

Abstract
A common feature of diverse brain disorders is the alteration of GABA-mediated inhibition because of aberrant, intracellular chloride homeostasis induced by changes in the expression and/or function of chloride transporters. Notably, pharmacological inhibition of the chloride importer NKCC1 is able to rescue brain-related core deficits in animal models of these pathologies and in some human clinical studies. Here, we show that reducing NKCC1 expression by RNA interference in the Ts65Dn mouse model of Down syndrome (DS) restores intracellular chloride concentration, efficacy of gamma-aminobutyric acid (GABA)-mediated inhibition, and neuronal network dynamics in vitro and ex vivo. Importantly, adeno-associated virus (AAV)-mediated, neuron-specific NKCC1 knockdown in vivo rescues cognitive deficits in diverse behavioral tasks in Ts65Dn animals. Our results highlight a mechanistic link between NKCC1 expression and behavioral abnormalities in DS mice and establish a molecular target for new therapeutic approaches, including gene therapy, to treat brain disorders characterized by neuronal chloride imbalance.
Keywords: Down syndrome, GABA, cognitive disabilities, gene therapy, RNA interference, chloride, inhibition, AAV, learning and memory
Graphical abstract
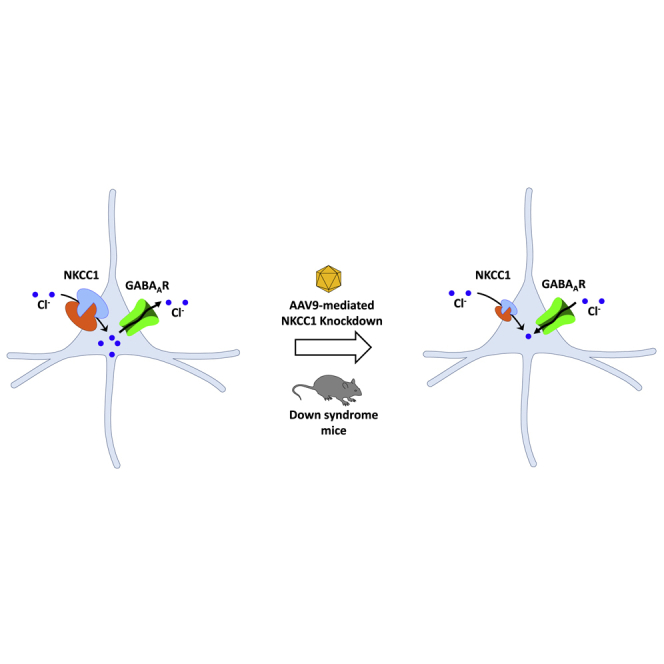
A common feature of diverse brain disorders is the alteration of GABA-mediated inhibition because of aberrant intracellular chloride homeostasis. Here, the authors show that reducing the expression of chloride importer NKCC1 by AAV-mediated RNA interference in a mouse model of Down syndrome restores GABA signaling and rescues associated cognitive deficits.
Introduction
Optimal neural network activity and brain functions critically depend on the correct balance between excitatory and inhibitory synaptic inputs. In most neurons of the adult brain, gamma-aminobutyric acid (GABA) is the main inhibitory neurotransmitter through chloride-permeable ionotropic GABAA receptors (GABAARs). In mature neurons, low intracellular chloride concentration ([Cl−]i) causes an inward (hyperpolarizing) flow of Cl− ions through GABAARs.1 However, relatively small changes in neuronal [Cl−]i are sufficient to switch the polarity of GABAergic responses from hyperpolarizing to depolarizing, as seen during development.2 Therefore, [Cl−]i must be tightly controlled in neurons to ensure appropriate GABAergic hyperpolarization and inhibition and to avoid catastrophic consequences on the excitatory/inhibitory balance and neural network activity.
Regulation of neuronal [Cl−]i is mainly controlled by the opposing action of two major cation/chloride cotransporters: the Cl− importer NKCC1 and the exporter KCC2.1,3 Interestingly, alteration of neuronal chloride homeostasis because of variations in the expression or transport activity of NKCC1 and/or KCC2 is a common feature of a wide range of brain disorders,3 including Down syndrome (DS),4 autism,5 Rett syndrome,6, 7, 8 fragile X syndrome,5,9,10 tuberous sclerosis,11 22q11.2 microdeletion syndrome,12 maternal immune activation,13 some forms of epilepsy,14, 15, 16, 17, 18 chronic pain,19 schizophrenia,20 and Huntington disease.21,22 Accordingly, pharmacological treatment with the U.S. Food and Drug Administration (FDA)-approved NKCC1 inhibitor bumetanide is able to rescue brain-related core deficits in rodent models of a number of these disorders as well as in clinical trials with autistic individuals,23,24 and some case studies on patients with other neurological disorders.3 In that regard, we found that cognitive deficits and altered GABAAR signaling in the Ts65Dn (Ts) mouse model of DS are possibly linked to the upregulation of NKCC1, which we also found increased in brain samples from individuals with DS.4 Indeed, in the brain of adult Ts65Dn mice, the neuronal [Cl−]i is increased, and the efficacy of GABAAR-mediated inhibition is impaired because GABAAR signaling is shifted from hyperpolarizing to depolarizing.4 Importantly, bumetanide treatment restores both GABAAR-mediated inhibition and cognitive performances in Ts65Dn mice, although a chronic, daily treatment is required.4
Despite the very large and fast-increasing literature in both animal models and patients that indicate positive outcomes with bumetanide treatment, there is not yet a strong, demonstrated direct link between NKCC1 inhibition and restoration of Cl− homeostasis, full GABAergic inhibitory signaling, and rescue of brain deficits. Moreover, bumetanide has strong diuretic activity, triggering ionic imbalances and potential ototoxicity.25,26 This hampers its use for clinical applications in lifelong treatments4,23 and may strongly jeopardize treatment compliance during years of treatment. Moreover, bumetanide was given systemically in most studies, and the suboptimal brain pharmacokinetic profile of the drug27 raises questions about its mechanism of action.28 Here, we demonstrate that adeno-associated virus (AAV)-mediated RNA interference (RNAi) to target (and reduce) neuronal NKCC1 expression rescues neuronal Cl− homeostasis, GABAergic transmission, and cognitive deficits in the Ts65Dn mouse model of DS. This sets the stage for the development of a gene-therapy approach to overcome the shortcomings of bumetanide treatment.
Results
Anti-NKCC1 amiR treatment restores intracellular chloride concentration in Ts65Dn neurons in culture
As a first step toward validating NKCC1 as a molecular target and developing a gene-therapy approach for DS, we established an RNAi strategy based on the use of artificial microRNAs (amiRs) expressed from RNA polymerase type II (Pol II) promoters.29 First, we generated lentiviral vectors (LVs) expressing two different NKCC1-specific amiRs (1 or 2), together with the enhanced green fluorescent protein (EGFP, to monitor transduction efficiency) under the control of the ubiquitous hPGK1 (human phosphoglycerate kinase 1) promoter (Figure 1A, left). Transduction efficiency for the different LVs was between 85% and 90% and was similar in both wild-type (WT) and Ts65Dn neuronal cultures (Figure S1A).
Figure 1.

Anti-NKCC1 amiR treatment rescues intracellular chloride concentration in Ts65Dn neurons in cultures
(A) Left: schematic representation of the lentiviral vector (LV) expressing either control or NKCC1 amiRs from the ubiquitous hPGK1 promoter. Right: representative immunofluorescent images of LV-transduced WT and Ts65Dn hippocampal cultures at 15 DIVs expressing EGFP (green) and immunostained for the neuronal marker MAP2 (red) and the astrocytic marker GFAP (blue). Arrows indicate EGFP and MAP2 double-positive neurons, whereas arrowheads indicate EGFP and GFAP double-positive astrocytes. Scale bar: 50 μm. (B) Left: representative immunoblot for NKCC1 in protein extracts from LV-transduced WT and Ts65Dn neurons at 15 DIVs. Right: quantification of NKCC1 protein (expressed as the percentage of WT neurons transduced with control amiR) showed increased NKCC1 expression in Ts65Dn compared with that of WT neurons. Expression of amiRs 1 and 2 induced significant NKCC1 knockdown in both WT and Ts65Dn neurons. Actin was used as an internal standard. (C) Left: schematic representation of the LVs expressing either control or NKCC1 amiRs from the neuron-specific hSyn promoter. Right: representative immunofluorescent images of LV-transduced WT and Ts65Dn hippocampal cultures at 15 DIVs expressing mCherry (red), and immunostained for the neuronal marker MAP2 (green) and the astrocytic marker GFAP (blue). Arrows indicate mCherry and MAP2 double-positive neurons, whereas arrowheads indicate GFAP+/mCherry-negative astrocytes. Scale bar: 50 μm. (D) Representative images of LV-transduced WT and Ts65Dn hippocampal neurons at 15 DIV (expressing mCherry; red) during imaging experiments with the chloride-sensitive dye MQAE (pseudo-colors). Fluorescent intensity of the dye (color-coded on the bottom) is inversely proportional to [Cl−]i. Scale bar: 100 μm. (E) Quantification of [Cl−]i with MQAE (expressed as the percentage of WT neurons transduced with control amiR) showed lower fluorescent intensity (i.e., greater [Cl−]i) in Ts65Dn compared with that of WT neurons. NKCC1 knockdown by amiRs 1 or 2 significantly increased MQAE fluorescent intensity (i.e., lower [Cl−]i) in both WT and Ts65Dn neurons. Data in (B) are means ± SEM; dots indicate values of individual samples (obtained from five independent neuronal cultures). Boxplots in (E) indicate median and 25th–75th percentiles, whiskers represent the 5th–95th percentiles, and numbers in parentheses indicate the number of analyzed samples (obtained from six independent neuronal cultures). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, Tukey post hoc test after two-way ANOVA.
To evaluate NKCC1 knockdown efficiency, we transduced primary cortical neurons from WT and Ts65Dn mice with LVs expressing either a control amiR or one of the two amiRs against NKCC1 at 7 days in vitro (DIVs). One week after infection, immunoblot analysis showed that NKCC1 was significantly upregulated in cultured trisomic cells compared with that of WT mice (Figure 1B), similar to what was previously reported for the brains of adult Ts65Dn mice and DS individuals.4 NKCC1-specific amiRs expressed EGFP and robustly reduced NKCC1 protein expression in both WT and Ts65Dn cultures (Figures 1B and S1B).
Next, we assessed whether NKCC1 knockdown resulted in changes of neuronal [Cl−]i. For that, we performed imaging experiments with the green fluorescent chloride-sensitive dye MQAE (N-(ethoxycarbonylmethyl)-6-methoxyquinolinium bromide) in primary hippocampal neurons at 15 DIVs. We expressed the NKCC1-specific amiRs together with the red fluorescent protein mCherry from the neuron-specific promoter hSyn (human synapsin; Figure 1C, left) to allow MQAE imaging selectively in neurons (Figure 1C, right). As previously reported for Ts65Dn brain slices,4 Ts65Dn-cultured neurons showed a significant decrease in MQAE fluorescence (mirror of a higher [Cl−]i) compared with that of WT cells (Figures 1D and 1E). However, NKCC1 knockdown by both amiRs strongly decreased [Cl−]i in Ts65Dn neurons and also slightly, but significantly, decreased [Cl−]i in WT cells (Figures 1D and 1E).
Anti-NKCC1 amiR treatment restores hyperpolarizing GABAA signaling in Ts65Dn neurons
Because high [Cl−]i is predictive of depolarizing, rather than hyperpolarizing, GABA responses,1 we tested whether the reduced [Cl−]i, consequence of NKCC1 knockdown, would rescue full hyperpolarizing and inhibitory GABA signaling in Ts65Dn neurons. For that, we recorded spontaneous firing of mature hippocampal neurons in cultures at 16–20 DIVs from WT and Ts65Dn mice. We used cell-attached patch-clamp, a method that preserves the endogenous [Cl−]i.4,30 First, we tested whether NKCC1 knockdown could restore endogenous GABAAR signaling in Ts65Dn neuronal cultures by bath-applying the GABAAR antagonist bicuculline (BIC; 10 μM). Consistent with previous findings in brain slices from Ts65Dn mice,4 we found that BIC application increased the average spike frequency in WT cells, as expected by inhibitory GABAergic signaling in mature neurons (Figures 2A and 2B). Conversely, blocking GABAAR signaling in Ts65Dn neurons transduced with control amiR elicited the opposite response, decreasing the average spike frequency of recorded Ts65Dn neurons (Figure 2B). Analysis of changes in activity (of at least 20%) of individual neurons that were active before or after BIC application, showed that spike frequency was, in fact, decreased in 70% of recorded Ts65Dn neurons transduced with control amiR and increased (as in WT) in the remaining 30% of cells (Figure 2C). Interestingly, similar analysis of individual WT neuron responses before and after BIC application showed that spike frequency was increased in 62% of cells but actually decreased in 23%. This highlighted the existence of heterogeneous responses to endogenous GABAAR signaling in WT neurons as well but with a somehow reversed proportion compared with that of Ts65Dn. Remarkably, knockdown of NKCC1 expression with both amiRs restored the physiological levels of inhibitory action of endogenous GABA in Ts65Dn neurons, as demonstrated by the increase in spiking frequency upon BIC application similar to that of WT neurons (Figures 2B and 2C).
Figure 2.

Anti-NKCC1 amiR treatment rescues inhibitory GABAergic signaling in Ts65Dn neurons in culture
(A) Left: schematic representation of the LV expressing either control or NKCC1 amiRs. Right: symbol legend for panels (B)–(E). (B) Quantification of the average spontaneous spiking activity recorded in cell-attached, patch-clamp configuration in LV-transduced hippocampal neurons (16–20 DIVs) from WT and Ts65Dn mice before and after bath application of the GABAAR antagonist bicuculline (10 μM). (C) Example traces and spiking frequency of single, active cells, from experiments in (B) for LV-transduced WT and Ts65Dn neurons before (baseline) and during bath application of bicuculline. (D) Quantification of the average spontaneous spiking activity recorded in cell-attached patch-clamp configuration in LV-transduced hippocampal neurons (16–20 DIVs) from WT and Ts65Dn mice before and after bath applications of increasing concentrations of GABA (1–200 μM). (E) Example traces and spiking frequency of single, active cells, from experiments in (D) for LV-transduced WT and Ts65Dn neurons before (baseline) and during bath application of GABA at 100 μM. Data shown in (B) and (D) are means ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, Tukey post hoc test after two-way RMs-ANOVA. In (C) and (E), the numbers in parentheses indicate the number of active cells recorded before and after drug application for each experimental group, and boxed numbers indicate the percentages of cells showing at least a 20% increase (↑) or decrease (↓) in spiking frequency. #p < 0.05, ##p < 0.01, ###p < 0.001, paired Student’s t test or Wilcoxon signed-rank test.
Next, to further strengthen the above results, we tested the effect of exogenous GABA application to WT and Ts65Dn hippocampal neuronal cultures at 16–20 DIVs. Also consistent with the previous findings in brain slices from Ts65Dn mice,4 we found that bath application of GABA at increasing concentrations (1–200 μM) dose-dependently increased the average spike frequency of recorded Ts65Dn neurons transduced with control amiR and abolished firing in nearly all WT neurons (Figure 2D). Interestingly, analysis of the changes in activity (of at least 20%) of individual cells that were active before or after GABA application (at 100 μM) showed that spike frequency was actually increased in 70% of recorded Ts65Dn neurons transduced with control amiR, whereas it was decreased (similar to WT cultures) in the remaining 30% of cells (Figure 2E). Importantly, when NKCC1 was knocked down, Ts65Dn neurons showed an almost-complete cessation of spontaneous firing in virtually all cells, which was similar to that of WT cells (Figures 2D and 2E).
Moreover, we also assessed possible differences in spontaneous firing between WT and Ts65Dn hippocampal neurons in culture by analyzing pooled baseline recordings of cells shown in Figures 2B and 2D. The percentage of active neurons in baseline recordings was similar among the different experimental groups (Figures S2A and S2B). In addition, spontaneous baseline-firing frequency was not significantly different between groups, although Ts65Dn neurons showed a tendency toward increased activity (Figure S2C). This likely reflects the high variability in firing activity shown by individual neurons in culture.
Altogether, these data show that altered GABAergic signaling in cultured Ts65Dn neurons can be fully rescued by NKCC1 knockdown, similar to results obtained with pharmacological NKCC1 inhibition in Ts65Dn acute brain slices.4
Anti-NKCC1 amiR treatment restores neuronal-correlated network dynamics and GABA-mediated network responses in Ts65Dn neurons
The above results confirmed the presence of a significantly increased population of neurons with depolarizing responses to GABA in hippocampal Ts65Dn cultures, as previously reported in acute hippocampal slices from adult Ts65Dn animals.4 However, the patch-clamp experiments could not disclose the effect of chloride dyshomeostasis on global neuronal network dynamics. Therefore, we next performed multi-electrode array (MEA) experiments—a method that allows the simultaneous recording of the firing activity from hundreds of neurons—on WT and Ts65Dn hippocampal cultures at 21 DIVs, transduced with either control or NKCC1 amiRs at 15 DIVs (Figure 3A). Although the basal mean firing rate (MFR) and mean bursting rate (MBR) were not significantly different in neurons transduced with control amiR (Figures S3A–S3E), we found that network dynamics were markedly altered in Ts65Dn neurons compared with that of WT neurons (Figure S3A). In particular, analysis of correlated network activity by spike time tiling coefficients (STTCs)31 showed that network synchronization was strongly reduced in Ts65Dn hippocampal cultures compared with that of WT cultures (Figures 3B and 3C). Interestingly, NKCC1 knockdown with both amiRs partially, but significantly, rescued network-correlated activity of Ts65Dn neuron and produced a slightly opposite effect on the WT network (Figures 3B and 3C). Altogether, these data indicate a role for [Cl−]i and GABAergic signaling in regulating neuronal network dynamics in DS, as previously shown for WT neurons.32, 33, 34
Figure 3.

Anti-NKCC1 amiR treatment rescues neuronal networks dynamics in Ts65Dn cultures
(A) Left: schematic representation of the LVs expressing either control or NKCC1 amiRs. Right: merged fluorescent and transmitted-light images showing MEA seeded with LV-transduced WT and Ts65Dn hippocampal neurons expressing EGFP (green) at 21 DIVs. Scale bar: 50 μm. (B) Examples of color-coded (top) correlation matrices quantified by STTCs analysis between each active electrode in representative MEAs seeded with LV-transduced WT and Ts65Dn hippocampal neurons. (C) Correlated firing activity of LV-transduced WT and Ts65Dn neuronal networks was quantified by measuring mean STTCs values for each electrode versus all other electrodes in MEAs seeded with LV-transduced WT and Ts65Dn neurons. (D) Beanplot showing the distribution of the percentage of MFR variation recorded by MEAs upon administration of the GABAARs antagonist BIC (20 μM) in LV-transduced WT and Ts65Dn hippocampal cultures. (E) Quantification of the percentage of MEA channels showing significant changes in MFR by bootstrap analysis in comparison with their basal conditions after BIC administration in LV-transduced WT and Ts65Dn hippocampal neurons. (F) Beanplot showing the distribution of the percentage of MFR variation recorded by MEAs upon GABA (20 μM) application in LV-transduced WT and Ts65Dn hippocampal cultures. (G) Quantification of the percentage of MEA channels showing significant changes in MFR by bootstrap analysis in comparison with their basal conditions after GABA administration in LV-transduced WT and Ts65Dn hippocampal neurons. Boxplots in (C) indicate median and 25th–75th percentiles, and whiskers represent the 5th–95the percentiles. In panel (D) and (F), short ticks represent the %MFRvar value for each channel; horizontal lines indicate median value of the corresponding distribution. Data shown in (E) and (G) are means ± SEM. The numbers in parentheses indicate the number of MEAs analyzed (obtained from nine independent neuronal cultures). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, Tukey post hoc test after two-way ANOVA.
Next, we evaluated the effect of NKCC1 knockdown on endogenous GABAergic signaling at the network level in Ts65Dn and WT hippocampal cultures by recording the firing rate with MEAs before and after bath application of the GABAAR antagonist BIC. We found that BIC application in Ts65Dn neuronal networks transduced with control amiR led to a significant alteration of GABAergic signaling with a distinguishably opposite outcome compared with WT neuronal networks (Figure S4A). To quantify that outcome, we calculated the percentage of MFR variation (%MFRvar; see Materials and methods) for each active MEA electrode before and after BIC application. We found that upon blocking endogenous GABAA signaling with BIC the distribution of the MFRvar for Ts65Dn cultures transduced with control amiR appeared shifted toward more-negative values compared with that of WT cultures. However, NKCC1 knockdown resulted in an MFRvar distribution of Ts65Dn neurons indistinguishable from the one in WT cultures, indicating the complete rescue of GABAA-mediated inhibition (Figure 3D). Next, we quantified the percentage of electrodes showing a significant change in MFR by bootstrap analysis.35 In WT cultures, we found that the firing was increased in 86% of active electrodes, confirming our data by patch-clamp recordings on single neurons. Notably, in Ts65Dn neuronal networks, blocking GABAA-mediated signaling determined the opposite behavior in 30% of active electrodes, which, in fact, showed a significant decrease in firing, whereas only 62% showed an increase in firing (Figures 3E, S4A, and S4B). Remarkably, in Ts65Dn cultures in which NKCC1 was knocked down, firing was increased in 80%–84% of active electrodes, similar to that of WT neurons (Figures 3E, S4A, and S4B), indicating a full recovery of GABAA-mediated inhibitory signaling.
Finally, we strengthened our data by assessing whether NKCC1 knockdown could also reestablish inhibitory responses to exogenously applied GABA at the network level. We found that the distribution of MFRvar after treatment with an intermediate concentration of GABA (20 μM) in Ts65Dn neuronal networks in culture appeared largely dispersed and shifted toward more-positive values compared with WT neuronal networks but, again, was restored upon NKCC1 knockdown (Figures 3F, S4C, and S4D). As expected, in WT hippocampal neurons transduced with control amiR, application of exogenous GABA elicited a sharp decrease of firing in virtually all (98%) active electrodes (Figures 3G, S4C, and S4D). Conversely, in Ts65Dn neuronal networks, application of GABA determined a decrease of in firing in only 75% of active electrodes, whereas 12% showed the reverse response by increasing firing (Figures 3G, S4C, and S4D). Remarkably, upon NKCC1 knockdown in Ts65Dn neurons, exogenous GABA caused a strong decrease in firing in 95%–98% of active electrodes, similar to that of WT cultures (Figures 3G, S4C, and S4D).
Therefore, these data indicate that NKCC1 knockdown completely restored GABA-mediated inhibitory signaling in Ts65Dn neurons at the network level as well.
Neuro-specific anti-NKCC1 amiR treatment in vivo restores inhibitory GABAA signaling in Ts65Dn hippocampal slices
To specifically address whether NKCC1 upregulation and consequent dysregulation of GABAergic signaling has a causative role in the cognitive deficits that are characteristic of Ts65Dn mice,4 we performed NKCC1 knockdown in vivo in the hippocampus, a brain region with a pivotal role in learning and memory. For that, we stereotaxically injected an AAV serotype 9 (AAV9) vector expressing EGFP (for visualization of infected neurons) and either a control amiR or one of the two anti-NKCC1 amiRs expressed from the neuro-specific hSyn promoter in the dorsal hippocampal CA1 area of adult (2–3 month old) Ts65Dn mice and WT littermates (Figure 4A). Immunohistochemistry analysis showed that EGFP was specifically expressed in more than 95% of NeuN+ neurons, but not in GFAP+ astrocytes or Iba1+ microglial cells (Figure 4B). qPCR bio-distribution analysis of viral vector genome copy number (VGCN) showed that AAV9 transduction was predominant in the hippocampus versus the overlying cortical tissue and almost undetectable in peripheral organs (Figure S5). Next, we assessed the degree of NKCC1 knockdown in vivo in the hippocampal CA1 region upon neuro-specific AAV9 delivery of amiRs. Immunoblot analysis showed that NKCC1 was significantly upregulated in Ts65Dn dorsal CA1 (Figures 4C and 4D) compared with that in the WT CA1 region. Neuro-specific expression of both anti-NKCC1 amiRs 1 and 2, indeed, reduced NKCC1 protein expression in Ts65Dn dorsal CA1 to levels comparable with that of WT samples (Figures 4C and 4D). However, these data clearly represent an underestimation of the effective neuronal knockdown efficiency in vivo. Indeed, NKCC1 is ubiquitously expressed in all cell types (e.g., astrocytes)36 and, thus, the western blot (WB) quantification performed on whole lysate detected NKCC1 expression from cell types other than neurons as well. Interestingly, NKCC1 knockdown by amiR 1 or amiR 2 did not affect KCC2 expression in the same samples in which we performed analysis of expression of NKCC1 (Figures 4E and 4F). This excludes possible secondary effects on the chloride exporter KCC2, which may underlie changes in [Cl−]i.
Figure 4.

Neuro-specific hippocampal AAV9 delivery reduced NKCC1 expression in Ts65Dn mice
(A) Left: schematic representation of the experimental timeline: 2–3 month-old WT and Ts65Dn mice were stereotaxically injected in the dorsal hippocampal CA1 region with AAV9 vectors and analyzed by immunohistochemistry 6 WPIs. Right: low-magnification images showing the expression of EGFP (green) in the dorsal CA1 hippocampal region in WT and Ts65Dn mice. Nuclei are counterstained with Hoechst (blue). Scale bar: 250 μm. (B) Top-left: representative confocal z stack projection images of AAV-transduced WT and Ts65Dn hippocampi expressing EGFP (green) and immunostained for the neuronal marker NeuN (red); nuclei are counterstained with Hoechst (blue). EGFP expression was restricted to NeuN+ neurons. Scale bar: 50 μm. Bottom-left: representative confocal z stack projection images showing EGFP expression (green) in the dorsal hippocampal CA1 region of AAV9-injected WT and Ts65Dn mice at 6 WPIs. Sections were immunostained with the astrocyte marker GFAP (blue) and the microglia marker Iba1 (red). EGFP expression was virtually absent in both astrocytes and microglia cells. Scale bar: 50 μm. Right: quantification of the percentage of EGFP and NeuN-double-positive (NeuN+-EGFP+) cells over the total EGFP+ cells in the hippocampal CA1 pyramidal layer of AAV9-injected WT and Ts65Dn mice. (C) Top: schematic representation of the experimental timeline: 2–3 month-old WT and Ts65Dn mice were stereotaxically injected in the dorsal hippocampal CA1 region with AAV9 vectors and analyzed by immunoblot at 4 WPIs. Bottom: representative immunoblot for NKCC1 in protein extracts from AAV9-transduced WT and Ts65Dn dorsal CA1 regions. (D) Quantification of NKCC1 protein (expressed as the percentage of WT neurons transduced with control amiR) showed increased NKCC1 expression in Ts65Dn compared with that of WT samples. Expression of amiRs 1 and 2 induced significant NKCC1 knockdown in the Ts65Dn dorsal CA1 region. (E) Representative immunoblot for KCC2 in protein extracts from AAV9-transduced WT and Ts65Dn dorsal CA1 regions. (F) Quantification of KCC2 protein, in the same samples as in (D), showed no significant difference in KCC2 expression in any of the experimental groups. Actin was used as an internal standard. Data in (B), (D), and (F) are means ± SEM, and dots indicate values of individual samples. ∗p < 0.05, ∗∗∗p < 0.001, Tukey post hoc test after two-way ANOVA.
We next evaluated whether NKCC1 knockdown could restore endogenous inhibitory GABAAR signaling in adult Ts65Dn mice. For that, we recorded spontaneous firing of CA1 pyramidal neurons in acute brain slices obtained from animals injected with AAV9-expressing amiRs at 2–3 months old and sacrificed 4 weeks later. We found that the percentage of spontaneously active CA1 neurons was significantly increased in the slice obtained from Ts65Dn animals receiving control amiR compared with that of WT animals (Figure 5B). Knockdown of NKCC1 with both amiRs showed a trend toward a decrease in the percentage of spontaneously active CA1 neurons in Ts65Dn mice, although that was not statistically significant (Figure 5B). In fact, we found that baseline firing frequency was significantly increased in Ts65Dn CA1 pyramidal neurons transduced with control amiR compared with those of WT animals, as previously reported.4 Interestingly, NKCC1 knockdown with both amiRs efficiently induced a significant decrease in spontaneous firing activity in Ts65Dn CA1 neurons to a level similar to that of WT neurons (Figure 5C). This was likely due to increased efficacy of GABAAR-mediated inhibition. Indeed, after blocking endogenous GABAAR signaling with BIC, CA1 neurons from WT animals exhibited increased spiking activity (Figure 5C), as predicted by inhibitory GABAergic signaling in adult animals. Conversely, CA1 neurons from Ts65Dn mice transduced with control amiR showed a decrease in firing frequency, again indicating an opposite action of the neurotransmitter in those animals. Interestingly, NKCC1 knockdown by both amiRs restored inhibitory GABAAR-mediated signaling in Ts65Dn CA1 neurons, as demonstrated by an increase in firing activity in response to BIC treatment, which was similar to the one in WT neurons (Figure 5C). Finally, when we assessed changes in activity (of at least 20%) of individual CA1 neurons that were active before or after BIC application, we found that spike frequency was decreased in 82% of Ts65Dn CA1 neurons transduced with control amiR and was increased in only 5% of cells (Figure 5D). Conversely, 100% of individual CA1 neuron from WT animals showed increased spike frequency in response to BIC application (as expected). Interestingly, knockdown of NKCC1 expression in CA1 neurons with both amiR 1 and 2 against NKCC1 almost completely rescued GABAAR-mediated inhibitory signaling by reestablishing the expected BIC-induced firing increase in 97% and 91% of Ts65Dn CA1 neurons, respectively (Figure 5D).
Figure 5.

Neuro-specific hippocampal AAV9 delivery in vivo restores inhibitory GABAergic signaling in acute brain slices from Ts65Dn mice
(A) Schematic representation of the experimental timeline and of the hippocampal region of acute brain slices, showing the site of electrophysiological recordings. Two-3-month-old WT and Ts65Dn mice were stereotaxically injected in the dorsal hippocampal CA1 region with AAV9 vectors and analyzed by electrophysiology 4 WPIs. (B) Percentage of spontaneously active CA1 hippocampal neurons during baseline recordings for the different experimental groups. Numbers inside the bars indicate the number of recorded cells (4–5 animals/each experimental group) that were either active or silent during baseline recordings. ∗p < 0.05, Fisher’s exact test with the Sidak adjustment for multiple comparisons. (C) Quantification of the means ± SEM spontaneous spiking activity recorded in cell-attached, patch-clamp configuration in AAV9-transduced hippocampal CA1 neurons from WT and Ts65Dn mice before and after bath application of the GABAAR antagonist bicuculline (20 μM). (D) Example traces and spiking frequency of single, active CA1 neurons, from experiments in (C), in WT and Ts65Dn brain slices before (baseline) and during bath application of bicuculline. Numbers in parentheses indicate the number of active cells either before or after bicuculline application that were recorded for each experimental group; boxed numbers indicate the percentages of cells showing at least a 20% increase (↑) or decrease (↓) in spiking frequency after bicuculline application. ∗∗∗p < 0.001, Tukey post hoc test after two-way RMs ANOVA; ##p < 0.01, ###p < 0.001, Wilcoxon signed-rank test.
Neuro-specific anti-NKCC1 amiR treatment in vivo rescues learning and memory in Ts65Dn mice
Next, we evaluated cognitive performance in AAV9-injected mice with a battery of hippocampus-dependent behavioral tests at 4 to 6 weeks post injection (WPI; Figure 6A). First, we used the T-maze test to assess short-term working memory. Consistent with previous studies,37 Ts65Dn mice injected with control AAV9 showed reduced alternation behavior in this test in comparison with that of WT animals. However, NKCC1 knockdown with either amiR 1 or 2 completely restored short-term working memory performance in Ts65Dn mice (Figure 6B). Next, we used the contextual fear conditioning (CFC) test to evaluate associative memory. Ts65Dn mice that received control AAV9 displayed a strong reduction in their freezing response compared with that of WT littermates upon re-exposure to the adverse contest 24 h after conditioning.4,38 Interestingly, NKCC1 knockdown completely restored associative learning in Ts65Dn mice (Figure 6C), without inducing changes in non-associative freezing (i.e., exposure to a new context) or altering sensitivity to the shock (Figure S6A). Furthermore, we used the novel object recognition (NOR) test to evaluate long-term discriminative memory. Consistent with the results from previous studies,4,39 Ts65Dn mice injected with control AAV9 were unable to discriminate the novel object after a retention period of 24 h. However, NKCC1 knockdown with either amiR 1 or 2 completely restored NOR ability in Ts65Dn mice (Figure 6D). Finally, we evaluated the effect of NKCC1 knockdown on long-term spatial memory with the object location (OL) test. Ts65Dn mice injected with control AAV9 could not identify the new object location in relation to the available spatial information 24 h after the acquisition phase.4,38 However, after NKCC1 knockdown with either of the amiRs, the performance of Ts65Dn was indistinguishable from that of WT mice, indicating a complete recovery of spatial memory in trisomic animals (Figure 6E). Importantly, the effect of NKCC1 knockdown on Ts65Dn mice in both OL and NOR tests was independent of object exploratory behavior or object preference (Figures S6B and S6C). Moreover, NKCC1 knockdown did not cause changes in motor performance, which could have interfered with the evaluation of cognitive functions in the behavioral tests (Figure S7A).
Figure 6.

Neuro-specific hippocampal anti-NKCC1 amiR treatment restores cognitive function in Ts65Dn mice
(A) Left: schematic representation of the experimental timeline: 2–3-month-old WT and Ts65Dn mice were stereotaxically injected in the dorsal hippocampal CA1 region with AAV9 vectors expressing EGFP and control or NKCC1 amiRs from the neuron-specific hSyn promoter. Mice were tested in behavioral experiments between 4 and 6 WPIs. Right: symbol legend for panels (B)–(E). (B) Top, schematic representation of the T-maze test. Bottom, quantification of correct alternations in AAV-injected WT and Ts65Dn mice. (C) Top, schematic representation of the contextual fear-conditioning test. Bottom, quantification of the freezing response in AAV-injected WT and Ts65Dn mice. (D) Top, schematic representation of the novel-object recognition test. Bottom, quantification of the discrimination index in AAV-injected WT and Ts65Dn mice. (E) Top, schematic representation of the object-location test. Bottom, quantification of the discrimination index in AAV-injected WT and Ts65Dn mice. For (A)–(E): symbols indicate values from single animals; lines report group means ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, Tukey post hoc test after two-way ANOVA.
Altogether, the results of our behavioral assessment point to a causative role for NKCC1 upregulation in DS cognitive disability and indicate NKCC1 knockdown as a potential gene-therapy strategy for the rescue of learning deficits in different memory domains in trisomic animals.
Long-term efficacy of in vivo anti-NKCC1 amiR treatment in Ts65Dn mice
As a further step in developing a gene-therapy approach for the treatment of DS-related cognitive disabilities, we also evaluated long-term effects of chronic NKCC1 knockdown (Figure 7A). We injected WT and Ts65Dn mice with AAV9 expressing control or either of the two NKCC1 amiRs as above and confirmed normal motor performance at 5–6 months post injection (MPI; Figure S7B). Then, we assessed cognitive behaviors at 5–6 MPI in the different memory domains, as above. Similar to what we observed at 4–6 WPI, long-term NKCC1 knockdown rescued working memory in the T-maze test and associative learning in the CFC test at 5–6 MPI as well (Figures 7B and 7C), without inducing changes in non-associative freezing or shock sensitivity (Figure S8A). Moreover, object-recognition and spatial memories in the NOR and OL tests were also rescued in Ts65Dn mice at 5–6 MPI (Figures 7D, 7E, S8B, and S8C).
Figure 7.

Long-term anti-NKCC1 amiR treatment restores cognitive function in Ts65Dn mice
(A) Left: schematic representation of the experimental timeline: 2–3-month-old WT and Ts65Dn mice were stereotaxically injected in the dorsal hippocampal CA1 region with AAV9 vectors expressing EGFP and control or NKCC1 amiRs. Mice were tested in behavioral experiments between 5 and 6 months later. Right: symbol legend for (B)–(E). (B) Top: schematic representation of the T-maze test. Bottom, quantification of correct alternations in AAV-injected WT and Ts65Dn mice. (C) Top: schematic representation of the contextual fear-conditioning test. Bottom, quantification of the freezing response in AAV-injected WT and Ts65Dn mice. (D) Top: schematic representation of the novel-object recognition test. Bottom, quantification of the discrimination index in AAV-injected WT and Ts65Dn mice. (E) Top: schematic representation of the object-location test. Bottom, quantification of the discrimination index in AAV-injected WT and Ts65Dn mice. For (A)–(E), symbols indicate values from single animals; lines report group means ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, Tukey post hoc test after two-way ANOVA.
Thus, our results indicate the efficacy of long-term, AAV9-mediated, neuro-specific NKCC1 knockdown in rescuing cognitive deficits in Ts65Dn mice.
Discussion
In a time when neuroscience drug discovery programs in the pharmaceutical industry are often dismissed, a pre-requisite to any treatment-discovery effort is to identify a clear molecular target responsible for the phenotypic manifestations of the disorder. A number of studies have evaluated the contribution of various human chromosome 21 (HSA21)-derived genes to DS-related cognitive impairment by genetically restoring disomy of some triplicated genes or pharmacologically reducing their activity. Most of these studies have been performed in Ts65Dn mice, the most widely used trisomic model of DS carrying a freely segregating extra chromosome that contains ∼55% of the mouse ortholog genes to HSA21.40 Interestingly, much of the work on HSA21 genes has focused on a small set of triplicated orthologs in Ts65Dn mice: the amyloid-precursor protein (App),41, 42, 43 the G protein-coupled inwardly rectifying potassium channel 2 (Girk2/Kcnj6),37,44 the dual-specificity tyrosine-phosphorylation-regulated kinase 1A (Dyrk1A)45, 46, 47, the basic helix-loop-helix (bHLH) transcription factors Olig1/Olig2,48 and the phosphoinositide phosphatase synaptojanin 1 (Synj1).49 These studies have clearly shown that specific DS-related phenotypes critically arise from the triplication of different HSA21 dosage-sensitive genes in Ts65Dn mice.
However, transcriptome profiling in brains of both Ts65Dn mice and DS people have shown global gene-expression dysregulation of hundreds of non-triplicated transcripts as a secondary consequence of trisomy.50, 51, 52 This greatly complicates the understanding of the neurophysiological alterations underlining cognitive disabilities in DS.53 For example, although HSA21 does not contain any gene encoding for GABA receptors, a large body of evidence has pointed to altered signaling from GABAARs, and excitatory/inhibitory imbalance as major players in cognitive impairment in Ts65Dn mice.39,48,54, 55, 56 In that framework, we previously found that intracellular chloride accumulation shifts GABAAR-mediated currents from hyperpolarizing to depolarizing and dampens the efficacy of GABA-mediated inhibition in adult Ts65Dn mice.4 Here, we have shown that knockdown of NKCC1, which is upregulated in the brains of both Ts65Dn mice and DS people,4 restores [Cl−]i along with inhibitory GABAAR-mediated signaling in Ts65Dn hippocampal neurons in culture by patch-clamp and MEA recordings. Interestingly, the extent of aberrant GABAergic signaling in acute hippocampal slices from Ts65Dn animals was somehow greater than the one seen in the corresponding hippocampal cultures (compare the results in Figure 5D to the one in Figure 2C). However, anti-NKCC1 amiRs where able to efficiently rescue endogenous inhibitory GABAARs activity in an acute brain slice from AAV9-injected Ts65Dn animals as well. Altogether, these data established a causative role for NKCC1 upregulation and chloride dyshomeostasis on the alterations of GABAergic inhibitory transmission in DS. Moreover, in line with the role for GABAergic signaling in controlling neuronal network dynamics in WT animals,32, 33, 34 our MEA recordings showed that NKCC1 knockdown rescued the correlated activity of the hippocampal neuronal network of in vitro Ts65Dn cultures. Therefore, our results further support previous reports indicating a role for secondary dysregulation of non-triplicated genes in establishing different manifestation of the pathology.57,58
Similar to what we found in Ts65Dn mature neurons, high levels of neuronal [Cl−]i (and, therefore, depolarizing GABAAR-mediated signaling) also occurs under physiological conditions during brain development in WT animals1. However, after the first postnatal weeks in mice, [Cl−]i is less because of high expression of the chloride exporter KCC2, and GABAAR-mediated signaling turns hyperpolarizing and inhibitory. Interestingly, we found that hippocampal delivery of NKCC1 amiRs in adult mice did not induce compensatory changes in KCC2 expression that could also affect [Cl−]i. Notably, previous studies have indicated that the GABAAR-mediated developmental polarity switch from depolarizing to hyperpolarizing is delayed in Ts65Dn mice during neuronal maturation in culture,59 highlighting the possibility of a neurodevelopmental origin for depolarizing GABA signaling in DS. Nevertheless, our data show that neuron-specific NKCC1 knockdown in the hippocampus of adult Ts65Dn animals is sufficient to fully rescue cognitive deficits in four different behavioral tasks, indicating that the temporal window for intervention aimed in restoring chloride homeostasis exceeds the developmental period and extends into adulthood. On the other hand, modification of neuronal [Cl−]i can also occur as a response to changes in neural network activity or intracellular signaling.60, 61, 62, 63 Understanding whether increased NKCC1 expression in DS is a primary event directly determined by some of the HSA21-triplicated genes or an adaptive response to complex pathophysiological mechanisms and/or changes in neuronal network dynamics will need further experimental investigations.
In the context of the fast-growing number of animal models of diverse brain diseases characterized by altered chloride homeostasis—by either NKCC1 upregulation and/or KCC2 downregulation—and both clinical and pre-clinical studies showing positive outcomes upon treatment with the NKCC1 inhibitor bumetanide,3 a causative, direct link between altered expression of the chloride transporters and behavioral outcomes in DS was still missing. Moreover, bumetanide’s suboptimal brain pharmacokinetic profile27 has, in fact, raised further questions about its mechanism of action in animals when given systemically and, thus, about the validity of NKCC1 as an effective brain molecular target of the drug.28,64 Although our data do not directly support the engagement of NKCC1 in the pharmacological action of bumetanide, we demonstrate here that neuronal NKCC1 knockdown is sufficient to fully rescue cognitive impairment, at least in adult animals of the Ts65Dn mouse model of DS. This highlights a causative role of NKCC1 upregulation in learning and memory deficits in adult Ts65Dn mice, thus also validating brain NKCC1 as a target for ameliorating cognitive disabilities in DS. Furthermore, our neuro-specific knockdown approach points to neurons as major players in the NKCC1-dependent cognitive impairment in DS mice. Nevertheless, we cannot exclude the possibility that other cell types that also express NKCC1 (e.g., glial cells) could still have a role in the overall cognitive impairment that characterizes DS.
In addition to establishing a causal link between NKCC1 upregulation and cognitive impairment in DS, our data also provide a proof-of-concept for a neuro-specific RNAi gene-therapy approach to restore hippocampus-dependent cognitive behaviors in adult animals, specifically in the brain, and without affecting peripheral organs (e.g., the kidney). This is particularly relevant in the context of the current clinical trials repurposing the strong diuretic bumetanide to treat brain disorders with impaired chloride homeostasis.3 Importantly, we achieved a comparable degree of long-term cognitive rescue with two different amiR sequences against NKCC1, underlining the specificity of our approach. Among the pathologies characterized by dysregulated chloride homeostasis, we identified DS as the most promising for testing an NKCC1-knockdown approach. This is because, in contrast with other brain disorders,5, 6, 7,10,14, 15, 16, 17,19,21,65 the Ts65Dn mouse model and people with DS present upregulation of brain NKCC1, with no changes in KCC2. This makes the mechanistic interpretation of our data more straightforward.4 We designed our experiments to establish a relationship between NKCC1 knockdown and rescue of cognitive performance in DS. However, we cannot exclude the possibility that some indirect effects of the experimental procedures may have had specific outcomes on Ts65Dn animals. Moreover, for translational applications, further studies will be needed to assess the efficacy of knockdown using vectors devoid of fluorescent markers.66 Finally, the fact that RNAi using amiRs expressed from Pol II promoters is safer than traditional short hairpin RNA (shRNA)-mediated knockdown through Pol III promoters67 but still retains its effects for months—as demonstrated here—strongly strengthens the translational value of our investigation. Interestingly, one earlier study has explored the value of AAV-mediated knockdown of the HSA21-triplicated gene Dyrk1A in the hippocampus of Ts65Dn mice with a ubiquitously expressed shRNA through the U6 Pol III promoter. However, spatial memory deficits were only partially ameliorated after Dyrk1A knockdown.47 Conversely, our strategy with amiRs expressed from the neuro-specific hSyn Pol II promoter showed a complete recovery of behavioral performances in different cognitive domains, including spatial memory, upon hippocampal NKCC1 knockdown in Ts65Dn mice. Finally, by demonstrating that neuronal NKCC1 is a valid molecular target for restoring chloride homeostasis in DS and possibly other neurological disorders, the stage is set for future efforts to discover selective inhibitors of brain NKCC1 without the unfavorable diuretic/ionic imbalance effect, which is due to inhibition of the kidney-expressed, cognate transporter NKCC2.68,69
In summary, our study has established a mechanistic link between upregulation of NKCC1 and cognitive impairment in DS and has proven that NKCC1 can be a molecular target for selective downregulation with either gene-therapy interventions and/or chemical drug inhibitors to rescue cognitive disabilities in individuals with DS. Finally, our results support the possibility that other brain disorders featuring altered chloride homeostasis may benefit from similar gene therapy and/or pharmacological approaches in adulthood.
Materials and methods
Primary neuronal culture
Primary neuronal cultures were prepared from WT and Ts65Dn pups at postnatal day 2 (P2), as previously described.70,71 In brief, brains were dissected under a stereomicroscope in ice-cold dissection buffer (DB) composed of Hank’s balanced-salt solution (HBSS; Gibco) supplemented with 6 mg/mL glucose, 3 mg/mL bovine serum albumin (BSA), 5.5 mM MgSO4, 5 μg/mL gentamycin, and 10 mM HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid), pH 7.4 (all from Sigma Pharmaceuticals). Cortical or hippocampal tissue was minced and then enzymatically digested with 0.25% trypsin in HBSS (Gibco) containing 0.6 mg/mL Deoxyribonuclease I (DNase; Sigma Pharmaceuticals) for 5 min at 37°C. Tissue chunks were washed in DB, incubated for 5 min in DB supplemented with 1 mg/mL of soybean-trypsin inhibitor (Sigma Pharmaceuticals), and mechanically dissociated in DB supplemented with 0.6 mg/mL DNase. Cells were passed through a 40-μm cell strainer and then centrifuged (110 × g for 7 min at 4°C) to remove cellular debris. Cells were plated on glass coverslips, 6-wells plates, or MEAs coated with poly-l-lysine (Sigma; 0.1 mg/mL in 100 mM borate buffer, pH 8.5) at a density of 250–500 cells/mm2. Neurons were maintained in a culture medium consisting of Neurobasal-A supplemented with 2% B-27, 1% GlutaMax, and 5 μg/mL gentamycin (all from Gibco) at 37°C in humidified atmosphere (95% air, 5% CO2).
RNAi
The target sequences for mouse NKCC1 (SLC12A2, GenBank: NM_009194) used to design amiRs for RNAi were selected with BLOCK-iT RNAi Designer (Invitrogen). The two amiR sequences against mouse NKCC1 used in the study (NKCC1 amiR 1: 5′-TTAACATGCAGCGGACTAATA-3′ and NKCC1 amiR 2: 5′-ATCACCAGCAGCACAATCTGA-3′) were first cloned into the pcDNA6.2-GW/EmGFP-miR plasmid using the BLOCK-iT Pol II miR RNAi kit (Invitrogen), as previously described.72,73 The expression cassette, thus, consisted of a 5′-miR flanking region, a target-specific stem-loop amiR sequence, and a 3′-miR flanking region. This amiR cassette can be expressed from the 3′ untranslated region (UTR) of any reporter gene under the control of an RNA polymerase type II promoter.29 As a negative control, we used the control amiR sequence from pcDNA6.2-GW/EmGFP-miR-neg-control plasmid (provided with the kit) containing a sequence that did not target any known vertebrate gene (control amiR: 5′-AAATGTACTGCGCGTGGAGAC-3′). The amiR expression cassettes were then sub-cloned in LV plasmids derived from pCCL.sin.cPPT.PGK.GFP.WPRE74 (a kind gift by L. Naldini, TIGET, San Raffaele Scientific Institute, Milan, Italy) downstream of the EGFP under the control of the human phosphoglycerate kinase promoter 1 (hPGK1) or downstream of the red fluorescent protein mCherry under the control of the neuro-specific hSyn promoter. Similarly, the amiR cassettes were cloned downstream of EGFP under the control of the hSyn promoter in an AAV vector plasmid derived from pAAV-hSyn-EGFP (Addgene, no. 50465).
Viral preparations
Vesicular stomatitis virus glycoprotein G (VSV-G)-pseudotyped third-generation LVs were produced by transient co-transfection into HEK293T cells using the calcium-phosphate-transfection method. Supernatants were collected, passed through a 0.45-μm filter and purified by ultracentrifugation, as previously described.72,74 LVs were titrated at concentrations ranging from 5 × 108 to 1 × 109 transducing units (TU)/mL by fluorescence-activated cell sorting (FACS) analysis on HEK293T or Neuro2a cells by the limiting-dilution method. Neuronal cultures were infected at 7 DIVs with 5–10 multiplicity of infection (MOI) and used for experiments between 15 and 20 DIVs. The efficiency of infection was estimated to range between 80% and 95% of cells. AAV9 used in this study was produced with a slight modification of the adenovirus-free transient-transfection methods previously described.75,76 Briefly, adherent HEK293 cells grown in roller bottles were transfected with three plasmids containing the adenovirus helper proteins, the AAV Rep and Cap genes, and the inverted terminal repeat (ITR)-flanked transgene expression cassette. Three days after transfection, cells were harvested, lysed by sonication, and treated with benzonase (Merck Millipore). Vectors were purified using two successive ultracentrifugation rounds in cesium-chloride density gradients. Full capsids were collected. The final product was formulated in sterile phosphate-buffered saline (PBS) containing 0.001% of pluronic F-68 (Sigma Pharmaceuticals), and stored at −80°C. Titers of the AAV vector stocks were determined by SDS-PAGE followed by SYPRO Ruby protein gel stain and band densitometry.
Biochemistry
Primary neurons were transduced at 7 DIV and used for WB analysis at 15 DIVs. For WB analysis of AAV-transduced animals, WT and Ts65Dn brains at 4 WPI were dissected in ice-cold PBS, and the dorsal CA1 region was isolated under a fluorescent stereomicroscope. All samples were lysed in ice-cold RIPA buffer (1% NP40, 0.5% deoxycholic acid, 0.1% SDS, 150 mM NaCl, 1 mM EDTA, and 50 mM Tris; pH 7.4) containing 1 mM PMSF, 10 mM NaF, 2 mM sodium orthovanadate, and 1% (v/v) protease and phosphatase inhibitor cocktails (Sigma Pharmaceuticals). Samples were clarified through centrifugation at 20,000 × g at 4°C, and the protein concentration was determined using the BCA kit (Pierce Chemical). For immunoblot analysis, protein extracts were prepared in lithium-dodecyl-sulfate (LDS) sample buffer (Thermo Fisher Scientific) containing 50 mM dithiothreitol (DTT). To avoid NKCC1 protein aggregation/precipitation, samples were not heat treated before loading.77,78 Equal amounts of proteins were run on 4%–12% Bis-Tris, NuPAGE (Thermo Fisher Scientific) or Criterion-XT (Bio-Rad) gels with MOPS (3-(N-morpholino)propanesulfonic acid) buffer and transferred overnight at 4°C onto nitrocellulose membranes (GE Healthcare) with Tris-glycine transfer buffer (25 mM Tris-base, 192 mM glycine, and 20% methanol). Membranes were probed with mouse anti-NKCC1 (clone T4c, Developmental Studies Hybridoma Bank; 1:4,000), rabbit anti-KCC2 (Millipore, catalog no. 07-432; 1:4,000), goat anti-GFP (Santa Cruz Biotechnology, catalog no. sc-5385; 1:1,000), and rabbit anti-actin (Sigma Pharmaceuticals, catalog no. A2066; 1:10,000), followed by horseradish peroxide (HRP)-conjugated goat anti-mouse, goat anti-rabbit, or rabbit anti-goat secondary antibodies (Thermo Fisher Scientific; 1:5,000). Chemiluminescent signals were revealed with SuperSignal West Pico substrate (Pierce Chemical) and digitally acquired on an LAS 4000 Mini imaging system (GE Healthcare). Band intensities were quantified using ImageQuant software (GE Healthcare). Specificity of the anti-NKCC1 and anti-KCC2 antibodies was previously verified on brain samples from NKCC1- and KCC2-deficient mice.4
MQAE intracellular chloride imaging
Imaging of intracellular Cl− in hippocampal neurons was performed with the fluorescent chloride-sensitive indicator MQAE, as previously described.4,69 MQAE dye detects Cl− ions via diffusion-limited collisional quenching, resulting in a concentration-dependent decrease of fluorescence emission after an increase in Cl− concentration.79 Therefore, a decrease in MQAE fluorescence is indicative of a higher [Cl−]i and vice versa. Hippocampal neurons at 15 DIVs transduced with LV-expressing amiRs and mCherry were loaded with 5 mM MQAE (Molecular Probes) for 30 min at 37°C. Coverslips were then transferred to a holding chamber and perfused (2 mL/min) with extracellular solution (NaCl 145 mM, KCl 5 mM, CaCl2 2 mM, MgCl2 1 mM, HEPES 10 mM, and d-glucose 5.5 mM; pH 7.4) at 25°C for 5 min before imaging. Images were taken with a Nikon A1 scanning confocal microscope equipped with a 20× air objective (numerical aperture [NA] 0.75). MQAE was excited with a 405-nm diode laser, and fluorescence collected with a 525/50-nm band-pass emission filter. For concomitant mCherry imaging, cells were excited with a 561-nm solid-state laser, and fluorescence collected with a 595/50-nm band-pass emission filter. All excitation and acquisition parameters (laser intensity, photomultiplier tube [PMT] offset, and gain) were keep constant throughout experiments. Image analysis was performed with NIS-Elements software (Nikon) by measuring the mean fluorescent intensity of regions of interest (ROIs) on the cell body of individual neurons expressing mCherry from six randomly selected fields for each coverslip. For each experiment, the average fluorescent intensity of all ROIs from a coverslip was normalized to the average fluorescent intensity of the control samples (WT neurons expressing control amiR) in the same experiment. Pseudo-color images shown in Figure 1D were generated with ImageJ software (https://rsbweb.nih.gov/ij/).
Electrophysiology
For measuring spontaneous action potential firing of LV-transduced hippocampal neurons, coverslips were mounted in a holding chamber and perfused (∼2–3 mL/min) with extracellular solution (as above). Cell-attached recordings were performed with glass micropipettes (resistance, 2–5 MΩ) filled with the same extracellular solution. After obtaining a gigaΩ seal, baseline spiking was recorded for 10 min; after which, cells were perfused with extracellular solution containing increasing concentrations of GABA (1–200 μM) or BIC (10 μM) and recorded for an additional 10 min after each addition.
For measuring spontaneous action-potential firing of CA1 hippocampal neurons in acute brain slices, mice were anesthetized with isoflurane and transcardially perfused with ice-cold cutting solution containing 220 mM sucrose, 7 mM MgCl2, 2.5 mM KCl, 1.25 mM NaH2PO4, 0.5 mM CaCl2, 25 mM NaHCO3, and 10 mM d-glucose (∼300 mOsm; pH 7.4; oxygenated with 95% O2 and 5% CO2). Brains were removed, and 270-μm-thick coronal slices were cut with a VT1000S vibratome (Leica Microsystem) in the same solution. Slices were allowed to recover for 20 min at 35°C in a solution containing 117 mM NaCl, 2.5 mM KCl, 1.25 mM NaH2PO4, 3 mM MgCl2, 0.5 mM CaCl2, 25 mM NaHCO3, and 10 mM glucose (∼310 mOsm; pH 7.4; oxygenated with 95% O2 and 5% CO2). All recordings were performed at room temperature in an holding chamber perfused with artificial cerebrospinal fluid (ACSF; ∼2–3 mL/min) composed of 117 mM NaCl, 3 mM KCl, 1.25 mM NaH2PO4, 1 mM MgCl2, 2 mM CaCl2, 25 mM NaHCO3, and 10 mM glucose (∼310 mOsm; pH 7.4; oxygenated with 95% O2 and 5% CO2). Cell-attached recordings were performed with glass micropipettes (resistance, 4–6 MΩ) filled with ACSF. After obtaining a gigaΩ seal, baseline spiking were recorded for 5 min; after which, cells were perfused with BIC (20 μM) and recorded for additional 10–15 min.
All cell-attached recordings were performed in the voltage-clamp mode at the holding potential (V0), providing an average baseline current of 0 pA, as previously described.4,30 Recordings were discarded if the seal resistance was <1 GΩ. Data, filtered at 0.1 Hz and 5 kHz and sampled at 25 kHz, were acquired with a patch-clamp amplifier (Multiclamp 700B, Molecular Devices), and analyzed with pClamp 10.2 software (Molecular Devices). For the analysis shown in Figures 2C, 2E, and 5D, we only considered cells that were active either before or after drug administration (i.e., neurons showing a firing activity greater than 0.01 Hz).
MEA recordings
Hippocampal neurons at 20–21 DIVs were grown on MEAs consisting of 60 TiN/SiN planar, round electrodes (30 μm diameter; 200 μm inter-electrode distance) divided into six separated wells (Multichannel Systems). Each well contained nine recording electrodes, arranged in a 3 × 3-square grid, and one big ground electrode. The activity of all cultures was recorded by means of the MEA60 System (Multichannel Systems). After 1,200× amplification, signals were sampled at 10 kHz and acquired through the data acquisition card and MC-Rack software (Multichannel Systems). To avoid any stress on the cell, recordings were performed on neuronal cultures maintained in their original medium.80, 81, 82 Cultures were transferred to the recording setup and acclimatized for 20 min before beginning the recordings to allow the cultures to adapt to the new environment and reach a stable level of activity.80, 81, 82 The recording setup was equipped with a controlled thermostat (Multichannel Systems) and a custom-made incubator chamber to maintain cells at 37°C in a humidified atmosphere consisting of 95% air and 5% CO2.80, 81, 82 Basal spontaneous network activity of cultures was then recorded for 30 min. After recording basal activity, BIC or GABA (both 20 μM; Sigma Pharmaceuticals) were applied, and activity was recorded for an additional 40 min. The drugs were added to the bath solution by directly pipetting in the medium. Because we noticed that mechanical perturbation from the pipette injection in the medium could cause a temporary instability in the firing rate, we discarded the first 10 min at the beginning of the 40-min recording phase.
Data analysis was performed offline with MATLAB software (MathWorks) using the custom software package SPYCODE.83 The steps in the analysis are described briefly below. Raw traces were high-pass filtered (>300 Hz) to isolate spike events from the low fluctuation of the signal (low field potential [LFP]). We computed the spike detection offline using a custom software, as previously described,84 to discriminate spike events from the noise. The algorithm is based on three parameters: (1) a differential threshold (DT) set independently for each channel as 8-fold the standard deviation (SD) of the noise, (2) the signal peak lifetime period (PLP) set to 2 ms, and (3) the refractory period set to 1 ms. To characterize the activity level of the recorded cultures, we computed the MFR for each MEA, which was defined as the mean number of spikes/s over the total recording time. We considered active electrodes as those showing a firing rate greater than 0.02 spikes/s. This low threshold guarantees that only electrodes that are not covered by cells and those with very few spikes are excluded and that all others are retained. Spontaneous bursting activity was detected using a custom burst-detection method.85, 86, 87 Briefly, the algorithm used two thresholds to detect burst events: (1) the maximum inter-spike interval (ISI) allowed for spikes within a burst (maxISI, set at 50 ms), and (2) the minimum number of consecutive spikes belonging to a burst (minSpikes, set at 4). We considered bursting electrodes as those showing a bursting rate greater than 0.2 burst/min. Once bursts were detected for each channel, we evaluated the MBR for each MEA, which was defined as mean number of burst/min over the entire recording.
To assess the synchronicity of the neuronal networks, we calculated the STTCs31 between each pair of spike trains recorded from all active channels (i.e., MFR higher than 0.02 spikes/s) in an MEA. For that analysis, we considered only MEAs that showed a percentage of bursting channels (i.e., MBR higher than 0.2 burst/min) equal to or greater than 50% of the active channels. Within these subset of recordings, we further excluded MEAs showing correlation values lower than 0.01 (STTCs correlation values range from 0 to 1) for more than 18 of 36 (50%) possible couples of connections (thus excluding MEAs with extremely low synchronicity). Statistical analysis was conducted by considering mean STTCs values for each electrode versus all other electrodes in the MEA.
To evaluate the effect of the drug administration on the firing-rate activity, we computed the %MFRvar for each active electrode as follows:
Statistically significant changes in firing rate induced by drug treatments (either GABA or BIC) with respect to the basal condition were assessed by bootstrap analysis.35 In brief, the peak train for each time segment (basal or drug) was divided into 1-min bins. The MFRs recorded during each 1-min bin for the two time segments for each well were randomly shuffled into two groups for 10,000 times. The differences between the means of the randomly shuffled groups produced a null distribution. For each channel, we computed the real difference between the basal and the drug values and assessed whether it fell outside the 95% confidence interval of the null distribution. The beanplots shown in Figure 3 were generated with R package (http://cran.r-project.org/web/packages/beanplot/index.html).
Animals and treatment
The Ts65Dn colony40 was maintained by crossing Ts65Dn females to C57BL/6JEi x C3SnHeSnJ (B6EiC3) F1 males (Jackson Laboratory, Bar Harbor, ME, USA). Animals were genotyped by PCR as previously described.88 A veterinarian was employed to monitor the health and comfort of the animals. Mice were housed in a temperature-controlled room with a 12:12-h dark/light cycle and ad libitum access to water and food. Eight to 12-week-old, male Ts65Dn and WT control littermates were randomly assigned to the different experimental groups (control amiR or NKCC1 amiRs 1 and 2). For stereotaxic surgery, animals were anesthetized with 2% isoflurane and positioned into a digital stereotaxic frame (Stoelting) over a heating pad (∼37°C). After exposing the skull, small holes were drilled bilaterally at the following coordinates relative to bregma:89 antero-posterior, −1.94 mm; lateral, ±1.20 mm. AAV9 injections into the dorsal CA1 hippocampal region of both hemispheres was performed at −1.3 mm from the cortical surface with pulled-glass capillaries connected to a Hamilton syringe mounted on a digital infusion micro-pump (Harvard Apparatus). For each injection site, mice received 1 μL (flow rate, 0.1 μl/min) of AAV9 suspensions (5 × 1012 viral genomes [vg]/mL). After completing the infusion, the capillary was maintained in place for 10 min, and the skin was sutured upon its removal. After surgery, the animals were maintained under a heat lamp until recovery. Separate cohorts of AAV-injected animals were used for biochemical analysis and behavioral testing.
Immunofluorescence and cell counting
Animals were deeply anesthetized and transcardially perfused with 4% paraformaldehyde in 100 mM phosphate buffer (PB), pH 7.4. Brains were collected, post-fixed for 24 h in the same fixative solution, washed in PB, cryo-preserved in 30% sucrose in PB, and stored at −80°C until use. Immunohistochemistry was performed on 30-μm free-floating coronal sections. Cultured neurons were fixed for 20 min with 4% paraformaldehyde in PB and then washed extensively in PBS. For immunofluorescence, samples were permeabilized in PBS containing 0.1% Triton X-100 (PBST), incubated for 1 h with blocking buffer (5% normal goat serum in PBST) and incubated overnight at 4°C with the following primary antibodies: mouse anti-NeuN (clone A60, Millipore, catalog no MAB377; 1:200); guinea pig anti-Map2 (SySy, catalog no188-004; 1:600); mouse anti-GFAP (clone GA5, Sigma Pharmaceuticals, catalog no G6171; 1:400); rabbit anti-Iba1 (Wako Pure Chemicals Industry, catalog no 019-19741; 1:400). Fluorophore conjugated (Alexa Fluor 488, Alexa Fluor 568, and Alexa Fluor 647), goat secondary antibodies (1:500; Thermo Fisher Scientific) were used for detection. Samples were counterstained with the nuclear dye Hoechst-33342 (Sigma Pharmaceuticals). Fluorescent images of brain slices were acquired with a Leica SP5 confocal scanning microscope equipped with a 40× oil-immersion objective, or with a Nikon Eclipse wide-field fluorescent microscope equipped with a 4× air objective and a DS-Ri1 digital camera (Nikon). Fluorescent images of neurons in culture were acquired with a Nikon A1 confocal scanning microscope equipped with a 20× air objective.
The percentage of EGFP+ cells in the neuronal culture was assessed in four randomly selected images for each coverslip. For each image, an ROI containing at least 20 cells was selected and the percentage of EGFP+ cells over total cells was counted with NIS-Elements software (Nikon).
The number of EGFP and NeuN-double-positive cells over total EGFP+ cells was counted with NIS-Elements software in the dorsal CA1 pyramidal layer of AAV9-injected WT and Ts65Dn animals at 6 WPIs. For each image, at least 20 EGFP+ cells were analyzed.
Behavioral testing
Behavioral tests were performed as previously described.4,38,90 Behavioral testing was performed at two time points after AAV9 stereotaxic delivery on separate groups of animals. One group of animals was evaluated between 4 and 6 WPIs and another between 5 and 6 MPIs. Except where otherwise stated, animals were subjected to behavioral tests in the following order: NOR test, T-maze spontaneous alternation test, OL test, CFC test. Animal behavior was video-recorded throughout the experimental sessions and was subsequently analyzed by a trained operator blinded to both genotype and treatment group. The NOR, OL, and T-maze tests were performed in dim-light illumination (12–14 Lux), whereas the CFC test was conducted with standard illumination (80 Lux). WT and Ts65Dn mice were always evaluated in parallel and with the same time schedule. To minimize olfactory cues from previous trials, all apparatus used were thoroughly cleaned with 70% ethanol and dried after each animal. Three days before starting the behavioral experiments, mice were handled once a day for 5 min each.
NOR test
This test measures the preference of mice for a novel object versus previously encountered familiar objects. The test was conducted in a gray acrylic arena (44 × 44 cm) during 3 consecutive days. On the first day, mice were habituated to the apparatus by freely exploring the empty arena for 15 min. The next day (acquisition session), three different objects were placed into the arena, and mice were allowed to explore for 15 min. Object preference was evaluated during this session. The objects used during the test were different in shape, color, size, and material. On the third day (trial session), one of the objects used during the acquisition session was replaced by a novel object, and the mice were allowed to freely explore the objects for 15 min. The position of the new and old objects was counterbalanced between mice and sessions. Exploratory behavior of the object was defined as a direct contact with the object by the animal mouth, nose, or paws or if the animal’s nose was within 1 cm of an object (sniffing); any indirect or accidental contact with the objects was not included in the scoring. The discrimination index was calculated as the difference between the percentages of time spent investigating the novel object and the time spent investigating the familiar objects: Discrimination index = (New object exploration time/Total exploration time × 100%) − (Familiar object exploration time/Total exploration time × 100%).
T-maze spontaneous alternation test
The test was conducted similarly to what has previously been described for Ts65Dn mice to measure short-term working memory.37 The T-maze apparatus was made of black, opaque, acrylic plastic and consisted of a start arm and two perpendicular goal arms, each equipped with a sliding door. At the beginning of the trial, a mouse was confined in the start chamber, and after 20 s, the sliding door was removed. After the mouse had entered (with all four limbs) in one of the two goal arms, the opposite goal arm was closed with the corresponding sliding door. The mouse was left to explore the remaining part of the apparatus. As the animal spontaneously returned to the starting location, the previously closed arm was opened. The mouse was then given the possibility to choose again one of the two goals arm. The procedure was repeated 11 times (for a total of 10 possible alternations). Entry into a goal arm opposite to the one chosen before was defined as a correct alternation, whereas entry into the arm visited before was scored as an incorrect alteration. Alternation score was defined as the percentage of correct alternations (i.e., left to right or right to left) over the total number of possible alternations.
OL test
This test measures spatial memory by evaluating the ability of mice to recognize the new location of familiar object with respect to spatial external cues. The test was conducted in a gray, acrylic arena (44 × 44 cm) during three consecutive days. On the first day, mice were habituated to the empty arena for 15 min. The next day, mice were exposed to two identical objects for 15 min (acquisition session). Object preference was evaluated during this session. On the third day (trial session), one of the objects was moved to a novel location, and the mice were allowed to explore the objects for 15 min. Exploratory behavior of the objects was defined as a direct contact with the object by the animal mouth, nose, or paws or if the animal’s nose was within 1 cm of an object (sniffing); any indirect or accidental contact with the objects was not included in the scoring. The discrimination index was calculated as the percentage of time spent investigating the object in the new location minus the percentage of time spent investigating the object in the old location: Discrimination index = (New OL exploration time/Total exploration time × 100%) − (Old OL exploration time/Total exploration time × 100%).
Motor activity
The distance traveled by mice in the empty arena during the habituation sessions of the NOR and OL tests was measured with ANY-maze tracking software (Stoelting).
CFC test
The fear-conditioning system (TSE System) consisted of a transparent, acrylic conditioning chamber (20 × 10 cm) equipped with a stainless-steel grid. Mice were held outside the experimental room in their home cages before testing and were individually transported to the conditioning apparatus in standard cages. For the conditioning session, mice were placed in the chamber, and 3 min later, they received one electric shock (2 s, 0.75 mA constant electric current) through the floor grid. Mice were removed 15 s after the shock. Twenty-four hours later, mice were placed in the same chamber for 3 min (context test), and after 2 h placed in a new context (black chamber with plastic black floor and vanilla odor) for additional 3 minutes. The freezing behavior was scored by a trained operator blinded to the experimental groups.
In behavioral experiments, we adopted the following exclusion criteria independent of genotype or treatment (before blinded code break). In the T-maze test, we excluded mice not concluding the 10 trials within 10 min of the test (3 of 129 mice). In the CFC test, we excluded mice showing very high non-associative freezing in the new context: more than 30 s freezing during the 3-min test (4 of 128 mice). In the NOR and OL tests, we excluded animals showing very low explorative behavior: less than 10 s of direct objects exploration during the 15-min test (2 of 120 mice for the OL test and 1 of 129 mice for NOR test).
After completing the behavioral assessment, animals were sacrificed for immunohistochemistry analysis or AAV9 bio-distribution studies.
VGCN analysis
Total DNA was extracted from tissues using the MagNA Pure 96 DNA and viral NA small volume kit (Roche Diagnostics), according to the manufacturer’s instructions. VGCN was measured by qPCR and normalized by the copies of Titin gene measured in each sample. qPCR was performed on a LightCycler 480 (Roche Diagnostics) using SybrGreen mix (Thermo Fisher Scientific) and the following specific primers: GFP forward 5′-GGTGAACTTCAAGATCCGCC-3′, GFP reverse 5′-CTTGTACAGCTCGTCCATGC-3′, Titin forward 5′-AAAACGAGCAGTGACGTGAGC-3′, Titin reverse 5′-TTCAGTCATGCTGCTAGCGC-3′.
Statistical analysis
Except were otherwise stated, the results are presented as the means ± SEM. Statistical analysis was performed using SigmaPlot (Systat) or GraphPad (Prism) software. Where appropriate, the statistical significance was assessed using the following parametric test: paired Student’s t test, two-way ANOVA, or two-way repeated-measures (RMs) ANOVA, followed by all pairwise Turkey’s post hoc test. In cases in which normal distribution or equal variance assumptions were not valid, statistical significance was evaluated with a Wilcoxon signed-rank test (non-parametric) or two-way ANOVA and two-way RMs-ANOVA on ranked, transformed data. Percentages of spontaneously active neurons were analyzed by Fischer’s exact test with Sidak adjustment for multiple comparisons. Assessments of significant correlations shown in Figure S1C were analyzed by a Pearson’s test. p < 0.05 was considered significant.
Ethical approval declaration
All animal experiments were performed in accordance with the guidelines established by the European Community Council Directive 2010/63/EU of September 22, 2010, and were approved by the Italian Ministry of Health (authorization no. 829/2015-PR to A.C.).
Acknowledgments
This work was supported by Telethon (grant TCP15021 to L.C.). This project received partial funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (grant no. 725563 to L.C.). We thank the IIT animal facility staff for their valuable work. We thank Fabio Benfenati (Italian Institute of Technology and University of Genova) for his support.
Author contributions
M.P. collected and analyzed the behavioral and immunohistochemistry data; M.P., M.A., and F.P. performed stereotaxic injections; S.N. collected and analyzed the electrophysiology data in cultured neurons; G.M. collected and analyzed the electrophysiology data in brain slices; M.A. collected and analyzed the biochemical data; I.C. collected and analyzed the multi electrode array data; M.A. and A.C. collected and analyzed the chloride-imaging data; M.A. and A.R. produced the LVs; M.A. and M.N. prepared primary neuronal cultures; F.M. and G.R. provided the AAV9 vectors and collected and analyzed the AAV bio-distribution data; A.C. conceived the study, designed the experiments, and wrote the manuscript. L.C. supervised the experiments and wrote the manuscript. All authors read and revised the manuscript.
Declaration of interests
A.C. and L.C. are named as co-inventors on granted patent US 9822368, EP 3083959, and JP 6490077. G.R., F.M., A.C., and L.C. are named as co-inventors on the patent application WO 2018/189225. A.C. and L.C. are named as co-inventors on patent application IT 102019000004929.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2021.05.023.
Contributor Information
Andrea Contestabile, Email: andrea.contestabile@iit.it.
Laura Cancedda, Email: laura.cancedda@iit.it.
Supplemental information
References
- 1.Ben-Ari Y. Excitatory actions of GABA during development: the nature of the nurture. Nat. Rev. Neurosci. 2002;3:728–739. doi: 10.1038/nrn920. [DOI] [PubMed] [Google Scholar]
- 2.Raimondo J.V., Richards B.A., Woodin M.A. Neuronal chloride and excitability—the big impact of small changes. Curr. Opin. Neurobiol. 2017;43:35–42. doi: 10.1016/j.conb.2016.11.012. [DOI] [PubMed] [Google Scholar]
- 3.Ben-Ari Y. NKCC1 chloride importer antagonists attenuate many neurological and psychiatric disorders. Trends Neurosci. 2017;40:536–554. doi: 10.1016/j.tins.2017.07.001. [DOI] [PubMed] [Google Scholar]
- 4.Deidda G., Parrini M., Naskar S., Bozarth I.F., Contestabile A., Cancedda L. Reversing excitatory GABAAR signaling restores synaptic plasticity and memory in a mouse model of Down syndrome. Nat. Med. 2015;21:318–326. doi: 10.1038/nm.3827. [DOI] [PubMed] [Google Scholar]
- 5.Tyzio R., Nardou R., Ferrari D.C., Tsintsadze T., Shahrokhi A., Eftekhari S., Khalilov I., Tsintsadze V., Brouchoud C., Chazal G. Oxytocin-mediated GABA inhibition during delivery attenuates autism pathogenesis in rodent offspring. Science. 2014;343:675–679. doi: 10.1126/science.1247190. [DOI] [PubMed] [Google Scholar]
- 6.Banerjee A., Rikhye R.V., Breton-Provencher V., Tang X., Li C., Li K., Runyan C.A., Fu Z., Jaenisch R., Sur M. Jointly reduced inhibition and excitation underlies circuit-wide changes in cortical processing in Rett syndrome. Proc. Natl. Acad. Sci. USA. 2016;113:E7287–E7296. doi: 10.1073/pnas.1615330113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tang X., Kim J., Zhou L., Wengert E., Zhang L., Wu Z., Carromeu C., Muotri A.R., Marchetto M.C., Gage F.H., Chen G. KCC2 rescues functional deficits in human neurons derived from patients with Rett syndrome. Proc. Natl. Acad. Sci. USA. 2016;113:751–756. doi: 10.1073/pnas.1524013113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lozovaya N., Nardou R., Tyzio R., Chiesa M., Pons-Bennaceur A., Eftekhari S., Bui T.T., Billon-Grand M., Rasero J., Bonifazi P. Early alterations in a mouse model of Rett syndrome: the GABA developmental shift is abolished at birth. Sci. Rep. 2019;9:9276. doi: 10.1038/s41598-019-45635-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.He Q., Arroyo E.D., Smukowski S.N., Xu J., Piochon C., Savas J.N., Portera-Cailliau C., Contractor A. Critical period inhibition of NKCC1 rectifies synapse plasticity in the somatosensory cortex and restores adult tactile response maps in fragile X mice. Mol. Psychiatry. 2019;24:1732–1747. doi: 10.1038/s41380-018-0048-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.He Q., Nomura T., Xu J., Contractor A. The developmental switch in GABA polarity is delayed in fragile X mice. J. Neurosci. 2014;34:446–450. doi: 10.1523/JNEUROSCI.4447-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Talos D.M., Sun H., Kosaras B., Joseph A., Folkerth R.D., Poduri A., Madsen J.R., Black P.M., Jensen F.E. Altered inhibition in tuberous sclerosis and type IIb cortical dysplasia. Ann. Neurol. 2012;71:539–551. doi: 10.1002/ana.22696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Amin H., Marinaro F., De Pietri Tonelli D., Berdondini L. Developmental excitatory-to-inhibitory GABA-polarity switch is disrupted in 22q11.2 deletion syndrome: a potential target for clinical therapeutics. Sci. Rep. 2017;7:15752. doi: 10.1038/s41598-017-15793-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Corradini I., Focchi E., Rasile M., Morini R., Desiato G., Tomasoni R., Lizier M., Ghirardini E., Fesce R., Morone D. Maternal immune activation delays excitatory-to-inhibitory gamma-aminobutyric acid switch in offspring. Biol. Psychiatry. 2018;83:680–691. doi: 10.1016/j.biopsych.2017.09.030. [DOI] [PubMed] [Google Scholar]
- 14.Nardou R., Yamamoto S., Chazal G., Bhar A., Ferrand N., Dulac O., Ben-Ari Y., Khalilov I. Neuronal chloride accumulation and excitatory GABA underlie aggravation of neonatal epileptiform activities by phenobarbital. Brain. 2011;134:987–1002. doi: 10.1093/brain/awr041. [DOI] [PubMed] [Google Scholar]
- 15.Huberfeld G., Wittner L., Clemenceau S., Baulac M., Kaila K., Miles R., Rivera C. Perturbed chloride homeostasis and GABAergic signaling in human temporal lobe epilepsy. J. Neurosci. 2007;27:9866–9873. doi: 10.1523/JNEUROSCI.2761-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pallud J., Le Van Quyen M., Bielle F., Pellegrino C., Varlet P., Cresto N., Baulac M., Duyckaerts C., Kourdougli N., Chazal G. Cortical GABAergic excitation contributes to epileptic activities around human glioma. Sci. Transl. Med. 2014;6:244ra89. doi: 10.1126/scitranslmed.3008065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Palma E., Amici M., Sobrero F., Spinelli G., Di Angelantonio S., Ragozzino D., Mascia A., Scoppetta C., Esposito V., Miledi R., Eusebi F. Anomalous levels of Cl- transporters in the hippocampal subiculum from temporal lobe epilepsy patients make GABA excitatory. Proc. Natl. Acad. Sci. USA. 2006;103:8465–8468. doi: 10.1073/pnas.0602979103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eftekhari S., Mehvari Habibabadi J., Najafi Ziarani M., Hashemi Fesharaki S.S., Gharakhani M., Mostafavi H., Joghataei M.T., Beladimoghadam N., Rahimian E., Hadjighassem M.R. Bumetanide reduces seizure frequency in patients with temporal lobe epilepsy. Epilepsia. 2013;54:e9–e12. doi: 10.1111/j.1528-1167.2012.03654.x. [DOI] [PubMed] [Google Scholar]
- 19.Ferrini F., Trang T., Mattioli T.A., Laffray S., Del’Guidice T., Lorenzo L.E., Castonguay A., Doyon N., Zhang W., Godin A.G. Morphine hyperalgesia gated through microglia-mediated disruption of neuronal Cl− homeostasis. Nat. Neurosci. 2013;16:183–192. doi: 10.1038/nn.3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim H.R., Rajagopal L., Meltzer H.Y., Martina M. Depolarizing GABAA current in the prefrontal cortex is linked with cognitive impairment in a mouse model relevant for schizophrenia. Sci. Adv. 2021;7:eaba5032. doi: 10.1126/sciadv.aba5032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dargaei Z., Bang J.Y., Mahadevan V., Khademullah C.S., Bedard S., Parfitt G.M., Kim J.C., Woodin M.A. Restoring GABAergic inhibition rescues memory deficits in a Huntington’s disease mouse model. Proc. Natl. Acad. Sci. USA. 2018;115:E1618–E1626. doi: 10.1073/pnas.1716871115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hsu Y.T., Chang Y.G., Liu Y.C., Wang K.Y., Chen H.M., Lee D.J., Yang S.S., Tsai C.H., Lien C.C., Chern Y. Enhanced Na+ -K+ -2Cl− cotransporter 1 underlies motor dysfunction in huntington’s disease. Mov. Disord. 2019;34:845–857. doi: 10.1002/mds.27651. [DOI] [PubMed] [Google Scholar]
- 23.Lemonnier E., Degrez C., Phelep M., Tyzio R., Josse F., Grandgeorge M., Hadjikhani N., Ben-Ari Y. A randomised controlled trial of bumetanide in the treatment of autism in children. Transl. Psychiatry. 2012;2:e202. doi: 10.1038/tp.2012.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lemonnier E., Villeneuve N., Sonie S., Serret S., Rosier A., Roue M., Brosset P., Viellard M., Bernoux D., Rondeau S. Effects of bumetanide on neurobehavioral function in children and adolescents with autism spectrum disorders. Transl. Psychiatry. 2017;7:e1056. doi: 10.1038/tp.2017.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pressler R.M., Boylan G.B., Marlow N., de Vries L.S., Blennow M., Chiron C., Cross J.H., Hallberg B., Hellström-Westas L., Jullien V., NEMO consortium (NEonatal seizure treatment with Medication Off-patent) Bumetanide for neonatal seizures-back from the cotside. Nat. Rev. Neurol. 2015;11:724. doi: 10.1038/nrneurol.2015.116. [DOI] [PubMed] [Google Scholar]
- 26.Delpire E., Lu J., England R., Dull C., Thorne T. Deafness and imbalance associated with inactivation of the secretory Na-K-2Cl co-transporter. Nat. Genet. 1999;22:192–195. doi: 10.1038/9713. [DOI] [PubMed] [Google Scholar]
- 27.Li Y., Cleary R., Kellogg M., Soul J.S., Berry G.T., Jensen F.E. Sensitive isotope dilution liquid chromatography/tandem mass spectrometry method for quantitative analysis of bumetanide in serum and brain tissue. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2011;879:998–1002. doi: 10.1016/j.jchromb.2011.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Puskarjov M., Kahle K.T., Ruusuvuori E., Kaila K. Pharmacotherapeutic targeting of cation-chloride cotransporters in neonatal seizures. Epilepsia. 2014;55:806–818. doi: 10.1111/epi.12620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chung K.H., Hart C.C., Al-Bassam S., Avery A., Taylor J., Patel P.D., Vojtek A.B., Turner D.L. Polycistronic RNA polymerase II expression vectors for RNA interference based on BIC/miR-155. Nucleic Acids Res. 2006;34:e53. doi: 10.1093/nar/gkl143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Perkins K.L. Cell-attached voltage-clamp and current-clamp recording and stimulation techniques in brain slices. J. Neurosci. Methods. 2006;154:1–18. doi: 10.1016/j.jneumeth.2006.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cutts C.S., Eglen S.J. Detecting pairwise correlations in spike trains: an objective comparison of methods and application to the study of retinal waves. J. Neurosci. 2014;34:14288–14303. doi: 10.1523/JNEUROSCI.2767-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mann E.O., Paulsen O. Role of GABAergic inhibition in hippocampal network oscillations. Trends Neurosci. 2007;30:343–349. doi: 10.1016/j.tins.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 33.Flossmann T., Kaas T., Rahmati V., Kiebel S.J., Witte O.W., Holthoff K., Kirmse K. Somatostatin interneurons promote neuronal synchrony in the neonatal hippocampus. Cell Rep. 2019;26:3173–3182.e5. doi: 10.1016/j.celrep.2019.02.061. [DOI] [PubMed] [Google Scholar]
- 34.Spoljaric I., Spoljaric A., Mavrovic M., Seja P., Puskarjov M., Kaila K. KCC2-Mediated Cl− Extrusion Modulates Spontaneous Hippocampal Network Events in Perinatal Rats and Mice. Cell Rep. 2019;26:1073–1081.e3. doi: 10.1016/j.celrep.2019.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Slomowitz E., Styr B., Vertkin I., Milshtein-Parush H., Nelken I., Slutsky M., Slutsky I. Interplay between population firing stability and single neuron dynamics in hippocampal networks. eLife. 2015;4:e04378. doi: 10.7554/eLife.04378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Markadieu N., Delpire E. Physiology and pathophysiology of SLC12A1/2 transporters. Pflugers Arch. 2014;466:91–105. doi: 10.1007/s00424-013-1370-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kleschevnikov A.M., Belichenko P.V., Faizi M., Jacobs L.F., Htun K., Shamloo M., Mobley W.C. Deficits in cognition and synaptic plasticity in a mouse model of Down syndrome ameliorated by GABAB receptor antagonists. J. Neurosci. 2012;32:9217–9227. doi: 10.1523/JNEUROSCI.1673-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Contestabile A., Greco B., Ghezzi D., Tucci V., Benfenati F., Gasparini L. Lithium rescues synaptic plasticity and memory in Down syndrome mice. J. Clin. Invest. 2013;123:348–361. doi: 10.1172/JCI64650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fernandez F., Morishita W., Zuniga E., Nguyen J., Blank M., Malenka R.C., Garner C.C. Pharmacotherapy for cognitive impairment in a mouse model of Down syndrome. Nat. Neurosci. 2007;10:411–413. doi: 10.1038/nn1860. [DOI] [PubMed] [Google Scholar]
- 40.Reeves R.H., Irving N.G., Moran T.H., Wohn A., Kitt C., Sisodia S.S., Schmidt C., Bronson R.T., Davisson M.T. A mouse model for Down syndrome exhibits learning and behaviour deficits. Nat. Genet. 1995;11:177–184. doi: 10.1038/ng1095-177. [DOI] [PubMed] [Google Scholar]
- 41.Salehi A., Faizi M., Colas D., Valletta J., Laguna J., Takimoto-Kimura R., Kleschevnikov A., Wagner S.L., Aisen P., Shamloo M., Mobley W.C. Restoration of norepinephrine-modulated contextual memory in a mouse model of Down syndrome. Sci. Transl. Med. 2009;1:7ra17. doi: 10.1126/scitranslmed.3000258. [DOI] [PubMed] [Google Scholar]
- 42.Belichenko P.V., Madani R., Rey-Bellet L., Pihlgren M., Becker A., Plassard A., Vuillermot S., Giriens V., Nosheny R.L., Kleschevnikov A.M. An anti-β-amyloid vaccine for treating cognitive deficits in a mouse model of Down syndrome. PLoS ONE. 2016;11:e0152471. doi: 10.1371/journal.pone.0152471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Salehi A., Delcroix J.D., Belichenko P.V., Zhan K., Wu C., Valletta J.S., Takimoto-Kimura R., Kleschevnikov A.M., Sambamurti K., Chung P.P. Increased App expression in a mouse model of Down’s syndrome disrupts NGF transport and causes cholinergic neuron degeneration. Neuron. 2006;51:29–42. doi: 10.1016/j.neuron.2006.05.022. [DOI] [PubMed] [Google Scholar]
- 44.Kleschevnikov A.M., Yu J., Kim J., Lysenko L.V., Zeng Z., Yu Y.E., Mobley W.C. Evidence that increased Kcnj6 gene dose is necessary for deficits in behavior and dentate gyrus synaptic plasticity in the Ts65Dn mouse model of Down syndrome. Neurobiol. Dis. 2017;103:1–10. doi: 10.1016/j.nbd.2017.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.García-Cerro S., Martínez P., Vidal V., Corrales A., Flórez J., Vidal R., Rueda N., Arbonés M.L., Martínez-Cué C. Overexpression of Dyrk1A is implicated in several cognitive, electrophysiological and neuromorphological alterations found in a mouse model of Down syndrome. PLoS ONE. 2014;9:e106572. doi: 10.1371/journal.pone.0106572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Neumann F., Gourdain S., Albac C., Dekker A.D., Bui L.C., Dairou J., Schmitz-Afonso I., Hue N., Rodrigues-Lima F., Delabar J.M. DYRK1A inhibition and cognitive rescue in a Down syndrome mouse model are induced by new fluoro-DANDY derivatives. Sci. Rep. 2018;8:2859. doi: 10.1038/s41598-018-20984-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Altafaj X., Martín E.D., Ortiz-Abalia J., Valderrama A., Lao-Peregrín C., Dierssen M., Fillat C. Normalization of Dyrk1A expression by AAV2/1-shDyrk1A attenuates hippocampal-dependent defects in the Ts65Dn mouse model of Down syndrome. Neurobiol. Dis. 2013;52:117–127. doi: 10.1016/j.nbd.2012.11.017. [DOI] [PubMed] [Google Scholar]
- 48.Chakrabarti L., Best T.K., Cramer N.P., Carney R.S., Isaac J.T., Galdzicki Z., Haydar T.F. Olig1 and Olig2 triplication causes developmental brain defects in Down syndrome. Nat. Neurosci. 2010;13:927–934. doi: 10.1038/nn.2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Voronov S.V., Frere S.G., Giovedi S., Pollina E.A., Borel C., Zhang H., Schmidt C., Akeson E.C., Wenk M.R., Cimasoni L. Synaptojanin 1-linked phosphoinositide dyshomeostasis and cognitive deficits in mouse models of Down’s syndrome. Proc. Natl. Acad. Sci. USA. 2008;105:9415–9420. doi: 10.1073/pnas.0803756105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Olmos-Serrano J.L., Kang H.J., Tyler W.A., Silbereis J.C., Cheng F., Zhu Y., Pletikos M., Jankovic-Rapan L., Cramer N.P., Galdzicki Z. Down syndrome developmental brain transcriptome reveals defective oligodendrocyte differentiation and myelination. Neuron. 2016;89:1208–1222. doi: 10.1016/j.neuron.2016.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lockstone H.E., Harris L.W., Swatton J.E., Wayland M.T., Holland A.J., Bahn S. Gene expression profiling in the adult Down syndrome brain. Genomics. 2007;90:647–660. doi: 10.1016/j.ygeno.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 52.Chrast R., Scott H.S., Papasavvas M.P., Rossier C., Antonarakis E.S., Barras C., Davisson M.T., Schmidt C., Estivill X., Dierssen M. The mouse brain transcriptome by SAGE: differences in gene expression between P30 brains of the partial trisomy 16 mouse model of Down syndrome (Ts65Dn) and normals. Genome Res. 2000;10:2006–2021. doi: 10.1101/gr.10.12.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Antonarakis S.E., Lyle R., Chrast R., Scott H.S. Differential gene expression studies to explore the molecular pathophysiology of Down syndrome. Brain Res. Brain Res. Rev. 2001;36:265–274. doi: 10.1016/s0165-0173(01)00103-5. [DOI] [PubMed] [Google Scholar]
- 54.Costa A.C., Grybko M.J. Deficits in hippocampal CA1 LTP induced by TBS but not HFS in the Ts65Dn mouse: a model of Down syndrome. Neurosci. Lett. 2005;382:317–322. doi: 10.1016/j.neulet.2005.03.031. [DOI] [PubMed] [Google Scholar]
- 55.Kleschevnikov A.M., Belichenko P.V., Villar A.J., Epstein C.J., Malenka R.C., Mobley W.C. Hippocampal long-term potentiation suppressed by increased inhibition in the Ts65Dn mouse, a genetic model of Down syndrome. J. Neurosci. 2004;24:8153–8160. doi: 10.1523/JNEUROSCI.1766-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Contestabile A., Magara S., Cancedda L. The GABAergic hypothesis for cognitive disabilities in Down syndrome. Front. Cell. Neurosci. 2017;11:54. doi: 10.3389/fncel.2017.00054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Navarro-Romero A., Vázquez-Oliver A., Gomis-González M., Garzón-Montesinos C., Falcón-Moya R., Pastor A., Martín-García E., Pizarro N., Busquets-Garcia A., Revest J.M. Cannabinoid type-1 receptor blockade restores neurological phenotypes in two models for Down syndrome. Neurobiol. Dis. 2019;125:92–106. doi: 10.1016/j.nbd.2019.01.014. [DOI] [PubMed] [Google Scholar]
- 58.Wang X., Zhao Y., Zhang X., Badie H., Zhou Y., Mu Y., Loo L.S., Cai L., Thompson R.C., Yang B. Loss of sorting nexin 27 contributes to excitatory synaptic dysfunction by modulating glutamate receptor recycling in Down’s syndrome. Nat. Med. 2013;19:473–480. doi: 10.1038/nm.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lysenko L.V., Kim J., Madamba F., Tyrtyshnaia A.A., Ruparelia A., Kleschevnikov A.M. Developmental excitatory-to-inhibitory GABA polarity switch is delayed in Ts65Dn mice, a genetic model of Down syndrome. Neurobiol. Dis. 2018;115:1–8. doi: 10.1016/j.nbd.2018.03.005. [DOI] [PubMed] [Google Scholar]
- 60.Heubl M., Zhang J., Pressey J.C., Al Awabdh S., Renner M., Gomez-Castro F., Moutkine I., Eugène E., Russeau M., Kahle K.T. GABAA receptor dependent synaptic inhibition rapidly tunes KCC2 activity via the Cl−-sensitive WNK1 kinase. Nat. Commun. 2017;8:1776. doi: 10.1038/s41467-017-01749-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lee H.H., Deeb T.Z., Walker J.A., Davies P.A., Moss S.J. NMDA receptor activity downregulates KCC2 resulting in depolarizing GABAA receptor-mediated currents. Nat. Neurosci. 2011;14:736–743. doi: 10.1038/nn.2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lagostena L., Rosato-Siri M., D’Onofrio M., Brandi R., Arisi I., Capsoni S., Franzot J., Cattaneo A., Cherubini E. In the adult hippocampus, chronic nerve growth factor deprivation shifts GABAergic signaling from the hyperpolarizing to the depolarizing direction. J. Neurosci. 2010;30:885–893. doi: 10.1523/JNEUROSCI.3326-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chen M., Wang J., Jiang J., Zheng X., Justice N.J., Wang K., Ran X., Li Y., Huo Q., Zhang J. APP modulates KCC2 expression and function in hippocampal GABAergic inhibition. eLife. 2017;6:e20142. doi: 10.7554/eLife.20142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Brandt C., Seja P., Töllner K., Römermann K., Hampel P., Kalesse M., Kipper A., Feit P.W., Lykke K., Toft-Bertelsen T.L. Bumepamine, a brain-permeant benzylamine derivative of bumetanide, does not inhibit NKCC1 but is more potent to enhance phenobarbital’s anti-seizure efficacy. Neuropharmacology. 2018;143:186–204. doi: 10.1016/j.neuropharm.2018.09.025. [DOI] [PubMed] [Google Scholar]
- 65.Hyde T.M., Lipska B.K., Ali T., Mathew S.V., Law A.J., Metitiri O.E., Straub R.E., Ye T., Colantuoni C., Herman M.M. Expression of GABA signaling molecules KCC2, NKCC1, and GAD1 in cortical development and schizophrenia. J. Neurosci. 2011;31:11088–11095. doi: 10.1523/JNEUROSCI.1234-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Coumans J.V., Gau D., Poljak A., Wasinger V., Roy P., Moens P. Green fluorescent protein expression triggers proteome changes in breast cancer cells. Exp. Cell Res. 2014;320:33–45. doi: 10.1016/j.yexcr.2013.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.McBride J.L., Boudreau R.L., Harper S.Q., Staber P.D., Monteys A.M., Martins I., Gilmore B.L., Burstein H., Peluso R.W., Polisky B. Artificial miRNAs mitigate shRNA-mediated toxicity in the brain: implications for the therapeutic development of RNAi. Proc. Natl. Acad. Sci. USA. 2008;105:5868–5873. doi: 10.1073/pnas.0801775105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lykke K., Töllner K., Feit P.W., Erker T., MacAulay N., Löscher W. The search for NKCC1-selective drugs for the treatment of epilepsy: Structure-function relationship of bumetanide and various bumetanide derivatives in inhibiting the human cation-chloride cotransporter NKCC1A. Epilepsy Behav. 2016;59:42–49. doi: 10.1016/j.yebeh.2016.03.021. [DOI] [PubMed] [Google Scholar]
- 69.Savardi A., Borgogno M., Narducci R., La Sala G., Ortega J.A., Summa M., Armirotti A., Bertorelli R., Contestabile A., De Vivo M., Cancedda L. Discovery of a small molecule drug candidate for selective NKCC1 inhibition in brain disorders. Chem. 2020;6:2073–2096. doi: 10.1016/j.chempr.2020.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Beaudoin G.M., 3rd, Lee S.H., Singh D., Yuan Y., Ng Y.G., Reichardt L.F., Arikkath J. Culturing pyramidal neurons from the early postnatal mouse hippocampus and cortex. Nat. Protoc. 2012;7:1741–1754. doi: 10.1038/nprot.2012.099. [DOI] [PubMed] [Google Scholar]
- 71.Valente P., Lignani G., Medrihan L., Bosco F., Contestabile A., Lippiello P., Ferrea E., Schachner M., Benfenati F., Giovedì S., Baldelli P. Cell adhesion molecule L1 contributes to neuronal excitability regulating the function of voltage-gated Na+ channels. J. Cell Sci. 2016;129:1878–1891. doi: 10.1242/jcs.182089. [DOI] [PubMed] [Google Scholar]
- 72.Pozzi D., Lignani G., Ferrea E., Contestabile A., Paonessa F., D’Alessandro R., Lippiello P., Boido D., Fassio A., Meldolesi J. REST/NRSF-mediated intrinsic homeostasis protects neuronal networks from hyperexcitability. EMBO J. 2013;32:2994–3007. doi: 10.1038/emboj.2013.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Thalhammer A., Contestabile A., Ermolyuk Y.S., Ng T., Volynski K.E., Soong T.W., Goda Y., Cingolani L.A. Alternative splicing of P/Q-Type Ca2+ channels shapes presynaptic plasticity. Cell Rep. 2017;20:333–343. doi: 10.1016/j.celrep.2017.06.055. [DOI] [PubMed] [Google Scholar]
- 74.Follenzi A., Ailles L.E., Bakovic S., Geuna M., Naldini L. Gene transfer by lentiviral vectors is limited by nuclear translocation and rescued by HIV-1 pol sequences. Nat. Genet. 2000;25:217–222. doi: 10.1038/76095. [DOI] [PubMed] [Google Scholar]
- 75.Ayuso E., Mingozzi F., Montane J., Leon X., Anguela X.M., Haurigot V., Edmonson S.A., Africa L., Zhou S., High K.A. High AAV vector purity results in serotype- and tissue-independent enhancement of transduction efficiency. Gene Ther. 2010;17:503–510. doi: 10.1038/gt.2009.157. [DOI] [PubMed] [Google Scholar]
- 76.Matsushita T., Elliger S., Elliger C., Podsakoff G., Villarreal L., Kurtzman G.J., Iwaki Y., Colosi P. Adeno-associated virus vectors can be efficiently produced without helper virus. Gene Ther. 1998;5:938–945. doi: 10.1038/sj.gt.3300680. [DOI] [PubMed] [Google Scholar]
- 77.Blaesse P., Guillemin I., Schindler J., Schweizer M., Delpire E., Khiroug L., Friauf E., Nothwang H.G. Oligomerization of KCC2 correlates with development of inhibitory neurotransmission. J. Neurosci. 2006;26:10407–10419. doi: 10.1523/JNEUROSCI.3257-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sagné C., Isambert M.F., Henry J.P., Gasnier B. SDS-resistant aggregation of membrane proteins: application to the purification of the vesicular monoamine transporter. Biochem. J. 1996;316:825–831. doi: 10.1042/bj3160825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Verkman A.S., Sellers M.C., Chao A.C., Leung T., Ketcham R. Synthesis and characterization of improved chloride-sensitive fluorescent indicators for biological applications. Anal. Biochem. 1989;178:355–361. doi: 10.1016/0003-2697(89)90652-0. [DOI] [PubMed] [Google Scholar]
- 80.Colombi I., Mahajani S., Frega M., Gasparini L., Chiappalone M. Effects of antiepileptic drugs on hippocampal neurons coupled to micro-electrode arrays. Front. Neuroeng. 2013;6:10. doi: 10.3389/fneng.2013.00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Frega M., Pasquale V., Tedesco M., Marcoli M., Contestabile A., Nanni M., Bonzano L., Maura G., Chiappalone M. Cortical cultures coupled to micro-electrode arrays: a novel approach to perform in vitro excitotoxicity testing. Neurotoxicol. Teratol. 2012;34:116–127. doi: 10.1016/j.ntt.2011.08.001. [DOI] [PubMed] [Google Scholar]
- 82.El Merhie A., El Merhie A., Ito D., Colombi I., Keshavan S., Mishra N., Miseikis V., Diaspro A., Coletti C., Chiappalone M. Single layer graphene functionalized MEA for enhanced detection of neuronal network development. Sens. Actuators B Chem. 2018;277:224–233. [Google Scholar]
- 83.Bologna L.L., Pasquale V., Garofalo M., Gandolfo M., Baljon P.L., Maccione A., Martinoia S., Chiappalone M. Investigating neuronal activity by SPYCODE multi-channel data analyzer. Neural Netw. 2010;23:685–697. doi: 10.1016/j.neunet.2010.05.002. [DOI] [PubMed] [Google Scholar]
- 84.Maccione A., Gandolfo M., Massobrio P., Novellino A., Martinoia S., Chiappalone M. A novel algorithm for precise identification of spikes in extracellularly recorded neuronal signals. J. Neurosci. Methods. 2009;177:241–249. doi: 10.1016/j.jneumeth.2008.09.026. [DOI] [PubMed] [Google Scholar]
- 85.Chiappalone M., Novellino A., Vajda I., Vato A., Martinoia S., van Pelt J. Burst detection algorithms for the analysis of spatio-temporal patterns in cortical networks of neurons. Neurocomputing. 2005;65:653–662. [Google Scholar]
- 86.Turnbull L., Dian E., Gross G. The string method of burst identification in neuronal spike trains. J. Neurosci. Methods. 2005;145:23–35. doi: 10.1016/j.jneumeth.2004.11.020. [DOI] [PubMed] [Google Scholar]
- 87.Chiappalone M., Vato A., Berdondini L., Koudelka-Hep M., Martinoia S. Network dynamics and synchronous activity in cultured cortical neurons. Int. J. Neural Syst. 2007;17:87–103. doi: 10.1142/S0129065707000968. [DOI] [PubMed] [Google Scholar]
- 88.Duchon A., Raveau M., Chevalier C., Nalesso V., Sharp A.J., Herault Y. Identification of the translocation breakpoints in the Ts65Dn and Ts1Cje mouse lines: relevance for modeling Down syndrome. Mamm. Genome. 2011;22:674–684. doi: 10.1007/s00335-011-9356-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Paxinos G., Franklin K.B.J. Academic Press; 2001. The Mouse Brain in Stereotaxic Coordinates. [Google Scholar]
- 90.Parrini M., Ghezzi D., Deidda G., Medrihan L., Castroflorio E., Alberti M., Baldelli P., Cancedda L., Contestabile A. Aerobic exercise and a BDNF-mimetic therapy rescue learning and memory in a mouse model of Down syndrome. Sci. Rep. 2017;7:16825. doi: 10.1038/s41598-017-17201-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.