

Abstract
The classic Th1/Th2 dogma has been significantly reshaped since the subsequent introduction of several new T helper cell subsets, among which the most intensively investigated during the last decade is the Th17 lineage that demonstrates critical pathogenic roles in autoimmunity and chronic inflammation – including the highly prevalent dry eye disease. In this review, we summarize current concepts of Th17-mediated disruption of ocular surface immune homeostasis that leads to autoimmune inflammatory dry eye disease, by discussing the induction, activation, differentiation, migration, and function of effector Th17 cells in disease development, highlighting the phenotypic and functional plasticity of Th17 lineage throughout the disease initiation, perpetuation and sustention. Furthermore, we emphasize the most recent advance in Th17 memory formation and function in the chronic course of dry eye disease, a major area to be better understood for facilitating the development of effective treatments in a broader field of autoimmune diseases that usually present a chronic course with recurrent episodes of flare in the target tissues or organs.
Keywords: Th17, Plasticity, Immunological memory, Dry eye disease
1. Introduction
Dry eye disease (DED) is a multifactorial condition of the ocular surface, characterized by chronic self-perpetuating inflammatory damages of the cornea, conjunctiva, and the covering tear film, along with the Meibomian and lacrimal glands contributing to the stability of the tear film, and neurosensory system innervating the ocular surface. DED is arguably the most common ophthalmologic condition for which patients visit eye care professionals, with estimated >16 million US adults having this disease and an increasing prevalence with age [1]. DED can be broadly classified into two categories based on the lack of aqueous tears or not – aqueous-deficient form (with reduced tear quantity, for example Sjögren’s syndrome) and evaporative form (with reduced tear quality, for example Meibomian gland dysfunction) [2,3], however, it is worth to be noted that the two types of DED are not mutually exclusive, and coexistence of deficiencies of tear quality and quantity is not uncommon in human patients [4], and thus the most recent Dry Eye Work Shop (DEWS II) report has advocated a continuum classification scheme to highlight such overlap between the two primary types of DED [5]. Nevertheless, current evidence has suggested that no matter how the disease is initially induced, characteristic features of both types may occur as disease progresses, and common pathophysiological processes are shared among various etiologies of DED.
It is well-known that continued inflammation plays a key role in DED pathohenesis [6] and lymphocytic infiltration (primarily CD4+ T cells with small numbers of B cells) in lacrimal glands is a hallmark of Sjögren’s DED [7,8]. In the past two decades, considerable attention has been devoted to defining the immunopathological changes at the ocular surface in both Sjögren’s and non-Sjögren’s DED via various animal models including the popular desiccating stress-induced mouse models that were similarly established by Pflugfelder group [9] and us [10] independently in early 2000s and have become a standard non-Sjögren’s DED model ever since. It is now recognized that the loss of immune homeostasis at the ocular surface and the resultant activation of CD4+ T cells are the key drivers leading to the disruptions of the tear film stability, corneal epithelial barrier, and corneal sensation [5,11]. Furthermore, a specific subset of CD4+ T cells – IL-17-secreting CD4+ T cells (Th17) has been revealed as the central player in causing autoimmune damage in DED supported by both experimental and clinical data [12–14]. In this review, we will update the most recent evidence demonstrating the pathogenic roles of autoimmunity in causing ocular surface inflammation in DED by highlighting the critical functions of both effector and memory Th17 cells in disease initiation, perpetuation, and chronicity.
2. Immune homeostasis of normal ocular surface
The ocular surface comprises of a continuous mucosal lining of cornea and conjunctiva, extending to the mucocutaneous junctions of the lid margins [15]. Despite being constantly exposed to outside environment, the ocular surface remains integrated and uninflamed while keeping its visual function. This ability to maintain immune homeostasis is mediated through both physical barrier function and active mechanisms, encompassing epithelium, stroma, nerve and resident immune cells in ocular surface. The avascular nature of normal cornea is essential for its transparency and the “immune privileged” status by creating an access barrier for circulating immune cells to enter the site [16]. To maintain the avascularity, corneal epithelium constitutively expresses soluble vascular endothelial growth factor receptor-1 (sVEGFR-1) to inhibit VEGF-A mediated new blood vessel formation (hemangiogenesis) [17], and VEGFR-3, serving as a trap, to prevent VEGF-C and VEGF-D mediated new blood and lymphatic vessels formation (lymphangiogenesis) [18].
Normal cornea has no T or B lymphocytes, but is endowed with a significant myeloid population characterized as CD11b+CD3−CD19−phenotypes, including the major CD11b+CD11c− macrophages/monocytes in the deep stroma, along with some CD11b+CD11c+ dendritic cells in the anterior stroma and a few CD11blo/−CD11c+ Langerhans cells in the epithelium [19–24]. These cells are collectively believed to be capable of functioning as antigen-presenting cells (APC) in adaptive immune response, and they are phenotypically “immature” characterized as low expression levels of MHC class II (MHC-II) along with absence of costimulatory molecules B7 (CD80 and CD86) and CD40. These immature APC predominantly reside at corneal periphery (throughout the entire stroma) and decrease in number gradually toward the corneal center (primarily in anterior stroma) [22,25]. The immature status of corneal resident APC, along with absence of lymphocytes in normal cornea contributes to the immune quiescence by not only avoiding effector cell activation but inducing immune tolerance [21,26].
Corneal epithelium also constitutively expresses a variety of critical immunoregulatory factors, including programmed death-ligand 1 (PD-L1), Fas ligand (FasL), pigment epithelial-derived factor (PEDF), and thrombospondin-1 (TSP-1) [27–30]. PD-L1 is a newer member of B7 family, and its ligation with the receptor programmed death (PD)-1 on activated T cells leads to suppression of T cells [31]. PD-L1 KO mice show spontaneously significant T cell infiltration in cornea [32]. In addition, PD-L1 actively inhibits corneal hemangiogenesis and thus contributes to corneal avascularity [33]. Similarly, FasL, a member of tumor necrosis factor (TNF) family, can lead to apoptotic cell death by binding to its receptor Fas that is expressed by a variety of cells and tissues, including T cells, and prevent neovascularization [34,35]. Two forms of FasL have been identified including the anti-inflammatory, soluble form (sFasL) that can antagonize the function of the other pro-inflammatory, membrane-bound form (mFasL) [36]. Although sFasL is the predominant form of FasL expressed in the retina of mice [37], it remains unclear what form(s) of FasL that is constitutively expressed in the cornea. Both PD-L1 and FasL constitutively expressed in corneal epithelium may trigger apoptosis of invading effector T cells that infiltrate in the cornea in response to inflammation. PEDF is a ubiquitously expressed glycoprotein belonging to the serine protease inhibitor family [38]. TSP-1 is a major activator of latent TGF-β, promoting the production of activated TGF-β that serves as a critical anti-inflammatory cytokine [39,40]. Normal corneal epithelium-derived PEDF and TSP-1 have both been shown to potently suppress APC activation [29,30]. Both factors exert anti-angiogenic function [38,41] with PEDF showing additional neurotropic roles [42].
The cornea is among the most densely innervated tissues, and corneal nerves play a crucial role for ocular surface homeostasis by protecting the cornea from irritants (through regulating tear secretion and the blink reflex) and by secreting a variety of neuropeptides that are essential for maintaining the epithelial and stromal cells [43]. Healthy innervation in cornea has been shown to regulate corneal epithelial cell and limbal stem cell survival [44,45], and suppress corneal neovascularization [46]. Corneal neuropathy has been observed in DED patients and animal models [47–50]. Specifically, among various nerve-derived factors, neuropeptides SP and vasoactive intestinal peptide (VIP) are constitutively secreted by nerve endings in normal cornea, with SP contributing to corneal epithelial homeostasis and VIP playing anti-inflammatory functions [51,52].
3. Induction of adaptive T cell immunity in dry eye disease
The immune homeostasis of ocular surface is disrupted in DED, mediated by both innate and adaptive immunity. Innate response serves as the immediate, non-specific reaction to various insults, and involve the mucosal barrier and innate immune cells as well as their secreted cytokines and chemokines. Among the innate cellular components, dendritic cells, monocytes, and macrophages, once activated, are capable of efficiently inducing the subsequent, specific adaptive immunity.
3.1. Immune response of ocular surface mucosal lining
Desiccating stress or hyperosmolar stress on ocular surface [15], either direct or consequent to lacrimal or Meibomian glands damage that results from extrinsic insults or intrinsic changes, can be the initiating factors driving the development of DED (Fig. 1). Stressed ocular surface epithelial cells quickly up-regulate the levels of activated mitogen-activated protein kinases (MAPK), including c-jun N-terminal kinases (JNK)-1/2, extracellular-regulated kinases (ERK)-1/2, and p38 in hours [53–55], which may subsequently activate downstream kinases and transcription factors such as NFκB, leading to early up-regulation of pro-inflammatory cytokines TNF-α, IL-1β, and IL-6 at the ocular surface [53,55–57]. In addition, desiccating stress can elicit the cellular signal of damage-associated molecular patterns (DAMPs), which activate corresponding pattern recognition receptors (PRR) such as toll-like receptor 4 (TLR4) – an essential innate immune activation mechanism α in cornea, demonstrated by translocation of TLR4 in epithelial cells (from cell inside to cell surface) and up-regulation in stroma cells [58]. TLR4 activation further leads to downstream caspase-8 activation facilitating the assembly and activation of inflammasomes NLRP12 and NLRC4, thereby promoting the activation and secretion of pro-inflammatory cytokines such as IL-1β [59]. In addition to TLR4-mediated pathway, increased reactive oxygen species (ROS) upon desiccating stress exposure also induce activation of caspase-8 and NLRP3 inflammasome, further promoting the production of bioactive IL-1β [60]. Both IL-1β and TNF-α have been shown to promote corneal expression of matrix metalloproteinase-9 (MMP-9) [53,61], a critical proteolytic enzyme cleaving epithelial basement membrane components and tight junction proteins (such as ZO-1 and occludin) and thus leading to corneal barrier disruption [53,62].
Figure 1. Th17-mediated autoimmunity in dry eye pathogenesis.
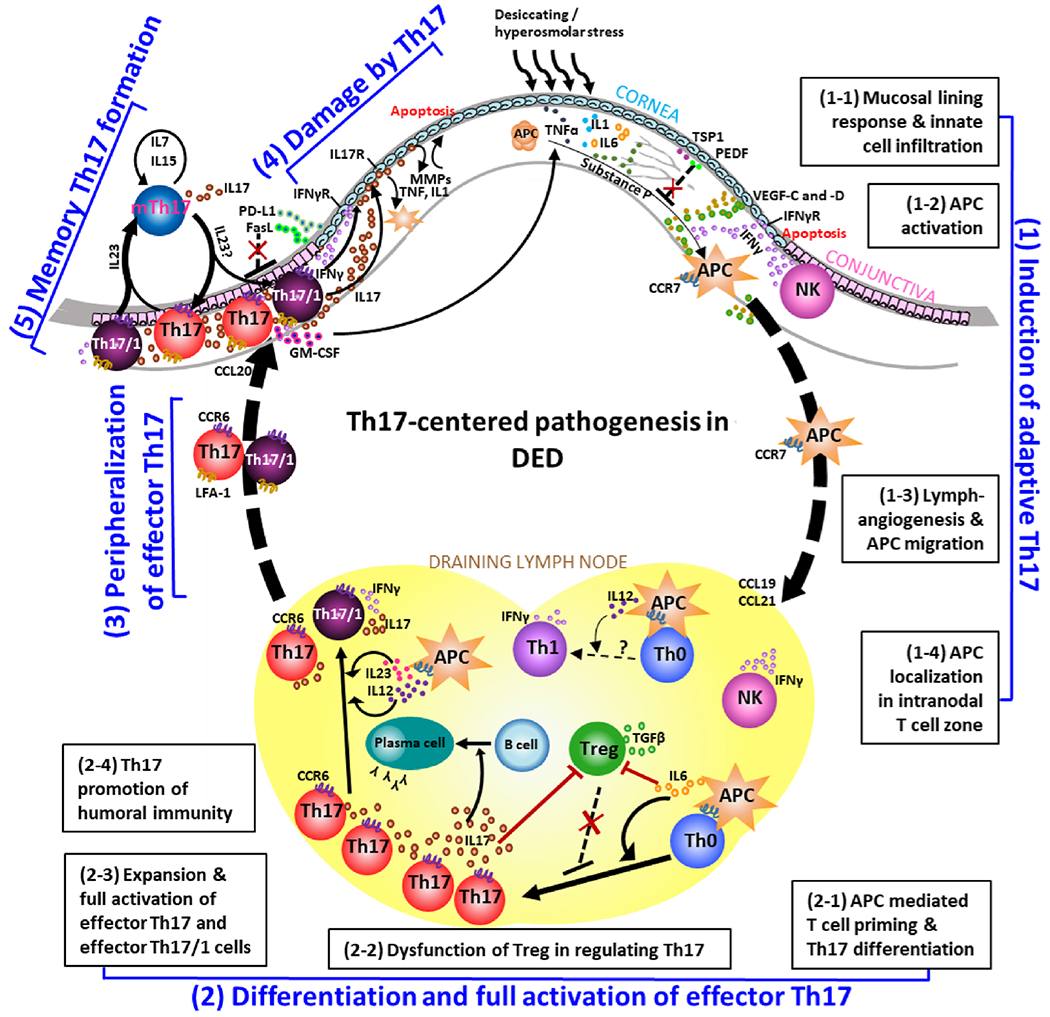
(1) Induction of adaptive Th17 immunity is initiated by the early activation of innate immunity in response to desiccating stress or hyperosmolar stress on ocular surface, including release of TNF-α, IL-1β, and IL-6 by mucosal lining cells, infiltration of monocytes, macrophages, NK cells (1-1), and activation of antigen-presenting cells (APC) (1-2), which subsequently egress via newly-formed lymphatic vessels (1-3) to draining lymph nodes, specifically intranodal T cell zone, guided by chemokine-receptor axis (1-4). (2) In the draining lymph nodes, APC prime naive T cells (Th0) and promote their differentiation into Th17 cells (2-1), a process accompanied by dysfunction of inflammation-limiting regulatory T cells (Treg) (2-2). Initially differentiated Th17 cells undergo clonal expansion and full activation by gaining effector function of producing both IL-17 and IFN-γ under the stimulation of IL-23 and IL-12 (2-3). Effector Th17 cells can further promote B cell differentiation to plasma cells (2-4). (3) Tissue-specific effector Th17 cells generated in the regional draining lymphoid compartment home to the ocular surface via chemokine-receptor axis and adhesion molecules. (4) Effector Th17 cells cause ocular surface epitheliopathy, inflammatory cascade, and neurosensory abnormalities via IL-17, IFN-γ, and GM-CSF. (5) With the resolution of acute inflammation, a small part of effector Th17 pool (both eTh17 and eTh17/1 cells) becomes long-lived memory Th17 cells (mTh17) which maintains chronic inflammation.
3.2. Innate immune cells recruitment and infiltration in ocular surface
Early infiltration of CD11b+ monocytes and macrophages in DED corneas is a hallmark consistently found in various animal models [56,58,63], and increased density and distribution of leukocytes in cornea have been documented in both Sjögren’s syndrome and non-Sjögren’s syndrome DED patients [64]. Influx of these inflammatory cells relies on the chemotactic gradient between the periphery and corneal tissues created by higher expression of specific chemokines on the ocular surface in DED, including macrophage inflammatory protein 1α (MIP-1α, or CCL3) and MIP-1β (CCL4) [65]. Chemokines are small molecular weight cytokines with chemoattractant properties that serve an essential function in immunity by coordinating the trafficking of specific immune cells between different anatomic sites [66]. In some cases, multiple chemokines may interact with the same chemokine receptor. Increased levels of these chemokines at ocular surface in DED can lead to tissue-specific recruitment of peripheral circulating monocytes that express corresponding specific receptors, including those expressing CCR1 for chemokine MIP-1α, and CCR5 for both chemokines MIP-1α and MIP-1β [67]. In addition, chemokine monocyte chemotactic protein 1 (MCP-1, or CCL2) and its receptor CCR2 are also involved in the recruitment of CD11b+ cells in DED [68]. In fact, the corneal infiltrating CD11b+ cells in DED have shown significant up-regulation of CCR1, CCR2, and CCR5 [69]. These infiltrated monocytes and macrophages become activated under the stimulation of environmental TNF-α and IL-1β in cornea, and can subsequently produce the same inflammatory cytokines by themselves, thus amplifying the ocular surface inflammation [58,68]. In addition, Natural Killer (NK) cells are also significantly increased in the conjunctiva shortly after desiccating stress, and these NK cells are actively secreting the inflammatory cytokine IFN-γ that directly disrupts integrality of the ocular surface barrier [70].
3.3. Ocular surface APC activation, migration, and engagement with lymphoid T cells
Besides direct tissue damages, the other important consequence of innate immune activation in DED is the maturation of ocular surface influxed and resident APC, including dendritic cells and Langerhans cells [64,71–73]. In normal healthy cornea, the preponderance of APC is in an “immature” status that is critical to maintain ocular surface immune quiescence and contributes to immune privilege of cornea [74]. However, in DED, significantly increased inflammatory cytokines IL-1β and TNF-α in the ocular surface overturn the microenvironmental anti-inflammatory mechanisms mediated by factors such as TSP-1 and PEDF [29,30] and promote resident and infiltrating APC to acquire “mature” phenotypes by up-regulating their MHC-II expression [56], which is required for APC presentation of antigenic epitopes to T cells. In addition, activated NK cells also promotes APC up-regulation of MHC-II and co-stimulators B7 [70], and depletion of NK cells has been shown to lead to decreased frequency and maturation of dendritic cells in DED [75]. APC maturing process is accompanied by the presumed “autoantigen” capture and processing. To date, the nature of the hypothesized ocular surface autoantigen(s) in DED remains unclear, although kallikrein-13 has been proposed as a putative candidate based on its expression in ocular surface tissues and reactivity with sera from DED mice [76,77]. Additionally, immunization of rats with a kallikrein family protein led to marked lymphocytic infiltration in the lacrimal glands [78].
These mature, antigen-bearing APC are essential for the induction of T cell immunity in DED, demonstrated by that depletion of ocular surface APC prevents T cell activation in DED [79]. For efficient activation of naive T cells that primarily reside in the T cell zone in the draining lymph nodes, mature APC have to first gain spatial proximity to the distantly located T cells. Thus, activated APC in ocular surface acquire the ability to the draining lymph nodes of the eye. This trafficking process is tightly regulated by the specific chemokine-receptor axis. Corneal mature MHC-II+ APC in DED show significantly enhanced expression of CCR7, a chemokine receptor guiding APC egress toward the draining lymph nodes, specifically intranodal T cell zone, through the guidance of enriched environmental CCR7 ligands – CCL19 and CCL21 [69,80]. Topical blockade of CCR7 at the ocular surface has been shown to sufficiently prevent the migration of corneal APC to draining lymph nodes and subsequent T cell activation in DED [69]. In addition to chemokines, ocular surface APC have to gain access to lymphoid compartment via afferent lymphatic vessels which are absent in normal uninflamed cornea. Interestingly, there is considerable and exclusive growth of lymphatic, but not blood, vessels in DED cornea, that starts early after DED induction from peripheral cornea and advances into central cornea with the progression of the disease. This selective corneal lymphangiogenesis in DED is dependent on lymphangiogenic-specific VEGF-C and VEGF-D and their receptor VEGFR-3 [81–83]. Co-localization of CCR7+ cells with CCL19/21 within corneal lymphatic vessels has been demonstrated in DED cornea [80]. Consistently, dendritic cells in the peripheral cornea where there are abundance of lymphatic vessels in DED display significantly increased migratory kinetics [84]. Once relocated to the appropriate anatomical compartment within the regional lymph nodes, APC are able to prime naive T cells via delivering three signals – antigen on MHC molecule, co-stimulation, and polarizing cytokines [85,86].
4. Activation and expansion of effector Th17 cells in the eye-draining lymphoid tissue
To date, substantial evidence demonstrate that CD4+ T cell are the predominant lymphocytes infiltrating in the ocular surface (primarily in the conjunctiva) [87,88] and plays a central pathogenic role in both Sjögren’s and non-Sjögren’s DED [89–92]. Recent advance in the field has shed considerable light on the precise mechanisms including which subsets of CD4+ T cells are activated and how they cause ocular surface inflammatory epitheliopathy via coordinating with an array of other key cellular and molecular factors (Fig. 1).
4.1. T cell activation and effector Th17 differentiation and plasticity
Regional eye-draining lymph nodes are the primary site of T cell activation in DED. Mature migratory CCR7+ APC in local draining lymph nodes provides antigenic peptide to cognate naive CD4+ T cells via its highly expressed MHC-II, along with co-stimulatory signals via B7 (CD80 and CD86) and CD40 molecules [69,70,88]. The primed T cells accordingly up-regulate activation markers of CD69 and CD154 (CD40 ligand, CD40L) [93]. Once the immune synapse between APC and T cells is formed, the environmental polarizing cytokines, primarily secreted by APC or infiltrating monocytes, are predominant factors determining the differentiation fate of primed CD4+ T cells. It is known that the ligation of CD154 with CD40 on APC promotes the production of IL-12 by APC [94], and increased levels of IL-12 in the milieu of draining lymph nodes of DED have been demonstrated [95]. In addition, CD4+ T cells also show up-regulation of IL-12 receptor (IL-12R) expression [93], and IL-12/IL-12R signaling promotes polarization of naive CD4+ T to IFN-γ-expressing Th1 cells, as evidenced by increased levels of the Th1 signature cytokine IFN-γ in DED lymph nodes, conjunctiva, and tears [93,96–99] despite no confirmation of the cellular source of IFN-γ in these studies. Subsequently, the concept of classical Th1 dominance in DED was revised with the characterization of a newly-defined CD4+ T cell subset – IL-17-expressing Th17 cells in DED [12,100–104]. Initial differentiation of naive T cells towards Th17 lineage is in general driven by the pro-inflammatory cytokine IL-6 in the presence of TGF-β [105,106], and increased expression of IL-6 in DED lymph nodes has been specifically noted [100,107]. Moreover, the heightened levels of IL-2 and IL-23 in the lymphoid compartment in the acute DED induction stage [95,108] further promotes differentiated Th17 cells to proliferate, expand, and gain full effector functions. Unlike most T cell differentiating and activating cytokines that are mainly produced by APC, the heightened IL-2 is principally secreted by activated effector T cells themselves and acts in an autocrine fashion. In DED, mature APC have been suggested to be the major microenvironmental source of IL-6, TGF-β, and IL-23 [57]. Fully activated Th17 cells are capable of secreting effector cytokines, including the signature cytokine IL-17 and others such as granulocyte-macrophage colony-stimulating factor (GM-CSF), exerting pathogenic function in DED [100,109].
Interestingly, unlike Th1 subset that is regarded as a terminally differentiated CD4+ T cell lineage and stays relatively stable, the Th17 cells show significant phenotypic and functional plasticity demonstrated by their ability of acquiring features of other lineages under stimulation of certain signals in the microenvironment [110]; this concept has led us to revisit the cellular source of the heightened IFN-γ expression in the late acute stage of DED (when NK cell response subsides while adaptive immunity emerges), which had been thought of dominantly from classic Th1 cells by default although no cellular characterization was done. In non-Sjögren’s DED, two subsets of effector Th17 cells have been identified by our group, including IFN-γ−IL-17+ “single-positive” Th17 and IFN-γ+IL-17+ “double-positive” Th17/1 [95]. In fact, quantitative analysis of IFN-γ+IL-17’CD4+ Th1 cells showed no numeric changes [95,96], suggesting that simple link of increased IFN-γ levels in DED to classic Th1 lineage without examining its cellular source is inaccurate; instead, the major source of IFN-γ in DED could be the double-positive Th17/1 cells in the disease progression stage while NK cells serve as the primary source in early disease induction phase when adaptive immunity has not been activated. Further studies have demonstrated that the increased IFN-γ in DED is indeed an integral part of Th17 immunity, as evidenced by the ability of single-positive Th17 cells to convert into double-positive Th17/1 cells via stimulation of environmental IL-12 and IL-23 in DED [95]. Importantly, double-positive Th17/1 cells are more pathogenic than single-positive Th17 cells, and Th17/1 are required to induce severe acute ocular surface inflammation in DED; in contrast, the classic IL-17−TFN-γ+ Th1 cells isolated from DED mice are unable to transfer the disease phenotype to normal animals [95]. Taken together, effector Th17 response characterized by their dynamic and coordinated production of various cytokines including IL-17, IFN-γ and GM-CSF, plays a major pathogenic role in DED. In Sjögren’s syndrome, significant increase of IL-17 in tears, serum, lacrimal glands and salivary glands have been demonstrated [102–104,111,112]. While pathogenic roles of Th17 and IL-17 in lacrimal gland inflammation remains unclear, limited experimental data focusing on salivary glands have suggested a critical role of Th17 immunity in sialoadenitis [91]. Overexpression of IL-17 alone in salivary glands in Sjögren’s syndrome-resistant mouse strain is sufficient to induce characteristic disease profiles [113]. Immunization of mice with type 3 muscarinic acetylcholine receptor (M3R) peptides has been shown to lead to increased serum levels of anti-M3R antibodies and low saliva volume, accompanied by CD4+ T cell and B cell infiltrates in the salivary glands [114]. Anti-M3R autoantibodies are believed to be a marker for Sjögren’s syndrome, and suggested to contribute to the glandular dysfunction in the disease [115–118]. Besides autoantibody formation, the M3R immunized mice have been shown to produce M3R-reactive T cells secreting both IL-17 and IFN-γ in the spleen, and adoptive transfer of splenocytes from these immunized mice into naive animals results in decreased saliva production and increased anti-M3R autoantibodies [114]. Treatment of the recipient animals with a RORγt (the master transcription factor for Th17 cells) antagonist can effectively reduce both IL-17 and IFN-γ levels in the adoptive transfer recipients [119]. Furthermore, IFN-γ+IL-17+ “double-positive” Th17/1 cells are reported to be detected in the salivary glands and lymph nodes from patients with Sjögren’s syndrome [120]. These data indicate a potential pathogenic role of double-positive Th17/1 cells in Sjögren’s syndrome; however, definitive evidence supporting the contribution of Th17 immunity including its plasticity to Sjögren’s syndrome, particularly to Sjögren’s dry eye, along with the critical questions of how and where those observed Th17 effectors are generated remain to be further explored.
4.2. Modulation of Th17 response
4.2.1. Regulatory T cells
The dominant players on the immunoregulatory arm that restrict and suppress excessive inflammation, including in autoimmunity, are specialized CD4+ regulatory T cells (Treg), characterized by their high expression of CD25 (the high affinity receptor subunit for IL-2 to maintain survival and proliferation of Treg) and Foxp3 (the master transcriptional factor for the development and function of Treg), which are in contrast to pro-inflammatory CD4+ effector T cell subsets Th1, Th2 and Th17. To date, two types of Treg have been defined, including the larger population that is developed during the normal process of T cell maturation in the thymus (termed “natural Treg”, nTreg or tTreg) and the smaller but more antigen-specific population that is induced in the periphery from naive T cells in the presence of IL-2 and TGF-β or after encounters with foreign antigens (termed “induced Treg”, iTreg or pTreg) [121,122]. Both subsets of Treg work in synchrony to maintain peripheral tolerance and immune homeostasis through dampening naive T cell priming or attenuating effector T cell function [123,124]. In DED the regulatory function of Treg is compromised demonstrated by their inability to suppress IL-17 production by effector CD4+ T cells despite that the number of Treg is minimally affected [96,100]. The characteristic phenotype of dysfunctional Treg is down-regulation of Foxp3 expression [125], which has been demonstrated in DED mice [100]. It remained to be elucidated whether other critical Treg function-associated molecules are altered in DED as well, including surface expression of cell contact-dependent co-inhibitory molecules CTLA-4 and GITR as well as secreted anti-inflammatory cytokines IL-10 and TGF-β by Treg. It is known that TGF-β is required for the generation of both Treg and Th17, therefore the presence or absence of the other cytokine IL-6 is a key determinant skewing the immune balance between the two reciprocally interconnected and functionally opposed cell subsets [126,127]. As increased IL-6 levels have been consistently observed in both ocular surface and draining lymphoid tissue in DED [53,55,56,100,107], it is plausible to attribute IL-6 as one of the critical factors leading to Treg dysfunction while promoting Th17 in DED. Significantly higher Th17/Treg ratio and lower Treg numbers in the peripheral blood of Sjögren’s syndrome patients than healthy subjects have been reported [128,129]. In addition, in vivo blockade of IL-17 effectively restores Treg function in DED [100], suggesting that IL-17 may also directly or indirectly contributes to Treg dysfunction. Nevertheless, the detailed mechanisms underlying Treg dysfunction in DED, especially their interaction with Th17-associated factors, have yet to be investigated. Furthermore, even though Treg show compromised function in DED, complete depletion of Treg using an anti-CD25 antibody or in CD25 knockout mice led to even worsened disease, while reconstitution of normal Treg in DED mice conferred the recipients resistance to disease induction [89,111,130,131], suggesting that Treg, probably primarily nTreg, play important regulatory roles in DED pathogenesis. In a pilot study in patients with Sjögren’s syndrome, low-dose IL-2 treatment has been demonstrated to successfully restore functional Treg and the Th17/Treg ratio in the periphery, along with the reductions in the use of glucocorticoid and disease-modifying anti-rheumatic drugs [129]. These promising results suggest that restoration of normal Treg function can be a novel therapeutic strategy in treating DED.
CD8+ regulatory T cells have also been implicated in DED pathogenesis by a study demonstrating that depletion of CD8+ T cells in mice promotes the generation of Th17 response and leads to a more severe ocular surface disease [132]. The precise subset is assumed to be CD8+CD103+ cells, which are presumably to limit the Th17 immunity in DED via suppressing the process of APC-mediated Th17 generation, but not restricting already activated Th17 cells [132].
4.2.2. Neuromediators
As described above, healthy corneal innervation with the physiological levels of neuropeptides at the ocular surface is essential to corneal epithelial homeostasis [133–135]. Substance P and calcitonin gene-related peptide (CGRP) are the two main neuropeptides expressed by corneal nerves [136], and they originate from sensory nerve [137]. Mice with genetic deletion of functional SP receptor – neurokinin-1 receptor (NK1R) develop corneal epitheliopathy [138]. In a mouse model of Sjögren’s syndrome, significantly reduced CGRP-containing corneal nerves is observed with the disease progression [139]. However, in DED mice and refractory surgery patients who subsequently developed dry eye symptoms, abnormally increased expression of SP has been noted [140,141], which in turn induces neurogenic inflammation and propagates inflammatory cascade in DED. The heightened levels of sensory nerve-derived SP promote corneal APC mobilization and maturation, which subsequently induces effector Th17 response in DED [140]. Elevated levels of SP in draining lymph nodes is accompanied by increased proportion of NK1R+ Treg among total Treg in DED, and NK1R+ Treg show significantly compromised regulatory function as compared to NK1R− Treg [142]. Co-culture of normal Treg with SP leads to the loss of function in Treg, demonstrating that SP contributes to Treg dysfunction via direct acts on Treg [142]. It remains to be determined whether SP also has a direct effect on Th17 differentiation and function besides via enhancing APC-mediated Th17 activation [140,143] and suppressing Treg-mediated Th17 restriction [142].
4.3. Th17-dependent humoral immunity
B cell is the other major cellular component of adaptive immunity, and B cell-mediated humoral immunity participates in disease pathogenicity by primarily differentiating into plasma cells that actively produce autoantibodies causing self-tissue damages. Autoreactive B cells are known crucial effectors mediating Sjögren’s syndrome evidenced by the presence of autoantibodies directed to ribonuclear proteins SS-A/Ro and SS-B/La, as well as to the M3R protein in both patients and animal models [112,115–118,144], along with the formation of ectopic germinal centers in exocrine glands in about one fourth patients [145,146]. However, the B cell-centric concept in the pathogenesis of Sjögren’s syndrome has been challenged by the failure of a recent clinical trial testing B cell depletion strategy with rituximab, in which no significant improvement in primary endpoints of dryness or pain has been achieved [147]. New evidence indicates that T cell immunity plays a critical pathogenic role in Sjögren’s syndrome by direct effector function or promotion of humoral immunity. Focal dominant CD4+ T cell infiltration over B cells in lacrimal glands has been reported in various animal models [7,112,116,148–151]. Similar infiltration pattern in the exocrine glands is also observed in patients, including larger CD4+ T cell population, particularly in the earlier stage of disease [152,153]. In Aire-deficient mouse model of Sjögren’s syndrome, after individual depletion of CD4+ T cells, CD8+ T cells, or B cells, only CD4+T cell-depleted group showed significant improvement of lacrimal gland pathology [154]. In contrast, in Id3 knockout mouse model, depletion of B cells led to significant disease improvement [155]. Furthermore, the glandular infiltrating CD4+ T cells are capable of secreting IL-17 [112], which promotes autoreactive B cell response and autoantibodies production in NOD mice [156], and is associated with ectopic germinal center formation in patients with primary Sjögren’s syndrome [157]. Taken together with the salivary gland evidence outlined in the “effector Th17” section above, Th17 immunity is suggested to play critical role in disease initiation and subsequent B cell activation in Sjögren’s syndrome [91,158,159]; these findings need to be further confirmed in studies focusing on lacrimal glands and ocular surface.
The role of B cell immunity in non-Sjögren’s DED along with its interaction with T cell immunity have not been extensively studied. In mice, only a long period (3 weeks) of desiccating stress has been shown to lead to increased serum antibodies against kallikrein-13 [77], suggesting that humoral immunity may participate in the pathogenesis of non-Sjögren’s DED in a similar temporal pattern (i.e. after the activation of T cell immunity) as indicated in Sjögren’s syndrome. Furthermore, adoptive transfer of serum or purified IgG collected from these DED mice to T cell-deficient nude mice has led to decreased tear production along with increased cytokines IL-1β, TNF-α, IFN-γ, and IL-17 in tears, as well as reduced conjunctival goblet cells [77], indicating that autoantibodies contribute to T cell-centered pathogenesis in non-Sjögren’s DED. Our recent work has shown that CD4+ T cells isolated from desiccation-induced DED mice promotes B cell proliferation and activation primarily via secreted IL-17, and activated B cells further up-regulate their own expression of IL-17 receptor, through which IL-17 further induces B cell class switching and differentiation to plasma cells [160], thus demonstrating a strong line of evidence of T cell-dependent B cell activation in DED pathology.
In addition to their major function of producing autoantibodies in autoimmunity, B cells may also serve as APC and secrete various inflammatory cytokines that facilitate T cell immunity; these areas have yet to be explored in DED pathogenesis.
5. Homing of effector Th17 cells to the ocular surface and the resulting tissue damage
Fully functional, tissue-specific effector Th17 cells generated in the regional draining lymphoid compartment subsequently migrate to the ocular surface, where they play pathogenic functions causing characteristic DED pathologies in the target tissues (Fig. 1), including (i) disruption of corneal epithelial barrier [161], (ii) decreased numbers and atrophy of conjunctival goblet cells [92,95,162], (iii) apoptosis of corneal and conjunctival epithelia [163], (iv) basal acinar cell proliferation and altered lipid production in Meibomian gland [164], and (v) nerve degeneration [49,165–170].
5.1. Infiltration of effector Th17 cells into ocular surface
At the ocular surface of DED, T cells primarily infiltrate in the conjunctiva [68,87,88,92]. To facilitate the ingress of effector T cells that are activated in the regional lymph nodes to the peripheral targeting tissues, T cells have to up-regulate specific chemokine receptors facilitating their interaction with blood vessel endothelium, and thus migrate to the target site via efferent blood vessels. These effector CD4+ T cells in DED preferentially up-regulate expressions of CCR5, CXCR3 and CCR6 [93,171,172], directing them to migrate towards ocular surface where higher levels of corresponding chemokines are present, including CCR5 ligands MIP-1α (CCL3), MIP-1β (CCL4), and regulated on activation, normal T-cell expressed and secreted (RANTES, or CCL5); CXCR3 ligands monokine induced by interferon-γ (MIG, or CXCL9), interferon-γ inducible protein 10 (IP-10, or CXCL10); and CCR6 ligand CCL20 [12,65,172–174]. Among these various T cell-associated chemokine/receptor axis, CCR6 is discretely expressed by Th17 cells, and CCR6/CCL20 axis has been shown functionally critical for the homing of effector Th17 cells to the ocular surface from the lymphoid compartment, demonstrated by significant abolishment of T cell infiltration in conjunctiva in the absence of CCR6 (knockout) or after topical neutralization of CCL20 in DED mice [171,172]. CCR2, a receptor expressed by monocytes, is also expressed by Th17 cells [175], particularly on the highly pathogenic IFN-γ+GM-CSF+ Th17 subset [176], and thus CCR2 also contributes to Th17 cell trafficking in DED as demonstrated by decreased T cell infiltration in conjunctiva after topical blockade of CCR2 in DED mice [68]. In addition to the requirement of guidance by chemokine-receptor axis, peripheralization of T cells is dependent on their additional expression of adhesion molecules, such as integrins, which facilitate their binding to extracellular matrix components. In DED, two important pairs including lymphocyte function-associated antigen-1(LFA-1, or integrin αLβ2) and its binding ligand intercellular adhesion molecule-1 (ICAM-1), as well as very late antigen-4 (VLA-4, integrin α4β1) and its binding ligands such as vascular cell adhesion molecule-1 (VCAM-1) have been demonstrated to mediate T cell journey to the ocular surface. Specifically, topical application of LFA-1 or VLA-4 small molecule antagonists to DED mice has been shown to lead to significant inhibition of T cell response in ocular surface [88,177]. Lifitegrast, a topical LFA-1 antagonist, has been recently approved by the FDA for DED patients therapy [178].
5.2. Th17-mediated ocular surface damage
Although predominantly infiltrated in the conjunctiva, effector Th17 cells cause entire ocular surface tissue damages in DED involving cornea, conjunctiva, and possibly eyelids, primarily through secreted cytokines. At the ocular surface of both DED animal models and patients, significantly increased levels of IFN-γ and IL-17 have been widely documented [12,56,100,179–182]; and our most recent identification of IFN-γ+IL-17+ “double-positive” Th17/1 cells in DED demonstrate that both these two critical cytokines are integral parts of Th17 immunity, and thus has reshaped the previous view on the pathogenic role of classic Th1 immunity in DED. In fact, there is significant infiltration of both IFN-γ−IL-17+ “single-positive” Th17 and IFN-γ+IL-17+ “double-positive” Th17/1, but not IFN-γ+IL-17− Th1, in DED conjunctiva [172,183].
Furthermore, a series of elegant adoptive transfer experiments have undoubtedly demonstrated that both single-positive Th17 and double-positive Th17/1 cells are pathogenic effectors in DED with Th17/1 particularly capable of exacerbating tissue destructions due to their ability of producing both IFN-γ and IL-17 [183]. Lack of new blood vessel formation in DED cornea [81] may limit the direct access of effector T cells to cornea, however, cytokines secreted by conjunctival infiltrating T cells can diffuse throughout the tear film and thus cause wide range of tissue damage in DED.
5.2.1. Ocular surface epitheliopathy
IL-17 has been shown to promote corneal epithelial production of both MMP-9 and MMP-3 by binding to its receptor IL-17RA that is constitutively expressed by ocular surface epithelia [100], resulting in corneal epithelial barrier disruption [12]. In vivo blockade of IL-17 in DED has been shown to effectively improve the corneal epitheliopathy [12,83]. IFN-γ can cause significant conjunctival epithelial squamous metaplasia characterized by loss of goblet cells and decrease of mucin production, accompanied with epithelial apoptosis [97,184]. Goblet cells are not only the primary source of mucins for tear film that contributes to the physical barrier [185], but also can directly induce immune tolerance via conditioning APC, and thus loss of conjunctival goblet cells may further exacerbate ocular surface inflammation in DED [186]. Topical neutralization of IFN-γ in DED has been shown to reduce the conjunctival goblet cell loss, and specific elimination of Th17-derived IFN-γ improves corneal epitheliopathy in DED [183].
5.2.2. Ocular surface inflammatory cascade
In addition to causing direct ocular surface tissue damage, Th17 propagates the pathological process and leads to a vicious cycle by amplifying the inflammation. IL-17 is able to efficiently induce endothelial and epithelial cells to secret IL-6, TNF-α, IL-1β and IL-8 [187,188], thus inducing a cytokine cascade at the ocular surface that can further recruit neutrophils and macrophages and activate APC, perpetuating tissue damages (including corneal nerves described in the next section) and promoting epitope spreading to a more diverse set of target antigens. In a mouse model of allergy-associated Meibomian gland dysfunction, Th17-mediated recruitment of neutrophils to conjunctiva was found to play a central role in causing obstruction of Meibomian gland in the eyelids, and blockade of Th17 immunity significantly reduced Meibomian gland plugging [189]. Furthermore, IL-17 is critical in promoting corneal lymphangiogenesis via inducing expression of pro-lymphangiogenic VEGF-D by corneal epithelial cells in DED [82], thus driving progressive ingrowth of lymphatic vessels that allows continuous trafficking of corneal APC to lymphoid compartment and enhances autoimmune response [190]. In addition, Th17-produced GM-CSF contributes to ocular surface inflammation in DED primarily through promoting ocular surface APC maturation and migration [109], and increased tear level of GM-CSF has been reported in DED patients [182].
5.2.3. Ocular surface neurosensory abnormalities
Cornea is the most densely innervated tissue in the body, and corneal sensory signals controlled by the trigeminal ganglion neurons are critical in maintaining the ocular surface homeostasis via regulating lacrimation, secretion of epitheliotrophic factors, and the blink reflex. Corneal degenerative neuropathy is a frequently encountered pathology in DED, associated with the ocular surface inflammation and corneal pain [191]. The up-regulated cytokines IL-1β, IL-6 and TNF-α at the ocular surface of DED sensitize various nociceptors expressed by sensory nerve terminals at the ocular surface, increase their activity and excitation, facilitate release of SP, and evoke nociceptive pain [192–195]. In addition to the peripheral pathway, central sensitization of trigeminal neurons is demonstrated as well involved in ocular pain sensation in DED [196]. However, with the progression of the ocular surface inflammation, structural damage of corneal nerves occurs evidenced by reduced intraepithelial nerve terminals in the epithelium as well as the decrease of their originating subbasal nerves in both pre-clinical models and patients [49,165–170], along with the reduced corneal sensitivity or hypoalgesia [49,50,165,167–169,197,198]. Furthermore, in chronic DED, altered gene expression and disturbed regeneration of corneal nerves (such as increased nerve tortuosity) [199–201] subsequent to acute nerve damage and chronic inflammation, may lead to persistent sensory hypersensitivity (hyperalgesia) or frequent spontaneous discharge (allodynia) [199], thus potentially contributing to chronic pain or discomfort in DED. The precise role of Th17 immunity in causing direct corneal nerve degeneration, regeneration, and the abnormal sensation, besides via indirectly enhancing the innate cytokine cascade, remains to be further investigated.
6. Th17 memory formation and chronic dry eye inflammation
6.1. Development and function of memory Th17 cells in chronic DED
Clinical DED often presents as a chronic disease, while the majority of mechanistic and therapeutic data derived from the pre-clinical models reflect an acute inflammatory process; and thus a major gap between experimental models and clinical setting has been identified. Accordingly, our recent studies have aimed to address this critical gap. We found that when acute DED mice induced by a period of desiccating stress was transferred to a normal-humidity environment, corneal epitheliopathy in mice gradually regressed to a lower level, but never normalized, for many months, despite recovery of tear deficiency, thus establishing a critical murine model of chronic DED [190]. Partial resolution of the acute severe corneal epitheliopathy occurred dominantly in the first week of deprivation of desiccating stress, during which period the previously expanded effector Th17 pool in acute disease dramatically contracted, accompanied by the emergence of a substantial memory Th17 population defined as CD4+CD44highCD62L−IL-17+ – a characteristic phenotype of “effector memory cells” distinct from the “central memory cells” [108]. Memory T or B cell-mediated “immunological memory” is a defining feature of adaptive immunity, and its broad benefits in host defense have been widely demonstrated in vaccine-induced and post-infection acquired protection, primarily via B cell or CD8+ T cell memory mechanisms. In contrast, the roles and biology of memory CD4+ T cells are more complex and less well understood due to the diverse subsets of effector T helper cells. A few recent studies in the field have implicated potentially detrimental outcomes mediated by memory Th17 cells in autoimmunity and chronic inflammation [202,203]. We next examined the functions of those memory Th17 cells in DED. It is known that two cardinal features of adaptive immune memory are the specificity of response and the enhanced amplitude and speed of the response. We thus adoptively transferred the memory cells isolated from chronic DED to healthy naive mice and found that recipients developed the similar clinical disease [190]. Furthermore, secondary challenge with a repeated short-term desiccating stress on chronic DED mice led to a more rapid and severe disease exacerbation than the primary challenge [204]. Taken together, these data demonstrate that memory Th17 cells play a specific, pathogenic role in causing autoimmune dry eye disease. We subsequently performed a series of in-depth investigations exploring the generation and maintenance mechanisms of memory Th17 cells in DED.
6.2. Generation of memory Th17 cells from effector precursors
Our study shows that the DED-specific memory Th17 cells emerging during the contraction phase of primary response are generated from a small fraction of surviving effector Th17 cells, consistent with the previously reported “linear” memory development pathway [205,206], driven by persisting environmental IL-23 stimulation and diminished IL-2 signaling at both ocular surface and regional draining lymph nodes (Fig. 2) [108]. Blockade of IL-23 specifically during the contraction phase effectively prevents the Th17 memory formation, and the IL-23-dependent linear conversion of effector Th17 to memory Th17 is through inhibition of T cell apoptosis [108]. Although early up-regulated IL-2 level in the expansion phase of primary response is critical for effector Th17 proliferation and is required for subsequent memory formation possibly due to its role in promoting specificity through clonal expansion and promoting more robust responses through early epigenetic reprogramming [108,207], persistent IL-2 stimulation on effector Th17 cells suppresses generation of memory cells through inducing cell apoptosis [108] and possible exhaustion via activating the aryl hydrocarbon receptor [208]. In fact, reduced IL-2 level has been observed in the periphery of Sjögren’s syndrome patients, which is linked to unrestrained Th17 generation via epigenetic modification of Il17a locus in CD4+ T cells [209]. Along with the chronic low-grade corneal epitheliopathy, memory Th17 cells persist for prolonged period in both conjunctiva and draining lymphoid tissues, allowing them constant contact with the ocular surface antigens; interestingly, they do not co-produce IFN-γ, a critical cytokine contributing to heightened inflammation in the acute disease [183]. Consistent with the cellular findings, only heightened levels of the cytokine IL-17, but not IFN-γ are present in the ocular surface in chronic DED [95,190]. Intriguingly, our ongoing work further shows that the major effector precursors of memory Th17 cells are IFN-γ-producing double-positive effector Th17/1 cells (manuscript submitted for peer-review). Although memory Th17 cells lose the capacity of secreting IFN-γ, they maintain the expression, at a lower level, of the transcriptional factor T-bet (the master regulator for IFN-γ expression) as their effector precursor Th17/1 cells, suggesting that transcriptional imprinting on T cells induced during the primary response via TCR ligation, cytokine cues, and others may play a determinant role in subsequent memory Th17 cell generation [210]. Indeed, low level T-bet expression has been shown to critically control the development and function of memory B cells, memory CD8+ T, and memory Th1 cells [211–215]. Although IFN-γ itself and IL-12 are known potent T-bet expression inducers, their roles in maintaining low level T-bet expression during memory formation, including memory Th17 pool, need to be further investigated, along with other possible factors. Taken together, transcriptional changes in T cells induced in the effector phase synergize with cytokine cues in the contraction phase to critically govern the generation of memory Th17 pool in DED.
Figure 2. Generation and maintenance of memory Th17 cells in dry eye disease.
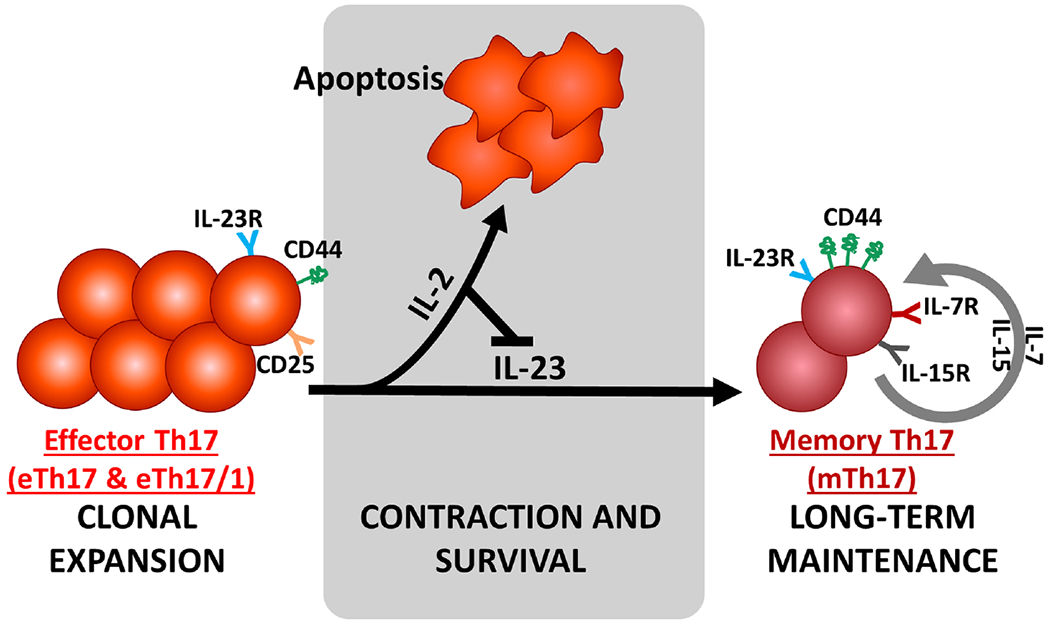
In the expansion phase, IL-2/CD25 signaling and IL-23/IL-23R signaling are critical for effector Th17 cell proliferation and full activation. During the contraction phase, persisting environmental IL-23 stimulation is required for driving the conversion of CD44low effector Th17 to CD44hi memory Th17, while persistent IL-2 stimulation on effector Th17 cells suppresses generation of memory cells through inducing cell apoptosis. The memory Th17 cell pool is maintained for long term by both IL-7 and IL-15 signaling, and mediates the chronic inflammation in dry eye disease.
6.3. Maintenance of memory Th17 cells and sustention of chronic inflammation
It is demonstrated that continuous production of IL-17 by memory Th17 cells sustains the chronic ocular surface inflammation in DED, and topical blockade of IL-17 significantly improves the disease severity [204]. The next question is why memory Th17 cells are long-lived in contrast to the short-lived effector Th17 cells. One major phenotypic change during the IL-23-driven conversion of effector Th17 to memory Th17 is the newly-acquired cell surface expressions of IL-7 receptor and IL-15 receptor [108], both of which are critical for the prolonged survival of memory Th17 cells in DED [204]. In addition to the antigen persistence, environmental cytokine signaling plays essential roles for the maintenance of memory T cells. In this regard, the two cytokines IL-7 and IL-15 provide key support to memory Th17 cell survival via maintaining robust cell proliferative and preventing cell apoptosis (Fig. 2) [204]. Specifically, both IL-7 and IL-15 provide survival signals via activating signal transducer and activator of transcription 5 (STAT5) pathway, and IL-15 provides additional proliferation signals via activating both STAT5 and protein kinase B (Akt) pathways. Topical neutralization of either cytokine efficiently abolishes pathogenic memory Th17 cells at both conjunctiva and draining lymphoid tissues, along with the significant and long-lasting DED amelioration [204]. These two cytokines, along with IL-2 which is fundamentally involved in memory Th17 generation as described above, belong to the same common-gamma chain receptor cytokine family. These pre-clinical data suggest that manipulation of local environmental factors supporting memory Th17 cells, especially the common-gamma chain family cytokines, can be a novel, specific, and safe strategy in the treatment of DED without eliciting systemic adverse effects that may result from jeopardizing protective immunity.
The memory Th17 cells are functionally pathogenic demonstrated by their ability of transferring DED clinical phenotypes to healthy naive mice [190]. Interestingly, re-challenge of chronic DED mice with a shorter period of desiccating stress than the primary challenge leads to a faster and more severe acute-on-chronic disease exacerbation, along with a robust effector response characterized by the efficient conversion of the small memory Th17 pool to an expanded effector pool that comprises both IL-17-producing effector Th17 cells and IFN-γ/IL-17 co-producing effector Th17/1 cells [95], suggesting that memory Th17 cell-mediated recall response may be responsible for the intermittent disease “flare” in clinical patients that is not only seen in DED but frequently seen in other autoimmune conditions. The distinct ability of these “antigen-experienced” memory T cells to mount a recall response from antigen-inexperienced naive T cells is rooted to their lower activation threshold and less co-stimulation dependence (instead, more dependent on the environment cues upon re-encounter with cognate antigen) [216,217], higher antigen-specificity [217,218], and circulation between lymphoid and peripheral tissues (thus allowing more efficient encounter with antigens in peripheral tissues) [219]. Specifically for memory Th17 cells, IL-23 has been found again an essential factor to achieve a fully functional recall response during the repeated antigen exposure in other inflammatory disorders, primarily through mediating memory-to-effector conversion [220,221]. In DED, memory Th17 cells are present in both regional lymphoid tissues and the peripheral ocular surface [204]. Abolishment of memory Th17 cells in chronic DED has been shown to efficiently diminish the recall response and prevent the disease flare [204].
7. Role of aging and microbiome on Th17 immunity in dry eye disease
7.1. Effects of aging
Age-related changes can result in a shift in the balance between protective and pathogenic immune responses, collectively termed “immunosenescence”. Advanced age has been shown a risk factor for autoimmune phenomena [222], and increased prevalence of DED in the elderly [1] and the spontaneous development of DED in aged animals [111,223–227] have been widely reported. Interestingly, in elderly wild-type mice showing DED-associated corneal epitheliopathy, there is no decreased, but paradoxically increased tear production, along with mild lymphocytic infiltration in lacrimal glands [228,229], suggesting that aging-associated higher susceptibility to DED is independent of tear deficiency but mainly attributed to an “imbalance” between pro-inflammatory and anti-inflammatory pathways in elderly, characterized as amplified T effector response and failed immunoregulatory mechanisms. Pre-clinical studies have shown increased matured APC in conjunctiva with enhanced antigen capturing capability in aged mice [230]. Relatedly, increased Th17 cells in the eye draining lymph nodes and dramatic up-regulation of IL-17 at the ocular surface have been identified in aged mice [228,230]. Furthermore, adoptive transfer of CD4+ T cells isolated from draining lymph nodes and spleen of aged mice to naive recipients effectively led to T cell infiltration in the ocular surface and DED signs in recipients [228]. Taken together, these data suggest a causal relationship between spontaneously activated CD4+ T cells (especially Th17 subset) and DED in the aged mice. More recently our group has further made an effort to dissect whether those systemically activated CD4+ T cells in the aged in fact represent a memory cell population which contributes to the chronic DED in the aged. Using our robust environmental desiccating stress model, we showed that in aged DED mice a significantly larger memory Th17 pool than in young counterparts was formed, and upon secondary exposure to desiccating stress, those aged developed more severe corneal epitheliopathy associated with heightened Th17 recall response; furthermore, depletion of the enlarged memory Th17 pool in the aged prevented the rechallenge-induced DED exacerbation, demonstrating the important role of memory Th17 cells in predisposing the aged to DED [231]. The precise underlying mechanisms by which immunoscenescence contributes to the expanded autoreactive memory Th17 pool remain to be elucidated, such as Treg-mediated regulation of immunological memory.
7.2. Effects of the microbiome
Commensal bacteria colonize all exposed body surfaces among which gut is the most densely populated organ; in contrast, ocular surface is a relatively sterile site with low bacterial load [15,232]. The inhabited bacterial communities are collectively referred to microbiota, and their collective genomes are referred to microbiome [15]. There has been recently increasing interest in studying the effect of microbiome on immune homeostasis in health and disease, and it has been demonstrated that gut microbiota impact not only intestinal diseases but also diseases in other sites, including the eye, a phenomenon called gut-eye axis [131,233]. In a spontaneous transgenic autoimmune uveitis (inflammation of uvea of the eye) model, gut microbiota are shown as a trigger of retina-specific autoreactive Th17 cell activation in the periphery (gut lamina propria) independent of retinal antigens, possibly via the antigenic mimic mechanisms (though the culprit microbe and its immunogenic antigen remain unclear); these Th17 cells are pathogenic in causing uveitis [234]. In contrast, in the ocular surface, gut microbiota seem to play protective roles. Depletion of commensal bacteria in wild-type mice in germ-free environment leads to spontaneous development of DED, and fecal microbiota transplant from conventional mice to these germ-free mice ameliorates DED severity and decreases CD4+ T cell infiltration [235]. Similar findings have been demonstrated in a transgenic Sjögren’s Syndrome DED model [236]. In addition, wild-type mice subjected to desiccating stress develop more severe DED when treated with oral antibiotics cocktail that induces intestinal dysbiosis [237]. The protective roles of gut microbiota in DED may be related to their induction of functional Treg [238–240] that restrict autoreactive Th17 immunity. These findings suggest fecal microbial transplant a potentially novel therapy for DED, and currently a clinical trial using this strategy to treat Sjögren’s Syndrome DED is ongoing (ClinicalTrials.gov Identifier: NCT03926286). Notably, serious adverse effects of fatal infections with multi-drug resistant organisms from fecal microbiota transplant have been recently reported in clinical trials. An alternative approach of modulating gut microbiome is oral supplement with probiotics, which are selective live microorganisms that can provide beneficial effects. In a mouse model of Sjögren’s Syndrome which is subject to gut microbiota depletion by antibiotic cocktail and germ-free environment, probiotic treatment significantly improves corneal barrier function and tear production, along with increased Treg population [241], indicating probiotics an appealing approach to treat autoimmune DED; nevertheless its definitive benefits in patients are to be determined by extensive human studies. Further studies are required to gain a better understanding on what commensal microbes and how they affect Th17 immunity in DED; answers to these questions may lead to more efficient microbiome-modulating therapies utilizing antibiotics, probiotics, or microbiota-derived metabolites (such as short-chain fatty acids).
8. Conclusions and perspectives
Accumulating experimental and clinical evidence demonstrate the central role of Th17-mediated pathogenesis in causing the persistent ocular surface inflammation in DED that leads to a myriad of ocular surface pathologies involving the tear film, epithelium, and nerves. Precise delineation on the generation and function of Th17 autoimmunity in other DED-associated target organs including lacrimal glands and Meibomian glands within the concept of ocular surface-local lymphoid compartment axis remained to be wrought. The understanding on the interplay between Th17 immunity and nervous system, angiogenesis, microbiome in DED pathophysiology is rapidly evolving with the use of novel animal tools, such as IL-17 reporter mice for tracking plastic Th17 cells. Given the potentially critical roles of T cell memory in autoimmune conditions that usually present with chronic and relapsing courses, including DED, specific immunomodulation that either directly targeting memory Th17 cells or indirectly restricting them via enhancing their limiting factors may represent novel and efficient approaches that can restore ocular surface homeostasis in autoimmune DED, and thus holds a promise in future translational and clinical studies.
Highlights.
Th17-mediated autoimmunity plays a central pathogenic role in dry eye disease.
Effector Th17 cells are highly plastic producing both IL-17 and IFN-γ, leading to ocular surface tissue damage and inflammatory cascade.
Effector Th17 cells derived memory Th17 cells are long-lived and maintain the chronic inflammation in dry eye disease.
Targeting immunological memory can be a promising strategy for treating Th17 cell-mediated autoimmunity.
Funding source:
This work was supported by NIH grant EY20889 and NIH core grant P30EY003790.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest: R.D. is consultant to Novartis, GSK, and Kala, and holds equity in Claris Biotherapeutics, Aramis Biosciences, GelMEDIX, and Kera Therapeutics. Massachusetts Eye and Ear owns intellectual property related to anti-inflammation of targeting memory Th17 cells and substance P in ocular immunoinflammatory diseases.
References
- [1].Farrand KF, Fridman M, Stillman IÖ, Schaumberg DA. Prevalence of Diagnosed Dry Eye Disease in the United States Among Adults Aged 18 Years and Older. Am J Ophthalmol 2017;182:90–8. 10.1016/j.ajo.2017.06.033. [DOI] [PubMed] [Google Scholar]
- [2].Lemp MA. Report of the National Eye Institute/Industry Workshop on Clinical Trials in Dry Eyes. CLAO J, vol. 21, 1995, p. 221–32. [PubMed] [Google Scholar]
- [3].Lemp MA, Baudouin C, Baum J, Dogru M, Foulks GN, Kinoshita S, et al. The definition and classification of dry eye disease: Report of the definition and classification subcommittee of the international Dry Eye Workshop (2007). Ocul. Surf, vol. 5, 2007, p. 75–92. 10.1016/sl542-0124(12)70081-2. [DOI] [PubMed] [Google Scholar]
- [4].Lemp MA, Crews LA, Bron AJ, Foulks GN, Sullivan BD. Distribution of aqueous-deficient and evaporative dry eye in a clinic-based patient cohort: A retrospective study. Cornea 2012;31:472–8. 10.1097/ICO.0b013e318225415a. [DOI] [PubMed] [Google Scholar]
- [5].Craig JP, Nichols KK, Akpek EK, Caffery B, Dua HS, Joo CK, et al. TFOS DEWS II Definition and Classification Report. Ocul Surf 2017;15:276–83. 10.1016/jjtos.2017.05.008. [DOI] [PubMed] [Google Scholar]
- [6].Wei Y, Asbell PA. The core mechanism of dry eye disease is inflammation. Eye Contact Lens 2014;40:248–56. 10.1097/ICL.0000000000000042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Jabs DA, Prendergast RA. Reactive lymphocytes in lacrimal gland and vasculitic renal lesions of autoimmune MRL/lpr mice express L3T4. J Exp Med 1987. 10.1084/jem.166.4.1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Matsumoto I, Tsubota K, Satake Y, Kita Y, Matsumura R, Murata H, et al. Common T cell receptor clonotype in lacrimal glands and labial salivary glands from patients with Sjögren’s syndrome. J Clin Invest 1996;97:1969–77. 10.1172/JCI118629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Dursun D, Wang M, Monroy D, Li DQ, Lokeshwar BL, Stern ME, et al. A mouse model of keratoconjunctivitis sicca. Investig Ophthalmol Vis Sci 2002;43:632–8. [PubMed] [Google Scholar]
- [10].Barabino S, Shen LL, Chen L, Rashid S, Rolando M, Dana MR. The controlled-environment chamber: A new mouse model of dry eye. Investig Ophthalmol Vis Sci 2005;46:2766–71. 10.1167/iovs.04-1326. [DOI] [PubMed] [Google Scholar]
- [11].Niederkorn JY, Stern ME, Pflugfelder SC, De Paiva CS, Corrales RM, Gao J, et al. Desiccating Stress Induces T Cell-Mediated Sjögren’s Syndrome-Like Lacrimal Keratoconjunctivitis. J Immunol 2006;176:3950–7. 10.4049/jimmunol.176.7.3950. [DOI] [PubMed] [Google Scholar]
- [12].De Paiva CS, Chotikavanich S, Pangelinan SB, Pitcher JD, Fang B, Zheng X, et al. IL-17 disrupts corneal barrier following desiccating stress. Mucosal Immunol 2009;2:243–53. 10.1038/mi.2009.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Chauhan SK, El Annan J, Ecoiffier T, Goyal S, Zhang Q, Saban DR, et al. Autoimmunity in Dry Eye Is Due to Resistance of Th17 to Treg Suppression. J Immunol 2009; 182:1247–52. 10.4049/jimmunol.182.3.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Luo D, Chen Y, Zhou N, Li T, Wang H. Blockade of Th17 response by IL-38 in primary Sjögren’s syndrome. Mol Immunol 2020. 10.1016/j.molimm.2020.09.006. [DOI] [PubMed] [Google Scholar]
- [15].Bron AJ, de Paiva CS, Chauhan SK, Bonini S, Gabison EE, Jain S, et al. TFOS DEWS II pathophysiology report. Ocul Surf 2017;15:438–510. 10.1016/jjtos.2017.05.011. [DOI] [PubMed] [Google Scholar]
- [16].Cursiefen C Immune privilege and angiogenic privilege of the cornea. Chem Immunol Allergy 2007;92:50–7. 10.1159/000099253. [DOI] [PubMed] [Google Scholar]
- [17].Ambati BK, Nozaki M, Singh N, Takeda A, Jani PD, Suthar T, et al. Corneal avascularity is due to soluble VEGF receptor-1. Nature 2006;443:993–7. 10.1038/nature05249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Cursiefen C, Chen L, Saint-Geniez M, Hamrah P, Jin Y, Rashid S, et al. Nonvascular VEGF receptor 3 expression by corneal epithelium maintains avascularity and vision. Proc Natl Acad Sci U S A 2006;103:11405–10. 10.1073/pnas.0506112103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Hamrah P, Liu Y, Zhang Q, Dana MR. The corneal stroma is endowed with a significant number of resident dendritic cells. Investig Ophthalmol Vis Sci 2003;44:581–9. 10.1167/iovs.02-0838. [DOI] [PubMed] [Google Scholar]
- [20].Hattori T, Chauhan SK, Lee H, Ueno H, Dana R, Kaplan DH, et al. Characterization of langerin-expressing dendritic cell subsets in the normal cornea. Investig Ophthalmol Vis Sci 2011;52:4598–604. 10.1167/iovs.10-6741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Hattori T, Takahashi H, Dana R. Novel insights into the immunoregulatory function and localization of dendritic cells. Cornea 2016;35:S49–54. 10.1097/ic0.0000000000001005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Hamrah P, Zhang Q, Liu Y, Dana MR. Novel characterization of MHC class II-negative population of resident corneal Langerhans cell-type dendritic cells. Investig Ophthalmol Vis Sci 2002;43:639–46. [PubMed] [Google Scholar]
- [23].Yamagami S, Ebihara N, Usui T, Yokoo S, Amano S. Bone marrow-derived cells in normal human corneal stroma. Arch Ophthalmol 2006;124:62–9. 10.1001/archopht.124.1.62. [DOI] [PubMed] [Google Scholar]
- [24].Yamagami S, Yokoo S, Usui T, Yamagami H, Amano S, Ebihara N. Distinct populations of dendritic cells in the normal human donor corneal epithelium. Investig Ophthalmol Vis Sci 2005;46:4489–94. 10.1167/iovs.05-0054. [DOI] [PubMed] [Google Scholar]
- [25].Barabino S, Chen Y, Chauhan S, Dana R. Ocular surface immunity: Homeostatic mechanisms and their disruption in dry eye disease. Prog Retin Eye Res 2012;31:271–85. 10.1016/j.preteyeres.2012.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Lutz MB, Schuler G. Immature, semi-mature and fully mature dendritic cells: Which signals induce tolerance or immunity? Trends Immunol 2002;23:445–9. 10.1016/S1471-4906(02)02281-0. [DOI] [PubMed] [Google Scholar]
- [27].Shen L, Jin Y, Freeman GJ, Sharpe AH, Dana MR. The Function of Donor versus Recipient Programmed Death-Ligand 1 in Corneal Allograft Survival. J Immunol 2007;179:3672–9. 10.4049/jimmunol.179.6.3672. [DOI] [PubMed] [Google Scholar]
- [28].Griffith TS, Brunner T, Fletcher SM, Green DR, Ferguson TA. Fas ligand-induced apoptosis as a mechanism of immune privilege. Science (80-) 1995;270:1189–92. 10.1126/science.270.5239.1189. [DOI] [PubMed] [Google Scholar]
- [29].Tan X, Chen Y, Foulsham W, Amouzegar A, Inomata T, Liu Y, et al. The immunoregulatory role of corneal epithelium-derived thrombospondin-1 in dry eye disease. Ocul Surf 2018;16:470–7. 10.1016/jjtos.2018.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Singh RB, Blanco T, Mittal SK, Taketani Y, Chauhan SK, Chen Y, et al. Pigment Epithelium-derived Factor secreted by corneal epithelial cells regulates dendritic cell maturation in dry eye disease. Ocul Surf 2020;18:460–9. 10.1016/jjtos.2020.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med 2000;192:1027–34. 10.1084/jem.192.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].El-Annan J, Goyal S, Zhang Q, Freeman GJ, Sharpe AH, Dana R. Regulation of T-cell chemotaxis by programmed Death-Ligand 1 (PD-L1) in dry eye-associated corneal inflammation. Investig Ophthalmol Vis Sci 2010;51:3418–23. 10.1167/iovs.09-3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Jin Y, Chauhan SK, Annan JEI, Sage PT, Sharpe AH, Dana R. A novel function for programmed death ligand-1 regulation of angiogenesis. Am J Pathol 2011;178:1922–9. 10.1016/j.ajpath.2010.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Suda T, Takahashi T, Golstein P, Nagata S. Molecular cloning and expression of the fas ligand, a novel member of the tumor necrosis factor family. Cell 1993;75:1169–78. 10.1016/0092-8674(93)90326-L. [DOI] [PubMed] [Google Scholar]
- [35].Ferguson TA, Griffith TS. A vision of cell death: Fas ligand and immune privilege 10 years later. Immunol Rev 2006;213:228–38. 10.Ill1/j.1600-065X.2006.00430.x. [DOI] [PubMed] [Google Scholar]
- [36].Hohlbaum AM, Moe S, Marshak-Rothstein A. Opposing effects of transmembrane and soluble Fas ligand expression on inflammation and tumor cell survival. J Exp Med 2000;191:1209–19. 10.1084/jem.191.7.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Krishnan A, Fei F, Jones A, Busto P, Marshak-Rothstein A, Ksander BR, et al. Overexpression of Soluble Fas Ligand following Adeno-Associated Virus Gene Therapy Prevents Retinal Ganglion Cell Death in Chronic and Acute Murine Models of Glaucoma. J Immunol 2016;197:4626–38. 10.4049/jimmunol.1601488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Dawson DW, Volpert OV, Gillis P, Crawford SE, Xu HJ, Benedict W, et al. Pigment epithelium-derived factor: A potent inhibitor of angiogenesis. Science (80-) 1999;285:245–8. 10.1126/science.285.5425.245. [DOI] [PubMed] [Google Scholar]
- [39].Crawford SE, Stellmach V, Murphy-Ullrich JE, Ribeiro SMF, Lawler J, Hynes RO, et al. Thrombospondin-1 is a major activator of TGF-β1 in vivo. Cell 1998;93:1159–70. 10.1016/S0092-8674(00)81460-9. [DOI] [PubMed] [Google Scholar]
- [40].Ribeiro SMF, Poczatek M, Schultz-Cherry S, Villain M, Murphy-Ullrich JE. The activation sequence of thrombospondin-1 interacts with the latency-associated peptide to regulate activation of latent transforming growth factor-β. J Biol Chem 1999;274:13586–93. 10.1074/jbc.274.19.13586. [DOI] [PubMed] [Google Scholar]
- [41].Cursiefen C, Masli S, Ng TF, Dana MR, Bornstein P, Lawler J, et al. Roles of thrombospondin-1 and −2 in regulating corneal and iris angiogenesis. Investig Ophthalmol Vis Sci 2004;45:1117–24. 10.1167/iovs.03-0940. [DOI] [PubMed] [Google Scholar]
- [42].Yabe T, Sanagi T, Yamada H. The Neuroprotective Role of PEDF: Implication for the Therapy of Neurological Disorders. Curr Mol Med 2010;10:259–66. 10.2174/156652410791065354. [DOI] [PubMed] [Google Scholar]
- [43].Dastjerdi MH, Dana R. Corneal nerve alterations in dry eye-associated ocular surface disease. Int Ophthalmol Clin 2009;49:11–20. 10.1097/110.0b013e31819242c9. [DOI] [PubMed] [Google Scholar]
- [44].Ferrari G, Chauhan SK, Ueno H, Nallasamy N, Gandolfi S, Borges L, et al. A novel mouse model for neurotrophic keratopathy: Trigeminal nerve stereotactic electrolysis through the brain. Investig Ophthalmol Vis Sci 2011;52:2532–9. 10.1167/iovs.10-5688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Ueno H, Ferrari G, Hattori T, Saban DR, Katikireddy KR, Chauhan SK, et al. Dependence of corneal stem/progenitor cells on ocular surface innervation. Investig Ophthalmol Vis Sci 2012;53:867–72. 10.1167/iovs.ll-8438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Ferrari G, Hajrasouliha AR, Sadrai Z, Ueno H, Chauhan SK, Dana R. Nerves and neovessels inhibit each other in the cornea. Investig Ophthalmol Vis Sci 2013;54:813–20. 10.1167/iovs.ll-8379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Kheirkhah A, Dohlman TH, Amparo F, Amoldner MA, Jamali A, Hamrah P, et al. Effects of corneal nerve density on the response to treatment in dry eye disease. Ophthalmology 2015;122:662–8. 10.1016/j.ophtha.2014.ll.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Benítez Del Castillo JM, Wasfy MAS, Fernandez C, Garcia-Sanchez J. An in vivo confocal masked study on corneal epithelium and subbasal nerves in patients with dry eye. Investig Ophthalmol Vis Sci 2004;45:3030–5. 10.1167/iovs.04-0251. [DOI] [PubMed] [Google Scholar]
- [49].Stepp MA, Pal-Ghosh S, Tadvalkar G, Williams A, Pflugfelder SC, de Paiva CS. Reduced intraepithelial corneal nerve density and sensitivity accompany desiccating stress and aging in C57BL/6 mice. Exp Eye Res 2018;169:91–8. 10.1016/j.exer.2018.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Guzmán M, Miglio M, Keitelman I, Shiromizu CM, Sabbione F, Fuentes F, et al. Transient tear hyperosmolarity disrupts the neuroimmune homeostasis of the ocular surface and facilitates dry eye onset. Immunology 2020;161:148–61. 10.1111/imm.13243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Suvas S Role of Substance P Neuropeptide in Inflammation, Wound Healing, and Tissue Homeostasis. J Immunol 2017;199:1543–52. 10.4049/jimmunol.1601751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Szliter EA, Lighvani S, Barrett RP, Hazlett LD. Vasoactive Intestinal Peptide Balances Pro- and Anti-Inflammatory Cytokines in the Pseudomonas aeruginosa-Infected Cornea and Protects against Corneal Perforation . J Immunol 2007;178:1105–14. 10.4049/jimmunol.178.2.1105. [DOI] [PubMed] [Google Scholar]
- [53].Luo L, Li DQ, Doshi A, Farley W, Corrales RM, Pflugfelder SC. Experimental dry eye stimulates production of inflammatory cytokines and MMP-9 and activates MAPK signaling pathways on the ocular surface. Investig Ophthalmol Vis Sci 2004;45:4293–301. 10.1167/iovs.03-1145. [DOI] [PubMed] [Google Scholar]
- [54].De Paiva CS, Corrales RM, Villarreal AL, Farley WJ, Li DQ, Stern ME, et al. Corticosteroid and doxycycline suppress MMP-9 and inflammatory cytokine expression, MAPK activation in the corneal epithelium in experimental dry eye. Exp Eye Res 2006;83:526–35. 10.1016/j.exer.2006.02.004. [DOI] [PubMed] [Google Scholar]
- [55].Luo L, Li DQ, Corrales RM, Pflugfelder SC. Hyperosmolar saline is a proinflammatory stress on the mouse ocular surface. Eye Contact Lens 2005;31:186–93. 10.1097/01.ICL.0000162759.79740.46. [DOI] [PubMed] [Google Scholar]
- [56].Rashid S, Jin Y, Ecoiffier T, Barabino S, Schaumberg DA, Dana MR. Topical omega-3 and omega-6 fatty acids for treatment of dry eye. Arch Ophthalmol 2008;126:219–25. 10.1001/archophthalmol.2007.61. [DOI] [PubMed] [Google Scholar]
- [57].Zheng X, de Paiva CS, Li DQ, Farley WJ, Pflugfelder SC. Desiccating stress promotion of Th17 differentiation by ocular surface tissues through a dendritic cell-mediated pathway. Investig Ophthalmol Vis Sci 2010;51:3083–91. 10.1167/iovs.09-3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Lee HS, Hattori T, Park EY, Stevenson W, Chauhan SK, Dana R. Expression of toll-like receptor 4 contributes to corneal inflammation in experimental dry eye disease. Investig Ophthalmol Vis Sci 2012;53:5632–40. 10.1167/iovs.12-9547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Chen H, Gan X, Li Y, Gu J, Liu Y, Deng Y, et al. NLRP12- and NLRC4-mediated corneal epithelial pyroptosis is driven by GSDMD cleavage accompanied by IL-33 processing in dry eye. Ocul Surf 2020;18:783–94. 10.1016/jjtos.2020.07.001. [DOI] [PubMed] [Google Scholar]
- [60].Zheng Q, Ren Y, Reinach PS, She Y, Xiao B, Hua S, et al. Reactive oxygen species activated NLRP3 inflammasomes prime environment-induced murine dry eye. Exp Eye Res 2014;125:1–8. 10.1016/j.exer.2014.05.001. [DOI] [PubMed] [Google Scholar]
- [61].Li DQ, Lokeshwar BL, Solomon A, Monroy D, Ji Z, Pflugfelder SC. Regulation of MMP-9 production by human corneal epithelial cells. Exp Eye Res 2001;73:449–59. 10.1006/exer.2001.1054. [DOI] [PubMed] [Google Scholar]
- [62].Pflugfelder SC, Farley W, Luo L, Chen LZ, De Paiva CS, Olmos LC, et al. Matrix metalloproteinase-9 knockout confers resistance to corneal epithelial barrier disruption in experimental dry eye. Am J Pathol 2005;166:61–71. 10.1016/S0002-9440(10)62232-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Zhang Z, Yang WZ, Zhu ZZ, Hu QQ, Chen YF, He H, et al. Therapeutic effects of topical doxycycline in a benzalkonium chloride-induced mouse dry eye model. Investig Ophthalmol Vis Sci 2014;55:2963–74. 10.1167/iovs.13-13577. [DOI] [PubMed] [Google Scholar]
- [64].Lin H, Li W, Dong N, Chen W, Liu J, Chen L, et al. Changes in corneal epithelial layer inflammatory cells in aqueous tear-deficient dry eye. Investig Ophthalmol Vis Sci 2010;51:122–8. 10.1167/iovs.09-3629. [DOI] [PubMed] [Google Scholar]
- [65].Yoon KC, De Paiva CS, Qi H, Chen Z, Farley WJ, Li DQ, et al. Expression of Th-1 chemokines and chemokine receptors on the ocular surface of C57BL/6 mice: Effects of desiccating stress. Investig Ophthalmol Vis Sci 2007;48:2561–9. 10.1167/iovs.07-0002. [DOI] [PubMed] [Google Scholar]
- [66].Mackay CR. Chemokines: Immunology’s high impact factors. Nat Immunol 2001;2:95–101. 10.1038/84298. [DOI] [PubMed] [Google Scholar]
- [67].Engl TN. CHEMOKINES—CHEMOTACTIC CYTOKINES THAT MEDIATE I INFLAMMATION A. N Engl J Med 1998. [DOI] [PubMed] [Google Scholar]
- [68].Goyal S, Chauhan SK, Zhang Q, Dana R. Amelioration of murine dry eye disease by topical antagonist to chemokine receptor 2. Arch Ophthalmol 2009;127:882–7. 10.1001/archophthalmol.2009.125. [DOI] [PubMed] [Google Scholar]
- [69].Kodati S, Chauhan SK, Chen Y, Dohlman TH, Karimian P, Saban D, et al. CCR7 is critical for the induction and maintenance of Th17 immunity in dry eye disease. Investig Ophthalmol Vis Sci 2014;55:5871–7. 10.1167/iovs.14-14481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Chen Y, Chauhan SK, Saban DR, Sadrai Z, Okanobo A, Dana R. Interferon-y-secreting NK cells promote induction of dry eye disease. J Leukoc Biol 2011;89:965–72. 10.1189/jlb.1110611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Lee HS, Amouzegar A, Dana R. Kinetics of Corneal Antigen Presenting Cells in Experimental Dry Eye Disease. BMJ Open Ophthalmol 2017; 1. 10.1136/bmjophth-2017-000078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Kheirkhah A, Rahimi Darabad R, Cruzat A, Hajrasouliha AR, Witkin D, Wong N, et al. Corneal epithelial immune dendritic cell alterations in subtypes of dry eye disease: A pilot in vivo confocal microscopic study. Investig Ophthalmol Vis Sci 2015. 10.1167/iovs.15-17433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Aggarwal S, Kheirkhah A, Cavalcanti BM, Cruzat A, Jamali A, Hamrah P. Correlation of corneal immune cell changes with clinical severity in dry eye disease: An in vivo confocal microscopy study. Ocul Surf 2021;19:183–9. 10.1016/jjtos.2020.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Hamrah P, Liu Y, Zhang Q, Dana MR. Alterations in corneal stromal dendritic cell phenotype and distribution in inflammation. Arch Ophthalmol 2003;121:1132–40. 10.1001/archopht.121.8.1132. [DOI] [PubMed] [Google Scholar]
- [75].Zhang X, Volpe EA, Gandhi NB, Schaumburg CS, Siemasko KF, Pangelinan SB, et al. NK cells promote Th-17 mediated corneal barrier disruption in dry eye. PLoS One 2012;7:e36822. 10.1371/journal.pone.0036822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Takada K, Takiguchi M, Konno A, Inaba M. Autoimmunity against a tissue kallikrein in IQI/Jic mice: A model for sjögren’s syndrome. J Biol Chem 2005;280:3982–8. 10.1074/jbc.M410157200. [DOI] [PubMed] [Google Scholar]
- [77].Stern ME, Schaumburg CS, Siemasko KF, Gao J, Wheeler LA, Grupe DA, et al. Autoantibodies contribute to the immunopathogenesis of experimental dry eye disease. Invest Ophthalmol Vis Sci 2012;53:2062–75. 10.1167/iovs.ll-9299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Jiang G, Ke Y, Sun D, Li H, Ihnen M, Jumblatt MM, et al. A new model of experimental autoimmune keratoconjunctivitis sicca (KCS) induced in Lewis rat by the autoantigen Klk1b22. Investig Ophthalmol Vis Sci 2009;50:2245–54. 10.1167/iovs.08-1949. [DOI] [PubMed] [Google Scholar]
- [79].Schaumburg CS, Siemasko KF, De Paiva CS, Wheeler LA, Niederkorn JY, Pflugfelder SC, et al. Ocular Surface APCs Are Necessary for Autoreactive T Cell-Mediated Experimental Autoimmune Lacrimal Keratoconjunctivitis. J Immunol 2011; 187:3653–62. 10.4049/jimmunol.1101442. [DOI] [PubMed] [Google Scholar]
- [80].Wang T, Li W, Cheng H, Zhong L, Deng J, Ling S. The Important Role of the Chemokine Axis CCR7-CCL19 and CCR7-CCL21 in the Pathophysiology of the Immuno-inflammatory Response in Dry Eye Disease. Ocul Immunol Inflamm 2019. 10.1080/09273948.2019.1674891. [DOI] [PubMed] [Google Scholar]
- [81].Goyal S, Chauhan SK, El Annan J, Nallasamy N, Zhang Q, Dana R. Evidence of corneal lymphangiogenesis in dry eye disease: A potential link to adaptive immunity? Arch Ophthalmol 2010;128:819–24. 10.1001/archophthalmol.2010.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Chauhan SK, Jin Y, Goyal S, Lee HS, Fuchsluger TA, Lee HK, et al. A novel prolymphangiogenic function for Th17/IL-17. Blood 2011;118:4630–4. 10.1182/blood-2011-01-332049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Goyal S, Chauhan SK, Dana R. Blockade of prolymphangiogenic vascular endothelial growth factor C in dry eye disease. Arch Ophthalmol 2012;130:84–9. 10.1001/archophthalmol.2011.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Jamali A, Seyed-Razavi Y, Chao C, Ortiz G, Kenyon B, Blanco T, et al. Intravital Multiphoton Microscopy of the Ocular Surface: Alterations in Conventional Dendritic Cell Morphology and Kinetics in Dry Eye Disease. Front Immunol 2020; 11. 10.3389/fimmu.2020.00742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Dana R Corneal antigen presentation: Molecular regulation and functional implications. Ocul Surf 2005;3:S169–72. 10.1016/sl542-0124(12)70248-3. [DOI] [PubMed] [Google Scholar]
- [86].Merad M, Sathe P, Helft J, Miller J, Mortha A. The dendritic cell lineage: Ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol 2013. 10.1146/annurev-immunol-020711-074950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Kunert KS, Tisdale AS, Stern ME, Smith JA, Gipson IK. Analysis of topical cyclosporine treatment of patients with dry eye syndrome: Effect on conjunctival lymphocytes. Arch Ophthalmol 2000; 118:1489–96. 10.1001/archopht.118.11.1489. [DOI] [PubMed] [Google Scholar]
- [88].Ecoiffier T, El Annan J, Rashid S, Schaumberg D, Dana R. Modulation of integrin α4 β1 (VLA-4) in dry eye disease. Arch Ophthalmol 2008;126:1695–9. 10.1001/archopht.126.12.1695. [DOI] [PubMed] [Google Scholar]
- [89].Niederkorn JY, Stern ME, Pflugfelder SC, De Paiva CS, Corrales RM, Gao J, et al. Desiccating Stress Induces T Cell-Mediated Sjögren’s Syndrome-Like Lacrimal Keratoconjunctivitis. J Immunol 2006;176:3950–7. 10.4049/jimmunol.176.7.3950. [DOI] [PubMed] [Google Scholar]
- [90].Fan NW, Dohlman TH, Foulsham W, McSoley M, Singh RB, Chen Y, et al. The role of Th17 immunity in chronic ocular surface disorders. Ocul Surf 2021;19:157–68. 10.1016/jjtos.2020.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Verstappen GM, Corneth OBJ, Bootsma H, Kroese FGM. Th17 cells in primary Sjögren’s syndrome: Pathogenicity and plasticity. J Autoimmun 2018;87:16–25. 10.1016/jjaut.2017.ll.003. [DOI] [PubMed] [Google Scholar]
- [92].Ouyang W, Wu Y, Lin X, Wang S, Yang Y, Tang L, et al. Role of CD4+ T Helper Cells in the Development of BAC-Induced Dry Eye Syndrome in Mice. Invest Ophthalmol Vis Sci 2021;62:25. 10.1167/iovs.62.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Annan J El, Chauhan SK, Ecoiffier T, Zhang Q, Saban DR, Dana R. Characterization of effector T cells in dry eye disease. Investig Ophthalmol Vis Sci 2009;50:3802–7. 10.1167/iovs.08-2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Grewal IS, Flavell RA. CD40 and CD154 in cell-mediated immunity. Annu Rev Immunol 1998; 16:111–35. 10.1146/annurev.immunol.l6.1.111. [DOI] [PubMed] [Google Scholar]
- [95].Chen Y, Chauhan SK, Shao C, Omoto M, Inomata T, Dana R. IFN-γ-Expressing Th17 Cells Are Required for Development of Severe Ocular Surface Autoimmunity. J Immunol 2017;199:1163–9. 10.4049/jimmunol.1602144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Chen Y, Chauhan SK, Lee HS, Stevenson W, Schaumburg CS, Sadrai Z, et al. Effect of desiccating environmental stress versus systemic muscarinic AChR blockade on dry eye immunopathogenesis. Investig Ophthalmol Vis Sci 2013;54:2457–64. 10.1167/iovs.l2-11121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].De Paiva CS, Villarreal AL, Corrales RM, Rahman HT, Chang VY, Farley WJ, et al. Dry eye-induced conjunctival epithelial squamous metaplasia is modulated by interferon-γ. Investig Ophthalmol Vis Sci 2007;48:2553–60. 10.1167/iovs.07-0069. [DOI] [PubMed] [Google Scholar]
- [98].Boehm N, Riechardt Al, Wiegand M, Pfeiffer N, Grus FH. Proinflammatory cytokine profiling of tears from dry eye patients by means of antibody microarrays. Investig Ophthalmol Vis Sci 2011;52:7725–30. 10.1167/iovs.ll-7266. [DOI] [PubMed] [Google Scholar]
- [99].Pflugfelder SC, de Paiva CS, Moore QL, Volpe EA, Li DQ, Gumus K, et al. Aqueous tear deficiency increases conjunctival interferon-γ (IFN-γ) expression and goblet cell loss. Investig Ophthalmol Vis Sci 2015;56:7545–50. 10.1167/iovs.15-17627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Chauhan SK, El Annan J, Ecoiffier T, Goyal S, Zhang Q, Saban DR, et al. Autoimmunity in Dry Eye Is Due to Resistance of Th17 to Treg Suppression. J Immunol 2009; 182:1247–52. 10.4049/jimmunol.182.3.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Chauhan SK, Dana R. Role of Th17 cells in the immunopathogenesis of dry eye disease. Mucosal Immunol 2009;2:375–6. 10.1038/mi.2009.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Lin X, Rui K, Deng J, Tian J, Wang X, Wang S, et al. Th17 cells play a critical role in the development of experimental Sjögren’s syndrome. Ann Rheum Dis 2015;74:1302–10. 10.1136/annrheumdis-2013-204584. [DOI] [PubMed] [Google Scholar]
- [103].Oh JY, Kim MK, Choi HJ, Ko JH, Kang EJ, Lee HJ, et al. Investigating the relationship between serum interleukin-17 levels and systemic immune-mediated disease in patients with dry eye syndrome. Korean J Ophthalmol 2011;25:73–6. 10.3341/kjo.2011.25.2.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Garbutcheon-Singh KB, Carnt N, Pattamatta U, Samarawickrama C, White A, Calder V. A Review of the Cytokine IL-17 in Ocular Surface and Corneal Disease. Curr Eye Res 2019;44:1–10. 10.1080/02713683.2018.1519834. [DOI] [PubMed] [Google Scholar]
- [105].Stockinger B, Veldhoen M. Differentiation and function of Th17 T cells. Curr Opin Immunol 2007;19:281–6. 10.1016/j.coi.2007.04.005. [DOI] [PubMed] [Google Scholar]
- [106].Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 2006;441:235–8. 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- [107].Sadrai Z, Stevenson W, Okanobo A, Chen Y, Dohlman TH, Hua J, et al. PDE4 inhibition suppresses IL-17-associated immunity in dry eye disease. Investig Ophthalmol Vis Sci 2012;53:3584–91. 10.1167/iovs.11-9110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Chen Y, Shao C, Fan NW, Nakao T, Amouzegar A, Chauhan SK, et al. The functions of IL-23 and IL-2 on driving autoimmune effector T-helper 17 cells into the memory pool in dry eye disease. Mucosal Immunol 2020. 10.1038/s41385-020-0289-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Dohlman TH, Ding J, Dana R, Chauhan SK. T cell–derived granulocyte-macrophage colony-stimulating factor contributes to dry eye disease pathogenesis by promoting CD11b+ Myeloid cell maturation and migration. Investig Ophthalmol Vis Sci 2017;58:1330–6. 10.1167/iovs.16-20789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Sundrud MS, Trivigno C. Identity crisis of Th17 cells: Many forms, many functions, many questions. Semin Immunol 2013;25:263–72. 10.1016/j.smim.2013.10.021. [DOI] [PubMed] [Google Scholar]
- [111].Rahimy E, Pitcher JD, Pangelinan SB, Chen W, Farley WJ, Niederkom JY, et al. Spontaneous autoimmune dacryoadenitis in aged CD25KO mice. Am J Pathol 2010;177:744–53. 10.2353/ajpath.2010.091116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Turpie B, Yoshimura T, Gulati A, Rios JD, Dartt DA, Masli S. Sjögren’s syndrome-like ocular surface disease in thrombospondin-1 deficient mice. Am J Pathol 2009;175:1136–47. 10.2353/ajpath.2009.081058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Nguyen CQ, Yin H, Lee BH, Carcamo WC, Chiorini JA, Peck AB. Pathogenic effect of interleukin-17A in induction of Sjögren’s syndrome-like disease using adenovirus-mediated gene transfer. Arthritis Res Ther 2010;12. 10.1186/ar3207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Iizuka M, Wakamatsu E, Tsuboi H, Nakamura Y, Hayashi T, Matsui M, et al. Pathogenic role of immune response to M3 muscarinic acetylcholine receptor in Sjögren’s syndrome-like sialoadenitis. J Autoimmun 2010. 10.1016/jjaut.2010.08.004. [DOI] [PubMed] [Google Scholar]
- [115].Yamamoto H, Sims NE, Macauley SP, Nguyen KHT, Nakagawa Y, Humphreys-Beher MG. Alterations in the secretory response of non-obese diabetic (NOD) mice to muscarinic receptor stimulation. Clin Immunol Immunopathol 1996;78:245–55. 10.1006/clin.1996.0036. [DOI] [PubMed] [Google Scholar]
- [116].Cha S, Nagashima H, Brown VB, Peck AB, Humphreys-Beher MG. Two NOD Idd-associated intervals contribute synergistically to the development of autoimmune exocrinopathy (Sjögren’s syndrome) on a healthy murine background. Arthritis Rheum 2002;46:1390–8. 10.1002/art.10258. [DOI] [PubMed] [Google Scholar]
- [117].Bacman S, Berra A, Sterin-Borda L, Borda E. Muscarinic acetylcholine receptor antibodies as a new marker of dry eye Sjögren syndrome. Investig Ophthalmol Vis Sci 2001;42:321–7. [PubMed] [Google Scholar]
- [118].Van Blokland SCA, Versnel MA. Pathogenesis of Sjögren’s syndrome: Characteristics of different mouse models for autoimmune exocrinopathy. Clin Immunol 2002; 103:111–24. 10.1006/clim.2002.5189. [DOI] [PubMed] [Google Scholar]
- [119].Tahara M, Tsuboi H, Segawa S, Asashima H, Iizuka-Koga M, Hirota T, et al. RORγt antagonist suppresses M3 muscarinic acetylcholine receptor-induced Sjögren’s syndrome-like sialadenitis. Clin Exp Immunol 2017;187:213–24. 10.1111/cei.12868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Nanke Y, Kobashigawa T, Yago T, Kawamoto M, Yamanaka H, Kotake S. Detection of IFN-γ+IL-17+ cells in salivary glands of patients with Sjögren’s syndrome and Mikulicz’s disease: Potential role of Th17·Th1 in the pathogenesis of autoimmune diseases. Japanese J Clin Immunol 2016. 10.2177/jsci.39.473. [DOI] [PubMed] [Google Scholar]
- [121].Piccirillo CA, Shevach EM. Naturally-occurring CD4+CD25+ immunoregulatory T cells: Central players in the arena of peripheral tolerance. Semin Immunol 2004;16:81–8. 10.1016/j.smim.2003.12.003. [DOI] [PubMed] [Google Scholar]
- [122].Kretschmer K, Apostolou I, Hawiger D, Khazaie K, Nussenzweig MC, von Boehmer H. Inducing and expanding regulatory T cell populations by foreign antigen. Nat Immunol 2005;6:1219–27. 10.1038/nil265. [DOI] [PubMed] [Google Scholar]
- [123].Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T Cells and Immune Tolerance. Cell 2008;133:775–87. 10.1016/j.cell.2008.05.009. [DOI] [PubMed] [Google Scholar]
- [124].Foulsham W, Marmalidou A, Amouzegar A, Coco G, Chen Y, Dana R. Review: The function of regulatory T cells at the ocular surface. Ocul Surf 2017;15:652–9. 10.1016/jjtos.2017.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Rubtsov YP, Niec RE, Josefowicz S, Li L, Darce J, Mathis D, et al. Stability of the regulatory T cell lineage in vivo. Science (80-) 2010;329:1667–71. 10.1126/science.1191996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Kimura A, Kishimoto T. IL-6: Regulator of Treg/Th17 balance. Eur J Immunol 2010;40:1830–5. 10.1002/eji.201040391. [DOI] [PubMed] [Google Scholar]
- [127].Noack M, Miossec P. Th17 and regulatory T cell balance in autoimmune and inflammatory diseases. Autoimmun Rev 2014;13:668–77. 10.1016/j.autrev.2013.12.004. [DOI] [PubMed] [Google Scholar]
- [128].Hao LR, Li XF, Gao C, Cao L, Han ZY, Gao H, et al. Th17/Treg cell level and clinical characteristics of peripheral blood of patients with Sjögren’s syndrome complicated with primary biliary cirrhosis. Med (United States) 2019;98. 10.1097/MD.0000000000015952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Miao M, Hao Z, Guo Y, Zhang X, Zhang S, Luo J, et al. Short-term and low-dose IL-2 therapy restores the Th17/Treg balance in the peripheral blood of patients with primary Sjögren’s syndrome. Ann Rheum Dis 2018;77:1838–40. 10.1136/annrheumdis-2018-213036. [DOI] [PubMed] [Google Scholar]
- [130].de Paiva CS, Hwang CS, Pitcher JD, Pangelinan SB, Rahimy E, Chen W, et al. Age-related T-cell cytokine profile parallels corneal disease severity in Sjögren’s syndrome-like keratoconjunctivitis sicca in CD25KO mice. Rheumatology 2010;49:246–58. 10.1093/rheumatology/kep357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Trujillo-Vargas CM, Schaefer L, Alam J, Pflugfelder SC, Britton RA, de Paiva CS. The gut-eye-lacrimal gland-microbiome axis in Sjögren Syndrome. Ocul Surf 2020;18:335–44. 10.1016/jjtos.2019.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Zhang X, Schaumburg CS, Coursey TG, Siemasko KF, Volpe EA, Gandhi NB, et al. CD8+ cells regulate the T helper-17 response in an experimental murine model of sjögren syndrome. Mucosal Immunol 2014;7:417–27. 10.1038/mi.2013.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Yamada M, Ogata M, Kawai M, Mashima Y, Nishida T. Substance P and its metabolites in normal human tears. Investig Ophthalmol Vis Sci 2002;43:2622–5. [PubMed] [Google Scholar]
- [134].Yamada M, Ogata M, Kawai M, Mashima Y, Nishida T. Substance P in human tears. Cornea, vol. 22, 2003. 10.1097/00003226-200310001-00007. [DOI] [PubMed] [Google Scholar]
- [135].Suvas S Role of Substance P Neuropeptide in Inflammation, Wound Healing, and Tissue Homeostasis. J Immunol 2017;199:1543–52. 10.4049/jimmunol.1601751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Hegarty DM, Tonsfeldt K, Hermes SM, Helfand H, Aicher SA. Differential localization of vesicular glutamate transporters and peptides in corneal afferents to trigeminal nucleus caudalis. J Comp Neurol 2010. 10.1002/cne.22414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Jones MA, Marfurt CF. Peptidergic innervation of the rat cornea. Exp Eye Res 1998;66:421–35. 10.1006/exer.1997.0446. [DOI] [PubMed] [Google Scholar]
- [138].Gaddipati S, Rao P, Jerome AD, Burugula BB, Gerard NP, Suvas S. Loss of Neurokinin-1 Receptor Alters Ocular Surface Homeostasis and Promotes an Early Development of Herpes Stromal Keratitis. J Immunol 2016;197:4021–33. 10.4049/jimmunol.1600836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Tatematsu Y, Khan Q, Blanco T, Bair JA, Hodges RR, Masli S, et al. Thrombospondin-1 is necessary for the development and repair of corneal nerves. Int J Mol Sci 2018; 19. 10.3390/ijmsl9103191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Yu M, Lee SM, Lee H, Amouzegar A, Nakao T, Chen Y, et al. Neurokinin-1 Receptor Antagonism Ameliorates Dry Eye Disease by Inhibiting Antigen-Presenting Cell Maturation and T Helper 17 Cell Activation. Am J Pathol 2020;190:125–33. 10.1016/j.ajpath.2019.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Chao C, Stapleton F, Zhou X, Chen S, Zhou S, Golebiowski B. Structural and functional changes in corneal innervation after laser in situ keratomileusis and their relationship with dry eye. Graefe’s Arch Clin Exp Ophthalmol 2015. 10.1007/s00417-015-3120-1. [DOI] [PubMed] [Google Scholar]
- [142].Taketani Y, Marmalidou A, Dohlman TH, Singh RB, Amouzegar A, Chauhan SK, et al. Restoration of Regulatory T-Cell Function in Dry Eye Disease by Antagonizing Substance P/Neurokinin-1 Receptor. Am J Pathol 2020;190:1859–66. 10.1016/j.ajpath.2020.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Cunin P, Caillon A, Corvaisier M, Garo E, Scotet M, Blanchard S, et al. The Tachykinins Substance P and Hemokinin-1 Favor the Generation of Human Memory Th17 Cells by Inducing IL-1β, IL-23, and TNF-Like 1A Expression by Monocytes. J Immunol 2011;186:4175–82. 10.4049/jimmunol.1002535. [DOI] [PubMed] [Google Scholar]
- [144].Routsias JG, Tzioufas AG. Sjögren’s syndrome - Study of autoantigens and autoantibodies. Clin Rev Allergy Immunol 2007;32:238–51. 10.1007/sl2016-007-8003-8. [DOI] [PubMed] [Google Scholar]
- [145].Risselada AP, Looije MF, Kruize AA, Bijlsma JWJ, Van Roon JAG. The Role of Ectopic Germinal Centers in the Immunopathology of Primary Sjögren’s Syndrome: A Systematic Review. Semin Arthritis Rheum 2013. 10.1016/j.semarthrit.2012.07.003. [DOI] [PubMed] [Google Scholar]
- [146].Kroese FGM, Abdulahad WH, Haacke E, Bos NA, Vissink A, Bootsma H. B-cell hyperactivity in primary Sjögren’s syndrome. Expert Rev Clin Immunol 2014. 10.1586/1744666X.2014.891439. [DOI] [PubMed] [Google Scholar]
- [147].Bowman SJ, Everett CC, O’Dwyer JL, Emery P, Pitzalis C, Ng WF, et al. Randomized Controlled Trial of Rituximab and Cost-Effectiveness Analysis in Treating Fatigue and Oral Dryness in Primary Sjögren’s Syndrome. Arthritis Rheumatol 2017. 10.1002/art.40093. [DOI] [PubMed] [Google Scholar]
- [148].Takahashi M, Ishimaru N, Yanagi K, Haneji N, Saito I, Hayashi Y. High incidence of autoimmune dacryoadenitis in male non-obese diabetic (NOD) mice depending on sex steroid. Clin Exp Immunol 1997;109:555–61. 10.1046/j.13652249.1997.4691368.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Törnwall J, Lane TE, Fox RI, Fox HS. T cell attractant chemokine expression initiates lacrimal gland destruction in nonobese diabetic mice. Lab Investig 1999;79:1719–26. [PubMed] [Google Scholar]
- [150].Jabs DA, Lee B, Whittum-Hudson JA, Prendergast RA. Th1 versus Th2 immune responses in autoimmune lacrimal gland disease in MRL/Mp mice. Investig Ophthalmol Vis Sci 2000;41:826–31. [PubMed] [Google Scholar]
- [151].Fujita H, Fujihara T, Takeuchi T, Saito I, Tsubota K. Lacrimation and salivation are not related to lymphocytic infiltration in lacrimal and salivary glands in MRL lpr/lpr mice. Adv. Exp. Med. Biol, vol. 438, 1998, p. 941–8. 10.1007/978-l-4615-5359-5_133. [DOI] [PubMed] [Google Scholar]
- [152].Voulgarelis M, Tzioufas AG. Pathogenetic mechanisms in the initiation and perpetuation of SjÖgren’s syndrome. Nat Rev Rheumatol 2010. 10.1038/nrrheum.2010.118. [DOI] [PubMed] [Google Scholar]
- [153].Skopouli FN, Fox PC, Galanopoulou V, Atkinson JC, Jaffe ES, Moutsopoulos HM. T cell subpopulations in the labial minor salivary gland histopathologic lesion of Sjogren’s syndrome. J Rheumatol 1991. [PubMed] [Google Scholar]
- [154].DeVoss JJ, LeClair NP, Hou Y, Grewal NK, Johannes KP, Lu W, et al. An Autoimmune Response to Odorant Binding Protein 1a Is Associated with Dry Eye in the Aire -Deficient Mouse . J Immunol 2010;184:4236–46. 10.4049/jimmunol.0902434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Hayakawa I, Tedder TF, Zhuang Y. B-lymphocyte depletion ameliorates Sjögren’s syndrome in Id3 knockout mice. Immunology 2007;122:73–9. 10.1111/j.1365-2567.2007.02614.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Voigt A, Esfandiary L, Wanchoo A, Glenton P, Donate A, Craft WF, et al. Sexual dimorphic function of IL-17 in salivary gland dysfunction of the C57BL/6.NOD-Aec1Aec2 model of Sjögren’s syndrome. Sci Rep 2016;6. 10.1038/srep38717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Reksten TR, Jonsson MV, Szyszko EA, Brun JG, Jonsson R, Brokstad KA. Cytokine and autoantibody profiling related to histopathological features in primary Sjögren’s syndrome. Rheumatology 2009. 10.1093/rheumatology/kepl49. [DOI] [PubMed] [Google Scholar]
- [158].Chivasso C, Sarrand J, Perret J, Delporte C, Soyfoo MS. The involvement of innate and adaptive immunity in the initiation and perpetuation of sjögren’s syndrome. Int J Mol Sci 2021;22:1–21. 10.3390/ijms22020658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Verstappen GM, Kroese FGM, Bootsma Η. T cells in primary Sjögren’s syndrome: targets for early intervention. Rheumatology 2019. 10.1093/rheumatology/kez004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Subbarayal B, Chauhan SK, Di Zazzo A, Dana R. IL-17 Augments B Cell Activation in Ocular Surface Autoimmunity. J Immunol 2016;197:3464–70. 10.4049/jimmunol.1502641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Wolffsohn JS, Arita R, Chalmers R, Djalilian A, Dogru M, Dumbleton K, et al. TFOS DEWS II Diagnostic Methodology report. Ocul Surf 2017;15:539–74. 10.1016/jjtos.2017.05.001. [DOI] [PubMed] [Google Scholar]
- [162].Fabiani C, Barabino S, Rashid S, Dana MR. Corneal epithelial proliferation and thickness in a mouse model of dry eye. Exp Eye Res 2009;89:166–71. 10.1016/j.exer.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Yeh S, Song XJ, Farley W, Li DQ, Stern ME, Pflugfelder SC. Apoptosis of ocular surface cells in experimentally induced dry eye. Investig Ophthalmol Vis Sci 2003;44:124–9. 10.1167/iovs.02-0581. [DOI] [PubMed] [Google Scholar]
- [164].Suhalim JL, Parfitt GJ, Xie Y, De Pavia CS, Pflugfelder SC, Shah TN, et al. Effect of desiccating stress on mouse meibomian gland function. Ocul Surf 2014;12:59–68. 10.1016/jjtos.2013.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Stepp MA, Pal-Ghosh S, Tadvalkar G, Williams AR, Pflugfelder SC, de Paiva CS. Reduced corneal innervation in the CD25 null model of Sjögren syndrome. Int J Mol Sci 2018;19. 10.3390/ijmsl9123821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Liu L, Dana R, Yin J. Sensory neurons directly promote angiogenesis in response to inflammation via substance P signaling. FASEB J 2020;34:6229–43. 10.1096/fj.201903236R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Benítez-Del-Castillo JM, Acosta MC, Wassfi MA, Díaz-Valle D, Gegúndez JA, Fernandez C, et al. Relation between corneal innervation with confocal microscopy and corneal sensitivity with noncontact esthesiometry in patients with dry eye. Investig Ophthalmol Vis Sci 2007. 10.1167/iovs.06-0127. [DOI] [PubMed] [Google Scholar]
- [168].Labbé A, Alalwani H, Van Went C, Brasnu E, Georgescu D, Baudouin C. The relationship between subbasal nerve morphology and corneal sensation in ocular surface disease. Investig Ophthalmol Vis Sci 2012;53:4926–31. 10.1167/iovs.11-8708. [DOI] [PubMed] [Google Scholar]
- [169].Labbé A, Liang Q, Wang Z, Zhang Y, Xu L, Baudouin C, et al. Corneal nerve structure and function in patients with non-sjögren dry eye: Clinical correlations. Investig Ophthalmol Vis Sci 2013. 10.1167/iovs.13-12370. [DOI] [PubMed] [Google Scholar]
- [170].Kheirkhah A, Satitpitakul V, Hamrah P, Dana R. Patients with Dry Eye Disease and Low Subbasal Nerve Density Are at High Risk for Accelerated Corneal Endothelial Cell Loss. Cornea, 2017. 10.1097/ic0.0000000000001057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Coursey TG, Gandhi NB, Volpe EA, Pflugfelder SC, De Paiva CS. Chemokine receptors CCR6 and CXCR3 are necessary for CD4+ T cell mediated ocular surface disease in experimental dry eye disease. PLoS One 2013;8:e78508. 10.1371/journal.pone.0078508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Dohlman TH, Chauhan SK, Kodati S, Hua J, Chen Y, Omoto M, et al. The CCR6/CCL20 axis mediates Th17 cell migration to the ocular surface in dry eye disease. Investig Ophthalmol Vis Sci 2013;54:4081–91. 10.1167/iovs.12-11216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Choi W, Li Z, Oh HJ, Im SK, Lee SH, Park SH, et al. Expression of CCR5 and its ligands CCL3, −4, and −5 in the tear film and ocular surface of patients with dry eye disease. Curr Eye Res 2012. 10.3109/02713683.2011.622852. [DOI] [PubMed] [Google Scholar]
- [174].Yoon KC, Park CS, You IC, Choi HJ, Lee KH, Im SK, et al. Expression of CXCL9, −10, −11, and CXCR3 in the tear film and ocular surface of patients with dry eye syndrome. Investig Ophthalmol Vis Sci 2010. 10.1167/iovs.09-3425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Sato W, Aranami T, Yamamura T. Cutting Edge: Human Th17 Cells Are Identified as Bearing CCR2 + CCR5 – Phenotype. J Immunol 2007;178:7525–9. 10.4049/jimmunol.178.12.7525. [DOI] [PubMed] [Google Scholar]
- [176].Kara EE, McKenzie DR, Bastow CR, Gregor CE, Fenix KA, Ogunniyi AD, et al. CCR2 defines in vivo development and homing of IL-23-driven GM-CSF-producing Th17 cells. Nat Commun 2015;6. 10.1038/ncomms9644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Guimaraes De Souza R, Yu Z, Stern ME, Pflugfelder SC, De Paiva CS. Suppression of Th1-Mediated Keratoconjunctivitis Sicca by Lifitegrast. J Ocul Pharmacol Ther 2018;34:543–9. 10.1089/jop.2018.0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Perez VL, Pflugfelder SC, Zhang S, Shojaei A, Haque R. Lifitegrast, a Novel Integrin Antagonist for Treatment of Dry Eye Disease. Ocul Surf 2016;14:207–15. 10.1016/j.jtos.2016.01.001. [DOI] [PubMed] [Google Scholar]
- [179].Corrales RM, Villarreal A, Farley W, Stern ME, Li DQ, Pflugfelder SC. Strain-related cytokine profiles on the murine ocular surface in response to desiccating stress. Cornea 2007;26:579–84. 10.1097/ICO.0b013e318033a729. [DOI] [PubMed] [Google Scholar]
- [180].Liu R, Gao C, Chen H, Li Y, Jin Y, Qi H. Analysis of Th17-associated cytokines and clinical correlations in patients with dry eye disease. PLoS One 2017. 10.1371/journal.pone.0173301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Tan X, Sun S, Liu Y, Zhu T, Wang K, Ren T, et al. Analysis of Th17-Associated cytokines in tears of patients with dry eye syndrome. Eye 2014;28:608–13. 10.1038/eye.2014.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Enríquez-de-Salamanca A, Castellanos E, Stern ME, Fernández I, Carreño E, García-Vázquez C, et al. Tear cytokine and chemokine analysis and clinical correlations in evaporative-type dry eye disease. Mol Vis 2010. [PMC free article] [PubMed] [Google Scholar]
- [183].Chen Y, Chauhan SK, Shao C, Omoto M, Inomata T, Dana R. IFN-γ-Expressing Th17 Cells Are Required for Development of Severe Ocular Surface Autoimmunity. J Immunol 2017;199:1163–9. 10.4049/jimmunol.1602144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Zhang X, Chen W, De Paiva CS, Corrales RM, Volpe EA, McClellan AJ, et al. Interferon-γ exacerbates dry eye-induced apoptosis in conjunctiva through dual apoptotic pathways. Investig Ophthalmol Vis Sci 2011;52:6279–85. 10.1167/iovs.10-7081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Floyd AM, Zhou X, Evans C, Rompala OJ, Zhu L, Wang M, et al. Mucin Deficiency Causes Functional and Structural Changes of the Ocular Surface. PLoS One 2012;7. 10.1371/journal.pone.0050704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Ko BY, Xiao Y, Barbosa FL, de Paiva CS, Pflugfelder SC. Goblet cell loss abrogates ocular surface immune tolerance. JCI Insight 2018;3. 10.1172/jci.insight.98222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].McGeachy MJ, Cua DJ. Th17 Cell Differentiation: The Long and Winding Road. Immunity 2008;28:445–53. 10.1016/j.immuni.2008.03.001. [DOI] [PubMed] [Google Scholar]
- [188].Ouyang W, Kolls JK, Zheng Y. The Biological Functions of T Helper 17 Cell Effector Cytokines in Inflammation. Immunity 2008;28:454–67. 10.1016/j.immuni.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Reyes NJ, Yu C, Mathew R, Kunnen CM, Kalnitsky J, Redfern RL, et al. Neutrophils cause obstruction of eyelid sebaceous glands in inflammatory eye disease in mice. Sci Transl Med 2018;10:eaas9164. 10.1126/scitranslmed.aas9164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Chen Y, Chauhan SK, Soo Lee H, Saban DR, Dana R. Chronic dry eye disease is principally mediated by effector memory Th17 cells. Mucosal Immunol 2014;7:38–45. 10.1038/mi.2013.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Siedlecki AN, Smith SD, Siedlecki AR, Hayek SM, Sayegh RR. Ocular pain response to treatment in dry eye patients: Ocular Pain Treatment Response. Ocul Surf 2020. 10.1016/jjtos.2019.12.004. [DOI] [PubMed] [Google Scholar]
- [192].Skrzypecki J, Tomasz H, Karolina C. Variability of Dry Eye Disease Following Removal of Lacrimal Glands in Rats. Adv. Exp. Med. Biol, vol. 1153, 2019, p. 109–15. 10.1007/5584_2019_348. [DOI] [PubMed] [Google Scholar]
- [193].Mecum NE, Cyr D, Malon J, Demers D, Cao L, Meng ID. Evaluation of corneal damage after lacrimal gland excision in male and female mice. Investig Ophthalmol Vis Sci 2019;60:3264–74. 10.1167/iovs.18-26457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Mecum NE, Demers D, Sullivan CE, Denis TE, Kalliel JR, Meng ID. Lacrimal gland excision in male and female mice causes ocular pain and anxiety-like behaviors. Sci Rep 2020;10. 10.1038/s41598-020-73945-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].Li F, Yang W, Jiang H, Guo C, Huang AJW, Hu H, et al. TRPV1 activity and substance P release are required for corneal cold nociception. Nat Commun 2019; 10. 10.1038/s41467-019-13536-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [196].Rahman M, Okamoto K, Thompson R, Katagiri A, Bereiter DA. Sensitization of trigeminal brainstem pathways in a model for tear deficient dry eye. Pain 2015;156:942–50. 10.1097/j.pain.0000000000000135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [197].Aicher SA, Hermes SM, Hegarty DM. Denervation of the lacrimal gland leads to corneal hypoalgesia in a novel rat model of aqueous dry eye disease. Investig Ophthalmol Vis Sci 2015;56:6981–9. 10.1167/iovs.15-17497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [198].Bourcier T, Acosta MC, Borderie V, Borrás F, Gallar J, Bury T, et al. Decreased corneal sensitivity in patients with dry eye. Investig Ophthalmol Vis Sci 2005;46:2341–5. 10.1167/iovs.04-1426. [DOI] [PubMed] [Google Scholar]
- [199].Tuisku IS, Konttinen YT, Konttinen LM, Tervo TM. Alterations in corneal sensitivity and nerve morphology in patients with primary Sjögren’s syndrome. Exp Eye Res 2008. 10.1016/j.exer.2008.03.002. [DOI] [PubMed] [Google Scholar]
- [200].Tuominen ISJ, Konttinen YT, Vesaluoma MH, Moilanen JAO, Helintö M, Tervo TMT. Corneal innervation and morphology in primary Sjögren’s syndrome. Investig Ophthalmol Vis Sci 2003. 10.1167/iovs.02-1260. [DOI] [PubMed] [Google Scholar]
- [201].Zhang M, Chen J, Luo L, Xiao Q, Sun M, Liu Z. Altered corneal nerves in aqueous tear deficiency viewed by in vivo confocal microscopy. Cornea 2005. 10.1097/01.ico.0000154402.01710.95. [DOI] [PubMed] [Google Scholar]
- [202].Kryczek I, Zhao E, Liu Y, Wang Y, Vatan L, Szeliga W, et al. Human TH17 cells are long-lived effector memory cells. Sci Transl Med 2011;3. 10.1126/scitranslmed.3002949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Muranski P, Borman ZA, Kerkar SP, Klebanoff CA, Ji Y, Sanchez-Perez L, et al. Th17 Cells Are Long Lived and Retain a Stem Cell-like Molecular Signature. Immunity 2011;35:972–85. 10.1016/j.immuni.2011.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [204].Chen Y, Chauhan SK, Tan X, Dana R. Interleukin-7 and -15 maintain pathogenic memory Th17 cells in autoimmunity. J Autoimmun 2017;77:96–103. 10.1016/jjaut.2016.ll.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [205].van Leeuwen EM, Sprent J, Surh CD. Generation and maintenance of memory CD4+ T Cells. Curr Opin Immunol 2009. 10.1016/j.coi.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [206].Pepper M, Jenkins MK. Origins of CD4+ effector and central memory T cells. Nat Immunol 2011. 10.1038/ni.2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Amadi-Obi A, Yu CR, Liu X, Mahdi RM, Clarke GL, Nussenblatt RB, et al. TH17 cells contribute to uveitis and scleritis and are expanded by IL-2 and inhibited by IL-27/STAT1. Nat Med 2007;13:711–8. 10.1038/nml585. [DOI] [PubMed] [Google Scholar]
- [208].Liu Y, Zhou N, Zhou L, Wang J, Zhou Y, Zhang T, et al. IL-2 regulates tumor-reactive CD8+ T cell exhaustion by activating the aryl hydrocarbon receptor. Nat Immunol 2021. 10.1038/s41590-020-00850-9. [DOI] [PubMed] [Google Scholar]
- [209].Luo J, Ming B, Zhang C, Deng X, Li P, Wei Z, et al. IL-2 inhibition of Th17 generation rather than induction of treg cells is impaired in primary Sjögren’s syndrome patients. Front Immunol 2018;9. 10.3389/fimmu.2018.01755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [210].Daniels MA, Teixeiro E. TCR signaling in T cell memory. Front Immunol 2015. 10.3389/fimmu.2015.00617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [211].Johnson JL, Rosenthal RL, Knox JJ, Myles A, Naradikian MS, Madej J, et al. The Transcription Factor T-bet Resolves Memory B Cell Subsets with Distinct Tissue Distributions and Antibody Specificities in Mice and Humans. Immunity 2020. 10.1016/j.immuni.2020.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [212].Mackay LK, Wynne-Jones E, Freestone D, Pellicci DG, Mielke LA, Newman DM, et al. T-box Transcription Factors Combine with the Cytokines TGF-β and IL-15 to Control Tissue-Resident Memory T Cell Fate. Immunity 2015. 10.1016/j.immuni.2015.11.008. [DOI] [PubMed] [Google Scholar]
- [213].Intlekofer AM, Takemoto N, Wherry EJ, Longworth SA, Northrup JT, Palanivel VR, et al. Effector and memory CD8+ T cell fate coupled by T-bet and eomesodermin. Nat Immunol 2005. 10.1038/nil268. [DOI] [PubMed] [Google Scholar]
- [214].Marshall HD, Chandele A, Jung YW, Meng H, Poholek AC, Parish IA, et al. Differential Expression of Ly6C and T-bet Distinguish Effector and Memory Th1 CD4 + Cell Properties during Viral Infection. Immunity 2011;35:633–46. 10.1016/j.immuni.2011.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [215].Pepper M, Linehan JL, Pagán AJ, Zell T, Dileepan T, Cleary PP, et al. Different routes of bacterial infection induce long-lived T H 1 memory cells and short-lived T H 17 cells. Nat Immunol 2010;11:83–9. 10.1038/ni.1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [216].MacLeod MKL, Kappler JW, Marrack P. Memory CD4 T cells: Generation, reactivation and re-assignment. Immunology 2010. 10.llll/j.1365-2567.2010.03260.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [217].Berard M, Tough DF. Qualitative differences between naïve and memory T cells. Immunology 2002. 10.1046/j.1365-2567.2002.01447.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [218].Blattman JN, Antia R, Sourdive DJD, Wang X, Kaech SM, Murali-Krishna K, et al. Estimating the precursor frequency of naive antigen-specific CD8 T cells. J Exp Med 2002. 10.1084/jem.20001021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [219].Woodland DL, Kohlmeier JE. Migration, maintenance and recall of memory T cells in peripheral tissues. Nat Rev Immunol 2009. 10.1038/nri2496. [DOI] [PubMed] [Google Scholar]
- [220].Cornelissen F, Asmawidjaja PS, Mus AMC, Cometh O, Kikly K, Lubberts E. IL-23 Dependent and Independent Stages of Experimental Arthritis: No Clinical Effect of Therapeutic IL-23p19 Inhibition in Collagen-induced Arthritis. PLoS One 2013. 10.1371/journal.pone.0057553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [221].Haines CJ, Chen Y, Blumenschein WM, Jain R, Chang C, Joyce-Shaikh B, et al. Autoimmune Memory T Helpera 17 Cell Function and Expansion Are Dependent on Interleukin-23. Cell Rep 2013;3:1378–88. 10.1016/j.celrep.2013.03.035. [DOI] [PubMed] [Google Scholar]
- [222].Goronzy JJ, Weyand CM. Immune aging and autoimmunity. Cell Mol Life Sci 2012;69:1615–23. 10.1007/s00018-012-0970-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [223].Kaswan R, Pappas C, Wall K, Hirsh SG. Survey of canine tear deficiency in veterinary practice. Adv. Exp. Med. Biol, vol. 438, 1998, p. 931–9. 10.1007/978-1-4615-5359-5132. [DOI] [PubMed] [Google Scholar]
- [224].Nien CJ, Paugh JR, Massei S, Wahlert AJ, Kao WW, Jester JV. Age-related changes in the meibomian gland. Exp Eye Res 2009;89:1021–7. 10.1016/j.exer.2009.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [225].Yoon CH, Ryu JS, Hwang HS, Kim MK. Comparative analysis of age-related changes in lacrimal glands and meibomian glands of a c57bl/6 male mouse model. Int J Mol Sci 2020;21:1–19. 10.3390/ijms21114169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [226].Coursey TG, Bian F, Zaheer M, Pflugfelder SC, Volpe EA, De Paiva CS. Age-related spontaneous lacrimal keratoconjunctivitis is accompanied by dysfunctional T regulatory cells. Mucosal Immunol 2017;10:743–56. 10.1038/mi.2016.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [227].Marko CK, Menon BB, Chen G, Whitsett JA, Clevers H, Gipson IK. Spdef null mice lack conjunctival goblet cells and provide a model of dry eye. Am J Pathol 2013;183:35–48. 10.1016/j.ajpath.2013.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [228].McClellan AJ, Volpe EA, Zhang X, Darlington GJ, Li DQ, Pflugfelder SC, et al. Ocular surface disease and dacryoadenitis in aging C57BL/6 mice. Am J Pathol 2014;184:631–43. 10.1016/j.ajpath.2013.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [229].De Silva MEH, Hill LJ, Downie LE, Chinnery HR. The effects of aging on corneal and ocular surface homeostasis in mice. Investig Ophthalmol Vis Sci 2019;60:2705–15. 10.1167/iovs.19-26631. [DOI] [PubMed] [Google Scholar]
- [230].Bian F, Xiao Y, Barbosa FL, de Souza RG, Hernandez H, Yu Z, et al. Age-associated antigen-presenting cell alterations promote dry-eye inducing Th1 cells. Mucosal Immunol 2019;12:897–908. 10.1038/s41385-018-0127-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [231].Foulsham W, Mittal SK, Taketani Y, Chen Y, Nakao T, Chauhan SK, et al. Aged Mice Exhibit Severe Exacerbations of Dry Eye Disease with an Amplified Memory Th17 Cell Response. Am J Pathol 2020;190:1474–82. 10.1016/j.ajpath.2020.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [232].Wan SJ, Sullivan AB, Shieh P, Metruccio MME, Evans DJ, Bertozzi CR, et al. IL-1R and MyD88 contribute to the absence of a bacterial microbiome on the healthy murine cornea. Front Microbiol 2018;9. 10.3389/fmicb.2018.01117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [233].Horai R, Caspi RR. Microbiome and autoimmune uveitis. Front Immunol 2019; 10. 10.3389/fimmu.2019.00232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [234].Horai R, Zárate-Bladés CR, Dillenburg-Pilla P, Chen J, Kielczewski JL, Silver PB, et al. Microbiota-Dependent Activation of an Autoreactive T Cell Receptor Provokes Autoimmunity in an Immunologically Privileged Site. Immunity 2015;43:343–53. 10.1016/j.immuni.2015.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [235].Wang C, Zaheer M, Bian F, Quach D, Swennes AG, Britton RA, et al. Sjögren-like lacrimal Keratoconjunctivitis in germ-free mice. Int J Mol Sci 2018; 19. 10.3390/ijmsl9020565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [236].Zaheer M, Wang C, Bian F, Yu Z, Hernandez H, de Souza RG, et al. Protective role of commensal bacteria in Sjögren Syndrome. J Autoimmun 2018;93:45–56. 10.1016/jjaut.2018.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [237].De Paiva CS, Jones DB, Stem ME, Bian F, Moore QL, Corbiere S, et al. Altered Mucosal Microbiome Diversity and Disease Severity in Sjögren Syndrome. Sci Rep 2016;6. 10.1038/srep23561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [238].Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 2013;504:446–50. 10.1038/naturel2721. [DOI] [PubMed] [Google Scholar]
- [239].Arpaia N, Campbell C, Fan X, Dikiy S, Van Der Veeken J, Deroos P, et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 2013;504:451–5. 10.1038/naturel2726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [240].Östman S, Rask C, Wold AE, Hultkrantz S, Telemo E. Impaired regulatory T cell function in germ-free mice. Eur J Immunol 2006;36:2336–46. 10.1002/eji.200535244. [DOI] [PubMed] [Google Scholar]
- [241].Kim J, Choi SH, Kim YJ, Jeong HJ, Ryu JS, Lee HJ, et al. Clinical effect of IRT-5 probiotics on immune modulation of autoimmunity or alloimmunity in the eye. Nutrients 2017;9. 10.3390/nu9111166. [DOI] [PMC free article] [PubMed] [Google Scholar]