

Abstract
N-Acetylgalactosamine (GalNAc) conjugated short interfering RNAs (siRNAs) are a leading RNA interference (RNAi) platform allowing targeted inhibition of disease-causing genes in hepatocytes. More than a decade of development has recently resulted in the first approvals for this class of drugs. While substantial effort has been made to improve nucleic acid modification patterns for better payload stability and efficacy, relatively little attention has been given to the GalNAc targeting ligand. In addition, the lack of an intrinsic endosomal release mechanism has limited potency. Here, we report a stepwise analysis of the structure activity relationships (SAR) of the components comprising these targeting ligands. We show that there is relatively little difference in biological performance between bi-, tri-, and tetravalent ligand structures while identifying other features that affect their biological activity more significantly. Further, we demonstrate that subcutaneous co-administration of a GalNAc-functionalized, pH responsive endosomal release agent markedly improved the activity and duration of effect for siRNA conjugates, without compromising tolerability, in non-human primates. These findings could address a significant bottleneck for future siRNA ligand conjugate development.
Keywords: ASGPr, GalNAc, siRNA, endosomal release
Graphical abstract
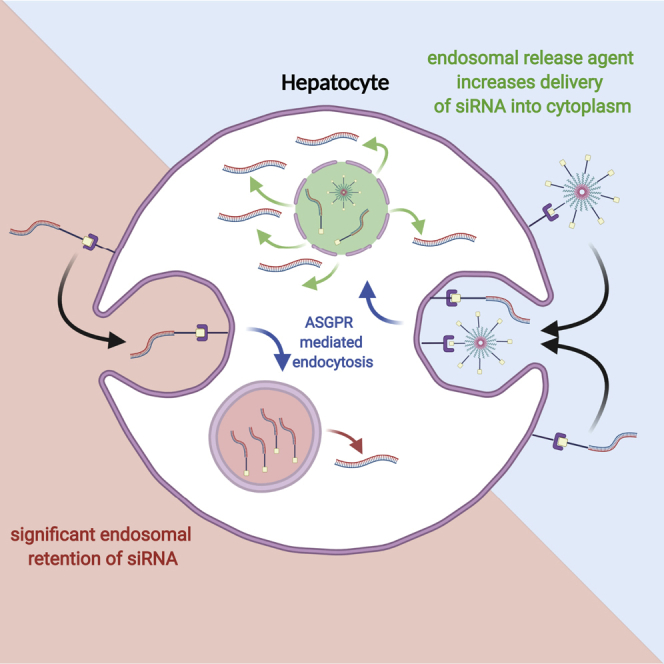
GalNAc-siRNA conjugates are a leading RNA interference platform; however, they lack an intrinsic endosomal release mechanism limiting potency. Subcutaneous co-administration of a GalNAc-functionalized, endosomal release agent markedly improves activity and duration of effect without compromising tolerability. Results translate well from rodents to non-human primates.
Introduction
Short interfering RNAs (siRNAs) enable target specific inhibition of disease-causing genes by harnessing naturally occurring RNA interference (RNAi) machinery inside cells.1 Sequence-dependent target recognition and degradation allows precise treatment for a variety of diseases with a clear and predictable mechanism of action. Recent approvals of siRNA-based drugs such as Onpattro and Givlaari further demonstrate the clinical potential of this promising class of therapeutics.2 A key challenge for RNAi modalities is delivery of siRNAs to target tissues. Multiple barriers, including nuclease degradation, immune recognition, transmembrane trafficking, and endosome escape, impede the effective function of exogenous siRNAs.1 The successful application of conjugated GalNAc as a targeting ligand has recently enabled development of effective siRNA delivery platforms for hepatocytes. Guiding uptake via the asialoglycoprotein receptor (ASGPR), GalNAc conjugates have exhibited superior duration of effect and a more favorable safety profile than other delivery technologies in certain contexts.3,4 ASGPR is highly expressed on the membrane surface of hepatocytes.5 Efficient endocytosis and short receptor recycling times (circa 15 min) make it an ideal entry point to target when accessing this liver cell type.6 Additionally, subcutaneous administration of GalNAc-siRNA is more practical than the intravenous route required by other platforms. Although several RNAi biotechnology companies have established GalNAc-siRNA conjugate platforms, a detailed investigation on the ligand SAR with in vivo testing has yet to be reported. In this study, we synthesized a variety of ligand structures to examine structural aspects of GalNAc ligand design: ligand valency, geometry, linker chemistry, and derivatization of the GalNAc sugar.
Additionally, while an effective strategy for targeting oligonucleotides to hepatocytes, GalNAc conjugates possess no intrinsic mechanism of endosomal escape, a significant rate-limiting step for siRNA function (it is estimated that less than 1% of delivered siRNAs escape across the endosomal lipid bilayer membrane).7 Therefore, increasing endosomal escape of GalNAc-siRNA should hypothetically improve potency and duration of effect. In this study, we identified a novel application for a polymer previously considered for intravenous administration with lipid nanoparticles (LNPs).8 We demonstrate that co-administration of this agent facilitated faster onset of gene silencing, greater potency, and longer duration of effect than with GalNAc-siRNA treatment alone. Importantly, there was no evidence of change in treatment tolerability, such as abnormal appearance, injection site reaction, weight loss, or systemic immune stimulation when the endosomal escape agent, a polymer micelle, was co-administered with the conjugate. The ability to maintain a subcutaneous route of administration is a key advantage of this novel GalNAc conjugate platform.
Results
Our studies comprised a systematic assessment of SAR of GalNAc-siRNA conjugates. We set out to evaluate structural features (Figure 1A) and their impact on biological performance. GalNAc ligands disclosed to date have most or all of these features: a tri-coordinate “linker” to connect siRNA, binding domain, and solid support during synthesis; a “tether” to provide spatial separation between the linker and binding domain; a “splitter,” or connection point for multiple individual binding moieties; “spacers,” which provide spatial separation of the individual binding moieties; and the binding moieties (or ligands) themselves.
Figure 1.

Schematic of GalNAc ligand, geometry and interaction with ASGPr
(A) GalNAc conjugate structural nomenclature. (B) Schematic representation of the individual ligand elements aligning with trimeric carbohydrate recognition domains of ASGPR.
We began with the linker. In addition to the basic requirement of having functional groups to connect siRNA, ligand, and (depending on synthetic strategy) the solid support during synthesis, we were interested to see whether the linker’s identity exerted any effect on potency. To this end, we set the targeting ligand as a trivalent GalNAc molecule (compound 1, Figure S1) and attached it to an siRNA targeting transthyretin (TTR) using assorted linkers (Table 1).
Table 1.
| Linker | Structure | % KDc day 7 | % KDc day 14 |
|---|---|---|---|
| 1 | 60 ± 12 | 27 ± 12 | |
| 2 | 59 ± 18 | 35 ± 9 | |
| 3 | 91 ± 2 | 74 ± 6 | |
| 4 | 91 ± 2 | 77 ± 6 | |
| 5 | 94 ± 1 | 85 ± 2 | |
| 6 | 72 ± 11 | 44 ± 17 | |
| 7 | 71 ± 6 | 44 ± 10 | |
| 8 | 82 ± 11 | 56 ± 25 |
All data shown as mean ± SEM.
Transthyretin
Small interfering ribonucleic acid
Percent reduction (knockdown) of TTR protein in plasma at indicated time point after 2 mg/mL subcutaneous injection in C57BL/6 female mice aged 6–8 weeks (n = 4 per group)
Using the previously reported 2-hydroxy proline (linker 3) as a benchmark,9 we designed and synthesized 7 other linker structures that would be compatible with conjugation and RNA synthesis (typically a hydroxyl for oligo attachment, a second hydroxyl for the solid support, and either a primary or secondary amine for the ligand). Resulting siRNA conjugates were subcutaneously administered to mice as a single 2 mg/kg dose, and “knockdown” (KD; i.e., gene silencing mediated reduction) of target TTR protein levels in plasma assessed 7 and 14 days later (Table 1). Best results were obtained with linker 5, which provided strongest and most durable reduction of TTR protein at both day 7 and 14.
Continuing studies with linker 5, we next turned to ligand valency, which has been widely reported to affect binding affinity with a hierarchy of tetra- > tri- > bi- > monoantennary galactosides.10, 11, 12 We synthesized GalNAc ligands with similar structures but different galactoside valencies and compared their binding affinity in competition with a biotinylated trivalent ligand (compound 2, Figure S2A). We confirmed the reported hierarchy, although the difference between tri- and tetravalent ligands was minimal and comparable to asialofetuin (ASF, a natural ligand for ASGPR).
To investigate whether the ligand binding affinity translated to siRNA conjugate activity, we coupled GalNAc ligands with different valencies to the same TTR siRNA using linker 5 and measured their activity in vivo (structures and results shown in Table 2). Interestingly, while minimal activity was observed for the monovalent ligand (compound 2), a significant level of target protein knockdown was observed with the bivalent ligand (compound 3), comparable to the trivalent and tetravalent ligands (compounds 4 and 5). These 3 compounds also exhibited similar duration of effect. Tetravalent compound 5 exhibited a slightly better activity profile overall. We further compared the phamacokinetic (PK) profiles of bivalent and tetravalent ligand conjugate siRNA in vivo (Figure S3). A faster blood clearance was observed in the tetravalent ligand conjugate dose animals than the bivalent ones but both conjugates were completely cleared within 3 h post injection. This early-stage PK profile difference did not lead to any activity difference even at the 24 h acute time point. We also noted that loading capacity on controlled pore glass (CPG) in preparation for nucleic acid synthesis decreased with increasing ligand size. Load concentrations for bi-, tri-, and tetravalent ligands were ∼0.4 mmol/g, 0.3 mmol/g, and 0.2 mmol/g, respectively. We reasoned that the decrease in ligand size allowed greater access to reactive sites on the CPG and therefore provided a higher support loading. This had a proportionate impact on siRNA-conjugate product yields.
Table 2.
| Compound | Structure | % KDc day 7 | % KDc day 14 |
|---|---|---|---|
| 3 | 17 ± 10 | 29 ± 13 | |
| 4 | 85 ± 5 | 74 ± 9 | |
| 5 | 90 ± 5 | 75 ± 11 | |
| 6 | 93 ± 1 | 89 ± 3 |
All data shown as mean ± SEM.
Transthyretin
Small interfering ribonucleic acid
Percent reduction (knockdown) of TTR protein in plasma at indicated time point after 2 mg/mL subcutaneous injection in C57BL/6 female mice aged 6–8 weeks (n = 4 per group)
Knowing that we could achieve satisfactory levels of target knockdown with a bivalent ligand and that these could be accessed in fewer synthetic steps than higher valency ligands, we used this scaffold to evaluate related analogs of GalNAc. These were selected based on literature reports of higher binding affinity than GalNAc in vitro.13,14 When incorporated into conjugates based on bivalent architecture (Figure S4), no discernible difference in target knockdown in vivo was observed as compared to GalNAc control (compound 6, Table 3).
Table 3.
| Compound | Structure | % KDc day 7 | % KDc day 15 |
|---|---|---|---|
| 7 | 89 ± 1 | 78 ± 4 | |
| 8 | 87 ± 4 | 65 ± 15 | |
| 9 | 87 ± 2 | 69 ± 6 | |
| 10 | 88 ± 4 | 80 ± 6 |
All data shown as mean ± SEM.
Transthyretin
Small interfering ribonucleic acid
Percent reduction (knockdown) of TTR protein in plasma at indicated time point after 2 mg/mL subcutaneous injection in C57BL/6 female mice aged 6–8 weeks (n = 4 per group)
As the tetravalent compound 5 had provided the most sustained target knockdown (Table 2) to this point, we next sought to vary the geometry of the GalNAc moieties in this ligand cluster, including length and bond angles of the spacers for the individual GalNAc saccharides. In contrast to the reported SAR with trivalent ligands,15 these changes did not have a pronounced effect, with all iterations furnishing high activity in vivo (Table 4).
Table 4.
| Compound | Structure | % KDc day 7 | % KDc day 14 |
|---|---|---|---|
| 11 | 90 ± 3 | 79 ± 4 | |
| 6 | 93 ± 1 | 89 ± 3 | |
| 12 | 91 ± 1 | 83 ± 3 | |
| 13 | 86 ± 3 | 76 ± 4 | |
| 14 | 88 ± 3 | 77 ± 4 |
All data shown as mean ± SEM.
Transthyretin
Small interfering ribonucleic acid
Percent reduction (knockdown) of TTR protein in plasma at indicated time point after 2 mg/mL subcutaneous injection in C57BL/6 female mice aged 6–8 weeks (n = 4 per group)
To further examine bivalent versus tetravalent GalNAc ligand presentation, we assessed TTR knockdown profile in dose response and repeat dose studies in mice. Consistent with the single dose study previously described (Table 2), we observed identical dose response profiles for bivalent and tetravalent GalNAc ligand conjugates (Figure 2A). These two conjugates also behaved similarly in the repeat dose study, where mice received a total of 6 doses over 18 weeks (Figures 2B and 2C). No loss of potency or body weight was observed upon subsequent re-administrations during the experiment.
Figure 2.

Activity and tolerability of GalNAc conjugates ± polymer micelle in mice
(A) Dose response study: C57BL/6 mice (n = 4) were treated with a single subcutaneous dose of either 1 or 3 mg/kg GalNAc-siRNA alone, or in combination with 10 mg/kg polymer (n.s. = p > 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, two-way ANOVA analysis). (B) Repeat dose study: C57BL/6 mice (n = 4) were treated with subcutaneous administrations of 2 mg/kg conjugate alone or in combination with 10 mg/kg polymer, every 3 weeks (as indicated by arrows), for a period of 18 weeks. TTR protein concentration in plasma was determined using ELISA and reported as a percentage of pre-dose saline control group mean (n.s. = p > 0.05, ∗∗p < 0.01, two-way ANOVA analysis). (C) Repeat dose study: C57BL/6 mice (n = 4) were monitored for body weight changes after repeated subcutaneous administrations of 2 mg/kg conjugate alone or in combination with 10 mg/kg polymer (n.s. = p > 0.05, two-way ANOVA analysis). (D) Polymer titration study: C57BL/6 mice (n = 5) were treated with a single subcutaneous dose of polymer micelle at the specified dose levels. At 6 h post-dose, mouse plasma samples were collected for determination of cytokine levels by ELISA (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001; one-way ANOVA analysis with Dunnett’s multiple comparison test when comparing with PBS control group). Data are presented as mean ± SEM.
Ligand-siRNA conjugates have no intrinsic mechanism of endosomal escape, compromising activity, as only a small percentage escapes and can interact with the RNA-induced silencing complex (RISC). We sought to rectify this by co-administration of a GalNAc-targeted polymer micelle promoting endosomal escape (activity confirmed by red blood cell (RBC) hemolysis assay (Figure S5).8 The composition of the endosomal release polymer is depicted in Figure S6, together with a description of its mechanism of action in relation to its chemical composition. In aqueous media, the polymer will spontaneously form a micelle structure with GalNAc orientated toward the solvent side. When administered subcutaneously, the presence of multiple GalNAc monosaccharides on the surface of the nanoparticle provides hepatocyte targeting via ASGPR uptake. Co-administration of the polymer micelle with the bivalent conjugate yielded significantly increased activity at all doses tested, in both the dose response (Figure 2A) and repeat dose studies (Figure 2B). This improvement was observed regardless of whether the conjugate and polymer micelle were dosed in the same vial or separate vials (Figure S7). Furthermore, we did not observe any loss in body weight upon repeated co-administrations of the conjugate and polymer (Figure 2C). We also conducted a dose titration study to determine the potential of the polymer micelle to induce an acute inflammatory response. At 6 h post-dose, dose-dependent increases in cytokines were observed (Figure 2D); however, administration of the polymer micelle at 10 mg/kg, which resulted in significantly increased potency and duration of activity (Figures 2A and 2B), did not result in significant increases in cytokines relative to the control and was generally well tolerated.
We advanced these delivery technologies into a non-human primate (NHP) model. We retained TTR as the model target gene but changed the siRNA sequence to account for species differences. Historically, GalNAc conjugate activity has translated well between rodents and primates, with a longer duration of effect often observed in primates.16 We observed this here too, with both the bivalent and tetravalent GalNAc conjugate groups exhibiting at least 3 months of sustained target protein knockdown after a single 3 mg/kg subcutaneous dose (Figure 3A). Both bivalent and tetravalent ligand conjugates reported similar maximal knockdown at day 28 and maintained more than 65% suppression at day 91 post-dose. The endosomal escape polymer micelle was co-administered subcutaneously in a third arm with the tetravalent GalNAc conjugate at one-fifth the dose (0.6 mg/kg) of the conjugate alone groups. Remarkably, the improvement in efficacy was more pronounced than in rodents, with plasma levels of the TTR target reduced to below the limits of quantitation, with the reduction maintained through to the pre-determined conclusion of the study at 3 months (Figure 3A). Importantly, these improvements did not alter the tolerability profile of the conjugates. No injection site reactions were observed; all animals appeared normal after treatment and survived to the end of the study. Furthermore, there was no notable increase in plasma levels of representative inflammatory biomarkers (Figure 3B). Additional tolerability data can be found in Table S1.
Figure 3.

Activity and tolerability of GalNAc conjugates ± polymer micelle in NHPs
(A) A single subcutaneous injection of bivalent or tetravalent GalNAc-siRNA was administered to male cynomolgus monkeys (Macaca fascicularis) at 3 mg/kg. In one treatment group, 0.6 mg/kg tetravalent GalNAc-siRNA was co-administered with 8.8 mg/kg polymer micelle. Blood samples were processed to serum and analyzed with a quantitative biomarker immunoassay. Serum TTR levels of each animal were normalized to its respective baseline level at pre-dose. Group average at each time point is represented as a percentage relative to its pre-dose group average (n.s = p > 0.05, ∗∗∗p < 0.001, two-way ANOVA analysis). (B) A single subcutaneous injection of 3 mg/kg tetravalent GalNAc-siRNA, 0.6 mg/kg tetravalent GalNAc-siRNA + 8.8 mg/kg polymer, or 8.8 mg/kg polymer alone was administered to male cynomolgus monkeys (Macaca fascicularis). Blood samples were collected at 6 h post-dose, processed to plasma, and analyzed with a quantitative, multiplexed inflammatory biomarker assay (n.s = p > 0.05, one-way ANOVA analysis). Data are presented as mean ± SEM (n = 3 per group).
Discussion
GalNAc-siRNA conjugates have demonstrated great potential as hepatocyte-specific RNAi therapeutics to address liver-expressed disease targets. An improved understanding of the GalNAc delivery platform will enhance future applications of the RNAi drug modality. Here, we investigated the key elements of GalNAc ligand structure to optimize conjugate activity. We further utilized a targeted endosome release agent to increase the potency and duration of effect for GalNAc-siRNAs while maintaining the practicality of a subcutaneous route of administration.
The binding of galactose to the then-unnamed ASGPR was first reported in 1968 by Ashwell and Morell, and not long after this, the first clinical exploration on delivery of non-glycoproteins to the liver using ASGPR targeting conjugates was conducted.17,18 ASGPR targeting platforms have since been applied to an array of biological modalities such as glycoproteins, small molecules, nucleoside analogs, plasmids, and antisense oligonucleotides (ASO).19, 20, 21, 22, 23, 24, 25, 26, 27 The first report of delivery of siRNA through GalNAc conjugation was in 2010 utilizing a trivalent GalNAc ligand developed in the 1990’s.24,28 In 2019, 50 years after the discovery of ASGPR and 20 years after the discovery of RNAi, Givlaari (Givosiran) became the first GalNAc-siRNA drug to receive clinical approval.
Several approaches have been reported for attaching GalNAc targeting ligands to therapeutic oligonucleotides (including ASO and siRNA). These include attachment through individual, successive nucleotides (either via nucleobase conjugation, 2′ ribose conjugation, or 3′ ribose conjugation),29 or through direct conjugation of a multivalent ligand at either the 5′ or 3′ end of the oligonucleotide. For the latter, trans-4-hydroxyprolinol has been identified as a utility moiety; its multiple chemical functionalities can be used to support solid phase synthesis of oligonucleotides through primary and secondary alcohols and simultaneously serve to attach the targeting ligand through the prolinol amine.9 We were interested to see whether the linker itself displayed any structure-activity relationship on the conjugate when evaluated in vivo (Table 1). We noticed that when the basic linker requirements listed above were incorporated, functional conjugates were indeed produced, yet they were not all equally active. Higher activity was observed when the nitrogen for ligand coupling was part of a ring structure (linkers 3, 4, and 5). If the nitrogen was outside the ring, irrespective of whether it was aromatic or not, potency suffered (linkers 1 and 7). Stereochemistry around the 5-membered ring did not appear to play a strong role (linker 3 versus 4). Acyclic motifs (linker 6 and 8) and one instance of using an ether instead of the usual amine to couple the ligand (linker 2) all exhibited inferior activity profiles. Linker 5 based on a C2 symmetric and stereospecific ([3S,4R]-3,4-dimethylpyrrolidine-3,4-diyl) dimethanol ring produced the greatest and most sustained knockdown in our TTR mouse model.
ASGPR is a calcium-dependent lectin receptor whose biological functions include the uptake and clearance (via clathrin-mediated endocytosis) of circulating glycoproteins.30 It is formed from two carbohydrate recognition domains into various homo and hetero oligomers, but the most abundant configuration is trimeric with the highest binding affinity to asialoorosomucoid ligand.31 This is in accordance with the reported observation that higher valency GalNAc ligands show higher affinity in vitro (tetraantennary > triantennary ≫ biantennary ≫ monoantennary),10, 11, 12,32,33 with stepwise improvements of orders of magnitude in size.34 These literature observations, primarily based on in vitro results, were independently corroborated by our own in vitro competitive binding studies. Interestingly, this hierarchy did not translate entirely to the in vivo activity of the corresponding siRNA conjugates. In fact, despite the bivalent ligand having almost 10-fold lower binding affinity, we observed comparable levels of resulting protein knockdown to trivalent or tetravalent conjugates (compounds 4 and 5). This disconnect between in vitro binding affinity and in vivo protein knockdown is perhaps not surprising. ASGPR binds various endogenous glycoproteins and trivalency is not a prerequisite for uptake and internalization. In addition, the binding afforded by bivalent ligands may simply be good enough to provide saturating levels of uptake. While we observed slightly faster clearance from the blood with the tetravalent versus bivalent ligands possibly due to more efficient binding of the higher valency ligand, both conjugates were fully cleared within 3 h and there was no difference in acute activity. These findings reinforced the requirement for appropriate in vivo testing of siRNA conjugate design. It is worth noting that while bivalent conjugates are simpler molecules to prepare than the tri- or tetravalent species, removal of one or more GalNAc groups through chemical instability or metabolic processes would render a bivalent compound inactive. This concern is mitigated by the additional GalNAc residues present in a tri- or tetravalent compound.
While significant investigation has been conducted over the years regarding the relationship between valency and spatial geometry of GalNAc ligands on ASGPR affinity, there are few reports on SAR of the monosaccharide motif itself. Endogenous glycopeptides with terminal GalNAc residues are the natural substrate of ASGPR; however, it is possible that analogs exist that have higher affinity than GalNAc itself. Two reports substantiate this scenario; Lobst and Drickramer35 showed that N-propionylgalactosamine had a higher affinity than GalNAc in a competitive binding assay using isolated rat hepatic lectin. In a broader SAR study, Mamidyala et al.13 determined the binding of various ligands to immobilized human ASGPR by surface plasmon resonance. This group identified several monosaccharide ligands with higher affinity than GalNAc for ASGPR. Trifluoroacetamide analogs were about 55 times more potent than GalNAc, and variations at the anomeric, C2, and C5 pyrannose positions were all well tolerated. We were not able to prepare multivalent trifluoroacetamide conjugates, as this motif (used as an amine protecting group in chemical synthesis) is not compatible with solid phase synthesis of oligonucleotides. However, the propanamide, C6 methoxyphenyltriazole, and 2,2-difluoropropaneamide (compounds 7, 8, and 9, Table 3) were all prepared as bivalent siRNA conjugates. When tested in vivo, there was essentially no difference in target protein knockdown between these analogs and the parent acetamide. As with our investigation into ligand valency, the discrepancy between binding affinity in vitro, and activity in vivo may simply reflect that beyond a certain degree, binding affinity is not rate-limiting for gene silencing activity. While receptor affinity is a component of conjugate activity, it is not the sole determinant. With the variations tested here, we were not able to improve upon GalNAc for activity in subsequent ligand iterations.
Multiple reports have detailed the optimal geometry of trivalent ASGPR ligands, specifying the distances and bond angles required to maximize ligand/receptor engagement (Figure 1B).15,36 Trivalent ligands with 20 Å spacers (corresponding to pentaethylene glycol) were reported to have 2,000-fold higher affinity than those with 4 Å spacers.37 We were interested to see whether this geometry and consequent SAR extended to our tetravalent system, and varied spacer length by approximately 12–20 Å using tri-, tetra-, or pentaethyleneglycol (compounds 10, 5, and 11, respectively). We also tested different splitter geometries; in addition to the default (repeating glutamic acid units), we prepared a homologated version (compound 12) with a repeating adipic acid motif that could potentially increase rotational freedom of the ligand and also a more rigid geometry with an aromatic splitter and 1,3,5 point of substitution (compound 13). Overall, tetravalent ligands appeared less sensitive to changes in spatial geometry than previously reported trivalent ligands. We hypothesized that the redundancy provided by the fourth GalNAc moiety increased the chances of engagement with the trimeric ASGPR permitting more freedom in the absolute ligand structure.
A major challenge in realizing the full potential of nucleic acid therapeutics is effective delivery to target tissues and cells. Delivery systems such as LNP are engineered to promote efficient endosomal escape,38 resulting in rapid onset of activity and high potency at low doses. GalNAc-siRNA conjugates, on the other hand, have slower onset of activity but also a much longer duration of action. Binding between the ligand and ASGPR alters the configuration of the receptor and triggers clathrin-mediated endocytosis. Endosomal acidification promotes the release of GalNAc-siRNA from ASGPR, and while the exact mechanism of their escape across the endosomal lipid bilayer remains unknown, enough siRNA does reach the cytoplasm to induce a robust RNAi response. However, the vast majority of delivered siRNA remains trapped in the endosome.3 High doses of GalNAc-siRNA are therefore normally required to render them sufficiently potent. In NHP, many reported studies dosed GalNAc-siRNA at 3 to 10 mg/kg to achieve 70% or higher target inhibition (Patents US20190078089A1, US20190078088A1),16,39, 40, 41 as compared to LNP-siRNA dosing at as low as 0.03 mg/kg for a similar suppression effect.42 Clinical doses of GalNAc-siRNAs range from 0.4 mg/kg to 3 mg/kg when targeting 70% or higher inhibition,43, 44, 45 while LNP-siRNAs are usually dosed at ∼0.3 mg/kg or lower.46, 47, 48
To overcome this limitation, we co-administered a GalNAc-targeted polymer micelle subcutaneously with the GalNAc-siRNA. The polymer micelle possesses a pH-responsive endosomal release functionality (as demonstrated in the RBC hemolysis assay), aiding release of the GalNAc-siRNA to improve potency. Other groups have tested similar strategies to improve the potency of both LNP and conjugates;8,49 however, in those cases, the candidates required intravenous administration, thereby eliminating one of the principal advantages of GalNAc conjugate technology. The polymer micelle we used is approximately 15 nm in size and comprises a GalNAc-targeted diblock polymer.8 We reasoned that the small size would mean the polymer micelle could be co-administered subcutaneously, and this was the case. Compared to the GalNAc conjugate alone, we observed a significant boost in potency, a more rapid onset of activity, and longer duration of effect, while preserving the benefit of subcutaneous administration. Furthermore, long-term repeat dosing in mice resulted in sustained gene silencing without loss of potency over time, suggesting that no neutralizing antibodies were generated against either the polymer micelle or the conjugate components. In NHPs (recognized as being highly predictive of clinical performance),50 we observed that a single low dose of GalNAc-siRNA (0.6 mg/kg) and polymer micelle resulted in maximal silencing. This was maintained for at least 3 months post-dose (to the end of the study observation period), compared to the standard GalNAc-siRNA alone where a rebound to baseline was evident within this time frame despite using a 5-fold higher dose of 3 mg/kg. Lower doses to achieve maximal knockdown and a longer duration of effect would mean reduced dosing frequency, lower costs of goods and increased patient convenience. It is important to recognize that nucleic acid drugs can stimulate cytokine release that may be accentuated by the delivery vehicle. Multicomponent systems, such as LNPs, have the potential to trigger an inflammatory response upon systemic administration. Interestingly, the polymer micelle not only improved endosomal escape, but also preserved the tolerability profile of subcutaneously administered GalNAc conjugates. There were no notable differences in appearance or behavior, no injection site reactions, or loss in body weight for the duration of the studies. Furthermore, co-administration of the polymer micelle with GalNAc-siRNA did not activate the innate immune response in mice or NHPs.
The GalNAc/ASGPR pairing is unique in its ability to provide for RNAi activity of conjugates without intrinsic or additional endosomal escape mechanisms, but reports of activity of RNAi conjugates via other receptors on extrahepatic cell types and tissues have been less forthcoming,51 with few entering the clinic, and none of those being systemically administered. The modular nature and chemistry of the endosomal release polymer discussed in this manuscript renders it amenable to being retargeted with other ligands, including motifs that would provide extrahepatic targeting. When paired with conjugates with a matched targeting motif, the potential to use RNAi to address diseases outside the hepatocyte could quickly be unlocked.
Materials and methods
Synthesis of GalNAc ligands
The ligands utilized in this manuscript were prepared in general using established organic chemistry techniques including purification by automated flash chromatography and product confirmation with a combination of analytical high-performance liquid chromatography (HPLC), liquid chromatography-mass spectrometry (LC-MS), MS, and proton nuclear magnetic resonance (NMR) spectroscopy as appropriate. Complete synthetic protocols for all ligands contained in this manuscript can be found in the previously published patents (WO2017177326, WO2018191278, WO2020093061, WO202093053, WO201951257).
Synthesis of GalNAc-conjugated siRNAs
Sense strand synthesis
Ligand succinate was loaded onto 1,000 long chain aminoalkyl CPG (control pore glass) using standard amide coupling chemistry. Loading was determined by DMTr assay @ UV/Vis 504 nm. The resulting GalNAc loaded CPG solid support was employed in automated oligonucleotide synthesis using standard procedures. Nucleotide deprotection followed by removal from the solid support (with concurrent galactosamine acetate deprotection) afforded the GalNAc-oligonucleotide conjugate.
Antisense strand synthesis and duplex formation
Antisense strands were prepared by automated oligonucleotide synthesis using standard procedures. Nucleotide deprotection followed by removal from the solid support afforded the deprotected antisense strand. Annealing of the sense and antisense strands using standard techniques afforded siRNA GalNAc conjugates.
The TTR siRNA sequence used for the mouse studies was described by Nair et al.9 The sequences are cited from the 5′ to 3′ end:
Sense strand: 5′-AsasCaGuGuUCUuGcUcUaUaA-3′
Antisense strand: 5′-usUsaUaGaGcAagaAcAcUgUususu-3′
2′-O-Methyl nucleotides are depicted in lower case, 2′-Fluoro nucleotides are depicted in upper case, and phosphorothioate linkers are depicted as “s.”
The TTR siRNA sequence used for the NHP studies was described in Patent Application WO2017/023660 as AD-65492. The sequence ID numbers of sense and antisense strand are 204 and 236, respectively. The sequences are cited from the 5′ to 3′ end:
Sense strand: 5′-usgsggauUfuCfAfUfguaaccaaga-3′
Antisense strand: 5′-usCfsuugGfuuAfcaugAfaAfucccasusc-3′
GalNAc biotin ligand preparation
Biotinylated ligands were prepared using biotin epsilon amino caproic acid (Sigma-Aldrich catalog no. 47868 coupled to appropriate valency clusters through a functional amine via HATU amide formation. The resultant ligands were purified by recrystallization from methanol.
GalNAc targeted polymer synthesis and polymer micelle preparation
The polymer was synthesized as previously described.8 The polymer was dissolved in 10 mM phosphate/200 mM sucrose PBS (pH 7), up to 40 mg/mL followed by successive filtration (3–4 times) through a 0.2-micron sterile filter. This produced stable polymer micelles with an approximate particle size of 15 nm diameter and polydispersity of <0.16 (Table S2)
Cell culture
HepG2 cells (ATCC, Manassas, VA, USA) were propagated as monolayers in 175 cm2 culture flasks at 37°C in a humidified atmosphere containing 5% CO2, using Minimum Essential Medium (MEM; Life Technologies, Burlington, ON) supplemented with 10% fetal bovine serum (FBS), 100 U/mL Penicillin, and 100 μg/mL Streptomycin, 2 mM L-glutamine, 1 mM sodium pyruvate, 1X nonessential amino acids, and 0.15% sodium bicarbonate. All components were from Life Technologies (Burlington, ON, Canada).
Receptor binding and uptake assay
Biotinylated GalNAc ligand was reconstituted in Tyrode buffer (containing 10 mM HEPES, 5.6 mM glucose, 10 mM KCl, 35 mM NaCl, 0.4 mM MgCl2, 1.0 mM CaCl2, and 0.1% BSA, pH 7.3) and was incubated with Alexa Fluor 488-labeled streptavidin (Invitrogen, Burlington, ON, Canada) in Tyrode buffer at 4°C overnight at a molar ratio of 4.5:1 of biotinylated ligands to streptavidin.
HepG2 cells were seeded into sterile 96-well plates (8 × 104 cells/well) in complete MEM media and cultured for 48 h at 37°C. After overnight incubation, the ligand-streptavidin complex was diluted to 400, 100, 25, and 6.25 nM of functionalized streptavidin in Tyrode buffer and was kept on ice. Prior to complex incubation, the cells were washed three times with ice cold Tyrode buffer and functionalized streptavidin was added to each well, which was incubated at 4°C for 1.5 h. After incubation, the cells were washed three times with ice-cold Tyrode buffer to remove any unbound ligand complex, before adding 37°C preheated complete MEM media and immediately incubating the cells in 37°C for 1 h to allow endocytosis of receptor bound ligand-complexes. After incubation, the cells were washed once with D-PBS and 50 μL of 0.25% Trypsin-EDTA (Invitrogen, Burlington, ON, Canada) was added to each well. After incubation at 37°C for 3–4 min, 100 μL of media was added to each well to inactivate the trypsin, and detachment of cells was accomplished with vigorous mixing. The cells were transferred to a V-bottom 96-well plate (Grenier Bio-one, Cat#651180), spun at 1,200 RPM for 5 min, and washed twice with D-PBS (Invitrogen, Burlington, ON, Canada) before resuspension in PBS containing Live/Dead Red stain (Invitrogen, Burlington, ON, Canada). After 30 min at 4°C, the cells were centrifuged, and the pellet resuspended in stain buffer (Invitrogen, Burlington, ON, Canada) prior to fluorescence-activated cell sorting (FACS) analysis.
Competitive binding assay
ASF (Sigma Aldrich, Oakville, ON), and the various GalNAc ligands were prepared at necessary concentrations in Tyrode buffer. These were incubated on HepG2 cells on ice for 1.5 h, followed by washing to remove unbound ASF or ligands before addition of the functionalized ligand B/streptavidin-AF488 (100 nM) complex & isolation of the HepG2 cells for FACS analysis as described below.
FACS analysis
Analyses were performed on a FACS-Canto II using the software FACS Diva (Becton Dickinson, San Jose, CA, USA). As a marker for viability, cells were stained with Live/Dead Red (Invitrogen, Burlington, ON, Canada). The forward scatter and side scatter gate were set to include all viable cells.
Approximately 10,000–15,000 cells were counted for each sample and binding/uptake was determined as increased intensity in green fluorescence at 488 nm detected in the FL1 channel. The mean fluorescence intensity (MFI) of cells incubated with functionalized streptavidin minus the MFI of cells incubated without functionalized streptavidin (free fluorophore only) was used to determine binding/uptake.
Preparation of GalNAc conjugates and endosomal release polymer for subcutaneous injection in mice and NHPs
GalNAc-siRNA conjugates were dissolved in sterile saline to 10 mg/mL for subcutaneous injection. The polymer used for co-injection was solubilized at 20 mg/mL in formulation buffer with agitation at 400 rpm for 1 h at room temperature and then stored overnight at 4°C. The di-block polymer was diluted to 6–10 mg/mL in sterile saline prior to injection. For mouse studies, GalNAc-siRNA and polymer were supplied in a single vial for injection or administered separately from two different vials (conjugate in one vial, polymer in second vial). For NHP studies, GalNAc-siRNA and polymer were provided in different vials for injection.
Assessment of GalNAc-siRNA and polymer micelle in mouse via subcutaneous administration
All animal-related procedures were conducted at Genevant Sciences Corporation, an accredited facility, according to written operating procedures, in accordance with Canadian Council on Animal Care (CCAC) Guidelines on Good Animal Practices and approved by the local Institutional Animal Care and Use Committee (IACUC). Rodent studies were performed under AUP #0618002.
C57BL/6 female mice aged 6–8 weeks (n = 4 per group) were dosed subcutaneously in the scapular region with vehicle control (saline) or GalNAc-siRNA (± polymer), using a volume of 10 mL/kg body weight. TTR protein levels were analyzed by a quantitative ELISA assay (Abnova, Taipei, Taiwan), as per the manufacturer’s instructions.
C57BL/6 female mice aged 6–8 weeks (n = 5 per group) were dosed subcutaneously in the scapular region with vehicle control (phosphate-buffered saline, pH 7.4) or polymer micelle at the specified doses (10, 20, 30, 40, 60, or 80 mg/kg), using a volume of 10 mL/kg body weight. Plasma cytokine levels were analyzed by a quantitative ELISA assay (BD Biosciences, San Jose, CA, USA), as per the manufacturer’s instructions.
Assessment of GalNAc-siRNA and polymer micelle in NHP via subcutaneous administration
NHP studies were conducted at Citoxlab USA (Stilwell, KS, USA) in non-naive male cynomolgus monkeys (Macaca fascicularis) of Chinese origin, between 3 and 6 years of age and between 4 and 8 kg in body weight (n = 3 per group). Animals were sourced from a Citoxlab USA approved vendor and housed in an accredited facility, in accordance with the USDA Animal Welfare Act (9 CFR, Parts 1, 2 and 3) and as described in the Guide for the Care and Use of Laboratory Animals. The NHP study protocol numbers were 2118-1263 and 2118-2103.
GalNAc-siRNA doses were subcutaneously administered equally at 2 dose sites of ≤2 mL per injection site into the scapular and mid-dorsal regions. Where applicable, a solution containing the polymer was administered equally at 2 additional dose sites in the scapular and mid-dorsal regions. Following dose administration, the area around the dose sites were marked to enable monitoring of any injection site reactions.
Serum samples were analyzed by Myriad RBM (Austin, TX, USA) for TTR protein concentration by DiscoveryMAP, a quantitative biomarker immunoassay, according to standard operating procedures. Plasma samples were analyzed by Myriad RBM (Austin, TX, USA) for cytokine levels using InflammationMAP, a quantitative, multiplexed inflammatory biomarker assay, according to standard operating procedures.
Acknowledgments
We thank Owen Daly for the creation of cartoons illustrated in Figure 1 and Hui Huang for conducting ligand binding studies outlined in the Supplemental information. Cartoons were created with BioRender.
Author contributions
R.J.H., K.L., X.Y., and J.H. designed the experiments and wrote this manuscript. R.J.H., A.D.M., M.C.W. synthesized the ligands. K.M., S.M., A.J., and L.P. conducted the rodent studies. K.L. provided experimental guidance to external contract research organizations for the nonhuman primate study. D.F. performed analytical testing of ligand conjugates. A.C.H.L., E.P.T., A.J., and J.H. provided experimental and strategic guidance.
Declaration of interests
The authors are employees or consultants of Genevant Sciences Corporation or Arbutus Biopharma Corporation as noted in the author affiliations and own shares or stock options in their respective companies.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2021.06.002.
Supplemental information
References
- 1.Hu B., Zhong L., Weng Y., Peng L., Huang Y., Zhao Y., Liang X.J. Therapeutic siRNA: state of the art. Signal Transduct. Target. Ther. 2020;5:101. doi: 10.1038/s41392-020-0207-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Second RNAi drug approved. (2020). Nat Biotechnol 38, 385. [DOI] [PubMed]
- 3.Brown C.R., Gupta S., Qin J., Racie T., He G., Lentini S., Malone R., Yu M., Matsuda S., Shulga-Morskaya S. Investigating the pharmacodynamic durability of GalNAc-siRNA conjugates. Nucleic Acids Res. 2020;48:11827–11844. doi: 10.1093/nar/gkaa670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Debacker A.J., Voutila J., Catley M., Blakey D., Habib N. Delivery of Oligonucleotides to the Liver with GalNAc: From Research to Registered Therapeutic Drug. Mol. Ther. 2020;28:1759–1771. doi: 10.1016/j.ymthe.2020.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Spiess M., Lodish H.F. Sequence of a second human asialoglycoprotein receptor: conservation of two receptor genes during evolution. Proc. Natl. Acad. Sci. USA. 1985;82:6465–6469. doi: 10.1073/pnas.82.19.6465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schwartz A.L., Fridovich S.E., Lodish H.F. Kinetics of internalization and recycling of the asialoglycoprotein receptor in a hepatoma cell line. J. Biol. Chem. 1982;257:4230–4237. [PubMed] [Google Scholar]
- 7.Springer A.D., Dowdy S.F. GalNAc-siRNA Conjugates: Leading the Way for Delivery of RNAi Therapeutics. Nucleic Acid Ther. 2018;28:109–118. doi: 10.1089/nat.2018.0736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Prieve M.G., Harvie P., Monahan S.D., Roy D., Li A.G., Blevins T.L., Paschal A.E., Waldheim M., Bell E.C., Galperin A. Targeted mRNA Therapy for Ornithine Transcarbamylase Deficiency. Mol. Ther. 2018;26:801–813. doi: 10.1016/j.ymthe.2017.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nair J.K., Willoughby J.L., Chan A., Charisse K., Alam M.R., Wang Q., Hoekstra M., Kandasamy P., Kel’in A.V., Milstein S. Multivalent N-acetylgalactosamine-conjugated siRNA localizes in hepatocytes and elicits robust RNAi-mediated gene silencing. J. Am. Chem. Soc. 2014;136:16958–16961. doi: 10.1021/ja505986a. [DOI] [PubMed] [Google Scholar]
- 10.Westerlind U., Westman J., Törnquist E., Smith C.I., Oscarson S., Lahmann M., Norberg T. Ligands of the asialoglycoprotein receptor for targeted gene delivery, part 1: Synthesis of and binding studies with biotinylated cluster glycosides containing N-acetylgalactosamine. Glycoconj. J. 2004;21:227–241. doi: 10.1023/B:GLYC.0000045095.86867.c0. [DOI] [PubMed] [Google Scholar]
- 11.Lee R.T., Lin P., Lee Y.C. New synthetic cluster ligands for galactose/N-acetylgalactosamine-specific lectin of mammalian liver. Biochemistry. 1984;23:4255–4261. doi: 10.1021/bi00313a037. [DOI] [PubMed] [Google Scholar]
- 12.Connolly D.T., Townsend R.R., Kawaguchi K., Bell W.R., Lee Y.C. Binding and endocytosis of cluster glycosides by rabbit hepatocytes. Evidence for a short-circuit pathway that does not lead to degradation. J. Biol. Chem. 1982;257:939–945. [PubMed] [Google Scholar]
- 13.Mamidyala S.K., Dutta S., Chrunyk B.A., Préville C., Wang H., Withka J.M., McColl A., Subashi T.A., Hawrylik S.J., Griffor M.C. Glycomimetic ligands for the human asialoglycoprotein receptor. J. Am. Chem. Soc. 2012;134:1978–1981. doi: 10.1021/ja2104679. [DOI] [PubMed] [Google Scholar]
- 14.Kolatkar A.R., Weis W.I. Structural basis of galactose recognition by C-type animal lectins. J. Biol. Chem. 1996;271:6679–6685. [PubMed] [Google Scholar]
- 15.Biessen E.A., Beuting D.M., Roelen H.C., van de Marel G.A., van Boom J.H., van Berkel T.J. Synthesis of cluster galactosides with high affinity for the hepatic asialoglycoprotein receptor. J. Med. Chem. 1995;38:1538–1546. doi: 10.1021/jm00009a014. [DOI] [PubMed] [Google Scholar]
- 16.Chan A., Liebow A., Yasuda M., Gan L., Racie T., Maier M., Kuchimanchi S., Foster D., Milstein S., Charisse K. Preclinical Development of a Subcutaneous ALAS1 RNAi Therapeutic for Treatment of Hepatic Porphyrias Using Circulating RNA Quantification. Mol. Ther. Nucleic Acids. 2015;4:e263. doi: 10.1038/mtna.2015.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morell A.G., Irvine R.A., Sternlieb I., Scheinberg I.H., Ashwell G. Physical and chemical studies on ceruloplasmin. V. Metabolic studies on sialic acid-free ceruloplasmin in vivo. J. Biol. Chem. 1968;243:155–159. [PubMed] [Google Scholar]
- 18.Rogers J.C., Kornfeld S. Hepatic uptake of proteins coupled to fetuin glycopeptide. Biochem. Biophys. Res. Commun. 1971;45:622–629. doi: 10.1016/0006-291x(71)90462-1. [DOI] [PubMed] [Google Scholar]
- 19.Baenziger J.U., Fiete D. Galactose and N-acetylgalactosamine-specific endocytosis of glycopeptides by isolated rat hepatocytes. Cell. 1980;22:611–620. doi: 10.1016/0092-8674(80)90371-2. [DOI] [PubMed] [Google Scholar]
- 20.Rensen P.C., van Leeuwen S.H., Sliedregt L.A., van Berkel T.J., Biessen E.A. Design and synthesis of novel N-acetylgalactosamine-terminated glycolipids for targeting of lipoproteins to the hepatic asialoglycoprotein receptor. J. Med. Chem. 2004;47:5798–5808. doi: 10.1021/jm049481d. [DOI] [PubMed] [Google Scholar]
- 21.Rohlff C., Watson S.A., Morris T.M., Skelton L., Jackman A.L., Page M.J. A novel, orally administered nucleoside analogue, OGT 719, inhibits the liver invasive growth of a human colorectal tumor, C170HM2. Cancer Res. 1999;59:1268–1272. [PubMed] [Google Scholar]
- 22.Fiume L., Mattioli A., Busi C., Balboni P.G., Barbanti-Brodano G., De Vries J., Altmann R., Wieland T. Selective inhibition of Ectromelia virus DNA synthesis in hepatocytes by adenine-9-beta-D-arabinofuranoside (ara-A) and adenine-9-beta-D-arabinofuranoside 5′-monophosphate (ara-AMP) conjugated to asialofetuin. FEBS Lett. 1980;116:185–188. doi: 10.1016/0014-5793(80)80639-9. [DOI] [PubMed] [Google Scholar]
- 23.Wu G.Y., Wu C.H. Evidence for targeted gene delivery to Hep G2 hepatoma cells in vitro. Biochemistry. 1988;27:887–892. doi: 10.1021/bi00403a008. [DOI] [PubMed] [Google Scholar]
- 24.Merwin J.R., Noell G.S., Thomas W.L., Chiou H.C., DeRome M.E., McKee T.D., Spitalny G.L., Findeis M.A. Targeted delivery of DNA using YEE(GalNAcAH)3, a synthetic glycopeptide ligand for the asialoglycoprotein receptor. Bioconjug. Chem. 1994;5:612–620. doi: 10.1021/bc00030a017. [DOI] [PubMed] [Google Scholar]
- 25.Wu G.Y., Wu C.H. Specific inhibition of hepatitis B viral gene expression in vitro by targeted antisense oligonucleotides. J. Biol. Chem. 1992;267:12436–12439. [PubMed] [Google Scholar]
- 26.Prakash T.P., Graham M.J., Yu J., Carty R., Low A., Chappell A., Schmidt K., Zhao C., Aghajan M., Murray H.F. Targeted delivery of antisense oligonucleotides to hepatocytes using triantennary N-acetyl galactosamine improves potency 10-fold in mice. Nucleic Acids Res. 2014;42:8796–8807. doi: 10.1093/nar/gku531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu R.Z., Graham M.J., Post N., Riney S., Zanardi T., Hall S., Burkey J., Shemesh C.S., Prakash T.P., Seth P.P. Disposition and Pharmacology of a GalNAc3-conjugated ASO Targeting Human Lipoprotein (a) in Mice. Mol. Ther. Nucleic Acids. 2016;5:e317. doi: 10.1038/mtna.2016.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Akinc A., Querbes W., De S., Qin J., Frank-Kamenetsky M., Jayaprakash K.N., Jayaraman M., Rajeev K.G., Cantley W.L., Dorkin J.R. Targeted delivery of RNAi therapeutics with endogenous and exogenous ligand-based mechanisms. Mol. Ther. 2010;18:1357–1364. doi: 10.1038/mt.2010.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matsuda S., Keiser K., Nair J.K., Charisse K., Manoharan R.M., Kretschmer P., Peng C.G., V Kel’in A., Kandasamy P., Willoughby J.L. siRNA conjugates carrying sequentially assembled trivalent N-acetylgalactosamine linked through nucleosides elicit robust gene silencing in vivo in hepatocytes. ACS Chem. Biol. 2015;10:1181–1187. doi: 10.1021/cb501028c. [DOI] [PubMed] [Google Scholar]
- 30.Spiess M. The asialoglycoprotein receptor: a model for endocytic transport receptors. Biochemistry. 1990;29:10009–10018. doi: 10.1021/bi00495a001. [DOI] [PubMed] [Google Scholar]
- 31.Huang X., Leroux J.C., Castagner B. Well-Defined Multivalent Ligands for Hepatocytes Targeting via Asialoglycoprotein Receptor. Bioconjug. Chem. 2017;28:283–295. doi: 10.1021/acs.bioconjchem.6b00651. [DOI] [PubMed] [Google Scholar]
- 32.Rensen P.C., Sliedregt L.A., Ferns M., Kieviet E., van Rossenberg S.M., van Leeuwen S.H., van Berkel T.J., Biessen E.A. Determination of the upper size limit for uptake and processing of ligands by the asialoglycoprotein receptor on hepatocytes in vitro and in vivo. J. Biol. Chem. 2001;276:37577–37584. doi: 10.1074/jbc.M101786200. [DOI] [PubMed] [Google Scholar]
- 33.Lee Y.C., Townsend R.R., Hardy M.R., Lönngren J., Arnarp J., Haraldsson M., Lönn H. Binding of synthetic oligosaccharides to the hepatic Gal/GalNAc lectin. Dependence on fine structural features. J. Biol. Chem. 1983;258:199–202. [PubMed] [Google Scholar]
- 34.Lee Y.C., Lee R.T. In: Carbohydrates in Chemistry and Biology. Ernst B., Hart G., Sináy P., editors. Wiley-VCH Verlag GmBh; Weinheim, Germany: 2008. Interactions of oligosaccharides and glycoproteins with hepatic carbohydrate receptors; pp. 549–561. [Google Scholar]
- 35.Iobst S.T., Drickamer K. Selective sugar binding to the carbohydrate recognition domains of the rat hepatic and macrophage asialoglycoprotein receptors. J. Biol. Chem. 1996;271:6686–6693. doi: 10.1074/jbc.271.12.6686. [DOI] [PubMed] [Google Scholar]
- 36.Schmidt K., Prakash T.P., Donner A.J., Kinberger G.A., Gaus H.J., Low A., Østergaard M.E., Bell M., Swayze E.E., Seth P.P. Characterizing the effect of GalNAc and phosphorothioate backbone on binding of antisense oligonucleotides to the asialoglycoprotein receptor. Nucleic Acids Res. 2017;45:2294–2306. doi: 10.1093/nar/gkx060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Valentijn A.R.P.M., van der Marel G.A., Sliedregt L.A.J.M., van Berkel T.J.C., Biessen E.A.L., van Boom J.H. Solid-phase synthesis of lysine-based cluster galactosides with high affinity for the asialoglycoprotein Receptor. Tetrahedron. 1997;53:759–770. [Google Scholar]
- 38.Heyes J., Palmer L., Bremner K., MacLachlan I. Cationic lipid saturation influences intracellular delivery of encapsulated nucleic acids. J. Control. Release. 2005;107:276–287. doi: 10.1016/j.jconrel.2005.06.014. [DOI] [PubMed] [Google Scholar]
- 39.Sehgal A., Barros S., Ivanciu L., Cooley B., Qin J., Racie T., Hettinger J., Carioto M., Jiang Y., Brodsky J. An RNAi therapeutic targeting antithrombin to rebalance the coagulation system and promote hemostasis in hemophilia. Nat. Med. 2015;21:492–497. doi: 10.1038/nm.3847. [DOI] [PubMed] [Google Scholar]
- 40.Liebow A., Li X., Racie T., Hettinger J., Bettencourt B.R., Najafian N., Haslett P., Fitzgerald K., Holmes R.P., Erbe D. An Investigational RNAi Therapeutic Targeting Glycolate Oxidase Reduces Oxalate Production in Models of Primary Hyperoxaluria. J. Am. Soc. Nephrol. 2017;28:494–503. doi: 10.1681/ASN.2016030338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu J., Qin J., Borodovsky A., Racie T., Castoreno A., Schlegel M., Maier M.A., Zimmerman T., Fitzgerald K., Butler J., Akinc A. An investigational RNAi therapeutic targeting Factor XII (ALN-F12) for the treatment of hereditary angioedema. RNA. 2019;25:255–263. doi: 10.1261/rna.068916.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Love K.T., Mahon K.P., Levins C.G., Whitehead K.A., Querbes W., Dorkin J.R., Qin J., Cantley W., Qin L.L., Racie T. Lipid-like materials for low-dose, in vivo gene silencing. Proc. Natl. Acad. Sci. USA. 2010;107:1864–1869. doi: 10.1073/pnas.0910603106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sardh E., Harper P., Balwani M., Stein P., Rees D., Bissell D.M., Desnick R., Parker C., Phillips J., Bonkovsky H.L. Phase 1 Trial of an RNA Interference Therapy for Acute Intermittent Porphyria. N. Engl. J. Med. 2019;380:549–558. doi: 10.1056/NEJMoa1807838. [DOI] [PubMed] [Google Scholar]
- 44.Machin N., Ragni M.V. An investigational RNAi therapeutic targeting antithrombin for the treatment of hemophilia A and B. J. Blood Med. 2018;9:135–140. doi: 10.2147/JBM.S159297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fitzgerald K., Frank-Kamenetsky M., Shulga-Morskaya S., Liebow A., Bettencourt B.R., Sutherland J.E., Hutabarat R.M., Clausen V.A., Karsten V., Cehelsky J. Effect of an RNA interference drug on the synthesis of proprotein convertase subtilisin/kexin type 9 (PCSK9) and the concentration of serum LDL cholesterol in healthy volunteers: a randomised, single-blind, placebo-controlled, phase 1 trial. Lancet. 2014;383:60–68. doi: 10.1016/S0140-6736(13)61914-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Adams D., Gonzalez-Duarte A., O’Riordan W.D., Yang C.C., Ueda M., Kristen A.V., Tournev I., Schmidt H.H., Coelho T., Berk J.L. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018;379:11–21. doi: 10.1056/NEJMoa1716153. [DOI] [PubMed] [Google Scholar]
- 47.El Dika I., Lim H.Y., Yong W.P., Lin C.C., Yoon J.H., Modiano M., Freilich B., Choi H.J., Chao T.Y., Kelley R.K. An Open-Label, Multicenter, Phase I, Dose Escalation Study with Phase II Expansion Cohort to Determine the Safety, Pharmacokinetics, and Preliminary Antitumor Activity of Intravenous TKM-080301 in Subjects with Advanced Hepatocellular Carcinoma. Oncologist. 2019;24:747–e218. doi: 10.1634/theoncologist.2018-0838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hu B., Weng Y., Xia X.H., Liang X.J., Huang Y. Clinical advances of siRNA therapeutics. J. Gene Med. 2019;21:e3097. doi: 10.1002/jgm.3097. [DOI] [PubMed] [Google Scholar]
- 49.Wong S.C., Klein J.J., Hamilton H.L., Chu Q., Frey C.L., Trubetskoy V.S., Hegge J., Wakefield D., Rozema D.B., Lewis D.L. Co-injection of a targeted, reversibly masked endosomolytic polymer dramatically improves the efficacy of cholesterol-conjugated small interfering RNAs in vivo. Nucleic Acid Ther. 2012;22:380–390. doi: 10.1089/nat.2012.0389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nair J.K., Attarwala H., Sehgal A., Wang Q., Aluri K., Zhang X., Gao M., Liu J., Indrakanti R., Schofield S. Impact of enhanced metabolic stability on pharmacokinetics and pharmacodynamics of GalNAc-siRNA conjugates. Nucleic Acids Res. 2017;45:10969–10977. doi: 10.1093/nar/gkx818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tai W. Current Aspects of siRNA Bioconjugate for In Vitro and In Vivo Delivery. Molecules. 2019;24:E2211. doi: 10.3390/molecules24122211. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.