

Abstract
Parkinson’s disease (PD) is characterized by Lewy bodies (composed predominantly of alpha-synuclein [aSyn]) and loss of pigmented midbrain dopaminergic neurons comprising the nigrostriatal pathway. Most PD patients show significant deficiency of gangliosides, including GM1, in the brain, and GM1 ganglioside appears to keep dopaminergic neurons functioning properly. Thus, supplementation of GM1 could potentially provide some rescuing effects. In this study, we demonstrate that intranasal infusion of GD3 and GM1 gangliosides reduces intracellular aSyn levels. GM1 also significantly enhances expression of tyrosine hydroxylase (TH) in the substantia nigra pars compacta of the A53T aSyn overexpressing mouse, following restored nuclear expression of nuclear receptor related 1 (Nurr1, also known as NR4A2), an essential transcription factor for differentiation, maturation, and maintenance of midbrain dopaminergic neurons. GM1 induces epigenetic activation of the TH gene, including augmentation of acetylated histones and recruitment of Nurr1 to the TH promoter region. Our data indicate that intranasal administration of gangliosides could reduce neurotoxic proteins and restore functional neurons via modulating chromatin status by nuclear gangliosides.
Keywords: alpha-synuclein, dopaminergic neuron, epigenetic regulation, ganglioside therapy, GD3 ganglioside, GM1 ganglioside, hualpha-Syn, mitochondrion, nurr1, Parkinson's disease, tyrosine hydroxylase
Graphical abstract
Itokazu et al. demonstrate that intranasally administered ganglioside GM1 or GD3 decreases the accretion of alpha-synuclein and GM1 upregulates epigenetically the expression of tyrosine hydroxylase via recruitment of Nurr1 transcription factor in the brain of a mouse model of Parkinson’s disease (PD). These observations suggest the possibility of using gangliosides as alternative therapeutics for PD.
Introduction
Parkinson’s disease (PD), of which cases have been growing in the last few decades, is the second most common neurodegenerative disease after Alzheimer’s disease (AD), with the prevalence of PD increasing with age from about 1% among individuals older than age 65 years to about 4% in those older than age 85 years. PD is characterized by neurotoxic aggregation of alpha-synuclein (aSyn) and progressive loss of nigrostriatal dopaminergic neurons. Among multiple proposed mechanisms for PD pathogenesis, the expression of aSyn has been reported to be regulated by glycosphingolipids (GSLs).1,2 aSyn also shows strong affinity to GSLs. Significant deficiency of gangliosides has been assumed as a major contributing factor of PD pathogenesis.1,2, 3, 4, 5, 6 Although it has been reported that GM1 binds to aSyn, leading to inhibition of aSyn fibril formation in vitro,7,8 which might be underlying mechanisms of the positive effects of GM1 in PD, the molecular mechanisms of physiological GM1 to maintain resilience in vivo remain obscure.
GSLs are unique amphipathic molecules containing a hydrophilic carbohydrate portion and a hydrophobic lipid component. Glucosylceramide (GlcCer) and lactosylceramide (LacCer) are common precursors of gangliosides. GlcCer synthase (GlcT, Figure 1)-deficient mice and LacCer synthase (GalT-1, Figure 1)-knockout (KO) mice show embryonic lethality.9, 10, 11 These observations indicate that GSLs are essential for development. Gangliosides, sialic acid-containing GSLs, are found in virtually all vertebrate cells but are particularly abundant in the nervous system.12, 13, 14 Ganglioside synthesis pathways (Figure 1) were initially delineated by Yu and Ando15 following their structural characterization of c-series gangliosides. GM3 synthase (GM3S, sialyltransferase-I [ST-I], or St3gal5; Figure 1) is the first critical enzyme for the synthesis of a-, b- and c-series gangliosides. Mutation of GM3S is associated with human autosomal recessive infantile-onset symptomatic epilepsy syndrome,16 Rett syndrome-like phenotype,17 and profound intellectual disability.17,18 An alteration of the GM2 synthase (GM2S, GalNAc-T, B4galnt1) gene has been reported in patients of hereditary spastic paraplegias.19 These studies clearly demonstrate that deletions of complex gangliosides are associated with human diseases. Both GM2S- and GM3S-deficient (double KO) mice, which lack all gangliosides, die soon after weaning at 3 weeks of age,20 and they exhibit sudden death from audiogenic seizures.21 Although mice deficient in some of these molecules show subtle phenotypic abnormalities compared with the wild-type (WT) animals in early development, the aberrant phenotypes of abnormal ganglioside expression become progressively more severe in the postnatal and adult stages, and in pathogenic conditions. For example, GD3 synthase (GD3S, sialyltransferase-II [ST-II], or St8Sia1)-KO mice show decreased postnatal neural stem cell (NSC) amplification, and impaired postnatal neurogenesis leads to depression-like behaviors.12,14,22, 23, 24, 25. GM2S-KO mice exhibit impaired movement and have virtually all of the neuropathological findings of PD.2,3,26
Figure 1.

Metabolic pathways and structure of glycosphingolipids, including gangliosides
Cer, ceramide; GalNAc-T, N-acetylgalactosaminyltransferase I (B4galnt1, GA2/GM2/GD2/GT2 synthase); GalT-I, galactosyltransferase I (B4galt6, lactosylceramide synthase); GalT-II, galactosyltransferase II (B3galt4, GA1/GM1/GD1b/GT1c synthase); GalT-III, galactosyltransferase III (Ugt8a, galactosylceramide synthase); GlcT, glucosyltransferase (Ugcg, glucosylceramide synthase); ST-I, sialyltransferase I (St3gal5, GM3/GM4 synthase); ST-II, sialyltransferase II (St8Sia1, GD3 synthase); ST-III, sialyltransferase III (St8Sia3, GT3 synthase); ST-IV, sialyltransferase IV (St3gal2, GM1b/GD1a/GT1b/GQ1c synthase); ST-V, sialyltransferase V (St8sia5, GD1c/GT1a/GQ1b/GP1c synthase); ST-VII, sialyltransferase VII (St6galnac6, GD1aα/GT1aα/GQ1bα/GP1cα synthase). Official symbols of genes are represented in italics in this legend. GM1, GD1a, GD1b, and GT1b are the most abundant ganglioside species in adult mammalian brain and neurons.
Ganglioside expression profiles are associated with pathogenic mechanisms of neurodegenerative diseases, such as AD, PD, and Huntington’s disease, amyotrophic lateral sclerosis, and multiple sclerosis.27,28 Although the beneficial effects of GM1 for PD intervention have been suggested and investigated,1,3, 4, 5, 6 the molecular mechanisms underlying the protective effects of GM1 in PD brains need to be further investigated. Gangliosides are crucial components in mediating neurotrophin support and other vital functions necessary for the long-term maintenance of neuronal viability, which is known to occur mainly in the membrane microdomains or lipid rafts. Activation of adult neurogenesis is initiated by neurotrophic factors whose signaling is dependent on their receptors located in the ganglioside-enriched membrane microdomains. For example, GD3 ganglioside modulates NSC self-renewal by interacting with epidermal growth factor (EGF) receptors and regulating EGF signaling, and GM1 ganglioside is necessary for the formation of the glial cell line-derived neurotrophic factor (GDNF)-receptor complex in dopaminergic neurons.1,22, 23, 24,29. Moreover, we discovered that GM1 augments a novel epigenetic gene regulation mechanism for neuronal cell lineage differentiation.30, 31, 32 Furthermore, we demonstrated that depletion of GD3 induces fewer numbers of mitochondria with less dendritic arborization and reduced spine formation.33 We proposed that nuclear lipid microdomains may modulate gene transcription and that mitochondrial lipid raft may control mitochondrial functions during neural cell differentiation and in the pathogenic mechanisms. So far, intracerebroventricular (i.c.v.) administration is the most reliable method to deliver gangliosides into the brain, whereas intranasal infusion, which has emerged during last few decades as a promising approach for drug delivery into the brain,34 has not been tried yet as a route of gangliosides for PD. We developed a more convenient noninvasive delivery procedure by intranasal infusion of gangliosides with success.
Hualpha-Syn(A53T) transgenic mice display an age-dependent phenotype including progressive motor deficits, intraneuronal inclusion bodies, and neuronal loss. This genetically modified mouse line is widely used for studying PD and synucleinopathies,35 and we refer to this mouse line as the A53T PD mouse. We tested the hypothesis that that intranasal infusion of gangliosides could diminish aSyn neurotoxicity and protect dopaminergic neurons. In this study, we successively demonstrated that intranasally administered gangliosides could effectively reduce aSyn and restored the expression of tyrosine hydroxylase (TH) in the substantia nigra pars compacta of the A53T PD mouse. Furthermore, the expression of a major protein component of the outer mitochondrial membrane, voltage-dependent anion channel 1 (VDAC1), was restored by administration of ganglioside GD3 and/or GM1 in the A53T PD mouse. To gain further insight into the molecular mechanisms underlying restoration of TH expression, we investigated changes in expression and activation of nuclear receptor related 1 (Nurr1, also known as NR4A2), an essential transcription factor for differentiation, maturation, and maintenance of midbrain dopaminergic neurons. We found that GM1 induced epigenetic activation of the TH gene by recruiting Nurr1, located on the promotor of the TH gene, to facilitate TH expression. Thus, the present study clearly supports the following sequence of events that the intranasal administered gangliosides, without histological damage, alleviate PD symptoms by reducing neurotoxic protein expression and restoring functional neurons via modulating the chromatin structure.
Results
Intranasal administration of gangliosides
Our previous study showed that i.c.v. infusion of GD3 and GM1 acted functionally in mouse brains.25 Although currently i.c.v. administration is the most reliable method to deliver gangliosides into the brain, non-invasive delivery procedures are safer and more convenient. We attempted intranasal ganglioside infusion to establish a novel treatment strategy for patients with PD. To examine whether gangliosides could be delivered into the brain tissues intranasally, we administered ganglioside GM1 (0.5 or 5 mg/kg/day) into nasal holes of 8-month-old GM2S-KO mice for 7 days. Each 6 μL of ganglioside solution was infused into the right and left nares twice (total 24 μL) in day-to-day treatment. The placebo group received only saline infusion. No adverse effects were observed during and after intranasal administration of gangliosides. Mice lacking GM2S do not express GalNAc-containing gangliosides, including GM1 (Figure 1). Analysis by thin-layer chromatography (TLC) of extracted lipids demonstrated dose-dependent uptake of GM1 into the GM2S-KO brains (Figure 2A). Intranasally administered GM1 was also confirmed and observed by confocal microscopy in various regions of the brain tissues including cortex and midbrain (Figures 2B–2E), olfactory bulb, subventricular zone (SVZ), hippocampus, and cerebellum (data not shown). This non-invasive ganglioside delivery system is an innovative and effective method.
Figure 2.

Intranasally administered GM1 was delivered to mouse brain
(A) GM1 (0.5 or 5 mg/kg/day) was intranasally infused into GM2S-KO mice for 7 days. GM1 bands on TLC were visualized with cholera toxin B (CtxB)-HRP (CtxB has been frequently used as a probe of GM1). Intranasally infused GM1 (5 mg/kg/day) could be delivered to the cortical tissue (B and C) and substantia nigra (D and E) in the midbrain of GM2S-KO mice. Purple indicates GM1; blue indicates nuclear DAPI; green indicates tyrosine hydroxylase (TH). Scale bars, 50 μm (B) and 5 μm (D).
Intranasally administered gangliosides reduce aSyn
Since the accumulation of aSyn is a major pathological hallmark of PD, we utilized a PD mouse model, which is known as Hualpha-Syn(A53T) transgenic line G2-3 (referred to as the A53T PD mouse).35 These transgenic mice display an age-dependent phenotype including progressive motor deficits, intraneuronal inclusion bodies, and neuronal loss. This line is widely used for studying PD and synucleinopathies. We intranasally administered gangliosides (5 mg/kg/day) GD3 and GM1 singularly, or sequentially (GD3 for 14 days and then GM1 for another 14 days), into 8-month-old A53T PD mice for 28 days (Figures 3, 4, and 5). The placebo group (PD and WT mice) received only saline infusion. Although GM1 extensively downregulated aSyn protein levels, even GD3 reduced levels of aSyn in treated brains, implying an unknown functional role of GD3 on removal of aSyn (Figures 3A, 3C, and 3F). Thus, intranasal administration of gangliosides dramatically reduced aSyn levels in A53T PD brains. We further investigated the level of phosphorylated serine 129 (phospho-S129) of aSyn, because accumulation of phosphorylated aSyn at S129 has been reported in the brain of patients suffering from PD.36 Nearly 90% of aSyn deposited in the Lewy bodies of synucleinopathy brain is phosphorylated at S129, yet 4% of aSyn in normal brain is phosphorylated.37 Intranasal ganglioside administration also decreased the phospho-S129 aSyn levels in A53T PD mouse brains (Figures 3B, 3D, and 3F). Additionally, aSyn presents different subcellular localizations in neurons, suggesting the specific roles in each organelle,38 and nuclear aSyn is reported to promote neurotoxicity of aSyn for the degeneration of neurons.39,40 Figure 3E shows that aSyn is heavily localized in the nuclear membrane in the A53T PD mouse, whereas intranasal GM1 treatment markedly reduced aSyn on the nuclear envelope. These results suggest that gangliosides may function as a scavenger of neurotoxic proteins such as aSyn.
Figure 3.

Intranasally administered gangliosides reduce neurotoxic aSyn in A53T PD mouse brain
Intranasally infused gangliosides (5 mg/kg/day for 28 days) removed aSyn and phospho-S129 aSyn (pS129-aSyn) in A53T PD mouse brain. (A, B, and E) Confocal micrographs of cerebral cortex in brain tissue. (A) Green indicates aSyn; (B) purple indicates pS129-aSyn. (C and D) Quantitation of aSyn (C) and pS129-aSyn levels (D) by image analysis. (E) aSyn is accumulated on the nuclear membrane, and GM1 infusion reduced nuclear aSyn. (F) Western blot analysis of aSyn and pS129-aSyn in the substantia nigra. Values were normalized to control levels and are means ± SE (n = 3 mice/group; two-way ANOVA with a Tukey’s multiple comparison test). ∗p < 0.05. Scale bars, 10 μm (B) and 5 μm (E).
Figure 4.

Intranasally administered GD3 and GM1 restored expression of VDAC1, a major component of the outer mitochondrial membrane known to regulate mitochondrial functions
(A) Intranasally infused GD3 and/or GM1 (5 mg/kg/day for 28 days) increased VDAC1 expression in dopaminergic neurons within the substantia nigra pars compacta of A53T PD mouse brain. Green indicates TH; red indicates VDAC1. Scale bar, 10 μm. (B) Western blot analysis of VDAC1 in the substantia nigra.
Figure 5.

Intranasally administered GM1 restored dopaminergic neurons in PD mouse brain
TH is the rate-limiting enzyme in the biosynthesis of dopamine, and it is regularly used as a marker for dopaminergic neurons. The transcription factor Nurr1 is critical in the development and maintenance of dopaminergic neurons, and Nurr1 is associated with PD. Intranasally infused GM1 (5 mg/kg/day for 28 days) increased the expressions of TH and Nurr1 at substantia nigra in A53T PD mouse brain. (A) Purple indicates TH; green indicates Nurr1; blue indicates nuclear DAPI. (B) Quantitation of TH levels by image analysis. (C) Nuclear localization of Nurr1 quantified by image analysis. Values were normalized to control levels and are means ± SE (n = 3 mice/group; two-way ANOVA with a Tukey’s multiple comparison test). ∗p < 0.05. Scale bar, 5 μm.
Intranasally administered gangliosides restore the expression of VDAC1
The mitochondrion is the main intracellular organelle for producing adenosine triphosphate (ATP). PD is primarily regarded as a disease of dopaminergic neurons of the substantia nigra pars compacta where mitochondrial dysfunction is evident. aSyn is also known to reveal mitochondrial localization in dopaminergic neurons41,42 and induce mitochondrial dysfunction.43 Disruption of mitochondrial homeostasis leads to its fragmentation and loss of membrane potential, enhancing the production of reactive oxygen species. Elevated oxidative conditions promotes phosphorylation of aSyn at S12944 and thus neurotoxic aggregation of aSyn would be caused. Our findings (Figure 3) showed the reduction of aSyn and its phosphorylation levels at S129 by intranasal ganglioside treatment, which points to possible roles of gangliosides in alleviation of critical damage on mitochondria. Recently, we found that GD3 regulates mitochondrial dynamics by interacting with dynamin related protein-1 (Drp1), and deficiency of GD3 resulted in a reduced number of mitochondria due to dysregulation of mitochondrial fission. Our study thus suggested that gangliosides have an important role to maintain normal mitochondrial activity.33 Based on these findings, we examined whether the intranasal administration of gangliosides influences mitochondrial status in PD brains by immunohistochemistry and immunoblots for VDAC1, a major component of the outer mitochondrial membrane known to regulate mitochondrial functions. VDAC1 was drastically downregulated in dopaminergic neurons of the A53T PD mouse brain (Figure 4). Intranasally administered GD3 and/or GM1 dramatically restored VDAC1 levels to those of WT levels in the A53T PD brain. This result suggests that intranasal infusion of gangliosides can be an effective method to recover mitochondrial activities in neurodegenerative diseases.
Intranasally administered gangliosides restore TH expression and Nurr1 nuclear localizations
Currently the most widely used intervention for the disease symptoms of PD is based on treatment with the dopamine precursor l-dopa. As PD progresses, l-dopa therapy leads to motor complications such as wearing “off” of the medication between doses and to typical writhing, twisting movements termed dyskinesias in many patients. For this reason, it would be desirable to develop alternative strategies, such as dopamine neuron augmentation. TH is the rate-limiting enzyme in the synthesis of dopamine. The expression level of TH is markedly reduced at substantia nigra pars compacta in A53T PD mouse brain (Figures 5A and 5B), and, in contrast, intranasally infused ganglioside GM1 dramatically restored TH expression in the A53T PD mouse. This result suggests that GM1 protects dopaminergic neurons from the cytotoxicity of aSyn in PD brain. Intriguingly, the expression and nuclear localization of Nurr1, a dopaminergic neuron-associated transcription factor involved in the expression of TH, were restored by intranasal administration of GM1 in the substantia nigra pars compacta of A53T PD mouse brains (Figures 5A and 5C). These findings suggest that GM1 may participate in regulating the expression of TH by modulating Nurr1 activity.
GM1 induces epigenetic activation of the TH gene via recruitment of Nurr1, an essential transcription factor for dopaminergic neurons
We further examined whether GM1 regulates TH gene expression. Quantitative PCR (qPCR) analyses of the mRNA level for TH expression were substantially decreased in the substantia nigra pars compacta of GM2S-KO mice. Interestingly, intranasal administration of GM1 for 28 days could restore the normal TH expression (Figure 6A). These data suggested that GM1 is an important regulatory factor in modulating TH gene expression. Next, we analyzed dopaminergic neuron-specific gene expression utilizing Neuro 2a cells after treatment with GM1 or GD3. TH expression was not detected in untreated cells, whereas GM1 dramatically increased the TH expression (Figure 6B). A chromatin immunoprecipitation (ChIP) assay showed that ectopic GM1 significantly induced epigenetic activation of the TH gene, including augmentation of acetylated histone H3 (Figure 6C). Moreover, GM1 remarkably recruited the dopaminergic neuron-associated transcription factor, Nurr1, on the TH promoter region (Figure 6D). This result demonstrates that GM1 promotes the interaction of Nurr1 with the TH gene promoter for activating its gene expression. In addition, GM1 also recruited Pitx3, a critical transcription factor for the survival of midbrain dopaminergic neurons (Figure 6E).
Figure 6.

Ectopic GM1 induces epigenetic activation of the TH gene via recruitment of dopaminergic transcription factor Nurr1
(A) Reduced TH expression in substantia nigra of GM2S-KO mouse (8 months old), and intranasal GM1 administration (5 mg/kg/day for 28 days) restored TH levels. (B–E) Neuro 2a cells were cultured in the presence of gangliosides (5 μM of GM1 or GD3) for 24 h. (B) mRNA analysis for TH expression. Enrichment of epigenetic markers and recruitment of transcription factors in the TH gene promoter were analyzed by ChIP assays with anti-AcH3 (C), anti-Nurr1 (D), or anti-Pitx3 (E), followed by qPCR analyses. (F) Photoactivatable and clickable GM1 (pacGM1) was tagged with TAMRA (carboxytetramethylrhodamine) on isolated nuclei on Neuro 2a cells. pacGM1 technique is able to prove that exogenous GM1 (red) can be delivered inside the nucleus. (G) Proximity ligation assay (PLA) using anti-GM1 (mouse antibody, MINUS) and acetylated histone H3 anti (rabbit antibody, PLUS) in isolated nuclei from adult mouse brain. Values were normalized to control levels and are means ± SE (n = 3-6 independent experiments/group; two-way ANOVA with a Tukey’s multiple comparison test). ∗p < 0.05. Scale bars, 5 μm.
It has been suggested that specialized microdomains exist on the nuclear envelope that are functionally similar to glycolipid-enriched lipid rafts on the plasma membrane surface.30,45 To further investigate the significance of nuclear GM1, cells were treated with photoactivatable and clickable GM1 (pacGM1). pacGM1 in isolated nuclei was visualized using click chemistry-mediated tagging with fluorophores. Figure 6F shows that exogenous pacGM1 is indeed delivered into the nucleus. Recently, proximity ligation assays (PLAs) have been developed to detect the formation of lipid-protein interaction by immunohistochemistry.46 Each PLA probe contains a unique short DNA strand attached to it. If the PLA probes are in close proximity (<40 nm), the DNA strands interact and generate circle-forming DNA used for enzymatic ligation. The ligated DNA is amplified via rolling circle amplification using a polymerase. A several hundredfold replication of the DNA circle labels complementary oligonucleotide probes that yield high intensity of fluorescence (red). Using this technology, we isolated nuclei from WT mice and performed PLAs to detect GM1 and the acetylated histone complex. The PLA signals in Figure 6G demonstrate that GM1 is indeed co-localized with acetylated histone H3, i.e., GM1 is localized on active chromatin in the nucleus (1.94 ± 0.297 PLA signals in the nucleus). The results of both experiments (Figures 6F and 6G) clearly indicate that GM1 is localized in the nucleus and that GM1 interacts with transcriptionally active histones. Nuclear GM1 modulates gene transcription to sustain functional neurons to ameliorate aSyn toxicity. Further experiments are in progress to elucidate the detailed mechanisms. Nevertheless, it is safe to conclude that intranasal gangliosides reduce aSyn toxicity and that GM1 epigenetically sustains dopaminergic neurons.
Discussion
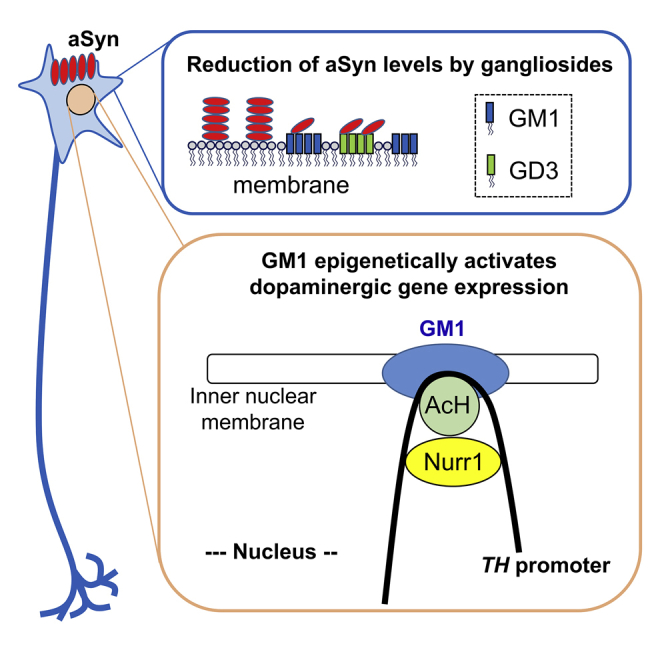
In this study, we clearly demonstrated that intranasally administrated gangliosides reduced aSyn levels and restored TH levels in A53T PD mice. Furthermore, we discovered a novel function of nuclear GM1 to epigenetically activate the TH gene promoter with recruitment of Nurr1 and acetylated histones to enhance TH expression (Figure 7).
Figure 7.

Schematic diagram of intranasal ganglioside therapy
Gangliosides prevent aSyn accumulation, and a supportive ganglioside composition will reduce aSyn neurotoxicity. GM1 strongly inhibits, and GD3 partially inhibits, aSyn accumulation. GM1 facilitates binding of acetylated histones (AcHs) and Nurr1 transcription factor on the TH promoter to increase TH expression via opening chromatin. The nuclear GM1-lipid domains may serve as a docking site at the nuclear periphery for specific active chromatins for dopaminergic neurons and for maintaining neuronal functions. Thus, ganglioside therapy is a two-pronged approach that effectively treats PD by decreasing cytotoxic aSyn and sustaining the function of dopaminergic neurons.
The accumulation of aSyn is thought to be a key step in the pathogenesis of PD. It has been reported that GM1 (but not asialo-GM1, GM2, or GM3) binds to aSyn with high affinity to stabilize an α-helical state and inhibit fibrillation.7,8 Importantly, aSyn has a ganglioside-binding domain,47 and this suggests that gangliosides may serve as a modulator or scavenger of aSyn. Previous research has shown the exciting possibility that gangliosides can modulate the effects of aSyn in neural cells, including reduction of the concentrations of cytotoxic aSyn.7,8,26 GM1 has been also reported to regulate internalization of aSyn into microglia through GM1-mediated endocytosis.48 A progressive imbalance of the cell membrane lipid composition is a physicochemical property altered with normal aging, and further disruptions to these processes are observed in neurodegenerative diseases, including PD and AD. It is well known that the ganglioside composition is altered in the brains of PD and AD patients; in particular, major “brain-type” gangliosides, such as GM1, are significantly decreased.26,49 Our results from this study and previous studies suggest that exogenous gangliosides may increase helical folding to inhibit accumulation of aSyn and to reduce neurotoxic aSyn levels.
Ledeen and colleagues50 observed a greatly elevated aSyn expression in substantia nigra pars compacta of GM2S-KO mice. Moreover, they demonstrated that the depletion of GM1 acquires characteristic symptoms of PD, including motor impairment, striatal dopamine depletion, selective loss of TH-expressing neurons, gastrointestinal dysfunction, cardiac pathology, and accumulation of aSyn, suggesting that the deficiency of GM1 correlates neuropathologically with PD in mice and humans.3,26 There are reports that intraperitoneal injection of GM1 is a promising treatment for PD patients and for use in animal models to protect dopaminergic neurons.6,51, 52, 53, 54 However, it remains unclear how the injected gangliosides, which typically exist in micellar form, could overcome the blood-brain barrier to enter the brain space.
Recently, Revunov et al.55 reported that less than 0.4% of an intravenously injection of a fluorinated ganglioside derivative, [18F]F-GM1, could enter a non-human primate (monkey) brain. The amount of chemicals exposed to the brain by intraperitoneal injection is typically less than that of intravenous injection. Since delivery of gangliosides is an extremely small amount even by intravenous administration, it is important to choose a more efficient route for ganglioside administration for therapy. Svennerholm et al.56 described that i.c.v. administration of GM1 ganglioside to AD patients appeared to stop the continuous deterioration of nerve processes and increased the turnover of transmitter substances. We confirmed that i.c.v.-infused GD3 and GM1 function as regulators of neurogenesis in mouse brains.25 The development of a safer, non-invasive, and more convenient procedure of ganglioside administration would be more acceptable for ganglioside treatment of neurodegenerative diseases. Intranasal delivery of cells to the brains in mouse models for neurodegenerative diseases has been reported with success.57 Intranasally infused GM1 was reported to improve cognitive function of rats with type 2 diabetes mellitus, even though it is not clear whether GM1 was delivered to the brain.58 In this study, we demonstrated that intranasally administered gangliosides could be delivered to the brain, including the substantia nigra, and functionally restored TH expression in dopaminergic neurons with a reduced aSyn level. In a rat experiment, GM1 was detected in the cerebrospinal fluid (CSF, 0.16 ± 0.03 μM) and plasma (3.88 ± 1.1 μM) after 30 min of intranasal infusion of 3.25 μmol of GM1.59 The CSF-to-plasma ratio of intranasally infused GM1 could be estimated to be about 4.12% ± 2.73%, and a 52% higher amount of GM1 in the CSF was found after intranasal administration than after intravenous injection, according to Kumbale et al.59 PD is characterized by pathological changes in nigral degeneration and striatal dopamine depletion, whereas PD is associated with hyposmia, urinary dysfunction, constipation, orthostatic hypotension, memory loss, depression, anxiety, pain, and sleep disturbances.60 For this reason, the peripheral nervous system needs to be targeted for treatment in addition to the central nervous system in PD. Intranasal administration can be a suitable route for efficient and non-invasive means for delivery of gangliosides to the brain as well as blood, reducing systemic exposure and potential undesirable side effects.
Every organelle in mammalian cells is surrounded by biological lipid membranes that define the individual cellular shape and help maintain cellular organization. Recently, membrane lipids have been revealed to be important not only for separating the cellular and the extracellular environments, but also for providing functional platforms for cellular molecules. For example, we have demonstrated that GD3 is the predominant ganglioside in NSCs,61 and GD3 is necessary to maintain the stemness of the cell. GD3 modulates self-renewal of NSCs by interacting with EGF receptors and regulates their downstream signaling.22 Furthermore, we discovered that GD3 is interacting with Drp1, a mitochondrial fission protein, thus regulating mitochondrial dynamics.33 It has also been reported that GM1 in plasma membranes are modulating GDNF receptors (RET and Gfa1) activities for their signaling on catecholaminergic neurons and the integrin-FAK signaling pathway to promote neurite outgrowth and cell migration.1,62, 63, 64 In the present study, we demonstrated that intranasal GM1 or GD3 administration restored the expression of the mitochondrial membrane protein VDAC1 in the brain of the A53T PD mouse. We have already found that GD3 binds to Drp1 and GM1 binds to other mitochondrial proteins (data not shown). These data suggest that GD3 and GM1 distinguishably interact with different mitochondrial proteins to regulate mitochondrial functions. We are currently investigating how gangliosides modulate mitochondrial activities during PD progression and resilience. Although the pathophysiology of PD is complex, mitochondrial dysfunction has been suggested to have an important role in PD progression. Dysfunctional mitochondrial dynamics are considered to contribute to neurodegenerative disorders, including AD and PD. Intriguingly, some pathogenic mutated genes identified in PD patients encode proteins important for mitochondrial fusion and fission dynamics. To cure the disease, targeting mitochondrial dysfunction in PD is an additional promising approach for the development of future therapies.
There are several lines of evidence revealing that the existence of GM1 in the nuclear membranes and the nuclear GM1 might exert biological functions such as calcium homeostasis and transcriptional regulation.30, 31, 32,65, 66, 67, 68 Recently, we demonstrated that nuclear GM1 binds to the NeuroD1 promoter region to enhance neuronal differentiation of NSCs. The nuclear envelope, in conjunction with the nuclear lamina and nuclear pore complexes, is a key structure to maintain chromatin architecture and cell-specific gene expression. In A53T PD mice, aSyn was found to accumulate on the nuclear membrane (Figure 3E), and this suggested that gene regulation in PD may be altered. Intranasal GM1 administration significantly reduced aSyn from the nuclear envelope and restored TH expression in murine brains (Figures 5 and 6A). We further demonstrated that GM1 augmented acetylation of histone H3 and recruited the dopaminergic transcriptional factor Nurr1 on the TH gene promoter (Figures 6C and 6D). In addition, we showed that exogenous pacGM1 can be detected in the nucleus and that nuclear GM1 binds with acetylated histone H3 (Figures 6F and 6G). Interestingly, GD3 decreased TH expression, Nurr1 nuclear localization, and recruitment of acetylated H3 and Pitx3 on the TH promoter (Figures 5 and 6). Our previous report clearly demonstrated that the GD3 increased expression of the NSC-associated transcription factor SRY (sex determining region Y)-box 2 (SOX2).25 Thus, it is likely that GD3 maintains stemness and inhibits further neuronal differentiation such as TH upregulation.
Since transcriptional activity of Nurr1 could be stimulated by GM1, it is possible that nuclear GM1 could modulate nuclear membrane and chromatin structure to enhance gene expression for augmenting dopaminergic neurons. Fundamental cellular processes are governed by changes in chromatin architecture that regulate neuronal gene expression during differentiation, pathogenesis, and resilience. Interestingly, our data suggest that nuclear gangliosides can modulate epigenetic gene expression for dopaminergic neurons. Ganglioside expression profiles are known to be closely associated with pathogenic mechanisms of neurodegenerative diseases in the central nervous system. Regulating gene expression by nuclear gangliosides is a novel mechanism to control cellular activity to rescue or protect neurons in PD and other neurodegenerative diseases.
Materials and methods
Antibodies
Mouse anti-aSyn (BD Biosciences, San Jose, CA, USA, 610787), rabbit anti-phospho-S129 aSyn (Abcam, Cambridge, MA, USA, ab51253), rabbit anti-TH (Millipore, St. Louis, MO, USA, AB152), mouse anti-TH (Invitrogen from Thermo Fisher Scientific, Rockford IL, USA, MA5-35009), goat anti-Nurr1 (R&D Systems, Minneapolis, MN, USA, AF2156), mouse anti-lamin B1 (Abcam, ab20396), rabbit anti-lamin B1 (Abcam, ab16048), rabbit anti-VDAC1 (Cell Signaling Technology, Danvers, MA, USA, D73D12), mouse anti-GM1 (TCI America, Portland, OR, USA, A2505), rabbit anti-actin (Sigma, St. Louis, MO, USA, A2066), rabbit anti-acetylated histone H3 (Millipore, 06-599), rabbit anti-PITX3 (Thermo Fisher Scientific, 382850), Alexa Fluor 488-conjugated goat anti-mouse immunoglobulin G (IgG) (Invitrogen, A28175), Alexa Fluor 488-conjugated goat anti-rabbit IgG (Invitrogen, A11008), Alexa Fluor 568-conjugated goat anti-rabbit IgG (Invitrogen, A11011), Alexa Fluor 647-conjugated goat anti-rabbit IgG (Invitrogen, A27040), Alexa Fluor 488-conjugated donkey anti-rabbit IgG (Invitrogen, R37118), Alexa Fluor 488-conjugated donkey anti-mouse IgG (Invitrogen, R37114), anti-mouse IgG-horseradish peroxidase (HRP) (Thermo Fisher Scientific, 45000692), and anti-rabbit IgG-HRP (Sigma-Aldrich, GENA934) antibodies were purchased.
Experimental models
All animal experiments were performed with animal protocols (references AUP 2009-0240 and 2014-0694) approved by the Institutional Animal Care and Use Committee (IACUC) at Augusta University (AU) according to the National Institutes of Health (NIH) guidelines. Animal protocols were approved for the described mice. For the PD model, mice expressing A53T mutant human aSyn under the murine prion promoter were utilized. This mouse line (B6.Cg-2310039L15RikTg(Prnp-SNCA∗A53T)23Mkle/J) (The Jackson Laboratory, Bar Harbor, ME, USA, stock no 006823) is referred to as the A53T PD mouse. C57B6/6 mice were obtained from The Jackson Laboratory (stock no. 000664). For GM2S-KO mice, the original GM2S-KO mice (GM2 synthase KO, B4galnt1-KO, B6;129S-B4galnt1tm1Rlp/Mmmh) (MMRRC stock no. 000036-MU) and their WT mates were kindly provided by Dr. Robert Ledeen (Rutgers New Jersey Medical School, NJ, USA) and crossed to generate the heterozygous mice. Then, the heterozygous male and female mice were mated, and PCR screening was performed for genotyping. Littermates were used as controls. All mice were housed in standard conditions with food and water provided ad libitum and maintained on a 12-h dark/12-h light cycle.
Mouse Neuro-2a neuroblastoma cells (ATCC, Manassas, VA, USA, CCL-131) were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum, 2 mM glutamine, and penicillin/streptomycin at 37°C, 5% CO2.
Intranasal ganglioside infusion
The acquisition, care, and experimental treatment of animals used in this study were in compliance with the NIH guidelines as published in the Guide for the Care and Use of Laboratory Animals. Gangliosides (GD3 or GM1; 5 mg/kg/day) were infused intranasally69 with a small pipette (each 6 μL into the right and left nares twice, or 24 μL per day) into 8-month-old WT (C57B6/J), A53T PD, and GM2S-KO mice daily for 7–28 days. GD3 and GM1 used in this study were isolated from either bovine buttermilk or brains in our laboratory by established procedures.70, 71, 72 Gangliosides, being amphipathic, were easily dissolved in saline. The placebo group received saline infusions. At first, to provide direct evidence for ganglioside administration into brain, mice totally deficient in GM1 (8-month-old GM2S-KO mice) were intranasally administered GM1 (0.5 or 5 mg/kg/day) for 7 days. Brain-delivered GM1 was analyzed by immunohistochemistry and TLC. After confirmation that ganglioside reached the brain of GM2S-KO mice by the intranasal route, 8-month-old A53T PD mice were utilized for ganglioside treatments (28 days). Animals were divided into five groups: (1) a WT with saline infusion group; (2) a A53T PD with saline infusion group; (3) a A53T PD with GD3 (5 mg/kg/day) infusion group; (4) a A53T PD with GM1 (5 mg/kg/day) infusion group; and (5) a GD3 infusion plus GM1 infusion group (GD3 at 5 mg/kg/day for 14 days, then GM1 at 5 mg/kg/day for the other 14 days). Each group consisted of n = 3–4 animals. Also, 5 mg/kg/day GM1 was intranasally administered to GM2S-KO mice for 28 days to analyze TH expression.
Immunohistochemistry
Mice were transcardially perfused with phosphate-buffered saline (PBS, pH 7.4) and 4% paraformaldehyde (PFA). The brains were removed and post-fixed with the same fixative overnight, followed by cryoprotection with 30% sucrose in PBS, and the solution was changed more than three times, at 4°C. After embedding in Tissue-Tek OCT compound (Sakura Finetek, Torrance, CA, USA), the brains were quickly frozen in liquid nitrogen. Cryosectioning was performed (20-μm-thick sections) using a cryostat (Leica, Wetzlar, Germany). For co-staining of GM1 and TH, sections were permeabilized with PBS containing 0.5% Triton X-100 for 5 min, followed by blocking with PBS containing 1% bovine serum albumin (BSA) for 30 min at room temperature, and then incubated with Alexa Fluor 594-conjugated cholera toxin B subunit (CtxB) (1:5,000, Invitrogen, C22842) and rabbit anti-TH antibody (1:500, Millipore, AB152) overnight. For co-immunostaining aSyn and phospho-S129 aSyn or lamin B1, antigen retrieval was performed by autoclave treatment in 10 mM citrate buffer (pH 3.0) at 121°C for 25 min, followed by permeabilization and blocking as described above. Then, sections were subjected to reaction with mouse anti-aSyn antibody (1:100, BD Biosciences, 610787) and rabbit anti-phospho-S129 aSyn antibody (1:100, Abcam, ab51253) or rabbit anti-lamin B1 antibody (1:100, Abcam, ab16048). For staining VDAC1 or co-staining Nurr1 and TH, microwave treatment was performed in pre-boiled 10 mM citrate buffer (pH 6.0) for 5 min, followed by incubation with rabbit anti-VDAC1 antibody (1:100, Cell Signaling Technology, D73D12) and mouse anti-TH antibody (1:100, Invitrogen, MA5-35009), or goat anti-Nurr1 antibody (1:100, R&D Systems, AF2156) and rabbit anti-TH antibody (1:500, Millipore, AB152). Each reaction with primary antibodies was followed by incubation with Alexa Fluor-conjugated secondary antibody for 2 h at room temperature, and then nuclei counterstaining was performed with 1 μg/mL 4′,6-diamidino-2-phenylindole (DAPI) (Thermo Fisher Scientific, D1306) for 30 min. After every incubation with antibodies or chemicals, sections were washed three times with PBS. Specimens were mounted with VectaMount (Vector Laboratories, Burlingame, CA, USA).
Microscopy and image processing
Confocal images were acquired using a Zeiss LSM 700 with a ×63 oil objective (Zeiss, Land Baden-Württemberg, Germany) with identical acquisition settings. Zen software was used for initial image acquisition of the fluorescent signals. Quantitative analyses of digital images were performed using Fiji (NIH, Bethesda, MD, USA). Serial z axis images were converted into stacks, and average intensities were measured within the indicative aSyn-, TH-, and Nurr1-stained subcellular or nuclear area (n = 3–4 mice, 7–12 regions per each condition). To generate unbiased data, the blinding procedures and randomized field approach for images were performed.
Ganglioside isolation and TLC
Gangliosides were isolated from cortical tissue from WT and GM2S-KO mice as previously described73 with some modifications.74 Briefly, total lipids were extracted from brain tissues with chloroform-methanol-water (30:60:8 [v/v]; solvent A) after cardiovascular perfusion with PBS. Then, the extracts were evaporated and dissolved in 1 mL of solvent A and applied to a diethylaminoethyl (DEAE)-Sephadex A-25 column (acetate form, 1-mL bed volume), followed by elution with 10 mL of solvent A to remove the neutral lipids. The acidic lipid fraction, containing gangliosides, was then eluted with 10 mL of chloroform-methanol-0.8 M sodium acetate in water (30:60:8 [v/v]; solvent B), followed by desalting using Sep-Pak cartridge column chromatography (Waters, Milford, MA, USA).75 Gangliosides were applied to a high-performance TLC (HPTLC, aluminum HPTLC silica gel; Merck, Darmstadt, Germany) plate and developed with the solvent system of chloroform-methanol-water containing 0.2% CaCl2·H2O (50:45:10 [v/v]). After developing the HPTLC plate described above, the plate was coated in a solution of n-hexane containing 0.02% poly(isobutyl methacrylate) for 1 min. After drying, the plate was then incubated in blocking buffer (1% BSA/1% polyvinylpyrrolidine) at room temperature for 30 min. Staining of GM1 on TLC plates was performed using a protocol for immunostaining of lipids except that HRP-labeled CtxB (Invitrogen, C34780) was used instead of antibodies.64 Plates were rinsed with washing buffer (PBS/1% Tween 20) and incubated with CtxB-HRP at 4°C overnight. After washing with washing buffer, signals were visualized with enhanced chemiluminescence reagent (PerkinElmer Life and Analytical Sciences, Boston, MA, USA).
Western blotting
The entire substantia nigra region was isolated by dissection under a SZX7 stereo microscope (Olympus, Tokyo, Japan). Tissue blocks were lysed in radioimmunoprecipitation assay (RIPA) buffer containing 50 mM Tris-HCl, 150 mM NaCl, 5 mM NaF, 1 mM Na3VO4, 1% Nonidet P-40 (NP-40), 0.5% sodium deoxycholate, and 1% SDS (pH 7.5), supplemented with a complete protease inhibitor cocktail (Roche Applied Science, Indianapolis, IN, USA). The protein concentrations were measured using a bicinchoninic acid protein assay kit (Thermo Fisher Scientific, Rockford IL, USA). Proteins (10 μg) were separated by SDS-PAGE (10% gel) under reducing conditions and transferred to polyvinylidene fluoride (PVDF) membranes. The membranes were probed with primary antibodies for aSyn, phospho-S129 aSyn, VDAC1, and actin, followed by appropriate secondary antibodies conjugated with HRP. Signals were visualized with Western Lightning western blot chemiluminescence reagent (PerkinElmer Life and Analytical Sciences, Waltham, MA, USA).
Gene expression analysis by qPCR
Total RNA samples were isolated from substantia nigra of mouse brains or cultured Neuro 2a cells using TRIzol reagent (Life Technologies from Thermo Fisher Scientific). Neuro 2a cells were untreated or treated with 5 μM GD3 or GM1 in B27-supplemented Neurobasal-A medium for 24 h. cDNAs were synthesized based on the total RNAs as templates using MultiScribe reverse transcriptase (Applied Biosystems, Waltham, MA, USA). qPCR was performed with run RT2 SYBR Green qPCR master mix (QIAGEN, Hilden, Germany) on the CFX96 system (Bio-Rad, Hercules, CA, USA). The relative expression levels of TH were normalized to the actin transcript level. The normalized value from control (WT + saline, or no treated cells) is defined as 1.0. The following primers were used: TH forward, 5′-CACTATGCCCACCCCCAG-3′, reverse: 5′-CGCCGTCCAATGAACCTT-3′; actin forward, 5′-CTAAGGCCAACCGTGAAAAGAT-3′, reverse, 5′-CACAGCCTGGATGGCTACGT-3′.
ChIP assay
ChIP assays were performed on cell lysates as previously described.31,32,76 Briefly, Neuro 2a cells, untreated or treated with 5 μM GD3 or GM1 for 24 h, were incubated in 1% PFA for 10 min at ambient temperature to crosslink the interacting partners. Cells with the cross-linked complexes were subjected to lysis by sonication with six 20-s pulses at the power scale 7 controlled by a sonicator (Sonic dismembrator model 100, Fisher Scientific). After centrifugation at 15,000 × g for 10 min, the supernatants were collected for the following experiments. Immunoprecipitation was carried out with the following specific antibodies bound on protein G-conjugated magnetic beads (Millipore): anti-acetylated histone H3 (Millipore), anti-Nurr1 (R&D Systems), or anti-PITX3 (Thermo Fisher Scientific) antibodies. The amounts of the co-precipitated DNA fragments of TH promoter (+0), the Nurr1 binding site on the TH promoter (TH-NBS), and the PITX3 binding site on TH promoter (TH-PBS) were analyzed by qPCR. After normalization against GAPDH, the value of no treated cells was defined as 1.0. The following primers were used: TH forward, 5′-TAAGAGGCCGCCTGCCTGGC-3′, reverse, 5′-GTCTCGTCCTATGGTTCGTC-3′; TH NBS forward, 5′-TCCAGGAGAACAGACGCCAGC-3′, reverse, 5′-GCCAGGCTGAAGGCAAGCACA-3′; TH PBS forward, 5′-TTCCATGAAAGCACAACTGGC-3′, reverse, 5′-CAGGGTCGGCTGCTGAGGAT-3′; GAPDH forward, 5′-ACCAGGGAGGGCTGCAGTCC, reverse, 5′-TCAGTTCGGAGCCCACACGC-3′.
Photoclick GM1 and isolation of nuclei from cultured cells
Cells were incubated with 1 μM pacGM1 (Avanti Polar Lipids, Birmingham, AL, USA, 900603) for 24 h. Cells were UV irradiated at 365 nm for 30 min on ice. Nuclei were isolated using modification of previously described procedures.77,78 The scraped cells in ice-cold PBS were pelleted at 250 × g, 4°C for 5 min. The pellets were suspended in 300 μL of TM buffer (20 mM Tris-Cl [pH 7.5], 1 mM MgCl2) supplemented with 0.1% Triton X-100 and incubated on ice for 30 min. Nuclei were recovered by centrifugation at 800 × g, 4°C. The pellet was homogenized in 2 M sucrose in TM buffer and centrifuged at 100,000 × g, 4°C for 25 min. To minimize contamination by other subcellular organelles, the ultracentrifuged pellet was homogenized in 0.32 M sucrose in TM buffer, overlaid on 2.2 M sucrose in TM buffer, and recentrifuged as before. The isolated nuclei were placed on poly-d-lysine-coated glass slides and fixed in 4% PFA in PBS for 20 min at room temperature. Nuclei were washed with PBS, and the click reaction was performed using the Invitrogen Click-iT cell reaction buffer kit (Thermo Fisher Scientific, C10269) using TAMRA (carboxytetramethylrhodamine)-azide-desthiobiotin (Click Chemistry Tools, Scottsdale, AZ, USA, 1110-5) as fluorophores following the manufacturers’ protocols. Zeiss LSM 700 confocal microscopy was performed as described above (see Microscopy and image processing).
PLAs on purified nuclei of adult mouse brain
Nuclei were isolated from WT mouse cerebrum (8-month-old mice) using modification of previously described procedures.77,79 Brain tissue was homogenized in 1.3 M sucrose in TM buffer, and the homogenate was centrifuged at 100,000 × g, 4°C for 25 min. The ultracentrifuged pellet was homogenized in 0.32 M sucrose in TM buffer, overlaid on 2.2 M sucrose in TM buffer, and recentrifuged as before. The isolated nuclei were fixed on cover glass as described above. Nonspecific binding sites were blocked with Duolink PLA blocking solution for 1 h at 37°C. The primary antibodies used were mouse anti-GM1 (1:100) and rabbit anti-acetylated histone H3 (1:100). Secondary PLA probes were anti-rabbit PLUS affinity-purified donkey anti-rabbit IgG (H+L) and anti-mouse MINUS affinity-purified donkey anti-mouse IgG (H+L), which were diluted 1:5 in 1× antibody diluent buffer and samples were incubated for 1 h at 37°C. After washing, ligation and amplification steps were performed following the manufacturer’s protocol. Zeiss LSM 700 confocal microscopy was performed as above (see Microscopy and image processing). A PLA Duolink in situ red starter kit mouse/rabbit was purchased from MilliporeSigma (DUO92101).
Statistical analysis
All statistical procedures were performed using GraphPad Prism 9 (GraphPad, San Diego, CA, USA). Normality and homogeneity of variances of datasets were checked by a Kolmogorov-Smirnov test and Brown-Forsythe test, respectively. When datasets passed these tests, a one-way ANOVA with a Tukey’s multiple comparison test was performed. In all cases, p values are shown in the figure legends, and p < 0.05 was regarded as significant. All graphs depict mean ± SEM.
Acknowledgments
This work was supported by a National Institute of Neurological Disorders and Stroke grant (R01 NS100839 to R.K.Y.) and by a Sheffield Memorial Grant of the CSRA Parkinson’s Disease Support Group (to R.K.Y.). We thank Dr. Toshio Ariga and Dr. Dongpei Li for excellent technical support. The authors also wish to acknowledge the excellent infrastructural support of the Department of Neuroscience and Regenerative Medicine (Chair Dr. Xin-Yun Lu), Medical College of Georgia at Augusta University, which made this investigation possible.
Author contributions
Y.I., T.F., J.C.M., and R.K.Y. provided conceptual framework and participated in the design of experiments and interpretation of the results; Y.I. and T.F. performed experiments and analyzed the data; Y.I. and T.F. prepared figures; Y.I., T.F., J.C.M., and R.K.Y. were actively involved in regular discussions that determined the research direction; R.K.Y. and Y.I. drafted the manuscript; and Y.I., T.F., J.C.M., and R.K.Y. approved the final version of manuscript.
Declaration of interests
The authors declare no competing interests.
Contributor Information
Yutaka Itokazu, Email: yitokazu@augusta.edu.
Robert K. Yu, Email: ryu@augusta.edu.
References
- 1.Hadaczek P., Wu G., Sharma N., Ciesielska A., Bankiewicz K., Davidow A.L., Lu Z.H., Forsayeth J., Ledeen R.W. GDNF signaling implemented by GM1 ganglioside; failure in Parkinson’s disease and GM1-deficient murine model. Exp. Neurol. 2015;263:177–189. doi: 10.1016/j.expneurol.2014.10.010. [DOI] [PubMed] [Google Scholar]
- 2.Ledeen R.W., Wu G. Gangliosides, α-synuclein, and Parkinson’s disease. Prog. Mol. Biol. Transl. Sci. 2018;156:435–454. doi: 10.1016/bs.pmbts.2017.12.009. [DOI] [PubMed] [Google Scholar]
- 3.Wu G., Lu Z.H., Seo J.H., Alselehdar S.K., DeFrees S., Ledeen R.W. Mice deficient in GM1 manifest both motor and non-motor symptoms of Parkinson’s disease; successful treatment with synthetic GM1 ganglioside. Exp. Neurol. 2020;329:113284. doi: 10.1016/j.expneurol.2020.113284. [DOI] [PubMed] [Google Scholar]
- 4.Schneider J.S., Gollomp S.M., Sendek S., Colcher A., Cambi F., Du W. A randomized, controlled, delayed start trial of GM1 ganglioside in treated Parkinson’s disease patients. J. Neurol. Sci. 2013;324:140–148. doi: 10.1016/j.jns.2012.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schneider J.S., Cambi F., Gollomp S.M., Kuwabara H., Brašić J.R., Leiby B., Sendek S., Wong D.F. GM1 ganglioside in Parkinson’s disease: Pilot study of effects on dopamine transporter binding. J. Neurol. Sci. 2015;356:118–123. doi: 10.1016/j.jns.2015.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schneider J.S., Aras R., Williams C.K., Koprich J.B., Brotchie J.M., Singh V. GM1 ganglioside modifies α-synuclein toxicity and is neuroprotective in a rat α-synuclein model of Parkinson’s disease. Sci. Rep. 2019;9:8362. doi: 10.1038/s41598-019-42847-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bartels T., Kim N.C., Luth E.S., Selkoe D.J. N-alpha-acetylation of α-synuclein increases its helical folding propensity, GM1 binding specificity and resistance to aggregation. PLoS ONE. 2014;9:e103727. doi: 10.1371/journal.pone.0103727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Martinez Z., Zhu M., Han S., Fink A.L. GM1 specifically interacts with α-synuclein and inhibits fibrillation. Biochemistry. 2007;46:1868–1877. doi: 10.1021/bi061749a. [DOI] [PubMed] [Google Scholar]
- 9.Kumagai T., Tanaka M., Yokoyama M., Sato T., Shinkai T., Furukawa K. Early lethality of β-1,4-galactosyltransferase V-mutant mice by growth retardation. Biochem. Biophys. Res. Commun. 2009;379:456–459. doi: 10.1016/j.bbrc.2008.12.078. [DOI] [PubMed] [Google Scholar]
- 10.Nishie T., Hikimochi Y., Zama K., Fukusumi Y., Ito M., Yokoyama H., Naruse C., Ito M., Asano M. β4-galactosyltransferase-5 is a lactosylceramide synthase essential for mouse extra-embryonic development. Glycobiology. 2010;20:1311–1322. doi: 10.1093/glycob/cwq098. [DOI] [PubMed] [Google Scholar]
- 11.Yamashita T., Wada R., Sasaki T., Deng C., Bierfreund U., Sandhoff K., Proia R.L. A vital role for glycosphingolipid synthesis during development and differentiation. Proc. Natl. Acad. Sci. USA. 1999;96:9142–9147. doi: 10.1073/pnas.96.16.9142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu R.K., Itokazu Y. Glycolipid and glycoprotein expression during neural development. Adv. Neurobiol. 2014;9:185–222. doi: 10.1007/978-1-4939-1154-7_9. [DOI] [PubMed] [Google Scholar]
- 13.Yu R.K., Nakatani Y., Yanagisawa M. The role of glycosphingolipid metabolism in the developing brain. J. Lipid Res. 2009;50(Suppl):S440–S445. doi: 10.1194/jlr.R800028-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yu R.K., Tsai Y.T., Ariga T., Yanagisawa M. Structures, biosynthesis, and functions of gangliosides—An overview. J. Oleo Sci. 2011;60:537–544. doi: 10.5650/jos.60.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu R.K., Ando S. Structures of some new complex gangliosides of fish brain. Adv. Exp. Med. Biol. 1980;125:33–45. doi: 10.1007/978-1-4684-7844-0_5. [DOI] [PubMed] [Google Scholar]
- 16.Simpson M.A., Cross H., Proukakis C., Priestman D.A., Neville D.C., Reinkensmeier G., Wang H., Wiznitzer M., Gurtz K., Verganelaki A. Infantile-onset symptomatic epilepsy syndrome caused by a homozygous loss-of-function mutation of GM3 synthase. Nat. Genet. 2004;36:1225–1229. doi: 10.1038/ng1460. [DOI] [PubMed] [Google Scholar]
- 17.Lee J.S., Yoo Y., Lim B.C., Kim K.J., Song J., Choi M., Chae J.H. GM3 synthase deficiency due to ST3GAL5 variants in two Korean female siblings: Masquerading as Rett syndrome-like phenotype. Am. J. Med. Genet. A. 2016;170:2200–2205. doi: 10.1002/ajmg.a.37773. [DOI] [PubMed] [Google Scholar]
- 18.Zunke F., Moise A.C., Belur N.R., Gelyana E., Stojkovska I., Dzaferbegovic H., Toker N.J., Jeon S., Fredriksen K., Mazzulli J.R. Reversible conformational conversion of α-synuclein into toxic assemblies by glucosylceramide. Neuron. 2018;97:92–107.e10. doi: 10.1016/j.neuron.2017.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boukhris A., Schule R., Loureiro J.L., Lourenço C.M., Mundwiller E., Gonzalez M.A., Charles P., Gauthier J., Rekik I., Acosta Lebrigio R.F. Alteration of ganglioside biosynthesis responsible for complex hereditary spastic paraplegia. Am. J. Hum. Genet. 2013;93:118–123. doi: 10.1016/j.ajhg.2013.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yamashita T., Wu Y.P., Sandhoff R., Werth N., Mizukami H., Ellis J.M., Dupree J.L., Geyer R., Sandhoff K., Proia R.L. Interruption of ganglioside synthesis produces central nervous system degeneration and altered axon-glial interactions. Proc. Natl. Acad. Sci. USA. 2005;102:2725–2730. doi: 10.1073/pnas.0407785102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kawai H., Allende M.L., Wada R., Kono M., Sango K., Deng C., Miyakawa T., Crawley J.N., Werth N., Bierfreund U. Mice expressing only monosialoganglioside GM3 exhibit lethal audiogenic seizures. J. Biol. Chem. 2001;276:6885–6888. doi: 10.1074/jbc.C000847200. [DOI] [PubMed] [Google Scholar]
- 22.Wang J., Yu R.K. Interaction of ganglioside GD3 with an EGF receptor sustains the self-renewal ability of mouse neural stem cells in vitro. Proc. Natl. Acad. Sci. USA. 2013;110:19137–19142. doi: 10.1073/pnas.1307224110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang J., Cheng A., Wakade C., Yu R.K. Ganglioside GD3 is required for neurogenesis and long-term maintenance of neural stem cells in the postnatal mouse brain. J. Neurosci. 2014;34:13790–13800. doi: 10.1523/JNEUROSCI.2275-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Itokazu Y., Wang J., Yu R.K. Gangliosides in nerve cell specification. Prog. Mol. Biol. Transl. Sci. 2018;156:241–263. doi: 10.1016/bs.pmbts.2017.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Itokazu Y., Li D., Yu R.K. Intracerebroventricular infusion of gangliosides augments the adult neural stem cell pool in mouse brain. ASN Neuro. 2019;11 doi: 10.1177/1759091419884859. 1759091419884859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu G., Lu Z.H., Kulkarni N., Ledeen R.W. Deficiency of ganglioside GM1 correlates with Parkinson’s disease in mice and humans. J. Neurosci. Res. 2012;90:1997–2008. doi: 10.1002/jnr.23090. [DOI] [PubMed] [Google Scholar]
- 27.Ariga T. Pathogenic role of ganglioside metabolism in neurodegenerative diseases. J. Neurosci. Res. 2014;92:1227–1242. doi: 10.1002/jnr.23411. [DOI] [PubMed] [Google Scholar]
- 28.Ariga T. The pathogenic role of ganglioside metabolism in Alzheimer’s disease-cholinergic neuron-specific gangliosides and neurogenesis. Mol. Neurobiol. 2017;54:623–638. doi: 10.1007/s12035-015-9641-0. [DOI] [PubMed] [Google Scholar]
- 29.Ledeen R.W., Wu G. The multi-tasked life of GM1 ganglioside, a true factotum of nature. Trends Biochem. Sci. 2015;40:407–418. doi: 10.1016/j.tibs.2015.04.005. [DOI] [PubMed] [Google Scholar]
- 30.Itokazu Y., Tsai Y.T., Yu R.K. Epigenetic regulation of ganglioside expression in neural stem cells and neuronal cells. Glycoconj. J. 2017;34:749–756. doi: 10.1007/s10719-016-9719-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tsai Y.T., Itokazu Y., Yu R.K. GM1 ganglioside is involved in epigenetic activation loci of neuronal cells. Neurochem. Res. 2016;41:107–115. doi: 10.1007/s11064-015-1742-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tsai Y.T., Yu R.K. Epigenetic activation of mouse ganglioside synthase genes: implications for neurogenesis. J. Neurochem. 2014;128:101–110. doi: 10.1111/jnc.12456. [DOI] [PubMed] [Google Scholar]
- 33.Tang F.L., Wang J., Itokazu Y., Yu R.K. Ganglioside GD3 regulates dendritic growth in newborn neurons in adult mouse hippocampus via modulation of mitochondrial dynamics. J. Neurochem. 2021;156:819–833. doi: 10.1111/jnc.15137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pires A., Fortuna A., Alves G., Falcão A. Intranasal drug delivery: How, why and what for? J. Pharm. Pharm. Sci. 2009;12:288–311. doi: 10.18433/j3nc79. [DOI] [PubMed] [Google Scholar]
- 35.Lee M.K., Stirling W., Xu Y., Xu X., Qui D., Mandir A.S., Dawson T.M., Copeland N.G., Jenkins N.A., Price D.L. Human α-synuclein-harboring familial Parkinson’s disease-linked Ala-53 → Thr mutation causes neurodegenerative disease with α-synuclein aggregation in transgenic mice. Proc. Natl. Acad. Sci. USA. 2002;99:8968–8973. doi: 10.1073/pnas.132197599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Anderson J.P., Walker D.E., Goldstein J.M., de Laat R., Banducci K., Caccavello R.J., Barbour R., Huang J., Kling K., Lee M. Phosphorylation of Ser-129 is the dominant pathological modification of α-synuclein in familial and sporadic Lewy body disease. J. Biol. Chem. 2006;281:29739–29752. doi: 10.1074/jbc.M600933200. [DOI] [PubMed] [Google Scholar]
- 37.Fujiwara H., Hasegawa M., Dohmae N., Kawashima A., Masliah E., Goldberg M.S., Shen J., Takio K., Iwatsubo T. α-Synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell Biol. 2002;4:160–164. doi: 10.1038/ncb748. [DOI] [PubMed] [Google Scholar]
- 38.Bernal-Conde L.D., Ramos-Acevedo R., Reyes-Hernández M.A., Balbuena-Olvera A.J., Morales-Moreno I.D., Argüero-Sánchez R., Schüle B., Guerra-Crespo M. Alpha-synuclein physiology and pathology: A perspective on cellular structures and organelles. Front. Neurosci. 2020;13:1399. doi: 10.3389/fnins.2019.01399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen V., Moncalvo M., Tringali D., Tagliafierro L., Shriskanda A., Ilich E., Dong W., Kantor B., Chiba-Falek O. The mechanistic role of α-synuclein in the nucleus: Impaired nuclear function caused by familial Parkinson’s disease SNCA mutations. Hum. Mol. Genet. 2020;29:3107–3121. doi: 10.1093/hmg/ddaa183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wong Y.C., Krainc D. α-Synuclein toxicity in neurodegeneration: Mechanism and therapeutic strategies. Nat. Med. 2017;23:1–13. doi: 10.1038/nm.4269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Devi L., Raghavendran V., Prabhu B.M., Avadhani N.G., Anandatheerthavarada H.K. Mitochondrial import and accumulation of α-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J. Biol. Chem. 2008;283:9089–9100. doi: 10.1074/jbc.M710012200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li W.W., Yang R., Guo J.C., Ren H.M., Zha X.L., Cheng J.S., Cai D.F. Localization of α-synuclein to mitochondria within midbrain of mice. Neuroreport. 2007;18:1543–1546. doi: 10.1097/WNR.0b013e3282f03db4. [DOI] [PubMed] [Google Scholar]
- 43.Luth E.S., Stavrovskaya I.G., Bartels T., Kristal B.S., Selkoe D.J. Soluble, prefibrillar α-synuclein oligomers promote complex I-dependent, Ca2+-induced mitochondrial dysfunction. J. Biol. Chem. 2014;289:21490–21507. doi: 10.1074/jbc.M113.545749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Perfeito R., Lázaro D.F., Outeiro T.F., Rego A.C. Linking alpha-synuclein phosphorylation to reactive oxygen species formation and mitochondrial dysfunction in SH-SY5Y cells. Mol. Cell. Neurosci. 2014;62:51–59. doi: 10.1016/j.mcn.2014.08.002. [DOI] [PubMed] [Google Scholar]
- 45.Lucki N.C., Sewer M.B. Nuclear sphingolipid metabolism. Annu. Rev. Physiol. 2012;74:131–151. doi: 10.1146/annurev-physiol-020911-153321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kong J.N., Zhu Z., Itokazu Y., Wang G., Dinkins M.B., Zhong L., Lin H.P., Elsherbini A., Leanhart S., Jiang X. Novel function of ceramide for regulation of mitochondrial ATP release in astrocytes. J. Lipid Res. 2018;59:488–506. doi: 10.1194/jlr.M081877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Di Scala C., Yahi N., Boutemeur S., Flores A., Rodriguez L., Chahinian H., Fantini J. Common molecular mechanism of amyloid pore formation by Alzheimer’s β-amyloid peptide and α-synuclein. Sci. Rep. 2016;6:28781. doi: 10.1038/srep28781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Park J.Y., Kim K.S., Lee S.B., Ryu J.S., Chung K.C., Choo Y.K., Jou I., Kim J., Park S.M. On the mechanism of internalization of alpha-synuclein into microglia: roles of ganglioside GM1 and lipid raft. J. Neurochem. 2009;110:400–411. doi: 10.1111/j.1471-4159.2009.06150.x. [DOI] [PubMed] [Google Scholar]
- 49.Seyfried T.N., Choi H., Chevalier A., Hogan D., Akgoc Z., Schneider J.S. Sex-related abnormalities in substantia nigra lipids in Parkinson’s disease. ASN Neuro. 2018;10 doi: 10.1177/1759091418781889. 1759091418781889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wu G., Lu Z.H., Kulkarni N., Amin R., Ledeen R.W. Mice lacking major brain gangliosides develop parkinsonism. Neurochem. Res. 2011;36:1706–1714. doi: 10.1007/s11064-011-0437-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Agnati L.F., Fuxe K., Calza L., Benfenati F., Cavicchioli L., Toffano G., Goldstein M. Gangliosides increase the survival of lesioned nigral dopamine neurons and favour the recovery of dopaminergic synaptic function in striatum of rats by collateral sprouting. Acta Physiol. Scand. 1983;119:347–363. doi: 10.1111/j.1748-1716.1983.tb07363.x. [DOI] [PubMed] [Google Scholar]
- 52.Schneider J.S., Roeltgen D.P., Rothblat D.S., Chapas-Crilly J., Seraydarian L., Rao J. GM1 ganglioside treatment of Parkinson’s disease: An open pilot study of safety and efficacy. Neurology. 1995;45:1149–1154. doi: 10.1212/wnl.45.6.1149. [DOI] [PubMed] [Google Scholar]
- 53.Schneider J.S., Sendek S., Daskalakis C., Cambi F. GM1 ganglioside in Parkinson’s disease: Results of a five year open study. J. Neurol. Sci. 2010;292:45–51. doi: 10.1016/j.jns.2010.02.009. [DOI] [PubMed] [Google Scholar]
- 54.Schneider J.S., Yuwiler A. GM1 ganglioside treatment promotes recovery of striatal dopamine concentrations in the mouse model of MPTP-induced parkinsonism. Exp. Neurol. 1989;105:177–183. doi: 10.1016/0014-4886(89)90117-9. [DOI] [PubMed] [Google Scholar]
- 55.Revunov E., Johnström P., Arakawa R., Malmquist J., Jucaite A., Defay T., Takano A., Schou M. First radiolabeling of a ganglioside with a positron emitting radionuclide: In vivo PET demonstrates low exposure of radiofluorinated GM1 in non-human primate brain. ACS Chem. Neurosci. 2020;11:1245–1249. doi: 10.1021/acschemneuro.0c00161. [DOI] [PubMed] [Google Scholar]
- 56.Svennerholm L., Bråne G., Karlsson I., Lekman A., Ramström I., Wikkelsö C. Alzheimer disease—Effect of continuous intracerebroventricular treatment with GM1 ganglioside and a systematic activation programme. Dement. Geriatr. Cogn. Disord. 2002;14:128–136. doi: 10.1159/000063604. [DOI] [PubMed] [Google Scholar]
- 57.Danielyan L., Beer-Hammer S., Stolzing A., Schäfer R., Siegel G., Fabian C., Kahle P., Biedermann T., Lourhmati A., Buadze M. Intranasal delivery of bone marrow-derived mesenchymal stem cells, macrophages, and microglia to the brain in mouse models of Alzheimer’s and Parkinson’s disease. Cell Transplant. 2014;23(Suppl 1):S123–S139. doi: 10.3727/096368914X684970. [DOI] [PubMed] [Google Scholar]
- 58.Sukhov I.B., Lebedeva M.F., Zakharova I.O., Derkach K.V., Bayunova L.V., Zorina I.I., Avrova N.F., Shpakov A.O. Intranasal administration of insulin and gangliosides improves spatial memory in rats with neonatal type 2 diabetes mellitus. Bull. Exp. Biol. Med. 2020;168:317–320. doi: 10.1007/s10517-020-04699-8. [DOI] [PubMed] [Google Scholar]
- 59.Kumbale R., Frey W.H., Wilson S., Rahman Y.E. GM1 delivery to the CSF via the olfactory pathway. Drug Deliv. 1999;6:23–30. [Google Scholar]
- 60.Tolosa E., Garrido A., Scholz S.W., Poewe W. Challenges in the diagnosis of Parkinson’s disease. Lancet Neurol. 2021;20:385–397. doi: 10.1016/S1474-4422(21)00030-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nakatani Y., Yanagisawa M., Suzuki Y., Yu R.K. Characterization of GD3 ganglioside as a novel biomarker of mouse neural stem cells. Glycobiology. 2010;20:78–86. doi: 10.1093/glycob/cwp149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Palazzo A.F., Eng C.H., Schlaepfer D.D., Marcantonio E.E., Gundersen G.G. Localized stabilization of microtubules by integrin- and FAK-facilitated Rho signaling. Science. 2004;303:836–839. doi: 10.1126/science.1091325. [DOI] [PubMed] [Google Scholar]
- 63.Wu G., Lu Z.H., Obukhov A.G., Nowycky M.C., Ledeen R.W. Induction of calcium influx through TRPC5 channels by cross-linking of GM1 ganglioside associated with α5β1 integrin initiates neurite outgrowth. J. Neurosci. 2007;27:7447–7458. doi: 10.1523/JNEUROSCI.4266-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Itokazu Y., Pagano R.E., Schroeder A.S., O’Grady S.M., Limper A.H., Marks D.L. Reduced GM1 ganglioside in CFTR-deficient human airway cells results in decreased β1-integrin signaling and delayed wound repair. Am. J. Physiol. Cell Physiol. 2014;306:C819–C830. doi: 10.1152/ajpcell.00168.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Parkinson M.E., Smith C.G., Garland P.B., van Heyningen S. Identification of cholera toxin-binding sites in the nucleus of intestinal epithelial cells. FEBS Lett. 1989;242:309–313. doi: 10.1016/0014-5793(89)80491-0. [DOI] [PubMed] [Google Scholar]
- 66.Rufini S., Lena A.M., Cadot B., Mele S., Amelio I., Terrinoni A., Desideri A., Melino G., Candi E. The sterile alpha-motif (SAM) domain of p63 binds in vitro monoasialoganglioside (GM1) micelles. Biochem. Pharmacol. 2011;82:1262–1268. doi: 10.1016/j.bcp.2011.07.087. [DOI] [PubMed] [Google Scholar]
- 67.Saito M., Sugiyama K. Characterization of nuclear gangliosides in rat brain: concentration, composition, and developmental changes. Arch. Biochem. Biophys. 2002;398:153–159. doi: 10.1006/abbi.2001.2725. [DOI] [PubMed] [Google Scholar]
- 68.Wu G., Xie X., Lu Z.H., Ledeen R.W. Sodium-calcium exchanger complexed with GM1 ganglioside in nuclear membrane transfers calcium from nucleoplasm to endoplasmic reticulum. Proc. Natl. Acad. Sci. USA. 2009;106:10829–10834. doi: 10.1073/pnas.0903408106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hanson L.R., Fine J.M., Svitak A.L., Faltesek K.A. Intranasal administration of CNS therapeutics to awake mice. J. Vis. Exp. 2013;(74) doi: 10.3791/4440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ariga T., Tao R.V., Lee B.C., Yamawaki M., Yoshino H., Scarsdale N.J., Kasama T., Kushi Y., Yu R.K. Glycolipid composition of human cataractous lenses. Characterization of Lewisx glycolipids. J. Biol. Chem. 1994;269:2667–2675. [PubMed] [Google Scholar]
- 71.Ledeen R.W., Yu R.K. Gangliosides: Structure, isolation, and analysis. Methods Enzymol. 1982;83:139–191. doi: 10.1016/0076-6879(82)83012-7. [DOI] [PubMed] [Google Scholar]
- 72.Ren S., Scarsdale J.N., Ariga T., Zhang Y., Klein R.A., Hartmann R., Kushi Y., Egge H., Yu R.K. O-acetylated gangliosides in bovine buttermilk. Characterization of 7-O-acetyl, 9-O-acetyl, and 7,9-di-O-acetyl GD3. J. Biol. Chem. 1992;267:12632–12638. [PubMed] [Google Scholar]
- 73.Yu R.K., Ledeen R.W. Gangliosides of human, bovine, and rabbit plasma. J. Lipid Res. 1972;13:680–686. [PubMed] [Google Scholar]
- 74.Ariga T., Macala L.J., Saito M., Margolis R.K., Greene L.A., Margolis R.U., Yu R.K. Lipid composition of PC12 pheochromocytoma cells: Characterization of globoside as a major neutral glycolipid. Biochemistry. 1988;27:52–58. doi: 10.1021/bi00401a010. [DOI] [PubMed] [Google Scholar]
- 75.Kubo H., Hoshi M. Elimination of silica gel from gangliosides by using a reversed-phase column after preparative thin-layer chromatography. J. Lipid Res. 1985;26:638–641. [PubMed] [Google Scholar]
- 76.Suzuki Y., Yanagisawa M., Ariga T., Yu R.K. Histone acetylation-mediated glycosyltransferase gene regulation in mouse brain during development. J. Neurochem. 2011;116:874–880. doi: 10.1111/j.1471-4159.2010.07042.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Petro K.A., Schengrund C.L. Membrane raft disruption promotes axonogenesis in n2a neuroblastoma cells. Neurochem. Res. 2009;34:29–37. doi: 10.1007/s11064-008-9625-9. [DOI] [PubMed] [Google Scholar]
- 78.Wu G., Lu Z.H., Ledeen R.W. Induced and spontaneous neuritogenesis are associated with enhanced expression of ganglioside GM1 in the nuclear membrane. J. Neurosci. 1995;15:3739–3746. doi: 10.1523/JNEUROSCI.15-05-03739.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Saito M., Fronda C.L., Yu R.K. Sialidase activity in nuclear membranes of rat brain. J. Neurochem. 1996;66:2205–2208. doi: 10.1046/j.1471-4159.1996.66052205.x. [DOI] [PubMed] [Google Scholar]