

Abstract
Shotgun-metagenomics may give valuable clinical information beyond the detection of potential pathogen(s). Identification of antimicrobial resistance (AMR), virulence genes and typing directly from clinical samples has been limited due to challenges arising from incomplete genome coverage. We assessed the performance of shotgun-metagenomics on positive blood culture bottles (n = 19) with periprosthetic tissue for typing and prediction of AMR and virulence profiles in Staphylococcus aureus. We used different approaches to determine if sequence data from reads provides more information than from assembled contigs. Only 0.18% of total reads was derived from human DNA. Shotgun-metagenomics results and conventional method results were consistent in detecting S. aureus in all samples. AMR and known periprosthetic joint infection virulence genes were predicted from S. aureus. Mean coverage depth, when predicting AMR genes was 209 ×. Resistance phenotypes could be explained by genes predicted in the sample in most of the cases. The choice of bioinformatic data analysis approach clearly influenced the results, i.e. read-based analysis was more accurate for pathogen identification, while contigs seemed better for AMR profiling. Our study demonstrates high genome coverage and potential for typing and prediction of AMR and virulence profiles in S. aureus from shotgun-metagenomics data.
Subject terms: Computational biology and bioinformatics, Microbiology, Diseases
Introduction
Staphylococcus aureus is an important opportunistic pathogen considered as the most common cause of periprosthetic infections (PIs)1–5. The emergence and spread of resistance pose an increasing threat to public health, in particular, methicillin-resistant S. aureus (MRSA)6. The success of S. aureus as a pathogen is in part due to its ability to develop resistance to a wide variety of antimicrobial compounds. Additionally, S. aureus can adapt to a biofilm mode of growth whereby infections become persistent and recurrent, particularly in association with prosthetic implants7.
Microbiological diagnosis of PJI is challenging. A variety of tools are available for facilitating the diagnosis of PJI, including emerging technologies such as metagenomic approaches8. The use of shotgun-metagenomics (SMg) for the analysis of clinical specimens has emerged as a promising approach for pathogen identification, antimicrobial resistance (AMR) identification and outbreak investigation in clinical microbiology laboratories. This approach has been used for the analysis of different types of clinical specimens, including samples related to PJI, e.g. synovial fluid9,10, sonication fluid11–14 and tissue15, mainly for the identification of pathogens.
The advantage of using direct material in SMg is mainly short turnaround time for identification of pathogens, and the use of e.g. Nanopore sequencing is very promising for rapid identification of pathogens within 4–24 h14,16,17. However, in these studies, extracted DNA is contaminated with a high concentration of human DNA while the bacterial DNA yield is very low, and thus, leading to a low number of bacterial reads. In a previous study, we showed that SMg performed directly on positive blood culture bottles (BCBs) inoculated with periprosthetic tissue (PT), is a convenient method to identify potential pathogens causing PJI18. However, beyond the identification of pathogens, SMg provides unlimited access to other clinically relevant genomic features such as antibiotic resistance, virulence genes profiles and strain-level typing19,20.
Currently, SMg is considered in its infancy for pathogen characterization, including inference of antibiotic susceptibility21. Challenges arise due to the diversity of drug resistance mechanisms, multidrug resistance, and incomplete genome coverage, leading to insufficient sequence reads for detection of ARGs22. However, there are some studies that show the potential of SMg for the detection of ARGs23–25 by comparing the genotype against the phenotype, or generating AMR profiles from SMg assemblies and comparing them with whole genome sequences (WGS) from isolates. The use of SMg on samples from bone and joint infections has been used where they could predict antibiotic susceptibility in 94.1% (monomicrobial) and 76.5% (polymicrobial) of the cases15. However, in these studies, the main obstacle has been a high background of genetic material mainly derived from the host, which generates very few bacterial reads. Similarly, when using SMg data for subtyping bacteria, one of the main challenges is missing loci. This problem arises when coverage is too low to guarantee the presence of a read containing a given sequence in the targeted genome26.
We previously showed that SMg on BCBs with PT resulted in acceptable high number of bacterial reads, genome coverage and genome sequencing depth18. Here, we wanted to assess the potential of SMg for the identification and typing of the most common cause of PJI, S. aureus, and the prediction of virulence and AMR directly from clinical samples.
Results
Sequencing data
SMg sequencing from the 19 samples resulted in a mean number of 3,990,292 reads per clinical sample (range 2,608,766–8,086,037). Sequencing of DNA from the sample spiked with S. aureus and the negative control produced 5,942,038 and 11,192,852 reads, respectively (Supplementary Table S1). Samples contained a lower proportion of reads classified either as human or horse or PhiX, while 98% of the reads did not map to any of the reference sequences used for the alignment (Supplementary Table S2 and Fig. S1).
After data preprocessing, a mean number of 3,700,731 reads remained for further taxonomical classification while from the negative control only 187,094 reads (1.67%) remained for taxonomical analyses (Supplementary Table S3). Kraken taxonomically classified a mean proportion of 98.36% reads, with 95.74% bacterial reads (Fig. 1 and Supplementary Table S4). Assembly with metaSPAdes yielded a mean number of 232 contigs (range 134–378), with a mean total size of 3.1 Mb (range 2.6–4.8 Mb) in the clinical samples and 213 contigs for a total length of 2.7 Mb in the sample spiked with S. aureus (Supplementary Table S5). The total number of base pairs was higher in polymicrobial samples than in monomicrobial ones (4.8 Mb vs 2.7 Mb, respectively, t-test and P-value < 0.0001). The mean of the “maximum contig size” was 262,574 bp (median 264,931 bp, maximum 425,306 bp) in the clinical samples and 218,856 bp in the spiked sample, and no significant difference was observed between polymicrobial and monomicrobial samples (184,461 bp and 283,495 bp, respectively, t-test and P-value = 0.149). Binning with MaxBin in polymicrobial samples grouped a mean number of 43 contigs in the bin assigned to S. aureus (range 39–51) with a mean total of 2.6 Mb (range 2.6–2.7 Mb) and a mean maximum length 297,040 bp (234,035–381,826 bp).
Figure 1.
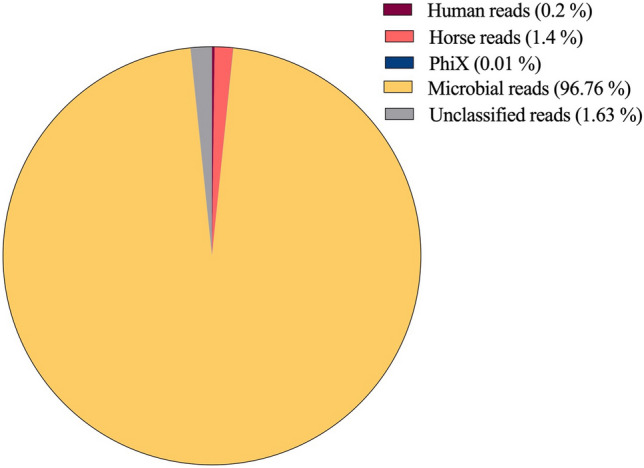
Proportion of reads taxonomically classified as human, horse, PhiX, microbial and unclassified.
Identification of S. aureus by SMg
The taxonomical classification was performed using Kraken on reads and assembled contigs. When the taxonomical classification from the reads identified multiple highly abundant species (polymicrobial samples), contigs were grouped by species into bins, and then used for taxonomical classification. Relative abundance (determined by Bracken) from the most abundant species in the samples varied depending on the selected approach. The bin classified as S. aureus was used for downstream analyses for pathogen characterization.
Staphylococcus aureus was identified in all the samples by SMg. S. aureus (19/19) and S. agalactiae (4/19) was identified from both the reads and contigs. S. aureus was the most abundant species identified by SMg, with exception of samples 7 and 9, where S. agalactiae was more abundant (86.4% and 85.8%, from the reads, respectively) (Table 1 and Supplementary Fig. S1).
Table 1.
Bacteria identified in the samples and in the positive control (PC) by MALDI-TOF from blood culture bottles (BCBs) and by Kraken from the shotgun-metagenomics (SMg) with their relative abundance (percentage in parentheses) determined by Bracken on the reads, contigs and bins.
| Sample ID | Patient no. | Microorganism(s) identified | ||||
|---|---|---|---|---|---|---|
| BCBs (MALDI-TOF) | Shotgun-metagenomics | |||||
| Reads | Contigs | Bins | ||||
| No | Taxonomy | |||||
| 1 | 1 | S. aureus | S. aureus (99.9%) | S. aureus (100%) | ||
| 2 | 2 | S. aureus | S. aureus (99.9%) | S. aureus (100%) | ||
| 3 | 3 | S. aureus | S. aureus (99.9%) | S. aureus (100%) | ||
| 4 | 4 | S. aureus | S. aureus (99.3%) | S. aureus (98.6%) | ||
| 5 | 5 | S. aureus | S. aureus (99.9%) | S. aureus (100%) | ||
| 6 | 6 | S. aureus |
S. aureus (90.9%) S. agalactiae (8.5%) |
S. aureus (60.6%) S. agalactiae (39.4%) |
1 | S. aureus (100%) |
| 2 | S. agalactiae (100%) | |||||
| 7 | 7 |
S. aureus S. agalactiae |
S. aureus (10.7%) S. agalactiae (86.4%) |
S. aureus (62.4%) S. agalactiae (37.6%) |
1 |
S. agalactiae (97%) S. aureus (2%) |
| 2 | S. aureus (100%) | |||||
| 8 | 8 | S. aureus |
S. aureus (97.6%) S. agalactiae (2.2%) |
S. aureus (51.5%) S. agalactiae (48.2%) |
1 | S. aureus (100%) |
| 2 | S. agalactiae (100%) | |||||
| 9 | 9 | S. aureus |
S. agalactiae (85.8%) S. aureus (11.4%) |
S. agalactiae (39.8%) S. aureus (60.3%) |
1 | S. agalactiae (94.8%) |
| 2 | S. aureus (100%) | |||||
| 10 | 10 | S. aureus | S. aureus (99.8%) | S. aureus (100%) | ||
| 11 | 11 | S. aureus | S. aureus (98.2%) | S. aureus (98.3%) | ||
| 12 | 12 | S. aureus | S. aureus (99.8%) | S. aureus (100%) | ||
| 13 | 13 | S. aureus | S. aureus (99.4%) | S. aureus (100%) | ||
| 14 | 14 | S. aureus | S. aureus (99.6%) | S. aureus (100%) | ||
| 15 | 15 | S. aureus | S. aureus (99.4%) | S. aureus (100%) | ||
| 16 | 16 | S. aureus | S. aureus (99.9%) | S. aureus (99.0%) | ||
| 17 | 17 | S. aureus | S. aureus (89.8%) | S. aureus (99.2%) | ||
| 18 | 2 | S. aureus | S. aureus (89.7%) | S. aureus (97.6%) | ||
| 19 | 1 | S. aureus | S. aureus (99.9%) | S. aureus (84.7%) | ||
| PC | NA | S. aureus | S. aureus (99.9%) | S. aureus (95.1%) | ||
We identified some reads assigned to S. aureus (10.3%) in the negative control. The reads were evaluated by mapping them against a S. aureus reference genome (accession number GCF_000144955.1). Results showed that 4% of the reads mapped to the reference genome and 6% of the reference genome was covered. When visualizing the mapping results, reads mapped with genetic areas belonging to coding sequences annotated as RNAs with a very low coverage depth, and they were not distributed all over the genomes. Also, other species were found in the NC but in a very low abundance except for the species Bacillus cereus (81.5%) which was the most abundant species found. (For further details on the taxa identified in the negative control, see Sanabria et al.27).
Staphylococcus aureus antibiotic resistance determinants by SMg
The presence of antibiotic resistance genes (ARGs) was determined by SMg from the reads and contigs for all the samples, and from the bins classified as S. aureus in polymicrobial samples (Sample 6, 7, 8 and 9). The presence of ARGs found in the reads, contigs and bins using the the NCBI bacterial antimicrobial resistance reference gene database as reference, were determined, and compared with the results obtained by the phenotypic antimicrobial susceptibility testing (AST) (Table 2, Supplementary Tables S6 and S7).
Table 2.
Antibiotic resistance genes (ARGs) detected in this study using different approaches (reads, contigs and bins) with the NCBI bacterial antimicrobial resistance reference gene database, chromosomal mutations detected using ResFinder, and antibiotic resistance phenotype observed by the conventional antibiotic susceptibility testing (AST).
| Sample no. | Conventional antibiotic susceptibility test | Chromosomal mutations | ARGs detected from shotgun-metagenomics | ||
|---|---|---|---|---|---|
| Reads | Contigs | Bins | |||
| 1 | Penicillin | blaZ, tet38 | fosB, blaZ, tet38 | ||
| 2 | Penicillin | blaZ, tet38 | blaZ, tet38 | ||
| 3 | grlA | tet38 | tet38 | ||
| 4 | Penicillin | fosB, blaZ, tet38 | |||
| 5 | grlA | tet38 | tet38 | ||
| 6 | Penicillin | grlA | tet38 | tet38 | |
| 7 | Penicillin | tetM, tet38 | fosB, tetM, tet38 | fosB, tet38 | |
| 8 | tet38, tetM | tet38 | |||
| 9 | Penicillin | tet38, tetM | fosB, tetM, tet38 | fosB, tet38 | |
| 10 | grlA | tet38 | |||
| 11 | fosB, tet38, blaZ | ||||
| 12 |
Penicillin Fusidic acid |
fosB, blaZ, tet38 | |||
| 13 | blaZ, tet38 | ||||
| 14 | Penicillin | blaZ | fosB, blaZ, tet38 | ||
| 15 | Penicillin | grlA | blaZ, tet38 | blaZ, tet38 | |
| 16 |
Penicillin Fusidic acid |
blaZ, tet38 | fosB, blaZ, tet38 | ||
| 17 | tet38 | ||||
| 18 |
Penicillin Fusidic acid |
grlA | blaZ, tet38 | ||
| 19 |
Penicillin Fusidic acid |
blaZ, tet38 | fosB, blaZ, tet38 | ||
| PC | tet38 | fosB, tet38 | |||
The mean coverage depth when predicting ARGs from the reads was 209x (Supplementary Table S6). We applied a threshold of a minimum 20 × coverage depth, 90% sequence identity and 90% sequence coverage for determining the presence of ARGs. In total, we were able to identify three different resistance genes in S. aureus (tet38, blaZ and fosB) in the 20 samples (including the spiked sample) by SMg. A higher number of ARGs were detected in the samples using the contigs approach compared with the reads approach (43 and 20 predictions in total, respectively).
The gene tet38 encoding the chromosomally encoded efflux pump of S. aureus was detected across all samples. The other genes detected, corresponded to the blaZ gene (35%), and the fosfomycin resistance gene fosB (50%). In the polymicrobial samples, the gene tetM was also detected, but it was not identified in the S. aureus bins.
Resistant phenotypes were observed only for penicillin in 12/19 samples (63.5%), and for fusidic acid in 2/19 (10.5%) samples (Table 2). The penicillin resistant phenotype could be explained by the presence of the blaZ gene in most of the samples. However, in samples 6, 7 and 9, the penicillin resistance phenotype could not be explained by the genotypic profile generated from the reads, contigs or bin. The fusidic acid resistance phenotype could not be explained by the results obtained genotypically. Chromosomal point mutations in the genes reported to induce resistance to fusidic acid (genes fusA and fusE) were not identified. Contrariwise, mutations in the grlA gene were observed in six samples (31.6%). Although, no ciprofloxacin resistance phenotype was observed in any of them (Table 2).
Virulence factors
Overall, a total of 73 genes coding for virulence factors (VFs) were found by SMg in S. aureus (Supplementary Table S8) and a mean of 55 virulence genes (range, 50–62) were detected per sample using the VFDB and thresholds of 90% identity and 90% sequence coverage. Toxins, adhesins and immune evasion molecules were among the genes predicted. Genes encoding 40 virulence factors were present in all the samples, e.g., the cap8 capsule genes (cap8A-G and cap8L-P), iron sequestration operon isdA-isdG, and exotoxins hla, hld, hlgA-C, among others. Additionally, in five samples the toxic shock syndrome toxin 1 (tsst-1) gene was detected.
Virulence genes recognized as belonging to S. agalactiae were removed from the analysis of polymicrobial samples. Several virulence genes known or proposed to play a role for the pathogenicity of S. aureus in PJI were identified from the metagenomes (Fig. 2 and Table 3). Prokka annotation tool was used to confirm the virulence genes presence or absence in the metagenomes. Figure 2 and Table 3 represent the Prokka results on the selected virulence genes found associated with PJI together with the results obtained when using the VFDB applying the thresholds (90% identity and 90% sequence coverage).
Figure 2.

Virulence genes predicted by SMg from S. aureus in PT samples in this study.
Table 3.
Prevalence of virulence genes known or proposed to play a role in S. aureus pathogenicity in PJI predicted from SMg in this study using 90% identity and 90% sequence coverage.
| Virulence gene | Product | Approach | |
|---|---|---|---|
| Prokka annotation | VFDB annotation | ||
| aur | Zinc metalloproteinase aureolysin | 100 | 50 |
| clfA | Clumping factor A fibrinogen-binding protein | 35 | 25 |
| cna | Collagen adhesin precursor | 70 | 10 |
| fnbA | Fibronectin-binding protein A | 100 | 25 |
| hld | Delta-hemolysin | 100 | 100 |
| hlgA | Gamma-hemolysin chain II precursor | 100 | 100 |
| hlgB | Beta-hemolysin | 100 | 100 |
| hlgC | Gamma-hemolysin component C | 100 | 100 |
| sdrD | Ser-Asp rich fibrinogen-binding bone sialoprotein-binding protein | 65 | 25 |
| sdrE | Ser-Asp rich fibrinogen-binding bone sialoprotein-binding protein | 75 | 25 |
| spa | Immunoglobulin G binding protein A precursor | 100 | 60 |
| sspA | Serine protease; V8 protease; glutamyl endopeptidase | 100 | 100 |
| sspB | Staphopain cysteine proteinase SspB | 100 | 100 |
| sspC | Staphostatin B | 100 | 100 |
| hly/hla | Alpha-hemolysin precursor | 100 | 100 |
MLST and cgMLST analysis
The S. aureus Multilocus sequence types (ST) were identified for all the samples at both core genome and whole genome (core genome + accessory genome genes) level (Fig. 3 and Supplementary Table S9). Typing from SMg data showed that S. aureus in our samples is of different lineages. S. aureus in the samples represent six different clonal complexes (CCs), and they belonged to different STs (ST45 (30%), ST121 (15%), ST30 (15%), ST22 (10%), ST5 (10%), ST15 (5%), ST243 (5%), ST7 (5%) and ST97 (5%)). With respect to polymicrobial samples 7 and 9, typing analyses from both the contigs and the bins classified S. aureus as belonging to the same sequence type (ST5). CCs could not be identified for six samples (23%). Samples belonging to the same patient (sample 1 and 18; sample 2 and 19) did not cluster together and did not belong to the same ST (Fig. 3 and Supplementary Table S9). The S. aureus isolates analyzed here represent eight phylogenetically diverse STs. cgMLST subdivided the samples into 13 different complex types (CT, 21,308–21,321) (Supplementary Table S9).
Figure 3.

Minimun-spanning tree based on cgMLST (a) and wgMLST (b) allelic profiles of S. aureus genomes obtained from SMg. Color nodes according to sequence type. The number in the connecting lines illustrates the number of targeted genes with differing alleles.
Discussion
Here, we assessed the performance of SMg for the prediction of ARGs, virulence gene determinants and typing of S. aureus from clinical PT samples on BCBs. We investigated if there was a difference in outcome from analyzing sequencing data from reads and contigs, and our data analysis followed different analytical approaches in order to identify the procedures that may give the most relevant and accurate information from our SMg data.
It is established that it is possible to analyze sequence data without assembly, but most analyses can be improved by constructing longer contiguous sequences (contigs) through assembly processes28. According to our results, the selection of approach to some degree depends on what type of information you require from the data. For instance, if the aim is pathogen identification, taxonomy from the reads is sufficient while if AMR is the focus, the contigs approach will provide a more comprehensive resistome resolution.
For identification of potential pathogen(s) causing PJI, we found that the relative abundance of the taxa was influenced by the approach used to analyze the data. This was evidenced by the differences in abundance obtained, especially in the polymicrobial samples. We observed that the proportion of contigs classified into a certain taxonomical level is also influenced by the genome size. Determining the taxa present and the relative level or number of cells of one taxon vs another (e.g., polymicrobial samples) in a clinical sample is important for identifying the pathogen(s) causing the infection. Our results suggest that analyzing reads provides a more trustworthy representation of the species in a clinical sample than analyzing contigs. Moreover, it is considered that the reads will describe more accurately the proper distribution of species in the sample and provide a more accurate and specific assignment of taxa than using the contigs29. This statement applies especially to quantitative community profiling and identification of organisms with close relatives in the database as in the case of clinical microbiology, where the focus is mainly the presence or absence of infectious pathogens, which are well studied and have many completed genomes in the reference databases30. In addition, it could also be errors in joining contigs from two closely related species. Specificity is lost when working with contigs as the quality of the assembly will depend strongly on the length and quality of the reads, sometimes misrepresenting the original sample29.
Interpretation of SMg results can be challenging, especially when distinguishing microorganisms actually present in the sample from contaminants31. In this study, S. agalactiae was detected in four samples by SMg, but it was only detected in one of these samples by the laboratory method (BCBs). S. agalactiae has been reported as one of the most common pathogens found together with S. aureus in polymicrobial PJIs32. This could suggest an increased sensitivity of SMg to detect bacteria, or alternatively a contamination problem in the laboratory workflow. These findings highlight the importance of the use of parallel negative controls (no-template control) and DNA spiked positive controls to exclude or identify sources of contamination and minimize false-positive results33. Bioinformatic tools have been developed to streamline identification of contaminants and/or cross-contamination, like e.g. Recentrifuge34 and Decontam35.
S. aureus has been studied extensively with a special focus on resistance and virulence. In this study, only resistance to penicillin (63.5%) and fusidic acid (10.5%) were observed by AST. No MRSA isolates were detected, neither by phenotypical nor genotypical testing. This result is not surprising, since Norway has a very low prevalence of MRSA36.
Although SMg is a promising approach, the in silico translation from genotype into phenotype relies on the knowledge about the genomic resistance determinants37. Our SMg analysis allowed the prediction of the genotypical resistance profile from S. aureus present in the samples. The ARG tet38 was detected in all samples. However, tetracycline resistant phenotypes were not observed, and the presence of tet38 is not enough to produce a resistant phenotype. The tet38 determinant is an inherent, chromosomally encoded efflux pump in S. aureus and resistance to tetracyclines is often associated with plasmid-mediated genes encoding active efflux pumps or proteins that protect ribosomes from drug action38.
In three of the metagenomic samples with phenotypic penicillin resistance, the blaZ gene was absent (25%). Similarly, fusidic acid resistance phenotype was observed in two samples (sample 12 and 16). The presence of the fusB gene or mutations in fusA (encoding EF-G) or fusE (encoding ribosome protein L6) genes known to cause fusidic acid resistance were not detected in the metagenomes. Therefore, the phenotype could not be explained by the results obtained genotypically. Overall, interpretation of cases where phenotyping reports resistance but WGS methods predict susceptibility is considered difficult39.
Sometimes, detection of ARGs may be challenging due to the variable location of the resistance genes, since some of them can be on a plasmid or integrated into the chromosome, as is the case of the blaZ gene and the fusB gene which has been found on S. aureus plasmid or on phage-related resistance islands integrated into the chromosome40. Bacterial isolates with plasmid-encoded copies may have very high (if multiple copies are carried) or very low (because of poor mapping to the reference) coverage in that region41,42. As a result, these regions may be rejected as low coverage when predicted from the reads, or as poor quality by the assembly tools when predicted from the contigs and bins since they fall outside the coverage level of the rest of the genome. This problem may be overcome in the future with long-read sequencing methods such as nanopore sequencing or alternative methods for de novo assembly.
Another possibility is the presence of small colony variants (SCV) being present phenotypically but overgrown in WGS culture and thus not represented in the sequence39. This phenomenon has been observed for resistance associated with fusidic acid43.
We observed that three of the five samples with phenotype-genotype disagreement for penicillin resistance were found to be polymicrobial by SMg (samples 6, 7 and 9). In samples 7 and 9, S. aureus was not the most abundant species present in the sample (< 12%) which may affect the prediction of ARGs by a lower sequence coverage.
Prediction of ARGs was done at the reads, contigs and bin level. The total number of different genes detected (3 ARGs) was influenced by the parameters selected to report a gene as present, as these parameters constitute a trade-off between specificity and depth44. We used strict parameters, and only ARGs that had ≥ 90% similarity and ≥ 90% sequence coverage to that of the reference were reported from the reads, contigs and bins. In addition, at the read level, only ARGs with at least 20 × coverage depth were considered as present. The selection of ARGs using stringent cutoffs (≥ 90% per read or contig) can increase the probability of targeting genes that are actually functional44. However, thresholds should be adapted for certain genes (e.g., blaZ, which can be chromosomally integrated or carried on plasmids)39.We consider that the high coverage depth (> 200 ×) is an advantage in our SMg approach.
Most of the tools developed for identifying ARGs from metagenomic reads can detect acquired ARGs, but are not able to identify point mutations associated with AMR45. The focus here was on acquired resistance since we have used the tools Groot or ABRicate with the NCBI bacterial antimicrobial resistance reference gene database. However, ResFinder was used to detect chromosomal point mutations associated with antimicrobial resistance from the contigs. In our study, more genes were detected at the contig level than at the read level. The detection of more ARGs from the contig-based approach may be explained by the fact that it is easier to reach a high sequence coverage of the gene (90%) from contigs (which are longer) than by reads that are shorter in length. We consider that prediction of ARGs at the contig level is the best approach when looking for ARGs. However, it is important to highlight that both approaches are valid for certain purposes, and both have their limitations. Read-based prediction of ARGs provides an advantage when dealing with metagenomic samples, as ARGs in less abundant organisms can be predicted despite low coverage, which may be missed by assembly-based methods owing to incomplete or poor assemblies46. Detecting ARGs from reads is more prone to false positive results because of sequencing errors present in single reads or from DNA contamination from other bacteria. A previous study comparing ARG detection from reads and contigs suggested using both approaches when the sequence coverage is set to a high percentage, since it is possible that ARGs may be separated into different contigs when the number of reads is too low during the assembly process23. The use of long-read sequencing can overcome this problem as shown in other studies17,47–49. However, there are some limitations for achieving enough bacterial reads to ensure high accuracy when predicting ARGs.
Strategies for predicting AMR phenotypes in polymicrobial samples present an interesting challenge50. We tested the binning approach for the prediction of ARGs in S. aureus and we found that it gives similar results as prediction of ARGs in monomicrobial samples for most of the genes with exception of the blaZ gene. This approach allowed us to separate the contigs belonging to S. agalactiae from the contigs belonging to S. aureus and predict the S. aureus resistance profile, even though there were not many contigs (mean, 44, 3 contigs; range 39–51). This means that AMR genotype predictions could be made from contigs that are binned in a metagenomic assembly, even when they belong to a species that is not in a high abundance. In this study, the binning strategy was no further evaluated. Conclusions from bins should be made with precaution since the binning process can lead to incorrect assumptions due to misbinning (the wrong assignment of a genome fragment from one organism to another), namely if the abundance of the species is very similar, which may lead to neglection of specific determinants51.
Apart from antibiotic resistance, virulence of a pathogen is also an important factor clinicians may be concerned about when considering appropriate treatment. Horizontally acquired virulence genes can directly contribute to an infection outbreak, and thus, early identification of virulence factors is important49,52. In our study, we identified some VFs that are known or proposed to play a role in S. aureus PJI, e.g. genes involved in colonization and attachment of host tissue or implanted biomaterials such as the adhesins clfA and fnbA that encode the fibrinogen and fibronectin-binding proteins, respectively53. We have observed that the database used, and the thresholds established to consider the presence of a VF play an important role in the results. The need for a continuously updated curated database is a key challenge for SMg sequencing methods. Thresholds should be adapted for certain genes to improve the prediction and for quality control39. Specific VFs may also require the use of different approaches to confirm the results, e.g. to detect genes that are highly polymorphic as the spa gene.
MLST was used for strain level typing including a contig-based approach, which means that we had sufficient depth for assembly from the metagenome. wgMLST demonstrated that S. aureus in the samples consisted of several lineages. Our MLST results were in accordance with results from the population-based Tromsø Staph and Skin Study, showing that CC30, CC45 and CC15 were the most common CCs in MSSA54. Additionally, the most common S. aureus lineages in PJI reported in a recent study, were CC30, CC45, CC5, CC15 and CC22. S. aureus in our study belonged to the same CCs32,54.
Our study has several limitations. First, the sample size was small (n = 19), and we only analyzed a limited number of polymicrobial samples. Second, S. aureus isolates were not whole genome sequenced for comparison and confirmation of ARG and VF profiles. Third, no clinical data about the patients were obtained making it difficult to classify the samples as true PJI. Fourth, we have used short-read sequencing which makes detection of ARGs on mobile genetic elements difficult. Long-read sequencing may overcome this limitation. Fifth, we only predicted acquired resistance, which made it difficult explaining the disagreement found between the phenotype and the genotype for penicillin resistance. Errors in sensitivity and specificity of ARG prediction can have different consequences for PJI treatment. False negatives (phenotypically resistant and SMg-susceptible) can lead to inadequate treatment of a resistant infection, increasing morbidity and mortality, whereas false positives (phenotypically susceptible and SMg-resistant) may lead to inappropriate antibiotic use and increase the risk of resistance development55. Sixth, we did not include blank negative controls to help identifying genuine sources of contamination coming from the environment or from the reagents. We did not calculate the turnaround time since it was not the focus of the study. However, other studies have estimated the time for SMg using the Illumina technology estimating the total time to 29 h (24–94) from sample extraction to identification and AMR profile56. Our protocol would probably also fall into similar turnaround time from BCB positivity to pathogen ID.
Our approach is primarily useful for those using SMg on specimens related to PJI cultured on BCBs, such as synovial fluid, sonication fluid and periprosthetic tissue. It can also be useful for further validation and standardization for the general use of BCBs inoculated with clinical sample. Today, the high costs of our SMg method cannot justify for application on a routine basis56. However, in this study, we obtained a high enough sequencing depth, making it possible to multiplex samples and thereby reducing costs considerably.
We do not believe that SMg can replace conventional culturing, but it can be a potential diagnostic tool to support conventional culture in cases when PJI diagnosis is challenging, e.g. fastidious/slow-growing microorganisms, polymicrobial infections, discrepancies between conventional methods, when culture-negative but with clinical sign of infection57, and in complicated infections with antibiotic resistant bacteria and long-term antibiotic treatment58. In practice, there are no real problems in identifying microorganisms from BCBs. The added value of SMg over classical blood culturing, is mainly that SMg, in addition to pathogen identification, allows extra information in one single procedure, e.g. detecting coinfections, predicting AMR, detecting virulence factors, and bacterial typing. It is still a long way until SMg can be used in the clinical microbiology laboratory. Our SMg approach, including enrichment of microbes using BCBs and human DNA depletion method, has been successful, enabling AMR-prediction, virulence gene detection and bacterial typing.
In conclusion, this study showed that SMg from BCBs inoculated with PT, allowed the identification of potential PJI pathogens and strain-level typing of S. aureus. We obtained S. aureus ARGs and virulence gene profiles from both monomicrobial and polymicrobial samples. However, the use of this approach for the detection of AMR to help guide clinical treatment needs to be further elucidated, due to the disagreement between the AMR phenotype and genotype. We conclude that the approach chosen for analyzing SMg data (reads, contigs or metagenomic assembled genomes) will have a key impact on the results. Precise AMR prediction is required for mainstream adoption of SMg into the clinical microbiology laboratory. Thus, several improvements are needed for AMR prediction using SMg, including a better understanding of the mechanisms underlying AMR and the procedures (including workflows, tools and databases) that may give the most relevant and accurate information when analyzing SMg data.
Methods
Ethics statement
This study was performed in accordance with the ethical guidelines established by UiT—The Arctic University of Norway. The project has been evaluated by the Regional Committee for Medical and Health Research Ethics, REC North, Norway (document no. 2016/1247/REK nord), concluding that ethical approval was not required. According to the Norwegian guidelines, informed consent of the patients is not needed and there were not ethical issues to consider due to the use of anonymous clinical samples and the development of methodological procedures.
Sample collection
Periprosthetic tissue samples (from hip, knee, elbow, ankle, and shoulder) routinely submitted to the Department of Microbiology and Infection Control at the University Hospital of North Norway (UNN), Tromsø, Norway, were included in this study. Nineteen positive BCBs inoculated with PT from 17 individual patients with suspicion of PJI were used in this study. Clinical samples were selected on the basis of being positive for S. aureus by the BCB method18, either monomicrobial (n = 18) or polymicrobial (n = 1). Samples were collected continuously over a 28-month period (August 2017–December 2019). Samples were anonymized and de-identified. All samples were taken from aerobic bottles (Bact/Alert® FA plus bottles, bioMérieux, Marcy l’Etoile, France).
Five of the 19 clinical samples in this study, in addition to one positive control (spiked sample, BCB inoculated with tissue spiked with S. aureus ATCC 2592) and one negative control (DNA not subjected to human DNA depletion, extracted from a BCB enriched with horse blood and inoculated with sterilized tissue from a donor with no suspicion of infection) were obtained from a sample collection in a previous study18,27. No blank negative control was included. For further details on the BCB sample preparation method and on the controls, see Sanabria et al.27. Bacterial identification was performed using matrix-assisted laser desorption ionization-time of flight mass spectrometry (MALDI-TOF® MS Bruker Daltonics—microflex™). An overview of all the samples included in this study, is shown in Table 1.
Antibiotic susceptibility testing
Antibiotic susceptibility testing (AST) of S. aureus isolates was performed by disc diffusion test according to EUCAST guidelines and the breakpoint table v.10.0 (2020)59. The antibiotics tested were penicillin (10 µg), trimethoprim-sulfamethoxazole (25 µg), cefoxitin (30 µg), fusidic acid (10 µg), clindamycin (2 µg), erythromycin (15 µg), linezolid (10 µg), tetracycline (30 µg), norfloxacin (10 µg), ciprofloxacin (5 µg), gentamicin (10 µg) and rifampicin (5 µg) (Oxoid, Basingstoke, UK).
DNA preparation
DNA was extracted and processed as previously described18. In short, all samples were pre-treated using MolYsis™ Basic5 kit (Molzym, Bremen, Germany) to deplete human and horse DNA before DNA extraction using the QIAamp BiOstic Bacteremia DNA Kit (Qiagen, Hilden, Germany). Total DNA concentration was measured using a Qubit dsDNA HS Assay Kit (Life Technologies, Carlsbad, CA, USA) and DNA quality was determined by Nanodrop.
Metagenomic sequencing
Sequencing libraries were prepared as previously described18, using the ThruPLEX® DNA-seq Kit (Rubicon Genomics, USA) following the manufacturer’s instructions. Approximately 100 ng of DNA was used as input for library preparation from the clinical and spiked samples. The sequencing process was performed at the Norwegian Sequencing Centre, Oslo, using a MiSeq sequencer (Illumina Inc., San Diego, CA, USA) with v2 chemistry and 500 cycles for 250 bp paired-end sequencing. Samples were multiplexed with four samples per lane.
Bioinformatic data analysis
Our SMg bioinformatics pipeline was created and optimized, based on publicly available tools and pipelines and tools from other SMg studies9,11–13,15,23,60,61. The bioinformatic analysis followed in this study is summarized in Fig. 4. The quality of the raw reads in fastq format was assessed using FastQC software v0.11.8 (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Optical duplicates were removed using the program Clumpify v38.36 from BBTools suite (https://jgi.doe.gov/data-and-tools/bbtools/) with default parameters. Adapter sequences were trimmed off and the poor-quality reads were removed using BBDuk of BBTools suite. The minimal read length and Phred score were set to 50 nucleotides and 20, respectively. In order to filter out known sources of contaminant host DNA, the reads mapped against the reference genomes of human GRCh38.p13 (GCF_000001405.39), horse (GCF_002863925.1) and the PhiX phage (Escherichia virus phiX174, GCF_000819615.1) aligning with Bowtie2 in the tool FastQ Screen v0.13.062. Unmapped reads were used in subsequent analyses.
Figure 4.

Workflow summarizing the bioinformatic analyses in this study, including (a) data preprocessing, (b) data analyses approaches and (c) data analyses and interpretation. ARG antimicrobial resistance gene, VF virulence factor, AMR antimicrobial resistance.
Data analyses in this study followed different approaches: using the preprocessed reads directly and using assembled contigs or bins for monomicrobial and polymicrobial samples, respectively. Reads were assembled into contigs using metaSPAdes63 from SPAdes v.3.14.064 and the quality of the assemblies was assessed using QUAST v5.0.265. Resultant contigs were annotated using Prokka v.1.13 (http://github.com/tseemann/prokka). Contigs can be grouped by species into discrete units, referred to as bins, which were predicted using the tool MaxBin v.2.2.766, for recovering the S. aureus genome from the metagenomic datasets in polymicrobial samples.
Species identification on reads, contigs and bins was performed using Kraken v1.1.167 (as illustrated in Fig. 4) and the 8 GB DustMasked MiniKraken database (as of Oct. 18, 2017) with default parameters. Species relative abundance was determined using Bracken68. The detection of antimicrobial resistance genes (ARGs) from the reads was determined using the tool Groot v.1.0.269 (https://github.com/will-rowe/groot). ARGs and virulence genes from the assembled contigs and bins were detected using ABRicate v0.8 (https://github.com/tseemann/abricate). For the detection of ARGs with Groot and ABRicate the NCBI bacterial antimicrobial resistance reference gene database (BioProject accession number PRJNA313047, as of April 24th, 2020) was used. Moreover, chromosomal point mutations associated with antimicrobial resistance were identified with ResFinder v.4.1 (Point Finder database as of Feb. 1, 2021). For detection of virulence genes, the virulence factor database (VFDB) was used70. The thresholds used for determining the presence of ARG genes and VFs were set as 90% identity and 90% sequence coverage. Additionally, for ARGs prediction from the reads, a coverage depth of at least 20 × was considered to report an ARG as present. Presence or absence of several VFs in the metagenomes, known or proposed to play a role in S. aureus pathogenicity in PJI were confirmed using the results from Prokka v.1.13. We used default parameters for all the bioinformatic tools unless it is stated.
Typing
The assembled contigs and the bins were imported into SeqSphere + software v.6.0.2 (Ridom GmbH, Münster, Germany) for a gene-by-gene allele calling comparison using the S. aureus species-specific scheme within SeqSphere + for a cgMLST scheme for comparison of the 1,816 core loci in S. aureus, and an accessory typing scheme (wgMLST) with 706 accessory loci. Loci that flagged as failed (i.e., found but bearing frameshifts, or a differing consensus sequence, or having too-low coverage) were considered absent. Phylogenetic trees were constructed in SeqSphere + using a minimum-spaning tree; missing values were pairwise ignored. The cluster-alert distance was set at a default of 24 allelic differences32.
Statistical analysis
Descriptive statistics for categorical variables were based on percentages and frequencies and was used to determine the prevalence of the ARGs and VFs, while continuous variables were based on means, medians, and interquartile ranges (IQRs) and was used to describe the results obtained from the sequencing data. In addition, t-test was used to evaluate if the differences between the total number of base pairs in polymicrobial and monomicrobial samples were statistically significant. The differences were considered statistically significant with p values < 0.0001. Data were analyzed utilizing GraphPad Prism software, version 8.3.0 (GraphPad Software Inc., CA, US).
Supplementary Information
{kind=link}
Acknowledgements
We thank the Department of Microbiology and Infection Control, University Hospital of North Norway, Tromsø, especially Lisbeth Engen Mortensen for help with the Antibiotic susceptibility testing (AST).
Author contributions
A.S., G.S.S., and A.M.H. initiated and planned the study design. A.S. performed the experiments and the bioinformatic analysis. A.S., G.S.S. and A.M.H. assisted in scientific and technical design of experiments. J.J. and E.H. contributed to the bioinformatic analysis. A.S., G.S.S. and A.M.H. analyzed and interpreted the results. A.S. prepared the first version of the manuscript. All authors reviewed the manuscript, gave inputs, and approved the submitted version.
Funding
This work was supported by grants from joint “Miljøstøtte” financed by Strategisk-HN05–14 (Helse Nord RFH) and Faculty of Health Sciences A20389 (2014–2017), and the National Graduate School in Infection Biology and Antimicrobials (Grant No. 249062). The publication charges for this article have been funded by a grant from the publication fund of UiT—The Arctic University of Norway. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Data availability
The preprocessed reads generated for this study for each sample can be found in the European Nucleotide Archive (ENA) repository (www.ebi.ac.uk/ena) under the project number PRJEB43858.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Adriana Maria Sanabria, Email: sannanita@gmail.com.
Anne-Merethe Hanssen, Email: anne-merethe.hanssen@uit.no.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-021-00383-7.
References
- 1.Li Z-L, et al. Identifying common pathogens in periprosthetic joint infection and testing drug-resistance rate for different antibiotics: A prospective, single center study in Beijing. Orthop. Surg. 2018;10:235–240. doi: 10.1111/os.12394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tande AJ, Patel R. Prosthetic joint infection. Clin. Microbiol. Rev. 2014;27:302–345. doi: 10.1128/CMR.00111-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tsai J-C, Sheng W-H, Lo W-Y, Jiang C-C, Chang S-C. Clinical characteristics, microbiology, and outcomes of prosthetic joint infection in Taiwan. J. Microbiol. Immunol. Infect. 2015;48:198–204. doi: 10.1016/j.jmii.2013.08.007. [DOI] [PubMed] [Google Scholar]
- 4.Peel TN, Buising KL, Choong PFM. Prosthetic joint infection: Challenges of diagnosis and treatment. ANZ J. Surg. 2011;81:32–39. doi: 10.1111/j.1445-2197.2010.05541.x. [DOI] [PubMed] [Google Scholar]
- 5.Benito N, et al. Time trends in the aetiology of prosthetic joint infections: A multicentre cohort study. Clin. Microbiol. Infect. 2016;22(732):e1–732.e8. doi: 10.1016/j.cmi.2016.05.004. [DOI] [PubMed] [Google Scholar]
- 6.Goudarzi M, Fazeli M, Goudarzi H, Azad M, Seyedjavadi SS. Spa typing of Staphylococcus aureus strains isolated from clinical specimens of patients with nosocomial infections in Tehran, Iran. Jundishapur J. Microbiol. 2016;9:685. doi: 10.5812/jjm.35685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Otto M. Staphylococcal biofilms. Curr. Top. Microbiol. Immunol. 2008;322:207–228. doi: 10.1007/978-3-540-75418-3_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vasoo S. Improving the diagnosis of orthopedic implant-associated infections: Optimizing the use of tools already in the box. J. Clin. Microbiol. 2018;56:e01379–e1418. doi: 10.1128/JCM.01379-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ivy MI, et al. Direct detection and identification of prosthetic joint infection pathogens in synovial fluid by metagenomic shotgun sequencing. J. Clin. Microbiol. 2018;56:9. doi: 10.1128/JCM.00402-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang C, et al. Preliminary assessment of nanopore-based metagenomic sequencing for the diagnosis of prosthetic joint infection. Int. J. Infect. Dis. 2020;97:54–59. doi: 10.1016/j.ijid.2020.05.044. [DOI] [PubMed] [Google Scholar]
- 11.Zhang C, et al. Value of mNGS in sonication fluid for the diagnosis of periprosthetic joint infection. Arthroplasty. 2019;1:9. doi: 10.1186/s42836-019-0006-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Street TL, et al. Molecular diagnosis of orthopedic-device-related infection directly from sonication fluid by metagenomic sequencing. J. Clin. Microbiol. 2017;55:2334–2347. doi: 10.1128/JCM.00462-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thoendel MJ, et al. Identification of prosthetic joint infection pathogens using a shotgun metagenomics approach. Clin. Infect. Dis. 2018;67:1333–1338. doi: 10.1093/cid/ciy303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sanderson ND, et al. Real-time analysis of nanopore-based metagenomic sequencing from infected orthopaedic devices. BMC Genomics. 2018;19:714. doi: 10.1186/s12864-018-5094-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ruppé E, et al. Clinical metagenomics of bone and joint infections: a proof of concept study. Sci. Rep. 2017;7:7718. doi: 10.1038/s41598-017-07546-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schmidt K, et al. Identification of bacterial pathogens and antimicrobial resistance directly from clinical urines by nanopore-based metagenomic sequencing. J. Antimicrob. Chemother. 2017;72:104–114. doi: 10.1093/jac/dkw397. [DOI] [PubMed] [Google Scholar]
- 17.Noone JC, Helmersen K, Leegaard TM, Skråmm I, Aamot HV. Rapid diagnostics of orthopaedic-implant-associated infections using nanopore shotgun metagenomic sequencing on tissue biopsies. Microorganisms. 2021;9:97. doi: 10.3390/microorganisms9010097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sanabria A, et al. Shotgun-metagenomics on positive blood culture bottles inoculated with prosthetic joint tissue: A proof of concept study. Front. Microbiol. 2020;11:1687. doi: 10.3389/fmicb.2020.01687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jacob JJ, Veeraraghavan B, Vasudevan K. Metagenomic next-generation sequencing in clinical microbiology. Indian J. Med. Microbiol. 2019;37:133–140. doi: 10.4103/ijmm.IJMM_19_401. [DOI] [PubMed] [Google Scholar]
- 20.Wilson MR, et al. Clinical metagenomic sequencing for diagnosis of meningitis and encephalitis. N. Engl. J. Med. 2019;380:2327–2340. doi: 10.1056/NEJMoa1803396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ellington MJ, et al. The role of whole genome sequencing in antimicrobial susceptibility testing of bacteria: Report from the EUCAST subcommittee. Clin. Microbiol. Infect. 2017;23:2–22. doi: 10.1016/j.cmi.2016.11.012. [DOI] [PubMed] [Google Scholar]
- 22.Watts GS, Hurwitz BL. Metagenomic next-generation sequencing in clinical microbiology. Clin. Microbiol. Newsl. 2020;42:53–59. doi: 10.1016/j.clinmicnews.2020.03.004. [DOI] [Google Scholar]
- 23.Couto N, et al. Critical steps in clinical shotgun metagenomics for the concomitant detection and typing of microbial pathogens. Sci. Rep. 2018;8:1. doi: 10.1038/s41598-018-31873-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hasman H, et al. Rapid whole-genome sequencing for detection and characterization of microorganisms directly from clinical samples. J. Clin. Microbiol. 2014;52:139–146. doi: 10.1128/JCM.02452-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Watts GS, et al. Identification and quantitation of clinically relevant microbes in patient samples: Comparison of three k-mer based classifiers for speed, accuracy, and sensitivity. PLOS Comput. Biol. 2019;15:e1006863. doi: 10.1371/journal.pcbi.1006863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Joseph SJ, et al. The single-species metagenome: Subtyping Staphylococcusaureus core genome sequences from shotgun metagenomic data. PeerJ. 2016;4:e2571–e2571. doi: 10.7717/peerj.2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sanabria A, et al. Culturing periprosthetic tissue in BacT/Alert® Virtuo blood culture system leads to improved and faster detection of prosthetic joint infections. BMC Infect. Dis. 2019;19:607. doi: 10.1186/s12879-019-4206-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sangwan N, Xia F, Gilbert JA. Recovering complete and draft population genomes from metagenome datasets. Microbiome. 2016;4:8. doi: 10.1186/s40168-016-0154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rodríguez-Brazzarola P, Pérez-Wohlfeil E, Díaz-del-Pino S, Holthausen R, Trelles O. Analyzing the differences between reads and contigs when performing a taxonomic assignment comparison. In: Rojas I, Ortuño F, editors. Metagenomics BT: Bioinformatics and Biomedical Engineering. Springer; 2018. pp. 450–460. [Google Scholar]
- 30.Breitwieser FP, Lu J, Salzberg SL. A review of methods and databases for metagenomic classification and assembly. Brief. Bioinform. 2019;20:1125–1136. doi: 10.1093/bib/bbx120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.d’Humières C, et al. The potential role of clinical metagenomics in infectious diseases: Therapeutic perspectives. Drugs. 2021;81:1453–1466. doi: 10.1007/s40265-021-01572-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wildeman P, et al. Genomic characterization and outcome of prosthetic joint infections caused by Staphylococcus aureus. Sci. Rep. 2020;10:5938. doi: 10.1038/s41598-020-62751-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shen J, McFarland AG, Young VB, Hayden MK, Hartmann EM. Toward accurate and robust environmental surveillance using metagenomics. Front. Genet. 2021;12:151. doi: 10.3389/fgene.2021.600111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martí JM. Recentrifuge: Robust comparative analysis and contamination removal for metagenomics. PLoS Comput. Biol. 2019;15:e100751. doi: 10.1371/journal.pcbi.1006967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Davis NM, Proctor DM, Holmes SP, Relman DA, Callahan BJ. Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome. 2018;6:226. doi: 10.1186/s40168-018-0605-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.N.-V. NORM/NORM-VET 2018. Usage of Antimicrobial Agents and Occurrence of Antimicrobial Resistance in Norway. (2018). ISSN: 1502–2307
- 37.Ruppé E, Cherkaoui A, Lazarevic V, Emonet S, Schrenzel J. Establishing genotype-to-phenotype relationships in bacteria causing hospital-acquired pneumonia: A prelude to the application of clinical metagenomics. Antibiotics. 2017;6:30. doi: 10.3390/antibiotics6040030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen C, Hooper DC. Effect of Staphylococcus aureus Tet38 native efflux pump on in vivo response to tetracycline in a murine subcutaneous abscess model. J. Antimicrob. Chemother. 2018 doi: 10.1093/jac/dkx432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mason A, et al. Accuracy of different bioinformatics methods in detecting antibiotic resistance and virulence factors from staphylococcus aureus whole-genome sequences. J. Clin. Microbiol. 2018;56:e01815–e1817. doi: 10.1128/JCM.01815-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.O’Neill AJ, Larsen AR, Skov R, Henriksen AS, Chopra I. Characterization of the epidemic European fusidic acid-resistant impetigo clone of Staphylococcus aureus. J. Clin. Microbiol. 2007;45:1505–1510. doi: 10.1128/JCM.01984-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gordon NC, et al. Prediction of Staphylococcus aureus antimicrobial resistance by whole-genome sequencing. J. Clin. Microbiol. 2014;52:1182–1191. doi: 10.1128/JCM.03117-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Olsen JE, Christensen H, Aarestrup FM. Diversity and evolution of blaZ from Staphylococcus aureus and coagulase-negative staphylococci. J. Antimicrob. Chemother. 2006;57:450–460. doi: 10.1093/jac/dki492. [DOI] [PubMed] [Google Scholar]
- 43.Norström T, Lannergård J, Hughes D. Genetic and phenotypic identification of fusidic acid-resistant mutants with the small-colony-variant phenotype in Staphylococcus aureus. Antimicrob. Agents Chemother. 2007;51:4438–4446. doi: 10.1128/AAC.00328-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lal Gupta C, Kumar Tiwari R, Cytryn E. Platforms for elucidating antibiotic resistance in single genomes and complex metagenomes. Environ. Int. 2020;138:105667. doi: 10.1016/j.envint.2020.105667. [DOI] [PubMed] [Google Scholar]
- 45.Uelze L, et al. Typing methods based on whole genome sequencing data. One Heal. Outlook. 2020;2:3. doi: 10.1186/s42522-020-0010-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Boolchandani M, D’Souza AW, Dantas G. Sequencing-based methods and resources to study antimicrobial resistance. Nat. Rev. Genet. 2019;20:356–370. doi: 10.1038/s41576-019-0108-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Noone JC, Ferreira FA, Aamot HV. Culture-independent genotyping, virulence and antimicrobial resistance gene identification of Staphylococcus aureus from orthopaedic implant-associated infections. Microorganisms. 2021;9:707. doi: 10.3390/microorganisms9040707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhou K, et al. Use of whole-genome sequencing to trace, control and characterize the regional expansion of extended-spectrum β-lactamase producing ST15 Klebsiella pneumoniae. Sci. Rep. 2016;6:20840. doi: 10.1038/srep20840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhou M, et al. Comprehensive pathogen identification, antibiotic resistance, and virulence genes prediction directly from simulated blood samples and positive blood cultures by nanopore metagenomic sequencing. Front. Genet. 2021;12:244. doi: 10.3389/fgene.2021.620009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nguyen M, Olson R, Shukla M, VanOeffelen M, Davis JJ. Predicting antimicrobial resistance using conserved genes. BioRxiv. 2020 doi: 10.1101/2020.04.29.068254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen LX, Anantharaman K, Shaiber A, Murat Eren A, Banfield JF. Accurate and complete genomes from metagenomes. Genome Res. 2020 doi: 10.1101/gr.258640.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brynildsrud OB, et al. Acquisition of virulence genes by a carrier strain gave rise to the ongoing epidemics of meningococcal disease in West Africa. Proc. Natl. Acad. Sci. 2018;115:5510–5515. doi: 10.1073/pnas.1802298115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Greene C, et al. Adhesion properties of mutants of Staphylococcus aureus defective in fibronectin-binding proteins and studies on the expression of fnb genes. Mol. Microbiol. 1995;17:1143–1152. doi: 10.1111/j.1365-2958.1995.mmi_17061143.x. [DOI] [PubMed] [Google Scholar]
- 54.Sangvik M, et al. Age- and gender-associated Staphylococcus aureus spa types found among nasal carriers in a general population: the Tromso Staph and Skin Study. J. Clin. Microbiol. 2011;49:4213–4218. doi: 10.1128/JCM.05290-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Su M, Satola SW, Read TD. Genome-based prediction of bacterial antibiotic resistance. J. Clin. Microbiol. 2018;57:3. doi: 10.1128/JCM.01405-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Govender KN, Street TL, Sanderson ND, Eyre DW. Metagenomic sequencing as a pathogen-agnostic clinical diagnostic tool for infectious diseases: A systematic review and meta-analysis of diagnostic test accuracy studies. J. Clin. Microbiol. 2021 doi: 10.1128/jcm.02916-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Peel TN, et al. Improved diagnosis of prosthetic joint infection by culturing periprosthetic tissue specimens in blood culture bottles. MBio. 2016;7:e01776. doi: 10.1128/mBio.01776-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Charretier Y, Lazarevic V, Schrenzel J, Ruppé E. Messages from the fourth international conference on clinical metagenomics. Microbes Infect. 2020;22:635–641. doi: 10.1016/j.micinf.2020.07.007. [DOI] [PubMed] [Google Scholar]
- 59.EUCAST. Testing breakpoint tables for interpretation of MICs and zone diameters. https://www.eucast.org/ast_of_bacteria/ (2020).
- 60.Kirstahler P, et al. Genomics-based identification of microorganisms in human ocular body fluid. Sci. Rep. 2018;8:4126. doi: 10.1038/s41598-018-22416-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Anson LW, et al. DNA extraction from primary liquid blood cultures for bloodstream infection diagnosis using whole genome sequencing. J. Med. Microbiol. 2018;67:347–357. doi: 10.1099/jmm.0.000664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wingett SW, Andrews S. FastQ Screen: A tool for multi-genome mapping and quality control. F1000 Res. 2018;7:1338. doi: 10.12688/f1000research.15931.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nurk S, Meleshko D, Korobeynikov A, Pevzner PA. metaSPAdes: A new versatile metagenomic assembler. Genome Res. 2017;27:824–834. doi: 10.1101/gr.213959.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bankevich A, et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012;19:455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gurevich A, Saveliev V, Vyahhi N, Tesler G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics. 2013;29:1072–1075. doi: 10.1093/bioinformatics/btt086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wu Y-W, Tang Y-H, Tringe SG, Simmons BA, Singer SW. MaxBin: An automated binning method to recover individual genomes from metagenomes using an expectation-maximization algorithm. Microbiome. 2014;2:26. doi: 10.1186/2049-2618-2-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Venter JC. Environmental genome shotgun sequencing of the Sargasso sea. Science. 2004;304:66–74. doi: 10.1126/science.1093857. [DOI] [PubMed] [Google Scholar]
- 68.Lu J, Breitwieser FP, Thielen P, Salzberg SL. Bracken: Estimating species abundance in metagenomics data. PeerJ Comput. Sci. 2017;3:e104. doi: 10.7717/peerj-cs.104. [DOI] [Google Scholar]
- 69.Rowe WPM, Winn MD. Indexed variation graphs for efficient and accurate resistome profiling. Bioinformatics. 2018;34:3601–3608. doi: 10.1093/bioinformatics/bty387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chen L. VFDB: A reference database for bacterial virulence factors. Nucleic Acids Res. 2004;33:D325–D328. doi: 10.1093/nar/gki008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The preprocessed reads generated for this study for each sample can be found in the European Nucleotide Archive (ENA) repository (www.ebi.ac.uk/ena) under the project number PRJEB43858.