

Abstract
Purpose:
To evaluate the safety and tolerability in phase I first-in-human combination therapy with pexidartinib, an inhibitor of colony-stimulating factor-1 receptor, and sirolimus, an mTOR inhibitor, to target tumor-associated macrophage (TAM) polarization in soft tissue sarcomas (STS).
Patients and Methods:
This multicenter phase I study used the time-to-event continual reassessment method (TITE-CRM) to study the combination of sirolimus, doses ranging from 2 to 6 mg, with pexidartinib, doses ranging from 600 to 1,000 mg, both provided continuously on a 28-day cycle, in patients with advanced sarcoma. A total of 24 patients [8 malignant peripheral nerve sheath tumor, 3 tenosynovial giant cell tumor (TGCT), 5 leiomyosarcoma, and 8 with other sarcoma subtypes] were enrolled. The median age was 46 years, 56% were male, and 61% had >2 prior lines of therapy.
Results:
The recommended phase II dose was 2 mg of sirolimus combined with 1,000 mg of pexidartinib daily. Of the 18 evaluable subjects, 5 experienced dose-limiting toxicities (2 elevated aspartate aminotransferase/alanine aminotransferase, 2 elevated sirolimus trough levels, and 1 grade 5 dehydration). Most common grade 2 or higher treatment-related adverse events included anemia, fatigue, neutropenia, and lymphopenia. Clinical benefit was observed in 12 of 18 (67%) evaluable subjects with 3 partial responses (all in TGCT) and 9 stable disease. Tissue staining indicated a decreased proportion of activated M2 macrophages within tumor samples with treatment.
Conclusions:
Pexidartinib can be safely administered with sirolimus. These findings support further investigation of this combination to determine clinical efficacy. Clinicaltrials.gov identifier NCT02584647.
Translational Relevance.
Pexidartinib is a novel potent small molecule inhibitor of colony-stimulating factor-1 receptor (CSF-1R) and targets polarization of the protumorigenic M2 tumor-associated macrophages (TAM) to M1. In our preclinical screen, we identified malignant peripheral nerve sheath tumor (MPNST) cell lines to express high levels of activated CSF-1R. Pexidartinib inhibited cell proliferation and tumor growth in an MPNST xenograft model. The addition of sirolimus to pexidartinib resulted in sustained tumor growth suppression and a profound decrease in TAMs compared with mice treated with pexidartinib alone. These results formed the rationale to conduct a first-in-human study to test this combination in heavily treated patients with soft tissue sarcomas (STS). This phase I study was conducted using the time-to-event continual reassessment method and demonstrated acceptable safety, tolerability, profound tumor response in tenosynovial giant cell tumor (TGCT) as expected, and prolonged tumor stabilization in pretreated patients with MPNST. TGCT is a rare nonaggressive sarcoma subtype involving the synovium, bursa, and tendon sheath for which pexidartinib monotherapy has been demonstrated to be effective previously. Analysis of tumor tissue from pretreatment biopsy and on-treatment resection from 2 patients with TGCT and 1 with TSC complex subunit 1 (TSC1)-mutated leiomyosarcoma using multiplex immunofluorescence identified a decline in activated M2 TAMs in all three on-treatment specimens, but not of M1 TAMs or CD8+ T cells. A phase II study testing the combination at the recommended phase II dose is ongoing with required pretreatment and on-treatment biopsies for correlative analysis, including M2 TAM infiltration.
Introduction
Although soft tissue sarcomas (STS) constitute less than 1% of all malignancies, there are greater than 60 distinct subtypes. In general, response to cytotoxic chemotherapy is poor and identifying treatments with meaningful clinical benefit in all subtypes is a daunting challenge (1, 2). Conversely, identifying shared underlying vulnerabilities within distinct STS subtypes and targeting them as a subgroup is another strategy to identify novel effective treatment options in this rare disease.
A study surveying tissue microarrays of 1,242 sarcoma specimens from 24 subtypes revealed that tumor-associated macrophages (TAM) outnumber tumor-infiltrating lymphocytes across a majority of sarcomas with M2-polarized macrophages (tumor-promoting phenotype) representing the majority of TAMs (3). Similarly, analysis of The Cancer Genome Atlas (TCGA) sarcoma data set using gene expression signatures found that M2-polarized TAMs represented a larger proportion of the immune infiltrate compared with M0- or M1- (tumoricidal phenotype) polarized TAMs (3). These results suggest that macrophage polarizing agents may represent a therapeutic opportunity in certain STS subtypes. Mouse and human neurofibromas, benign precursors to malignant peripheral nerve sheath tumors (MPNST), demonstrate substantial TAMs where they account for almost half of neurofibroma cells (4).
Colony-stimulating factor-1 receptor (CSF-1R) is expressed by many monocytes, macrophages, and certain tumor cells, and expression of CSF-1R is associated with poor prognosis in certain cancers (5). The ligand to CSF-1R, colony-stimulating factor-1 (CSF-1) plays a critical role in differentiation, maturation, survival of mononuclear phagocytic cells, activation of the CSF-1/CSF-1R pathway, and contributes to the conversion of TAMs from an M1 to M2 phenotype (6).
We previously screened STS cell lines for potential vulnerability to the CSF-1R axis and found that multiple MPNST cell lines express high levels of activated CSF-1R (7). In a xenograft model of MPNST, we previously demonstrated that oral administration of pexidartinib, a novel small molecule inhibitor of CSF-1R, KIT, and platelet-derived growth factor receptor (PDGFR), blocked MPNST cell proliferation in vitro and tumor growth in vivo. mTOR plays a key role in cell survival and proliferation through AKT signaling. The addition of sirolimus, an mTOR inhibitor, to pexidartinib in our xenograft MPNST model resulted in sustained antitumor activity and decreased TAMs in harvested tumor tissue, compared with tumors harvested from mice treated with pexidartinib alone (7).
Pexidartinib was approved by the FDA as monotherapy for symptomatic tenosynovial giant cell tumor (TGCT) associated with severe morbidity or functional limitations and not amenable to improvement with surgery (8). In contrast, the European Medicines Agency's advisory group recommended against pexidartinib for patients with TGCT citing limited symptomatic improvement and concern for hepatotoxicity. To date, pexidartinib either alone or in combination has not been evaluated in MPNSTs. This study is the first-in-human combination of pexidartinib with sirolimus and aims to target TAM-M2 polarization based on our preclinical findings. We conducted a multi-institutional phase I study using the time-to-event continual reassessment method (TITE-CRM) to evaluate pexidartinib and sirolimus in heavily pretreated patients with advanced STS. The TITE-CRM was selected as it has improved performance compared with the conventional 3+3 design at identifying the true MTD, treating fewer patients at suboptimal doses, allowing the specification of a fixed sample size for the trial, and increased flexibility in the specification of the target toxicity rate (9). Moreover, the TITE-CRM can use the patient's partial information before a complete follow-up is achieved while allowing for a longer toxicity evaluation window beyond the first cycle to account for late-onset toxicities. Thus, we can conduct the trial in a continuous fashion without having patients being turned away due to waiting time. The primary aims of this study were to determine safety, preliminary toxicity, and the recommended phase II doses (RP2D) of the combination of pexidartinib and sirolimus.
Patients and Methods
Patient eligibility
Eligible patients included those with unresectable pathologically confirmed sarcoma, who were either intolerant to or had progressed on standard of care therapy, with evaluable disease as per RECIST version 1.1 at baseline, with an Eastern Cooperative Oncology Group (ECOG) performance 0 or 1, and adequate organ and bone marrow function. Patients were excluded if they were unable to swallow capsules or if they suffered from hepatobiliary disease. The study was approved by the Columbia University Irving Medical Center and Washington University institutional review board and was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice. Each patient provided written informed consent.
Study design
This phase I, multicenter, open-label clinical trial (ClinicalTrials.gov identifier: NCT02584647) used the TITE-CRM to evaluate safety and to estimate the MTD of a first-in-human combination with pexidartinib (PLX3397) and sirolimus in patients with advanced sarcoma.
Enrolled patients were treated with combination therapy at one of five predefined dose levels with sirolimus doses ranging from 2 to 6 mg orally once daily and pexidartinib doses ranging from 600 to 1,000 mg total orally daily. The dose combinations were ordered as seen in Fig. 1A. Treatment was initiated at dose level 2 with 2 mg sirolimus oral daily and 400 mg pexidartinib oral twice daily. The MTD was defined as the drug combination associated with a target probability of dose-limiting toxicity (DLT) of 0.25. A DLT was defined as any grade 4 or higher hematologic toxicity or grade 3 or higher nonhematologic toxicity according to the National Cancer Terminology Criteria for Adverse Events (version 4.03) within 8 weeks. Grade 3 neutropenia with fever, grade 3 thrombocytopenia with significant bleeding, and any circumstance resulting in a dose reduction, which included dose-level reduction of sirolimus for persistently elevated trough levels, were also considered DLTs. All patients were required to receive at least 80% of the planned doses of sirolimus and pexidartinib during the DLT period to be considered evaluable for DLT, after which time dose interruptions were allowed for up to 21 days.
Figure 1.
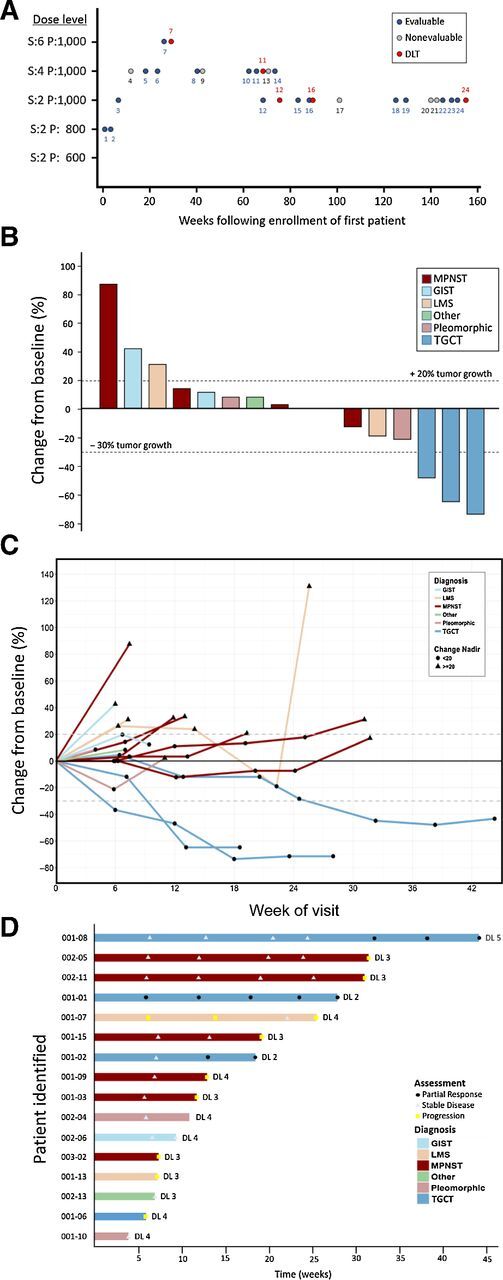
TITE-CRM and antitumor activity. A, TITE-CRM was used to estimate the RP2D with the probability of DLT of 0.25. Treatment was initiated at dose level 2. Total daily doses of sirolimus (S) and pexidartinib (P) are indicated on the y-axis in milligrams. Evaluable (blue circle) and nonevaluable patients (gray circle) enrolled sequentially are depicted. Patients who experienced a DLT are depicted separately at the time of the event (red circle). B, Maximal change of tumor size from baseline assessed by an independent radiologist per RECIST version 1.1 (N = 16). Percent change from baseline represents the maximal decrease or minimal increase in target lesion(s). C, Change in individual tumor burden over time from baseline as assessed by RECIST 1.1 (N = 16). D, Exposure and duration of response per RECIST 1.1 (N = 16). Patients with reduction of target lesion(s) of greater than 30% ( ), or those between 30% reduction and 20% increase (
), or those between 30% reduction and 20% increase ( ), or those with equal or greater than 20% (
), or those with equal or greater than 20% ( ) are depicted. DL, dose level.
) are depicted. DL, dose level.
Confirmed radiologic responses were evaluated by an independent radiologist using RECIST 1.1 every 6 weeks. Patients, whose disease progressed by RECIST 1.1 but appeared to derive clinical benefit, as determined by the investigator, were allowed to continue on study as per immune-related RECIST (irRECIST). Change in tumor size was used to evaluate overall response rate (ORR) and progression-free survival (PFS). The pharmacokinetic (PK) profile of pexidartinib and sirolimus was determined using drug-specific assays.
Statistical analysis
The TITE-CRM used an empiric dose-toxicity model, a linear weight function, and a normal prior distribution on the parameter with mean 0 and variance of 1.34 (10). The dose toxicity model was calibrated such that the method will eventually select a dose that yields between 18% and 32% DLT (11). We imposed a minimum of 2 weeks of observation between successive patients. Patients who discontinued treatment because of disease progression or death without experiencing a DLT prior to 4 weeks and those who received less than 80% of planned doses were replenished (12). On the basis of simulation studies, a sample size of 24 was initially proposed to obtain probability of correct selection above 60% across a range of possible scenarios. The protocol was amended on 27, 2017, after enrollment of 17 patients, to recruit 18 patients, to increase probability of correct selection above 53%.
The MTD was estimated using TITE-CRM based on a target toxicity probability of 0.25 (10). The proportion of DLT was reported along with the final estimates of the probability for DLT based on the TITE-CRM and its 90% probability interval by dose level. Baseline characteristics for evaluable patients were summarized. Median and range for continuous variable, frequency, and percentage for categorical variable were reported. Adverse events were reported by type with frequency and percentage for all grade 3 or higher events and for events where more than 20% of patients experienced grade 1 or 2 events. ORR was estimated for the entire cohort and for those assigned to the estimated MTD along with 95% confidence interval (CI). Overall survival and PFS were estimated for the entire cohort, for patients treated at the estimated MTD, and for patients with MPNST using the Kaplan–Meier estimator. Median OS and PFS were reported with 95% CI if applicable.
Macrophage and cytotoxic T-lymphocyte analysis in paired tumor specimens
Staining and imaging
Tumor specimens were stained using Opal Multiplex Kits, according to the manufacturer's protocol (Perkin-Elmer), for CD8 (clone 4B11; Leica, PA0183), CD68 (clone KP1; BioGenex, Am416-5M), HLA-DR (EP96; BioSB, BSB6797), CD163 (clone OTI2G1; Abcam, ab156769), CD206 (clone 15-2; Abcam, ab64693), and CSF1R (clone EPR20754; Abcam, ab229188). Specimens underwent serial staining with primary and secondary antibodies followed by the use of a covalently bound tyramide signal amplification process (13, 14).
Multispectral imaging
Prior to multiplex staining, hematoxylin and eosin (H&E)-stained sections of tumor were verified by two independent pathologists with expertise in sarcoma to identify representative areas for multispectral image capture. Representative tumor areas were scanned at 20× magnification with the VECTRA platform (Perkin-Elmer) and factored equally into the analysis for each patient. Images from each single-stained and unstained slide was used to create a multispectral library in inForm (14). All images were analyzed using inForm software. DAPI counterstaining was used to differentiate cellular and nuclear compartments, with each associated cytosolic or membrane bound protein detected via presence of a specific stain (CSF1R, CD206, CD8, CD68, CD163, and HLA-DR).
Image analysis
All images were analyzed using inForm software. DAPI counterstaining (Supplementary Fig. S2 online only) was used to differentiate cellular and nuclear compartments, with each associated cytosolic or membrane bound protein detected via presence of a specific stain (CSF1R, CD206, CD8, CD68, CD163, and HLA-DR). Cell segmentation was performed using the minimum DAPI signal to accurately locate all cells and adjusted on the basis of splitting, to avoid hypersegmentation or hyposegmentation of each nucleus, and adjusting sizes of nuclei to fit both tumor and immune cells (Supplementary Figs. S2B and S2C). Following this, tissue segmentation was performed by highlighting examples of tumor and nontumor tissue, and through an iterative process, a computer algorithm capable of "learning" each tissue type, based morphology, was created (Supplementary Fig. S2D). Cells were phenotyped using the phenotyping step of InForm software. Approximately 10 representative cells for each base variable were chosen to train the phenotyping algorithm: tumor (red), cytotoxic T cells (magenta), and other (blue; Supplementary Fig. S2E). Finally, each scoreable variable (CSF1R, CD206, CD163, HLA-DR) was scored for intensity again using the InForm software (Supplementary Fig. S2F).
qmIF quantification methods
Batch-processed output files from InForm were analyzed with an in-house Python script. Cells were categorized by the base phenotype determined by InForm, as well as a binary ± for each scored marker (e.g., CD206, HLA-DR) depending on if the intensity exceeded the score threshold. Only cells found in the tumor compartment (as defined by InForm tissue segmentation) were used. The counts of each cell type for each image were then divided by the total number of cells to get a cellular proportion for each image. These images were grouped by patient and time point (relative to treatment course), taking the mean proportion across replicate images.
Results
Patient population
From November 04, 2015 to September 26, 2018, a total of 24 patients with advanced sarcoma were enrolled in the study. Six patients were considered nonevaluable (nonadherence: 2; withdrawal of consent on day 4: 1; progression of disease prior to week 4: 2; serious adverse event unlikely to be drug related: 1; of these, one patient died within 4 weeks due to disease progression). Among the 18 evaluable patients, 2 had no tumor assessment and 1 had tumor assessment after DLT. Baseline characteristics are listed in Table 1. Notably, most patients had >2 prior lines of systemic therapy. Toxicity data from 1 nonevaluable patient was used for determination of the subsequent patient's dose determination as per the TITE-CRM design.
Table 1.
Baseline characteristics of clinical trial participants (N = 18).
| Characteristics | Number of patients (range of %) |
|---|---|
| Median age, years (range) | 46.2 (21–68) |
| Sex | |
| Female | 8 (44.4) |
| Male | 10 (56.6) |
| Race | |
| Caucasian | 10 (55.6) |
| Hispanic | 4 (22.2) |
| African American | 2 (11.1) |
| Asian | 2 (11.1) |
| Sarcoma subtype | |
| Malignant peripheral nerve sheath tumors | 6 (33.3) |
| Pigmented villonodular synovitis | 3 (16.7) |
| Pleiomorphic sarcoma | 2 (11.1) |
| Leiomyosarcoma | 2 (11.1) |
| Gastrointestinal stromal tumor | 2 (11.1) |
| Spindle cell sarcoma | 1 (5.6) |
| Epithelioid angiomyolipoma | 1 (5.6) |
| Hemangiopericytoma | 1 (5.6) |
| Number of prior therapies | |
| 0–1 | 7 (38.9) |
| >2 | 11 (61.1) |
Treatment-related toxicity
Overall, the combination pexidartinib and sirolimus had acceptable toxicity with 5 of 18 evaluable patients experiencing DLTs (Supplementary Table S1). DLTs included elevated aspartate aminotransferase (AST)/alanine aminotransferase (ALT) in 2 patients, elevated sirolimus trough levels in 2 patients, and grade 5 dehydration and cardiac arrest in 1 patient who refused supportive therapy. Elevated AST/ALT and sirolimus trough levels normalized on dose interruption and/or reduction. All 18 evaluable patients experienced treatment-related adverse events (TRAE). Grade 3 and 4 TRAEs occurred in 7 (39%) patients and 1 (5.6%) patient, respectively (Table 2). One patient experienced a grade 3 elevation in AST/ALT at dose level 5 that normalized after a brief interruption in treatment. Given clinical benefit, the data safety review committee approved resumption of treatment in this patient who was rechallenged at dose level 4 at which AST/ALT remained ≤ grade 1. Two patients required dose reductions (11%), 7 required dose interruptions (39%) of which 3 were due to elevated sirolimus trough levels (17%), and 11 (61%) patients died. Of the 11 patients who died, 1 experienced grade 5 dehydration and the remaining 10 patients expired due to disease progression.
Table 2.
Number of patients with maximum-grade adverse events (N = 18).
| TRAEs | Grade 1 | Grade 2 | Grade 3 | Grade 4 | Grade 5 |
|---|---|---|---|---|---|
| Aspartate aminotransferase increased | 14 (77.8%) | 1 (5.6%) | 1 (5.6%) | ||
| Anemia | 6 (33.3%) | 7 (38.9%) | 2 (11.1%) | ||
| Fatigue | 6 (33.3%) | 5 (27.8%) | 2 (11.1%) | ||
| Alanine aminotransferase increased | 8 (44.4%) | 1 (5.6%) | 2 (11.1%) | ||
| Neutrophil count decreased | 5 (27.8%) | 1 (5.6%) | 1 (5.6%) | ||
| White blood cell decreased | 5 (27.8%) | 5 (27.8%) | |||
| Alkaline phosphatase increased | 5 (27.8%) | 1 (5.6%) | |||
| Anorexia | 4 (22.2%) | 1 (5.6%) | 1 (5.6%) | ||
| Diarrhea | 5 (27.8%) | 1 (5.6%) | |||
| Hypertension | 2 (11.1%) | 3 (16.7%) | 1 (5.6%) | ||
| Lymphocyte count decreased | 3 (16.7%) | 4 (22.2%) | |||
| Nausea | 5 (27.8%) | 1 (5.6%) | |||
| Platelet count decreased | 4 (22.2%) | 1 (5.6%) | |||
| Creatinine increased | 3 (16.7%) | 1 (5.6%) | |||
| Dysgeusia | 3 (16.7%) | 1 (5.6%) | |||
| Skin and subcutaneous tissue disorders | 5 (27.8%) | ||||
| Dehydration | 1 (5.6%) | 1 (5.6%) | |||
| Upper respiratory infection | 1 (5.6%) | 1 (5.6%) | |||
| Cardiac arrest | 1 (5.6%) |
Using the TITE-CRM design, we determined that dose level 3 (1,000 mg oral total daily dose of pexidartinib and 2 mg oral daily of sirolimus) to be the RP2D with an estimated toxicity probability of 0.267 with 90% probability interval (0.109–0.456; Supplementary Table S1). The proportion of DLT at each dose level was reported with the final estimates of the probability for DLT based on the TITE-CRM with 90% probability interval as shown in Supplementary Table S1. The PK profile indicated expected increased levels of pexidartinib and sirolimus with time. In addition, increased sirolimus doses did not significantly affect pexidartinib levels as depicted in Supplementary Fig. S1 and Supplementary Table S2.
Antitumor activity
For all the evaluable patients by July 26, 2019, the last date of follow-up, no patients remained on therapy. Eleven (61%) and 3 (17%) patients discontinued therapy due to disease progression and intolerance, respectively. In all, 12 of 18 evaluable patients (66.7%; 95% CI, 41.15%–85.64%) experienced clinical benefit [3 partial response (PR) and 9 stable disease (SD); Fig. 1B]. A durable (≥18 weeks) reduction in target lesions compared with baseline was observed in 3 (16.7%) patients, all of whom had TGCT (Fig. 1). All 3 patients with localized TGCT (001–01, 001–02, 001–08), which at presentation were not amenable to surgery, underwent limb sparing tumor resections following the combination therapy for 164, 119, and 363 days, respectively. One of the 3 patients experienced disease recurrence 9 months after surgery (001–01), likely due to incomplete disease removal. The other 2 patients (001–02 and 001–08) with TGCT remain disease free at last follow-up for 5 and 3.5 years, respectively. Nine patients (50%) achieved SD, of whom 4 (22%) had confirmed SD and 2 (11.1%) were durable (MPNST; ≥18 weeks). In all, 12 of 18 patients (66.7%; 95% CI, 41.15%–85.64%) experienced clinical benefit (3 PR and 9 SD; Fig. 1). Patient 001–07 remained on study despite progressive disease due to clinical benefit. This patient was found to have SD on the third scheduled assessment but ultimately progressed on follow-up scans and came off of study (Fig. 1C). Median progression-free survival (mPFS) and overall survival (mOS) for all evaluable patients was 11.6 weeks (95% CI, 6–24.57) and 35.9 weeks (95% CI, noncalculable), respectively (Fig. 2A and B). mPFS at the estimated MTD (dose level 3) for all evaluable patients and those with MPNST was 11.9 weeks (95% CI, 4.29–30) and 18.6 weeks (95% CI, noncalculable), whereas mOS at dose level 3 for all evaluable patients and those with MPNST was 35.9 weeks (95% CI, noncalculable) and 145.1 weeks (95% CI, noncalculable), respectively (Fig. 2C–F). Of the 6 MPNST patients, 1 was NF1-associated (002–11), 4 were sporadic, and NF1 status of patient 003–02 was not known. mPFS and mOS for all who received treatment (24 patients) was 9.7 weeks (95% CI, 6–18.57) and 34.6 weeks (95% CI, 10.9–86.3), respectively; mPFS and mOS at dose level 3 for all who received treatment (12 patients) was 10.9 weeks (95% CI, 4.3–22.1) and 34.6 weeks (95% CI, 7.3–145.1), respectively.
Figure 2.
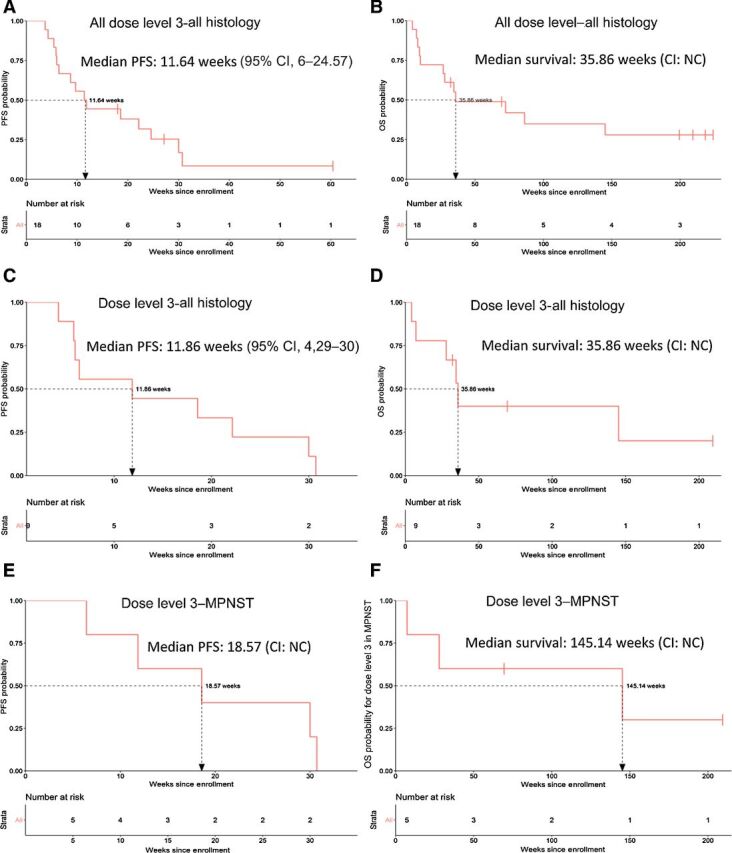
PFS and overall survival. Kaplan–Meier curves by RECIST version 1.1 for PFS and overall survival of 18 evaluable patients with advanced sarcoma who were previously treated (A and B); 9 patients at dose level 3 (C and D); and 5 patients with MPNST treated at dose level 3 (E and F). Probability of survival is shown at indicated time points and number of patients at risk at indicated time points are shown below the x-axis. NC, noncalculable.
Macrophage and cytotoxic T-lymphocyte tumor infiltration
In an effort to determine the effects of the combination of pexidartinib and sirolimus therapy on TAMs within the sarcoma tumor microenvironment, we applied quantitative multiplex immunofluorescence (qmIF) on available pre- and on-treatment tumor tissue. Three patients on study with TGCT achieved tumor response permitting tumor resection. Patient 001–07, diagnosed with TSC complex subunit 1 (TSC1)-mutated leiomyosarcoma, experienced a malignant small bowel obstruction on week 11 for which she underwent palliative small bowel resection and tumor specimens were collected for analysis. Presence of tumor in each biopsy specimen was confirmed with H&E staining and verified by an expert sarcoma pathologist. Of the 4 patients with available paired pre- and on-treatment tissue specimens, resected tumor tissue from patient 001–08 (TGCT) failed to identify neoplastic cells and was not used for TAM quantification. Each tumor tissue was stained with antibodies specific for CD8 (cytotoxic T lymphocytes, CTL), CSF-1R and CD163 (tumor cells and macrophages), CD68 (M1 and M2 macrophages), HLA-DR (activated M1 and M2 macrophages), CD206 (M2 macrophages), and DAPI (nuclear stain; Supplementary Figs. S2A–S2F). Representative paired pre- and on-treatment tissue from patient 001–01 is depicted in Fig. 3A. Quantification using the mIF panel revealed a decrease in mean cellular proportion of activated M2 macrophages in all on-treatment tumor tissues compared with their corresponding pretreatment biopsies (Fig. 3B and C). Although proportion of activated total macrophages also decreased in all on-treatment tumor tissues, the magnitude of the decline was less compared with that observed for activated M2 macrophages. Moreover, the therapy did not appear to affect M1 or CTL populations (Supplementary Figs. S2G–S2J).
Figure 3.
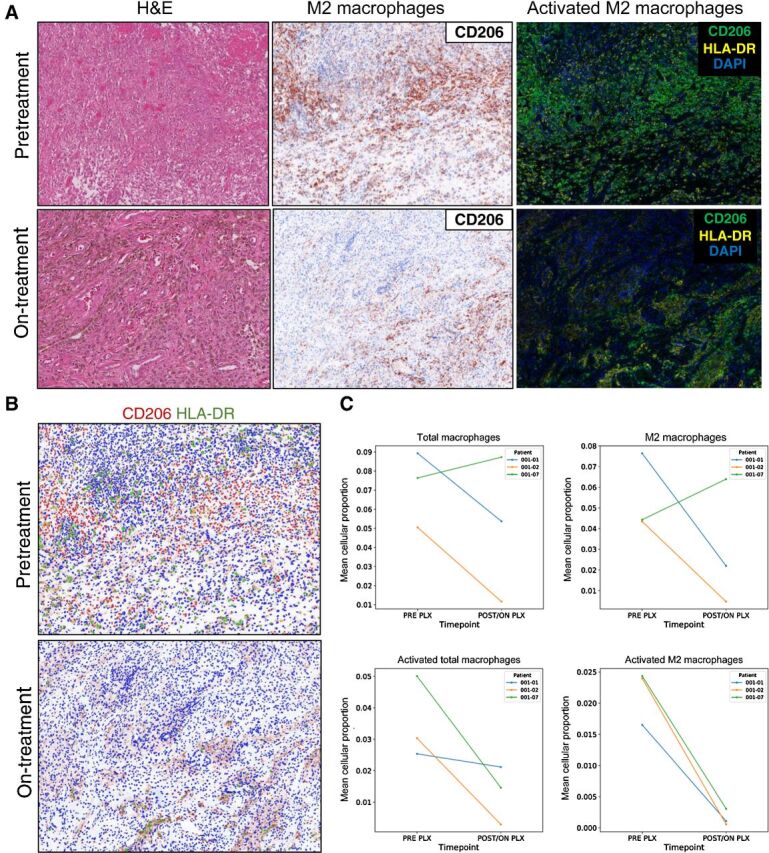
Activated M2 macrophages in tumor tissue. Treatment with pexidartinib and sirolimus resulted in a decrease in both CD206 expressing cells and CD206 and HLA-DR coexpressing cells. A, Pretreatment (top row) and on-treatment (bottom row) sections obtained from a patient with TGCT stained with H&E (left column), antisera against CD206 (middle column), and qmIF staining of both CD206 and HLA-DR (right column), with pexidartinib plus sirolimus. B, Scoring of pre- and on-treatment tumor tissue from a patient with TGCT for CD206 and HLA-DR using inForm (Perkin-Elmer) scoring of CD206 (red) and HLA-DR (green). C, Graphs depict non-statistically significant trends in total and activated macrophages (left column) and total and activated M2 macrophages (right column).
Discussion
This phase I trial of the combination of pexidartinib and sirolimus demonstrated feasibility and an acceptable safety profile. Pexidartinib and sirolimus resulted in profound and durable tumor responses in patients with TGCT which was previously demonstrated with pexidartinib alone (8). Our preclinical data indicated that the combination of pexidartinib and sirolimus would result in both greater and more durable responses than pexidartinib alone, even after discontinuing drug therapy (8). Though the study was not powered or designed to examine this question, the prolonged benefit seen in 2 patients with TGCT who were resected and remain off drug now for over 3.5 and 5 years may indicate a benefit that exceeds pexidartinib alone. This will require future clinical investigation. Pexidartinib in combination with sirolimus resulted in prolonged tumor stabilization (≥18 weeks) in 1 of 2 patients with leiomyosarcoma (LMS) and 3 of 6 patients with MPNST, all of whom had prior treatments. The clinical benefit observed in MPNST is encouraging and mirrors our preclinical studies in MPNST models, which predicted efficacy with this combination. Acknowledging the limitations of cross-study comparisons and the small number of patients with MPNST treated in our study, these results compare favorably to other studies targeting MPNSTs. Combination therapy targeting mTOR and VEGF, with everolimus and bevacizumab, respectively, in advanced MPNST patients demonstrated a clinical benefit rate of 12% with only 3 of 25 patients achieving SD for 4 months or longer (15). Similarly, combination therapy with sirolimus and ganetespib, an Hsp90 inhibitor, in phase I resulted in a sustained partial response in one patient, who had LMS, of the 10 evaluable patients (16). None of the 10 patients with MPNST in the phase II portion achieved clinical benefit, as defined by the study protocol. The median duration on the phase II trial was 8 weeks (4–16 weeks), although one patient experienced sustained stable disease by RECIST for 32 weeks after being allowed to continue therapy despite progression per WHO criteria (16). The mPFS in our study for the evaluable 18 patients was 11.6 weeks and was 11.9 weeks for the 9 patients treated at dose level 3, the RP2D (Fig. 2). In comparison, although limited by a small number of patients and preliminary data from a phase I study, the 5 subjects with MPNST treated at dose level 3 experienced a mPFS of 18.6 weeks which is encouraging and is consistent with our preclinical experiments which predicted activity in this histology.
The prolonged mOS (35.9 weeks) observed in our patient population is also encouraging and was influenced by the 3 TGCT subjects, all of whom had a profound response and proceeded to curative surgeries. Two of these patients remain disease free, whereas one patient experienced local disease recurrence. This patient went on to respond to another anti-CSF-1R investigational agent but discontinued treatment due to poor tolerance. This patient was rechallenged with pexidartinib as standard of care monotherapy and experienced further decrease in size of the left knee mass and continues to derive benefit. The fact that this patient continued to respond to subsequent CSF-1R–targeted therapies suggests that the recurrence was likely due to residual disease rather than the development of resistance to the CSF-1R pathway.
Dosing at the RP2D of pexidartinib (1,000 mg total daily dose and sirolimus 2 mg daily) had an acceptable toxicity profile. Most patients developed liver function abnormalities. AST and ALT were increased in 89% and 61.1%, and of these 5.6% and 11.1% were grade 3, respectively. The proportion of patients experiencing elevated AST and ALT is similar to that reported in the randomized study (ENLIVEN) of 1,000 mg loading dose daily for 2 weeks followed by 800 mg of total daily dose of pexidartinib versus placebo in TGCT (17). Monotherapy pexidartinib resulted in an increase in AST and ALT in 88% and 60% of patients, respectively, and of which 12% and 18% were grade 3 or higher. These results were comparable to those reported in our study and suggest that the addition of sirolimus to pexidartinib does not increase the risk of hepatotoxicity. Other common adverse events (>20%) included fatigue, anemia, neutropenia, thrombocytopenia, elevated alkaline phosphatase, anorexia, nausea, diarrhea, and changes in skin and hair color. These AEs have been previously attributed to pexidartinib or sirolimus individually and no new unexpected AEs were identified.
The tumor microenvironment (TME) is plastic and prone to modulation by neoplastic and stromal cells where inherent signals are manipulated to enhance tumor growth. Reprogramming of the CSF-1/CSF-1R axis by tumor cells has been implicated in homing monocytes to the TME, and their differentiation to the pro-oncogenic M2-polarized macrophages (3). As expected, our interrogation of the macrophage population within paired samples from 2 patients with TGCT and 1 patient with LMS revealed a decline in the activated M2-polarized macrophages in all three paired tumor samples. The total number of macrophages also declined in on-treatment tumor issue from patients with TGCT but not in the patient with LMS. There was no association with treatment and M1-polarized macrophages or CTLs suggesting a treatment-specific effect on M2 macrophages. Although the number of samples limit statistical significance, the decline in activated M2 macrophages within the TME in all available paired samples suggests suppression of the CSF-1/CSF-1R axis. However, with the study design we are unable to determine individual contributions of pexidartinib and sirolimus to the overall decline in activated M2-polarized macrophages. Given the prolonged disease stabilization in MPNST and the correlative analysis, a multicenter phase 2 study testing pexidartinib in combination with sirolimus at the RP2D in patients with MPNST was activated and is currently accruing patients (NCT02584647).
In conclusion, the combination of pexidartinib and sirolimus was generally well tolerated and the RP2D for pexidartinib was 1,000 mg oral total daily dose and sirolimus was 2 mg oral daily. Correlative studies suggest a decline in the activated tumor-associated M2-polarized macrophage population indicating that this combination may hold potential to reprogram TAM polarization.
Authors' Disclosures
G.A. Manji reports grants from Plexxikon and FDA during the conduct of the study; G.A. Manji also reports grants from Merck, Roche/Genentech, and Regeneron, as well as grants and personal fees from BioLine Rx, CEND Pharm, and Arcus Biosciences outside the submitted work. B.A. Van Tine reports other support from Daiichi Sankyo during the conduct of the study. B.A. Van Tine also reports consultancy from Ayala, Bayer, Adaptimmune Limited, Apexigen Inc., Cytokinetics Inc., Daiichi Sankyo, Deciphere Pharm., Epizyme, GSK, and ADRx; research grants from Merek, GSK, Tracon, and Pfizer; speaker fees from Novartis and Lilly; and licensing from Accuronix Therapeutics. B.A. Van Tine also reports a patent “Patent on the Use of ME1 as a Biomarker, Patent on ALEXT3102”. S.M. Lee reports grants from Plexxikon and FDA during the conduct of the study; S.M. Lee also reports grants from Merck and Roche/Genentech, as well as personal fees from PTC Therapeutics outside the submitted work. A.C. Hirbe reports other support from Springworks Therapeutics and AstraZeneca, as well as grants from Tango Therapeutics outside the submitted work. R. Rabadan is the founder of Genotwin and a member of the SAB of AimedBio; neither of these activities have relation to the work in this manuscript. G.K. Schwartz reports grants from FDA and other support from Plexxikon during the conduct of the study, as well as other support from Pfizer outside the submitted work. No disclosures were reported by the other authors.
Supplementary Material
Pharmacokinetics
Multiplex Immunofluorescence
DLT Model Estimates
Pharmacokinetic parameters
Acknowledgments
This research was supported by the FDA under award number 1R01FD005745 to G.A. Manji and G.K. Schwartz, and Plexxikon, Inc. FDA-R01 (1R01FD005745 to G.A. Manji and G.K. Schwartz) and the study was partly funded by Plexxikon, Inc.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
This article is featured in Highlights of This Issue, p. 5441
Footnotes
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
Authors' Contributions
G.A. Manji: Conceptualization, resources, data curation, formal analysis, supervision, funding acquisition, investigation, writing–original draft, writing–review and editing. B.A. Van Tine: Resources, investigation, writing–review and editing. S.M. Lee: Conceptualization, resources, data curation, formal analysis, funding acquisition, validation, investigation, methodology, writing–review and editing. A.G. Raufi: Data curation, investigation, writing–original draft, writing–review and editing. I. Pellicciotta: Conceptualization, data curation, formal analysis, validation, investigation, writing–review and editing. A.C. Hirbe: Investigation, writing–review and editing. J. Pradhan: Investigation, writing–review and editing. A. Chen: Investigation, writing–review and editing. R. Rabadan: Data curation, investigation, writing–original draft, writing–review and editing. G.K. Schwartz: Conceptualization, resources, formal analysis, funding acquisition, investigation, methodology, writing–review and editing.
References
- 1. Seddon B, Strauss SJ, Whelan J, Leahy M, Woll PJ, Cowie F, et al. Gemcitabine and docetaxel versus doxorubicin as first-line treatment in previously untreated advanced unresectable or metastatic soft-tissue sarcomas (GeDDiS): a randomised controlled phase 3 trial. Lancet Oncol 2017;18:1397–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Worden FP, Taylor JMG, Biermann JS, Sondak VK, Leu KM, Chugh R, et al. Randomized phase II evaluation of 6 g/m2 of ifosfamide plus doxorubicin and granulocyte colony-stimulating factor (G-CSF) compared with 12 g/m2 of ifosfamide plus doxorubicin and G-CSF in the treatment of poor-prognosis soft tissue sarcoma. J Clin Oncol 2005;23:105–12. [DOI] [PubMed] [Google Scholar]
- 3. Dancsok AR, Gao D, Lee AF, Steigen SE, Blay J-Y, Thomas DM, et al. Tumor-associated macrophages and macrophage-related immune checkpoint expression in sarcomas. Oncoimmunology 2020;9:1747340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Prada CE, Jousma E, Rizvi TA, Wu J, Dunn RS, Mayes DA, et al. Neurofibroma-associated macrophages play roles in tumor growth and response to pharmacological inhibition. Acta Neuropathol 2013;125:159–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhang S-Y, Song X-Y, Li Y, Ye L-L, Zhou Q, Yang W-B. Tumor-associated macrophages: a promising target for a cancer immunotherapeutic strategy. Pharmacol Res 2020;161:105111. [DOI] [PubMed] [Google Scholar]
- 6. Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, Luo J, et al. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res 2014;74:5057–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Patwardhan PP, Surriga O, Beckman MJ, de Stanchina E, Dematteo RP, Tap WD, et al. Sustained inhibition of receptor tyrosine kinases and macrophage depletion by PLX3397 and rapamycin as a potential new approach for the treatment of MPNSTs. Clin Cancer Res 2014;20:3146–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tap WD, Wainberg ZA, Anthony SP, Ibrahim PN, Zhang C, Healey JH, et al. Structure-guided blockade of CSF1R kinase in tenosynovial giant-cell tumor. N Engl J Med 2015;373:428–37. [DOI] [PubMed] [Google Scholar]
- 9. Iasonos A, Wilton AS, Riedel ER, Seshan VE, Spriggs DR. A comprehensive comparison of the continual reassessment method to the standard 3 + 3 dose escalation scheme in Phase I dose-finding studies. Clin Trials 2008;5:465–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cheung YK, Chappell R. Sequential designs for phase I clinical trials with late-onset toxicities. Biometrics 2000;56:1177–82. [DOI] [PubMed] [Google Scholar]
- 11. Lee SM, Ying Kuen C. Model calibration in the continual reassessment method. Clin Trials 2009;6:227–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Biard L, Cheng B, Manji GA, et al. A simulation study of approaches for handling disease progression in dose-finding clinical trials. J Biopharm Stat 2021:31;156–67. [DOI] [PubMed] [Google Scholar]
- 13. Bobrow MN, Shaughnessy KJ, Litt GJ. Catalyzed reporter deposition, a novel method of signal amplification. II. Application to membrane immunoassays. J Immunol Methods 1991;137:103–12. [DOI] [PubMed] [Google Scholar]
- 14. Stack EC, Wang C, Roman KA, Hoyt CC. Multiplexed immunohistochemistry, imaging, and quantitation: a review, with an assessment of Tyramide signal amplification, multispectral imaging and multiplex analysis. Methods 2014;70:46–58. [DOI] [PubMed] [Google Scholar]
- 15. Widemann BC, Lu Y, Reinke D, Okuno SH, Meyer CF, Cote GM, et al. Targeting sporadic and neurofibromatosis type 1 (NF1) related refractory malignant peripheral nerve sheath tumors (MPNST) in a phase II study of everolimus in combination with bevacizumab (SARC016). Sarcoma 2019;2019:7656747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kim A, Lu Y, Okuno SH, Reinke D, Maertens O, Perentesis J, et al. Targeting refractory sarcomas and malignant peripheral nerve sheath tumors in a phase I/II study of sirolimus in combination with ganetespib (SARC023). Sarcoma 2020;2020:5784876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tap WD, Gelderblom H, Palmerini E, Desai J, Bauer S, Blay J-Y, et al. Pexidartinib versus placebo for advanced tenosynovial giant cell tumour (ENLIVEN): a randomised phase 3 trial. Lancet 2019;394:478–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Pharmacokinetics
Multiplex Immunofluorescence
DLT Model Estimates
Pharmacokinetic parameters