotty L.E., Casazza J.P., Kuruppu J., Migueles S.A., Connors M. Expression of CD57 defines replicative senescence and antigen-induced apoptotic death of CD8+ T cells. Blood. 2003;101:2711–2720. doi: 10.1182/blood-2002-07-2103. [DOI] [PubMed] [Google Scholar]
- 30.Elsaesser H., Sauer K., Brooks D.G. IL-21 is required to control chronic viral infection. Science. 2009;324:1569–1572. doi: 10.1126/science.1174182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fröhlich A., Kisielow J., Schmitz I., Freigang S., Shamshiev A.T., Weber J., Marsland B.J., Oxenius A., Kopf M. IL-21R on T cells is critical for sustained functionality and control of chronic viral infection. Science. 2009;324:1576–1580. doi: 10.1126/science.1172815. [DOI] [PubMed] [Google Scholar]
- 32.Yi J.S., Du M., Zajac A.J. A vital role for interleukin-21 in the control of a chronic viral infection. Science. 2009;324:1572–1576. doi: 10.1126/science.1175194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alvarez-Fernández C., Escribà-Garcia L., Vidal S., Sierra J., Briones J. A short CD3/CD28 costimulation combined with IL-21 enhance the generation of human memory stem T cells for adoptive immunotherapy. J. Transl. Med. 2016;14:214. doi: 10.1186/s12967-016-0973-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tominaga K., Yoshimoto T., Torigoe K., Kurimoto M., Matsui K., Hada T., Okamura H., Nakanishi K. IL-12 synergizes with IL-18 or IL-1beta for IFN-gamma production from human T cells. Int. Immunol. 2000;12:151–160. doi: 10.1093/intimm/12.2.151. [DOI] [PubMed] [Google Scholar]
- 35.Papadakis K.A., Prehn J.L., Landers C., Han Q., Luo X., Cha S.C., Wei P., Targan S.R. TL1A synergizes with IL-12 and IL-18 to enhance IFN-gamma production in human T cells and NK cells. J. Immunol. 2004;172:7002–7007. doi: 10.4049/jimmunol.172.11.7002. [DOI] [PubMed] [Google Scholar]
- 36.Szabo S.J., Kim S.T., Costa G.L., Zhang X., Fathman C.G., Glimcher L.H. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell. 2000;100:655–669. doi: 10.1016/s0092-8674(00)80702-3. [DOI] [PubMed] [Google Scholar]
- 37.Joshi N.S., Cui W., Chandele A., Lee H.K., Urso D.R., Hagman J., Gapin L., Kaech S.M. Inflammation directs memory precursor and short-lived effector CD8(+) T cell fates via the graded expression of T-bet transcription factor. Immunity. 2007;27:281–295. doi: 10.1016/j.immuni.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.DeBlaker-Hohe D.F., Yamauchi A., Yu C.R., Horvath-Arcidiacono J.A., Bloom E.T. IL-12 synergizes with IL-2 to induce lymphokine-activated cytotoxicity and perforin and granzyme gene expression in fresh human NK cells. Cell. Immunol. 1995;165:33–43. doi: 10.1006/cimm.1995.1184. [DOI] [PubMed] [Google Scholar]
- 39.Zeng R., Spolski R., Finkelstein S.E., Oh S., Kovanen P.E., Hinrichs C.S., Pise-Masison C.A., Radonovich M.F., Brady J.N., Restifo N.P. Synergy of IL-21 and IL-15 in regulating CD8+ T cell expansion and function. J. Exp. Med. 2005;201:139–148. doi: 10.1084/jem.20041057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gattinoni L., Speiser D.E., Lichterfeld M., Bonini C. T memory stem cells in health and disease. Nat. Med. 2017;23:18–27. doi: 10.1038/nm.4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Faustino-Rocha A., Oliveira P.A., Pinho-Oliveira J., Teixeira-Guedes C., Soares-Maia R., da Costa R.G., Colaço B., Pires M.J., Colaço J., Ferreira R., Ginja M. Estimation of rat mammary tumor volume using caliper and ultrasonography measurements. Lab Anim. (NY) 2013;42:217–224. doi: 10.1038/laban.254. [DOI] [PubMed] [Google Scholar]
- 42.Vizcardo R., Klemen N.D., Islam S.M.R., Gurusamy D., Tamaoki N., Yamada D., Koseki H., Kidder B.L., Yu Z., Jia L. Generation of Tumor Antigen-Specific iPSC-Derived Thymic Emigrants Using a 3D Thymic Culture System. Cell Rep. 2018;22:3175–3190. doi: 10.1016/j.celrep.2018.02.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Leishman A.J., Gapin L., Capone M., Palmer E., MacDonald H.R., Kronenberg M., Cheroutre H. Precursors of functional MHC class I- or class II-restricted CD8alphaalpha(+) T cells are positively selected in the thymus by agonist self-peptides. Immunity. 2002;16:355–364. doi: 10.1016/s1074-7613(02)00284-4. [DOI] [PubMed] [Google Scholar]
- 44.Pobezinsky L.A., Angelov G.S., Tai X., Jeurling S., Van Laethem F., Feigenbaum L., Park J.H., Singer A. Clonal deletion and the fate of autoreactive thymocytes that survive negative selection. Nat. Immunol. 2012;13:569–578. doi: 10.1038/ni.2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wencker M., Turchinovich G., Di Marco Barros R., Deban L., Jandke A., Cope A., Hayday A.C. Innate-like T cells straddle innate and adaptive immunity by altering antigen-receptor responsiveness. Nat. Immunol. 2014;15:80–87. doi: 10.1038/ni.2773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moran A.E., Holzapfel K.L., Xing Y., Cunningham N.R., Maltzman J.S., Punt J., Hogquist K.A. T cell receptor signal strength in Treg and iNKT cell development demonstrated by a novel fluorescent reporter mouse. J. Exp. Med. 2011;208:1279–1289. doi: 10.1084/jem.20110308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ma A., Koka R., Burkett P. Diverse functions of IL-2, IL-15, and IL-7 in lymphoid homeostasis. Annu. Rev. Immunol. 2006;24:657–679. doi: 10.1146/annurev.immunol.24.021605.090727. [DOI] [PubMed] [Google Scholar]
- 48.Dubois S., Waldmann T.A., Müller J.R. ITK and IL-15 support two distinct subsets of CD8+ T cells. Proc. Natl. Acad. Sci. USA. 2006;103:12075–12080. doi: 10.1073/pnas.0605212103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kennedy M.K., Glaccum M., Brown S.N., Butz E.A., Viney J.L., Embers M., Matsuki N., Charrier K., Sedger L., Willis C.R. Reversible defects in natural killer and memory CD8 T cell lineages in interleukin 15-deficient mice. J. Exp. Med. 2000;191:771–780. doi: 10.1084/jem.191.5.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lodolce J.P., Boone D.L., Chai S., Swain R.E., Dassopoulos T., Trettin S., Ma A. IL-15 receptor maintains lymphoid homeostasis by supporting lymphocyte homing and proliferation. Immunity. 1998;9:669–676. doi: 10.1016/s1074-7613(00)80664-0. [DOI] [PubMed] [Google Scholar]
- 51.Hinrichs C.S., Spolski R., Paulos C.M., Gattinoni L., Kerstann K.W., Palmer D.C., Klebanoff C.A., Rosenberg S.A., Leonard W.J., Restifo N.P. IL-2 and IL-21 confer opposing differentiation programs to CD8+ T cells for adoptive immunotherapy. Blood. 2008;111:5326–5333. doi: 10.1182/blood-2007-09-113050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hémar A., Subtil A., Lieb M., Morelon E., Hellio R., Dautry-Varsat A. Endocytosis of interleukin 2 receptors in human T lymphocytes: distinct intracellular localization and fate of the receptor alpha, beta, and gamma chains. J. Cell Biol. 1995;129:55–64. doi: 10.1083/jcb.129.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Villarino A.V., Tato C.M., Stumhofer J.S., Yao Z., Cui Y.K., Hennighausen L., O’Shea J.J., Hunter C.A. Helper T cell IL-2 production is limited by negative feedback and STAT-dependent cytokine signals. J. Exp. Med. 2007;204:65–71. doi: 10.1084/jem.20061198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kasaian M.T., Whitters M.J., Carter L.L., Lowe L.D., Jussif J.M., Deng B., Johnson K.A., Witek J.S., Senices M., Konz R.F. IL-21 limits NK cell responses and promotes antigen-specific T cell activation: a mediator of the transition from innate to adaptive immunity. Immunity. 2002;16:559–569. doi: 10.1016/s1074-7613(02)00295-9. [DOI] [PubMed] [Google Scholar]
- 55.Alves N.L., Arosa F.A., van Lier R.A. IL-21 sustains CD28 expression on IL-15-activated human naive CD8+ T cells. J. Immunol. 2005;175:755–762. doi: 10.4049/jimmunol.175.2.755. [DOI] [PubMed] [Google Scholar]
- 56.Spolski R., Leonard W.J. Interleukin-21: basic biology and implications for cancer and autoimmunity. Annu. Rev. Immunol. 2008;26:57–79. doi: 10.1146/annurev.immunol.26.021607.090316. [DOI] [PubMed] [Google Scholar]
- 57.Ozaki K., Spolski R., Feng C.G., Qi C.F., Cheng J., Sher A., Morse H.C., 3rd, Liu C., Schwartzberg P.L., Leonard W.J. A critical role for IL-21 in regulating immunoglobulin production. Science. 2002;298:1630–1634. doi: 10.1126/science.1077002. [DOI] [PubMed] [Google Scholar]
- 58.Fink P.J. The biology of recent thymic emigrants. Annu. Rev. Immunol. 2013;31:31–50. doi: 10.1146/annurev-immunol-032712-100010. [DOI] [PubMed] [Google Scholar]
- 59.Hogquist K.A., Xing Y., Hsu F.C., Shapiro V.S. T Cell Adolescence: Maturation Events Beyond Positive Selection. J. Immunol. 2015;195:1351–1357. doi: 10.4049/jimmunol.1501050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fulton R.B., Hamilton S.E., Xing Y., Best J.A., Goldrath A.W., Hogquist K.A., Jameson S.C. The TCR’s sensitivity to self peptide-MHC dictates the ability of naive CD8(+) T cells to respond to foreign antigens. Nat. Immunol. 2015;16:107–117. doi: 10.1038/ni.3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Williams L.R., Quinn L.L., Rowe M., Zuo J. Induction of the Lytic Cycle Sensitizes Epstein-Barr Virus-Infected B Cells to NK Cell Killing That Is Counteracted by Virus-Mediated NK Cell Evasion Mechanisms in the Late Lytic Cycle. J. Virol. 2015;90:947–958. doi: 10.1128/JVI.01932-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chang J.T., Segal B.M., Nakanishi K., Okamura H., Shevach E.M. The costimulatory effect of IL-18 on the induction of antigen-specific IFN-gamma production by resting T cells is IL-12 dependent and is mediated by up-regulation of the IL-12 receptor beta2 subunit. Eur. J. Immunol. 2000;30:1113–1119. doi: 10.1002/(SICI)1521-4141(200004)30:4<1113::AID-IMMU1113>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 63.Walsh M.C., Pearce E.L., Cejas P.J., Lee J., Wang L.S., Choi Y. IL-18 synergizes with IL-7 to drive slow proliferation of naive CD8 T cells by costimulating self-peptide-mediated TCR signals. J. Immunol. 2014;193:3992–4001. doi: 10.4049/jimmunol.1400396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Twohig J.P., Marsden M., Cuff S.M., Ferdinand J.R., Gallimore A.M., Perks W.V., Al-Shamkhani A., Humphreys I.R., Wang E.C. The death receptor 3/TL1A pathway is essential for efficient development of antiviral CD4+ and CD8+ T-cell immunity. FASEB J. 2012;26:3575–3586. doi: 10.1096/fj.11-200618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Migone T.S., Zhang J., Luo X., Zhuang L., Chen C., Hu B., Hong J.S., Perry J.W., Chen S.F., Zhou J.X. TL1A is a TNF-like ligand for DR3 and TR6/DcR3 and functions as a T cell costimulator. Immunity. 2002;16:479–492. doi: 10.1016/s1074-7613(02)00283-2. [DOI] [PubMed] [Google Scholar]
- 66.Teixeiro E., Daniels M.A., Hamilton S.E., Schrum A.G., Bragado R., Jameson S.C., Palmer E. Different T cell receptor signals determine CD8+ memory versus effector development. Science. 2009;323:502–505. doi: 10.1126/science.1163612. [DOI] [PubMed] [Google Scholar]
- 67.Gattinoni L., Klebanoff C.A., Restifo N.P. Paths to stemness: building the ultimate antitumour T cell. Nat. Rev. Cancer. 2012;12:671–684. doi: 10.1038/nrc3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Eyquem J., Mansilla-Soto J., Giavridis T., van der Stegen S.J., Hamieh M., Cunanan K.M., Odak A., Gönen M., Sadelain M. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature. 2017;543:113–117. doi: 10.1038/nature21405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Roth T.L., Puig-Saus C., Yu R., Shifrut E., Carnevale J., Li P.J., Hiatt J., Saco J., Krystofinski P., Li H. Reprogramming human T cell function and specificity with non-viral genome targeting. Nature. 2018;559:405–409. doi: 10.1038/s41586-018-0326-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Legut M., Dolton G., Mian A.A., Ottmann O.G., Sewell A.K. CRISPR-mediated TCR replacement generates superior anticancer transgenic T cells. Blood. 2018;131:311–322. doi: 10.1182/blood-2017-05-787598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gornalusse G.G., Hirata R.K., Funk S.E., Riolobos L., Lopes V.S., Manske G., Prunkard D., Colunga A.G., Hanafi L.A., Clegg D.O. HLA-E-expressing pluripotent stem cells escape allogeneic responses and lysis by NK cells. Nat. Biotechnol. 2017;35:765–772. doi: 10.1038/nbt.3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Adachi K., Kano Y., Nagai T., Okuyama N., Sakoda Y., Tamada K. IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor. Nat. Biotechnol. 2018;36:346–351. doi: 10.1038/nbt.4086. [DOI] [PubMed] [Google Scholar]
- 73.Kawana-Tachikawa A., Tomizawa M., Nunoya J., Shioda T., Kato A., Nakayama E.E., Nakamura T., Nagai Y., Iwamoto A. An efficient and versatile mammalian viral vector system for major histocompatibility complex class I/peptide complexes. J. Virol. 2002;76:11982–11988. doi: 10.1128/JVI.76.23.11982-11988.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Brockman M.A., Tanzi G.O., Walker B.D., Allen T.M. Use of a novel GFP reporter cell line to examine replication capacity of CXCR4- and CCR5-tropic HIV-1 by flow cytometry. J. Virol. Methods. 2006;131:134–142. doi: 10.1016/j.jviromet.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 75.Folks T.M., Clouse K.A., Justement J., Rabson A., Duh E., Kehrl J.H., Fauci A.S. Tumor necrosis factor alpha induces expression of human immunodeficiency virus in a chronically infected T-cell clone. Proc. Natl. Acad. Sci. USA. 1989;86:2365–2368. doi: 10.1073/pnas.86.7.2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Terahara K., Yamamoto T., Mitsuki Y.Y., Shibusawa K., Ishige M., Mizukoshi F., Kobayashi K., Tsunetsugu-Yokota Y. Fluorescent Reporter Signals, EGFP, and DsRed, Encoded in HIV-1 Facilitate the Detection of Productively Infected Cells and Cell-Associated Viral Replication Levels. Front. Microbiol. 2012;2:280. doi: 10.3389/fmicb.2011.00280. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.